Back to Journals » ImmunoTargets and Therapy » Volume 4
Alzheimer’s disease and immunotherapy: what is wrong with clinical trials?
Authors Kohyama K, Matsumoto Y
Received 30 September 2014
Accepted for publication 11 November 2014
Published 8 January 2015 Volume 2015:4 Pages 27—34
DOI https://doi.org/10.2147/ITT.S49923
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Michael Shurin
Kuniko Kohyama,1 Yoh Matsumoto2–4
1Department of Brain Development and Neural Regeneration, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan; 2Department of Sensory and Motor Systems, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan; 3Immunotherapy Development Inc., Saitama, Japan; 4Geriatric Health Services Facility “Asahigaoka”, Saitama, Japan
Abstract: Alzheimer’s disease (AD) is characterized by progressive neurodegeneration and is the most common cause of dementia. Immunotherapy has recently been regarded as a potential treatment for AD. This stems from the fact that the clinical and pathological findings from the active AD vaccine trial suggests that such vaccine therapy may be effective for AD. However, this trial was halted because of the occurrence of meningoencephalitis in some patients. Avoiding excessive immune reaction is necessary for the success of vaccine therapy. For this purpose, adjuvant-free vaccine therapies (eg, passive immunization or DNA vaccines) are currently under investigation. However, the results of clinical trials employing both active and passive anti-amyloid-beta immunotherapy have been unsatisfactory. In this article, we will analyze the reasons for the limited efficacy of currently available immunotherapies and discuss the effectiveness of new vaccine therapies. Finally, we will speculate on the possibility of its clinical application.
Keywords: Aβ peptide vaccine, amyloid-beta, amyloid cascade theory, immunotherapy, monoclonal antibody, tau
Introduction
Alzheimer’s disease (AD) is the most common cause of age-related cognitive decline. Currently, more than 18 million people worldwide are affected with AD and patient numbers are rapidly increasing with the aging of society.1,2 Although its pathological features and the risk factors for onset have been examined in detail, the cause of the disease remains unclear and a radical treatment has not been developed. There has been recent focus on vaccine therapy as a cure for AD by targeting the underlying cause, which is based on the amyloid cascade hypothesis (ACH). Circulating anti-amyloid-beta (Aβ) antibodies are expected to prevent de novo Aβ development and reduce existing deposits of harmful Aβ in the brain. However, recent anti-Aβ immunotherapies employing peptide vaccines and humanized monoclonal antibodies (mAbs) have revealed unsatisfactory results3,4 because they failed to improve cognitive decline and to extend life span (Table 1). The results suggest that tau pathology is a critical factor for AD in addition to Aβ. The wide range of immunotherapy options available and proposed shall be addressed now.
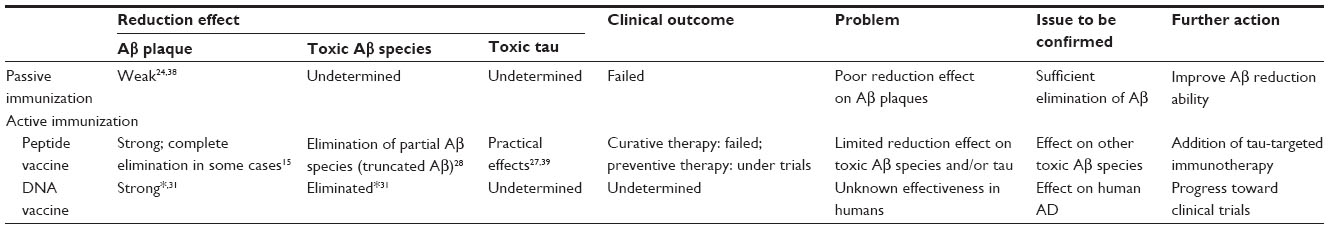 | Table 1 Effectiveness of Aβ-based immunotherapies |
In this report, we will introduce the current status of AD immunotherapies and their limitations. Furthermore, we will analyze why these strategies have not been effective and propose an improved strategy based on an assumption. A number of excellent review articles have recently been published, from which readers can obtain detailed information on each clinical analysis.
