Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 17
An Update on the Role and Potential Molecules in Relation to Ruminococcus gnavus in Inflammatory Bowel Disease, Obesity and Diabetes Mellitus
Authors Hong J ![]() , Fu T, Liu W, Du Y, Bu J, Wei G, Yu M, Lin Y, Min C, Lin D
, Fu T, Liu W, Du Y, Bu J, Wei G, Yu M, Lin Y, Min C, Lin D
Received 22 December 2023
Accepted for publication 27 February 2024
Published 11 March 2024 Volume 2024:17 Pages 1235—1248
DOI https://doi.org/10.2147/DMSO.S456173
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Konstantinos Tziomalos
Jinni Hong,1,2 Tingting Fu,1,2 Weizhen Liu,1,2 Yu Du,1,2 Junmin Bu,1,2 Guojian Wei,1,2 Miao Yu,1,2 Yanshan Lin,1,2 Cunyun Min,1,2 Datao Lin3
1Department of Traditional Chinese Medicine, Guangdong Provincial People’s Hospital (Guangdong Academy of Medical Sciences), Southern Medical University, Guangzhou, Guangdong, 510080, People’s Republic of China; 2Guangdong Provincial Institute of Geriatric, Guangzhou, Guangdong, 510080, People’s Republic of China; 3Department of Parasitology, Zhongshan School of Medicine, Sun Yat-Sen University, Guangzhou, Guangdong, 510080, People’s Republic of China
Correspondence: Jinni Hong, Department of Traditional Chinese Medicine, Guangdong Provincial People’s Hospital (Guangdong Academy of Medical Sciences), Southern Medical University, Guangzhou, Guangdong, 510080, People’s Republic of China, Tel/Fax +86 02083827812, Email [email protected] Datao Lin, Department of Parasitology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, Guangdong, 510080, People’s Republic of China, Tel/Fax +86 02087330118, Email [email protected]
Abstract: Ruminococcus gnavus (R. gnavus) is a gram-positive anaerobe commonly resides in the human gut microbiota. The advent of metagenomics has linked R. gnavus with various diseases, including inflammatory bowel disease (IBD), obesity, and diabetes mellitus (DM), which has become a growing area of investigation. The initial focus of research primarily centered on assessing the abundance of R. gnavus and its potential association with disease presentation, taking into account variations in sample size, sequencing and analysis methods. However, recent investigations have shifted towards elucidating the underlying mechanistic pathways through which R. gnavus may contribute to disease manifestation. In this comprehensive review, we aim to provide an updated synthesis of the current literature on R. gnavus in the context of IBD, obesity, and DM. We critically analyze relevant studies and summarize the potential molecular mediators implicated in the association between R. gnavus and these diseases. Across numerous studies, various molecules such as methylation-controlled J (MCJ), glucopolysaccharides, ursodeoxycholic acid (UDCA), interleukin(IL)-10, IL-17, and capric acid have been proposed as potential contributors to the link between R. gnavus and IBD. Similarly, in the realm of obesity, molecules such as hydrogen peroxide, butyrate, and UDCA have been suggested as potential mediators, while glycine ursodeoxycholic acid (GUDCA) has been implicated in the connection between R. gnavus and DM. Furthermore, it is imperative to emphasize the necessity for additional studies to evaluate the potential efficacy of targeting pathways associated with R. gnavus as a viable strategy for managing these diseases. These findings have significantly expanded our understanding of the functional role of R. gnavus in the context of IBD, obesity, and DM. This review aims to offer updated insights into the role and potential mechanisms of R. gnavus, as well as potential strategies for the treatment of these diseases.
Keywords: Ruminococcus gnavus, inflammatory bowel disease, obesity, diabetes mellitus
Graphical Abstract:

Background
The human gut microbiota is a complex ecosystem comprising bacteria viruses, fungi and other microbial and eukaryotic species.1 With bacteria being the predominant taxonomic group, the microbiota comprises around 100 trillion microorganisms, classified into at least 1000 different species.2 The medicinal fungus Hericium erinaceus as been shown to ameliorate gastrointestinal disorders through modulation of inflammatory processes.3,4 The gut microbiota grows within the mucin layer and plays a crucial role in maintaining host homeostasis by offering protection to the gut barrier, shaping the immune system, and regulating human metabolism, nutrient absorption, and drug metabolism.5–9
Perturbations in gut microbiota have been implicated in the development of multiple diseases, including inflammatory bowel disease (IBD), obesity, and diabetes mellitus (DM).10 In IBD patients, the most prominent changes in the microbiota were the decreased diversity in bacteria species associated with decreased abundance of Bacteroidetes and Firmicutes,11,12 alongside an increase in the abundance of Proteobacteria.11 In addition, lower abundance of Faecalibacterium prausnitzii and Roseburia were recorded in patients with Crohn’s disease (CD) and ulcerative colitis (UC).13 There was also an enhancement of proteobacteria, Neisseriacaea corrodens, Pasteurellaceae, Veillonella parvula and E. coli in patients with CD.14 Yeast and fungal taxa have also increased in CD patients such as Cyberlindnera jadinii, Clavispora lusitaniae, Candida albicans, Saccharomyces cerevisiae and Kluyveromyces marxianus.12 A systematic review on gut microbiota analysed sixty studies and concluded that the Proteobacteria phylum was most frequently associated with IBD15 and obesity.16 As seen in the animal model of obesity, the increased ratio Firmicutes to Bacteroidetes observed in individuals with obesity becomes a predisposing condition to excessive production of metabolites from nondigestible polysaccharides.17–20 Observational studies in humans have shown that the genera of opportunistic pathogens were enriched in type 2 diabetes mellitus (T2DM) patients, while the genera Bifidobacterium, Bacteroides, Faecalibacterium, and A. muciniphila were negatively associated with T2DM.21 Gut microbiota alternation in diseases have become a growing area of investigation.22
Ruminococcus gnavus (R. gnavus; family Lachnospiraceae) was first identified in 1974 as a strict anaerobe in the gut of healthy individuals.23 It was reported to be one of the 57 species present in more than 90% of healthy North American and European adults, and in 65% of publicly available metagenomes of gut microbiota from healthy adults from China, Ethiopia, Spain, USA, and Sweden.24,25 But recent evidence indicated that a disproportionate representation in the composition of R. gnavus can impact the pathophysiology of certain diseases, including IBD, obesity, DM, neurological disorders, and cancer.26 While initial research generally examined the abundance of R. gnavus and its relation to disease presentation with various sample size, methods of sequencing and analysis, there has recently been a more prominent shift towards understanding the mechanisms by which R. gnavus can lead to disease manifestation.27 Clarifying these mechanisms can inform the development of novel therapies and optimise clinical practice.22 Therefore, through a critical review of current literature, we aim to provide an updated overview of the significant advancements made and summarize the potential mechanisms or the role of molecules in relation to R. gnavus in IBD, obesity, and DM.
R. gnavus and IBD
Role of R. gnavus in IBD
IBD is a complex immune-mediated inflammatory condition that affects the gastrointestinal tract, encompassing CD and UC.28 It has been sporadically observed since ancient times, and has emerged as a growing problem in newly industrialized countries.29–31 The prevalence of IBD has been increasing over the past decades in most regions.32 Accumulating evidence suggests that perturbations in the microbiota, particularly commensal flora, and host defensive responses at the mucosal frontier has been implicated in the initiation and pathogenesis of IBD.33–35 An increasing number of studies show a positive association between R. gnavus and IBD, although a causal relationship remains to be demonstrated.36 Active IBD often coincides with an increase in the abundance of R. gnavus, goes from an average of 0.1% in healthy controls (HC) to 69% in IBD patients with active disease.37 This increase in R. gnavus abundance is associated with a decrease in Akkermansia, and this microbial shift has been proposed as a biomarker for mucosal integrity in IBD.38–40 A study by Christine et al, analyzing fecal samples from children under 18 years old, found that the fecal microbiota profiles differentiated between IBD and non-IBD symptomatic children when compared to healthy children. However, both IBD and non-IBD symptomatic patients displayed similar dysbiosis.41 Similarly, there was a more remarkable increase in the abundance of R. gnavus in the macroscopically and histologically normal intestinal epithelium of IBD, when compared to HC.38
R. gnavus has been implicated in the pathogenesis of CD.42 By sequencing colonic biopsies from CD patients (undergoing colonoscopy or surgical resection) and HC with 16S rDNA, and detecting fecal samples with whole metagenome shotgun sequencing (WMGS), R. gnavus was remarkably enriched in CD patients when compared to HC.43 Additionally, when comparing fecal samples from CD patients and their unaffected relatives, Joossens et al, found a higher abundance of R. gnavus in CD patients.44 In IBD patients, the most distinctive feature between CD and UC was a significantly higher abundance of R. gnavus in CD.45 Interestingly, studies on twins concordant or discordant for CD or UC have also demonstrated an increase in R. gnavus in CD patients.46 A study of 56 mucosal microbiome samples from 28 Chinese UC patients and their healthy family partners showed an increase in R. gnavus in the mucosal microbiome of UC patients.47 Consistently, furthermore, in UC patients who underwent fecal microbiota transplantation (FMT), the abundance of R. gnavus was found to be higher in donors of failed FMT and decreased after FMT.48,49
Pouchitis, a common complication following restorative proctocolectomy with ileal pouch-anal anastomosis for UC, is associated with increased levels of R. gnavus compared to UC and CD.50–52 Additionally, in patients with Clostridium difficile infection (CDI), which can be a complication of IBD, R. gnavus has been found to be more abundant compared to HC and IBD patients without CDI.53,54
However, it is worth mentioning that some studies have reported decreased or even absent levels of R. gnavus in IBD patients, including those with UC and CD.55,56 In a cohort study conducted in Korea, R. gnavus was found to be significantly more abundant in CD patients with a better prognosis, suggesting it may serve as a biomarker for favorable outcomes in CD.57 Overall, the role of R. gnavus in the context of IBD remains unclear, but studies suggest that it plays a significant role in modulating the gut microbiota at the mucosal level. For a comprehensive summary of studies investigating the relationship between R. gnavus and IBD, please refer to Supplementary Table 1.
