Back to Journals » Journal of Inflammation Research » Volume 18
Advances in the Understanding of Mitochondrial Inflammatory Regulation in the Pathogenesis of Alzheimer’s Disease
Authors Zhang RZ, Li L, Ma C, Dou TT, Wei YT, Yan XK
Received 29 July 2025
Accepted for publication 8 October 2025
Published 23 October 2025 Volume 2025:18 Pages 14475—14491
DOI https://doi.org/10.2147/JIR.S557000
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Dharmappa Krishnappa
Ren-Zhen Zhang,1,* Li Li,1,* Cui Ma,1 Ting-Ting Dou,1 Yu-Ting Wei,1 Xing-Ke Yan1,2
1School of Acupuncture-Moxibustion and Tuina, Gansu University of Traditional Chinese Medicine, Lanzhou, People’s Republic of China; 2Gansu Provincial Traditional Chinese Medicine Research Center, Lanzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xing-Ke Yan, Acupuncture and moxibustion and Massage College of Gansu University of Traditional Chinese Medicine, 35 Dingxi East Road, Lanzhou, People’s Republic of China, Email [email protected]
Abstract: Mitochondria, beyond serving as the powerhouse of the cell, play a pivotal role in the regulation of inflammatory responses. Mitochondrial dysfunction-induced immune activation and chronic inflammation are deeply implicated in the pathogenesis of Alzheimer’s disease (AD), influencing its onset and progression through multiple inflammatory pathways. This review summarizes the involvement of several mitochondrial-related mechanisms in AD, including the release of mitochondrial DNA (mtDNA), signal transduction via mitochondrial antiviral-signaling protein (MAVS), the accumulation of mitochondrial damage-associated molecular patterns (DAMPs), the regulation of mitophagy, and the activation of the cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway. These insights aim to shed new light on mitochondrial inflammation as a regulatory mechanism in AD and to explore its potential as a therapeutic target.
Keywords: Alzheimer’s disease, mitochondrion, neuroinflammation, inflammatory mechanism, review
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by impairments in memory and cognitive functions.1 Epidemiological research shows that the number of AD patients continues to rise, and it is expected that the global number of patients will reach 114 million by 2050, posing a huge threat to the quality of life, social and economic burden of patients.2 Although the deposition of Aβ plaques and excessive phosphorylation of tau protein are considered classic pathological features of AD,3 recent studies have continuously shown that mitochondrial dysfunction and inflammatory response also play a central role in the occurrence and development of the disease.4 Mitochondrial and bioenergetic dysfunctions are recognized as key contributors to the onset and progression of AD. Amyloid-beta (Aβ) disrupts mitochondrial integrity and energy metabolism, thereby initiating a cascade of mitochondrial disturbances.5 Mitochondrial dysfunction is considered an early hallmark of AD and plays a central role in its pathophysiological mechanisms.6,7 Inflammation, a fundamental physiological response to infection and tissue injury, can become pathological when prolonged or excessive, contributing to a wide range of diseases, including autoimmune disorders, metabolic syndromes, and cancers.8
Damaged mitochondrial energy metabolism can lead to reduced ATP production and increased levels of reactive oxygen species (ROS), exacerbating oxidative stress and ultimately damaging the structure and function of neurons.9 Mitochondria, beyond their classical role in cellular energy production, are increasingly recognized as regulators of inflammatory signaling.10 In the pathogenesis of AD, inflammatory signals promote the sustained activation of microglia and astrocytes, producing a large amount of pro-inflammatory cytokines and forming a chronic neuroinflammatory environment.11 Long term inflammation not only exacerbates synaptic loss and neuronal death, but also interacts with Aβ deposition and abnormal phosphorylation of tau protein, driving disease progression.12,13 Therefore, the interaction between mitochondrial dysfunction and inflammatory response forms a vicious cycle, which is considered an important mechanism driving the onset and development of AD.
A growing body of evidence indicates that mitochondria not only support immune responses via energy supply but also engage directly in immune modulation through various molecular pathways.14–16 A pivotal mechanism in the mitochondrial-immune interface is the release of mtDNA, which acts as a damage-associated molecular pattern (DAMP) to activate innate immune responses.17 MAVS, a critical adaptor in mitochondrial signaling, orchestrates antiviral immune responses. Furthermore, DAMPs exert broad effects on inflammatory pathways, while mitophagy—a selective form of autophagy—plays a crucial role in removing damaged mitochondria and modulating inflammation.18–20 The cGAS-STING pathway, a recently identified regulator of interferon signaling, has emerged as a key player in diverse cellular processes, including autophagy, translation, metabolic homeostasis, cell adhesion, DNA damage repair, senescence, and cell death, all tightly linked to mitochondrial function.21 Mitochondria are not only “victims” but also “regulators” in the regulation of inflammatory response in AD. Mitochondrial dysfunction interacts with abnormal activation of inflammatory pathways, forming a “mitochondrial inflammatory axis” that drives neuronal damage and cognitive decline. Thoroughly elucidating this mechanism not only helps to understand the complex pathological process of AD, but also provides a theoretical basis for developing novel therapeutic strategies targeting mitochondrial inflammatory pathways. Taken together, this review outlines the diverse mitochondrial mechanisms involved in inflammatory regulation, aiming to provide novel insights into the inflammatory pathogenesis of AD and potential avenues for therapeutic intervention.
The Relationship Between mtDNA Release and AD
mtDNA, the genetic material housed within the mitochondrial matrix, is typically sequestered by the organelle’s double-membrane structure under physiological conditions. However, in response to cellular injury or stress, mtDNA can be released into the cytoplasm, where it acts as a DAMP that is directly recognized by receptors of the innate immune system, triggering inflammatory responses.22 Multiple mechanisms are involved in mtDNA release, including mitochondrial permeability transition, apoptosis, and necrosis.23 These processes are activated under various stressors and pathological states, ultimately resulting in the extrusion of mtDNA and subsequent activation of immune signaling. Aberrant or excessive release of mtDNA has been linked to the induction of chronic neuroinflammation, a pathological hallmark that contributes to the initiation and progression of AD. (Figure 1).
 |
Figure 1 Schematic diagram of the molecular mechanism of mtDNA release. |
Mitochondrial Permeability Transition Mechanism
MPT refers to a sudden increase in the permeability of the mitochondrial membrane, typically triggered by calcium overload, oxidative stress, and other cellular stressors. This phenomenon is primarily mediated by the opening of the mitochondrial permeability transition pore (mPTP) located on the inner mitochondrial membrane. Opening of the mPTP disrupts the mitochondrial membrane potential, abolishes the asymmetric distribution of solutes across the membrane, and leads to mitochondrial swelling and rupture of the outer membrane. Consequently, mtDNA and other mitochondrial components are released into the cytosol, initiating innate immune responses.24 A key regulator of mPTP is Cyclophilin D (CypD), a mitochondrial matrix-resident peptidyl-prolyl isomerase that modulates pore opening through interaction with the adenine nucleotide translocator (ANT) on the inner mitochondrial membrane.25 The CypD–ANT interaction is a critical step in mPTP activation, altering membrane permeability. The voltage-dependent anion channel (VDAC), located on the outer membrane, is also a component of the mPTP complex. Its interaction with ANT influences mitochondrial membrane potential and the transport of metabolites, further affecting mPTP dynamics.26 Calcium ions (Ca²⁺) are major inducers of mPTP opening. Elevated intracellular Ca²⁺ levels enhance the CypD–ANT interaction, increasing inner membrane permeability. Under oxidative stress, ROS oxidize mitochondrial membrane proteins, further promoting mPTP opening.27 ATP depletion also facilitates mPTP activation; when energy stores are exhausted, mitochondria fail to maintain membrane potential, leading to osmotic imbalance, matrix swelling, outer membrane rupture, and the cytosolic release of mtDNA and other DAMPs—events that precipitate immune activation, inflammation, and cell death.28
Disruption of mitochondrial Ca²⁺ homeostasis and aberrant mPTP opening represent upstream signaling events in the mitochondrial dysfunction cascade that underlies AD pathology.29 Dysregulated Ca²⁺ signaling plays a central role: both Aβ and hyperphosphorylated tau impair mitochondrial calcium balance in neuronal and glial cells. Elevated Ca²⁺ levels drive mitochondrial dysfunction by increasing ROS production, impairing ATP synthesis, and altering membrane permeability.The mPTP, a high-conductance channel tightly regulated by CypD, maintains mitochondrial Ca²⁺ homeostasis. Aβ aggregates and other neurotoxins interact with CypD, inducing mitochondrial depolarization via ROS accumulation and Ca²⁺ overload-mediated mPTP opening.30,31 Studies have shown that in aged AD model mice, reduced CypD expression enhances calcium buffering capacity and limits mPTP opening. In contrast, hippocampal overexpression of CypD impairs mitochondrial function, leading to ATP depletion, increased mPTP opening, and cognitive deficits such as memory loss.32 Tau deficiency has also been shown to preserve mitochondrial integrity by reducing mPTP opening through CypD-dependent mechanisms, thereby potentially preventing age-related cognitive decline. Moreover, elevated CypD expression has been observed in neurons of the temporal cortex and hippocampus—but not the cerebellum—of mAPP mice exposed to high Aβ levels.33,34 This region-specific upregulation correlates with age-dependent disturbances in calcium handling, increased mitochondrial swelling, mPTP activation, and cytochrome c release, underscoring the mechanistic link between Aβ toxicity, mPTP dysregulation, and AD progression.