Amyloid cascade hypothesis
AD is pathologically characterized by senile plaque, neurofibrillary tangle, and neuronal death.5 AD pathogenesis is generally explained based on the ACH, one of the most convincing theories. According to this theory, the disorder first starts with Aβ accumulation and deposition. Subsequent Aβ oligomerization alters neuronal cell homeostasis and may enhance tau phosphorylation, leading to the formation of neurofibrillary tangles. The end result of this process is widespread neuronal cell dysfunction, including cell death and signal transmission deficits, ultimately leading to dementia. Familiar AD-related mutations, such as the Swedish (K595N/M596L), British (H6R), and Dutch (E22Q) mutations, are strong grounds for this hypothesis.
If the pathological mechanisms of AD are thoroughly clarified, studies of rational drug and therapy design will be rapidly developed.6–8 However, the ACH has been both supported and challenged by several important facts, which will be discussed later in this report.
Anti-Aβ immunotherapy in animal models
Anti-Aβ immunotherapy has been developed based on the ACH. Using PDAPP transgenic mice, certain model of familial early-onset AD, Schenk et al demonstrated that monthly inoculation with an Aβ vaccine consisting of synthetic Aβ peptide in complete Freund’s adjuvant could lead to high anti-Aβ antibody titers and dramatic reductions in Aβ deposition.9 Even in cases wherein Aβ deposition had started, the vaccine was able to reverse amyloid deposit formation. In addition, neuritic plaques and astrocytic reactions observed in model mice were decreased by the vaccine administration. These results were repeatedly reproduced in a variety of other mouse models. Subsequent studies revealed an important finding, demonstrating that Aβ clearance following immunization protected Tg2576 or TgCRND8 mice from developing memory deficits.10,11
Aβ clearance and memory improvement in PDAPP mice were also observed after passive administration of antibodies against Aβ.12,13 Peripheral administration of antibodies against the Aβ peptide was sufficient to reduce the amyloid burden. Despite relatively modest serum levels, passively administered antibodies were able to enter the central nervous system, bind Aβ molecules, suppress oligomerization, decorate plaques, and induce clearance of preexisting Aβ deposits.12
Anti-Aβ immunotherapy and human clinical trials
Trials of therapeutic vaccination
The first vaccine clinical trials for AD patients were started with an Aβ peptide vaccine (AN1792). However, the Phase II-A study was halted in 2002 because 6% of the patients developed meningoencephalitis.14 It was suggested that vaccination with Aβ peptide in a Th1-type adjuvant (QS-21) may induce T-cell responses against Aβ, which, in turn, would result in the development of meningoencephalitis. Later, autopsy of an AD patient who received this vaccine revealed an apparent clearance of Aβ plaques from large areas of the neocortex, as well as a decrease in plaque-associated astrocyte clusters and neuritic dystrophy.15 These results suggested that vaccine therapy is potentially effective for human AD if excessive immune reactions are minimized to avoid unwanted neuroinflammation. To control harmful T-cell responses, alternative vaccination approaches using different routes, adjuvants, and immunogens were developed, following this initial trial. A short Aβ immunogen (Aβ1-15) containing antibody epitopes, but lacking the T-cell-reactive sites residing in full-length Aβ1-42, induced Aβ-specific antibody production in the absence of Aβ-specific cellular immune responses in wild-type mice.16 Furthermore, it significantly reduced Aβ plaques in AD model mice.17 After further improvement of peptide vaccines, two Phase I clinical trials of active immunization demonstrated minimum side effects using ACC-001 (Elan Corp and Wyeth), which contains Aβ1-7 derivatives, and CAD106 (Novartis), which consists of an Aβ fragment coupled to a carrier.18 Although these results are promising, active immunization still carries the risk of meningoencephalitis because of the requirement of adjuvants for peptide vaccination.
Passive transfer of anti-Aβ antibodies is an alternative strategy, which is as effective as active immunization in the mouse model of AD. In this therapy, the risk of meningoencephalitis can be minimized because the antibodies are administrated without adjuvant. Phase II and Phase III clinical trials with humanized anti-Aβ mAbs (bapineuzumab and solanezumab) were started in 2005.19 However, both mAbs did not give expected results. Bapineuzumab showed no clinical benefit compared to placebo and solanezumab slowed cognitive decline in patients with mild, but not moderate, AD.20,21
In 2009, a Phase I trial of ACI-24 (peptide vaccine containing repeated N-terminal Aβ sequence) combined with florbetaben, a positron emission tomography (PET) tracer, started in Europe. It was the first trial to monitor Aβ deposition in patients’ brains in parallel with vaccination. This new approach is expected to provide useful information for choosing good candidates among AD patients with ongoing treatment.
