Back to Journals » Journal of Inflammation Research » Volume 16
Non-Coding RNA in Microglia Activation and Neuroinflammation in Alzheimer’s Disease
Authors He C, Li Z, Yang M, Yu W, Luo R, Zhou J, He J, Chen Q, Song Z, Cheng S
Received 16 June 2023
Accepted for publication 12 September 2023
Published 21 September 2023 Volume 2023:16 Pages 4165—4211
DOI https://doi.org/10.2147/JIR.S422114
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tara Strutt
Chunxiang He,1,2 Ze Li,1,2 Miao Yang,1,2 Wenjing Yu,1,2 Rongsiqing Luo,1,2 Jinyong Zhou,1,2 Jiawei He,1,2 Qi Chen,1,2 Zhenyan Song,1,2 Shaowu Cheng1,2
1School of Integrated Chinese and Western Medicine, Hunan University of Chinese Medicine, Changsha, Hunan, People’s Republic of China; 2Key Laboratory of Hunan Province for Integrated Traditional Chinese and Western Medicine on Prevention and Treatment of Cardio-Cerebral Diseases, College of Integrated Traditional Chinese and Western Medicine, Hunan University of Chinese Medicine, Changsha, Hunan, People’s Republic of China
Correspondence: Zhenyan Song; Shaowu Cheng, Tel +86 073188458257, Email [email protected]; [email protected]
Abstract: Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by complex pathophysiological features. Amyloid plaques resulting from extracellular amyloid deposition and neurofibrillary tangles formed by intracellular hyperphosphorylated tau accumulation serve as primary neuropathological criteria for AD diagnosis. The activation of microglia has been closely associated with these pathological manifestations. Non-coding RNA (ncRNA), a versatile molecule involved in various cellular functions such as genetic information storage and transport, as well as catalysis of biochemical reactions, plays a crucial role in microglial activation. This review aims to investigate the regulatory role of ncRNAs in protein expression by directly targeting genes, proteins, and interactions. Furthermore, it explores the ability of ncRNAs to modulate inflammatory pathways, influence the expression of inflammatory factors, and regulate microglia activation, all of which contribute to neuroinflammation and AD. However, there are still significant controversies surrounding microglial activation and polarization. The categorization into M1 and M2 phenotypes may oversimplify the intricate and multifaceted regulatory processes in microglial response to neuroinflammation. Limited research has been conducted on the role of ncRNAs in regulating microglial activation and inducing distinct polarization states in the context of neuroinflammation. Moreover, the regulatory mechanisms through which ncRNAs govern microglial function continue to be refined. The current understanding of ncRNA regulatory pathways involved in microglial activation remains incomplete and may be influenced by spatial, temporal, and tissue-specific factors. Therefore, further in-depth investigations are warranted. In conclusion, there are ongoing debates and uncertainties regarding the activation and polarization of microglial cells, particularly concerning the categorization into M1 and M2 phenotypes. The study of ncRNA regulation in microglial activation and polarization, as well as its mechanisms, is still in its early stages and requires further investigation. However, this review offers new insights and opportunities for therapeutic approaches in AD. The development of ncRNA-based drugs may hold promise as a new direction in AD treatment.
Keywords: Alzheimer’s disease, non-coding RNA, neuroinflammation, microglia activation, miRNA, circRNA, lncRNA
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by intricate pathophysiological features, affecting 47 million individuals worldwide, with a projected increase to 131 million by 2050.1 The rising global population of individuals aged 60 and above, accounting for 17.9% of the total population, and a dementia prevalence rate of approximately 4.2% by the end of 2018,2 pose significant challenges in healthcare and medical services, particularly concerning neurodegenerative diseases such as AD. Cognitive impairment is a pathological condition associated with AD, and it is mainly due to the loss of neurotransmitter acetylcholine (ACh) from the neurons of the central nervous system. The ACh is not only acting as a parasympathetic neurotransmitter but also strengthening the synaptogenesis of active neurons to modulate memory and learning in humans Oxidative stress is a significant contributor to memory impairments, resulting from an imbalance in antioxidant enzymes and excessive production of reactive oxygen species (ROS). An acetylcholinesterase (AChE) inhibitor exhibiting potent antioxidant properties has emerged as a promising therapeutic candidate for the treatment of dementia by enhancing learning and memory.3,4 The primary neuropathological criteria for AD diagnosis involve the presence of neuritic plaques with extracellular amyloid deposits and neurofibrillary tangles consisting of intracellular hyperphosphorylated tau accumulation.5 Besides, mitochondrial dysfunction along with mitophagy significantly contributes to the accumulation of Aβ fibrils and hyperphosphorylated tau protein tangles which lead to synaptic dysfunctions and cognitive impairments such as memory loss through reactive oxygen species (ROS)-mediated pathway.6 These pathological hallmarks are typically accompanied by heightened neuroinflammation, which represents an attempt to counteract the underlying pathology.7 Notably, neuroinflammation is closely intertwined with the regulatory role of microglia, the immune cells within the brain.8,9 Similarly, emerging evidence suggests that astroglia also play a vital role in neuroinflammation and AD progression. While both microglia and astroglia contribute to neuroinflammation and the progression of AD, their specific roles may differ. Microglia are known to be involved in the clearance of amyloid beta plaques through phagocytosis and are considered key players in the immune response within the brain. Astroglia, on the other hand, is thought to be more involved in the regulation of neuronal homeostasis, synaptic plasticity, and the maintenance of the blood-brain barrier integrity.10 This review explores the impact of non-coding RNAs (ncRNAs) on the pathogenesis of AD by regulating microglia activation. It suggests that ncRNAs play a role in AD progression by modulating microglia activation in the context of neuroinflammation, amyloid β-protein (Aβ) deposition, and neurofibrillary tangles. Initially, a concise overview of microglia activation and its importance in AD is provided, along with an introduction to ncRNAs. Subsequently, the regulatory roles of different types of ncRNAs, including microRNAs (miRNAs), Circular RNAs (circRNAs), and long non-coding RNAs (lncRNAs), in microglia activation are reviewed, along with their underlying mechanisms. Finally, the review discusses the opportunities and challenges associated with using ncRNA-mediated regulation of microglia activation in future AD research and drug development. Overall, this review primarily focuses on investigating the role of ncRNAs in microglia activation and neuroinflammation in AD, offering potential novel directions for the diagnosis and treatment of AD.
Microglia Activation and Pathological Changes of AD
Microglia, as pivotal immune cells within the brain, actively survey the environment and perform essential roles in central nervous system tissue maintenance, injury response, and pathogen defense.8,11 They also contribute to the development and refinement of neural circuits through the phagocytosis and elimination of unwanted neurons and synapses.12 Microglia activation serves as a critical link in the neuroinflammatory response.13 Enhanced microglial activation and the accumulation of inflammatory mediators are observed in the pathogenesis of various neurodegenerative diseases.14,15 Similar to macrophages, activated microglia exhibit two consecutive functional states: the classical activation (M1) and alternative activation (M2) states. Classical microglia release pro-inflammatory cytokines such as TNF-α, IL-1β, TLR2, TLR416 superoxide, NO, and ROS, whereas M2 microglia release anti-inflammatory cytokines such as IL-4, IL-13, IL-10, Arg-1, and TGF-β to counteract the pro-inflammatory response.17–19 Classical microglial activation drives the neuroinflammatory response and exerts deleterious effects on neurons, while the alternative activation state, which is beneficial, plays a crucial role in tissue maintenance and repair.20 The balance between these phenotypes significantly influences disease progression in the central nervous system (CNS).
Microglia Activation and Neuroinflammation
Neuroinflammation plays a pivotal role in the intricate pathogenesis of AD, characterized by excessive microglial activation and the subsequent release of numerous inflammatory factors21 (Figure 1). As frontline defenders in the CNS, activated microglia undergo morphological, genetic, and functional changes. While they release inflammatory mediators, excessive activation can lead to harm and contribute to the progression of neurodegenerative diseases.22 Modulating microglial polarization and ameliorating neuroinflammation have emerged as novel therapeutic approaches for AD treatment.23–25 This “double-edged sword” nature of microglia has spurred extensive research on the neuroprotective effects of modulating microglial function. For instance, TREM2 is one of the most critical AD risk genes found in microglia. The toxic activity of Aβ species in neurons is reduced because of its compaction by TREM2 into dense plaques. Thus, this Trem2-dependent compaction of Aβ into dense plaques shows neuroprotective activity.26 TREM2 has also been shown to regulate the transition of microglia from the M1 to the M2 phenotype, thereby reducing neuroinflammation in AD.27,28 Besides, multiple molecular pathways, including STAT, NF-κB and interferon regulatory factor (IRF), are involved in the regulation of the M1/M2 phenotypic transitions.29–31 Interleukin-10 (IL-10) is a key cytokine that induces M2 polarization by suppressing M1-associated cytokine production and promoting the expression of M2 markers. IL-10 deficiency may promote the polarization of microglia into M1-prone phenotype under pro-inflammatory conditions.32 KLF4 is an anti-inflammation transcriptional regulator, which has been reported to play a key role in regulating microglial polarization. KLF4 was found to cooperate with STAT6 to induce an M2 genetic program and inhibit M1 targets via sequestration of coactivators required for NF-κB activation.33 Another research also suggested that intra-nuclear SphK2-S1P axis might facilitate the transformation of microglial polarization from the M1 phenotype to the M2 phenotype, by inhibiting KLF4 to interact with HDAC1 and suppressing KLF4 deacetylation.34 Flibanserin (Flib), a 5HT1A agonist, can modulate microglia phenotype switching from M1 to M2 via PI3K/AKT/KLF4 signaling.35 Low-intensity pulsed ultrasound (LIPUS) treatment prevented M1 polarization of microglia and enhanced or sustained M2 polarization by regulating M1/M2 polarization through STAT1/STAT6/PPARγ signaling pathways.36 Therefore, modulating both polarization states could effectively impact neuroinflammation.
 |
Figure 1 The mechanisms of neuroinflammation mediated by microglia activation in AD. |
Microglia Activation and Aβ Deposition
Microglia have emerged as a critical cell type in the context of neurodegenerative diseases.37,38 In AD, microglial activation plays a significant role in promoting the phagocytosis and clearance of Aβ, consequently reducing amyloid plaque deposition.39,40 Notably, the activation and distribution patterns of microglial cells exhibit correlations with the amount and distribution of amyloid deposits in brain regions such as the parietal, frontal, and temporal cortices. Therefore, the level of microglial activation changes in accordance with the increase in amyloid accumulation.41 In a study conducted by Fan et al, a substantial increase in microglial activation was observed in most AD participants, which began at a high baseline level and continued to rise over time. Moreover, this increase in microglial activation was found to be associated with amyloid accumulation and a decline in cerebral metabolic rate in specific brain regions over time.42 Aβ oligomers activate pattern recognition receptors on microglia, triggering inflammatory responses and morphological changes.43 Activated microglia engage in the phagocytosis of damaged cells and Aβ aggregates.44 TLR2, TLR4, and the NF-κB pathway play pivotal roles in microglial activation and neurodegeneration in AD.45,46 The NF-κB pathway upregulates the expression of β-secretase 1 (BACE1) and promotes amyloid precursor protein (APP) splicing, leading to Aβ generation during microglial activation.47 Besides, transcription factor EB (TFEB) is also an important agent that plays a vital role in redox-dependent and autophagy regulation and is activated by several different stimuli such as cytokines, lipopolysaccharide (LPS), and oxidative stress during inflammatory events in neurodegeneration. TFEB enhances lysosomal biogenesis and contributes to an increased Aβ clearance and reduced Aβ generation in both astrocytes and neurons.48 However, the ability of Aβ to activate microglia in vivo has not been definitively demonstrated, and several studies have observed an absence of microglial activation in human brains with very high Aβ loads.49 Interleukin-33 (IL-33) reduces soluble Aβ levels and amyloid plaque deposition by promoting microglial recruitment and enhancing Aβ phagocytosis.50 Transforming the phenotype of microglia from an inflammatory state to an anti-inflammatory state in AD mice has been shown to mitigate the detrimental effects caused by Aβ aggregation and facilitate its clearance.51
Microglia Activation and Neurofibrillary Tangles
Tau, a highly soluble hydrophilic protein, undergoes detachment from microtubules and accumulates, forming intracellular hyperphosphorylated aggregates or inclusions, such as neurofibrillary tangles (NFTs) observed in AD brains. These structures disrupt cellular function, leading to neuronal cell death and neurodegeneration.52 TFEB promotes lysosomal exocytosis and subsequent astroglia uptake of tau and TFEB-mediated glial uptake of extracellular tau prevents the cell-to-cell transfer of the NFT-like pathology.48 Microglia, through the engulfment of synapses potentially via a complement-dependent process, can induce synapse loss. Additionally, they can exacerbate tau pathology and release inflammatory factors that directly cause neuronal damage or activate neurotoxic astrocytes.53 In the early stages of tau degeneration, specific pro-inflammatory cytokines including IL-1, IL-6, and TNF-α, as well as the chemokine fractalkine (CX3CL1), can modify the patterns of tau phosphorylation. Moreover, these cytokines can impact the function and structure of tau proteins.54 Microglia-specific neuroinflammation accelerates tau pathology and contributes to neurodegeneration. For instance, disrupting the anti-inflammatory CX3CL1 receptor CX3CR1 and enhancing the pro-inflammatory activation of microglia can increase tangle formation.55 Studies by Maphis et al56 demonstrated that the absence of microglial fractalkine receptor CX3CR1 hastened tau pathology and resulted in memory impairment. Utilizing hTauCx3cr1(−/−) mice, they further established that changes in microglial morphology can modify the brain microenvironment, induce tau pathology in a cell-autonomous manner, and facilitate the spread of misfolded tau proteins between anatomically connected brain regions. Furthermore, reactive microglia alone are sufficient to drive neuronal tau phosphorylation/aggregation, leading to the propagation of tau pathology in the brain.57,58 Additionally, p38MAPK is implicated in the pathogenesis of AD as it promotes tau phosphorylation,59,60 thereby reducing synaptic plasticity61 and activating microglia to release pro-inflammatory factors.62
Non-Coding RNA and Microglia Activation and Neuroinflammation
In addition to mRNAs, there exist various RNA species known as ncRNAs, which include intron RNAs, miRNAs, lncRNAs, circRNAs, and extracellular RNAs. Unlike mRNAs, ncRNAs do not have the clear potential to encode proteins or peptides, hence their classification as non-coding.63 Functionally, ncRNAs can be categorized into housekeeping ncRNAs and regulatory ncRNAs. Housekeeping ncRNAs, such as transfer (t)RNA, ribosomal (r)RNA, and small nuclear (sn)RNA, are essential components involved in everyday cellular maintenance. On the other hand, regulatory ncRNAs are expressed in specific cell types and exhibit regulatory functions in response to developmental cues, internal conditions, and environmental stimuli.64 Besides, PIWI-interacting RNAs (piRNAs) are ncRNAs with 24–32 nts that interact with piwi proteins and function in a complex to regulate cellular activities through RNA silencing. Recently, specific dysregulated piRNAs have been reported to be associated with the function of AD-related biological pathways, playing important roles in apoptosis and oxidative stress and in regulating Aβ levels in individuals with AD. Kim et al have reported that the piwi-piRNA pathway may govern neuronal function in many animals, affecting axonal regeneration and memory loss.65 However, piRNAs are poorly conserved, even between closely related species, and are tissue specific. Therefore, relatively little knowledge is available on the potential roles of piRNAs in species and/or the brain, even in neurodegenerative diseases.66 Similarly, there is limited research on the role of piRNAs in microglia activation and neuroinflammation in AD. Therefore, this review focuses on regulatory ncRNAs, including miRNAs, circRNAs, and lncRNAs.
The presence of thousands of unique ncRNA sequences within cells has shifted the understanding of ncRNAs from being considered useless transcription products to being recognized as functional regulatory molecules. It has been discovered that ncRNAs play crucial roles in various cellular processes, such as chromatin remodeling, transcription, post-transcriptional modifications, and signal transduction. Furthermore, they have been found to have critical regulatory functions in development and disease processes. Notably, ncRNAs have emerged as important regulators of oncogenic drivers and tumor suppressors across different types of cancers.67 Substantial efforts have been dedicated to targeting these ncRNAs for therapeutic purposes. In the past five years alone, more than 100 antisense oligonucleotide-based therapies targeting ncRNAs have undergone Phase I clinical trials, with a quarter of them advancing to Phase II/III trials.68
miRNA in Microglia Activation and Neuroinflammation
miRNAs, which are short RNA molecules ranging from 19 to 25 nucleotides in length, play a crucial role in post-transcriptional gene silencing. A single miRNA can target multiple mRNAs, thereby influencing the expression of functionally interconnected genes.69 Ongoing research aims to further understand the mechanisms underlying miRNA-mediated gene silencing.70 These miRNAs predominantly regulate gene expression by binding to the 3’-untranslated region (UTR) of mRNA molecules in a Dicer-dependent manner, resulting in the repression of target gene expression.71 Their targets are not limited to mRNAs but also include lncRNAs, pseudogenes, and circRNAs. Moreover, miRNAs can be encapsulated in exosomes or microvesicles and released into the extracellular environment, facilitating long-distance cell-cell communication.72
The intricate regulation of miRNA expression occurs at multiple levels and is influenced by factors such as cell type, the physiological state of the organism, and various external stimuli. The biogenesis of miRNAs is tightly controlled in terms of temporal and spatial aspects, and dysregulation of miRNA expression is associated with numerous human diseases, particularly cancer.73,74 The regulatory targets and functions of miRNAs have also been identified in various human diseases, including neurodegeneration, autoimmune diseases, cancer, and stroke.75 In the context of microglial activation, miRNAs participate in the regulation of microglia-mediated neuroinflammation by targeting relevant cellular signaling pathways, such as the NF-κB signaling pathway.76 Notable examples include miR-155, miR-146a, and miR-124, which are involved in microglial activation. Profiling miRNA expression using techniques like miRNA profiling has become a widely utilized approach for analyzing miRNA expression patterns in different tissues and diseases. It holds great potential for identifying new therapeutic targets, developing biomarkers, and predicting drug responses in personalized medicine. In the diagnosis and prognosis of various conditions, including AD and cancer, miRNA profiling offers promising applications.77–79 Table 1 summarizes the mechanisms of miRNAs in microglia-mediated neuroinflammation, while Figure 2 provides a visual representation of these mechanisms.
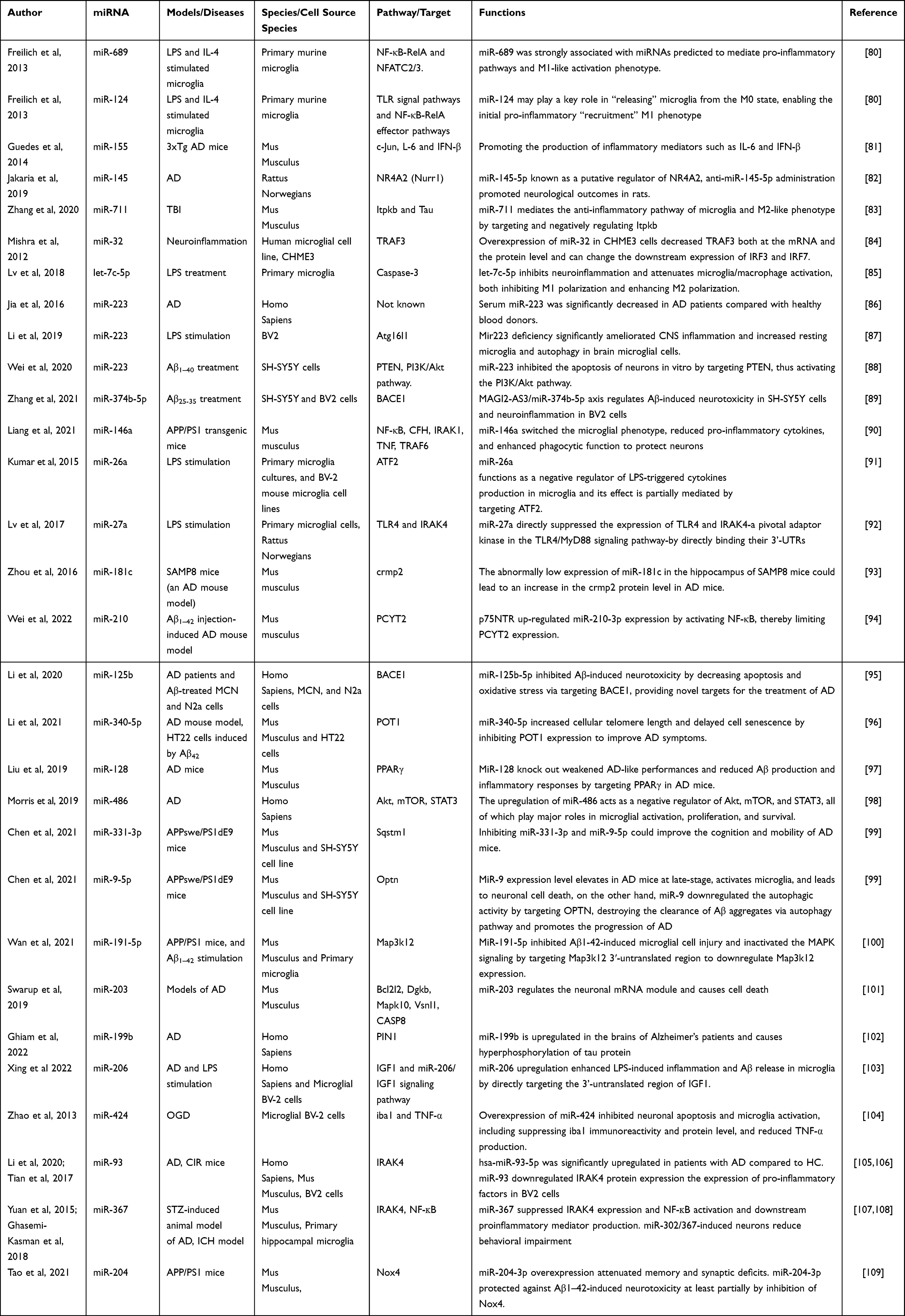 |
Table 1 Summary of the Mechanisms of miRNAs in Microglia-Mediated Neuroinflammation |
 |
Figure 2 Regulatory role of several miRNAs in microglia activation and potential mechanisms. |
miR-689
miR-689 emerges as a highly significant miRNA predicted to play a key role in mediating pro-inflammatory pathways and promoting an M1-like activation phenotype. Decreased expression of miR-689 is associated with the disinhibition of several canonical inflammatory pathways. Notably, miR-689 has the potential to modulate the transcriptional networks of various pro-inflammatory pathways, including NF-κB-RelA and NFATC2/3. Stimulation of cultured microglia with pro-inflammatory signals such as lipopolysaccharide (LPS) leads to a reduction in miR-689 expression. The downregulation of miR-689 serves as a trigger, releasing microglia from their resting (M0) state and promoting the activation of canonical TLR signaling pathways and NF-κB-RelA effector pathways, thus facilitating the initial pro-inflammatory “recruitment” of the M1 phenotype.80
miR-711
miR-711 represents a highly relevant miRNA associated with the modulation of anti-inflammatory pathways and the promotion of an M2-like activation phenotype. The cytokine interleukin-4 (IL-4) is known to selectively induce M2 activation and has been found to stimulate the expression of miR-711. Notably, a reduction in miR-711 levels may exert regulatory effects on inflammatory signaling pathways and the peroxisome proliferator-activated receptor-gamma (PPAR-γ) pathway. By mediating the anti-inflammatory pathway and facilitating an M2-like activation phenotype in microglia, miR-711 plays a critical role in modulating microglial function.80
Moreover, miR-711 has been demonstrated to target and inhibit the activity of 1,4,5-trisphosphate 3-kinase B (Itpkb), leading to the repression of Tau phosphorylation and an increase in the M2/M1 ratio. In a notable study, the administration of microglia-derived extracellular vesicles (EVs) loaded with miR-711 was found to effectively alleviate neurodegenerative changes and cognitive dysfunction in AD. These EVs mediated the hyperphosphorylation of the Tau protein by targeting the Itpkb pathway.83
miR-32
miR-32 has emerged as a key regulator of microglia-mediated neuroinflammation, contributing to the process of neurodegeneration.110 Tumor necrosis factor-receptor-associated factor 3 (TRAF3) has been identified as a direct target of miR-32. Overexpression of miR-32 in CHME3 cells resulted in a decrease in both the mRNA and protein levels of TRAF3. Notably, TRAF3 levels play a crucial role in regulating the expression of interferon regulatory factor 3 (IRF3) and IRF7 in microglial cells. Upon overexpression of miR-32 and subsequent application of anti-miR-32, the expression levels of IRF3 and IRF7 showed an inverse relationship with TRAF3 expression levels. Thus, miR-32 functions by suppressing TRAF3 expression and subsequently modulating the downstream expression of IRF3 and IRF7.84
Furthermore, the inhibition of miR-32-5p has been shown to ameliorate the production of inflammatory cytokines in microglia stimulated with lipopolysaccharide LPS. This effect is attributed to miR-32’s direct repression of dual specificity phosphatase 5 (Dusp5), a protein known to be involved in neuropathic pain and neuroinflammation. These findings provide evidence that miR-32 plays a regulatory role in microglial inflammatory responses.111
miR-145
miR-145 plays a potential role in regulating the differentiation of peripheral monocytes/macrophages and promoting the M2-skewing phenotype.80 Specifically, miR-145-5p has been shown to directly bind to the 3’-UTR) of the mRNA encoding Nurr1, leading to the inhibition of Nurr1-mediated microglial activation and subsequent alleviation of neuronal injury.112 Nurr1, also known as NR4A2, is expressed in microglia and astrocytes, and it possesses the ability to suppress the expression of proinflammatory mediators, thereby offering protection against inflammation-induced neuronal death. Dysregulated expression of NR4A2 has been implicated in the progression of AD, and activation of this protein holds the potential to enhance cognitive function. miR-145-5p has been identified as a key regulator of NR4A2, and in an experimental model of middle cerebral artery occlusion/reperfusion, the administration of anti-miR-145-5p resulted in improved neurological outcomes in rats. Given the significant involvement of NR4A2 in neuroinflammation and neuronal cell death, particularly in the context of neurodegenerative disorders, targeting this molecule holds promise for neuroprotective therapy.82
miR-155
miR-155, the most significantly upregulated miRNA, plays a regulatory role in the signal transducer and activator of transcription 3 (STAT3) signaling pathway, thereby enabling the late-phase response to M1-skewing stimulation. Stimulation of cultured microglia with pro-inflammatory signals, such as LPS, leads to an increase in miR-155 expression.80 The expression of miRNA-155 is dependent on TLR4/NF-κB pathways, its expression is increased by TLR4 ligands such as TNF-α, IL-1β, interferons, and LPS. Upon activation of the TLR4 receptor, proinflammatory signaling cascades cause translocation of NF-κB into the nucleus. This activation increases miRNA-155 expression and contributes to the regulation of the strength and duration of inflammation. The inhibition of miRNA-155 expression is substantially reversed after Nrf2 knockdown. Remarkably, there is competition between Nrf2 and NF-κB at DNA binding level. Notably, miR-155 acts as a pro-inflammatory miRNA in microglia through both TLR4/NF-κB pathways and Nrf2 signaling pathway.113 Activation of NF-κB can promote M1 polarization and inhibit the anti-inflammatory M2 phenotype in microglia. While activation of Nrf2 can promote M2 polarization and inhibit the pro-inflammatory M1 phenotype in microglia.