Molecules in Relation to R. gnavus in IBD
Methylation-Controlled J (MCJ)
MCJ functions as an endogenous negative regulator of mitochondrial respiration.58 Its absence can lead to increased activity of complex I and adenosine-triphosphate (ATP) synthesis, as well as the formation of respiratory supercomplexes, which helps to limit the production of reactive oxygen species (ROS).58–60 In MCJ-deficient colitis-induced mice, it has been observed that R. gnavus levels are increased, along with higher expression of myeloid differentiation primary response gene (Myd) 88, toll-like receptor (TLR) 9, and immunoglobulin A (IgA).61 R. gnavus is known to be highly IgA coated, regardless of the specific IgA target.62 Furthermore, gene expression analyses in UC patients have shown decreased levels of MCJ and higher expression of tissue inhibitor of metalloproteinase 3 (TIMP3). This leads to the inhibition of tumor necrosis factor (TNF) α converting enzyme (TACE) activity, which in turn inhibits the shedding of TNF from the cell membrane in the colon.61 Anti-TNFs have been the first line of biologic therapies for IBD and have remained a cornerstone of IBD treatment in clinical practice over the past two decades, despite the emergence of numerous new therapies.63 Therefore, the absence of MCJ may contribute to an altered gut microbiota composition, such as increased levels of R. gnavus, and dysregulated immune responses, which could have implications for the pathogenesis and treatment of IBD.
Glucorhamnan and Ursodeoxycholic Acid
Glucorhamnan is primarily composed of a repeating unit of five sugars, with a linear backbone consisting of three rhamnose units and a short side chain made up of two glucose units.64 R. gnavus has the ability to synthesize and secrete a complex glucorhamnan polysaccharide, which has been found to strongly induce the secretion of TNF-α through TLR4 activation.64,65
In addition to its polysaccharide synthesis capabilities, R. gnavus has also been reported as a producer of ursodeoxycholic acid (UDCA). It possesses an enzyme that can degrade 7-keto lithocholic acid (LCA) into UDCA.66 Administration of UDCA has been shown to increase colonic LCA levels and inhibit caspase-3 cleavage.67 Abnormal apoptosis in intestinal epithelial cells (IECs) can disrupt the integrity of the intestinal barrier, leading to bacterial infiltration and triggering an inflammatory cascade.68 The dysregulated apoptosis also stimulates the production of TNF-α and interferon-gamma (IFN-γ), both of which further promote apoptosis.68 Moreover, UDCA has been found to activate the epidermal growth factor receptor (EGFR)/ Raf-1/ extracellular regulating kinase (ERK) signaling pathway in colorectal cancer.67
IL-10 and IL-17
Stimulation of mesenteric lymph node cells from IL-10−/− mice with R. gnavus lysate has been found to result in higher production of IL-17, indicating that R. gnavus can induce the secretion of IL-17.69,70 IL-17 is a cytokine that is thought to contribute to the development of IBD, which expressed at higher levels in IBD patients compared to healthy controls.71,72 Additionally, the non-inflamed mucosa of IBD patients exhibits significantly lower IL-17 levels compared to inflamed mucosa.73 Furthermore, IL-17 has been shown to play a role in the progression of colorectal cancer through various signaling pathways, including signal transducer and activator of transcription 3 (STAT3), nuclear factor kappa B (NF-κB), and mitogen-activated protein kinase (MAPK) pathways.74,75
Tryptamine
In patients with IBD, R. gnavus has been found to be highly abundant and positively correlated with tryptamine levels.65 Specifically, R. gnavus is capable of producing tryptamine, which can affect gut motility through several mechanisms. First, tryptamine activates the serotonin receptor 4 (SR4) or 5-hydroxytryptamine receptor 4 (5-HT4R), leading to an increase in intracellular cyclic adenosine monophosphate (cAMP) concentration. This elevation in cAMP levels can enhance gut motility. Second, tryptamine can also increase fluid secretion by acting on the cystic fibrosis transmembrane regulator (CFTR). The activation of CFTR promotes the secretion of fluids into the gut, affecting gut motility.76–78
Caprylic Acid
Caprylic acid, also known as octanoic acid, is a medium-chain fatty acid (MCFA) that possesses antibacterial and antiviral properties.79 In the context of IBD, caprylic acid has been found to be enriched in non-IBD controls and negatively associated with the abundance of R. gnavus, which is consistent with previous observations of MCFA depletion in IBD.79,80 This negative association suggests that R. gnavus may either take up and metabolize caprylic acid, leading to its depletion, or caprylic acid may possess inhibitory effects on the growth of R. gnavus.
Relative studies have indicated that MCFA can induce the production of host defense peptides (HDP) and activate TLR4.81,82 Activation of TLR4 triggers the activation of NF-κB and MAPK pathways through the interaction between TLR4’s toll-IL-1 receptor domain and Myd88.83,84 Furthermore, TLR4 can mediate the production of interleukin(IL)-6 and IL-10.85,86 and influences the balance of T helper (Th) 1 and Th17 cells.87 Figure 1 provides a summary of the potential molecules in relation to R. gnavus in IBD.
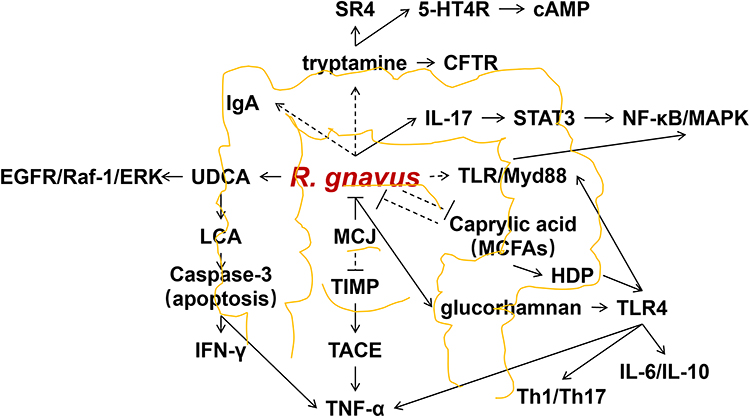 |
Figure 1 Molecules in relation to R. gnavus in IBD. Abbreviations: R. gnavus, Ruminococcus gnavus; IL, Interleukin; SR4, serotonin receptor 4; 5-HT4R, 5-hydroxytryptamine receptor 4; cAMP, cyclic adenosine monophosphate; CFTR, cystic fibrosis transmembrane regulator; STAT, signal transduction and activator of transcription; NF-κB, nuclear factors- kappa B; MAPK, mitogen-activated protein kinase; IgA, immunoglobulin A; MCFA, medium-chain fatty acid; HDP, host defense peptides; TLR, toll-like receptor; Myd, myeloid differentiation primary response gene; Th, T helper; TNF, tumor necrosis factor; TACE, tumor necrosis factor α converting enzyme; TIMP, tumor necrosis factor α converting enzyme inhibitor tissue inhibitor of metalloproteinase; MCJ, Methylation-controlled J; UDCA, ursodeoxycholic acid; LCA, lithocholic acid; EGFR, epidermal growth factor receptor; ERK, extracellular regulating kinase; IFN-γ, interferon-gamma. |
R. gnavus and Obesity
Role of R. gnavus in Obesity
According to the World Obesity Atlas 2023, over 4 billion adults and nearly 3 million children will be overweight or obese by 2035, a significant global health concern.88 R. gnavus has been found to be significantly more abundant in obese individuals and decreases with weight loss.89,90 Similar findings have been observed in obese dogs and rats.91 Furthermore, there is a positive association between R. gnavus and body mass index (BMI),92 as well as a strong correlation with fat mass.93 However, a cross-sectional study involving 236 children reported a significantly negative relationship between the abundance of R. gnavus and BMI status, body fat distribution, and leptin levels.94,95 A summary of studies investigating the relationship between R. gnavus and obesity can be found in Supplementary Table 2.