Trials of preventive vaccination
To overcome the difficulties mentioned above, new Aβ immunotherapies are focused on the prevention and very early treatment of AD. The details of prevention trials are well summarized in a review.22 A pharmaceutical company is currently conducting a secondary prevention trial (API, the Alzheimer’s Prevention Initiative) in individuals from a large Colombian family with a mutant PS1 gene, which is associated with familial AD. Participants 30 years of age and older are included in this study, which will test Genentech’s crenezumab mAb therapy. Crenezumab, a humanized mAb that binds soluble, oligomeric, and fibrillar Aβ, appears to have greater advantages compared to other mAbs because soluble oligomer is now regarded as a strong promoting factor of AD. Nevertheless, the Phase II trial of crenezumab recently gave modest results; the high-dose group of mild AD patients showed partial efficacy in cognitive function by exploratory data analysis. Further studies are necessary for final conclusion.
Recently, DIAN, the Dominantly Inherited Alzheimer Network, started other prevention and early-treatment trials. DIAN conducted a collaborative trial with a pharmaceutical company and the Alzheimer’s Association in individuals with familial AD. Furthermore, solanezumab and gantenerumab are under investigation. Additional Aβ passive immunotherapies using mAbs that recognize protofibrils (BAN2401) or plaque-associated fibrillar Aβ (BIIB037) are currently under investigation.
ACH: is it really wrong?
The results obtained in several clinical trials have not been satisfactory,14,19–21 which raises concern that the ACH is incorrect, leading some researchers to propose modifications to or denial of the hypothesis. Before arriving at a conclusion, we must examine the following issues regarding the results of clinical trials.
Are passive immunotherapies really effective in removing Aβ deposits?
Almost all the clinical trials using mAbs against different epitopes within the Aβ1-42 molecule failed to halt or improve cognitive decline. However, it remains unclear whether or not the treatment sufficiently eliminates Aβ deposits, including plaques and toxic species. Roher et al reported an autopsy case of a bapineuzumab-treated patient and demonstrated that the antibody treatment did not significantly reduce the number of Aβ plaques23 in mild-to-moderate dementia due to AD. In addition, PET imaging with Pittsburgh compound B (11C-PiB) of bapineuzumab-treated patients showed that reduction in the positive signal was marginal.24 Together, these results suggest that passive immunotherapy has weak Aβ reduction effects compared with active immunotherapy (described in the following sections). Under such conditions, it is difficult to evaluate the efficacy of passive immunization. In other words, whether unsatisfactory results are obtained from poor treatment efficacy (such as insufficient administration, timing of treatment, or mAb inefficiency, including its epitope specificity) or from limitations of the treatment itself (eg, failure despite successful reduction of targeted Aβ species; the strategic limitation) remains undetermined.
One reason for the low efficacy of anti-Aβ mAb treatment may be attributable to the nature of the antibody. Because an mAb is directed at only a single epitope of the Aβ molecule, several toxic Aβ species that do not possess the epitope would escape from attack by the mAb. Rosenblum presumed that the failure of some trials including bapineuzumab may be due to the mAb’s inability to significantly reduce loads of toxic Aβ oligomers.25
Are active immunotherapies really effective in removing toxic Aβ species?
Compared with passive immunotherapy, active immunotherapy appears to be more effective in Aβ reduction. Several autopsy reports have demonstrated that some Aβ vaccine (AN1792)-treated patients showed complete disappearance of Aβ plaques.15,26,27 Furthermore, AN1792 vaccination induced antibodies against a wide variety of Aβ species.28 However, AN1792 treatment did not halt AD progression.26 The most important point is that it remains undetermined whether or not toxic Aβ species are sufficiently removed by the treatment. Boche et al reasoned that failure of AN1792 to halt cognitive decline was because reduction of aggregated tau was limited in neuronal processes but not in the cell body.27 Owing to its ability to induce production of various antibodies by suitable design, active vaccine treatment may include large potential for beneficial effects; therefore, it should be investigated intensely.