Furthermore, miR-155 is upregulated following Japanese encephalitis virus (JEV) infection, and it exerts inhibitory effects on the expression of Pellino E3 ubiquitin ligases (PELI1) while upregulating the expression of TNF receptor-associated factor 3 (TRAF3), a negative regulator of NF-κB p65 activity. This leads to the inhibition of NF-κB p65 activation and the suppression of pro-inflammatory response and microglial polarization, ultimately facilitating viral replication.114 The upregulation of miR-155 in macrophages can also be induced by interferon (IFN)-γ/β through autocrine and paracrine pathways of TNF-α.115 Moreover, miR-155 promotes the expression of TNF-α, highlighting its crucial role in the regulation of the innate immune response.116
In the context of AD, miR-155 levels are significantly upregulated in the brains of 3xTg AD mice, coinciding with the activation of microglia and astrocytes. This effect is attributed to the miR-155-dependent downregulation of suppressor of cytokine signaling 1 (SOCS-1). Studies by Guedes et al81,117 have suggested that miR-155 and c-Jun are early upregulated in 3xTg AD mice as well as Aβ-activated microglia and astrocytes, leading to the production of inflammatory mediators such as IL-6 and IFN-β.118 These miRNAs are collectively referred to as “inflamma-miRs”.119 Moreover, inhibiting the expression of miR-155 can attenuate the upregulation of TNF-α, IL-1β, IL-6, and their receptors, and restore impaired learning ability in AD rats.120 Targeted regulation of miR-155 expression may hold promise as a strategy to modulate neuroinflammation in AD, as silencing c-Jun reduces the levels of miR-155 in Aβ-activated microglia and astrocytes.121
miR-146a
MiR-146a plays a significant role as a regulator of innate inflammatory responses and is implicated in cell death and survival. It is predominantly expressed in microglia, the major source of miR-146a in the central nervous system. The absence of miR-146a has differential effects on microglial function and proteome, and it may play an important role in gene regulation within active multiple sclerosis lesions.122
Overexpression of microglia-specific miR-146a has been shown to reduce cognitive deficits in learning and memory, attenuate neuroinflammation, decrease Aβ levels, ameliorate plaque-associated neuritic pathology, and prevent neuronal loss in APP/PS1 transgenic mice. Additionally, miR-146a induces a shift in microglial phenotype, reducing the production of pro-inflammatory cytokines and enhancing phagocytic function, thereby protecting neurons both in vitro and vivo.90
MiR-146a, which is sensitive to NF-κB signaling, exhibits high complementarity to the 3’-untranslated region of complement factor H (CFH), a crucial repressor of brain inflammatory responses. Upregulation of miR-146a coupled with downregulation of CFH has been observed in the brains of individuals with AD as well as in interleukin-1, Aβ, and/or oxidatively stressed human neural (HN) cells in primary culture. The upregulation of miR-146a downregulates CFH expression and may modulate CFH gene expression to regulate inflammation in AD brains and stressed HN cell models.123
MiR-146a binds to interleukin-1 receptor-associated kinase 1 (IRAK1) and TNF receptor-associated factor 6 (TRAF6) and negatively regulates the signaling pathway involving these proteins.124 Studies by Yang et al125 have demonstrated that upregulation of miR-146a in microglia enhances tolerance to Aβ and LPS, resulting in decreased Aβ clearance. MiR-146a is crucial for inducing TLR tolerance in macrophages and is present in EVs that can circulate throughout the body. This process helps maintain immune balance and prevents excessive immune activation and chronic inflammation, which are associated with various diseases. Upregulation of miR-146a induces TLR tolerance, modulates the expression of inflammatory genes associated with AD risk, and reduces the release of pro-inflammatory cytokines, thereby alleviating AD-related neuroinflammation in BV2 microglia in response to LPS treatment.126
Presenilin 2 (PS2), a membrane-associated protease implicated in AD pathogenesis, may contribute to neurodegeneration by influencing microglial pro-inflammatory behavior. PS2 plays a significant role in suppressing the pro-inflammatory response in microglia. MiR-146, a negative regulator of monocyte pro-inflammatory response, is constitutively downregulated in PS2 knockout microglia. This downregulation of miR-146 leads to higher expression levels of its target protein, IRAK1, and increased NF-κB transcriptional activity in PS2 knockout microglia.127
The interaction between miR-155 and miR-146 contributes to microglial activation in diseases, and both miRNAs are crucial in the process of microglial inflammation. MiR-146a acts as a negative regulator of inflammation by inhibiting NF-κB transcriptional activity, whereas miR-155 normally enhances microglia-mediated pro-inflammatory responses.128 A recent study identified the presence of miR-155 and miR-146a, two critical inflammation-related miRNAs that modulate microglial phenotype.129 The study demonstrated that injection of miR-146a-containing exosomes inhibited endotoxin-induced inflammation in mice, while miR-155 promoted it.
The upregulation of inflammatory-associated miR-155, miR-146a, and miR-124 by senescence-associated secretory phenotype (SASP) from senescent cells showed a time-dependent increase and an inverse correlation with their respective targets (SOCS-1, IRAK1, and C/EBP-α).130 In vitro, studies have shown increased senescence-associated β-galactosidase (SA-β-gal) activity and upregulated miR-146a expression in 16-day-old microglia cultures, which further increased upon Aβ treatment in 2-day-old microglia. Additionally, Aβ downregulated miR-155 and miR-124 and altered the phenotype of microglia subpopulations. Simultaneous expression of M1 and M2 markers was observed after Aβ treatment, but at lower levels in the in vitro aged microglia.131
In AD, brain-enriched miRNAs including miR-34a, miR-146a, and miR-155 are upregulated and target the mRNA 3’UTR of sirtuin 1 (SIRT1), leading to the downregulation of SIRT1 expression.132 MiR-146a, miR-132, and miR-155 were found to be upregulated in cells treated with LPS, which activates the TLR signaling pathway and promotes NF-κB and AP-1 transcription factor activation, resulting in increased cytokine release.133,134 Several research studies have implicated miRNAs such as miR-21, miR-146a, and miR-155 in the regulation of inflammation.135 In both short post-mortem AD brains and stressed primary human neuronal-glial (HNG) cells, there is consistent upregulation of brain-enriched miRNAs regulated by the pro-inflammatory transcription factor NF-κB, including miR-9, miR-34a, miR-125b, miR-146a, and miR-155.136
miRNA let-7c-5p
MiRNA let-7c-5p is a key player in the modulation of neuroinflammation and microglia activation, exerting inhibitory effects on both M1 polarization and M2 polarization. Additionally, let-7c-5p plays a crucial role in regulating the cytokine-dependent tissue microenvironment, facilitating the transition of microglia from the pro-inflammatory M1 phenotype to the anti-inflammatory M2 phenotype. Overexpression of let-7c-5p effectively suppresses microglia pro-inflammatory responses induced by LPS and hypoxia-glycemia, leading to a reduction in the release of inflammatory mediators such as IL-6, iNOS, TNF-α, and COX-2.
In a mouse model of traumatic brain injury, let-7c-5p downregulates the expression of cysteine aspartate-proteinase-3 (Caspase-3), which is potentially targeted by let-7c-5p. Notably, the activation of protein kinase C-δ (PKC-δ) is implicated in mediating the role of Caspase-3 in microglia activation. Remarkably, let-7c-5p overexpression effectively inhibits neuroinflammation, mitigates microglia activation, and improves the overall neurological prognosis in mice with traumatic brain injury.85
These findings underscore the significance of let-7c-5p in the regulation of neuroinflammation and microglia activation. Targeting the signaling pathways influenced by let-7c-5p holds great potential for the development of therapeutic strategies aimed at attenuating neuroinflammatory responses and restoring microglial homeostasis.
miR-223
Serum miR-223 exhibits a strong positive correlation with mini-mental state examination (MMSE) scores in patients with AD. Notably, it demonstrates superior performance as a potential biomarker for AD evaluation, displaying a higher receiver operating characteristic (ROC) score compared to other miRNAs. Moreover, the combination of serum miR-223 with miR-125b improves the sensitivity and specificity of AD prediction, highlighting the potential value of serum miR-223 as a diagnostic biomarker for AD.86 Furthermore, serum exosomal miR-223 demonstrates promise as a biomarker for diagnosing dementia and assessing disease progression. The level of miR-223 shows significant correlations with MMSE scores, Clinical Dementia Rating (CDR) scores, magnetic resonance spectroscopy (MRS) spectral ratios, as well as serum concentrations of IL-1β, IL-6, TNF-α, and CRP.137 The expression levels of let-7g-5p, miR-126-3p, miR-142-3p, miR-146a-5p, and miR-223-3p are correlated with the severity of AD. In patients with severe dementia, significantly lower concentrations of these EV-derived miRNAs (let-7g-5p, P = 0.039; miR-126-3p, P = 0.057; miR-142-3p, P = 0.027; miR-146a, P = 0.0062; miR-223-3p, P = 0.047; miR-26b, P = 0.0049) were observed compared to healthy controls. The downregulation of these specific miRNAs may serve as biomarkers reflecting the severity of AD.138
Oxidative stress plays a major role in AD pathogenesis. Notably, miR-223 overexpression has been shown to enhance cell viability, inhibit cell apoptosis, reduce ROS levels, enhance superoxide dismutase (SOD) activity, and decrease malondialdehyde (MDA) content. These effects are partly mediated by the direct targeting and inhibition of FOXO3 expression. Additionally, miR-223 activates TXNIP transcription through the FOXO3/TXNIP axis. The study highlights the neuroprotective role of miR-223 against oxidative stress injury and its augmentation of the neuroprotective effects of estradiol.139 Additionally, exosomal miR-223 derived from mesenchymal stem cells (MSCs) exerts a protective effect against cell apoptosis in an AD model by targeting the PTEN- PI3K/Akt pathway. The expression levels of PTEN are inversely correlated with miR-223 expression.140 The downregulation of Hp1bp3 is suggested to be a relevant driver of aging and AD-related phenotypes by Neuner et al Moreover, mir-223 deficiency leads to the downregulation of several immune-related genes. The upregulation of a large number of immune-related genes after Hp1bp3 knockdown may be partially attributed to the observed upregulation of mmu-mir-223-3p.141
miRNA-223 functions as an anti-inflammatory miRNA in microglial cells by directly targeting the NLRP3 protein. Upregulation of miRNA-223 expression in microglia contributes to debris clearance through phagocytosis and CNS remyelination. The functional effect of miRNA-223 deficiency was examined by transfecting a miRNA-223 inhibitor into microglial cells. Antagonizing miRNA-223 function significantly reverses the effect of Sulforaphane (SFN) on NLRP3 inflammasome activation. The mRNA levels of IL-1β, IL-18, and NLRP3 are increased, and NLRP3 protein levels are substantially elevated in SFN-pretreated cells with inhibition of miRNA-223.113 In the context of experimental autoimmune encephalomyelitis (EAE), a study observed that miR-223 deficiency significantly ameliorates CNS inflammation, demyelination, and clinical symptoms. This effect is accompanied by increased autophagy in resting microglia and brain microglia. Mechanistically, miR-223 targets Atg16l1, and its overexpression reduces Atg16l1 expression in BV2 cells, leading to decreased autophagy levels in microglia and an increase in activated microglia levels.87
miR-374b-5p
miR-374b-5p has been identified as a direct regulator of BACE1 through its binding to the 3’-UTR of the BACE1 mRNA. Upon induction of Aβ25–35, the expression of lncRNA MAGI2-AS3 was significantly upregulated, while miR-374b-5p expression was markedly downregulated in both SH-SY5Y and BV2 cells. Knockdown of MAGI2-AS3 resulted in elevated miR-374b-5p expression, whereas cells overexpressing MAGI2-AS3 exhibited significantly reduced miR-374b-5p expression in both cell lines. These findings suggest that MAGI2-AS3 may function as a sponge for miR-374b-5p in SH-SY5Y and BV2 cells. The MAGI2-AS3/miR-374b-5p axis plays a regulatory role in Aβ-induced neurotoxicity in SH-SY5Y cells and neuroinflammation in BV2 cells. Consequently, targeting the MAGI2-AS3/miR-374b-5p axis may offer potential biomarkers and therapeutic targets for AD.89
miR-26a
In the context of microglia activation, there exists a reciprocal relationship between TLR4 expression, which is upstream of NF-kB signaling, and miRNA regulation. On one hand, stimulation of TLR4 leads to a significant reduction in the expression of miR-26a in microglia. MiR-26a directly targets activating transcription factor (ATF) 2, and its overexpression results in the inhibition of ATF2 expression. Consequently, the production of inflammatory cytokines such as TNF-a and IL-6 is significantly reduced, thereby attenuating microglia activation induced by LPS.91
Notably, miR-26a-5p exhibits low expression levels in AD mice. Overexpression of miR-26a-5p has been shown to inhibit Tau phosphorylation and Aβ accumulation. This is achieved through the negative regulation of DYRK1A, a target of miR-26a-5p, via direct targeting of its 3’UTR. In vivo, studies have demonstrated that increased miR-26a-5p levels result in the downregulation of Aβ40, Aβ42, p-APP, and p-Tau levels in AD mice by decreasing DYRK1A expression. The overexpression of miR-26a-5p can effectively suppress Tau phosphorylation and Aβ accumulation by downregulating DYRK1A levels in AD mice.142 Similarly, decreased expression of brain-derived neurotrophic factor (BDNF) and increased levels of miR-26a/b have been observed in mice at 5 months of age. Targeting miR-26a/b may prove beneficial in mitigating Tau and Aβ pathology in individuals with Down syndrome (DS), who are more susceptible to AD.143 Regarding the hippocampus, the low vitamin D (LVD) group exhibited elevated BDNF levels, while miR-26a expression was significantly decreased compared to the APP/PS1 group. Additionally, miR-26a was found to be downregulated in the hippocampus of the high vitamin D (HVD) group. The reduced expression of miR-26a correlates with decreased Aβ plaques, tau phosphorylation, and neuroinflammation.144 Therefore, it is plausible that miR-26a may target BDNF and inhibit its expression, thereby exacerbating AD pathology.
The miR-26a-5p/PTGS2 axis represents a crucial target for investigating the pathogenesis of AD, as PTGS2 is a direct target gene of miR-26a-5p. In AD patients and AD model cells, miR-26a-5p is upregulated while PTGS2 is downregulated. Furthermore, the expression of miR-26a-5p promotes the proliferation of AD model cells, and PTGS2 is involved in regulating miR-26a-5p, capable of reversing its effect on cell proliferation. Targeting the miR-26a-5p/PTGS2 axis holds potential therapeutic implications for AD.145 MiR-26a has been found to directly target phosphatase and tensin homolog (PTEN). Through the suppression of PTEN expression, miR-26a promotes neurite outgrowth, highlighting its importance in neuronal development and morphogenesis. Thus, miR-26a has the potential to serve as a therapeutic target for individuals with AD.146
miR-27a
In patients with dementia attributed to AD, a decrease in the level of hsa-miR-27a-3p within the CSF has been observed. Notably, low levels of hsa-miR-27a-3p are accompanied by elevated CSF tau levels and diminished CSF β-amyloid levels. This investigation underscores hsa-miR-27a-3p as a potential candidate biomarker for AD.147 Patients afflicted with AD at mild and moderate-to-severe stages commonly exhibit lower MMSE and MoCA scores, as well as diminished levels of various biomarkers such as serum miR-27a-3p, cerebrospinal fluid (CSF) miR-27a-3p, Aβ42 levels, and the Aβ42/Aβ40 ratio when compared to healthy individuals. Notably, a positive correlation between serum and CSF miR-27a-3p levels, along with a negative correlation between serum miR-27a-3p levels and standardized uptake value ratio (SUVR) and CSF BACE1 levels, has been elucidated. Additionally, the levels of NEAT1 and miR-27a-3p in both serum and CSF of AD patients consistently display expression trends that align with disease progression, exhibiting a negative correlation. These findings imply the involvement of this correlation in AD pathogenesis, specifically about the extent of Aβ deposition within the brain.148 However, an alternative study has suggested the upregulation of miR-27a-5p in AD.149
Notably, the upregulation of miR-27a-3p has been shown to confer a multitude of beneficial effects, including the reduction of cellular apoptosis, promotion of cell activity, downregulation of amyloid protein, BACE1 protein, APP protein, Tau protein, and its phosphorylation, as well as upregulation of caspase 3 protein and its lysate protein. Importantly, it has been established that miR-27a-3p serves as a target gene of the lncRNA NEAT1. Cognitive dysfunction induced by AD in rats was observed to be ameliorated upon downregulation of NEAT1, further highlighting the therapeutic potential of this regulatory axis.150 Furthermore, miR-27a exerts negative regulation on SOX8 expression, a high mobility group-box transcription factor that holds crucial importance in the early development of embryos, particularly in gender determination. Activation of SOX8 subsequently leads to an upregulation of β-catenin expression, effectively suppressing apoptosis and neuroinflammation in AD.151
Microglial activation, triggered by LPS stimulation, elicits a notable decline in miR-27a levels. This particular microRNA assumes a critical role in governing the secretion of pro-inflammatory cytokines, including IL-6, IL-1β, TNF-α, and NO, by directly targeting the genes TLR4 and IRAK4. By curbing the microglial inflammatory response induced by LPS, miR-27a actively contributes to the reduction of microglia activation.92 Functionally, miR-27a exerts its regulatory influence on TLR4 and IRAK4 activity in microglia, thereby modulating the production of inflammatory cytokines in LPS-activated microglia. This regulatory mechanism is achieved through direct binding of miR-27a to the 3’-UTRs of TLR4 and IRAK4, effectively interfering with their expression. The resultant downregulation of TLR4 and IRAK4 expression subsequently leads to diminished production of downstream inflammatory mediators. Consequently, miR-27a emerges as a pivotal player in the precise regulation of inflammatory responses in microglia.92
miR-181c
Both miR-27a and miR-181c demonstrate the ability to suppress microglial activation by targeting TLR4. Additionally, miR-181c exerts an additional inhibitory effect on the production of inflammatory mediators through the NF-κB pathway. The regulation of TLR4 expression by these microRNAs holds promise for the mitigation of neuroinflammatory disorders.152 Investigations have identified MeCP2 and X-linked inhibitors of apoptosis as mRNA targets of miR-181. Knockdown of miR-181 has been shown to enhance the production of pro-inflammatory cytokines (TNF-α, IL-6, IL-1β, IL-8) and high-mobility group box 1 (HMGB1) upon LPS stimulation. Conversely, overexpression of miR-181 results in a significant increase in the expression of the anti-inflammatory cytokine IL-10. Considering the involvement of miR-181 in inflammatory events and CNS injury, novel therapeutic strategies for CNS disorders characterized by an inflammatory component, such as AD, may be devised.153
Bioinformatic analysis has revealed potential regulatory roles of miR-181c in axon guidance, MAPK signaling, dorsoventral axis formation, and long-term depression. Through binding to specific sites within the 3’-UTR of collapsin response mediator protein 2 (crmp2), overexpression of miR-181c leads to the downregulation of crmp2 protein abundance at the post-transcriptional level. These findings suggest that crmp2 is a target of miR-181c and that the abnormally low expression of miR-181c in the hippocampus of SAMP8 mice contributes to an increase in crmp2 protein levels in AD mice, potentially contributing to the pathogenesis of AD.93 Significantly elevated levels of miR-92a-3p, miR-181c-5p, and miR-210-3p have been detected in the plasma of both individuals with MCI and AD. MCI patients who progress to AD exhibit higher plasma levels of these miRNAs. These findings propose that plasma miR-92a-3p, miR-181c-5p, and miR-210-3p represent distinct molecular signatures that may serve as valuable biomarkers for AD.154
miR-210
Recent studies have proposed that the upregulation of miR-122-5p, miR-210-3p, and miR-590-5p in the plasma or plasma EVs of individuals with positive amyloid-beta positron emission tomography (Aβ-PET) results in increased Aβ production. This effect is achieved through the activation of beta-cleavage of amyloid precursor protein and the inhibition of ADAM10, BDNF, and JAG1 expression. These findings suggest the involvement of these miRNAs in amyloidogenesis during the onset and progression of AD.155 Li et al demonstrated that miR-210 contributes to microglial M1 activation by partially targeting SIRT1, resulting in decreased deacetylation of the NF-κB subunit p65 and enhanced NF-κB signaling activity.76 Activation of p75NTR induces upregulation of miR-210-3p expression through NF-κB activation, leading to the suppression of PCYT2 expression. The p75NTR-mediated NF-κB/miR-210-3p/PCYT2 axis contributes to cognitive dysfunction in AD.94
miR-125b
A20, a ubiquitin-editing enzyme, plays a critical role in inhibiting the NF-κB pathway. However, miR-125b has been identified as a direct suppressor of A20 expression, resulting in enhanced NF-κB function in microglia. Notably, this effect is contingent on the expression of the P2X7 receptor.156 Furthermore, the addition of miR-34a-5p or miR-125b-5p has been shown to attenuate Aβ-induced apoptosis and oxidative stress. BACE1 has been identified as a target of miR-34a-5p and miR-125b-5p, and restoring BACE1 weakened the impact of miR-34a-5p or miR-125b-5p on Aβ-induced neurotoxicity. By reducing apoptosis and oxidative stress through the targeting of BACE1, miR-34a-5p and miR-125b-5p offer novel therapeutic targets for AD treatment.95 Moreover, a study revealed significantly lower expression of miR-125b in the serum of AD patients compared to healthy controls. ROC analysis demonstrated that miR-125b exhibited high specificity (up to 68.3%) and sensitivity (80.8%). Additionally, miR-125b levels were found to be correlated with MMSE scores in AD patients. These findings suggest that miR-125b holds potential as a valuable non-invasive biomarker for AD diagnosis and monitoring.157
Consistent results were observed in SH-SY5Y cells and APP/PS1 transgenic mouse models.158 Notably, melatonin receptor 2 (MT2) expression was dramatically reduced in the dendritic compartment following exposure to Aβ oligomers. Activation of MT2 prevented Aβ-induced disruption of dendritic complexity and spine density. Importantly, MT2 activation decreased cAMP levels, leading to the inactivation of the transcription factor CCAAT/enhancer-binding protein α (C/EBPα), which subsequently suppressed miR-125b expression and elevated the expression of its target, GluN2A. The cAMP-C/EBPα-miR-125b-GluN2A signaling pathway is crucial for the neuroprotective effects of MT2 activation in Aβ-induced dendritic injuries and learning/memory impairments, offering a novel therapeutic target for AD synaptopathy.159
Furthermore, the downregulation of miR-125b has been identified as a key event in the neurotoxic effects of Aβ treatment in cortical neurons. Treatment with 17β-estradiol protects neurons from Aβ-induced neurotoxicity by increasing miR-125b expression, which in turn decreases the expression of the pro-apoptotic proteins Bak1 and p53 at both the gene and protein levels. These findings suggest miR-125b as a novel neuroprotective miRNA in AD.160 However, recent research has not observed differential expression of miR-125b-5p in either the superior temporal gyrus (STG) or the entorhinal cortex (EC). Therefore, it is suggested that miR-125b-5p and miR-501-3p may have less relevance in AD pathogenesis than previously hypothesized.161
miR-340-5p
Moreover, miRNAs play a crucial role in the regulation of microglial activation during brain and spinal cord injury. In the context of spinal cord injury, miR-340-5p has been shown to exert a significant influence on microglial function both in vitro and in vivo in rat models. Specifically, miR-340-5p targets p38, and overexpression of miR-340-5p leads to reduced expression of p38, thereby inhibiting the activation of the p38MAPK signaling pathway and suppressing microglial activation as well as inflammation levels.162
In the hippocampus of the senescence-accelerated mouse prone-8 (SAMP8) model of AD, miR-340 was found to be downregulated, while BACE1 was upregulated when compared to senescence-accelerated mice/resistant-1 (SAMR1) mice. This observation suggests a negative correlation between miR-340 and BACE1 in SAMP8 mice. Furthermore, miR-340 directly binds to BACE1, and overexpression of miR-340 in SH-SY5Y/APPswe cells leads to decreased expression of BACE1. Consequently, miR-340 reduces the accumulation of amyloid-beta and suppresses cell apoptosis by targeting BACE1. The downregulation of miR-340 in AD and its ability to reduce amyloid-beta accumulation through the modulation of BACE1 expression highlight its potential as a therapeutic target for AD.163
Additionally, inhibiting the expression of protection of telomere 1 (POT1) has been shown to improve the symptoms of AD in mice. This inhibition leads to a reduction in Aβ1-42 deposition while increasing telomere length and telomerase activity. Interestingly, miR-340-5p expression levels were found to increase following this intervention, which in turn alleviated cellular senescence and improved AD symptoms. The upregulation of miR-340-5p enhances cellular telomere length and delays cell senescence by inhibiting POT1 expression, ultimately resulting in the amelioration of AD symptoms in mice.96
miR-128
miR-128 has emerged as a key regulator of microglial viability, with the ability to modulate the expression of M1 and M2 markers. It downregulates M1 markers CD86 and CD32 while upregulating M2 markers Arg1 and CD206. Additionally, miR-128 reduces the secretion of inflammatory cytokines, exerting anti-inflammatory effects. These regulatory effects of miR-128 are mediated through the P38 pathway.164
In the context of AD, miR-128 shows significant upregulation in serum samples of AD patients compared to controls. Moreover, the upregulation of miR-128 is negatively correlated with MMSE scores. Serum levels of miR-128 in AD patients also positively correlate with the levels of inflammatory cytokines IL-1β and TNF-α in the serum. These findings indicate that serum miR-128 holds promise as a candidate diagnostic biomarker in AD patients, exhibiting good diagnostic performance both independently and in combination with other factors. Furthermore, it may serve as a potential therapeutic target for neuroinflammation in AD.165 In AD patient plasma and Aβ-treated MCN and N2a cells, miR-128 is upregulated while PPARγ is downregulated. Inhibition of miR-128 decreases Aβ-mediated toxicity by inactivating NF-κB in MCN and N2a cells, with PPARγ identified as a target of miR-128. Upregulation of PPARγ attenuates Aβ-mediated toxicity by inactivating NF-κB. Additionally, PPARγ knockdown reverses the effect of anti-miR-128 in MCN and N2a cells. Hence, the inhibition of miR-128 upregulates PPARγ, inactivates NF-κB, and reduces Aβ-mediated toxicity, presenting a novel potential target for AD treatment.166 miR128 can also target TFEB, resulting in lower expression and decreases in TFEB transcripts and their nuclear localization, and significant reduction of lysosomal enzymes and Aβ degradative capacity in AD patients.48
In AD mice, miR-128 interacts with the 3’UTR of STIM2 and inhibits its translation. Silencing miR-128 or disrupting its binding to STIM2 leads to increased STIM2 expression, subsequently restoring synaptic function and memory precision. These findings suggest that miR-128 could be a therapeutic target for AD, offering a means to restore impaired synaptic function.167 The expression of miR-128 is upregulated, while that of PPARγ is downregulated in the cerebral cortex of AD mice. PPARγ has been identified as a target of miR-128. Notably, miR-128 knockout or PPARγ upregulation inhibits AD-like performances, amyloid plaque formation, Aβ generation, APP amyloidogenic processing, and inflammatory responses in AD mice. Furthermore, the beneficial effects of miR-128 knockout are reversed by a PPARγ inhibitor. These results suggest that miR-128 knockout attenuates AD-like performances, and reduces Aβ production and inflammatory responses by targeting PPARγ in AD mice.97 Furthermore, miR-128 has been shown to regulate the expression of synaptic proteins SNAP-25 and Syt1, which are critical for synaptic transmission. Decreased miR-128 expression in primary hippocampal cultures from 5xFAD mice leads to increased neuronal network activity and excitability. Thus, miR-128 plays a significant role in synaptic functioning and plasticity by modulating the expression and function of synaptic proteins.168
In conclusion, miR-128 has emerged as a potential biomarker and therapeutic target for neurodegenerative diseases such as AD and MCI.169,170 Its ability to regulate microglial viability, modulate inflammatory responses, restore synaptic function, and influence AD-related pathologies highlights its potential in the diagnosis and treatment of these conditions.