The Apoptotic Pathway
Apoptosis is a form of programmed cell death essential for maintaining tissue homeostasis and eliminating damaged or abnormal cells. In the intrinsic, mitochondria-mediated apoptotic pathway, the release of mitochondrial factors plays a direct role in transducing and executing apoptotic signals, tightly linking energy metabolism to cell fate under both physiological and pathological conditions.35 Upon exposure to cellular stress or pro-apoptotic stimuli, the balance among Bcl-2 family proteins becomes disrupted. Pro-apoptotic proteins such as Bax and Bak oligomerize in the mitochondrial outer membrane, forming pores that increase membrane permeability. In contrast, anti-apoptotic proteins like Bcl-2 and Bcl-xL inhibit this process by binding to and neutralizing pro-apoptotic counterparts, thereby preventing pore formation. When pro-apoptotic signals prevail, mitochondrial outer membrane permeabilization (MOMP) occurs.36,37 The induction of MOMP facilitates the release of intermembrane mitochondrial proteins, most notably cytochrome c, into the cytosol. Once in the cytoplasm, cytochrome c associates with apoptotic protease activating factor-1 (Apaf-1) to form the apoptosome, initiating a cascade of caspase activation that culminates in apoptosis.38,39
In the context of AD, progressive loss of functional neurons is a well-documented pathological hallmark. Apoptosis contributes significantly to this neuronal degeneration, with programmed cell death observed even in the early stages of AD.40 Mitochondria are central mediators of this apoptotic process. Dysfunctional mitochondria act as primary triggers: impaired mitochondrial function leads to increased MOMP, facilitating the release of apoptogenic proteins into the cytosol and the subsequent initiation of apoptotic signaling pathways.Moreover, defective ATP production exacerbates neuronal vulnerability to apoptosis. Energy deprivation heightens sensitivity to apoptotic stimuli and accelerates the execution of programmed cell death in neurons.41 These findings underscore the critical role of mitochondrial dysfunction in apoptosis and its contribution to neurodegeneration in Alzheimer’s disease.
Necrotic Cell Death
Necrosis is a form of non-programmed cell death typically triggered by acute injury or pathological conditions, characterized by loss of membrane integrity and uncontrolled release of intracellular contents.42 Unlike apoptosis, necrosis often elicits a strong inflammatory response. The cellular debris released from necrotic cells functions as DAMPs, which can activate pattern recognition receptors (PRRs) of the innate immune system, thereby provoking robust immune and inflammatory responses.43 Under severe cellular stress conditions—such as oxidative stress, calcium overload, and energy depletion—the mPTP opens, leading to dissipation of the mitochondrial membrane potential and swelling of the organelle. This disrupts both the inner and outer mitochondrial membranes, ultimately resulting in their rupture and the release of mitochondrial contents, including mtDNA and other DAMPs, into the cytosol.44 Energy failure and excess ROS production are key contributors to necrotic cell death. Elevated intracellular Ca²⁺ levels promote mitochondrial calcium overload and mPTP opening. In the absence of sufficient ATP, cells cannot maintain ionic gradients or membrane stability, further exacerbating mitochondrial and plasma membrane rupture. ROS amplify the damage by oxidizing mitochondrial membrane proteins and lipids, increasing permeability and membrane breakdown.Mitochondria-derived DAMPs activate immune responses through a variety of PRRs.45 Upon mitochondrial damage or cell rupture, mtDNA is released into the cytosol or extracellular matrix, where it serves as a potent immunostimulatory molecule. It can activate innate immune pathways such as Toll-like receptor 9 (TLR9) and the cGAS-STING axis, triggering downstream inflammatory cascades.46,47 Additionally, extracellular ATP acts as a danger signal by activating the P2X7 receptor, leading to the assembly and activation of the NLRP3 inflammasome, which promotes the release of pro-inflammatory cytokines including interleukin-1β and interleukin-18.48
One of the hallmark features of AD is the persistent presence of chronic neuroinflammation in the brain, where mtDNA release is considered a potential initiating factor. Studies have reported a sustained decrease of mtDNA in microglia within the hippocampus during AD progression, while a modest increase was observed in microglia from the cerebellum, suggesting regional variation in mitochondrial stress.47 In experimental models, the injection of isolated mitochondria or mtDNA into the hippocampus of C57BL/6 mice led to elevated levels of neuroinflammatory markers after seven days. These included reductions in Trem2 mRNA, and increases in GFAP, CSF1R, phosphorylated NF-κB, phosphorylated AKT, along with heightened expression of App mRNA, APP protein, and Aβ1-42.49 These findings confirm that extracellular mitochondria and their components, especially mtDNA and mtDNA-associated proteins, can trigger neuroinflammation and modulate AD-relevant biomarkers. Mitochondrial DAMPs thus represent a mechanistic link between mitochondrial injury and inflammation in AD pathogenesis.
MAVS-Mediated Antiviral Signaling and Its Relationship with AD
MAVS plays a crucial role in the host’s innate immune defense against viral infections, primarily through the retinoic acid-inducible gene I (RIG-I)-like receptor (RLR) signaling pathway.50 Located on the outer mitochondrial membrane, MAVS functions as a key adaptor that integrates upstream viral RNA recognition with downstream activation of antiviral and pro-inflammatory signaling cascades. The N-terminal caspase activation and recruitment domain (CARD) of MAVS enables its interaction with the CARD domains of RLRs such as RIG-I and MDA5, while the C-terminal transmembrane region anchors it to the mitochondrial membrane, ensuring proper localization for signal transduction.51,52 During viral infection, RIG-I and MDA5 detect viral double-stranded RNA in the cytoplasm and undergo conformational changes that expose their CARD domains. These activated RLRs then interact with MAVS via CARD–CARD binding, initiating a signaling cascade on the mitochondrial surface.53 Activated MAVS assembles into a multiprotein complex that includes adaptor proteins and kinases such as TNF receptor-associated factors (TRAFs) and TANK-binding kinase 1 (TBK1).54 TBK1 phosphorylates transcription factors IRF3 and IRF7, which translocate to the nucleus to induce the expression of type I interferons and other antiviral genes.55 In parallel, MAVS also activates the NF-κB pathway. This involves the stimulation of the IKK complex, leading to the degradation of IκB and the nuclear translocation of NF-κB. Once in the nucleus, NF-κB promotes the transcription of pro-inflammatory cytokines such as IL-6 and TNF-α, establishing a robust antiviral state and limiting viral propagation.56 However, while the MAVS pathway is essential for antiviral defense, its chronic or aberrant activation has been implicated in neuroinflammatory conditions, including AD. Sustained MAVS signaling can lead to persistent inflammation within the central nervous system, exacerbating neuronal dysfunction and contributing to AD progression (Figure 2).
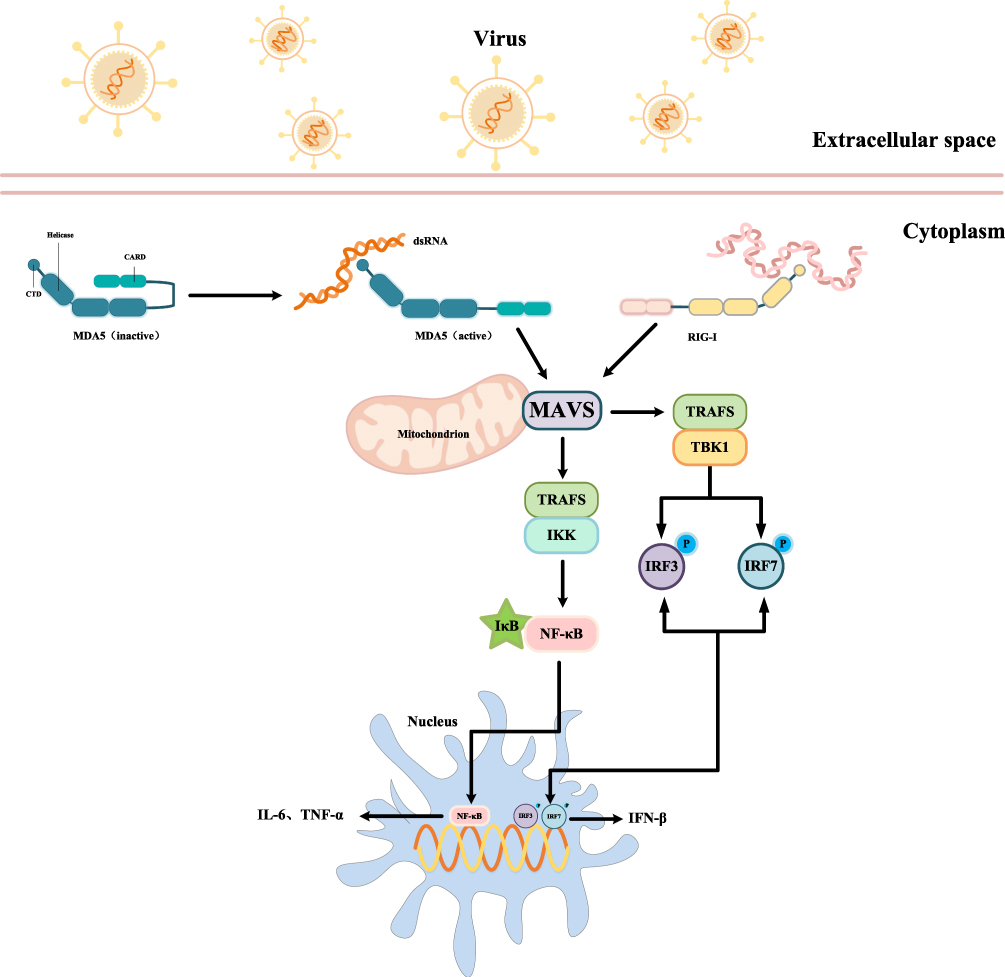 |
Figure 2 MAVS mediated antiviral signaling transduction mechanism. |
Emerging evidence highlights the role of Evolutionarily Conserved Signaling Intermediate in Toll pathways (ECSIT), a signaling adaptor that modulates NF-κB activation and enhances RIG-I/MAVS-mediated antiviral immunity. ECSIT also plays a critical role in mitochondrial physiology, influencing the assembly of mitochondrial complex I (CI), the production of mitochondrial mROS, the maintenance of mitochondrial membrane potential (MMP), and overall mitochondrial quality control.57 These functions directly intersect with key pathogenic mechanisms of AD.Notably, studies have shown that mice deficient in MAVS exhibit improved cognitive outcomes, suggesting that the suppression of MAVS-dependent immune signaling can alleviate neurodegeneration.58 Although MAVS is traditionally recognized for its role in antiviral defense, its involvement in mitochondrial homeostasis, inflammatory signaling, and apoptosis positions it as a potential contributor to AD pathology. Aberrant MAVS activation may promote neuroinflammation, intensify mitochondrial dysfunction, and accelerate neuronal apoptosis, thereby driving disease progression.