Dual pathway hypothesis: modification of the ACH
The unsatisfactory results of anti-Aβ immunotherapy proposed a modification to ACH. One of the major alternatives is the dual pathway hypothesis.29 In this theory, it is hypothesized that Aβ and tau can be linked by separated mechanisms driven by a common upstream driver. They postulated the apolipoprotein E (ApoE) genotype and glycogen synthase kinase 3 (GSK3) as candidates. Immunotherapy targeting these molecules is currently under investigation (see following sections). Yoshiyama et al speculated that tangle pathology, but not senile plaque formation, represents the earliest stages of pathology in sporadic AD.30 In other words, tau pathology starts independently before Aβ deposition. Although which theory is more likely is undetermined, it is clear that immunotherapy targeting both Aβ and tau should be considered.
Immunotherapy targeting both toxic Aβ and tau, with special reference to DNA vaccines
If anti-Aβ immunotherapy leads to complete removal of Aβ deposits and toxic Aβ species but does not improve cognitive decline in AD, we should select one of the two following options. First, anti-Aβ immunotherapy should be used only in prevention trials as described above. Second, immunotherapy should be directed to both toxic Aβ species and tau. Tau, the component protein of neurofibrillary tangle, is currently being focused as a dominant factor of AD. Before describing the tau-targeting therapy, we wish to introduce our newly developed anti-Aβ DNA vaccine designated YM3711.31
We developed several DNA vaccines and evaluated their Aβ production and secretion abilities, after which we chose an IgL-Aβ×4-Fc-IL-4 vaccine (YM3711) for further studies. YM3711 was used to vaccinate mice, rabbits, and monkeys, and its abilities to stimulate anti-Aβ antibody production and reduce Aβ were determined. It was found that YM3711 vaccination induced significantly higher levels of antibodies to AD-related