Molecules in Relation to R. gnavus in Obesity
Haptoglobin (Hp)
Hp is an inflammation marker that plays a role in the interaction between obesity and inflammation.96 Hp expression in white adipose tissue (WAT) is increased in obesity rodents,97 and the level of Hp was found to be significantly correlated with the abundance of R. gnavus in individuals of normal weight and overweight.98
Hp binds to free hemoglobin (Hb) and inhibits oxidative tissue damage by reducing the release of heme iron and the generation of reactive oxygen species (ROS).99 Hp has the ability to attract monocytes/macrophages through interaction with chemokine receptor 2 (CCR2), which is mediated by MAPK phosphorylation.100 It has been observed that the MAPK p38 inhibitor down-regulates the phosphorylation of CCAAT/enhancer binding protein (C/EBP)-α, peroxisome proliferator-activated receptor-γ (PPAR-γ), STAT 3, fatty acid synthase (FAS), perilipin A, as well as its downstream effector activating transcription factor-2 (ATF-2).101 Hp levels increase in WAT with weight gain, activate macrophages, and lead to the release of TNF-α and IL-6.102–104
Butyrate
R. gnavus was initially characterized in 1976 as a bacterium capable of producing acetate, formate, ethanol, and a small quantity of lactate, but not butyrate, when cultured in peptone-yeast extract glucose broth.26 However, a recent investigation on the dynamics of butyrate levels during the transition from an infant-like to an adult-like gut microbiota revealed a negative correlation between butyrate levels and the abundance of R. gnavus.105 Given the observational nature of this study, further research focusing on the direct relationship between butyrate levels and R. gnavus is warranted.
Butyrate has been shown to play a role in energy homeostasis mediated by the gut microbiota, and it is found in higher amounts in obese individuals compared to lean individuals.106 Butyrate is involved in the thermogenesis of adipose tissue.106 Brown adipose tissue (BAT) is a key site of thermogenesis and energy expenditure, and butyrate can stimulate thermogenesis by up-regulating uncoupling protein(UCP) 1 and uncoupling mitochondrial respiration from ATP synthesis.107 Additionally, butyrate has been shown to activate peroxisome proliferator-activated receptor coactivator-1α (PGC-1α) through UCP1.108 Butyrate supplementation has been found to increase PGC-1α expression in BAT and is positively associated with GPCR43, suggesting a role for GPCR43 in activating PGC-1α.109 Moreover, butyrate stimulates PGC-1α activity by activating AMP-activated protein kinase (AMPK) and inhibiting histone deacetylases (HDACs).110 The knockout of lysine-specific demethylase 1 (LSD1) has been shown to block the butyrate-induced increase in thermogenesis and energy expenditure in BAT, suggesting LSD1 as a potential mediator of butyrate-induced thermogenesis.111,112 Further studies have found that butyrate can activate LSD1 to increase UCP1 expression, and these effects are independent of AMPK.111 Germ-free mice have been reported to have impaired thermogenic capacity in BAT due to decreased expression of UCP1.113 Consistent with this, depletion of gut microbiota has been found to impair the thermogenesis of BAT, and butyrate supplementation partially rescues thermogenesis function.113 This indicates that butyrate improves the function of BAT by regulating gut microbiota dysbiosis in obese mice.114,115 Similar to BAT, beige adipocytes in WAT release energy as heat, and their thermogenesis is mediated by UCP1.116 Butyrate supplementation has been shown to increase the expression of UCP1 and beige adipocyte markers to mediate thermogenesis in WAT, requiring GPCR43, GPCR41, and LSD1.111 Butyrate acts as a ligand for metabolite-sensing GPCRs, mainly GPCR43, GPCR41, and GPCR109a.117,118 Butyrate is the only SCFA that can bind to GPCR109a and regulate body energy expenditure to maintain metabolic homeostasis.118 These mechanisms may be associated with the effects of butyrate on obesity.
Studies have shown that butyrate supplementation can lead to a reduction in body weight by decreasing lipogenesis in the liver and adipose tissue.119 This effect is mediated by the activation of the PPARγ-mediated AMPK-acetyl-CoA carboxylase (ACC) pathway, which increases the expression of the glucagon-like peptide-1 receptor (GLP-1R).119,120 Butyrate has also been found to modify histone acetylation on the promoter of the beta3-adrenergic receptors (β3AR) gene and activate cAMP-dependent protein kinase A (PKA), leading to the phosphorylation of adipose triglyceride lipase (ATGL) and hormone-sensitive lipase(HSL).121 Additionally, butyrate supplementation can reverse the reduction of HDACs.122
Increasing fatty acid oxidation (FAO) can help reduce fat accumulation and improve obesity.123 Butyrate has been shown to induce the expression of carnitine palmitoyltransferase-1 (CPT-1) in liver and adipose tissue,109 which is the rate-limiting enzyme of FAO.124 This effect is mediated by the AMPK/histone deacetylase inhibitor (HDACi)-PGC-1α pathway.110 Additionally, as an HDACi, butyrate activates AMPK by enhancing the expression of adiponectin receptors (adipoR)-1/2.125 Butyrate also increases the expression of cytochrome c oxidase (COX-1), UCP2, and UCP3 in skeletal muscle.126 Furthermore, butyrate has been found to significantly increase the levels of gut hormones such as glucagon-like peptide-1 (GLP-1) and polypeptide YY (PYY) in the colon and plasma, mediated by recombinant free fatty acid receptor 3 (FFAR3).127
Ursodeoxycholic Acid (UDCA)
R. gnavus has been reported to produce UDCA.128 In a mouse model of diet-induced obesity, UDCA treatment altered the profiles of bile acids and fatty acids.128 UDCA down-regulated sterol regulatory element-binding protein 1c (SREBP1c), a transcription factor that promotes the expression of lipogenic genes such as fatty acid synthase (FAS) and SCD1.129 Furthermore, UDCA reversed the inhibition of factors involved in FAO, including PPAR-α, CPT-1A, acyl-CoA oxidase (Aco), and down-regulated the expression of genes involved in fatty acid uptake in the liver, such as fatty acid transporter protein (FATP) and cluster of differentiation 36 (CD36).129 These findings indicate that UDCA alters the profile of free fatty acids (FFAs) by inhibiting lipogenesis, promoting FAO, and reducing fatty acid uptake in adipose tissue.
Moreover, UDCA down-regulated the phosphorylation of NF-κB and STAT3 by negatively regulating the expression of suppressor of cytokine signaling (SOCS)-1 and SOCS3 signaling pathways.129 These changes were accompanied by a decrease in angiogenesis, as evidenced by the down-regulation of vasoactive endothelial growth factor (VEGF), vascular cell adhesion molecule (VCAM), and transforming growth factor (TGF)-β expression.129 Additionally, UDCA significantly up-regulated adipose tissue browning, which was associated with the up-regulation of silent mating type information regulation 2 homolog (SIRT)-1-PGC1-α signaling in epididymal adipose tissue.129 Figure 2 provides a summary of the potential molecules in relation to R. gnavus in obesity.