The Role of DAMPs Accumulation in AD
DAMPs are endogenous molecules released by stressed, damaged, or necrotic cells that can activate the innate immune system and initiate inflammatory responses.59 They serve as key mediators in a variety of pathological conditions, including tissue injury, infection, and chronic inflammation.DAMPs are structurally diverse and originate from multiple cellular compartments. Common examples include mtDNA, high mobility group box 1 (HMGB1), heat shock proteins (HSPs), uric acid, and extracellular ATP.60 Once released into the extracellular space or cytosol, DAMPs are recognized by PRRs, which initiate downstream signaling pathways that culminate in inflammation. Major PRR families involved in DAMP sensing include Toll-Like Receptors (TLRs), nucleotide-binding oligomerization domain-like receptors (NLRs), and receptors for advanced glycation end-products (RAGE).61 In the context of AD, the chronic accumulation and release of DAMPs have been implicated in the persistent activation of neuroinflammatory pathways. These molecules play a pivotal role in initiating early inflammatory responses and driving the downstream pathological cascades characteristic of AD. Persistent engagement of PRRs by DAMPs in the central nervous system can sustain microglial activation, promote the production of pro-inflammatory cytokines, and exacerbate synaptic and neuronal damage. This DAMP–PRR axis represents a critical link between mitochondrial dysfunction, cellular stress, and the inflammatory processes underlying AD progression (Figure 3).
 |
Figure 3 DAMPs mediated inflammatory response mechanism. |
TLRs
TLRs are one of the earliest discovered classes of PRRs and play a central role in the innate immune system. TLRs recognize both pathogen-associated molecular patterns (PAMPs) and endogenous DAMPs, initiating immune responses by activating downstream signaling pathways.62 TLR9, in particular, is localized primarily on endosomal membranes and is capable of detecting mtDNA released from damaged or necrotic cells. Upon recognizing mtDNA, TLR9 signals through the MyD88-dependent pathway to activate NF-κB and interferon regulatory factor 3 (IRF3), thereby inducing the production of type I interferons (eg, IFN-α) and pro-inflammatory cytokines such as TNF-α and IL-6.63 While the inflammatory responses initiated by TLR activation serve as a protective mechanism against cellular damage, their persistent or excessive activation can lead to chronic inflammation and subsequent tissue injury.Among the TLR family members, TLR4 is another key receptor involved in innate immunity. Best known as the canonical sensor for bacterial lipopolysaccharide (LPS), TLR4 can also detect endogenous DAMPs such as HMGB1 and HSPs, highlighting the complexity and versatility of TLR-mediated inflammatory regulation.64
In AD, hallmark pathological features include extracellular Aβ plaque deposition and intracellular neurofibrillary tangles formed by hyperphosphorylated tau protein, both of which contribute to neuronal dysfunction and degeneration.8,65 Within the central nervous system, TLRs are expressed in both neurons and glial cells, where they detect DAMPs released from damaged or dying cells.66 Dysregulated TLR activity has been implicated not only in AD but also in other neurodegenerative diseases, including stroke, amyotrophic lateral sclerosis (ALS), and Parkinson’s disease.67 In the context of AD, TLR signaling pathways may amplify disease progression through inflammation-mediated mechanisms. For instance, TLR4 has been shown to mediate the neurotoxic effects of DAMPs associated with neuronal damage in AD, thereby worsening neuroinflammation and tissue injury.68 While stimulation of TLR9 has demonstrated beneficial effects on microglial function in AD mouse models, prolonged or excessive activation can provoke chronic inflammation and promote neurodegeneration.69 Furthermore, Aβ itself can activate the TLR4–NF-κB axis, promoting microglial activation and the release of inflammatory cytokines such as IL-1β, IL-6, and TNF-α, all of which contribute to the neuroinflammatory environment characteristic of AD.70 These findings underscore the dual role of TLRs in neuroprotection and neurodegeneration and highlight their relevance as potential therapeutic targets in the modulation of AD-related inflammation.
NLRs
NLRs are a family of cytoplasmic pattern recognition receptors that play a critical role in detecting intracellular DAMPs and modulating inflammatory responses.71 Among them, the NLR family pyrin domain-containing 3 (NLRP3) inflammasome is one of the best-characterized inflammasome complexes. It assembles in certain types of myeloid cells upon sensing microbial toxins or host-derived danger signals, leading to the activation of pro-inflammatory signaling cascades.72 The NLRP3 inflammasome is capable of responding to both PAMPs and endogenous DAMPs such as mtDNA and ATP. These molecules activate NLRP3 and promote the assembly of a multiprotein complex that recruits and activates caspase-1. Active caspase-1 cleaves pro-inflammatory cytokine precursors pro-IL-1β and pro-IL-18 into their mature, bioactive forms, which are subsequently secreted and drive robust inflammatory responses.73 Dysregulation of NLRP3 inflammasome activity has been implicated in a wide range of chronic inflammatory diseases, including atherosclerosis, gout, and metabolic syndrome.74 Therefore, NLRP3 is not only important in acute immune responses but also centrally involved in the pathogenesis of chronic diseases.
In the context of AD, overexpression and hyperactivation of the NLRP3 inflammasome have been documented in the brains of both human AD patients and animal models.75 NLRP3 has emerged as a key contributor to AD pathophysiology, particularly in mediating neuroinflammation and associated neurodegeneration. The inflammasome is activated in response to aggregated Aβ and hyperphosphorylated tau, triggering chronic inflammatory signaling and pyroptosis—a form of inflammatory cell death—ultimately accelerating cognitive decline.75 Mitochondrial dysfunction, a hallmark of AD, contributes to NLRP3 activation through the generation of mitochondrial reactive oxygen species (mtROS) and the release of mtDNA, both of which serve as potent DAMPs.76 In turn, excessive NLRP3 activation amplifies inflammatory damage within the brain. Notably, mitophagy—the selective autophagic clearance of damaged mitochondria—can suppress NLRP3 activation. Enhancing mitophagy has been shown to protect neurons and alleviate pathology in AD mouse models by limiting the activation of the NLRP3 inflammasome.77 These findings underscore the importance of NLRP3 as a molecular bridge linking mitochondrial stress to neuroinflammation in AD, and they highlight the inflammasome as a promising target for therapeutic intervention.
RAGE
The RAGE is a member of the PRR family that recognizes various DAMPs and plays a critical role in the regulation of chronic inflammation. RAGE is a multiligand receptor capable of binding to several endogenous DAMPs, including HMGB1 and HSPs, thereby activating downstream inflammatory signaling pathways.78 In addition to sensing DAMPs, RAGE can also act as a receptor for certain PAMPs, such as bacterial endotoxins, respiratory viruses, and microbial DNA.One of the main downstream pathways activated by RAGE is the NF-κB signaling cascade. RAGE-induced activation of NF-κB leads to the upregulation of pro-inflammatory cytokines (eg, TNF-α, IL-6, IL-1β), chemokines (eg, CCL2), adhesion molecules (eg, ICAM-1), and other mediators involved in inflammation. Notably, because RAGE activation typically occurs under conditions of chronic inflammation, the NF-κB activation it mediates is persistent rather than transient. This sustained inflammatory signaling contributes to the progression of chronic inflammatory diseases, including diabetes,79 atherosclerosis,80 neurodegenerative disorders,81 and various cancers.82,83 Additionally, chronic RAGE–NF-κB pathway activation has been associated with tissue injury and fibrosis, which further aggravate disease pathology.84,85
HMGB1 is believed to play a critical role in the onset and progression of neurodegenerative diseases, including AD.86 The receptor for advanced glycation end products (RAGE) contributes to the pathophysiological changes of AD by acting as a mediator of inflammation and an inducer of oxidative stress. When HMGB1 binds to RAGE, it triggers a cascade of downstream events that initiate neuroinflammatory responses, ultimately leading to the characteristic features of AD.87 Studies have demonstrated that suppression of HMGB1 or RAGE expression can attenuate neuroinflammation, reduce Aβ production and deposition, inhibit microglial activation, and halt the progression of AD.87 Moreover, inhibition of HMGB1-RAGE–mediated inflammatory responses has been shown to effectively improve memory deficits in AD mouse models.88
Mitophagy-Mediated Inflammatory Regulation and Its Relevance to AD
Mitophagy, a specialized form of autophagy, selectively eliminates dysfunctional or superfluous mitochondria and plays a pivotal role in mitochondrial quality control, cellular homeostasis, and the regulation of inflammatory responses. By removing damaged mitochondria, mitophagy prevents the release of mitochondrial DAMPs, thereby modulating inflammatory signaling.15,20 Impairment of mitophagy leads to the accumulation of defective mitochondria and subsequent DAMP release, which can trigger neuroinflammation and exacerbate the neurodegenerative processes characteristic of AD (Figure 4).