molecules, including AβpE3-42 (pyroglutamate-modified Aβ3-42) and ABri (amyloid Bri or British amyloid peptide), as well as Aβ1-42 (Figure 1). Importantly, YM3711 significantly reduced not only these monomeric amyloid species, as shown in microphotographs (Figure 2A and B) and quantitative analysis (Figure 2C and D), but also superstructural Aβ molecules (Figure 2E and F) in the brains of model mice. Binding (Figure 3B) and competition (Figure 3C) assays using purified YM3711 protein products (Figure 3A) (YM3711P; analog of Aβ oligomer) clearly demonstrated that a large portion of the antibodies induced by YM3711 vaccination was directed at conformational epitopes of the oligomeric Aβ and its complexes. As a result, YM3711-based therapy has an advantage of reducing overall toxic amyloid proteins. Of special note is that DNA vaccines can be used for active immunization without the need for adjuvant, which is a risk factor for inducing meningoencephalitis. This underscores the benefits of studying such vaccines further.
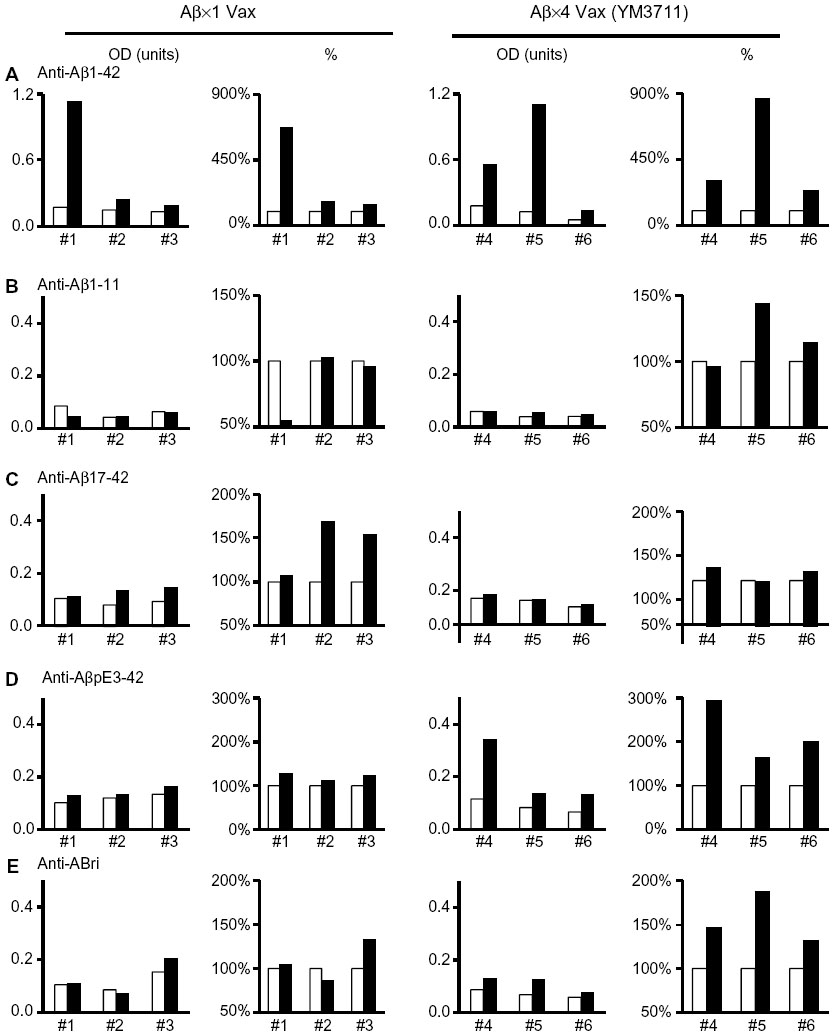 | Figure 1 YM3711 vaccination induced antibodies not only against Aβ species but also against an unrelated amyloidogenic peptide. |
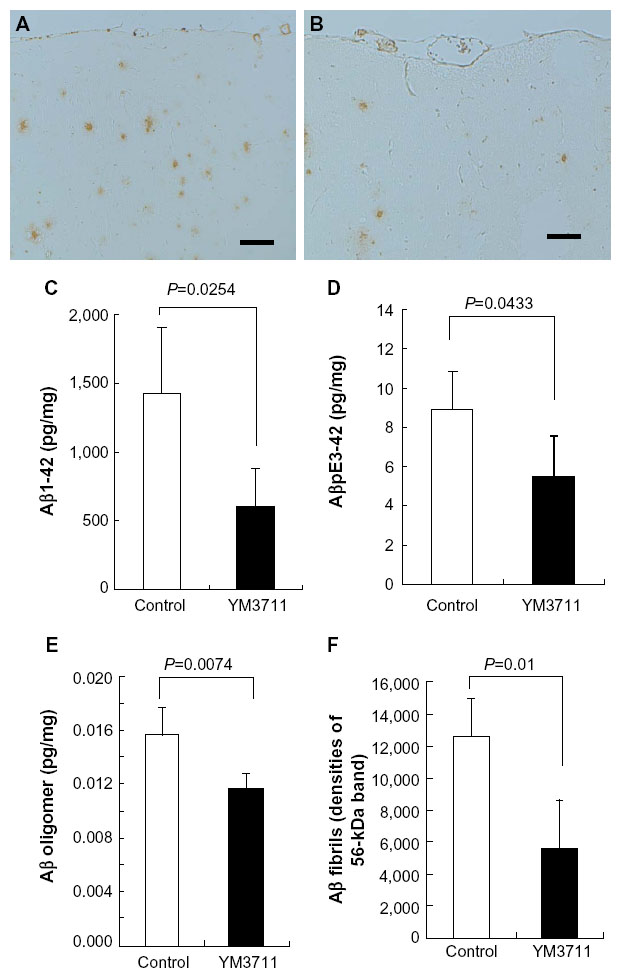 | Figure 2 YM3711 vaccination effectively reduced not only Aβ1-42 but other Aβ species as well. |
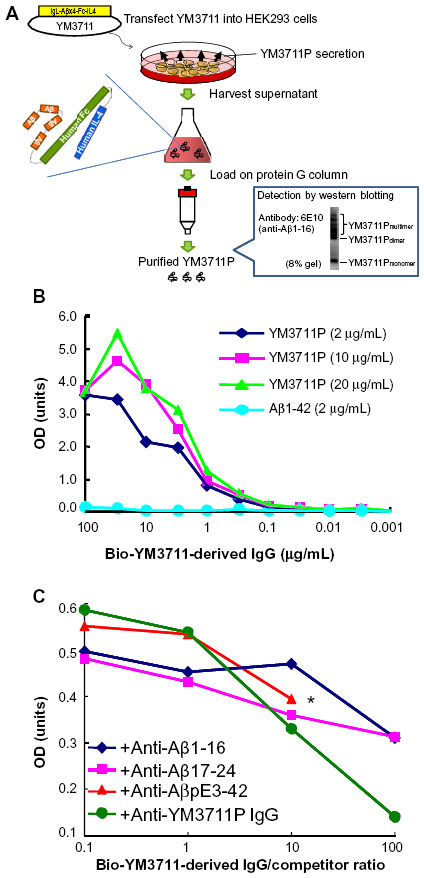 | Figure 3 Antibodies induced by YM3711 vaccination recognized conformational epitopes of Aβ constructs more intensely than linear ones. |
Similar to anti-Aβ immunotherapy, anti-tau immunotherapy using tau peptide vaccines or anti-tau mAbs have been developed and employed in model mice. Precise information is provided in the following review articles.3,32,33 Even when considering tau-targeted therapy, anti-Aβ immunotherapy should be continued in combination, even during the later stages of AD because Aβ oligomers are always neurotoxic in combination with toxic tau.