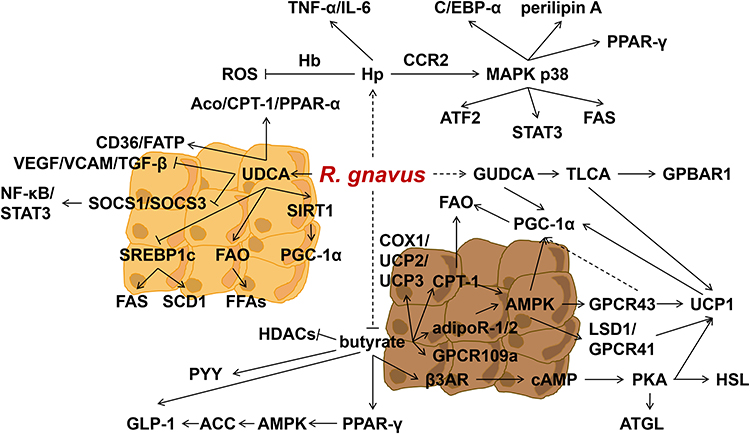 |
Figure 2 Molecules in relation to R. gnavus in obesity and DM. Abbreviations: R. gnavus, Ruminococcus gnavus; Hp, Haptoglobin; Hb, hemoglobin; ROS, reactive oxygen species; TNF-α, tumor necrosis factor alpha; IL-6, Interleukin-6; CCR2, chemokine (C-C motif) receptor 2; AMPK, AMP-activated protein kinase; MAPK, mitogen-activated protein kinase; C/ EBP, CCAAT/ enhancer binding protein; PPAR-γ, peroxisome proliferator-activated receptor-gamma; ATF-2, activating transcription factor-2; STAT, signal transduction and activator of transcription 3; UDCA, ursodeoxycholic acid; Aco, acyl-CoA oxidase; SREBP1c, sterol regulatory element-binding protein 1c; FFA, free fatty acid; CPT-1, carnitine palmitoyltransferase-1; GUDCA, glycine ursodeoxycholic acid; GPBAR, G-protein-coupled bile acid receptor; FAS, fatty acid synthase; HDACs, histone deacetylases; UCP, uncoupling protein; COX, cytochrome c oxidase; GLP-1, glucagon-like peptide-1; ACC, AMP-activated protein kinase to phosphorylate acetyl-CoA carboxylase; PKA, cAMP-dependent protein kinase A; ATGL, adipose triglyceride lipase; CD36, cluster of differentiation 36; HSL, hormone-sensitive lipase; β3AR, beta3-adrenergic receptors; FAO, fatty acid oxidation; GPCR, G protein-coupled receptors; PGC-1α, peroxisome proliferator-activated receptor coactivator-1α; LSD 1, lysinespecificdemethylase1; TCLA, taurolithocholic acid; cAMP, cyclic adenosine monophosphate; adipoR-1/2, adiponectin receptors-1/2; PYY, polypeptide YY; FATP, fatty acid transport protein; FAS, fatty acid synthase; SCD1, stearoyl-CoA desaturase-1; FFAs, free fatty acids; SIRT, silent mating type information regulation 2 homolog; SOCS, cytokine signaling; STAT, signal transduction and activator of transcription; NF-κB, nuclear factors- kappa B; VEGF, vasoactive endothelial growth factor; VCAM, vascular cell adhesion molecule; TGF-β, transforming growth factor beta. |
R. gnavus and Diabetes Mellitus
Role of R. gnavus in Diabetes Mellitus
Compared with HC, R. gnavus showed significantly higher abundance in patients with type 2 diabetes mellitus (T2DM) and prediabetes (PreDM).130,131 Specifically, R. gnavus was found to be positively associated with the incidence of T2DM.132 However, some studies have reported a reduction of R. gnavus in adults with PreDM-insulin resistance (IR), while it was found to be more enriched in a cluster characterized by moderate levels of blood glucose, severe IR, and hyperlipidemia. This was observed when compared to participants with the lowest blood glucose levels or participants with high blood glucose and insulin deficiency,133 Supplementary Table 3 provides a summary of studies examining the relationship between R. gnavus and DM.
Molecules in Relation to R. gnavus in Diabetes Mellitus
R. gnavus was found to have a positive correlation with glycine ursodeoxycholic acid (GUDCA) levels, which have been shown to improve metabolism by promoting fat thermogenesis134 Mammalian adipose tissue consists of WAT, BAT, and beige or brite adipose tissue.135 WAT is characterized by single-locular lipid droplets and few mitochondria, while BAT has abundant mitochondria and multi-locular lipid droplets, and is highly vascularized.136 Beige fat serves as an intermediary between BAT and WAT and can adapt to various environmental and pharmacological stimuli.136 Notably, BAT possesses a unique capacity, distinct from other mitochondria-rich tissues, to induce energy futile cycling through the protein UCP1.137 UCP1 can dissipate the mitochondrial membrane potential, resulting in the wastage of energy as heat.137 This process leads to increased oxidation of glucose and fat to maintain ATP production.137 However, in individuals with obesity and T2DM, the metabolic activity of BAT is suppressed.138,139 GUDCA has the ability to regulate bile acid metabolism and induce the production of taurolithocholic acid (TLCA), as well as activate the g-protein-coupled bile acid receptor (GPBAR)-1 and UCP1.140 The deletion of the β3AR receptor-cAMP-PKA-UCP1 cascade has been shown to reduce basal metabolic rate, making mice more prone to obesity.141 GUDCA tends to increase the levels of PGC-1α, although without a significant difference. Figure 2 provides a summary of the potential molecules in relation to R. gnavus in DM.
Conclusions
R.gnavus was initially described as an important component of the human gut microbiota by Moore et al, in 1976 and was classified in the genus Ruminococcus within the family Ruminococcaceae. However, with the introduction of 16S rRNA analysis, it was reclassified as a species belonging to the phylum Firmicutes, class Clostridia, Clostridium cluster XIVa, and family Lachnospiraceae.142 Across studies, gut dysbiosis, characterized by an imbalance in the abundance of R. gnavus, has been proposed to be associated with the development of inflammatory and metabolic diseases including IBD, obesity and DM. In this review, we provide an updated synthesis of the literature regarding R. gnavus in relation to IBD, obesity and DM in human subjects, and explore potential molecular mechanisms linking R. gnavus to these diseases through in vivo experiments. Despite variability in human metagenomic studies in terms of sample size, sequencing and analysis methods, and taxonomic classification, R. gnavus has been consistently reported to be more abundant in IBD, obesity, and DM compared to healthy individuals in most studies, although a few studies have reported contradictory results.
Regarding IBD, several molecules such as MCJ, glucorhamnan, UDCA, IL-10, IL-17, tryptamine, and caprylic acid, along with their related mechanisms, have been suggested as potential molecular mediators in the association between R. gnavus and IBD. In the context of obesity, molecules such as Hp, butyrate, and UDCA are proposed to be potential mediators in relation to R. gnavus, while GUDCA is implicated in the association between R. gnavus and DM. However, due to limited direct mechanistic studies on R. gnavus in these diseases, the proposed molecular relationships between R. gnavus and the diseases are predominantly derived from a combination of three types of research: animal studies investigating R. gnavus and molecules, correlation studies between R. gnavus and certain molecules in observational studies, and downstream effects of related molecules in disease pathogenesis. The latter two types of studies provide circumstantial evidence rather than direct proof, leading us to refer to these molecules as potential mediators in the interaction between R. gnavus and these diseases. This represents a limitation of our study.
Furthermore, it should be noted that the R. gnavus strain used in the research studies was ATCC 29149, which is strain-specific and may differ from R. gnavus as a species. Therefore, further intensive investigations are warranted to elucidate the direct effects of R. gnavus on the molecular pathways involved in IBD, obesity, and DM. It is essential to conduct additional research to determine whether targeting R. gnavus-related pathways could be an effective strategy in managing these conditions.
Abbreviations
R. gnavus, Ruminococcus gnavus; IBS, irritable bowel syndrome; IBD, inflammatory bowel disease; DM, diabetes mellitus; NGS, next generation sequencing; WMGS, whole metagenome shotgun sequencing; CD, Crohn’s disease; UC, ulcerative colitis; HC, healthy controls; FMT, fecal microbiota transplantation; CDI, Clostridium difficile infection; MCJ, Methylation-controlled J; SR4, serotonin receptor 4; ATP, adenosine-triphosphate; ROS, reactive oxygen species; Myd, myeloid differentiation primary response gene; TLR, toll-like receptor; IgA, immunoglobulin A; TNF, tumor necrosis factor; TACE, tumor necrosis factor α converting enzyme; TIMP3, tumor necrosis factor α converting enzyme inhibitor tissue inhibitor of metalloproteinase 3; UDCA, ursodeoxycholic acid; LCA, lithocholic acid; TCLA, taurolithocholic acid; IECs, intestinal epithelial cells; IFN-γ, interferon gamma; BAs, bile acids; EGFR, epidermal growth factor receptor; ERK, extracellular regulating kinase; STAT, signal transduction and activator of transcription; CCR2, chemokine (C-C motif) receptor 2; NF-κB, nuclear factors- kappa B; MAPK, mitogen-activated protein kinase; GPCR, epithelial G protein-coupled receptors; 5-HT4, 5-hydroxytryptamine receptor 4; cAMP, cyclic adenosine monophosphate; CFTR, cystic fibrosis transmembrane regulator; MCFA, medium-chain fatty acid; HDP, host defense peptides; IL, Interleukin; BMI, body mass index; Hp, Haptoglobin; WAT, white adipose tissue; Hb, hemoglobin; ROS, reactive oxygen species; C/EBP, CCAAT/ enhancer-binding protein; PPAR, peroxisome proliferator-activated receptor; FAS, fatty acid synthase; ATF-2, activating transcription factor-2; MCP-1, monocyte chemotactic protein 1; BAT, brown adipose tissue; UCP1, uncoupling protein-1; PGC-1α, peroxisome proliferator-activated receptor coactivator-1α; AMPK, AMP-activated protein kinase; GLP-1R, glucagon-like peptide-1 receptor; ACC, AMP-activated protein kinase to phosphorylate acetyl-CoA carboxylase; ATGL, adipose triglyceride lipase; HSL, hormone-sensitive lipase; β3AR, beta3-adrenergic receptors; PKA, protein kinase A; LPL, lipoprotein lipase; HDAC, histone deacetylase; HDACi, histone deacetylase inhibitor; FAO, fatty acid oxidation; CPT-1, carnitine palmitoyltransferase-1; adipoR, adiponectin receptors; COX-1, cytochrome c oxidase; FFAR3, free fatty acid receptor 3; SREBP1c, sterol regulatory element-binding protein 1c; SCD1, stearoyl-CoA desaturase-1; Aco, acyl-CoA oxidase; CD36, cluster of differentiation 36; PYY, polypeptide YY; FFAs, free fatty acids; FATP, fatty acid transport protein; CD 36, cluster of differentiation 36; SOCS, cytokine signaling; VEGF, vasoactive endothelial growth factor; VCAM, vascular cell adhesion molecule; SIRT, silent mating type information regulation 2 homolog; TGF, transforming growth factor; T2DM, type 2 diabetes mellitus; PreDM, prediabetes; IR, insulin resistance; GDM, gestational diabetes mellitus; HFD, high-fat diet; GUDCA, glycine ursodeoxycholic acid; TUDCA, tauroursodeoxycholic acid; TLCA, taurolithocholic acid; GPBAR, G-protein-coupled bile acid receptor; LSD 1, lysinespecificdemethylase1.