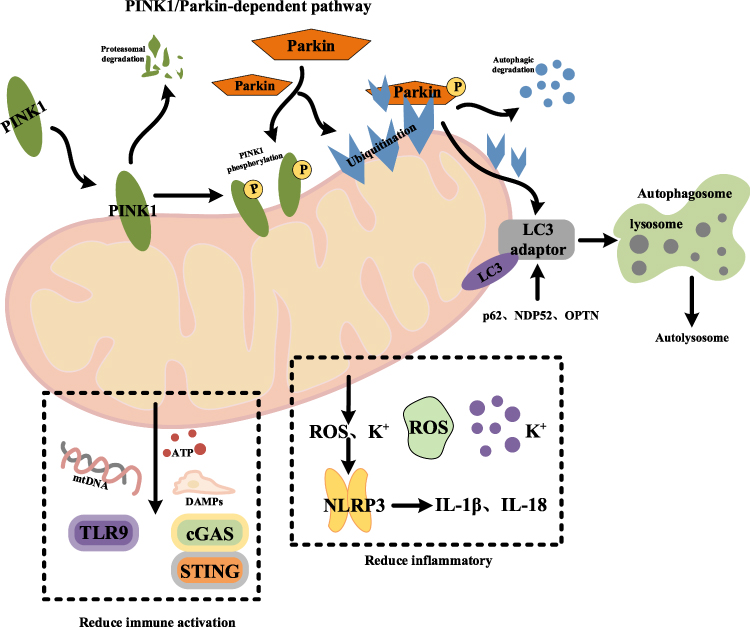 |
Figure 4 Regulatory mechanism of mitochondrial autophagy inflammation. |
Mitophagy
Mitophagy is initiated by the recognition of damaged mitochondria, a process primarily orchestrated by the PINK1/Parkin pathway. Under normal conditions, PINK1 is rapidly imported into healthy mitochondria and degraded by inner membrane proteases.15 However, when the mitochondrial membrane potential is lost, PINK1 accumulates on the outer mitochondrial membrane and recruits cytosolic Parkin. Upon activation, Parkin ubiquitinates numerous outer membrane proteins on the dysfunctional mitochondria, serving as a signal for selective autophagic clearance. Ubiquitinated mitochondria are subsequently recognized by autophagy adaptor proteins, including p62, NDP52, and OPTN, which bind to LC3 on the autophagosomal membrane via their LC3-interacting regions (LIRs). This interaction facilitates the sequestration of damaged mitochondria into autophagosomes. The autophagosomes then fuse with lysosomes to form autolysosomes, where mitochondrial components are degraded and recycled.89
Recent studies have demonstrated that moxibustion can prevent and delay the onset of AD, potentially through activation of the PINK1/Parkin signaling pathway. This activation regulates the expression of key autophagic factors, such as LC3-II, enhances mitophagic activity in the hippocampus, improves neuronal ultrastructure, and ultimately ameliorates cognitive deficits.90 Similarly, acupuncture has been shown to improve cognitive impairment and enhance learning and memory in APP/PS1 transgenic mice. Mechanistically, acupuncture activates mitophagy by promoting PINK1/Parkin-mediated ubiquitination of mitochondrial outer membrane proteins and facilitating the fusion of autophagosomes with lysosomes in the hippocampus. This process reduces hippocampal amyloid-β (Aβ) aggregation and deposition, thereby improving synaptic plasticity and cognitive function in APP/PS1 mice.91 In vitro evidence further supports these findings. Li HM using an Aβ-induced injury model in rat pheochromocytoma (PC12) cells, demonstrated that ginsenosides enhance PINK1/Parkin-dependent mitophagy, mitigating mitochondrial damage and protecting neuronal cells.92 Wang T reported that cadmium triggers mitophagy via AMP-activated protein kinase (AMPK) and PINK1/Parkin-dependent pathways, which facilitates the clearance of damaged mitochondria and suppresses neuroinflammation.93 Moreover, Wang NB found that β-asarone improves learning and memory in AD rat models by modulating PINK1/Parkin-mediated mitophagy.94
Clearance of DAMPs
An essential function of mitophagy is to prevent the release of DAMPs by selectively eliminating dysfunctional mitochondria. If damaged mitochondria are not promptly cleared, they can release mtDNA, ATP, and other DAMPs, which in turn activate multiple inflammatory signaling pathways.95 Released mtDNA acts as a potent DAMP, triggering immune responses by activating the TLR9 and cGAS-STING pathways. This activation leads to the production of type I interferons and proinflammatory cytokines. By degrading defective mitochondria, mitophagy effectively reduces mtDNA release and suppresses TLR9- and cGAS-STING-mediated immune activation.96,97 This process is particularly critical for preventing chronic inflammation and the development of autoimmune diseases. Additionally, damaged mitochondria release ATP, which not only serves as a key mediator of energy metabolism but also functions as a crucial regulator of inflammatory responses.98
Aberrant mitophagy is frequently observed during the onset and progression of AD.99 In pyramidal neurons of AD patients, immunoreactivity for mitochondrial markers localized to autophagic vacuoles—such as lipoic acid and cytochrome oxidase-1—is significantly increased, accompanied by elevated levels of mitophagic degradation products and mtDNA, suggesting enhanced mitophagic activity during the disease course.100 KUKREJA in a study using mtDNA-mutant mice, demonstrated that mitochondrial dysfunction exacerbates Aβ pathology and triggers neurodegeneration.101 Notably, increased mitophagy has been detected in neurons of AD patients even prior to overt histopathological alterations or clinical manifestations, which may represent a compensatory response secondary to mitochondrial dysfunction.102 While mitochondrial dysfunction accelerates AD progression, mitophagy alleviates disease pathology by eliminating defective mitochondria. Paradoxically, excessive mitophagy in AD has also been implicated in the release of mtDNA, activation of the cGAS-STING pathway, and subsequent promotion of neuroinflammation and neuronal death.103
Inhibition of Inflammasome Activation
Mitophagy not only suppresses inflammatory responses by clearing DAMPs but also directly regulates inflammasome activation within cells. The activation of the NLRP3 inflammasome depends critically on mitochondrial ROS and potassium (K⁺) efflux, both of which play key roles in inflammasome assembly and activation.76 Damaged mitochondria are a major intracellular source of ROS, and excessive ROS production can trigger the assembly and activation of the NLRP3 inflammasome, leading to the secretion of pro-inflammatory cytokines IL-1β and IL-18.104 Through mitophagy, cells can timely remove mitochondria that excessively produce ROS, thereby reducing ROS levels and inhibiting NLRP3 inflammasome activation.105 Moreover, mitophagy helps maintain intracellular ion homeostasis by eliminating damaged mitochondria, preventing K⁺ efflux-induced aberrant inflammasome activation. This mechanism is critical for suppressing chronic inflammation mediated by inflammasomes.