miR-124
M2 microglia-derived exosome-mediated miR-124 has been shown to target and downregulate ubiquitin-specific protease 14 (USP14), thereby reducing ischemic brain injury and promoting neuronal survival.171 The neuroprotective effects of miR-124 have been observed in promoting neuronal survival and inducing M2-like polarization of microglia, particularly during the first week of treatment. Notably, the presence of Arg1+ microglia was positively correlated with functional improvement during miR-124 treatment within the same period.172 MiR-124 is believed to play a crucial role in transitioning microglia from the resting M0 state. Exposure of cultured microglia to pro-inflammatory signaling, such as LPS, leads to decreased miR-124 expression. Additionally, IL-4, a cytokine known to promote selective activation of M2-type microglia, inhibits miR-124 expression. Since miR-124 is inhibited by both types of activation signals, it is thought to contribute to the promotion of microglial quiescence. Reductions in miR-124 expression release microglia from the resting (M0) state and facilitate canonical TLR signaling pathways and NF-κB-RelA effector pathways, consequently enabling the initial pro-inflammatory “recruitment” of the M1 phenotype.80
In vivo and in vitro studies have demonstrated that neuron-derived exosomes facilitate functional behavioral recovery by suppressing the activation of M1 microglia and A1 astrocytes. Among the miRNAs identified in neuron-derived exosomes, miR-124-3p was found to be the most enriched. Further investigation revealed that MYH9 serves as the downstream target gene of miR-124-3p. Several experiments were conducted to confirm the involvement of the miR-124-3p/MYH9 axis, ultimately suggesting the potential involvement of the PI3K/AKT/NF-κB signaling cascades in the modulation of microglia by exosomal miR-124-3p. Collectively, these findings indicate that exosomal miR-124-3p suppresses MYH9 by directly targeting its 3’-UTR, thereby modulating the PI3K/AKT/NF-κB pathway. Thus, the transmission of miR-124-3p through exosomes derived from neurons acts as a protective mechanism against traumatic spinal cord injury by inhibiting the activation of neurotoxic microglia and astrocytes.173 In vitro, studies have revealed that cocaine inhibits the levels of miR-124 in microglia, and a similar downregulation of miR-124 was observed in microglia isolated from mice treated with cocaine. The decrease in miR-124 expression is likely attributable to cocaine-induced DNA methylation in the promoter region of miR-124 precursors, resulting in microglia activation in the brain.174 In the context of APP Swedish SH-SY5Y (SWE) cells, inhibition of miR-124 favored an IFNγ-induced inflammatory signature characterized by upregulation of RAGE/HMGB1/iNOS/IL-1β and downregulation of IL-10/ARG-1. Conversely, the introduction of miR-124 mimics reduced microglia activation, downregulating TNF-α/iNOS expression and deactivating extracellular MMP-2/MMP-9 levels.175
miR-486
ANK1 is up-regulated in laser-captured microglia in the brains of individuals with AD. Specifically, ANK1 exhibits a significant 4-fold upregulation in AD microglia, while no such upregulation is observed in neurons or astrocytes from the same individuals. This indicates that the expression of ANK1 in AD brains is primarily confined to these glial cells.176 Furthermore, ANK1 serves as a host gene for miR-486, which generates two mature miRNAs: miR-486-3p and miR-486-5p. However, the transcription of miR-486 can be inhibited by ANK1 hypermethylation. The upregulation of miR-486 plays a crucial role in microglial activation, proliferation, and survival by acting as a negative regulator of Akt, mTOR, and STAT3.98
miR-331-3p and miR-9-5p
miR-331-3p and miR-9-5p exhibit distinct expression patterns in different stages of AD mice. In the early stage, these miRNAs are decreased, while in the late stage, they are increased. The downregulation of miR-331-3p and miR-9-5p is associated with higher autophagic activity and no significant accumulation of Aβ in early-stage AD mice. Conversely, the upregulation of miR-331-3p and miR-9-5p is associated with lower autophagic activity and significant accumulation of Aβ in late-stage AD mice. These findings suggest that miR-331-3p and miR-9-5p could serve as potential biomarkers to distinguish between the early and late stages of AD.99,177 In AD mice at the late stage, higher expression levels of miR-9 are observed, which leads to increased activation of microglia and a lower number of neuronal cells. MiR-9 overexpression in late-stage AD mice promotes microglial activation and neuronal cell death. Additionally, miR-9 downregulates autophagic activity by targeting OPTN, impairing the clearance of Aβ aggregates through the autophagy pathway. This, in turn, contributes to the progression of AD.99,177
Furthermore, miR-9 has been reported to promote microglial activation and neuronal cell death by targeting MCPIP1.178,179 Additionally, miR-9 is upregulated and targets the mRNA 3’UTR of SIRT1, resulting in the downregulation of SIRT1 expression in AD.132
miR-191-5p
In both APP/PS1 transgenic mice and the Aβ1-42-treated microglia AD model, a significant finding emerged regarding the role of miR-191-5p. It was discovered that miR-191-5p directly targeted the 3’UTR of Map3k12, leading to the downregulation of Map3k12 expression. This interaction had profound implications as miR-191-5p demonstrated the ability to inhibit Aβ1-42-induced microglial cell injury and effectively deactivate the MAPK signaling pathway by suppressing Map3k12 expression. Consequently, miR-191-5p exhibited a remarkable capacity to alleviate Aβ1-42-induced microglial cell injury by specifically targeting Map3k12, thereby impeding the activation of the MAPK signaling pathway in microglia.100
miR-203
Emerging evidence from recent studies has shed light on the regulatory role of miR-203 in microglia and its implications in neuronal injury. Notably, miR-203 has been identified as a direct targeting molecule for MyD88 in microglia. Experimental manipulations involving the overexpression of miR-203 or the knockdown of MyD88 have shown promising outcomes in the repression of NF-κB signaling and subsequent mitigation of microglial activation. Consequently, these interventions have demonstrated the potential to ameliorate neuronal injury by attenuating microglial-mediated processes.180
Furthermore, miR-203 has exhibited robust upregulation in transgenic mice, particularly in disease-affected regions and the frontal cortex, in both tau-positive and tau-negative frontotemporal dementia (FTD). Intriguingly, miR-203 appears to function as a driver of the neurodegeneration-associated transcriptional program in the nucleus accumbens shell (NAS). Moreover, overexpression of miR-203 in neurons has been linked to alterations in apoptotic pathways, as evidenced by increased Casp8 protein expression. Additionally, the overexpression of miR-203 in the cortex of one-month-old Tg4510 Tau transgenic mice has resulted in the downregulation of predicted targets of miR-203, specifically genes associated with the NAS module. This manipulation has also led to a significant increase in CASP8 protein expression, activation of apoptotic pathways, and altered expression of genes involved in calcium signaling and neuroactive ligand receptors, further highlighting the intricate role of miR-203 in neurodegenerative processes.101
miR-199b
MiR-199b has emerged as a key player in modulating microglial activation by targeting the IKKβ-NF-κB signaling pathway. Through its inhibitory effects on this pathway, miR-199b effectively suppresses the production of pro-inflammatory cytokines, positioning it as a promising therapeutic target for neuroinflammatory disorders.181
Furthermore, investigations have revealed intriguing associations between miR-199b-5p and AD as well as miR-199a-5p and both AD and diabetes mellitus (DM). In the context of AD, hsa-miR-199b-5p is upregulated, leading to the downregulation of PIN1, a protein involved in regulating the phosphorylation state of Tau. Consequently, hyperphosphorylation of Tau occurs, which is a hallmark of AD pathology. Similarly, miR-199a-5p, which is expressed in the brain, has been implicated in both AD and DM. Notably, this miRNA exerts regulatory control over GLUT4, a glucose transporter critical for hippocampal memory function. The upregulation of miR-199a-5p observed in the prefrontal cortex of AD patients and the plasma of individuals with diabetes disrupts the regulation of GLUT4, leading to impaired insulin uptake by neurons and subsequent insulin resistance. This mechanism provides a potential link between AD and DM, shedding light on the intricate interplay between these two conditions.102,182
miR-206
miR-206 has recently emerged as a compelling candidate involved in the modulation of microglial inflammation associated with AD. A recent study has shed light on the role of miR-206 in promoting inflammation in microglial cells and triggering the release of amyloid-β, a hallmark protein in AD. The underlying mechanism involves the direct binding of miR-206 to the 3’UTR of Insulin-like growth factor 1 (IGF1).
However, a subsequent recovery experiment revealed a fascinating aspect of this regulatory pathway. It was observed that exposure to IGF1 could counteract the inflammatory effects induced by miR-206 in microglia. This suggests a potential regulatory role for the miR-206/IGF1 signaling pathway in AD pathogenesis, whereby IGF1 can mitigate the inflammatory response triggered by miR-206.103
These findings provide valuable insights into the intricate molecular mechanisms governing AD-associated microglial inflammation. Furthermore, they underscore the therapeutic potential of targeting the miR-206/IGF1 pathway as a promising approach for preventing or treating AD. By understanding and modulating this pathway, it may be possible to intervene in the neuroinflammatory processes that contribute to AD pathology. Such interventions hold promise for future therapeutic strategies in the fight against AD.
miR-424
In recent research, miR-424 has emerged as a notable negative regulator of microglial activation and neuronal apoptosis. The study has demonstrated that miR-424 exerts its regulatory effects by inhibiting the expression of ionized calcium-binding adaptor molecule 1 (iba1), leading to a reduction in the secretion of the pro-inflammatory cytokine TNF-α. In vitro experiments have further confirmed the ability of miR-424 to suppress the activity of BV2 microglial cells, highlighting its potential as a modulator of microglial function.104
Moreover, miRNA-424 has shown promise as a candidate for enhancing cytoprotection and reducing inflammation in retinal disorders. Studies have indicated that overexpressing miR-424 in EVs, known as FEE424, can significantly enhance neuroprotection and anti-inflammatory functions in retinal cells in vitro. This suggests that targeting miR-424 could serve as a potential therapeutic strategy for retinal disorders, offering a means to boost cytoprotective mechanisms and alleviate inflammation.183
Additionally, hsa-miR-424-5p has exhibited differential expression patterns when comparing samples from individuals with AD, MCI, and vascular dementia (VaD). This suggests that miR-424 may play similar roles in repressing microglial activation and neuronal apoptosis in cerebral ischemia as it does in AD pathology.105
Taken together, these findings highlight the regulatory role of miR-424 in microglial activation, neuronal apoptosis, and inflammation. The potential therapeutic implications of targeting miR-424 in retinal disorders and cerebral ischemia warrant further investigation and may pave the way for novel treatment strategies for these conditions.
miR-93
A pilot study conducted on patients with AD has revealed a significant upregulation of hsa-miR-93-5p compared to healthy controls. Furthermore, differential expression of hsa-miR-93-5p was observed when comparing AD samples to those from individuals with MCI and vascular dementia (VaD). These findings suggest that hsa-miR-93-5p, along with the expression of phosphorylated tau at serine 396 (P-S396-tau) in extracellular vesicles (EVs), could potentially serve as a combined protein and miRNA signature to distinguish between HC, MCI, VaD, and patients with sporadic AD.105
Studies have indicated that miR-93 possesses anti-inflammatory effects in cerebral ischemia-reperfusion (CIR) mice by attenuating inflammatory responses and cell apoptosis. Moreover, miR-93 acts as an inhibitor of the expression of IRAK4 and other pro-inflammatory genes in microglia, thereby highlighting its potential as a therapeutic target for CIR and other inflammatory conditions within the central nervous system.106
Similarly, miR-93 may exert inhibitory effects on inflammatory responses and cell apoptosis in AD by targeting the IRAK4 signaling pathway. By modulating this pathway, miR-93 holds promise as a therapeutic intervention to mitigate inflammatory processes and cellular damage associated with AD.
Collectively, these findings underscore the potential of hsa-miR-93-5p as a biomarker for AD and its therapeutic implications in both cerebral ischemia and AD-related inflammation. Further investigations are warranted to elucidate the precise mechanisms through which miR-93 regulates inflammatory processes and to explore its therapeutic potential in treating central nervous system disorders.
miR-367
miR-367 has been identified as a key regulator in the suppression of IRAK4 expression by directly binding to its 3’-untranslated region. This interaction leads to the inhibition of NF-κB activation and subsequent production of proinflammatory mediators. In microglia, the knockdown of IRAK4 resulted in a significant decrease in its expression, leading to the inhibition of NF-κB activation and downstream production of proinflammatory mediators. These findings highlight the crucial role of the miR-367/IRAK4 pathway in microglial activation and neuroinflammation.108
Moreover, the therapeutic potential of miR-367 extends beyond its impact on microglia. In an experimental model of AD, miR-302/367-induced neurons demonstrated the ability to alleviate behavioral impairment. This suggests that miR-367 not only targets microglia but also exerts neuroprotective functions in neurons.107
Taken together, these findings emphasize the significance of the miR-367 pathway in regulating microglial activation and neuroinflammation. Additionally, the ability of miR-367 to exert neuroprotective effects in neurons further highlights its potential as a therapeutic target for neuroinflammatory conditions, including AD. Further studies are warranted to elucidate the precise mechanisms underlying the neuroprotective functions of miR-367 and its therapeutic implications for various neurodegenerative disorders.
miR-204
A recent study has revealed that the modulation of microglia-related neuroinflammation in mice can be achieved by inhibiting miR-204 or overexpressing SIRT1. In response to LPS, inhibiting miR-204 or increasing the expression of SIRT1 resulted in reduced inflammation and proliferation, while promoting apoptosis in mouse microglial cells. This study highlights the regulatory role of miR-204 in modulating SIRT1 and its potential in inhibiting microglia-related neuroinflammation in mice.184
In the context of AD, miR-204-3p was found to be downregulated in the hippocampus and plasma of 6-month-old APP/PS1 mice. Overexpression of miR-204-3p in these mice attenuated memory and synaptic deficits. Furthermore, miR-204-3p overexpression led to decreased levels of amyloid plaques and oxidative stress in the hippocampus of APP/PS1 mice. The study identified nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (Nox4) as a target of miR-204-3p, and inhibition of Nox4 by GLX351322 protected neuronal cells against neurotoxicity induced by Aβ1-42. These findings suggest that miR-204-3p attenuates memory deficits and oxidative stress in APP/PS1 mice by targeting Nox4. Therefore, the overexpression of miR-204-3p and/or inhibition of Nox4 could be potential therapeutic strategies for treating AD.109
These discoveries shed light on the role of miR-204 and its potential therapeutic implications in neuroinflammation and AD. Further investigations are necessary to fully understand the underlying molecular mechanisms and to explore the translational potential of targeting miR-204 and Nox4 in the development of AD treatments.
Other miRNA Related to Microglia Activation
The activation of human Toll-like receptor 7 (hTLR7) was significantly induced by miR-6888-3p, miR-4288, and miR-5701, which ranked high-5 in terms of their effectiveness. Additionally, miR-374b-3p and miR-130b-5p showed a trend toward receptor activation compared to the control group. Notably, miR-9-5p elicited significant hTLR7 activation, while exposure to miR-30a-3p, miR-30e-3p, miR-375-3p, and miR-381-5p resulted in NF-kB activation and microglial activation.185
Several miRNAs, including miRNA-219a-2-3, miR10527, miR-329, and miR-578, were found to target genes with key roles in the TLR pathway. Among these, miR10527 was predicted to target four genes primarily involved in protein kinase signaling during inflammation. The first target of miR-10527 downstream of TLR4 was tumor necrosis-associated factor 6 (TRAF6), which acts as a mediator of NF-kB and MAPK pathway activation, leading to the release of proinflammatory cytokines. The other three upregulated miRNAs, miRNA-219a-2-3, miR-10527, and miR-329, directly targeted MAPK8. MAPK8 is responsible for mediating the proinflammatory actions of microglia. Additionally, these miRNAs were found to target MAP2K1and MAPK1, which are involved in the MAPK pathway.186
These findings highlight the regulatory role of specific miRNAs in modulating the activation of hTLR7, NF-kB pathway, and MAPK signaling in microglia. Understanding the intricate interplay between miRNAs and the TLR pathway can provide valuable insights into the mechanisms underlying microglial activation and neuroinflammation. Further research is warranted to elucidate the precise molecular mechanisms and explore the therapeutic potential of targeting these miRNAs in the context of neuroinflammatory disorders.
miRNA as a Biomarker in Microglia Activation and Neuroinflammation
Inflamma-miRNAs, such as miR-124, miR-155, and miR-146a, play crucial roles in regulating microglial polarization by targeting specific signaling molecules.187,188 Among these, miR-155 showed increased expression in the cortex, supporting its potential as a biomarker for neurodegenerative disorders. Target genes downregulated by miR-155 in the cortex included Foxo3, Runx2, and CEBPβ at 3 months, and Foxo3, Runx2, and Socs1 at 9 months. These genes are implicated not only in cell survival but also in amyloid-beta pathology and microglia/astrocyte dysfunction.189
Furthermore, miR-485-3p was found to be elevated in LPS-induced activated microglia BV2 cells. Knockdown of miR-485-3p inhibited the release of pro-inflammatory cytokines, indicating its role in regulating the inflammatory response. FBXO protein 45 (FBXO45) was identified as a potential target of miR-485-3p, mediating its function. The upregulated expression of miR-485-3p in Parkinson’s disease (PD) suggests its potential as a diagnostic biomarker for PD. Notably, reducing the expression of miR-485-3p can inhibit inflammatory responses in BV2 cells, highlighting miR-485-3p as a potential therapeutic target for PD-associated neuroinflammation.190
Moreover, miR-21 has been identified as a consistent biomarker present in SHSwe cells, their released exosomes, recipient CHME3 microglia, and derived exosomes. This finding enhances our understanding of neuron-microglia communication and exosome-mediated neuroinflammation in AD. Importantly, miR-21 emerges as a promising biomarker and potential therapeutic target for intervention in AD.130
These studies shed light on the critical roles of specific miRNAs in regulating microglial polarization, neuroinflammation, and the pathogenesis of neurodegenerative disorders. The identification of these miRNAs as potential biomarkers and therapeutic targets provide new avenues for the development of diagnostic tools and therapeutic interventions in neurological diseases. Further investigations are warranted to elucidate the underlying mechanisms and validate the efficacy of targeting these miRNAs in preclinical and clinical settings.