Data Sharing Statement
Data available on request from the first authors.
Acknowledgments
The authors acknowledged Guangdong Provincial People’s Hospital and Professor Zhongdao Wu’s Lab in Sun Yat-sen University for the academic supports.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was funded by the National Natural Science Foundation of China (No. 82202560), the Guangdong Basic and Applied Basic Research Foundation (No.2022A1515110655), the Traditional Chinese Medicine Bureau of Guangdong Province (Nos. 20231003 and 20223001), the Natural Science Foundation of Guangdong Province (Nos. 2023A1515011458 and 2021A1515220050), the Science and Technology Projects in Guangzhou (No. 2024A04J4314), the National Parasitic Resource Center of China (No. NPRC-2019-194-30), and the Science and Technology Program of Guangzhou (No. SL2022A04J00042).
Disclosure
The authors declare no competing interests in this work.
References
1. Mohamed AA, Al-Ramadi BK, Fernandez-Cabezudo MJ. Interplay between Microbiota and gammadelta T Cells: insights into Immune Homeostasis and Neuro-Immune Interactions. Int J Mol Sci. 2024;25(3):1747. doi:10.3390/ijms25031747
2. He M, Shi B. Gut microbiota as a potential target of metabolic syndrome: the role of probiotics and prebiotics. Cell Biosci. 2017;7(1):54. doi:10.1186/s13578-017-0183-1
3. Gravina AG, Pellegrino R, Auletta S, et al. Hericium erinaceus, a medicinal fungus with a centuries-old history: evidence in gastrointestinal diseases. World J Gastroenterol. 2023;29(20):3048–3065. doi:10.3748/wjg.v29.i20.3048
4. Gravina AG, Pellegrino R, Palladino G, et al. Hericium erinaceus, in combination with natural flavonoid/alkaloid and B(3)/B(8) vitamins, can improve inflammatory burden in Inflammatory bowel diseases tissue: an ex vivo study. Front Immunol. 2023;14:1215329. doi:10.3389/fimmu.2023.1215329
5. Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature. 2016;535(7610):75–84. doi:10.1038/nature18848
6. Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336(6086):1268–1273. doi:10.1126/science.1223490
7. D’Aversa F, Tortora A, Ianiro G, Ponziani FR, Annicchiarico BE, Gasbarrini A. Gut microbiota and metabolic syndrome. Intern Emerg Med. 2013;8 Suppl 1:S11–S15. doi:10.1007/s11739-013-0916-z
8. Valdes AM, Walter J, Segal E, Spector TD. Role of the gut microbiota in nutrition and health. BMJ. 2018;361:k2179. doi:10.1136/bmj.k2179
9. Portincasa P, Bonfrate L, Khalil M, et al. Intestinal Barrier and Permeability in Health, Obesity and NAFLD. Biomedicines. 2021;10(1):83. doi:10.3390/biomedicines10010083
10. Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90(3):859–904. doi:10.1152/physrev.00045.2009
11. Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin J Gastroenterol. 2018;11(1):1–10. doi:10.1007/s12328-017-0813-5
12. Lane ER, Zisman TL, Suskind DL. The microbiota in inflammatory bowel disease: current and therapeutic insights. J Inflamm Res. 2017;10:63–73. doi:10.2147/JIR.S116088
13. Sokol H, Seksik P, Furet JP, et al. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15(8):1183–1189. doi:10.1002/ibd.20903
14. Zhu W, Winter MG, Byndloss MX, et al. Precision editing of the gut microbiota ameliorates colitis. Nature. 2018;553(7687):208–211. doi:10.1038/nature25172
15. Rizzatti G, Lopetuso LR, Gibiino G, Binda C, Gasbarrini A. Proteobacteria: a Common Factor in Human Diseases. Biomed Res Int. 2017;2017:9351507. doi:10.1155/2017/9351507
16. Xu Z, Jiang W, Huang W, Lin Y, Chan F, Ng SC. Gut microbiota in patients with obesity and metabolic disorders - a systematic review. Genes Nutr. 2022;17(1):2. doi:10.1186/s12263-021-00703-6
17. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–1031. doi:10.1038/nature05414
18. Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–484. doi:10.1038/nature07540
19. Tanase DM, Gosav EM, Neculae E, et al. Role of Gut Microbiota on Onset and Progression of Microvascular Complications of Type 2 Diabetes (T2DM). Nutrients. 2020;12(12):3719. doi:10.3390/nu12123719
20. Sikalidis AK, Maykish A. The Gut Microbiome and Type 2 Diabetes Mellitus: discussing a Complex Relationship. Biomedicines. 2020;8(1):8. doi:10.3390/biomedicines8010008
21. Gurung M, Li Z, You H, et al. Role of gut microbiota in type 2 diabetes pathophysiology. Ebiomedicine. 2020;51:102590. doi:10.1016/j.ebiom.2019.11.051
22. Al BZ, Nitert MD, Mousa A, Naderpoor N. The Gut Microbiota and Inflammation: an Overview. Int J Environ Res Public Health. 2020;17(20):56.
23. Moore WE, Holdeman LV. Human fecal flora: the normal flora of 20 Japanese-Hawaiians. Appl Microbiol. 1974;27(5):961–979. doi:10.1128/am.27.5.961-979.1974
24. Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. doi:10.1038/nature08821
25. Kraal L, Abubucker S, Kota K, Fischbach MA, Mitreva M. The prevalence of species and strains in the human microbiome: a resource for experimental efforts. PLoS One. 2014;9(5):e97279. doi:10.1371/journal.pone.0097279
26. Crost EH, Coletto E, Bell A, Juge N. Ruminococcus gnavus: friend or foe for human health. Fems Microbiol Rev. 2023;47(2). doi:10.1093/femsre/fuad014
27. Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, Knight R. Current understanding of the human microbiome. Nat Med. 2018;24(4):392–400. doi:10.1038/nm.4517
28. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28(1):573–621. doi:10.1146/annurev-immunol-030409-101225
29. Ng SC, Shi HY, Hamidi N, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390(10114):2769–2778. doi:10.1016/S0140-6736(17)32448-0
30. Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142(1):46–54, e30. doi:10.1053/j.gastro.2011.10.001
31. Mulder DJ, Noble AJ, Justinich CJ, Duffin JM. A tale of two diseases: the history of inflammatory bowel disease. J Crohns Colitis. 2014;8(5):341–348. doi:10.1016/j.crohns.2013.09.009
32. Alatab S. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 2020;5(1):17–30. doi:10.1016/S2468-1253(19)30333-4
33. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448(7152):427–434. doi:10.1038/nature06005
34. Gionchetti P, Rizzello F, Helwig U, et al. Prophylaxis of pouchitis onset with probiotic therapy: a double-blind, placebo-controlled trial. Gastroenterology. 2003;124(5):1202–1209. doi:10.1016/S0016-5085(03)00171-9
35. Sutherland L, Singleton J, Sessions J, et al. Double blind, placebo controlled trial of metronidazole in Crohn’s disease. Gut. 1991;32(9):1071–1075. doi:10.1136/gut.32.9.1071
36. Liu S, Zhao W, Lan P, Mou X. The microbiome in inflammatory bowel diseases: from pathogenesis to therapy. Protein Cell. 2021;12(5):331–345. doi:10.1007/s13238-020-00745-3
37. Hall AB, Yassour M, Sauk J, et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 2017;9(1):103. doi:10.1186/s13073-017-0490-5
38. Png CW, Linden SK, Gilshenan KS, et al. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol. 2010;105(11):2420–2428. doi:10.1038/ajg.2010.281
39. Vigsnaes LK, Brynskov J, Steenholdt C, Wilcks A, Licht TR. Gram-negative bacteria account for main differences between faecal microbiota from patients with ulcerative colitis and healthy controls. Benef Microbes. 2012;3(4):287–297. doi:10.3920/BM2012.0018
40. Berry D, Reinisch W. Intestinal microbiota: a source of novel biomarkers in inflammatory bowel diseases? Best Pract Res Clin Gastroenterol. 2013;27(1):47–58. doi:10.1016/j.bpg.2013.03.005
41. Olbjorn C, Cvancarova SM, Thiis-Evensen E, et al. Fecal microbiota profiles in treatment-naive pediatric inflammatory bowel disease - associations with disease phenotype, treatment, and outcome. Clin Exp Gastroenterol. 2019;12:37–49. doi:10.2147/CEG.S186235