The release of mtDNA is often accompanied by mitochondrial dysfunction and increased oxidative stress. Under conditions of mitochondrial damage, excessive production of ROS exacerbates oxidative stress, leading to cellular injury and is closely associated with the formation of β-amyloid plaques and tau protein phosphorylation.106 Studies have shown that levels of NOX4, a major source of ROS, are significantly elevated in damaged astrocytes from both AD patients and APP/PS1 transgenic mouse models. The upregulation of NOX4 impairs mitochondrial respiration and ATP production in astrocytes, thereby promoting mitochondrial metabolic dysfunction.107 Furthermore, ROS generation has been observed in the vicinity of amyloid plaques in APP/PS1 transgenic mouse brains. Real-time tracking of individual neurons revealed that localized oxidative stress around plaques contributes to long-range toxicity and selective neuronal death in AD.108 Additional in vivo studies demonstrated that Aβ plaque deposition and direct application of soluble Aβ oligomers to the brain exacerbate mitochondrial oxidative stress in neurons. This oxidative stress can be mitigated by blocking mitochondrial Ca²⁺ influx or by treatment with the mitochondria-targeted antioxidant SS31.109
Activation of the cGAS-STING Signaling Pathway in AD
The cGAS-STING signaling pathway represents a critical innate immune mechanism by which cells detect cytosolic DNA. It senses DNA within the cytoplasm and subsequently activates the expression of type I interferons and other antiviral genes. This pathway plays pivotal roles in antiviral immunity, tumor immunosurveillance, and autoimmune diseases, providing cellular protection against diverse DNA-containing pathogens and regulating pathological immune responses triggered by ectopic self-DNA localization, including mtDNA in the cytosol and extranuclear chromatin.110 Upon activation, cGAS catalyzes the synthesis of the second messenger 2’3’-cGAMP from ATP and GTP, which is essential for downstream signal transduction.21 cGAS serves as the primary cytosolic DNA sensor; its recognition and binding to cytosolic DNA induce conformational changes that activate its enzymatic function. The stimulator of interferon genes (STING), an adaptor protein localized on the endoplasmic reticulum membrane, binds cGAMP, undergoes conformational rearrangement, and translocates to the Golgi apparatus.111 At the Golgi, STING forms oligomeric complexes that recruit and activate downstream kinases such as TBK1 and IKK. TBK1 phosphorylates and activates the transcription factor IRF3, which dimerizes and translocates into the nucleus to induce the expression of type I interferon genes, including IFN-β.111 Additionally, STING activates the IKK complex, promoting IκB degradation and the release of NF-κB, which translocates into the nucleus to stimulate the production of pro-inflammatory cytokines such as IL-6 and TNF-α.112 Aberrant activation of the cGAS-STING pathway, often triggered by cytosolic mtDNA, drives chronic neuroinflammation in AD, exacerbating neuronal damage (Figure 5).
 |
Figure 5 Activation mechanism of cGAS-STING signaling pathway. |
Excessive stimulation of the cGAS-STING pathway and the associated neuroinflammation have been implicated in the development and progression of various neurodegenerative diseases.113 In AD, mitochondrial dysfunction leads to the leakage of mtDNA into the cytoplasm. MtDNA acts as a potent activator of cGAS; upon sensing this extranuclear mtDNA, cGAS activates STING, initiating inflammatory responses. The activation of the mtDNA–cGAS–STING axis highlights the complex interplay between mitochondrial dysfunction and neuroinflammation in AD.114 Chung S demonstrated that the cGAS-STING pathway is activated in AD mouse models, resulting in microglial dysfunction and sterile inflammation that exacerbate AD pathology. Aβ and tau protein induce mitochondrial stress, promoting the release of DNA into the cytoplasm of microglia and activating the cGAS-STING pathway. Activation of this pathway enhances microglial NLRP3 inflammasome activation, pro-inflammatory responses, and type I interferon signaling. Inhibition of STING activity effectively modulates microglial function and ameliorates multiple pathological features of AD.115 Furthermore, treatment with TSG (tetrahydroxystilbene glucoside), a natural active compound derived from Polygonum multiflorum, has been shown to alleviate neuroinflammation via the cGAS-STING pathway, significantly improving cognitive deficits in APP/PS1 mice. This effect is partially mediated through the cGAS-STING-dependent pathway, as interference with cGAS-STING inhibitors induces NLRP3 inflammasome activation, thereby suppressing neuroinflammation.114
Conclusion and Future Perspectives
This review systematically elucidates the pivotal role of mitochondria-mediated inflammatory mechanisms in the onset and progression of AD. Mitochondrial dysfunction not only disrupts energy metabolism but also triggers chronic neuroinflammation through multiple pathways, including the release of mtDNA, MAVS signaling, accumulation of DAMPs, impaired mitophagy, and aberrant activation of the cGAS-STING signaling pathway. The sustained inflammatory state provokes a cascade of immune responses within the central nervous system, exacerbating neuronal damage and cognitive decline. Under AD-related stress, increased mitochondrial membrane permeability causes mtDNA to leak into the cytosol. Acting as a double-stranded DNA (dsDNA) ligand, mtDNA directly activates cGAS to synthesize cGAMP, thereby activating STING. The activated STING is then cleared via the lysosomal pathway to prevent sustained signaling.110 Interrelated to this is mitochondrial autophagy, which reduces the production of DAMPs from the source by clearing depolarized mitochondria and relies on lysosomal pathways to promote the degradation of activated STING, thereby activating the cGAS STING pathway; to this end, it is necessary to further advance the mechanism research of mitochondrial inflammation regulation from prospective clinical trials to true clinical applications, and at the same time develop strategies based on disease staging, strengthen individualized evaluation and dynamic adjustment.103 Therefore, as a central hub for inflammation regulation, mitochondria’s involvement in AD extends beyond structural impairment to encompass immunomodulatory functions, underscoring the potential of targeting mitochondrial inflammatory pathways as a promising strategy for therapeutic intervention and clinical translation.116,117
The regulation of mitochondrial inflammation provides a clear clinical diagnosis and treatment approach for Alzheimer’s disease. Research has found that changes in cellular free mitochondrial DNA in cerebrospinal fluid/plasma have been suggested by multiple studies to be associated with AD and can serve as an auxiliary indicator for monitoring inflammation axis activity upstream of mtDNA release.118 In terms of treatment, enhancing mitochondrial autophagy and lysosomal function is expected to reduce the supply of DAMPs and cytoplasmic mtDNA; research has shown that Urolithin A can promote mitochondrial autophagy and improve mitochondrial function, and has the potential to be used as a combination therapy.119 However, Alzheimer’s disease may exhibit different inflammatory features in mitochondrial function at different stages (early, advanced, and late). In the early stages, before significant clinical symptoms appear, mitochondrial dysfunction has already begun to manifest,120,121 with decreased energy metabolism, decreased ATP synthesis, increased ROS, accompanied by decreased mitochondrial function and accumulation of mtDNA damage,122 These changes constitute the “premonitory” initiation of inflammation in the disease process - mitochondrial abnormalities may first appear in metabolic stress or other forms, providing a basis for subsequent inflammatory responses.123 As the disease progresses, damaged mitochondria begin to release mtDAMPs, which activate microglia and astrocytes through the NLRP3 or cGAS STING signaling pathways, triggering sustained immune responses in the nervous system. At the same time, inflammatory mediators feedback and inhibit mitochondrial function, forming a clear vicious cycle of function.124,125 In the late stage, mitochondrial function is further lost, chronic inflammation persists, and the process of neurodegeneration intensifies. During this period, the interaction between mitochondria and inflammation has become highly out of control, which is a key driving factor for cognitive decline and the expansion of nerve damage.126 To this end, it is necessary to further advance the mechanism research of mitochondrial inflammation regulation from prospective clinical trials to true clinical applications, and at the same time develop strategies based on disease staging, strengthen individualized evaluation and dynamic adjustment.
Although current studies have revealed the multifaceted associations between mitochondrial inflammation and AD, several critical issues remain unresolved: (1) most mechanistic investigations are confined to animal models or cellular systems, lacking direct validation in AD patients; (2) the specific interactions between mitochondrial inflammation and classical AD pathological markers such as Aβ and tau remain unclear; (3) the differential roles of pathways including mitophagy, MAVS, and cGAS-STING across distinct brain regions and cell types have not been fully elucidated, limiting the evaluation of their potential as precise therapeutic targets. Future research should focus on several key areas: first, clarifying the dynamic changes of mitochondria-associated inflammatory signaling pathways at various stages of AD and elucidating their causal relationships with canonical pathological markers; second, developing and validating the efficacy and safety of interventions targeting mtDNA release, MAVS inhibition, or blockade of the cGAS-STING pathway; third, employing multi-omics and single-cell analyses to explore the spatial and cellular specificity of mitochondrial inflammation expression patterns and regulatory networks across different brain regions. Therefore, an in-depth investigation of the “mitochondria-inflammation axis” will enhance understanding of AD pathogenesis and progression, offering precise targets and technical support for novel intervention strategies. This will provide robust theoretical and experimental foundations for developing mitochondria-targeted anti-inflammatory therapies, thereby facilitating the translation from fundamental research to clinical applications.
Consent for Publication
All authors have given final approval of the version and agreed with the publication of this study here.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Gansu Provincial Higher Education Institutions Scientific Research Project (Project No.: 2018C-18); the Special Open Project of Gansu Provincial Traditional Chinese Medicine Research Center in 2024 (zyzx-2024-zx03, zyzx-2024-zx09); the 2024 University Teacher Innovation Fund Project (2024A-082); the Gansu University of Traditional Chinese Medicine Talent Introduction and Research Launch Fund Project (2024YJRC-06).
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Rao RV, Subramaniam KG, Gregory J, et al. Rationale for a multi-factorial approach for the reversal of cognitive decline in Alzheimer’s disease and MCI: a review. Int J Mol Sci. 2023;24(2):1659.