Treatment Based on miRNA
Quercetin, a natural compound, has been shown to exert neuroprotective effects in learning and memory by normalizing the expression levels of miR-146a, miR-9, TNF-α, NF-κB, AβPP, BACE1, and Bax in the hippocampus. This modulation of pathological inflammation suggests a miRNA/NF-κB-dependent anti-inflammatory mechanism underlying the neuroprotection provided by quercetin.191 Studies suggest that quercetin may also play a role in neuroinflammation and cognitive function by reducing Aβ plaques and tau phosphorylation, regulating the expression of multiple miRNAs, and increasing BDNF protein levels. In a vitamin D-deficient mouse model of AD, quercetin supplementation led to a decrease in hippocampal miR-26a and miR-125b levels, as well as an increase in miR-132 expression, suggesting that quercetin improves tau phosphorylation by modulating miRNA expression levels. The regulatory effects of quercetin on miRNA expression may be relevant to AD development and may exhibit region-specific effects in the brain.192
Berberine (Ber), another natural compound, has shown protective effects against AD in animal models. In cells treated with Aβ, Ber treatment or overexpression of miR-188 increased cell proliferation while decreasing caspase-3 activity and the apoptotic rate. In Aβ-induced BV2 and N2a cells, miR-188 was downregulated, while NOS1 was upregulated and served as a target of miR-188. Ber may promote cell proliferation and inhibit apoptosis through the miR-188/NOS1 pathway.193
Furthermore, dipeptidyl vinyl sulfone (VS) at low levels has been found to prevent cell death and reduce microglial phagocytosis in response to Aβ treatment. VS also suppressed the Aβ-induced expression of inflammatory mediators, including matrix metalloproteinase (MMP)-2, MMP-9, high-mobility group box protein-1 (HMGB1), nod-like receptor protein 3 (NLRP3) inflammasome, and interleukin (IL)-1β in microglia. Interestingly, VS coincubation prevented the increased expression of two critical inflammation-related microRNAs, miR-155 and miR-146a, in microglia upon Aβ treatment. These findings suggest that VS holds promise as a potential therapeutic strategy for AD and warrants further investigation using improved cellular and animal models.194
In summary, miRNAs play a significant role in regulating microglial activation and neuroinflammation by modulating related pathways and the release of inflammatory mediators. Multiple signaling pathways, such as the Caspase and NF-κB signaling pathways,195 PI3K/AKT signaling pathway,140 TLR signaling pathway,196 B cell receptor signaling,197 JAK/STAT signaling,198 and p38MAPK signaling pathway,162 are involved in miRNA-mediated microglial activation and the regulation of inflammatory mediator production and release. Therefore, several miRNAs have been identified to play a role in AD progression, and targeting these miRNAs may offer potential therapeutic avenues. miRNAs not only serve as biomarkers for disease diagnosis but also represent potential therapeutic targets for AD, highlighting their multifaceted roles in the pathology of AD.
circRNA in Microglia Activation and Neuroinflammation
circRNAs are a subset of endogenous competitive endogenous RNAs (ceRNAs) that exert their regulatory effects through the sequestration of miRNAs, thereby influencing subsequent transcriptional regulation.199 Additionally, circRNAs can act as sponges for RNA-binding proteins (RBPs) and promote the translation of target mRNAs.200 Emerging evidence suggests that circRNAs not only facilitate gene transcription but also serve as dynamic protein scaffolds, facilitating the assembly of protein complexes.201 Owing to their unique biological properties, circRNAs are increasingly recognized as valuable biomarkers, particularly in the context of tumors and CNS diseases.202–205
In a groundbreaking study, Ashton Curry-Hyde et al conducted a comprehensive analysis of the circRNA transcriptome in human brain glial cells, including astrocytes, microglia, and oligodendrocytes. Using stringent criteria, they identified distinct circRNA profiles in each glial cell type, suggesting diverse roles of circRNAs in the brain. Specifically, 265, 239, and 442 unique circRNAs were identified in astrocytes, microglia, and oligodendrocytes, respectively. Notably, the most abundant circRNAs in these glial cell types were derived from genes that also expressed low levels of linear RNAs, indicating a preference for post-splicing mechanisms rather than traditional splicing activity mediated by the spliceosome.206 Importantly, circRNAs function as miRNA sponges to buffer miRNA-mediated repression of mRNA targets through competitive endogenous RNA networks in microglia.207 Furthermore, circRNAs can directly bind to target genes. For instance, the glioblastoma-associated microglia-derived exosomal circKIF18A was found to bind to FOXC2, promoting its stability and nuclear translocation.208 Additionally, circ_0000518 was shown to promote macrophage/microglial M1 polarization through the FUS/CaMKKβ/AMPK pathway.209 The mechanisms underlying circRNA-mediated regulation of microglia-mediated neuroinflammation are summarized in Table 2 and illustrated in Figure 3.
 |
Table 2 Summary of the Mechanisms of circRNAs in Microglia-Mediated Neuroinflammation |
 |
Figure 3 Regulatory role of several circRNA in microglia activation and potential mechanisms. |
circHivep2
circRNAs primarily regulate microglia activation through their ability to sequester miRNAs. A specific circRNA, circHivep2, has been identified as a regulator of microglia activation and inflammation in KA epileptogenic mice by modulating the miR-181a-5p/SOCS2 signaling pathway. The miR-181a-5p molecule exhibits a dual role in neuroinflammation. On one hand, circHivep2 acts as a negative regulator of miR-181a-5p, thereby suppressing the expression of pro-inflammatory cytokines and microglia activation, thus preventing neuroinflammation. This regulatory mechanism involves the direct interaction of TDP-43 with circHivep2, which influences the biogenesis of miR-181a-5p. By inhibiting the interaction between pri-miR-181a-5p and TDP-43, circHivep2 reduces the levels of miR-181a-5p. On the other hand, miR-181a-5p itself can promote pro-inflammatory signaling by inhibiting the expression of SOCS2. In cases where circHivep2 fails to impede the interaction between miR-181a-5p and SOCS2, miR-181a-5p may further activate pro-inflammatory pathways.210
circPTK2
circPTK2 exerts its regulatory role in microglia by inhibiting the expression of miR-29b in models of OGD. circPTK2 directly binds to miR-29b, leading to the suppression of JNK2/STAT3 signaling by inducing the expression of SOCS-1. The JAK2/STAT3 signaling pathway is known to regulate the production of IL-1β in microglia under hypoxic conditions. Therefore, circPTK2 modulates hypoxia-induced IL-1β production in microglia through the miR-29b-SOCS-1-JAK2/STAT3-IL-1β signaling pathway, which ultimately regulates neuronal apoptosis triggered by microglia activation under OGD conditions.217
Furthermore, induction of microglia with LPS resulted in the release of pro-inflammatory cytokines, upregulation of high mobility group box 1 (HMGB1) and circPTK2, and downregulation of miR-181c-5p. It was found that miR-181c-5p is a target of circPTK2 and directly binds to HMGB1. Overexpression of miR-181c-5p mimic partially reversed the effects of LPS and HMGB1 overexpression, leading to decreased levels of TNF-α, IL-1β, and HMGB1, as well as inhibited apoptosis. Similarly, silencing circPTK2 had similar effects to up-regulating miR-181c-5p. Moreover, the knockdown of circPTK2 restored the cognitive functions in mice with sepsis induced by Cecal Ligation and Puncture (CLP) and increased the survival rate of CLP mice. These findings suggest that circPTK2 regulates microglia activation and hippocampal neuronal apoptosis through the miR-181c-5p-HMGB1 signaling pathway.211
circ-Epc1
High-throughput sequencing analysis has revealed the significant role of circular RNA circ-Epc1 in hypoxia-pretreated adipose-derived stem cell (ADSC) exosomes, particularly in their capacity to enhance cognitive functions. It has been identified that circ-Epc1 exerts its effects through the regulation of downstream targets, including TREM2, and miR-770-3p acts as a crucial mediator in this regulatory pathway. Specifically, circ-Epc1 is believed to modulate the expression of TREM2 by acting as a sponge for miR-770-3p.
In the context of LPS treatment, the effects of circ-Epc1 on M2 microglia were reversed upon the overexpression of miR-770-3p or downregulation of TREM2. In vivo, experiments further demonstrated that ADSC exosomes containing circ-Epc1 improved cognitive function, reduced neuronal damage, and induced a shift in hippocampal microglia polarization from the M1 to the M2 stage. These findings collectively underscore the therapeutic potential of hypoxia-pretreated ADSC exosomes in improving cognition in an AD mouse model, achieved through the delivery of circ-Epc1 and modulation of microglial M1/M2 polarization dynamics.212
circTREM2_1
Genetic variations in the TREM2 gene, which is primarily expressed in microglia, have been widely recognized as risk factors for AD. Urdánoz-Casado et al conducted a comprehensive analysis of the genetic structure of TREM2 and put forth a hypothesis suggesting the involvement of alternative splicing of exon 4 in the generation of circular RNAs derived from TREM2 (circTREM2s). These circTREM2s are proposed to play a role in the pathogenesis of AD. One specific circRNA transcript originating from TREM2, referred to as circTREM2_1, encompasses nearly all exons of the TREM2 gene, including exons 2, 3, 4, and 5. Notably, exon 4 holds particular significance for TREM2 functionality as it encodes the transmembrane domain critical for its cellular activity. Additionally, circTREM2_1 comprises a substantial portion of the cytoplasmic region and a small segment of the extracellular domain of the Trem2 protein. Intriguingly, the expression levels of circTREM2_1, but not TREM2 mRNA, demonstrated a negative correlation with the accumulation of Aβ deposits in the entorhinal cortex of AD patients.213
circ_0004381
circ_0004381, an intriguing circular RNA, is upregulated in hippocampal neurons treated with Aβ1-42, a hallmark peptide in AD. Notably, suppressing circ_0004381 expression exhibited notable protective effects against Aβ1-42-induced neuronal apoptosis, oxidative stress, and mitochondrial dysfunction in hippocampal neurons. Additionally, the knockdown of circ_0004381 facilitated the polarization of microglia towards the M2 anti-inflammatory phenotype, while inhibiting the production of inflammatory factors by microglia. Moreover, in male transgenic mice carrying the APP/PS1 mutations, circ_0004381 knockdown led to improved cognitive function.
Mechanistically, circ_0004381 was found to regulate the expression of presenilin-1 (PSEN1) by acting as a sponge for miR-647. Inhibition of miR-647 attenuated the effects observed upon circ_0004381 knockdown, suggesting its involvement in mediating the cellular outcomes. Collectively, the knockdown of circ_0004381 alleviated hippocampal neuronal damage, promoted microglial M2 polarization, and enhanced cognitive function in AD model mice through the miR-647/PSEN1 axis.214 These findings shed light on the potential of circ_0004381 as a therapeutic target for AD and provide valuable insights into its underlying molecular mechanisms.
ciRS-7
CircRNA for miRNA-7, also known as ciRS-7 or CDR1as, has emerged as a significant risk factor associated with AD. CDR1as acts as a sponge, effectively sequestering miR-7 and impairing its normal function in the human brain. The interaction between CDR1as and miR-7 leads to a reduction in miR-7 expression levels, which in turn results in the upregulation of UBE2A activity—an important target implicated in the pathology of AD.215 This regulatory mechanism involving CDR1as and miR-7 sheds light on the intricate molecular processes underlying AD development and provides insights into potential therapeutic strategies targeting this interaction.
circ_0005835
The significant involvement of circ_0005835 in AD pathogenesis has been firmly established. Studies have demonstrated the upregulation of circ_0005835 in both AD patients and cell models, resulting in heightened neuroinflammation specifically in BV2 cells. Conversely, the downregulation of circ_0005835 exhibited a suppressive effect on neuroinflammation by acting as a sponge for miR-576-3p in BV2 cells. Notably, the expression of serum miR-576-3p was found to be reduced in AD patients, and it displayed a negative correlation with circ_0005835 expression. These findings underscore the crucial role of circ_0005835 in AD development through its regulatory influence on miR-576-3p expression.216 This knowledge advances our understanding of the molecular mechanisms underlying AD and highlights the potential of circ_0005835 as a therapeutic target for the disease.
circ-AXL
The circular RNA AXL, also known as circ-AXL (accession number circ_0002945), has emerged as a significant factor associated with the risk and severity of AD. Experimental investigations using cellular AD models have demonstrated that the overexpression of circ-AXL leads to increased apoptosis rates, diminished neurite outgrowth, and elevated levels of inflammatory cytokines. Notably, circ-AXL was found to exert a negative regulatory effect on miR-328 while positively regulating BACE1. Further, luciferase reporter gene assays have confirmed the direct binding of circ-AXL to miR-328, whereas miR-328 directly binds to BACE1. Importantly, the knockdown of miR-328 attenuated the impact of circ-AXL knockdown in cellular AD models. These findings suggest that circ-AXL could serve as a promising therapeutic target in AD, potentially through the modulation of miR-328-mediated BACE1 signaling pathway.218 This knowledge sheds light on the intricate molecular mechanisms underlying AD pathogenesis and highlights the therapeutic potential of targeting circ-AXL for effective AD treatment strategies.
Other circRNAs
circPSEN1 has emerged as a pivotal regulatory factor located at the apex of the dysregulated amyloid-beta pathway, which contributes to the neuroinflammatory state observed in AD. Notably, dysregulation of circPSEN1 has been observed in all cases of autosomal dominant AD (ADAD), irrespective of the specific mutation involved.219 A recent study aimed to differentiate AD from other types of dementia by measuring blood circRNAs. The findings revealed a panel of six circRNAs (hsa_circ_0077001, hsa_circ_0022417, hsa_circ_0014356, hsa_circ_0014353, hsa_circ_0074533 upregulated; hsa_circ_0089894 downregulated; all P < 0.05) that displayed AD-specific dysregulation, thus demonstrating their potential as promising biomarkers for AD.220 Another pilot study involving five participants with AD and five healthy controls (HC) investigated circRNAs using the Arraystar Human Circular RNA Microarray V2.0. The results confirmed the upregulation of hsa_circRNA_050263, hsa_circRNA_403959, and hsa_circRNA_003022 in both AD and MCI subjects compared to HCs, while the downregulation of hsa_circRNA_102049 and hsa_circRNA_102619 was observed only in participants with AD. Notably, hsa_circRNA_403959 exhibited significantly higher expression levels in MCI subjects compared to both AD and HC individuals. Although further studies are required to fully characterize the expression profiles and functions of circulating circRNAs, these findings suggest that circRNAs hold promise as novel biomarkers for both preclinical and clinical stages of AD.221
In summary, aside from their role as biomarkers in neurodegenerative diseases, circRNAs are involved in the sequestration of miRNAs, thereby regulating microglia activation and participating in the modulation of neuroinflammation in AD. These insights into the regulatory functions of circRNAs contribute to our understanding of the complex pathogenesis of AD and offer potential avenues for the development of therapeutic interventions.
LncRNA in Microglia Activation and Neuroinflammation
LncRNAs represent a class of RNA molecules exceeding 200 base pairs in length that lack protein-coding capacity.222 They exert critical roles in the development and functioning of the central nervous system, as well as in the pathogenesis of CNS disorders.223 The functionality of lncRNAs often relies on their interactions with one or more proteins, while RNA-binding proteins commonly exhibit the ability to bind multiple lncRNAs. To investigate protein-lncRNA interactions on a large scale, several experimental techniques have been devised, each offering unique advantages and limitations.224,225 Furthermore, lncRNAs can function as molecular sponges for miRNAs, thereby diminishing their regulatory effects on target mRNAs. The incorporation of lncRNA-miRNA interactions into miRNA functional analyses is crucial for comprehending the roles of ncRNAs in disease processes, developmental stages, and tissue-specific mechanisms.226 Notably, lncRNAs have been implicated in the regulation of microglial gene expression during neuroinflammation. They can modulate gene expression either through direct interactions with transcription factors or histone-modifying enzymes or by acting as competing endogenous RNAs (ceRNAs) that sequester downstream miRNAs, consequently influencing signaling pathways associated with microglial polarization.227 The mechanisms by which lncRNAs participate in microglia-mediated neuroinflammation are summarized in Table 3 and depicted in Figure 4.
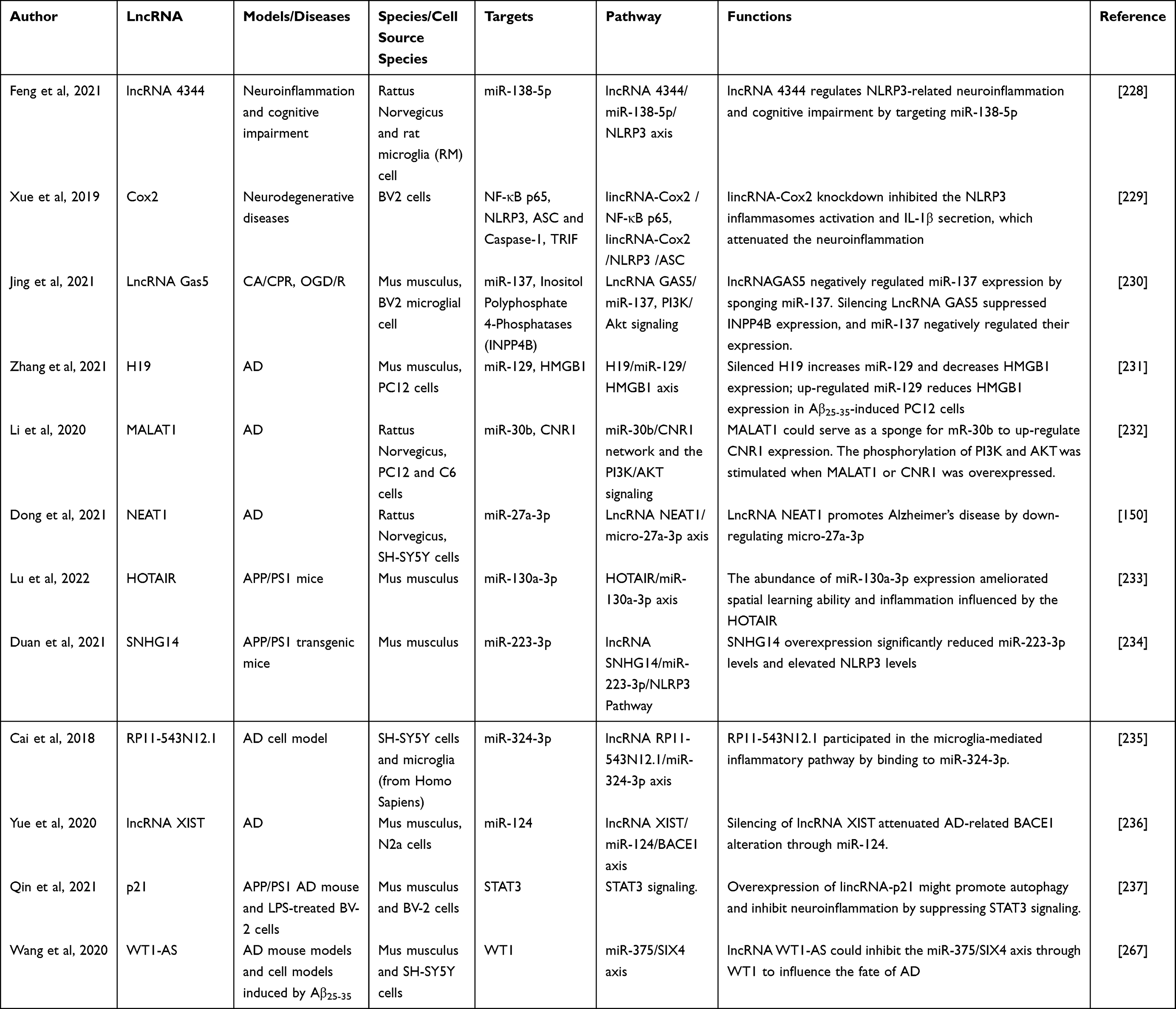 |
Table 3 Summary of the Mechanisms of lncRNAs in Microglia-Mediated Neuroinflammation |
 |
Figure 4 Regulatory role of several lncRNAs in microglia activation and potential mechanisms. |
LncRNA 4344
In cognitive impairment, neuroinflammation mediated by the NLRP3 pathway plays a significant role. Elevated expression of lncRNA 4344 and NLRP3 was observed in the hippocampal tissues and microglial cells of rats treated with LPS, a known inflammatory stimulus. Experimental findings demonstrated that overexpression of lncRNA 4344 resulted in increased neuronal apoptosis, upregulation of NLRP3 expression, as well as enhanced expression of downstream inflammatory genes such as caspase-1, IL-1β, and IL-18. Conversely, the silencing of lncRNA 4344 led to a reduction in inflammatory injuries. Further investigation revealed that miR-138-5p, which exhibited downregulated expression, serves as a direct target of lncRNA 4344. The lncRNA 4344/miR-138-5p/NLRP3 axis was found to regulate cognitive behavior, microglial activation, and the expression of inflammation-related factors during LPS-induced cognitive impairment in rats.228
lincRNA-Cox2
In a similar vein, LincRNA-Cox2 has been identified as an early primary gene that is regulated by NF-κB signaling in macrophages and microglia. Upon stimulation by LPS, LincRNA-Cox2 transcripts assemble into SWI/SNF complexes, which mediate chromatin remodeling and facilitate the transcription of late inflammatory genes in macrophages. Knockdown of LincRNA-Cox2 was found to attenuate LPS-induced transcription of late primary response genes. These findings indicate that LincRNA-Cox2 exerts a regulatory effect on the transcription of NF-κB-regulated late-primary response genes in innate immune cells, thereby influencing microglial polarization.238
Furthermore, another study suggested that LincRNA-Cox2 can interact with NF-κB p65 and enhance its nuclear translocation and transcriptional activity, thereby modulating the expression of Nlrp3 and Asc, key components of the NLRP3 inflammasome. Knockdown of LincRNA-Cox2 was shown to inhibit NLRP3 inflammasome activation by reducing the expression of Nlrp3 and Asc following LPS stimulation.229 Additionally, knockdown of LincRNA-Cox2 resulted in the inhibition of inflammasome activation, prevention of caspase-1 activation triggered by LincRNA-Cox2, reduced secretion of IL-1β, and attenuated cleavage of the TIR-domain-containing adapter-inducing interferon-β (TRIF). This, in turn, enhanced TRIF-mediated autophagy. As autophagic dysfunction is closely associated with inflammation, infection, and neurodegeneration, the regulatory role of lincRNA-Cox2 in autophagy and neuroinflammation may be of significance in the context of AD.229 Therefore, it is plausible that lincRNA-Cox2 plays a crucial role in AD by modulating autophagy and neuroinflammation, adding to its potential relevance in the pathogenesis of the disease.
LncRNA Gas5
The long non-coding RNA Growth Arrest-Specific 5 (Gas5) has demonstrated its significance in various aspects of microglia function and disease pathology. In multiple sclerosis (MS) patients, Gas5 is highly expressed in amoeboid (M1-type polarized) microglia. Functional experiments involving overexpression and repression of Gas5 have revealed its inhibitory effect on the transcription of TRF4, a key factor involved in the M2-type polarization of microglia. This repression occurs through the recruitment of the polycomb repressor complex 2 (PRC2), ultimately leading to the inhibition of M2 polarization in microglia.239
Similarly, elevated levels of Gas5 have been observed in patients with AD, and these levels are negatively correlated with the MMSE score, a measure of cognitive impairment. Moreover, a negative correlation between Gas5 expression and total hippocampal volume, as well as a positive correlation between hippocampal volume and MMSE score, has been established in AD patients. These findings indicate that Gas5 may serve as a valuable indicator of AD progression, either alone or in conjunction with hippocampal volume measurements.240
Gas5 exerts its effects through various molecular mechanisms. It acts as a molecular sponge for miR-223-3p, thereby suppressing its expression and leading to the upregulation of NLRP3, a key component of the inflammatory response in microglia.241 Gas5 also influences the expression of INPP4B by sponging miR-137, which subsequently suppresses PI3K/Akt signaling. This regulatory axis involving Gas5, miR-137, INPP4B, and PI3K/Akt signaling is thought to contribute to the communication between astrocytes and microglia and plays a role in cerebral ischemia/reperfusion injury.230
Gas5 exhibits its regulatory function by interacting with miR-146a-5p, which targets the 3’ non-coding region of notch1. Through this interaction, Gas5 inhibits M2 polarization and promotes M1 polarization in microglia after OGD/R by upregulating Notch1 expression.242 Additionally, Gas5 is involved in several other pathways, including the Gas5/miR-1192/STAT5A/AQP4 axis,243 the Gas5/miR-128-3p/Bax/Akt/GSK-3β axis,244 the Gas5/miR-532-5p/PI3K/AKT apoptosis pathway in myocardial ischemia-reperfusion injury,245 and the Gas5/miR-26a/EGR1 axis, which activates the PI3K/AKT pathway.246 Furthermore, Gas5 has been implicated in ischemic stroke progression by acting as a ceRNA for miR-137, thereby modulating the Notch1 signaling pathway.247
In summary, Gas5 plays a multifaceted role in microglia function and disease progression. It affects microglia polarization, neuroinflammation, apoptosis, and signaling pathways through its interactions with various miRNAs and target genes. The diverse regulatory mechanisms of Gas5 highlight its potential as a therapeutic target and diagnostic marker in neurological disorders.
LncRNA H19
The long non-coding RNA H19 has been implicated in promoting neuroinflammation and glial cell activation in various neurological conditions. Studies have shown that H19 drives HDAC1-dependent M1 microglia polarization, leading to increased inflammatory responses. Knockdown of H19 inhibits microglia polarization and inflammatory cytokine production, while overexpression of H19 has the opposite effect.248 Similarly, H19 overexpression induces the activation of hippocampal astrocytes and microglia, resulting in the release of pro-inflammatory cytokines. In contrast, H19 knockdown inhibits glial cell activation induced by persistent epilepsy. These findings indicate that H19 promotes hippocampal glial cell activation by regulating the JAK/STAT pathway.249
H19 has also been found to play a role in epileptogenesis. It promotes hippocampal glial cell activation during this process. Overexpression of let-7b, a microRNA, inhibits glial cell activation, inflammatory response, and seizures by targeting STAT3. However, H19 competitively binds to let-7b, inhibiting its expression and reversing the inhibitory effect on glial cell activation.250
Furthermore, in AD, H19 and HMGB1 levels are elevated, while miR-129 expression is decreased. Silencing H19 increases miR-129 expression and decreases HMGB1 expression. Upregulation of miR-129 reduces HMGB1 expression in cells induced by Aβ25-35, a peptide associated with AD. Suppression of H19 or elevation of miR-129 not only reduces ROS levels and upregulates matrix metalloproteinase (MMP) expression but also promotes cell cycle progression and inhibits apoptosis in Aβ25-35-induced cells. H19 binds to miR-129, and miR-129 targets the 3ʹ-UTR region of HMGB1 mRNA. These findings suggest that silencing H19 and upregulating miR-129 can enhance cell viability and suppress apoptosis in AD-related cellular models, providing potential therapeutic implications.231
In summary, H19 is involved in promoting neuroinflammation, glial cell activation, and pathological processes in neurological disorders such as epilepsy and AD. Its interactions with specific molecules and signaling pathways highlight its regulatory role in these conditions, and targeting H19 may hold promise for future therapeutic interventions.
LncRNA MALAT1
A study investigating lncRNA MALAT1 revealed its involvement in brain ischemia/reperfusion (I/R) injury and neuroinflammation. In a diabetic rat model of brain I/R injury, MALAT1 was found to be highly expressed, exacerbating brain I/R injury by promoting microglia activation and inflammatory responses.251 Similarly, MALAT1 was highly expressed in the brains of MPTP-induced PD model mice and LPS/ATP-induced microglia. MALAT1 was shown to promote neuroinflammation by recruiting EZH2 to the NRF2 promoter, thereby repressing NRF2 expression. Knockdown or repression of MALAT1 led to upregulation of NRF2, suppressing inflammatory vesicle activation and ROS production. Thus, MALAT1 epigenetically repressed NRF2, inducing inflammatory vesicle activation and ROS production in PD mouse models and microglia models.252
Furthermore, MALAT1 expression was found to be elevated in rats with spinal cord injury (SCI) compared to healthy rats. This upregulated MALAT1 triggered the activation of the IKKβ/NF-κB signaling pathway and promoted the production of pro-inflammatory cytokines in LPS-treated microglial cells. This process was facilitated by the downregulation of miR-199b.253
In the context of AD, lncRNA MALAT1 and CNR1 were found to be poorly expressed, while miR-30b was highly expressed in Aβ25-35-induced rat models and cells. Overexpression of MALAT1 or CNR1 reduced neuronal injury in the rat hippocampus and increased viability while decreasing apoptosis in injured PC12 and C6 cells. It also decreased the secretion of pro-inflammatory factors IL-6 and TNF-α but increased IL-10 production. However, overexpression of miR-30b reversed these trends. MALAT1 acted as a sponge for miR-30b, leading to the upregulation of CNR1 expression. Overexpression of MALAT1 or CNR1 stimulated the phosphorylation of PI3K and AKT. Thus, MALAT1 was found to promote neuronal recovery in AD through the miR-30b/CNR1 network and activation of the PI3K/AKT signaling pathway.232
In summary, lncRNA MALAT1 plays a role in various neurological conditions, including brain I/R injury, Parkinson’s disease, spinal cord injury, and Alzheimer’s disease. Its dysregulation contributes to microglia activation, neuroinflammation, and neuronal injury. Understanding the mechanisms underlying MALAT1’s functions may provide insights into the development of novel therapeutic strategies for these neurological disorders.