42. Lloyd-Price J, Arze C, Ananthakrishnan AN, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569(7758):655–662. doi:10.1038/s41586-019-1237-9
43. Prindiville T, Cantrell M, Wilson KH. Ribosomal DNA sequence analysis of mucosa-associated bacteria in Crohn’s disease. Inflamm Bowel Dis. 2004;10(6):824–833. doi:10.1097/00054725-200411000-00017
44. Joossens M, Huys G, Cnockaert M, et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut. 2011;60(5):631–637. doi:10.1136/gut.2010.223263
45. Cipcic PH, Baresic A, Panek M, et al. Gut microbiota in mucosa and feces of newly diagnosed, treatment-naive adult inflammatory bowel disease and irritable bowel syndrome patients. Gut Microbes. 2022;14(1):2083419. doi:10.1080/19490976.2022.2083419
46. Willing BP, Dicksved J, Halfvarson J, et al. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139(6):1844–1854. doi:10.1053/j.gastro.2010.08.049
47. Li W, Sun Y, Dai L, et al. Ecological and network analyses identify four microbial species with potential significance for the diagnosis/treatment of ulcerative colitis (UC). Bmc Microbiol. 2021;21(1):138. doi:10.1186/s12866-021-02201-6
48. Ren R, Gao X, Shi Y, et al. Long-Term Efficacy of Low-Intensity Single Donor Fecal Microbiota Transplantation in Ulcerative Colitis and Outcome-Specific Gut Bacteria. Front Microbiol. 2021;12:742255. doi:10.3389/fmicb.2021.742255
49. Fuentes S, Rossen NG, van der Spek MJ, et al. Microbial shifts and signatures of long-term remission in ulcerative colitis after faecal microbiota transplantation. Isme J. 2017;11(8):1877–1889. doi:10.1038/ismej.2017.44
50. Machiels K, Sabino J, Vandermosten L, et al. Specific members of the predominant gut microbiota predict pouchitis following colectomy and IPAA in UC. Gut. 2017;66(1):79–88. doi:10.1136/gutjnl-2015-309398
51. Gao X, Huang D, Yang LS, et al. Identification of gut microbiome and transcriptome changes in ulcerative colitis and pouchitis. Scand J Gastroenterol. 2022;57(8):942–952. doi:10.1080/00365521.2022.2047221
52. Dubinsky V, Reshef L, Rabinowitz K, et al. Dysbiosis in Metabolic Genes of the Gut Microbiomes of Patients with an Ileo-anal Pouch Resembles That Observed in Crohn’s Disease. Msystems. 2021;6(2). doi:10.1128/mSystems.00984-20
53. Yu S, Ge X, Xu H, et al. Gut microbiome and mycobiome in inflammatory bowel disease patients with Clostridioides difficile infection. Front Cell Infect Microbiol. 2023;13:1129043. doi:10.3389/fcimb.2023.1129043
54. Sokol H, Jegou S, McQuitty C, et al. Specificities of the intestinal microbiota in patients with inflammatory bowel disease and Clostridium difficile infection. Gut Microbes. 2018;9(1):55–60. doi:10.1080/19490976.2017.1361092
55. Al-Amrah H, Saadah OI, Mosli M, et al. Composition of the gut microbiota in patients with inflammatory bowel disease in Saudi Arabia: a pilot study. Saudi J Gastroenterol. 2023;29(2):102–110. doi:10.4103/sjg.sjg_368_22
56. Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15(3):382–392. doi:10.1016/j.chom.2014.02.005
57. Shin SY, Kim Y, Kim WS, et al. Compositional changes in fecal microbiota associated with clinical phenotypes and prognosis in Korean patients with inflammatory bowel disease. Intest Res. 2023;21(1):148–160. doi:10.5217/ir.2021.00168
58. Hatle KM, Gummadidala P, Navasa N, et al. MCJ/DnaJC15, an endogenous mitochondrial repressor of the respiratory chain that controls metabolic alterations. Mol Cell Biol. 2013;33(11):2302–2314.
59. Navasa N, Martin I, Iglesias-Pedraz JM, et al. Regulation of oxidative stress by methylation-controlled J protein controls macrophage responses to inflammatory insults. J Infect Dis. 2015;211(1):135–145. doi:10.1093/infdis/jiu389
60. Barbier-Torres L, Iruzubieta P, Fernandez-Ramos D, et al. The mitochondrial negative regulator MCJ is a therapeutic target for Acetaminophen-induced liver injury. Nat Commun. 2017;8(1):2068. doi:10.1038/s41467-017-01970-x
61. Pascual-Itoiz MA, Pena-Cearra A, Martin-Ruiz I, et al. The mitochondrial negative regulator MCJ modulates the interplay between microbiota and the host during ulcerative colitis. Sci Rep. 2020;10(1):572. doi:10.1038/s41598-019-57348-0
62. Bunker JJ, Drees C, Watson AR, et al. B cell superantigens in the human intestinal microbiota. Sci Transl Med. 2019;11(507). doi:10.1126/scitranslmed.aau9356
63. Baumgart DC, Le Berre C. Newer Biologic and Small-Molecule Therapies for Inflammatory Bowel Disease. N Engl J Med. 2021;385(14):1302–1315. doi:10.1056/NEJMra1907607
64. Henke MT, Kenny DJ, Cassilly CD, Vlamakis H, Xavier RJ, Clardy J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc Natl Acad Sci U S A. 2019;116(26):12672–12677. doi:10.1073/pnas.1904099116
65. Haynie T, Gubler S, Drees C, et al. Synthesis of the pentasaccharide repeating unit from Ruminococcus gnavus and measurement of its inflammatory properties. Rsc Adv. 2021;11(24):14357–14361. doi:10.1039/D1RA01918J
66. Lee JY, Arai H, Nakamura Y, Fukiya S, Wada M, Yokota A. Contribution of the 7beta-hydroxysteroid dehydrogenase from Ruminococcus gnavus N53 to ursodeoxycholic acid formation in the human colon. J Lipid Res. 2013;54(11):3062–3069. doi:10.1194/jlr.M039834
67. Im E, Martinez JD. Ursodeoxycholic acid (UDCA) can inhibit deoxycholic acid (DCA)-induced apoptosis via modulation of EGFR/Raf-1/ERK signaling in human colon cancer cells. J Nutr. 2004;134(2):483–486. doi:10.1093/jn/134.2.483
68. Gunther C, Neumann H, Neurath MF, Becker C. Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium. Gut. 2013;62(7):1062–1071. doi:10.1136/gutjnl-2011-301364
69. Deng Z, Wang S, Wu C, Wang C. IL-17 inhibitor-associated inflammatory bowel disease: a study based on literature and database analysis. Front Pharmacol. 2023;14:1124628. doi:10.3389/fphar.2023.1124628
70. Eun CS, Mishima Y, Wohlgemuth S, et al. Induction of bacterial antigen-specific colitis by a simplified human microbiota consortium in gnotobiotic interleukin-10-/- mice. Infect Immun. 2014;82(6):2239–2246. doi:10.1128/IAI.01513-13
71. Magyari L, Kovesdi E, Sarlos P, Javorhazy A, Sumegi K, Melegh B. Interleukin and interleukin receptor gene polymorphisms in inflammatory bowel diseases susceptibility. World J Gastroenterol. 2014;20(12):3208–3222. doi:10.3748/wjg.v20.i12.3208
72. Seiderer J, Elben I, Diegelmann J, et al. Role of the novel Th17 cytokine IL-17F in inflammatory bowel disease (IBD): upregulated colonic IL-17F expression in active Crohn’s disease and analysis of the IL17F p.His161Arg polymorphism in IBD. Inflamm Bowel Dis. 2008;14(4):437–445. doi:10.1002/ibd.20339
73. Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52(1):65–70. doi:10.1136/gut.52.1.65
74. Gu C, Wu L, Li X. IL-17 family: cytokines, receptors and signaling. Cytokine. 2013;64(2):477–485. doi:10.1016/j.cyto.2013.07.022
75. De Simone V, Franze E, Ronchetti G, et al. Th17-type cytokines, IL-6 and TNF-alpha synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene. 2015;34(27):3493–3503. doi:10.1038/onc.2014.286
76. Bhattarai Y, Williams BB, Battaglioli EJ, et al. Gut Microbiota-Produced Tryptamine Activates an Epithelial G-Protein-Coupled Receptor to Increase Colonic Secretion. Cell Host Microbe. 2018;23(6):775–785. doi:10.1016/j.chom.2018.05.004
77. Vich VA, Hu S, Andreu-Sanchez S, et al. Faecal metabolome and its determinants in inflammatory bowel disease. Gut. 2023.