2. Ferri CP, Prince M, Brayne C, et al. Alzheimer’s disease international. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366(9503):2112–2117. doi:10.1016/S0140-6736(05)67889-0
3. Lin S, Zhan Y, Wang R, et al. Decoding neuroinflammation in Alzheimer’s disease: a multi-omics and AI-driven perspective for precision medicine. Front Immunol. 2025;16:1616899. doi:10.3389/fimmu.2025.1616899
4. Dhapola R, Hota SS, Sarma P, et al. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. Inflammopharmacology. 2021;29(6):1669–1681. doi:10.1007/s10787-021-00889-6
5. Swerdlow RH. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J Alzheimers Dis. 2018;62(3):1403–1416. doi:10.3233/JAD-170585
6. Perez Ortiz JM, Swerdlow RH. Mitochondrial dysfunction in Alzheimer’s disease: role in pathogenesis and novel therapeutic opportunities. Br J Pharmacol. 2019;176(18):3489–3507. doi:10.1111/bph.14585
7. Wood H. Mitochondrial dysfunction manifests in the early stages of Alzheimer disease. Nat Rev Neurol. 2020;16(5):242.
8. Roe K. An inflammation classification system using cytokine parameters. Scand J Immunol. 2021;93(2):e12970. doi:10.1111/sji.12970
9. Xie W, Zhu T, Zhou P, et al. Notoginseng leaf triterpenes ameliorates mitochondrial oxidative injury via the NAMPT-SIRT1/2/3 signaling pathways in cerebral ischemic model rats. J Ginseng Res. 2023;47(2):199–209. doi:10.1016/j.jgr.2020.11.004
10. Andrieux P, Chevillard C, Cunha-Neto E, et al. Mitochondria as a cellular hub in infection and inflammation. Int J Mol Sci. 2021;22(21):11338.
11. Twarowski B, Herbet M. Inflammatory processes in Alzheimer’s disease—pathomechanism, diagnosis and treatment: a review. Int J Mol Sci. 2023;24(7):6518. doi:10.3390/ijms24076518
12. Spangenberg EE, Green KN. Inflammation in Alzheimer’s disease: lessons learned from microglia-depletion models. Brain Behav Immun. 2017;61:1–11. doi:10.1016/j.bbi.2016.07.003
13. Gaikwad S, Senapati S, Haque MA, et al. Senescence, brain inflammation, and oligomeric tau drive cognitive decline in Alzheimer’s disease: evidence from clinical and preclinical studies. Alzheimers Dement. 2024;20(1):709–727. doi:10.1002/alz.13490
14. De Gaetano A, Solodka K, Zanini G, et al. Molecular mechanisms of mtDNA-mediated inflammation. Cells. 2021;10(11):2898. doi:10.3390/cells10112898
15. Onishi M, Yamano K, Sato M, et al. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021;40(3):e104705. doi:10.15252/embj.2020104705
16. Picca A, Calvani R, Coelho-Junior HJ, et al. Cell death and inflammation: the role of mitochondria in health and disease. Cells. 2021;10(3):537. doi:10.3390/cells10030537
17. Riley JS, Tait SW. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020;21(4):e49799. doi:10.15252/embr.201949799
18. Liu B, Gao C. Regulation of MAVS activation through post-translational modifications. Curr Opin Immunol. 2018;50:75–81. doi:10.1016/j.coi.2017.12.002
19. Ren Z, Ding T, Zuo Z, et al. Regulation of MAVS expression and signaling function in the antiviral innate immune response. Front Immunol. 2020;11:1030. doi:10.3389/fimmu.2020.01030
20. Xu Y, Shen J, Ran Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy. 2020;16(1):3–17. doi:10.1080/15548627.2019.1603547
21. Chen C, Xu P. Cellular functions of cGAS-STING signaling. Trends Cell Biol. 2023;33(8):630–648.
22. Banoth B, Cassel SL. Mitochondria in innate immune signaling. Transl Res. 2018;202:52–68. doi:10.1016/j.trsl.2018.07.014
23. Bernardi P, Pavlov E. Mitochondrial permeability transition. Cells. 2022;11(23):3866. doi:10.3390/cells11233866
24. Ouyang W, Wang S, Yan D, et al. The cGAS-STING pathway-dependent sensing of mitochondrial DNA mediates ocular surface inflammation. Signal Transduct Target Ther. 2023;8(1):371. doi:10.1038/s41392-023-01624-z
25. Elrod JW, Molkentin JD. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ J. 2013;77(5):1111–1122. doi:10.1253/circj.CJ-13-0321
26. Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: another view. Biochimie. 2002;84(2–3):153–166. doi:10.1016/S0300-9084(02)01375-5
27. Carraro M, Bernardi P. Measurement of membrane permeability and the mitochondrial permeability transition. Methods Cell Biol. 2020;155:369–379.
28. Mnatsakanyan N, Beutner G, Porter GA, et al. Physiological roles of the mitochondrial permeability transition pore. J Bioenerg Biomembr. 2017;49(1):13–25. doi:10.1007/s10863-016-9652-1
29. Calvo-Rodriguez M, Hou SS, Snyder AC, et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat Commun. 2020;11(1):2146. doi:10.1038/s41467-020-16074-2
30. Calvo-Rodriguez M, Bacskai BJ. Mitochondria and calcium in Alzheimer’s disease: from cell signaling to neuronal cell death. Trends Neurosci. 2021;44(2):136–151. doi:10.1016/j.tins.2020.10.004
31. Zhang Q, Song Q, Yu R, et al. Nano-brake halts mitochondrial dysfunction cascade to alleviate neuropathology and rescue Alzheimer’s cognitive deficits. Adv Sci. 2023;10(7):e2204596.
32. Jara C, Cerpa W, Tapia-Rojas C, et al. Tau deletion prevents cognitive impairment and mitochondrial dysfunction age associated by a mechanism dependent on cyclophilin-D. Front Neurosci. 2021;14:586710. doi:10.3389/fnins.2020.586710
33. Du H, Guo L, Fang F, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14(10):1097–1105. doi:10.1038/nm.1868
34. Du H, Yan SS. Mitochondrial permeability transition pore in Alzheimer’s disease: cyclophilin D and amyloid beta. Biochim Biophys Acta. 2010;1802(1):198–204. doi:10.1016/j.bbadis.2009.07.005
35. Abate M, Festa A, Falco M, et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin Cell Dev Biol. 2020;98:139–153. doi:10.1016/j.semcdb.2019.05.022
36. Dadsena S, King LE, García-Sáez AJ. Apoptosis regulation at the mitochondria membrane level. Biochim Biophys Acta Biomembr. 2021;1863(12):183716. doi:10.1016/j.bbamem.2021.183716
37. Gillies LA, Kuwana T. Apoptosis regulation at the mitochondrial outer membrane. J Cell Biochem. 2014;115(4):632–640. doi:10.1002/jcb.24709
38. Bender CE, Fitzgerald P, Tait SW, et al. Mitochondrial pathway of apoptosis is ancestral in metazoans. Proc Natl Acad Sci USA. 2012;109(13):4904–4909. doi:10.1073/pnas.1120680109
39. Norberg E, Orrenius S, Zhivotovsky B. Mitochondrial regulation of cell death: processing of apoptosis-inducing factor (AIF). Biochem Biophys Res Commun. 2010;396(1):95–100. doi:10.1016/j.bbrc.2010.02.163
40. Fang EF, Scheibye-Knudsen M, Chua KF, et al. Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol. 2016;17(5):308–321. doi:10.1038/nrm.2016.14
41. Zhao S, Zhao J, Zhang T, et al. Increased apoptosis in the platelets of patients with Alzheimer’s disease and amnestic mild cognitive impairment. Clin Neurol Neurosurg. 2016;143:46–50. doi:10.1016/j.clineuro.2016.02.015
42. Dhuriya YK, Sharma D. Necroptosis: a regulated inflammatory mode of cell death. J Neuroinflammation. 2018;15(1):199. doi:10.1186/s12974-018-1235-0
43. Krysko DV, Agostinis P, Krysko O, et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011;32(4):157–164. doi:10.1016/j.it.2011.01.005
44. Roos D, Seeger R, Puntel R, et al. Role of calcium and mitochondria in MeHg-mediated cytotoxicity. J Biomed Biotechnol. 2012;2012:248764. doi:10.1155/2012/248764
45. Criddle DN, Gerasimenko JV, Baumgartner HK, et al. Calcium signalling and pancreatic cell death: apoptosis or necrosis? Cell Death Differ. 2007;14(7):1285–1294. doi:10.1038/sj.cdd.4402150
46. Tao G, Liao W, Hou J, et al. Advances in crosstalk among innate immune pathways activated by mitochondrial DNA. Heliyon. 2024;10(1):e24029. doi:10.1016/j.heliyon.2024.e24029
47. Strobel S, Grünblatt E, Heinsen H, et al. Astrocyte- and microglia-specific mitochondrial DNA deletions levels in sporadic Alzheimer’s disease. J Alzheimers Dis. 2019;67(1):149–157. doi:10.3233/JAD-180661
48. Li R, Wang J, Li R, et al. ATP/P2X7-NLRP3 axis of dendritic cells participates in the regulation of airway inflammation and hyper-responsiveness in asthma by mediating HMGB1 expression and secretion. Exp Cell Res. 2018;366(1):1–15. doi:10.1016/j.yexcr.2018.03.002
49. Wilkins HM, Koppel SJ, Weidling IW, et al. Extracellular mitochondria and mitochondrial components act as damage-associated molecular pattern molecules in the mouse brain. J Neuroimmune Pharmacol. 2016;11(4):622–628. doi:10.1007/s11481-016-9704-7
50. Zhang W, Wang G, Xu ZG, et al. Lactate is a natural suppressor of RLR signaling by targeting MAVS. Cell. 2019;178(1):176–189.e15. doi:10.1016/j.cell.2019.05.003
51. Wu B, Hur S. How RIG-I like receptors activate MAVS. Curr Opin Virol. 2015;12:91–98. doi:10.1016/j.coviro.2015.04.004
52. Wu B, Huoh YS, Hur S. Measuring monomer-to-filament transition of MAVS as an in vitro activity assay for RIG-I-like receptors. Methods Mol Biol. 2016;1390:131–142.