LncRNA NEAT1
The transcription factor YY1 induces the expression of the long non-coding RNA Nuclear Enriched Abundant Transcript 1 (NEAT1), which activates the Wnt/β-catenin signaling pathway in microglia subjected to oxygen-glucose deprivation/reoxygenation (OGD/R). This activation leads to upregulation of inflammatory factors such as IL-6, IL-1, and TNF-α. Inhibition of NEAT1 reduces the levels of these inflammatory factors, while administration of a Wnt/β-catenin signaling pathway activator reverses the response induced by NEAT1 inhibition, indicating that NEAT1 can activate the Wnt/β-catenin signaling pathway to promote microglial cell activation.254
Plasma lncRNA profiling has identified BC200 and NEAT1 as potential blood-based biomarkers for late-onset AD, as their levels in the plasma of AD patients were significantly higher compared to the control group.255 Serum NEAT1 level in AD patients showed a positive correlation with markers of disease progression, including standardized uptake value ratio (SUVR), cerebrospinal fluid NEAT1, and β-secretase 1 (BACE1).148
In a rat model of AD-induced cognitive dysfunction, downregulation of lncRNA NEAT1 improved cognitive function. The levels of NEAT1 and miR-27a-3p showed a negative correlation. Dual luciferase reporter gene assays and RNA pull-down experiments confirmed that miR-27a-3p was the target gene of NEAT1. Downregulation of NEAT1 or upregulation of miR-27a-3p reduced cell apoptosis, increased cell activity, downregulated amyloid protein, BACE1 protein, APP protein, Tau protein, and its phosphorylation, and upregulated caspase 3 protein and its lysate protein. Thus, NEAT1 promotes AD by downregulating miR-27a-3p.148,150
Furthermore, NEAT1 interacts with NEDD4L and promotes the ubiquitination and degradation of PTEN-induced putative kinase 1 (PINK1), impairing PINK1-dependent autophagy. Overall, lncRNA NEAT1 plays a role in promoting the pathogenesis of AD and represents a promising novel target for pharmacological intervention.256
In summary, NEAT1 exerts regulatory effects in AD by activating the Wnt/β-catenin signaling pathway, serving as a potential blood-based biomarker, influencing disease progression markers, modulating miR-27a-3p expression, and interfering with PINK1-dependent autophagy. Understanding the mechanisms underlying NEAT1’s involvement in AD pathology may pave the way for the development of therapeutic strategies targeting this lncRNA.
LncRNA HOTAIR
LncRNA HOTAIR is highly expressed in activated microglia, and its interference leads to the inhibition of microglial activation and the release of inflammatory factors by promoting Nrdp1-mediated ubiquitination of Myeloid differentiation factor-88 adaptor protein (MYD88) protein.257 During cuprizone-induced demyelination, Sulfasalazine (SF) prevents microglia from shifting towards a pro-inflammatory M1-like phenotype through the ceRNA effect of miR-136-5p and lncRNA HOTAIR. The downregulation of lncRNA HOTAIR by SF reduces the sequestration of miR-136-5p, leading to enhanced regulation of AKT2 by miR-136-5p. This, in turn, reduces AKT2 expression and inhibits NF-κB activation, ultimately preventing microglia from adopting a pro-inflammatory M1-like phenotype.258
Additionally, HOTAIR may function as a ceRNA of miR-129-5p. MiR-129-5p counteracts the impact of silenced HOTAIR on cell viability, apoptosis, inflammation, and oxidative stress. Moreover, in vivo intervention of HOTAIR reverses the influence of ISO on cognition and oxidative stress by binding to miR-129-5p. Thus, the downregulation of HOTAIR contributes to the recovery of the ISO-injured HT22 cell model by regulating miR-129-5p, resulting in improved viability, reduced apoptosis, inflammation, and oxidative stress.259
In SK-N-SH cells treated with MPP+, a neurotoxic compound, the expression of HOTAIR and ATG10 genes increases, while miR-874-5p decreases in a dose- and time-dependent manner. Knockdown of HOTAIR reduces MPP+-induced neuronal damage. HOTAIR promotes MPP+-induced neuronal injury by sequestering miR-874-5p and regulating ATG10 expression. Depletion of HOTAIR attenuates MPP+-induced inflammation in SK-N-SH cells. Thus, targeting the HOTAIR-miR-874-5p-ATG10 pathway holds therapeutic potential for treating neuroinflammation and neurodegenerative diseases.260
The expression of HOTAIR is increased in patients with AD. Exercise has been shown to ameliorate cognitive impairment and reduce the relative serum expression of HOTAIR. Exercise is an independent indicator of HOTAIR expression.261 Similarly, another study suggests that exercise can mitigate learning ability deficits, reduce inflammation, and inhibit HOTAIR expression in AD mice. The elevated expression of HOTAIR suppresses the beneficial effects of voluntary exercise (VE) on IL-6, IL-1β, and TNF-α levels, as well as cognitive function and inflammation. MiR-130a-3p acts as a ceRNA of HOTAIR, and its abundance ameliorates spatial learning ability and inflammation affected by HOTAIR.233
In summary, lncRNA HOTAIR plays a role in microglial activation, demyelination, neuronal injury, and AD pathology. Its ceRNA interactions with miRNAs contribute to these processes by regulating downstream target genes. Understanding the mechanisms underlying HOTAIR’s involvement in neuroinflammation and neurodegenerative diseases may pave the way for the development of targeted therapeutic strategies.
LncRNA SNHG14
LncRNA SNHG14 exhibits high expression levels in ischemic brain tissue and BV-2 cells. Knockdown of SNHG14 significantly inhibits hypoxia-induced activation of BV-2 cells, while overexpression of SNHG14 promotes BV-2 cell activation, as indicated by increased production of TNF-α and NO, as well as enhanced apoptosis of neuronal cells. The elevation of PLA2G4A in BV-2 cells induced by SNHG14 overexpression is reversed by the miR-145-5p mimic, leading to reduced levels of TNF-α and NO. Thus, SNHG14 enhances the expression of PLA2G4A by inhibiting miR-145-5p, thereby activating microglia.262
In an animal model of AD, AVE0991, a nonpeptide analog of Ang-(1-7), suppresses astrocytic NLRP3 inflammasome-mediated neuroinflammation in a manner dependent on lncRNA SNHG14. SNHG14 functions as a sponge for miR-223-3p, while NLRP3 is a direct target of miR-223-3p. Moreover, miR-223-3p is involved in the AVE0991-induced suppression of astrocytic NLRP3 inflammasome. Overexpression of SNHG14 significantly reduces the levels of miR-223-3p, while simultaneously increasing the levels of NLRP3. These findings suggest that the Ang-(1-7) analog AVE0991 inhibits astrocyte-mediated neuroinflammation through the SNHG14/miR-223-3p/NLRP3 pathway, providing neuroprotection in APP/PS1 mice.234
In summary, lncRNA SNHG14 plays a crucial role in the activation of microglia and astrocytes, contributing to neuroinflammation in ischemic conditions and Alzheimer’s disease. Its interactions with miRNAs, such as miR-145-5p and miR-223-3p, regulate the expression of target genes involved in inflammatory processes. Understanding the mechanisms underlying SNHG14-mediated neuroinflammation may open avenues for developing therapeutic strategies targeting this lncRNA to mitigate neuroinflammatory responses and potentially provide neuroprotection.
LncRNA RP11-543N12.1
Through chip data analysis, significant differences in the expression levels of several lncRNAs, including RP11-414H23.3, RP11-642D21.1, ZBTB20-AS1, RP11-354P11.2, RP1-77H15.1, RP11-121G22.3, and RP11-543N12.1, were confirmed between the AD cell model and control cells. Notably, RP11-543N12.1 exhibited relatively high and stable expression within AD cell models. Co-culturing microglia with lncRNA RP11-543N12.1- or miR-324-3p-overexpressing SH-SY5Y cells resulted in increased expression levels of TNF-α, IL-6, and NO. This finding suggests that RP11-543N12.1 is involved in the inflammatory response mediated by microglial cells by binding to miR-324-3p and positively regulating its expression. Furthermore, RP11-543N12.1 targeted miR-324-3p to suppress proliferation and promote apoptosis in the AD cell model, highlighting the potential of RP11-543N12.1 and miR-324-3p as biomarkers and therapeutic targets for AD.235
These findings provide valuable insights into the role of specific lncRNAs, particularly RP11-543N12.1, in the pathogenesis of AD. The interaction between RP11-543N12.1 and miR-324-3p appears to modulate microglial-mediated inflammation and cellular processes related to proliferation and apoptosis. Expanding our knowledge of the regulatory mechanisms involving lncRNAs and miRNAs may contribute to the development of novel diagnostic approaches and therapeutic strategies for AD.
LncRNA XIST
The expression of X–inactive specific transcript (XIST), a lncRNA, is upregulated in microglial cells treated with LPS as well as in damaged spinal cord tissues in rats. XIST acts as a miR-27a sponge, suppressing its expression. Removal of XIST prevents microglial apoptosis and reduces inflammatory injury caused by spinal cord injury through increased expression of miR-27a and decreased expression of Smurf1.263
In patients with breast cancer brain metastases, decreased expression of lncRNA XIST leads to increased secretion of exosomal microRNA-503. This, in turn, significantly enhances STAT3 phosphorylation and reduces NF-κB p65 subunit phosphorylation, thereby triggering the M1-M2 transition in microglia. The M1-M2 switch upregulates the expression of the immunosuppressive cytokine PD-L1 in microglia, suppressing T cell proliferation and autoimmunity while promoting tumor metastasis.264
Furthermore, the expression of lncRNA XIST is elevated in AD mice and cell models. Knockdown of XIST alleviates Aβ-induced neuronal inflammation and damage. XIST exhibits a negative correlation with NEP expression and regulates NEP partly through epigenetic mechanisms by binding with EZH2 in AD mice. By epigenetically repressing NEP, XIST induces Aβ accumulation and neuroinflammation in AD.265
Another study suggests that silencing of XIST negatively regulates miR-124 and positively regulates BACE1 expression in N2a cells. This effect is attenuated by co-transfection with anti-miR-124 oligodeoxyribonucleotide (AMO-124). Silencing of XIST reverses the impact of H2O2 on miR-124, BACE1, and Aβ1-42 expression in N2a cells, which is then re-reversed by co-transfection with AMO-124. Consequently, silencing XIST attenuates AD-related BACE1 alterations through miR-124, positioning XIST as a potential therapeutic target for AD treatment.236
These findings shed light on the intricate involvement of XIST in various pathological processes, such as microglial activation, tumor metastasis, and AD-related neuroinflammation and neurodegeneration. Understanding the regulatory mechanisms and functional roles of XIST and its interaction with miRNAs provides valuable insights into the development of novel therapeutic strategies for neurodegenerative diseases and cancer metastasis.
LincRNA-p21
LincRNA-p21, a transcript induced by p53 in LPS-treated BV2 cells, functions as a ceRNA for the miR-181 family. By acting as a ceRNA, lincRNA-p21 protects PKC-δ from suppression by the miR-181 family, leading to increased PKC-δ levels. This competitive binding with the miR-181 family results in elevated PKC-δ levels, which, in turn, contribute to increased expression of p53 and lincRNA-p21. This regulatory loop promotes microglia activation and inflammation.266
The expression of lincRNA-p21 is reduced in LPS-treated BV-2 cells and APP/PS1 AD mice, indicating its involvement in AD. LincRNA-p21 promotes autophagy and suppresses STAT3 signaling as well as LPS-induced activation of BV-2 cells. Bilobalide, a potential treatment for AD, improves learning and memory capabilities in mice by suppressing neuroinflammation. This effect is believed to be mediated by an increase in lincRNA-p21 levels, which inhibit STAT3 signaling. Furthermore, lincRNA-p21 also promotes autophagy, but its effect is blocked by a STAT3 inhibitor, suggesting that the beneficial effects of lincRNA-p21 are likely mediated through the STAT3 pathway.237
These findings highlight the regulatory role of lincRNA-p21 in microglia activation, inflammation, and autophagy, as well as its potential involvement in the pathogenesis of AD. Understanding the molecular mechanisms underlying lincRNA-p21-mediated regulation provides valuable insights for the development of novel therapeutic approaches targeting neuroinflammation and autophagy dysregulation in AD.
LncRNA WT1-AS
The expression of lncRNA WT1-AS was markedly reduced in AD. It was found that WT1-AS plays a crucial role in inhibiting oxidative stress-induced injury (OSI) and apoptosis by suppressing the expression of Wilms tumor 1 (WT1). Mechanistically, WT1 was identified as a direct target of miR-375, and WT1 could bind to the promoter region of miR-375 to promote its expression. Moreover, miR-375 was demonstrated to bind to the mRNA of SIX4, a transcription factor, and overexpression of miR-375 significantly reduced the expression of SIX4. These findings confirmed that WT1-AS exerts its regulatory function by inhibiting the expression of miR-375, which in turn influences the downstream SIX4 mRNA.
Further investigations revealed that WT1-AS functions by inhibiting the miR-375/SIX4 axis, thereby suppressing OSI and apoptosis, and this regulatory mechanism is mediated through the transcription factor WT1. Therefore, WT1-AS acts as a modulator of the miR-375/SIX4 axis through its interaction with WT1, influencing the pathological processes associated with AD and ultimately improving learning and memory abilities in AD mice.267
These findings shed light on the intricate regulatory network involving WT1-AS, miR-375, SIX4, and WT1 in AD pathology. Understanding the molecular mechanisms underlying these interactions provides potential avenues for therapeutic interventions aimed at modulating the fate of AD and ameliorating cognitive decline.
Other lncRNAs Associated with Microglia-Mediated Neuroinflammation
LncRNA Gm4419 was found to be significantly upregulated in microglia subjected to OGD/R treatment. Elevated levels of Gm4419 were observed to promote the phosphorylation of IkBa by physically binding to it. This, in turn, resulted in increased nuclear levels of NF-κB, subsequently activating the production of pro-inflammatory cytokines TNF-α, IL-1β, and IL-6.268
Following bilateral chronic constriction injury (BCCI), the expression of linc00311 and lncRNA-AK141205 was upregulated in spinal cord microglia. Inhibition of linc00311 and lncRNA-AK141205 expression led to downregulation of pSTAT3, COX-2, CCl-2, IL-1β, IL-6, and TNF-α in microglia, thereby inactivating the signaling pathway of STAT3 both in vivo and in vitro. Consequently, the inhibition of these factors resulted in reduced STAT3 activation and decreased production of pro-inflammatory cytokines, offering potential therapeutic targets for attenuating microglia-mediated neuroinflammation.269
LncRNA colorectal neoplasia differentially expressed (CRNDE) exhibited high expression levels in serum, amniotic fluid, and brain tissue of offspring rats with LPS-induced intrauterine infection. Downregulation of CRNDE through the use of shRNA inhibited the activation of astrocytes and microglia, as well as the secretion of pro-inflammatory factors in offspring rats. Furthermore, this downregulation improved spatial learning memory and mitigated brain histopathological changes, such as apoptosis, in brain-injured neonatal rats.270
In the context of traumatic brain injury (TBI), activation of microglia and astrocytes triggers innate immune and host defense responses. Administration of 7.5% hypertonic saline (HS) was found to regulate monocyte phenotype and improve intracranial pressure and coagulation fibrinolytic homeostasis through lncRNA2448-11 and lncRNA1403. These two lncRNAs positively regulated the expression of IL-6, IL-1, and TNF-α.271
The expression of LncRNA-1810034E14Rik was significantly downregulated in microglia stimulated with LPS or subjected to OGD. Overexpression of LncRNA-1810034E14Rik inhibited microglia activation, suppressed p65 phosphorylation, and reduced the expression of inflammatory cytokines. Similarly, overexpression of this lncRNA in the infarcted cortex area after middle cerebral artery occlusion (MCAO) reversed the increased levels of inflammatory cytokines. These findings highlight the potential of LncRNA-1810034E14Rik as a therapeutic target for controlling microglia-mediated neuroinflammation.272
LncRNA Ftx exhibited significantly decreased expression in spinal cord injury (SCI) mice tissues and LPS-stimulated BV2 cells. LPS induction of BV2 cells led to increased expression of miR-382-5p, which subsequently targeted the 3’UTR of Neuregulin-1 (Nrg1), resulting in decreased Nrg1 expression. Ftx acted as a regulator by targeting miR-382-5p and repressing its expression, as well as competing with Nrg1 for miR-382-5p binding. This led to the suppression of miR-382-5p’s repression of Nrg1 expression. Overexpression of Ftx increased Nrg1 expression, subsequently attenuating the inflammatory factors iNOS, IL-6, TNF-α, and IL-1β in LPS-treated BV2 cells. These findings suggest that upregulation of Ftx could be a potential therapeutic approach for alleviating microglial inflammation response in neuroinflammatory diseases.273
In a recent study, exosomes derived from hypoxia-treated adipose-derived stem cells were found to contain high levels of lncRNA Gm37494. These exosomes were able to promote a shift in microglia polarization from the pro-inflammatory M1 phenotype to the anti-inflammatory M2 phenotype by reducing the levels of miR-130b-3p and increasing the expression of PPARγ. This suggests that exosomes containing lncRNA Gm37494 have the potential to reduce inflammation in neurodegenerative diseases by promoting the neuroprotective M2 microglia polarization.274
LEF1-AS1, a type of lncRNA, was upregulated in microglia cells treated with LPS. Microglia cells treated with LPS showed apoptosis and increased expression of TNF-α and IL-6 when RAMP3 expression was enhanced or when a miR-222-5p inhibitor was used. LEF1-AS1 acted as a competing endogenous RNA (ceRNA) by sponging miR-222-5p. Knocking down LEF1-AS1 through siRNA increased miR-222-5p expression, reduced microglia apoptosis, and promoted their viability through the miR-222-5p-RAMP3 axis.275
LncRNA-F630028O10Rik was found to upregulate the expression of Col1a1 by acting as a sponge for miR-1231-5p. This increase in Col1a1 expression, in turn, enhanced microglial pyroptosis by activating the PI3K/AKT pathway. STAT1 was identified as the upstream transcriptional factor of lncRNA-F630028O10Rik, induced by the damage-responsive TLR4/MyD88 signaling pathway.276
Furthermore, LncRNAs TUG1 and lncSNHG15 were found to have dual effects on microglia activation, promoting one phenotype while suppressing the expression of the other. LncRNA TUG1 knockdown led to the conversion of microglia from an M1-like to an M2-like phenotype, resulting in decreased production of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6) and increased release of anti-inflammatory cytokines (IL-10). This effect was reversed by miR-145a-5p inhibitors. LncRNA TUG1 and miR-145a-5p exhibited a negative interaction, regulating microglia polarization and inflammatory cytokine production through the NF-κB pathway in the early stages of hypoxic brain injury.277
Palbociclib, a CDK6 inhibitor, was found to regulate the lncRNA SNHG15/CDK6/miR-627 pathway to overcome temozolomide resistance and reduce M2 polarization in glioblastoma multiforme-associated microglia. LncSNHG15 levels showed a positive correlation with oncogenes/markers such as EGFR, CDK6, SOX-2, and β-catenin. Silencing lncSNHG15 significantly decreased M2 polarization and promoted M1 polarization.278
These studies collectively demonstrate the significant role of lncRNAs in microglia activation. LncRNAs exert regulatory effects on microglia activation through direct targeting of specific factors and by acting as miRNA sponges. The JAK/STAT pathway, NF-κB signaling pathway, and Wnt/β-catenin signaling pathway are key mechanisms through which lncRNAs regulate microglia activation by directly targeting their associated factors. Additionally, lncRNAs play a role in regulating the transition between different phenotypes of microglia, further highlighting their importance in modulating microglia-mediated neuroinflammatory responses.
Discussion
NcRNAs have emerged as critical players in the pathogenesis of AD, unraveling the intricate relationship between ncRNAs, microglia, neuroinflammation, and AD progression. Microglia plays a pivotal role in the activation and regulation of neuroinflammation, a process linked to the pathogenesis of AD. Notably, miRNAs have been identified as key regulators of microglial activation in AD. MiR-155,113 miR-124,175 and miR-146a125 have been shown to modulate neuroinflammation in AD by targeting crucial signaling molecules involved in microglia activation. Dysregulation of these miRNAs has been associated with the production of pro-inflammatory cytokines and impaired clearance of Aβ plaques. Manipulating the expression of these miRNAs holds the potential for ameliorating microglia-mediated neuroinflammation in AD. CircRNAs have recently gained attention due to their unique circular structure and regulatory functions. CircPTK2 regulates microglia activation and hippocampal neuronal apoptosis through the miR-181c-5p-HMGB1 signaling pathway.211 Additionally, circ-Epc1 is believed to modulate the expression of TREM2 by acting as a sponge for miR-770-3p.212 Alterations in circRNA expression have been observed in AD, and their roles in regulating microglia activation and neuroinflammation are being unraveled. LncRNAs have also been implicated in microglial dysfunction and subsequent neuroinflammation in AD. LincRNA-Cox2 can interact with NF-κB p65 and enhance its nuclear translocation and transcriptional activity, resulting in the upregulation of pro-inflammatory cytokines.229 Targeting dysregulated lncRNAs holds the potential for therapeutically attenuating neuroinflammation and related neurodegenerative processes in AD. In addition to direct regulation of protein expression and modulation through miRNA sponge mechanisms, lncRNAs also exhibit a variety of regulatory mechanisms. Superimposed over the genomic and epigenomic programs, lncRNAs create an additional regulatory dimension: by interacting with the proteins and nucleic acids that regulate gene expression in the nucleus and cytoplasm, lncRNAs help establish robust, nimble, and specific transcriptional and post-transcriptional control.279 Furthermore, the regulatory effects of lncRNAs exhibit spatiotemporal and tissue-specific specificity. The cumulative evidence involving them in almost every cellular activity renders the assessment of their subcellular localization essential to fully understanding their biology. Their subcellular localization is critical to their function.280 Therefore, there is still much to be researched regarding the involvement of lncRNAs in microglial activation and neuroinflammation. Furthermore, the regulatory mechanisms of ncRNAs are highly complex, as they not only regulate microglial activation and neuroinflammation but also have an impact on other biological processes. For example, miR-223 targets Atg16l1, and its overexpression reduces Atg16l1 expression in BV2 cells, leading to decreased autophagy levels in microglia and an increase in activated microglia levels.87 LncRNA H19 not only drives HDAC1-dependent M1 microglia polarization, leading to increased inflammatory responses,248 but also inhibits excessive mitophagy by limiting Pink1 mRN A translation, thus alleviating this cardiac defect that occurs during obesity.281 Therefore, it is crucial to exercise caution when selecting these ncRNAs as therapeutic targets.
Understanding the intricate interplay between ncRNAs, microglia, neuroinflammation, and AD offers promising opportunities for developing novel therapeutic strategies. Targeting specific ncRNAs involved in microglial activation and neuroinflammation could potentially modulate the inflammatory response and mitigate the neurodegenerative processes associated with AD progression. However, several challenges remain. Overcoming the delivery barriers of ncRNAs to the central nervous system, such as the blood-brain barrier, necessitates innovative strategies like nanoparticle-based drug delivery systems. Additionally, the off-target effects and long-term consequences of manipulating ncRNA expression must be thoroughly evaluated to ensure proper regulation of the immune system in the brain. Besides, Considering the vast and continuously expanding family of ncRNAs, it is important to recognize that the same ncRNA can target different genes, and multiple ncRNAs can target the same gene. This presents both opportunities and challenges for future research. Comprehensive studies are necessary when targeting specific ncRNAs to overcome and minimize potential disadvantages while harnessing their benefits.