78. Williams BB, Van Benschoten AH, Cimermancic P, et al. Discovery and characterization of gut microbiota decarboxylases that can produce the neurotransmitter tryptamine. Cell Host Microbe. 2014;16(4):495–503. doi:10.1016/j.chom.2014.09.001
79. De Preter V, Machiels K, Joossens M, et al. Faecal metabolite profiling identifies medium-chain fatty acids as discriminating compounds in IBD. Gut. 2015;64(3):447–458. doi:10.1136/gutjnl-2013-306423
80. Franzosa EA, Sirota-Madi A, Avila-Pacheco J, et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol. 2019;4(2):293–305. doi:10.1038/s41564-018-0306-4
81. Wang J, Lu J, Xie X, et al. Blend of organic acids and medium chain fatty acids prevents the inflammatory response and intestinal barrier dysfunction in mice challenged with enterohemorrhagic Escherichia coli O157:H7. Int Immunopharmacol. 2018;58:64–71. doi:10.1016/j.intimp.2018.03.014
82. Martinez-Vallespin B, Vahjen W, Zentek J. Effects of medium-chain fatty acids on the structure and immune response of IPEC-J2 cells. Cytotechnology. 2016;68(5):1925–1936. doi:10.1007/s10616-016-0003-1
83. Zuany-Amorim C, Hastewell J, Walker C. Toll-like receptors as potential therapeutic targets for multiple diseases. Nat Rev Drug Discov. 2002;1(10):797–807. doi:10.1038/nrd914
84. Zhou M, Xu W, Wang J, et al. Boosting mTOR-dependent autophagy via upstream TLR4-MyD88-MAPK signalling and downstream NF-kappaB pathway quenches intestinal inflammation and oxidative stress injury. Ebiomedicine. 2018;35:345–360. doi:10.1016/j.ebiom.2018.08.035
85. Xiong T, Zheng X, Zhang K, et al. Ganluyin ameliorates DSS-induced ulcerative colitis by inhibiting the enteric-origin LPS/TLR4/NF-kappaB pathway. J Ethnopharmacol. 2022;289:115001. doi:10.1016/j.jep.2022.115001
86. Xue HH, Li JJ, Li SF, et al. Phillygenin Attenuated Colon Inflammation and Improved Intestinal Mucosal Barrier in DSS-induced Colitis Mice via TLR4/Src Mediated MAPK and NF-kappaB Signaling Pathways. Int J Mol Sci. 2023;24(3):2238. doi:10.3390/ijms24032238
87. Candelli M, Franza L, Pignataro G, et al. Interaction between Lipopolysaccharide and Gut Microbiota in Inflammatory Bowel Diseases. Int J Mol Sci. 2021;22(12):6242. doi:10.3390/ijms22126242
88. Silva S, de Lemos M, Dos SJO, et al. Overweight during development dysregulates cellular metabolism and critical genes that control food intake in the prefrontal cortex. Physiol Behav. 2024;276:114453. doi: 10.1016/j.physbeh.2023.114453
89. Palmas V, Pisanu S, Madau V, et al. Gut microbiota markers associated with obesity and overweight in Italian adults. Sci Rep. 2021;11(1):5532. doi:10.1038/s41598-021-84928-w
90. Jie Z, Yu X, Liu Y, et al. The Baseline Gut Microbiota Directs Dieting-Induced Weight Loss Trajectories. Gastroenterology. 2021;160(6):2029–2042. doi:10.1053/j.gastro.2021.01.029
91. Petriz BA, Castro AP, Almeida JA, et al. Exercise induction of gut microbiota modifications in obese, non-obese and hypertensive rats. Bmc Genomics. 2014;15(1):511. doi:10.1186/1471-2164-15-511
92. Verdam FJ, Fuentes S, de Jonge C, et al. Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity (Silver Spring). 2013;21(12):E607–E615. doi:10.1002/oby.20466
93. Grahnemo L, Nethander M, Coward E, et al. Cross-sectional associations between the gut microbe Ruminococcus gnavus and features of the metabolic syndrome. Lancet Diabetes Endocrinol. 2022;10(7):481–483. doi:10.1016/S2213-8587(22)00113-9
94. Wei Y, Liang J, Su Y, et al. The associations of the gut microbiome composition and short-chain fatty acid concentrations with body fat distribution in children. Clin Nutr. 2021;40(5):3379–3390. doi:10.1016/j.clnu.2020.11.014
95. Yan H, Qin Q, Chen J, et al. Gut Microbiome Alterations in Patients With Visceral Obesity Based on Quantitative Computed Tomography. Front Cell Infect Microbiol. 2021;11:823262. doi:10.3389/fcimb.2021.823262
96. Wan BN, Zhou SG, Wang M, Zhang X, Ji G. Progress on haptoglobin and metabolic diseases. World J Diabetes. 2021;12(3):206–214. doi:10.4239/wjd.v12.i3.206
97. Chiellini C, Bertacca A, Novelli SE, et al. Obesity modulates the expression of haptoglobin in the white adipose tissue via TNFalpha. J Cell Physiol. 2002;190(2):251–258. doi:10.1002/jcp.10061
98. Zacarias MF, Collado MC, Gomez-Gallego C, et al. Pregestational overweight and obesity are associated with differences in gut microbiota composition and systemic inflammation in the third trimester. PLoS One. 2018;13(7):e0200305. doi:10.1371/journal.pone.0200305
99. Bowman BH, Kurosky A. Haptoglobin: the evolutionary product of duplication, unequal crossing over, and point mutation. Adv Hum Genet. 1982;12:189–261, 453–454.
100. Maffei M, Funicello M, Vottari T, et al. The obesity and inflammatory marker haptoglobin attracts monocytes via interaction with chemokine (C-C motif) receptor 2 (CCR2). Bmc Biol. 2009;7(1):87. doi:10.1186/1741-7007-7-87
101. Perumal NL, Mufida A, Yadav AK, et al. Suppression of Lipid Accumulation in the Differentiation of 3T3-L1 Preadipocytes and Human Adipose Stem Cells into Adipocytes by TAK-715, a Specific Inhibitor of p38 MAPK. Life. 2023;13(2). doi:10.3390/life13020412
102. Baumann H, Morella KK, Wong GH. TNF-alpha, IL-1 beta, and hepatocyte growth factor cooperate in stimulating specific acute phase plasma protein genes in rat hepatoma cells. J Immunol. 1993;151(8):4248–4257. doi:10.4049/jimmunol.151.8.4248
103. Castell JV, Gomez-Lechon MJ, David M, Hirano T, Kishimoto T, Heinrich PC. Recombinant human interleukin-6 (IL-6/BSF-2/HSF) regulates the synthesis of acute phase proteins in human hepatocytes. FEBS Lett. 1988;232(2):347–350. doi:10.1016/0014-5793(88)80766-X
104. Chiellini C, Santini F, Marsili A, et al. Serum haptoglobin: a novel marker of adiposity in humans. J Clin Endocrinol Metab. 2004;89(6):2678–2683. doi:10.1210/jc.2003-031965
105. Nilsen M, Madelen SC, Leena AI, et al. Butyrate Levels in the Transition from an Infant- to an Adult-Like Gut Microbiota Correlate with Bacterial Networks Associated with Eubacterium Rectale and Ruminococcus Gnavus. Genes. 2020;11(11):1245. doi:10.3390/genes11111245
106. Zhang L, Liu C, Jiang Q, Yin Y. Butyrate in Energy Metabolism: there Is Still More to Learn. Trends Endocrinol Metab. 2021;32(3):159–169. doi:10.1016/j.tem.2020.12.003
107. Harms M, Seale P. Brown and beige fat: development, function and therapeutic potential. Nat Med. 2013;19(10):1252–1263. doi:10.1038/nm.3361
108. Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92(6):829–839. doi:10.1016/S0092-8674(00)81410-5
109. Lu Y, Fan C, Li P, Lu Y, Chang X, Qi K. Short Chain Fatty Acids Prevent High-fat-diet-induced Obesity in Mice by Regulating G Protein-coupled Receptors and Gut Microbiota. Sci Rep. 2016;6(1):37589. doi:10.1038/srep37589
110. Gao Z, Yin J, Zhang J, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. 2009;58(7):1509–1517. doi:10.2337/db08-1637
111. Wang D, Liu CD, Li HF, et al. LSD1 mediates microbial metabolite butyrate-induced thermogenesis in brown and white adipose tissue. Metabolism. 2020;102:154011. doi:10.1016/j.metabol.2019.154011
112. Sambeat A, Gulyaeva O, Dempersmier J, et al. LSD1 Interacts with Zfp516 to Promote UCP1 Transcription and Brown Fat Program. Cell Rep. 2016;15(11):2536–2549. doi:10.1016/j.celrep.2016.05.019
113. Li B, Li L, Li M, et al. Microbiota Depletion Impairs Thermogenesis of Brown Adipose Tissue and Browning of White Adipose Tissue. Cell Rep. 2019;26(10):2720–2737. doi:10.1016/j.celrep.2019.02.015