In addition, slightly different approaches are in consideration. ApoE is one of the candidates for a common upstream driver that links Aβ and tau with separate deposition mechanisms. If this is the case, then ApoE-targeted immunotherapy would be effective in decreasing toxic Aβ and tau. Kim et al34 and Liao et al35 demonstrated that both preventive and therapeutic administration of an anti-ApoE mAb, HJ6.3, decreases Aβ accumulation. Furthermore, Liu et al reported that blocking the ApoE/Aβ interaction with a synthetic peptide ameliorates Aβ and tau pathology in triple-transgenic mice.36 Goni et al took a different approach to target both Aβ and tau.37 They immunized TgSwDI or triple transgenic mice (PS1M146V, tauP301L and APPK670N/M671L) with polymerized ABri, whose amino acid sequence is not homologous to either Aβ or tau, and observed increased titers of both anti-Aβ and anti-tau antibodies. These findings suggest that the raised antibodies are directed toward the conformational epitope(s) formed by Aβ and tau accumulation. Interestingly, this immunotherapy ameliorated Aβ and tau pathology in triple-transgenic mice.37 Although these two types of immunotherapies are fascinating, it remains undetermined whether or not they influence molecules other than Aβ and tau, which may produce adverse effects.
Conclusion
It is clear that the results of clinical trials with anti-Aβ active and passive immunization were unsatisfactory. However, the reasons for this lack of clarity appear to be slightly different between the two approaches. In this review article, we attempted to clarify this difference and suggest that active immunization would be more effective than passive immunization as stated. A combined active immunization approach targeting both Aβ and tau should first be evaluated to find effective immunotherapy against AD.
Disclosure
Yoh Matsumoto is an inventor of DNA vaccines. The authors report no other conflicts of interest in this work.
References
Fuller JP, Stavenhagen JB, Teeling JL. New roles for Fc receptors in neurodegeneration-the impact on immunotherapy for Alzheimer’s disease. Front Neurosci. 2014;8:235. | |
Braidy N, Poljak A, Marjo C, et al. Metal and complementary molecular bioimaging in Alzheimer’s disease. Front Aging Neurosci. 2014;6:138. | |
Panza F, Frisardi V, Solfrizzi V, et al. Immunotherapy for Alzheimer’s disease: from anti-beta-amyloid to tau-based immunization strategies. Immunotherapy. 2012;4(2):213–238. | |
Tayeb HO, Murray ED, Price BH, Tarazi FI. Bapineuzumab and solanezumab for Alzheimer’s disease: is the ‘amyloid cascade hypothesis’ still alive? Expert Opin Biol Ther. 2013;13(7):1075–1084. | |
Katzman R, Saitoh T. Advances in Alzheimer’s disease. FASEB J. 1991;5(3):278–286. | |
Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12(10):383–388. | |
Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. | |
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. | |
Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400(6740):173–177. | |
Morgan D, Diamond DM, Gottschall PE, et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408(6815):982–985. | |
Janus C, Pearson J, McLaurin J, et al. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature. 2000;408(6815):979–982. | |
Bard F, Cannon C, Barbour R, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6(8):916–919. | |
DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2001;98(15):8850–8855. | |
Orgogozo JM, Gilman S, Dartigues JF, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61(1):46–54. | |
Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9(4):448–452. | |
Maier M, Seabrook TJ, Lemere CA. Developing novel immunogens for an effective, safe Alzheimer’s disease vaccine. Neurodegener Dis. 2005;2(5):267–272. | |