There are differences in ncRNA between M1 and M2 microglia (Table 4). However, it is important to acknowledge that microglial polarization, as traditionally defined by the M1/M2 activation states, has been challenged by recent research findings. Microglia activation in AD is a complex and dynamic process that involves highly heterogeneous microglial populations.282 To better understand the stage-specific responses of microglia in human AD, longitudinal transcriptomic and functional studies in different age groups are necessary. Integration of single-cell RNA sequencing with functional and histological data can aid in identifying distinct microglial phenotypes within transcriptionally classified microglial clusters.283
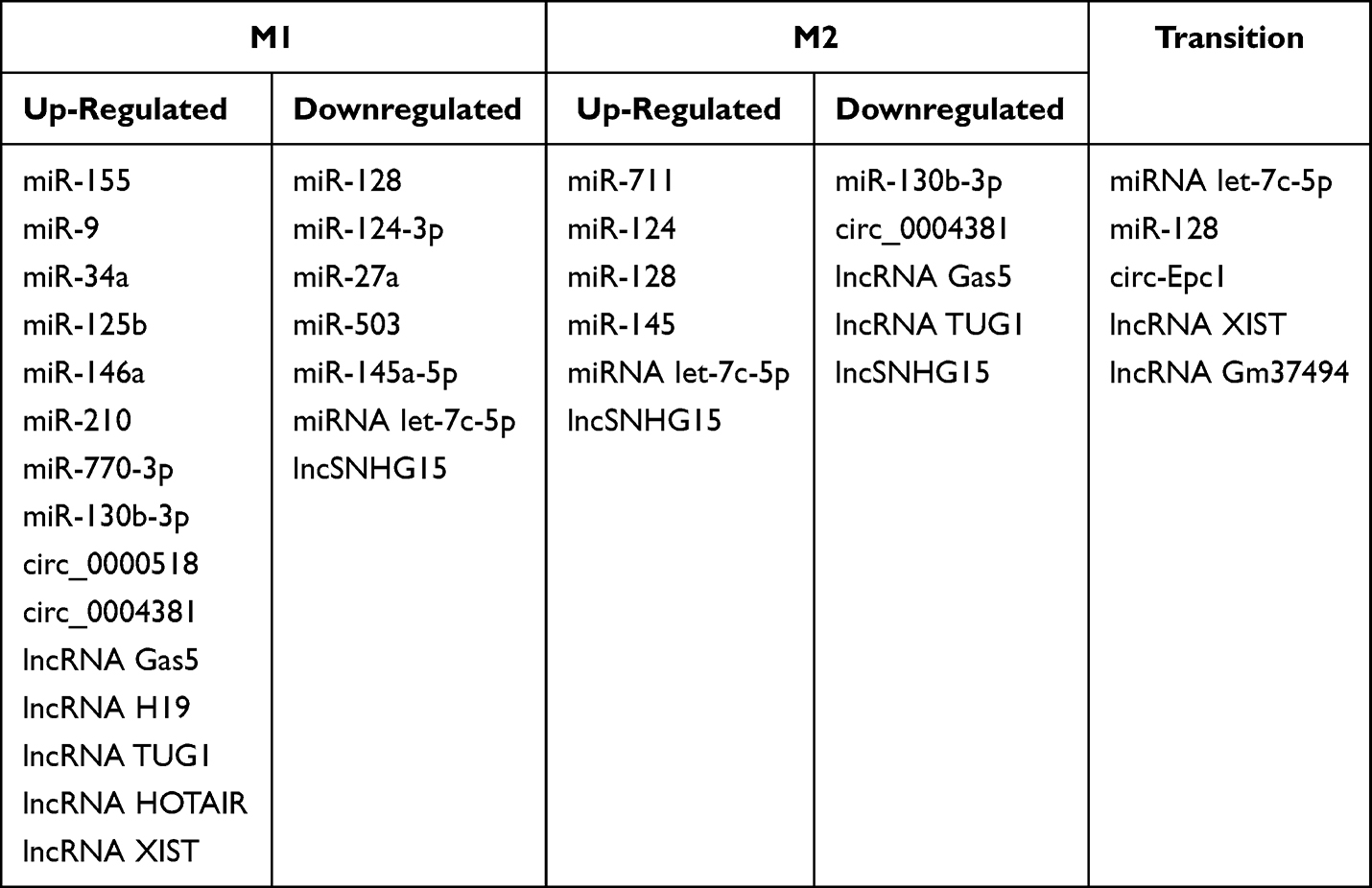 |
Table 4 The Differences of ncRNA Between M1 and M2 Microglia |
Furthermore, the context-dependent and highly plastic nature of microglia activation in AD should be considered. While some studies suggest that inhibiting microglia activation may be beneficial, other research highlights the protective effects of microglia activation. Therefore, careful consideration of the timing and extent of microglia inhibition or activation is crucial for the development of effective therapeutic approaches for AD. Future studies should focus on identifying specific microglial phenotypes and their associated therapeutic targets. Factors such as species, ontogeny, sex, genetic background, age, spatial location, and environmental influences, including nutrition, microbiota, pathogens, and drugs, collectively influence microglial states at multiple levels. These factors determine the functional properties of microglia, including epigenomic, transcriptomic, proteomic, metabolomic, ultrastructural, and phenomic characteristics.284 Understanding the regulation of microglia in such complexity is vital for the development of effective therapeutic strategies for neurodegenerative diseases.
Conclusion and Prospect
In conclusion, ncRNAs play a critical role in the regulation of microglia activation and neuroinflammation. They exert their effects by modulating gene expression and signaling pathways, thereby controlling inflammatory responses. Understanding the interplay between ncRNAs and microglia activation holds great promise for the development of novel therapies for neuroinflammatory diseases. miRNAs primarily regulate protein expression by inhibiting target mRNA transcription, translation, or inducing target mRNA degradation.74,285 This regulation affects key signaling pathways involved in microglia activation, including the PI3K/AKT pathway,140 JAK/STAT pathway,198 Caspase signaling pathway, NF-κB signaling pathway,195 and p38MAPK signaling pathway,162 which in turn regulate the production and release of inflammatory mediators. Additionally, circRNAs exert their regulatory effects on microglia activation mainly through sponge-like adsorption of miRNAs.207 Meanwhile, lncRNAs can regulate microglia activation by directly interacting with target proteins or acting as miRNA sponges, similar to circRNAs.226 Interestingly, lncRNAs can have dual effects on microglia activation, promoting or inhibiting activation and even facilitating the transition between different microglial phenotypes.227
Advancements in sequencing technology, genomics, transcriptomics, and RNAomics have greatly expanded our understanding of ncRNAs, providing researchers with new avenues of exploration and offering potential solutions for overcoming diseases and improving human health. NcRNAs not only serve as biomarkers (Table 5) but also represent important targets for the prevention and treatment of AD. Recent discoveries of the extensive regulatory networks formed by non-coding RNAs offer alternative targets and strategies to amplify the production of a specific protein. Khorkova et al discuss the growing range of RNA-targeted therapies in development that aim to boost gene expression, including nucleic acid-based therapeutics targeting the complex regulatory network of non-coding RNA species.286 The regulatory effects of these ncRNAs on genes and proteins, whether acting alone or in combination, directly or indirectly, have significant implications for microglia activation and the regulation of different polarization states. This sheds new light on the mechanisms underlying microglial actions in AD and enables in-depth studies of their functional mechanisms.
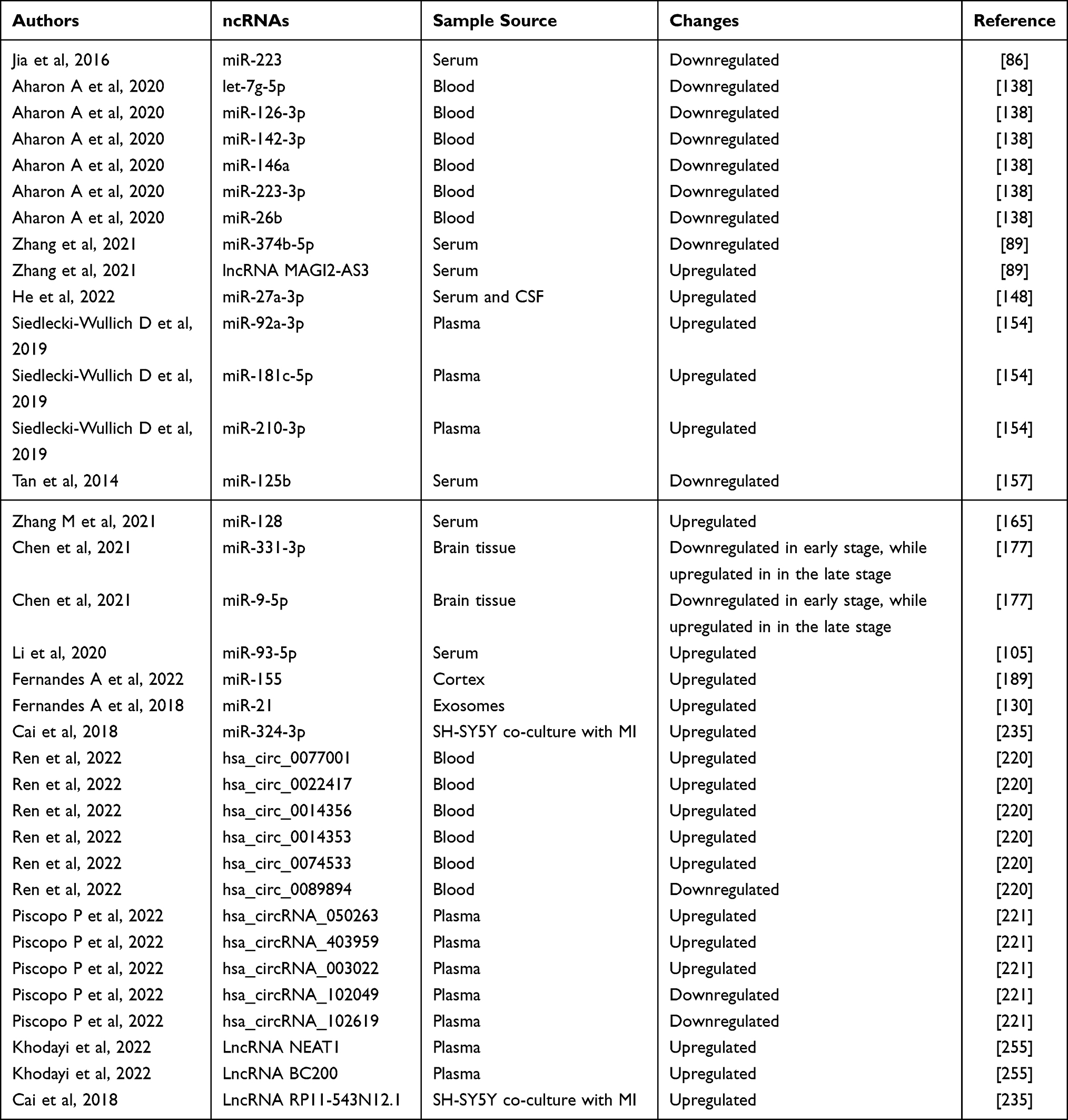 |
Table 5 Summary of the ncRNAs as biomarkers in microglia-mediated neuroinflammation |
In summary, the study of ncRNAs provides new insights into the mechanisms underlying microglia activation in AD. Further in-depth investigations are needed to establish the causal relationships among altered ncRNA expression and microglial activation and neuroinflammation in AD, ultimately paving the way for improved diagnostic tools and therapeutic interventions.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 82074046, 82004184); the Hunan Provincial “Shennong Talent” Project; the Natural Science Foundation of Hunan Province for Excellent Youth Project (Grant No.2023JJ20033); the Science and Technology Innovation Program of Hunan Province (Grant No.2023RC3166); Postgraduate Scientific Research Innovation Project of Hunan Province (Grant No.QL20220185); the Key Project for the Double First-Class Discipline of Integrating Traditional Chinese and Western Medicine in Hunan Province (Grant No.2021ZXYJH12).
Disclosure
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
1. Arvanitakis Z, Shah RC, Bennett DA. Diagnosis and Management of Dementia: Review. JAMA. 2019;322(16):1589–1599. doi:10.1001/jama.2019.4782
2. Jia J, Wei C, Chen S, et al. The cost of Alzheimer’s disease in China and re-estimation of costs worldwide. Alzheimers Dement. 2018;14(4):483–491. doi:10.1016/j.jalz.2017.12.006
3. Srivastava P, Tripathi PN, Sharma P, et al. Design and development of some phenyl benzoxazole derivatives as a potent acetylcholinesterase inhibitor with antioxidant property to enhance learning and memory. Eur J Med Chem. 2019;163:116–135. doi:10.1016/j.ejmech.2018.11.049
4. Tripathi PN, Srivastava P, Sharma P, et al. Biphenyl-3-oxo-1,2,4-triazine linked piperazine derivatives as potential cholinesterase inhibitors with anti-oxidant property to improve the learning and memory. Bioorg Chem. 2019;85:82–96. doi:10.1016/j.bioorg.2018.12.017
5. Long JM, Holtzman DM. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell. 2019;179(2):312–339. doi:10.1016/j.cell.2019.09.001
6. Rai SN, Singh C, Singh A, Singh MP, Singh BK. Mitochondrial Dysfunction: a Potential Therapeutic Target to Treat Alzheimer’s Disease. Mol Neurobiol. 2020;57(7):3075–3088. doi:10.1007/s12035-020-01945-y
7. Kloske CM, Wilcock DM. The Important interface between apolipoprotein E and neuroinflammation in Alzheimer’s disease. Front Immunol. 2020;11:754. doi:10.3389/fimmu.2020.00754
8. Colonna M, Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol. 2017;35:441–468. doi:10.1146/annurev-immunol-051116-052358
9. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. 2018;21(10):1359–1369. doi:10.1038/s41593-018-0242-x
10. Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener. 2020;9(1):42. doi:10.1186/s40035-020-00221-2
11. Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. J Cell Biol. 2018;217(2):459–472. doi:10.1083/jcb.201709069
12. Frost JL, Schafer DP. Microglia: Architects of the Developing Nervous System. Trends Cell Biol. 2016;26(8):587–597. doi:10.1016/j.tcb.2016.02.006
13. Gaire BP. Microglia as the Critical Regulators of Neuroprotection and Functional Recovery in Cerebral Ischemia. Cell Mol Neurobiol. 2022;42(8):2505–2525. doi:10.1007/s10571-021-01145-9
14. Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69. doi:10.1038/nrn2038
15. Lavisse S, Goutal S, Wimberley C, et al. Increased microglial activation in patients with Parkinson disease using [18F]-DPA714 TSPO PET imaging. Parkinsonism Relat Disord. 2021;82:29–36. doi:10.1016/j.parkreldis.2020.11.011
16. Bairamian D, Sha S, Rolhion N, et al. Microbiota in neuroinflammation and synaptic dysfunction: a focus on Alzheimer’s disease. Mol Neurodegener. 2022;17(1):19. doi:10.1186/s13024-022-00522-2
17. Shen H, He Z, Pei H, Zhai L, Guan Q, Wang G. Aurantiamide promotes M2 polarization of microglial cells to improve the cognitive ability of mice with Alzheimer’s disease. Phytother Res. 2023;37(1):101–110. doi:10.1002/ptr.7597
18. Tang Y, Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol. 2016;53(2):1181–1194. doi:10.1007/s12035-014-9070-5
19. Xie L, Liu Y, Zhang N, et al. Electroacupuncture Improves M2 Microglia Polarization and Glia Anti-inflammation of Hippocampus in Alzheimer’s Disease. Front Neurosci. 2021;15:689629. doi:10.3389/fnins.2021.689629
20. Jha MK, Lee WH, Suk K. Functional polarization of neuroglia: Implications in neuroinflammation and neurological disorders. Biochem Pharmacol. 2016;103:1–16. doi:10.1016/j.bcp.2015.11.003
21. Saito T, Saido TC. Neuroinflammation in mouse models of Alzheimer’s disease. Clin Exp Neuroimmunol. 2018;9(4):211–218. doi:10.1111/cen3.12475
22. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–934. doi:10.1016/j.cell.2010.02.016
23. Gupta N, Shyamasundar S, Patnala R, et al. Recent progress in therapeutic strategies for microglia-mediated neuroinflammation in neuropathologies. Expert Opin Ther Targets. 2018;22(9):765–781. doi:10.1080/14728222.2018.1515917
24. Yang Z, Liu B, Yang LE, Zhang C. Platycodigenin as Potential Drug Candidate for Alzheimer’s Disease via Modulating Microglial Polarization and Neurite Regeneration. Molecules. 2019;24(18). doi:10.3390/molecules24183207
25. Yao K, Zu HB. Microglial polarization: novel therapeutic mechanism against Alzheimer’s disease. Inflammopharmacology. 2020;28(1):95–110. doi:10.1007/s10787-019-00613-5
26. Rai SN, Chaturvedi VK, Singh BK, Singh MP. Commentary: Trem2 Deletion Reduces Late-Stage Amyloid Plaque Accumulation, Elevates the Aβ42:Aβ40 Ratio, and Exacerbates Axonal Dystrophy and Dendritic Spine Loss in the PS2APP Alzheimer’s Mouse Model. Front Aging Neurosci. 2020;12:219. doi:10.3389/fnagi.2020.00219
27. Ruganzu JB, Zheng Q, Wu X, et al.. TREM2 overexpression rescues cognitive deficits in APP/PS1 transgenic mice by reducing neuroinflammation via the JAK/STAT/SOCS signaling pathway. Exp Neurol. 2020:113506. doi:10.1016/j.expneurol.2020.113506
28. Xu Q, Xu W, Cheng H, Yuan H, Tan X. Efficacy and mechanism of cGAMP to suppress Alzheimer’s disease by elevating TREM2. Brain Behav Immun. 2019;81:495–508. doi:10.1016/j.bbi.2019.07.004
29. Gaojian T, Dingfei Q, Linwei L, et al. Parthenolide promotes the repair of spinal cord injury by modulating M1/M2 polarization via the NF-κB and STAT 1/3 signaling pathway. Cell Death Discov. 2020;6(1):97. doi:10.1038/s41420-020-00333-8
30. Qi S, Al Mamun A, Ngwa C, et al. X chromosome escapee genes are involved in ischemic sexual dimorphism through epigenetic modification of inflammatory signals. J Neuroinflammation. 2021;18(1):70. doi:10.1186/s12974-021-02120-3
31. Wu H, Zheng J, Xu S, et al. Mer regulates microglial/macrophage M1/M2 polarization and alleviates neuroinflammation following traumatic brain injury. J Neuroinflammation. 2021;18(1):2. doi:10.1186/s12974-020-02041-7
32. Laffer B, Bauer D, Wasmuth S, et al. Loss of IL-10 Promotes Differentiation of Microglia to a M1 Phenotype. Front Cell Neurosci. 2019;13:430. doi:10.3389/fncel.2019.00430
33. Liao X, Sharma N, Kapadia F, et al. Krüppel-like factor 4 regulates macrophage polarization. J Clin Invest. 2011;121(7):2736–2749. doi:10.1172/JCI45444
34. Ji J, Wang J, Yang J, et al. The Intra-nuclear SphK2-S1P Axis Facilitates M1-to-M2 Shift of Microglia via Suppressing HDAC1-Mediated KLF4 Deacetylation. Front Immunol. 2019;10:1241. doi:10.3389/fimmu.2019.01241
35. El-Deeb NK, El-Tanbouly DM, Khattab MA, El-Yamany MF, Mohamed AF. Crosstalk between PI3K/AKT/KLF4 signaling and microglia M1/M2 polarization as a novel mechanistic approach towards flibanserin repositioning in parkinson’s disease. Int Immunopharmacol. 2022;112:109191. doi:10.1016/j.intimp.2022.109191
36. Hsu CH, Pan YJ, Zheng YT, Lo RY, Yang FY. Ultrasound reduces inflammation by modulating M1/M2 polarization of microglia through STAT1/STAT6/PPARγ signaling pathways. CNS Neurosci Ther. 2023. doi:10.1111/cns.14333
37. Dräger NM, Sattler SM, Huang CT, et al. A CRISPRi/a platform in human iPSC-derived microglia uncovers regulators of disease states. Nat Neurosci. 2022;25(9):1149–1162. doi:10.1038/s41593-022-01131-4
38. Ryan KJ, White CC, Patel K, et al. A human microglia-like cellular model for assessing the effects of neurodegenerative disease gene variants. Sci Transl Med. 2017;9(421). doi:10.1126/scitranslmed.aai7635
39. Anwar S, Rivest S. Alzheimer’s disease: microglia targets and their modulation to promote amyloid phagocytosis and mitigate neuroinflammation. Expert Opin Ther Targets. 2020;24(4):331–344. doi:10.1080/14728222.2020.1738391
40. Lee J, Kim DE, Griffin P, et al. Inhibition of REV-ERBs stimulates microglial amyloid-beta clearance and reduces amyloid plaque deposition in the 5XFAD mouse model of Alzheimer’s disease. Aging Cell. 2020;19(2):e13078. doi:10.1111/acel.13078
41. Parbo P, Ismail R, Hansen KV, et al. Brain inflammation accompanies amyloid in the majority of mild cognitive impairment cases due to Alzheimer’s disease. Brain. 2017;140(7):2002–2011. doi:10.1093/brain/awx120
42. Fan Z, Okello AA, Brooks DJ, Edison P. Longitudinal influence of microglial activation and amyloid on neuronal function in Alzheimer’s disease. Brain. 2015;138(Pt 12):3685–3698. doi:10.1093/brain/awv288
43. Chiarini A, Armato U, Hu P, Dal Prà I. Danger-Sensing/Patten Recognition Receptors and Neuroinflammation in Alzheimer’s Disease. Int J Mol Sci. 2020;21(23). doi:10.3390/ijms21239036
44. Tejera D, Heneka MT. In Vivo Phagocytosis Analysis of Amyloid Beta. Methods Mol Biol. 2019;2034:287–292. doi:10.1007/978-1-4939-9658-2_21
45. Balducci C, Frasca A, Zotti M, et al. Toll-like receptor 4-dependent glial cell activation mediates the impairment in memory establishment induced by β-amyloid oligomers in an acute mouse model of Alzheimer’s disease. Brain Behav Immun. 2017;60:188–197. doi:10.1016/j.bbi.2016.10.012
46. Zusso M, Lunardi V, Franceschini D, et al. Ciprofloxacin and levofloxacin attenuate microglia inflammatory response via TLR4/NF-kB pathway. J Neuroinflammation. 2019;16(1):148. doi:10.1186/s12974-019-1538-9
47. Jha NK, Jha SK, Kar R, Nand P, Swati K, Goswami VK. Nuclear factor-kappa β as a therapeutic target for Alzheimer’s disease. J Neurochem. 2019;150(2):113–137. doi:10.1111/jnc.14687
48. Rai SN, Tiwari N, Singh P, et al. Therapeutic potential of vital transcription factors in Alzheimer’s and Parkinson’s disease with particular emphasis on transcription factor eb mediated autophagy. Front Neurosci. 2021;15:777347. doi:10.3389/fnins.2021.777347
49. Streit WJ, Xue QS, Tischer J, Bechmann I. Microglial pathology. Acta Neuropathol Commun. 2014;2:142. doi:10.1186/s40478-014-0142-6
50. Fu AK, Hung KW, Yuen MY, et al. IL-33 ameliorates Alzheimer’s disease-like pathology and cognitive decline. Proc Natl Acad Sci U S A. 2016;113(19):E2705–E2713. doi:10.1073/pnas.1604032113
51. Ren C, Li D, Zhou Q, Hu X. Mitochondria-targeted TPP-MoS2 with dual enzyme activity provides efficient neuroprotection through M1/M2 microglial polarization in an Alzheimer’s disease model. Biomaterials. 2020;232:119752. doi:10.1016/j.biomaterials.2019.119752
52. Perea JR, Llorens-Martín M, Ávila J, Bolós M. The Role of Microglia in the Spread of Tau: Relevance for Tauopathies. Front Cell Neurosci. 2018;12:172. doi:10.3389/fncel.2018.00172
53. Uddin MS, Lim LW. Glial cells in Alzheimer’s disease: From neuropathological changes to therapeutic implications. Ageing Res Rev. 2022;78:101622. doi:10.1016/j.arr.2022.101622
54. Zilka N, Kazmerova Z, Jadhav S, et al. Who fans the flames of Alzheimer’s disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J Neuroinflammation. 2012;9:47. doi:10.1186/1742-2094-9-47
55. Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68(1):19–31. doi:10.1016/j.neuron.2010.08.023
56. Maphis NM, Jiang S, Binder J, et al. Whole Genome Expression Analysis in a Mouse Model of Tauopathy Identifies MECP2 as a Possible Regulator of Tau Pathology. Front Mol Neurosci. 2017;10:69. doi:10.3389/fnmol.2017.00069
57. Maphis N, Xu G, Kokiko-Cochran ON, et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 2015;138(Pt 6):1738–1755. doi:10.1093/brain/awv081
58. Wang C, Xiong M, Gratuze M, et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron. 2021;109(10):1657–1674.e7. doi:10.1016/j.neuron.2021.03.024
59. Lin G, Zhu F, Kanaan NM, et al. Clioquinol Decreases Levels of Phosphorylated, Truncated, and Oligomerized Tau Protein. Int J Mol Sci. 2021;22(21). doi:10.3390/ijms222112063
60. Sun Y, Xiao Q, Luo C, et al. High-glucose induces tau hyperphosphorylation through activation of TLR9-P38MAPK pathway. Exp Cell Res. 2017;359(2):312–318. doi:10.1016/j.yexcr.2017.07.032
61. Beamer E, Corrêa S. The p38MAPK-MK2 Signaling Axis as a Critical Link Between Inflammation and Synaptic Transmission. Front Cell Dev Biol. 2021;9:635636. doi:10.3389/fcell.2021.635636
62. Liu Q, Zhang Y, Liu S, et al. Cathepsin C promotes microglia M1 polarization and aggravates neuroinflammation via activation of Ca2+-dependent PKC/p38MAPK/NF-κB pathway. J Neuroinflammation. 2019;16(1):10. doi:10.1186/s12974-019-1398-3
63. Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat Rev Drug Discov. 2017;16(3):167–179. doi:10.1038/nrd.2016.117
64. Idda ML, Munk R, Abdelmohsen K, Gorospe M. Noncoding RNAs in Alzheimer’s disease. Wiley Interdiscip Rev RNA. 2018;9(2). doi:10.1002/wrna.1463