114. Gao F, Lv YW, Long J, et al. Butyrate Improves the Metabolic Disorder and Gut Microbiome Dysbiosis in Mice Induced by a High-Fat Diet. Front Pharmacol. 2019;10:1040. doi:10.3389/fphar.2019.01040
115. Fang W, Xue H, Chen X, Chen K, Ling W. Supplementation with Sodium Butyrate Modulates the Composition of the Gut Microbiota and Ameliorates High-Fat Diet-Induced Obesity in Mice. J Nutr. 2019;149(5):747–754. doi:10.1093/jn/nxy324
116. Wu J, Bostrom P, Sparks LM, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150(2):366–376. doi:10.1016/j.cell.2012.05.016
117. Brown AJ, Goldsworthy SM, Barnes AA, et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem. 2003;278(13):11312–11319. doi:10.1074/jbc.M211609200
118. Singh N, Gurav A, Sivaprakasam S, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40(1):128–139. doi:10.1016/j.immuni.2013.12.007
119. den Besten G, Bleeker A, Gerding A, et al. Short-Chain Fatty Acids Protect Against High-Fat Diet-Induced Obesity via a PPARgamma-Dependent Switch From Lipogenesis to Fat Oxidation. Diabetes. 2015;64(7):2398–2408. doi:10.2337/db14-1213
120. Xu J, Chen P, Wu D, et al. The novel GLP-1/GIP dual agonist DA3-CH improves rat type 2 diabetes through activating AMPK/ACC signaling pathway. Aging (Albany NY). 2023;15(20):11152–11161. doi:10.18632/aging.205118
121. Jia Y, Hong J, Li H, et al. Butyrate stimulates adipose lipolysis and mitochondrial oxidative phosphorylation through histone hyperacetylation-associated beta(3) -adrenergic receptor activation in high-fat diet-induced obese mice. Exp Physiol. 2017;102(2):273–281. doi:10.1113/EP086114
122. Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr. 2003;133(7 Suppl):2485S–2493S. doi:10.1093/jn/133.7.2485S
123. Serra D, Mera P, Malandrino MI, Mir JF, Herrero L. Mitochondrial fatty acid oxidation in obesity. Antioxid Redox Signal. 2013;19(3):269–284. doi:10.1089/ars.2012.4875
124. Schlaepfer IR, Joshi M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology. 2020;161(2). doi:10.1210/endocr/bqz046
125. Hong J, Jia Y, Pan S, et al. Butyrate alleviates high fat diet-induced obesity through activation of adiponectin-mediated pathway and stimulation of mitochondrial function in the skeletal muscle of mice. Oncotarget. 2016;7(35):56071–56082. doi:10.18632/oncotarget.11267
126. Betz MJ, Enerback S. Targeting thermogenesis in brown fat and muscle to treat obesity and metabolic disease. Nat Rev Endocrinol. 2018;14(2):77–87. doi:10.1038/nrendo.2017.132
127. Lin HV, Frassetto A, Kowalik EJ, et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS One. 2012;7(4):567.
128. Zhang Y, Zheng X, Huang F, et al. Ursodeoxycholic Acid Alters Bile Acid and Fatty Acid Profiles in a Mouse Model of Diet-Induced Obesity. Front Pharmacol. 2019;10:842. doi:10.3389/fphar.2019.00842
129. Chen YS, Liu HM, Lee TY. Ursodeoxycholic Acid Regulates Hepatic Energy Homeostasis and White Adipose Tissue Macrophages Polarization in Leptin-Deficiency Obese Mice. Cells-Basel. 2019;8(3).
130. Kulkarni P, Devkumar P, Chattopadhyay I. Could dysbiosis of inflammatory and anti-inflammatory gut bacteria have an implications in the development of type 2 diabetes? A pilot investigation. BMC Res Notes. 2021;14(1):52. doi:10.1186/s13104-021-05466-2
131. Allin KH, Tremaroli V, Caesar R, et al. Aberrant intestinal microbiota in individuals with prediabetes. Diabetologia. 2018;61(4):810–820. doi:10.1007/s00125-018-4550-1
132. Ruuskanen MO, Erawijantari PP, Havulinna AS, et al. Gut Microbiome Composition Is Predictive of Incident Type 2 Diabetes in a Population Cohort of 5572 Finnish Adults. Diabetes Care. 2022;45(4):811–818. doi:10.2337/dc21-2358
133. Zhang X, Zhao A, Sandhu AK, Edirisinghe I, Burton-Freeman BM. Red Raspberry and Fructo-Oligosaccharide Supplementation, Metabolic Biomarkers, and the Gut Microbiota in Adults with Prediabetes: a Randomized Crossover Clinical Trial. J Nutr. 2022;152(6):1438–1449. doi:10.1093/jn/nxac037
134. Wang Y, Ye X, Ding D, Lu Y. Characteristics of the intestinal flora in patients with peripheral neuropathy associated with type 2 diabetes. J Int Med Res. 2020;48(9):1220736358.
135. Cheng L, Wang J, Dai H, et al. Brown and beige adipose tissue: a novel therapeutic strategy for obesity and type 2 diabetes mellitus. Adipocyte. 2021;10(1):48–65. doi:10.1080/21623945.2020.1870060
136. Townsend KL, Tseng YH. Of mice and men: novel insights regarding constitutive and recruitable brown adipocytes. Int J Obes Suppl. 2015;5(Suppl 1):S15–20. doi:10.1038/ijosup.2015.5
137. Tsakiridis EE, Morrow MR, Desjardins EM, et al. Effects of the pesticide deltamethrin on high fat diet-induced obesity and insulin resistance in male mice. Food Chem Toxicol. 2023;176:113763. doi:10.1016/j.fct.2023.113763
138. Leitner BP, Huang S, Brychta RJ, et al. Mapping of human brown adipose tissue in lean and obese young men. Proc Natl Acad Sci U S A. 2017;114(32):8649–8654. doi:10.1073/pnas.1705287114
139. Blondin DP, Labbe SM, Noll C, et al. Selective Impairment of Glucose but Not Fatty Acid or Oxidative Metabolism in Brown Adipose Tissue of Subjects With Type 2 Diabetes. Diabetes. 2015;64(7):2388–2397. doi:10.2337/db14-1651
140. Chen B, Bai Y, Tong F, et al. Glycoursodeoxycholic acid regulates bile acids level and alters gut microbiota and glycolipid metabolism to attenuate diabetes. Gut Microbes. 2023;15(1):2192155. doi:10.1080/19490976.2023.2192155
141. Bachman ES, Dhillon H, Zhang CY, et al. betaAR signaling required for diet-induced thermogenesis and obesity resistance. Science. 2002;297(5582):843–845. doi:10.1126/science.1073160
142. Liu C, Finegold SM, Song Y, Lawson PA. Reclassification of Clostridium coccoides, Ruminococcus hansenii, Ruminococcus hydrogenotrophicus, Ruminococcus luti, Ruminococcus productus and Ruminococcus schinkii as Blautia coccoides gen. nov., comb. nov., Blautia hansenii comb. nov., Blautia hydrogenotrophica comb. nov., Blautia luti comb. nov., Blautia producta comb. nov., Blautia schinkii comb. nov. and description of Blautia wexlerae sp. nov., isolated from human faeces. Int J Syst Evol Microbiol. 2008;58(Pt 8):1896–1902. doi:10.1099/ijs.0.65208-0
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Association Between Obesity and COVID-19 Disease Severity in Saudi Population
Alqahtani FY, Aleanizy FS, Mohamed RAEH, Al-Maflehi N, Alrfaei BM, Almangour TA, Alkhudair N, Bawazeer G, Shamlan G, Alanazi MS
Diabetes, Metabolic Syndrome and Obesity 2022, 15:1527-1535
Published Date: 16 May 2022
Acne Comorbidities
Wang Y, Zhu M, Wu S, Zheng H
Clinical, Cosmetic and Investigational Dermatology 2022, 15:2415-2420
Published Date: 10 November 2022
Prevalence of Obesity in Newly Onset Diabetes Mellitus and Its Relationship with Uric Acid: An Indian Cross-Sectional Study
Singh SK, Singh R, Singh SK, Iquebal MA, Jaiswal S, Rai PK
International Journal of General Medicine 2023, 16:1217-1226
Published Date: 6 April 2023
Metabolic Syndrome and Tendon Disease: A Comprehensive Review
Lai C, Li R, Tang W, Liu J, Duan XD, Bao D, Liu H, Fu S
Diabetes, Metabolic Syndrome and Obesity 2024, 17:1597-1609
Published Date: 9 April 2024
Semaglutide Effects on Metabolic Outcomes in Diabetes Mellitus Patients — Real World Study
Balcázar-Valencia CM, García-Ramos AF, Osorio-Toro LM, Ordoñez-Guzmán YA, Buitrago-Gómez N, Cabarcas-López WF, Vizcaino-Guerrero CJ, Daza-Arana JE, Ramírez-Rincón A, Restrepo-Erazo K
Diabetes, Metabolic Syndrome and Obesity 2024, 17:1667-1673
Published Date: 10 April 2024