Maier M, Seabrook TJ, Lazo ND, et al. Short amyloid-beta (Abeta) immunogens reduce cerebral Abeta load and learning deficits in an Alzheimer’s disease mouse model in the absence of an Abeta-specific cellular immune response. J Neurosci. 2006;26(18):4717–4728. | |
Masters CL, Beyreuther K. Alzheimer’s centennial legacy: prospects for rational therapeutic intervention targeting the Abeta amyloid pathway. Brain. 2006;129(pt 11):2823–2839. | |
Salloway S, Sperling R, Gilman S, et al; Bapineuzumab 201 Clinical Trial Investigators. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73(24):2061–2070. | |
Salloway S, Sperling R, Brashear HR. Phase 3 trials of solanezumab and bapineuzumab for Alzheimer’s disease. N Engl J Med. 2014;370(15):1460. | |
Doody RS, Thomas RG, Farlow M, et al; Alzheimer’s Disease Cooperative Study Steering Committee, Solanezumab Study Group. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):311–321. | |
Panza F, Solfrizzi V, Imbimbo BP, Tortelli R, Santamato A, Logroscino G. Amyloid-based immunotherapy for Alzheimer’s disease in the time of prevention trials: the way forward. Expert Rev Clin Immunol. 2014;10(3):405–419. | |
Roher AE, Maarouf CL, Daugs ID, et al. Neuropathology and amyloid-beta spectrum in a Bapineuzumab immunotherapy recipient. J Alzheimers Dis. 2011;24:315–325. | |
Rinne JO, Brooks DJ, Rossor MN, et al. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9(4):363–372. | |
Rosenblum WI. Why Alzheimer trials fail: removing soluble oligomeric beta amyloid is essential, inconsistent, and difficult. Neurobiol Aging. 2014;35(5):969–974. | |
Holmes C, Boche D, Wilkinson D, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372(9634):216–223. | |
Boche D, Donald J, Love S, et al. Reduction of aggregated tau in neuronal processes but not in the cell bodies after Abeta42 immunisation in Alzheimer’s disease. Acta Neuropathol. 2010;120(1):13–20. | |
Nicoll JA, Barton E, Boche D, et al. Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol. 2006;65(11):1040–1048. | |
Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer’s disease: a dual pathway hypothesis. Neuron. 2008;60(4):534–542. | |
Yoshiyama Y, Lee VM, Trojanowski JQ. Therapeutic strategies for tau mediated neurodegeneration. J Neurol Neurosurg Psychiatry. 2013;84(7):784–795. | |
Matsumoto Y, Niimi N, Kohyama K. Development of a new DNA vaccine for Alzheimer disease targeting a wide range of abeta species and amyloidogenic peptides. PLoS One. 2013;8(9):e75203. | |
Rosenmann H. Immunotherapy for targeting tau pathology in Alzheimer’s disease and tauopathies. Curr Alzheimer Res. 2013;10(3):217–228. | |
Sigurdsson EM. Tau immunotherapy and imaging. Neurodegener Dis. 2014;13(2–3):103–106. | |
Kim J, Eltorai AE, Jiang H, et al. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Abeta amyloidosis. J Exp Med. 2012;209(12):2149–2156. | |
Liao F, Hori Y, Hudry E, et al. Anti-ApoE antibody given after plaque onset decreases Abeta accumulation and improves brain function in a mouse model of Abeta amyloidosis. J Neurosci. 2014;34(21):7281–7292. | |
Liu S, Breitbart A, Sun Y, et al. Blocking the apolipoprotein E/amyloid beta interaction in triple transgenic mice ameliorates Alzheimer’s disease related amyloid beta and tau pathology. J Neurochem. 2014;128(4):577–591. | |
Goñi F, Herline K, Peyser D, et al. Immunomodulation targeting of both Abeta and tau pathological conformers ameliorates Alzheimer’s disease pathology in TgSwDI and 3xTg mouse models. J Neuroinflammation. 2013;10:150. | |
Roher AE, Cribbs DH, Kim RC, et al. Bapineuzumab alters abeta composition: implications for the amyloid cascade hypothesis and anti-amyloid immunotherapy. PLoS One. 2013;8(3):e59735. | |
Boche D, Denham N, Holmes C, Nicoll JA. Neuropathology after active Abeta42 immunotherapy: implications for Alzheimer’s disease pathogenesis. Acta Neuropathol. 2010;120(3):369–384. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.