65. Kim KW. PIWI Proteins and piRNAs in the Nervous System. Mol Cells. 2019;42(12):828–835. doi:10.14348/molcells.2019.0241
66. Wakisaka KT, Imai Y. The Dawn of pirna research in various neuronal disorders. Front Biosci. 2019;24(8):1440–1451. doi:10.2741/4789
67. Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer. 2018;18(1):5–18. doi:10.1038/nrc.2017.99
68. Adams BD, Parsons C, Walker L, Zhang WC, Slack FJ. Targeting noncoding RNAs in disease. J Clin Invest. 2017;127(3):761–771. doi:10.1172/JCI84424
69. Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018;141(4):1202–1207. doi:10.1016/j.jaci.2017.08.034
70. Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16(7):421–433. doi:10.1038/nrg3965
71. Guo Y, Hong W, Wang X, et al. MicroRNAs in Microglia: How do MicroRNAs Affect Activation, Inflammation, Polarization of Microglia and Mediate the Interaction Between Microglia and Glioma. Front Mol Neurosci. 2019;12:125. doi:10.3389/fnmol.2019.00125
72. Chen L, Heikkinen L, Wang C, Yang Y, Sun H, Wong G. Trends in the development of miRNA bioinformatics tools. Brief Bioinform. 2019;20(5):1836–1852. doi:10.1093/bib/bby054
73. Gulyaeva LF, Kushlinskiy NE. Regulatory mechanisms of microRNA expression. J Transl Med. 2016;14(1):143. doi:10.1186/s12967-016-0893-x
74. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15(8):509–524. doi:10.1038/nrm3838
75. Vasudeva K, Munshi A. miRNA dysregulation in ischaemic stroke: Focus on diagnosis, prognosis, therapeutic and protective biomarkers. Eur J Neurosci. 2020;52(6):3610–3627. doi:10.1111/ejn.14695
76. Li B, Dasgupta C, Huang L, Meng X, Zhang L. MiRNA-210 induces microglial activation and regulates microglia-mediated neuroinflammation in neonatal hypoxic-ischemic encephalopathy. Cell Mol Immunol. 2020;17(9):976–991. doi:10.1038/s41423-019-0257-6
77. Gullett JM, Chen Z, O’Shea A, et al. MicroRNA predicts cognitive performance in healthy older adults. Neurobiol Aging. 2020;95:186–194. doi:10.1016/j.neurobiolaging.2020.07.023
78. Pritchard CC, Cheng HH, Tewari M. MicroRNA profiling: approaches and considerations. Nat Rev Genet. 2012;13(5):358–369. doi:10.1038/nrg3198
79. Vishnoi A, Rani S. MiRNA Biogenesis and Regulation of Diseases: An Overview. Methods Mol Biol. 2017;1509:1–10. doi:10.1007/978-1-4939-6524-3_1
80. Freilich RW, Woodbury ME, Ikezu T. Integrated expression profiles of mRNA and miRNA in polarized primary murine microglia. PLoS One. 2013;8(11):e79416. doi:10.1371/journal.pone.0079416
81. Guedes JR, Custódia CM, Silva RJ, de Almeida LP, Pedroso de lima MC, Cardoso AL. Early miR-155 upregulation contributes to neuroinflammation in Alzheimer’s disease triple transgenic mouse model. Hum Mol Genet. 2014;23(23):6286–6301. doi:10.1093/hmg/ddu348
82. Jakaria M, Haque ME, Cho DY, Azam S, Kim IS, Choi DK. Molecular insights into NR4A2(Nurr1): an emerging target for neuroprotective therapy against neuroinflammation and neuronal cell death. Mol Neurobiol. 2019;56(8):5799–5814. doi:10.1007/s12035-019-1487-4
83. Zhang Y, Xu C, Nan Y, Nan S. Microglia-Derived Extracellular Vesicles Carrying miR-711 Alleviate Neurodegeneration in a Murine Alzheimer’s Disease Model by Binding to Itpkb. Front Cell Dev Biol. 2020;8:566530. doi:10.3389/fcell.2020.566530
84. Mishra R, Chhatbar C, Singh SK. HIV-1 Tat C-mediated regulation of tumor necrosis factor receptor-associated factor-3 by microRNA 32 in human microglia. J Neuroinflammation. 2012;9:131. doi:10.1186/1742-2094-9-131
85. Lv J, Zeng Y, Qian Y, Dong J, Zhang Z, Zhang J. MicroRNA let-7c-5p improves neurological outcomes in a murine model of traumatic brain injury by suppressing neuroinflammation and regulating microglial activation. Brain Res. 2018;1685:91–104. doi:10.1016/j.brainres.2018.01.032
86. Jia LH, Liu YN. Downregulated serum miR-223 servers as biomarker in Alzheimer’s disease. Cell Biochem Funct. 2016;34(4):233–237. doi:10.1002/cbf.3184
87. Li Y, Zhou D, Ren Y, et al. Mir223 restrains autophagy and promotes CNS inflammation by targeting ATG16L1. Autophagy. 2019;15(3):478–492. doi:10.1080/15548627.2018.1522467
88. Wei H, Xu Y, Chen Q, Chen H, Zhu X, Li Y. Mesenchymal stem cell-derived exosomal miR-223 regulates neuronal cell apoptosis. Cell Death Dis. 2020;11(4):290. doi:10.1038/s41419-020-2490-4
89. Zhang J, Wang R. Deregulated lncRNA MAGI2-AS3 in Alzheimer’s disease attenuates amyloid-β induced neurotoxicity and neuroinflammation by sponging miR-374b-5p. Exp Gerontol. 2021;144:111180. doi:10.1016/j.exger.2020.111180
90. Liang C, Zou T, Zhang M, et al. MicroRNA-146a switches microglial phenotypes to resist the pathological processes and cognitive degradation of Alzheimer’s disease. Theranostics. 2021;11(9):4103–4121. doi:10.7150/thno.53418
91. Kumar A, Bhatia HS, de Oliveira AC, Fiebich BL. microRNA-26a modulates inflammatory response induced by toll-like receptor 4 stimulation in microglia. J Neurochem. 2015;135(6):1189–1202. doi:10.1111/jnc.13364
92. Lv YN, Ou-Yang AJ, Fu LS. MicroRNA-27a Negatively Modulates the Inflammatory Response in Lipopolysaccharide-Stimulated Microglia by Targeting TLR4 and IRAK4. Cell Mol Neurobiol. 2017;37(2):195–210. doi:10.1007/s10571-016-0361-4
93. Zhou H, Zhang R, Lu K, et al. Deregulation of miRNA-181c potentially contributes to the pathogenesis of AD by targeting collapsin response mediator protein 2 in mice. J Neurol Sci. 2016;367:3–10. doi:10.1016/j.jns.2016.05.038
94. Wei Z, Yang C, Feng K, et al. p75NTR enhances cognitive dysfunction in a mouse Alzheimer’s disease model by inhibiting microRNA-210-3p-mediated PCYT2 through activation of NF-κB. Int J Biol Macromol. 2023;225:404–415. doi:10.1016/j.ijbiomac.2022.11.078
95. Li P, Xu Y, Wang B, Huang J, Li Q. miR-34a-5p and miR-125b-5p attenuate Aβ-induced neurotoxicity through targeting BACE1. J Neurol Sci. 2020;413:116793. doi:10.1016/j.jns.2020.116793
96. Li X, Zhang J, Yang Y, Wu Q, Ning H. MicroRNA-340-5p increases telomere length by targeting telomere protein POT1 to improve Alzheimer’s disease in mice. Cell Biol Int. 2021;45(6):1306–1315. doi:10.1002/cbin.11576
97. Liu Y, Zhang Y, Liu P, et al. MicroRNA-128 knockout inhibits the development of Alzheimer’s disease by targeting PPARγ in mouse models. Eur J Pharmacol. 2019;843:134–144. doi:10.1016/j.ejphar.2018.11.004
98. Morris G, Berk M, Maes M, Puri BK. Could Alzheimer’s Disease Originate in the Periphery and If So How So. Mol Neurobiol. 2019;56(1):406–434. doi:10.1007/s12035-018-1092-y
99. Chen ML, Hong CG, Yue T, et al. Erratum: Inhibition of miR-331-3p and miR-9-5p ameliorates Alzheimer’s disease by enhancing autophagy: Erratum. Theranostics. 2021;11(20):9774. doi:10.7150/thno.67227
100. Wan W, Liu G, Li X, et al. MiR-191-5p alleviates microglial cell injury by targeting Map3k12 (mitogen-activated protein kinase kinase kinase 12) to inhibit the MAPK (mitogen-activated protein kinase) signaling pathway in Alzheimer’s disease. Bioengineered. 2021;12(2):12678–12690. doi:10.1080/21655979.2021.2008638
101. Swarup V, Hinz FI, Rexach JE, et al. Identification of evolutionarily conserved gene networks mediating neurodegenerative dementia. Nat Med. 2019;25(1):152–164. doi:10.1038/s41591-018-0223-3
102. Ghiam S, Eslahchi C, Shahpasand K, Habibi-Rezaei M, Gharaghani S. Exploring the role of non-coding RNAs as potential candidate biomarkers in the cross-talk between diabetes mellitus and Alzheimer’s disease. Front Aging Neurosci. 2022;14:955461. doi:10.3389/fnagi.2022.955461
103. Xing H, Guo S, Zhang Y, Zheng Z, Wang H. Upregulation of microRNA-206 enhances lipopolysaccharide-induced inflammation and release of amyloid-β by targeting insulin-like growth factor 1 in microglia. Mol Med Rep. 2016;14(2):1357–1364. doi:10.3892/mmr.2016.5369
104. Zhao H, Wang J, Gao L, et al. MiRNA-424 protects against permanent focal cerebral ischemia injury in mice involving suppressing microglia activation. Stroke. 2013;44(6):1706–1713. doi:10.1161/STROKEAHA.111.000504
105. Li F, Xie XY, Sui XF, Wang P, Chen Z, Zhang JB. Profile of pathogenic proteins and MicroRNAs in plasma-derived extracellular vesicles in Alzheimer’s disease: a Pilot Study. Neuroscience. 2020;432:240–246. doi:10.1016/j.neuroscience.2020.02.044
106. Tian F, Yuan C, Hu L, Shan S. MicroRNA-93 inhibits inflammatory responses and cell apoptosis after cerebral ischemia reperfusion by targeting interleukin-1 receptor-associated kinase 4. Exp Ther Med. 2017;14(4):2903–2910. doi:10.3892/etm.2017.4874
107. Ghasemi-Kasman M, Shojaei A, Gol M, Moghadamnia AA, Baharvand H, Javan M. miR-302/367-induced neurons reduce behavioral impairment in an experimental model of Alzheimer’s disease. Mol Cell Neurosci. 2018;86:50–57. doi:10.1016/j.mcn.2017.11.012
108. Yuan B, Shen H, Lin L, Su T, Zhong L, Yang Z. MicroRNA367 negatively regulates the inflammatory response of microglia by targeting IRAK4 in intracerebral hemorrhage. J Neuroinflammation. 2015;12(1):206. doi:10.1186/s12974-015-0424-3
109. Tao W, Yu L, Shu S, et al. miR-204-3p/Nox4 Mediates Memory Deficits in a Mouse Model of Alzheimer’s Disease. Mol Ther. 2021;29(1):396–408. doi:10.1016/j.ymthe.2020.09.006
110. Niranjan R. Recent advances in the mechanisms of neuroinflammation and their roles in neurodegeneration. Neurochem Int. 2018;120:13–20. doi:10.1016/j.neuint.2018.07.003
111. Yan T, Zhang F, Sun C, et al. miR-32-5p-mediated Dusp5 downregulation contributes to neuropathic pain. Biochem Biophys Res Commun. 2018;495(1):506–511. doi:10.1016/j.bbrc.2017.11.013
112. Xie X, Peng L, Zhu J, et al. miR-145-5p/Nurr1/TNF-α Signaling-Induced Microglia Activation Regulates Neuron Injury of Acute Cerebral Ischemic/Reperfusion in Rats. Front Mol Neurosci. 2017;10:383. doi:10.3389/fnmol.2017.00383
113. Tufekci KU, Ercan I, Isci KB, et al. Sulforaphane inhibits NLRP3 inflammasome activation in microglia through Nrf2-mediated miRNA alteration. Immunol Lett. 2021;233:20–30. doi:10.1016/j.imlet.2021.03.004
114. Rastogi M, Singh SK. Japanese Encephalitis Virus exploits microRNA-155 to suppress the non-canonical NF-κB pathway in human microglial cells. Biochim Biophys Acta Gene Regul Mech. 2020;1863(11):194639. doi:10.1016/j.bbagrm.2020.194639
115. O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104(5):1604–1609. doi:10.1073/pnas.0610731104
116. Pedersen IM, Otero D, Kao E, et al. Onco-miR-155 targets SHIP1 to promote TNFalpha-dependent growth of B cell lymphomas. EMBO Mol Med. 2009;1(5):288–295. doi:10.1002/emmm.200900028
117. Guedes J, Cardoso AL, Pedroso de Lima MC. Involvement of microRNA in microglia-mediated immune response. Clin Dev Immunol. 2013;2013:186872. doi:10.1155/2013/186872
118. Teter B, Morihara T, Lim GP, et al. Curcumin restores innate immune Alzheimer’s disease risk gene expression to ameliorate Alzheimer pathogenesis. Neurobiol Dis. 2019;127:432–448. doi:10.1016/j.nbd.2019.02.015
119. Quinn SR, O’Neill LA. A trio of microRNAs that control Toll-like receptor signalling. Int Immunol. 2011;23(7):421–425. doi:10.1093/intimm/dxr034
120. Liu D, Zhao D, Zhao Y, Wang Y, Zhao Y, Wen C. Inhibition of microRNA-155 Alleviates Cognitive Impairment in Alzheimer’s Disease and Involvement of Neuroinflammation. Curr Alzheimer Res. 2019;16(6):473–482. doi:10.2174/1567205016666190503145207
121. Aloi MS, Prater KE, Sopher B, Davidson S, Jayadev S, Garden GA. The pro-inflammatory microRNA miR-155 influences fibrillar β-Amyloid1-42 catabolism by microglia. Glia. 2021;69(7):1736–1748. doi:10.1002/glia.23988
122. Martin NA, Hyrlov KH, Elkjaer ML, et al. Absence of miRNA-146a Differentially Alters Microglia Function and Proteome. Front Immunol. 2020;11:1110. doi:10.3389/fimmu.2020.01110
123. Lukiw WJ, Zhao Y, Cui JG. An NF-kappaB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J Biol Chem. 2008;283(46):31315–31322. doi:10.1074/jbc.M805371200
124. Williams AE, Perry MM, Moschos SA, Larner-Svensson HM, Lindsay MA. Role of miRNA-146a in the regulation of the innate immune response and cancer. Biochem Soc Trans. 2008;36(Pt 6):1211–1215. doi:10.1042/BST0361211
125. Yang J, Malone F, Go M, et al. Lipopolysaccharide-Induced Exosomal miR-146a Is Involved in Altered Expression of Alzheimer’s Risk Genes Via Suppression of TLR4 Signaling. J Mol Neurosci. 2021;71(6):1245–1255. doi:10.1007/s12031-020-01750-1
126. Mai H, Fan W, Wang Y, et al. Intranasal Administration of miR-146a Agomir Rescued the Pathological Process and Cognitive Impairment in an AD Mouse Model. Mol Ther Nucleic Acids. 2019;18:681–695. doi:10.1016/j.omtn.2019.10.002
127. Jayadev S, Case A, Alajajian B, Eastman AJ, Möller T, Garden GA. Presenilin 2 influences miR146 level and activity in microglia. J Neurochem. 2013;127(5):592–599. doi:10.1111/jnc.12400
128. Gaudet AD, Fonken LK, Watkins LR, Nelson RJ, Popovich PG. MicroRNAs: Roles in Regulating Neuroinflammation. Neuroscientist. 2018;24(3):221–245. doi:10.1177/1073858417721150
129. Cardoso AL, Guedes JR, de Lima MC. Role of microRNAs in the regulation of innate immune cells under neuroinflammatory conditions. Curr Opin Pharmacol. 2016;26:1–9. doi:10.1016/j.coph.2015.09.001
130. Fernandes A, Ribeiro AR, Monteiro M, Garcia G, Vaz AR, Brites D. Secretome from SH-SY5Y APPSwe cells trigger time-dependent CHME3 microglia activation phenotypes, ultimately leading to miR-21 exosome shuttling. Biochimie. 2018;155:67–82. doi:10.1016/j.biochi.2018.05.015
131. Caldeira C, Cunha C, Vaz AR, et al. Key Aging-Associated Alterations in Primary Microglia Response to Beta-Amyloid Stimulation. Front Aging Neurosci. 2017;9:277. doi:10.3389/fnagi.2017.00277
132. Cieślik M, Czapski GA, Wójtowicz S, et al. Alterations of Transcription of Genes Coding Anti-oxidative and Mitochondria-Related Proteins in Amyloid β Toxicity: Relevance to Alzheimer’s Disease. Mol Neurobiol. 2020;57(3):1374–1388. doi:10.1007/s12035-019-01819-y
133. Ghosh S, Dass J. Study of pathway cross-talk interactions with NF-κB leading to its activation via ubiquitination or phosphorylation: A brief review. Gene. 2016;584(1):97–109. doi:10.1016/j.gene.2016.03.008
134. Strickson S, Emmerich CH, Goh E, et al. Roles of the TRAF6 and Pellino E3 ligases in MyD88 and RANKL signaling. Proc Natl Acad Sci U S A. 2017;114(17):E3481–E3489. doi:10.1073/pnas.1702367114
135. Liang Y, Wang L. Inflamma-MicroRNAs in Alzheimer’s disease: from disease pathogenesis to therapeutic potentials. Front Cell Neurosci. 2021;15:785433. doi:10.3389/fncel.2021.785433
136. Zhao Y, Bhattacharjee S, Jones BM, Hill J, Dua P, Lukiw WJ. Regulation of neurotropic signaling by the inducible, NF-kB-sensitive miRNA-125b in Alzheimer’s disease (AD) and in primary human neuronal-glial (HNG) cells. Mol Neurobiol. 2014;50(1):97–106. doi:10.1007/s12035-013-8595-3
137. Wei H, Xu Y, Xu W, et al. Serum Exosomal miR-223 Serves as a Potential Diagnostic and Prognostic Biomarker for Dementia. Neuroscience. 2018;379:167–176. doi:10.1016/j.neuroscience.2018.03.016
138. Aharon A, Spector P, Ahmad RS, et al. Extracellular Vesicles of Alzheimer’s Disease Patients as a Biomarker for Disease Progression. Mol Neurobiol. 2020;57(10):4156–4169. doi:10.1007/s12035-020-02013-1
139. Pan Q, Ma J, Guo K. miR-223 Enhances the Neuroprotection of Estradiol Against Oxidative Stress Injury by Inhibiting the FOXO3/TXNIP Axis. Neurochem Res. 2022;47(7):1865–1877. doi:10.1007/s11064-021-03490-z
140. Wei H, Xu Y, Chen Q, Chen H, Zhu X, Li Y. Correction: mesenchymal stem cell-derived exosomal miR-223 regulates neuronal cell apoptosis. Cell Death Dis. 2020;11(6):431. doi:10.1038/s41419-020-2583-0
141. Neuner SM, Ding S, Kaczorowski CC. Knockdown of heterochromatin protein 1 binding protein 3 recapitulates phenotypic, cellular, and molecular features of aging. Aging Cell. 2019;18(1):e12886. doi:10.1111/acel.12886
142. Liu Y, Wang L, Xie F, et al. Overexpression of miR-26a-5p Suppresses Tau Phosphorylation and Aβ Accumulation in the Alzheimer’s Disease Mice by Targeting DYRK1A. Curr Neurovasc Res. 2020;17(3):241–248. doi:10.2174/1567202617666200414142637
143. Chaves J, Machado FT, Almeida MF, Bacovsky TB, Ferrari M. microRNAs expression correlates with levels of APP, DYRK1A, hyperphosphorylated Tau and BDNF in the hippocampus of a mouse model for Down syndrome during ageing. Neurosci Lett. 2020;714:134541. doi:10.1016/j.neulet.2019.134541
144. Lv M, Yang S, Cai L, Qin LQ, Li BY, Wan Z. Effects of quercetin intervention on cognition function in APP/PS1 Mice was affected by vitamin D status. Mol Nutr Food Res. 2018;62(24):e1800621. doi:10.1002/mnfr.201800621
145. Xie T, Pei Y, Shan P, et al. Identification of miRNA-mRNA Pairs in the Alzheimer’s Disease Expression Profile and Explore the Effect of miR-26a-5p/PTGS2 on Amyloid-β Induced Neurotoxicity in Alzheimer’s Disease Cell Model. Front Aging Neurosci. 2022;14:909222. doi:10.3389/fnagi.2022.909222
146. Li B, Sun H. MiR-26a promotes neurite outgrowth by repressing PTEN expression. Mol Med Rep. 2013;8(2):676–680. doi:10.3892/mmr.2013.1534
147. Sala Frigerio C, Lau P, Salta E, et al. Reduced expression of hsa-miR-27a-3p in CSF of patients with Alzheimer disease. Neurology. 2013;81(24):2103–2106. doi:10.1212/01.wnl.0000437306.37850.22
148. He L, Chen Z, Wang J, Feng H. Expression relationship and significance of NEAT1 and miR-27a-3p in serum and cerebrospinal fluid of patients with Alzheimer’s disease. BMC Neurol. 2022;22(1):203. doi:10.1186/s12883-022-02728-9
149. Su L, Li R, Zhang Z, Liu J, Du J, Wei H. Identification of altered exosomal microRNAs and mRNAs in Alzheimer’s disease. Ageing Res Rev. 2022;73:101497. doi:10.1016/j.arr.2021.101497
150. Dong LX, Zhang YY, Bao HL, Liu Y, Zhang GW, An FM. LncRNA NEAT1 promotes Alzheimer’s disease by down regulating micro-27a-3p. Am J Transl Res. 2021;13(8):8885–8896.