53. Bruns AM, Horvath CM. LGP2 synergy with MDA5 in RLR-mediated RNA recognition and antiviral signaling. Cytokine. 2015;74(2):198–206. doi:10.1016/j.cyto.2015.02.010
54. Fang R, Jiang Q, Zhou X, et al. MAVS activates TBK1 and IKKε through TRAFs in NEMO dependent and independent manner. PLoS Pathog. 2017;13(11):e1006720. doi:10.1371/journal.ppat.1006720
55. Jiang M, Österlund P, Fagerlund R, et al. MAP kinase p38α regulates type III interferon (IFN-λ1) gene expression in human monocyte-derived dendritic cells in response to RNA stimulation. J Leukoc Biol. 2015;97(2):307–320. doi:10.1189/jlb.2A0114-059RR
56. Seth RB, Sun L, Ea CK, et al. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122(5):669–682. doi:10.1016/j.cell.2005.08.012
57. Zhang T, Fan J, Wen X, et al. ECSIT: biological function and involvement in diseases. Int Immunopharmacol. 2024;143(Pt 3):113524. doi:10.1016/j.intimp.2024.113524
58. Sankowski R, Strohl JJ, Huerta TS, et al. Endogenous retroviruses are associated with hippocampus-based memory impairment. Proc Natl Acad Sci USA. 2019;116(51):25982–25990. doi:10.1073/pnas.1822164116
59. Marchi S, Guilbaud E, Tait SWG, et al. Mitochondrial control of inflammation. Nat Rev Immunol. 2023;23(3):159–173. doi:10.1038/s41577-022-00760-x
60. Patel S. Danger-associated molecular patterns (DAMPs): the derivatives and triggers of inflammation. Curr Allergy Asthma Rep. 2018;18(11):63. doi:10.1007/s11882-018-0817-3
61. Zhu Y, Deng J, Nan ML, et al. The interplay between pattern recognition receptors and autophagy in inflammation. Adv Exp Med Biol. 2019;1209:79–108.
62. Donnelly CR, Chen O, Ji RR. How do sensory neurons sense danger signals? Trends Neurosci. 2020;43(10):822–838. doi:10.1016/j.tins.2020.07.008
63. Saber MM, Monir N, Awad AS, et al. TLR9: a friend or a foe. Life Sci. 2022;307:120874. doi:10.1016/j.lfs.2022.120874
64. Paudel YN, Angelopoulou E, Akyuz E, et al. Role of innate immune receptor TLR4 and its endogenous ligands in epileptogenesis. Pharmacol Res. 2020;160:105172. doi:10.1016/j.phrs.2020.105172
65. Tiwari S, Atluri V, Kaushik A, et al. Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. Int J Nanomed. 2019;14:5541–5554. doi:10.2147/IJN.S200490
66. Fiebich BL, Batista CRA, Saliba SW, et al. Role of microglia TLRs in neurodegeneration. Front Cell Neurosci. 2018;12:329. doi:10.3389/fncel.2018.00329
67. Paschon V, Takada SH, Ikebara JM, et al. Interplay between exosomes, microRNAs and toll-like receptors in brain disorders. Mol Neurobiol. 2016;53(3):2016–2028. doi:10.1007/s12035-015-9142-1
68. Calvo-Rodriguez M, García-Rodríguez C, Villalobos C, et al. Role of toll like receptor 4 in Alzheimer’s disease. Front Immunol. 2020;11:1588. doi:10.3389/fimmu.2020.01588
69. Austad SN, Ballinger S, Buford TW, et al. Targeting whole body metabolism and mitochondrial bioenergetics in the drug development for Alzheimer’s disease. Acta Pharm Sin B. 2022;12(2):511–531. doi:10.1016/j.apsb.2021.06.014
70. Wu L, Xian X, Xu G, et al. Toll-like receptor 4: a promising therapeutic target for Alzheimer’s disease. Mediators Inflamm. 2022;2022:7924199. doi:10.1155/2022/7924199
71. Shao BZ, Xu ZQ, Han BZ, et al. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. 2015;6:262. doi:10.3389/fphar.2015.00262
72. Yu JW, Lee MS. Mitochondria and the NLRP3 inflammasome: physiological and pathological relevance. Arch Pharm Res. 2016;39(11):1503–1518. doi:10.1007/s12272-016-0827-4
73. Biasizzo M, Kopitar-Jerala N. Interplay between NLRP3 inflammasome and autophagy. Front Immunol. 2020;11:591803.
74. de Zoete MR, Palm NW, Zhu S, et al. Inflammasomes. Cold Spring Harb Perspect Biol. 2014;6(12):a016287. doi:10.1101/cshperspect.a016287
75. Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674. doi:10.1038/nature11729
76. Kelley N, Jeltema D, Duan Y, et al. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20(13):3328. doi:10.3390/ijms20133328
77. Fan L, Zhaohong X, Xiangxue W, et al. Melatonin ameliorates the progression of Alzheimer’s disease by inducing TFEB nuclear translocation,promoting mitophagy, and regulating NLRP3 inflammasome activity. Biomed Res Int. 2022;2022(1):8099459. doi:10.1155/2022/8099459
78. Hudson BI, Lippman ME. Targeting RAGE signaling in inflammatory disease. Annu Rev Med. 2018;69(1):349–364. doi:10.1146/annurev-med-041316-085215
79. Khalid M, Alkaabi J, Khan MAB, et al. Insulin signal transduction perturbations in insulin resistance. Int J Mol Sci. 2021;22(16):8590. doi:10.3390/ijms22168590
80. Shu M, Cheng W, Jia X, et al. AGEs promote atherosclerosis by increasing LDL transcytosis across endothelial cells via RAGE/NF-κB/Caveolin-1 pathway. Mol Med. 2023;29(1):113. doi:10.1186/s10020-023-00715-5
81. Rodríguez-Giraldo M, González-Reyes RE, Ramírez-Guerrero S, et al. Astrocytes as a therapeutic target in Alzheimer’s disease-comprehensive review and recent developments. Int J Mol Sci. 2022;23(21):13630. doi:10.3390/ijms232113630
82. Li X, Wang M, Gong T, et al. A S100A14-CCL2/CXCL5 signaling axis drives breast cancer metastasis. Theranostics. 2020;10(13):5687–5703. doi:10.7150/thno.42087
83. Zhang J, Shao S, Han D, et al. High mobility group box 1 promotes the epithelial-to-mesenchymal transition in prostate cancer PC3 cells via the RAGE/NF-κB signaling pathway. Int J Oncol. 2018;53(2):659–671. doi:10.3892/ijo.2018.4420
84. Ma YM, Zhao LJ, Liu MR, et al. The mechanism of synergistic prevention and treatment of acute lung injury by multiple components of Ma Huang Sheng Ma Tang based on RAGE/NF-κB signaling pathway. Chin J Trad Chin Med. 2021;46:5693–5700.
85. Abdelfattah AM, Mahmoud SS, El-Wafaey DI, et al. Diacerein ameliorates cholestasis-induced liver fibrosis in rat via modulating HMGB1/RAGE/NF-κB/JNK pathway and endoplasmic reticulum stress. Sci Rep. 2023;13(1):11455. doi:10.1038/s41598-023-38375-4
86. Gaikwad S, Puangmalai N, Bittar A, et al. Tau oligomer induced HMGB1 release contributes to cellular senescence and neuropathology linked to Alzheimer’s disease and frontotemporal dementia. Cell Rep. 2021;36(3):109419. doi:10.1016/j.celrep.2021.109419
87. Paudel YN, Angelopoulou E, Piperi C, et al. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s disease (AD): from risk factors to therapeutic targeting. Cells. 2020;9(2):383. doi:10.3390/cells9020383
88. Han Y, Chen R, Lin Q, et al. Curcumin improves memory deficits by inhibiting HMGB1-RAGE/TLR4-NF-κB signalling pathway in APPswe/PS1dE9 transgenic mice hippocampus. J Cell Mol Med. 2021;25(18):8947–8956. doi:10.1111/jcmm.16855
89. Lu Y, Li Z, Zhang S, et al. Cellular mitophagy: mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics. 2023;13(2):736–766. doi:10.7150/thno.79876
90. Shi KJ. Study on the mechanism of reverse moxibustion in regulating cognitive dysfunction in Alzheimer’s disease rats based on mitochondrial autophagy PINK1/Parkin signaling pathway. Anhui Univ Trad Chin Med. 2023.
91. Fan JQ. Study on regulating mitochondrial autophagy and improving cognitive function in APP/PS1 mice by regulating the divine acupuncture method. Guangzhou Univ Tradl Chin Med. 2023.
92. Li HM, Jiang YX, Huang PL, et al. Ginsenoside Rg1 enhances mitochondrial autophagy and protects Aβ-damaged PC12 cells by activating PINK1/Parkin. Chin J Trad Chin Med. 2022;47:484–491.
93. Wang T, Zhu Q, Cao B, et al. Cadmium induces mitophagy via AMP-activated protein kinases activation in a PINK1/Parkin-dependent manner in PC12 cells. Cell Prolif. 2020;53(6):e12817. doi:10.1111/cpr.12817
94. Wang NB, Wang ZF, Han YF, et al. β-Asarone activates PINK1/Parkin mediated mitochondrial autophagy to improve cognitive and memory dysfunction in APP/PS1 mice. J Hunan Univ Trad Chin Med. 2021;41:1178–1187.
95. Peeri M, Amiri S. Protective effects of exercise in metabolic disorders are mediated by inhibition of mitochondrial-derived sterile inflammation. Med Hypotheses. 2015;85(6):707–709. doi:10.1016/j.mehy.2015.10.026
96. Gao Y, Wang Y, Liu H, et al. Mitochondrial DNA from hepatocytes induces upregulation of interleukin-33 expression of macrophages in nonalcoholic steatohepatitis. Dig Liver Dis. 2020;52(6):637–643. doi:10.1016/j.dld.2020.03.021
97. Liu Z, Wang M, Wang X, et al. XBP1 deficiency promotes hepatocyte pyroptosis by impairing mitophagy to activate mtDNA-cGAS-STING signaling in macrophages during acute liver injury. Redox Biol. 2022;52:102305. doi:10.1016/j.redox.2022.102305
98. Sharma J, Parsai K, Raghuwanshi P, et al. Emerging role of mitochondria in airborne particulate matter-induced immunotoxicity. Environ Pollut. 2021;270:116242.
99. Q CAI, Y JEONGY. Mitophagy in Alzheimer’s disease and other age-related neurodegenerative diseases. Cells. 2020;9(1):150.
100. Moreirap I, Siedlaks L, Wang X, et al. Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy. 2007;3(6):614–615. doi:10.4161/auto.4872
101. Kukreja L, Kujoth GC, Prolla TA, Van Leuven F, Vassar R. Increased mtDNA mutations with aging promotes amyloid accumulation and brain atrophy in the APP/Ld transgenic mouse model of Alzheimer’s disease. Mol Neurodegener. 2014;9(1):16. doi:10.1186/1750-1326-9-16
102. Kerr JS, Adriaanse BA, Greig NH, et al. Mitophagy and Alzheimer’s Disease: cellular and molecular mechanisms. Trends Neurosci. 2017;40(3):151–166. doi:10.1016/j.tins.2017.01.002
103. Kalani K, Chaturvedi P, Chaturvedi P, et al. Mitochondrial mechanisms in Alzheimer’s disease: quest for therapeutics. Drug Discov Today. 2023;28(5):103547. doi:10.1016/j.drudis.2023.103547
104. Zheng X, Wan J, Tan G. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in diabetic retinopathy. Front Immunol. 2023;14:1151185. doi:10.3389/fimmu.2023.1151185
105. Zhao Y, Qiu C, Wang W, et al. Cortistatin protects against intervertebral disc degeneration through targeting mitochondrial ROS-dependent NLRP3 inflammasome activation. Theranostics. 2020;10(15):7015–7033. doi:10.7150/thno.45359
106. Dhiman A, Handa M, Ruwali M, et al. Recent trends of natural based therapeutics for mitochondria targeting in Alzheimer’s disease. Mitochondrion. 2022;64:112–124. doi:10.1016/j.mito.2022.03.006
107. Park MW, Cha HW, Kim J, et al. NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer’s diseases. Redox Biol. 2021;41:101947. doi:10.1016/j.redox.2021.101947
108. Xie H, Hou S, Jiang J, et al. Rapid cell death is preceded by amyloid plaque-mediated oxidative stress. Proc Natl Acad Sci USA. 2013;110(19):7904–7909. doi:10.1073/pnas.1217938110
109. Calvo-Rodriguez M, Kharitonova EK, Snyder AC, et al. Real-time imaging of mitochondrial redox reveals increased mitochondrial oxidative stress associated with amyloid β aggregates in vivo in a mouse model of Alzheimer’s disease. Mol Neurodegener. 2024;19(1):6. doi:10.1186/s13024-024-00702-2
110. Kim J, Kim H-S, Chung JH. Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp Mol Med. 2023;55(3):510–519. doi:10.1038/s12276-023-00965-7
111. Gui X, Yang H, Li T, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. 2019;567(7747):262–266. doi:10.1038/s41586-019-1006-9
112. Balka KR, Louis C, Saunders TL, et al. TBK1 and IKKε act redundantly to mediate STING-induced NF-κB responses in myeloid cells. Cell Rep. 2020;31(1):107492. doi:10.1016/j.celrep.2020.03.056
113. Govindarajulu M, Ramesh S, Beasley M, et al. Role of cGAS–sting signaling in Alzheimer’s disease. Int J Mol Sci. 2023;24(9):8151. doi:10.3390/ijms24098151
114. Gao D, Hao J-P, Li B-Y, et al. Tetrahydroxy stilbene glycoside ameliorates neuroinflammation for Alzheimer’s disease via cGAS-STING. Eur J Pharmacol. 2023;953:175809. doi:10.1016/j.ejphar.2023.175809
115. Chung S, Jeong J-H, Park J-C, et al. Blockade of STING activation alleviates microglial dysfunction and a broad spectrum of Alzheimer’s disease pathologies. Exp Mol Med. 2024;56(9):1936–1951. doi:10.1038/s12276-024-01295-y
116. Wang W, Zhao F, Ma X, et al. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol Neurodegener. 2020;15(1):30. doi:10.1186/s13024-020-00376-6
117. Wilkins HM, Swerdlow RH. Mitochondrial links between brain aging and Alzheimer’s disease. Transl Neurodegener. 2021;10(1):33. doi:10.1186/s40035-021-00261-2
118. Cervera-Carles L, Alcolea D, Estanga A, et al. Cerebrospinal fluid mitochondrial DNA in the Alzheimer’s disease continuum. Neurobiol Aging. 2017;53:
119. Singh A, D’Amico D, Andreux PA, et al. Urolithin A improves muscle strength, exercise performance, and biomarkers of mitochondrial health in a randomized trial in middle-aged adults. Cell Rep Med. 2022;3(5):100633. doi:10.1016/j.xcrm.2022.100633
120. Reddy PH, Oliver DM. Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells. 2019;8(5):488. doi:10.3390/cells8050488
121. Li Y, Xia X, Wang Y, et al. Mitochondrial dysfunction in microglia: a novel perspective for pathogenesis of Alzheimer’s disease. J Neuroinflammation. 2022;19(1):248. doi:10.1186/s12974-022-02613-9
122. Wang S, Liao Z, Zhang Q, et al. Mitochondrial dysfunction in Alzheimer’s disease: a key frontier for future targeted therapies. Front Immunol. 2025;15:1484373. doi:10.3389/fimmu.2024.1484373
123. Pszczołowska M, Walczak K, Miśków W, et al. Mitochondrial disorders leading to Alzheimer’s disease-perspectives of diagnosis and treatment. Geroscience. 2024;46(3):2977–2988. doi:10.1007/s11357-024-01118-y
124. Galizzi G, Di Carlo M. Mitochondrial DNA and Inflammation in Alzheimer’s Disease. Curr Issues Mol Biol. 2023;45(11):8586–8606. doi:10.3390/cimb45110540
125. Vontell RT, de Rivero Vaccari JP, Sun X, et al. Identification of inflammasome signaling proteins in neurons and microglia in early and intermediate stages of Alzheimer’s disease. Brain Pathol. 2023;33(4):e13142. doi:10.1111/bpa.13142
126. Mary A, Barale S, Eysert F, et al. Hampered AMPK-ULK1 cascade in Alzheimer’s disease (AD) instigates mitochondria dysfunctions and AD-related alterations which are alleviated by metformin. Alzheimers Res Ther. 2025;17(1):127. doi:10.1186/s13195-025-01772-0
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Protective Effects of Pioglitazone on Cognitive Impairment and the Underlying Mechanisms: A Review of Literature
Alhowail A, Alsikhan R, Alsaud M, Aldubayan M, Rabbani SI
Drug Design, Development and Therapy 2022, 16:2919-2931
Published Date: 31 August 2022
Non-Coding RNA in Microglia Activation and Neuroinflammation in Alzheimer’s Disease
He C, Li Z, Yang M, Yu W, Luo R, Zhou J, He J, Chen Q, Song Z, Cheng S
Journal of Inflammation Research 2023, 16:4165-4211
Published Date: 21 September 2023
Advances in the Understanding of the Correlation Between Neuroinflammation and Microglia in Alzheimer’s Disease
Yan H, Wang W, Cui T, Shao Y, Li M, Fang L, Feng L
ImmunoTargets and Therapy 2024, 13:287-304
Published Date: 12 June 2024
Prevention and Treatment Strategies for Alzheimer’s Disease: Focusing on Microglia and Astrocytes in Neuroinflammation
Zhang S, Gao Z, Feng L, Li M
Journal of Inflammation Research 2024, 17:7235-7259
Published Date: 13 October 2024
Jiawei Qifuyin Enhances Immunity and Improves Cognitive Impairment in APP/PS1 Mice Through Modulation of Neuroinflammatory Pathways
Cheng J, Li W, Wang L, Gao Y, Ma Y, Zhou M, Yang T, Yue C, Yan L, Lyu Y
Journal of Inflammation Research 2024, 17:9021-9040
Published Date: 18 November 2024