151. Hu Y, Wu L, Jiang L, et al. Notoginsenoside R2 reduces Aβ25-35-induced neuronal apoptosis and inflammation via miR-27a/SOX8/β-catenin axis. Hum Exp Toxicol. 2021;40(12_suppl):S347–S358. doi:10.1177/09603271211041996
152. Zhang L, Li YJ, Wu XY, Hong Z, Wei WS. MicroRNA-181c negatively regulates the inflammatory response in oxygen-glucose-deprived microglia by targeting Toll-like receptor 4. J Neurochem. 2015;132(6):713–723. doi:10.1111/jnc.13021
153. Hutchison ER, Kawamoto EM, Taub DD, et al. Evidence for miR-181 involvement in neuroinflammatory responses of astrocytes. Glia. 2013;61(7):1018–1028. doi:10.1002/glia.22483
154. Siedlecki-Wullich D, Català-Solsona J, Fábregas C, et al. Altered microRNAs related to synaptic function as potential plasma biomarkers for Alzheimer’s disease. Alzheimers Res Ther. 2019;11(1):46. doi:10.1186/s13195-019-0501-4
155. Mankhong S, Kim S, Moon S, et al. Circulating micro-RNAs differentially expressed in Korean Alzheimer’s patients with brain Aβ accumulation activate amyloidogenesis. J Gerontol A Biol Sci Med Sci. 2022. doi:10.1093/gerona/glac106
156. Parisi C, Napoli G, Amadio S, et al. MicroRNA-125b regulates microglia activation and motor neuron death in ALS. Cell Death Differ. 2016;23(3):531–541. doi:10.1038/cdd.2015.153
157. Tan L, Yu JT, Liu QY, et al. Circulating miR-125b as a biomarker of Alzheimer’s disease. J Neurol Sci. 2014;336(1–2):52–56. doi:10.1016/j.jns.2013.10.002
158. Hong H, Li Y, Su B. Identification of Circulating miR-125b as a Potential Biomarker of Alzheimer’s Disease in APP/PS1 Transgenic Mouse. J Alzheimers Dis. 2017;59(4):1449–1458. doi:10.3233/JAD-170156
159. Tang H, Ma M, Wu Y, et al. Activation of MT2 receptor ameliorates dendritic abnormalities in Alzheimer’s disease via C/EBPα/miR-125b pathway. Aging Cell. 2019;18(2):e12902. doi:10.1111/acel.12902
160. Micheli F, Palermo R, Talora C, Ferretti E, Vacca A, Napolitano M. Regulation of proapoptotic proteins Bak1 and p53 by miR-125b in an experimental model of Alzheimer’s disease: protective role of 17β-estradiol. Neurosci Lett. 2016;629:234–240. doi:10.1016/j.neulet.2016.05.049
161. Dobricic V, Schilling M, Schulz J, et al. Differential microRNA expression analyses across two brain regions in Alzheimer’s disease. Transl Psychiatry. 2022;12(1):352. doi:10.1038/s41398-022-02108-4
162. Qian Z, Chang J, Jiang F, et al. Excess administration of miR-340-5p ameliorates spinal cord injury-induced neuroinflammation and apoptosis by modulating the P38-MAPK signaling pathway. Brain Behav Immun. 2020;87:531–542. doi:10.1016/j.bbi.2020.01.025
163. Tan X, Luo Y, Pi D, Xia L, Li Z, Tu Q. MiR-340 reduces the accumulation of amyloid-β through targeting BACE1 (β-site Amyloid Precursor Protein Cleaving Enzyme 1) in Alzheimer’s disease. Curr Neurovasc Res. 2020;17(1):86–92. doi:10.2174/1567202617666200117103931
164. Yang Z, Xu J, Zhu R, Liu L. Down-Regulation of miRNA-128 Contributes to Neuropathic Pain Following Spinal Cord Injury via Activation of P38. Med Sci Monit. 2017;23:405–411. doi:10.12659/msm.898788
165. Zhang M, Han W, Xu Y, Li D, Xue Q. Serum miR-128 serves as a potential diagnostic biomarker for Alzheimer’s disease. Neuropsychiatr Dis Treat. 2021;17:269–275. doi:10.2147/NDT.S290925
166. Geng L, Zhang T, Liu W, Chen Y. Inhibition of miR-128 Abates Aβ-Mediated Cytotoxicity by Targeting PPAR-γ via NF-κB Inactivation in Primary Mouse Cortical Neurons and Neuro2a Cells. Yonsei Med J. 2018;59(9):1096–1106. doi:10.3349/ymj.2018.59.9.1096
167. Deng M, Zhang Q, Wu Z, et al. Mossy cell synaptic dysfunction causes memory imprecision via miR-128 inhibition of STIM2 in Alzheimer’s disease mouse model. Aging Cell. 2020;19(5):e13144. doi:10.1111/acel.13144
168. Shvarts-Serebro I, Sheinin A, Gottfried I, et al. miR-128 as a Regulator of Synaptic Properties in 5xFAD Mice Hippocampal Neurons. J Mol Neurosci. 2021;71(12):2593–2607. doi:10.1007/s12031-021-01862-2
169. Nguyen T, Kumar M, Fedele E, Bonanno G, Bonifacino T. MicroRNA Alteration, Application as Biomarkers, and Therapeutic Approaches in Neurodegenerative Diseases. Int J Mol Sci. 2022;23(9). doi:10.3390/ijms23094718
170. Sheinerman KS, Tsivinsky VG, Crawford F, Mullan MJ, Abdullah L, Umansky SR. Plasma microRNA biomarkers for detection of mild cognitive impairment. Aging. 2012;4(9):590–605. doi:10.18632/aging.100486
171. Song Y, Li Z, He T, et al. M2 microglia-derived exosomes protect the mouse brain from ischemia-reperfusion injury via exosomal miR-124. Theranostics. 2019;9(10):2910–2923. doi:10.7150/thno.30879
172. Hamzei Taj S, Kho W, Riou A, Wiedermann D, Hoehn M. MiRNA-124 induces neuroprotection and functional improvement after focal cerebral ischemia. Biomaterials. 2016;91:151–165. doi:10.1016/j.biomaterials.2016.03.025
173. Jiang D, Gong F, Ge X, et al. Neuron-derived exosomes-transmitted miR-124-3p protect traumatically injured spinal cord by suppressing the activation of neurotoxic microglia and astrocytes. J Nanobiotechnology. 2020;18(1):105. doi:10.1186/s12951-020-00665-8
174. Guo ML, Periyasamy P, Liao K, et al. Cocaine-mediated downregulation of microglial miR-124 expression involves promoter DNA methylation. Epigenetics. 2016;11(11):819–830. doi:10.1080/15592294.2016.1232233
175. Garcia G, Fernandes A, Stein F, Brites D. Protective Signature of IFNγ-Stimulated Microglia Relies on miR-124-3p Regulation From the Secretome Released by Mutant APP Swedish Neuronal Cells. Front Pharmacol. 2022;13:833066. doi:10.3389/fphar.2022.833066
176. Mastroeni D, Sekar S, Nolz J, et al. Correction: ANK1 is up-regulated in laser captured microglia in Alzheimer’s brain; the importance of addressing cellular heterogeneity. PLoS One. 2018;13(1):e0191382. doi:10.1371/journal.pone.0191382
177. Chen ML, Hong CG, Yue T, et al. Inhibition of miR-331-3p and miR-9-5p ameliorates Alzheimer’s disease by enhancing autophagy. Theranostics. 2021;11(5):2395–2409. doi:10.7150/thno.47408
178. Yang L, Chao J, Kook YH, Gao Y, Yao H, Buch SJ. Involvement of miR-9/MCPIP1 axis in PDGF-BB-mediated neurogenesis in neuronal progenitor cells. Cell Death Dis. 2013;4:e960. doi:10.1038/cddis.2013.486
179. Yao H, Ma R, Yang L, et al. MiR-9 promotes microglial activation by targeting MCPIP1. Nat Commun. 2014;5:4386. doi:10.1038/ncomms5386
180. Yang Z, Zhong L, Zhong S, Xian R, Yuan B. miR-203 protects microglia mediated brain injury by regulating inflammatory responses via feedback to MyD88 in ischemia. Mol Immunol. 2015;65(2):293–301. doi:10.1016/j.molimm.2015.01.019
181. Zhou HJ, Wang LQ, Xu QS, et al. Downregulation of miR-199b promotes the acute spinal cord injury through IKKβ-NF-κB signaling pathway activating microglial cells. Exp Cell Res. 2016;349(1):60–67. doi:10.1016/j.yexcr.2016.09.020
182. McNay EC, Pearson-Leary J. GluT4: A central player in hippocampal memory and brain insulin resistance. Exp Neurol. 2020;323:113076. doi:10.1016/j.expneurol.2019.113076
183. Mathew B, Acha LG, Torres LA, et al. MicroRNA-based engineering of mesenchymal stem cell extracellular vesicles for treatment of retinal ischemic disorders: engineered extracellular vesicles and retinal ischemia. Acta Biomater. 2023;158:782–797. doi:10.1016/j.actbio.2023.01.014
184. Li L, Sun Q, Li Y, et al. Overexpression of SIRT1 Induced by Resveratrol and Inhibitor of miR-204 Suppresses Activation and Proliferation of Microglia. J Mol Neurosci. 2015;56(4):858–867. doi:10.1007/s12031-015-0526-5
185. Raden M, Wallach T, Miladi M, et al. Structure-aware machine learning identifies microRNAs operating as Toll-like receptor 7/8 ligands. RNA Biol. 2021;18(sup1):268–277. doi:10.1080/15476286.2021.1940697
186. Markoutsa E, Mayilsamy K, Gulick D, Mohapatra SS, Mohapatra S. Extracellular vesicles derived from inflammatory-educated stem cells reverse brain inflammation-implication of miRNAs. Mol Ther. 2022;30(2):816–830. doi:10.1016/j.ymthe.2021.08.008
187. Brites D. Regulatory function of microRNAs in microglia. Glia. 2020;68(8):1631–1642. doi:10.1002/glia.23846
188. Ponomarev ED, Veremeyko T, Weiner HL. MicroRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS. Glia. 2013;61(1):91–103. doi:10.1002/glia.22363
189. Fernandes A, Caldeira C, Cunha C, Ferreiro E, Vaz AR, Brites D. Differences in Immune-Related Genes Underlie Temporal and Regional Pathological Progression in 3xTg-AD Mice. Cells. 2022;11(1). doi:10.3390/cells11010137
190. Lin X, Wang R, Li R, Tao T, Zhang D, Qi Y. Diagnostic Performance of miR-485-3p in Patients with Parkinson’s Disease and its Relationship with Neuroinflammation. Neuromolecular Med. 2022;24(2):195–201. doi:10.1007/s12017-021-08676-w
191. Konovalova J, Gerasymchuk D, Parkkinen I, Chmielarz P, Domanskyi A. Interplay between MicroRNAs and Oxidative Stress in Neurodegenerative Diseases. Int J Mol Sci. 2019;20(23). doi:10.3390/ijms20236055
192. Zhang Z, Yi P, Yi M, et al. Protective effect of quercetin against H2O2-induced oxidative damage in PC-12 Cells: comprehensive analysis of a lncRNA-associated ceRNA network. Oxid Med Cell Longev. 2020;2020:6038919. doi:10.1155/2020/6038919
193. Chen M, Li L, Liu C, Song L. Berberine attenuates Aβ-induced neuronal damage through regulating miR-188/NOS1 in Alzheimer’s disease. Mol Cell Biochem. 2020;474(1–2):285–294. doi:10.1007/s11010-020-03852-1
194. Falcão AS, Carvalho LA, Lidónio G, et al. Dipeptidyl Vinyl Sulfone as a novel chemical tool to inhibit HMGB1/NLRP3-inflammasome and Inflamma-miRs in Aβ-mediated microglial inflammation. ACS Chem Neurosci. 2017;8(1):89–99. doi:10.1021/acschemneuro.6b00250
195. Amjad N, Yang R, Li L, et al. Decrease of miR-19b-3p in Brain Microvascular Endothelial Cells Attenuates Meningitic Escherichia coli-Induced Neuroinflammation via TNFAIP3-Mediated NF-κB Inhibition. Pathogens. 2019;8(4). doi:10.3390/pathogens8040268
196. Paschon V, Takada SH, Ikebara JM, et al. Interplay Between Exosomes, microRNAs and Toll-Like Receptors in Brain Disorders. Mol Neurobiol. 2016;53(3):2016–2028. doi:10.1007/s12035-015-9142-1
197. Borbet TC, Hines MJ, Koralov SB. MicroRNA regulation of B cell receptor signaling. Immunol Rev. 2021;304(1):111–125. doi:10.1111/imr.13024
198. Zhang M, Ye Y, Cong J, et al. Regulation of STAT3 by miR-106a is linked to cognitive impairment in ovariectomized mice. Brain Res. 2013;1503:43–52. doi:10.1016/j.brainres.2013.01.052
199. Kristensen LS, Andersen MS, Stagsted L, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20(11):675–691. doi:10.1038/s41576-019-0158-7
200. Abdelmohsen K, Panda AC, Munk R, et al. Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol. 2017;14(3):361–369. doi:10.1080/15476286.2017.1279788
201. Du WW, Yang W, Liu E, Yang Z, Dhaliwal P, Yang BB. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016;44(6):2846–2858. doi:10.1093/nar/gkw027
202. Hanan M, Soreq H, Kadener S. CircRNAs in the brain. RNA Biol. 2017;14(8):1028–1034. doi:10.1080/15476286.2016.1255398
203. Li R, Jiang J, Shi H, Qian H, Zhang X, Xu W. CircRNA: a rising star in gastric cancer. Cell Mol Life Sci. 2020;77(9):1661–1680. doi:10.1007/s00018-019-03345-5
204. Meng S, Zhou H, Feng Z, et al. CircRNA: functions and properties of a novel potential biomarker for cancer. Mol Cancer. 2017;16(1):94. doi:10.1186/s12943-017-0663-2
205. Rybak-Wolf A, Stottmeister C, Glažar P, et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol Cell. 2015;58(5):870–885. doi:10.1016/j.molcel.2015.03.027
206. Curry-Hyde A, Gray L, Chen BJ, Ueberham U, Arendt T, Janitz M. Cell type-specific circular RNA expression in human glial cells. Genomics. 2020. doi:10.1016/j.ygeno.2020.09.042
207. Wu T, Li Y, Liang X, Liu X, Tang M. Identification of potential circRNA-miRNA-mRNA regulatory networks in response to graphene quantum dots in microglia by microarray analysis. Ecotoxicol Environ Saf. 2021;208:111672. doi:10.1016/j.ecoenv.2020.111672
208. Jiang Y, Zhao J, Xu J, et al. Glioblastoma-associated microglia-derived exosomal circKIF18A promotes angiogenesis by targeting FOXC2. Oncogene. 2022;41(26):3461–3473. doi:10.1038/s41388-022-02360-4
209. Jiang F, Liu X, Cui X, et al. Circ_0000518 Promotes Macrophage/Microglia M1 Polarization via the FUS/CaMKKβ/AMPK Pathway to Aggravate Multiple Sclerosis. Neuroscience. 2022;490:131–143. doi:10.1016/j.neuroscience.2021.12.012
210. Xiaoying G, Guo M, Jie L, et al. CircHivep2 contributes to microglia activation and inflammation via miR-181a-5p/SOCS2 signalling in mice with kainic acid-induced epileptic seizures. J Cell Mol Med. 2020. doi:10.1111/jcmm.15894
211. Li M, Hu J, Peng Y, Li J, Ren R. CircPTK2-miR-181c-5p-HMGB1: a new regulatory pathway for microglia activation and hippocampal neuronal apoptosis induced by sepsis. Mol Med. 2021;27(1):45. doi:10.1186/s10020-021-00305-3
212. Liu H, Jin M, Ji M, Zhang W, Liu A, Wang T. Hypoxic pretreatment of adipose-derived stem cell exosomes improved cognition by delivery of circ-Epc1 and shifting microglial M1/M2 polarization in an Alzheimer’s disease mice model. Aging. 2022;14(7):3070–3083. doi:10.18632/aging.203989
213. Urdánoz-Casado A, de Gordoa JS, Robles M, et al. Profile of TREM2-Derived circRNA and mRNA Variants in the Entorhinal Cortex of Alzheimer’s Disease Patients. Int J Mol Sci. 2022;23(14). doi:10.3390/ijms23147682
214. Li N, Zhang D, Guo H, Yang Q, Li P, He Y. Inhibition of circ_0004381 improves cognitive function via miR-647/PSEN1 axis in an Alzheimer disease mouse model. J Neuropathol Exp Neurol. 2022;82(1):84–92. doi:10.1093/jnen/nlac108
215. Akhter R. Circular RNA and Alzheimer’s Disease. Adv Exp Med Biol. 2018;1087:239–243. doi:10.1007/978-981-13-1426-1_19
216. Xu X, Gu D, Xu B, Yang C, Wang L. Circular RNA circ_0005835 promotes promoted neural stem cells proliferation and differentiate to neuron and inhibits inflammatory cytokines levels through miR-576-3p in Alzheimer’s disease. Environ Sci Pollut Res Int. 2022;29(24):35934–35943. doi:10.1007/s11356-021-17478-3
217. Wang H, Li Z, Gao J, Liao Q. Circular RNA circPTK2 regulates oxygen-glucose deprivation-activated microglia-induced hippocampal neuronal apoptosis via miR-29b-SOCS-1-JAK2/STAT3-IL-1β signaling. Int J Biol Macromol. 2019;129:488–496. doi:10.1016/j.ijbiomac.2019.02.041
218. Li Y, Han X, Fan H, et al. Circular RNA AXL increases neuron injury and inflammation through targeting microRNA-328 mediated BACE1 in Alzheimer’s disease. Neurosci Lett. 2022;776:136531. doi:10.1016/j.neulet.2022.136531
219. Chen HH, Eteleeb A, Wang C, et al. Circular RNA detection identifies circPSEN1 alterations in brain specific to autosomal dominant Alzheimer’s disease. Acta Neuropathol Commun. 2022;10(1):29. doi:10.1186/s40478-022-01328-5
220. Ren Z, Chu C, Pang Y, Cai H, Jia L. A circular RNA blood panel that differentiates Alzheimer’s disease from other dementia types. Biomark Res. 2022;10(1):63. doi:10.1186/s40364-022-00405-0
221. Piscopo P, Manzini V, Rivabene R, et al. A Plasma Circular RNA Profile Differentiates Subjects with Alzheimer’s Disease and Mild Cognitive Impairment from Healthy Controls. Int J Mol Sci. 2022;23(21). doi:10.3390/ijms232113232
222. Cabili MN, Trapnell C, Goff L, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25(18):1915–1927. doi:10.1101/gad.17446611
223. Clark BS, Blackshaw S. Long non-coding RNA-dependent transcriptional regulation in neuronal development and disease. Front Genet. 2014;5:164. doi:10.3389/fgene.2014.00164
224. Ferrè F, Colantoni A, Helmer-Citterich M. Revealing protein-lncRNA interaction. Brief Bioinform. 2016;17(1):106–116. doi:10.1093/bib/bbv031
225. Li JH, Liu S, Zheng LL, et al. Discovery of Protein-lncRNA Interactions by Integrating Large-Scale CLIP-Seq and RNA-Seq Datasets. Front Bioeng Biotechnol. 2014;2:88. doi:10.3389/fbioe.2014.00088
226. Paraskevopoulou MD, Hatzigeorgiou AG. Analyzing MiRNA-LncRNA Interactions. Methods Mol Biol. 2016;1402:271–286. doi:10.1007/978-1-4939-3378-5_21
227. Gao X, Cao Z, Tan H, et al. LncRNA, an Emerging Approach for Neurological Diseases Treatment by Regulating Microglia Polarization. Front Neurosci. 2022;16:903472. doi:10.3389/fnins.2022.903472
228. Feng X, Zhan F, Luo D, et al. LncRNA 4344 promotes NLRP3-related neuroinflammation and cognitive impairment by targeting miR-138-5p. Brain Behav Immun. 2021;98:283–298. doi:10.1016/j.bbi.2021.08.230
229. Xue Z, Zhang Z, Liu H, et al. lincRNA-Cox2 regulates NLRP3 inflammasome and autophagy mediated neuroinflammation. Cell Death Differ. 2019;26(1):130–145. doi:10.1038/s41418-018-0105-8
230. Jing W, Tuxiu X, Xiaobing L, et al. LncRNA GAS5/miR-137 Is a Hypoxia-Responsive Axis Involved in Cardiac Arrest and Cardiopulmonary Cerebral Resuscitation. Front Immunol. 2021;12:790750. doi:10.3389/fimmu.2021.790750
231. Zhang YY, Bao HL, Dong LX, Liu Y, Zhang GW, An FM. Silenced lncRNA H19 and up-regulated microRNA-129 accelerates viability and restrains apoptosis of PC12 cells induced by Aβ25-35 in a cellular model of Alzheimer’s disease. Cell Cycle. 2021;20(1):112–125. doi:10.1080/15384101.2020.1863681
232. Li L, Xu Y, Zhao M, Gao Z. Neuro-protective roles of long non-coding RNA MALAT1 in Alzheimer’s disease with the involvement of the microRNA-30b/CNR1 network and the following PI3K/AKT activation. Exp Mol Pathol. 2020;117:104545. doi:10.1016/j.yexmp.2020.104545
233. Lu J, Liu L, Chen J, et al. The Involvement of lncRNA HOTAIR/miR-130a-3p Axis in the Regulation of Voluntary Exercise on Cognition and Inflammation of Alzheimer’s Disease. Am J Alzheimers Dis Other Demen. 2022;37:15333175221091424. doi:10.1177/15333175221091424
234. Duan R, Wang SY, Wei B, et al. Angiotensin-(1-7) Analogue AVE0991 Modulates Astrocyte-Mediated Neuroinflammation via lncRNA SNHG14/miR-223-3p/NLRP3 Pathway and Offers Neuroprotection in a Transgenic Mouse Model of Alzheimer’s Disease. J Inflamm Res. 2021;14:7007–7019. doi:10.2147/JIR.S343575
235. Cai M, Wang YW, Xu SH, et al. Regulatory effects of the long non‑coding RNA RP11‑543N12.1 and microRNA‑324‑3p axis on the neuronal apoptosis induced by the inflammatory reactions of microglia. Int J Mol Med. 2018;42(3):1741–1755. doi:10.3892/ijmm.2018.3736
236. Yue D, Guanqun G, Jingxin L, et al. Silencing of long noncoding RNA XIST attenuated Alzheimer’s disease-related BACE1 alteration through miR-124. Cell Biol Int. 2020;44(2):630–636. doi:10.1002/cbin.11263
237. Qin YR, Ma CQ, Wang DP, et al. Bilobalide alleviates neuroinflammation and promotes autophagy in Alzheimer’s disease by upregulating lincRNA-p21. Am J Transl Res. 2021;13(4):2021–2040.
238. Hu G, Gong AY, Wang Y, et al. LincRNA-Cox2 Promotes Late Inflammatory Gene Transcription in Macrophages through Modulating SWI/SNF-Mediated Chromatin Remodeling. J Immunol. 2016;196(6):2799–2808. doi:10.4049/jimmunol.1502146
239. Sun D, Yu Z, Fang X, et al. LncRNA GAS5 inhibits microglial M2 polarization and exacerbates demyelination. EMBO Rep. 2017;18(10):1801–1816. doi:10.15252/embr.201643668
240. Chen X, Ren G, Li Y, et al. Level of LncRNA GAS5 and hippocampal volume are associated with the progression of Alzheimer’s disease. Clin Interv Aging. 2022;17:745–753. doi:10.2147/CIA.S363116
241. Xu W, Zhang L, Geng Y, Liu Y, Zhang N. Long noncoding RNA GAS5 promotes microglial inflammatory response in Parkinson’s disease by regulating NLRP3 pathway through sponging miR-223-3p. Int Immunopharmacol. 2020;85:106614. doi:10.1016/j.intimp.2020.106614
242. Zhang H, Lu M, Zhang X, et al. Isosteviol Sodium Protects against Ischemic Stroke by Modulating Microglia/Macrophage Polarization via Disruption of GAS5/miR-146a-5p sponge. Sci Rep. 2019;9(1):12221. doi:10.1038/s41598-019-48759-0
243. Jiang Z, Liu M, Huang D, Cai Y, Zhou Y. Silencing of long noncoding RNA GAS5 blocks experimental cerebral ischemia-reperfusion injury by restraining AQP4 expression via the miR-1192/STAT5A axis. Mol Neurobiol. 2022;59(12):7450–7465. doi:10.1007/s12035-022-03045-5
244. Wang L, Zhang Z, Wang H. Downregulation of lncRNA GAS5 prevents mitochondrial apoptosis and hypoxic-ischemic brain damage in neonatal rats through the microRNA-128-3p/Bax/Akt/GSK-3β axis. Neuroreport. 2021;32(17):1395–1402. doi:10.1097/WNR.0000000000001730
245. Han Y, Wu N, Xia F, Liu S, Jia D. Long non‑coding RNA GAS5 regulates myocardial ischemia‑reperfusion injury through the PI3K/AKT apoptosis pathway by sponging miR‑532‑5p. Int J Mol Med. 2020;45(3):858–872. doi:10.3892/ijmm.2020.4471
246. Wu Y, Rong W, Jiang Q, Wang R, Huang H. Downregulation of lncRNA GAS5 Alleviates Hippocampal Neuronal Damage in Mice with Depression-Like Behaviors Via Modulation of MicroRNA-26a/EGR1 Axis. J Stroke Cerebrovasc Dis. 2021;30(3):105550. doi:10.1016/j.jstrokecerebrovasdis.2020.105550
247. Chen F, Zhang L, Wang E, Zhang C, Li X. LncRNA GAS5 regulates ischemic stroke as a competing endogenous RNA for miR-137 to regulate the Notch1 signaling pathway. Biochem Biophys Res Commun. 2018;496(1):184–190. doi:10.1016/j.bbrc.2018.01.022
248. Wang J, Zhao H, Fan Z, et al. Long Noncoding RNA H19 Promotes Neuroinflammation in Ischemic Stroke by Driving Histone Deacetylase 1-Dependent M1 Microglial Polarization. Stroke. 2017;48(8):2211–2221. doi:10.1161/STROKEAHA.117.017387
249. Han CL, Ge M, Liu YP, et al. LncRNA H19 contributes to hippocampal glial cell activation via JAK/STAT signaling in a rat model of temporal lobe epilepsy. J Neuroinflammation. 2018;15(1):103. doi:10.1186/s12974-018-1139-z
250. Han CL, Liu YP, Guo CJ, et al. The lncRNA H19 binding to let-7b promotes hippocampal glial cell activation and epileptic seizures by targeting Stat3 in a rat model of temporal lobe epilepsy. Cell Prolif. 2020;53(8):e12856. doi:10.1111/cpr.12856
251. Wang LQ, Zhou HJ. LncRNA MALAT1 promotes high glucose-induced inflammatory response of microglial cells via provoking MyD88/IRAK1/TRAF6 signaling. Sci Rep. 2018;8(1):8346. doi:10.1038/s41598-018-26421-5
252. Cai LJ, Tu L, Huang XM, et al. LncRNA MALAT1 facilitates inflammasome activation via epigenetic suppression of Nrf2 in Parkinson’s disease. Mol Brain. 2020;13(1):130. doi:10.1186/s13041-020-00656-8
253. Zhou HJ, Wang LQ, Wang DB, et al. Long noncoding RNA MALAT1 contributes to inflammatory response of microglia following spinal cord injury via the modulation of a miR-199b/IKKβ/NF-κB signaling pathway. Am J Physiol Cell Physiol. 2018;315(1):C52–C61. doi:10.1152/ajpcell.00278.2017
254. Han D, Zhou Y. YY1-induced upregulation of lncRNA NEAT1 contributes to OGD/R injury-induced inflammatory response in cerebral microglial cells via Wnt/β-catenin signaling pathway. Vitro Cell Dev Biol Anim. 2019;55(7):501–511. doi:10.1007/s11626-019-00375-y
255. Khodayi M, Khalaj-Kondori M, Hoseinpour Feizi MA, Jabarpour Bonyadi M, Talebi M. Plasma lncRNA profiling identified BC200 and NEAT1 lncRNAs as potential blood-based biomarkers for late-onset Alzheimer’s disease. EXCLI J. 2022;21:772–785. doi:10.17179/excli2022-4764
256. Huang Z, Zhao J, Wang W, Zhou J, Zhang J. Depletion of LncRNA NEAT1 Rescues Mitochondrial Dysfunction Through NEDD4L-Dependent PINK1 Degradation in Animal Models of Alzheimer’s Disease. Front Cell Neurosci. 2020;14:28. doi:10.3389/fncel.2020.00028
257. Cheng S, Zhang Y, Chen S, Zhou Y. LncRNA HOTAIR Participates in Microglia Activation and Inflammatory Factor Release by Regulating the Ubiquitination of MYD88 in Traumatic Brain Injury. J Mol Neurosci. 2020. doi:10.1007/s12031-020-01623-7
258. Duan C, Liu Y, Li Y, et al. Sulfasalazine alters microglia phenotype by competing endogenous RNA effect of miR-136-5p and long non-coding RNA HOTAIR in cuprizone-induced demyelination. Biochem Pharmacol. 2018;155:110–123. doi:10.1016/j.bcp.2018.06.028
259. Wang Y, Zhao S, Li G, Wang D, Jin Y. Neuroprotective Effect of HOTAIR Silencing on Isoflurane-Induced Cognitive Dysfunction via Sponging microRNA-129-5p and Inhibiting Neuroinflammation. Neuroimmunomodulation. 2022;29(4):369–379. doi:10.1159/000521014
260. Zhao J, Li H, Chang N. LncRNA HOTAIR promotes MPP+-induced neuronal injury in Parkinson’s disease by regulating the miR-874-5p/ATG10 axis. EXCLI J. 2020;19:1141–1153. doi:10.17179/excli2020-2286
261. Lu J, Liu L, Chen J, et al. LncRNA HOTAIR in exercise-induced neuro-protective function in Alzheimer’s disease. Folia Neuropathol. 2022;60(4):414–420. doi:10.5114/fn.2022.118961
262. Qi X, Shao M, Sun H, Shen Y, Meng D, Huo W. Long non-coding RNA SNHG14 promotes microglia activation by regulating miR-145-5p/PLA2G4A in cerebral infarction. Neuroscience. 2017;348:98–106. doi:10.1016/j.neuroscience.2017.02.002
263. Zhao Q, Lu F, Su Q, et al. Knockdown of long noncoding RNA XIST mitigates the apoptosis and inflammatory injury of microglia cells after spinal cord injury through miR-27a/Smurf1 axis. Neurosci Lett. 2020;715:134649. doi:10.1016/j.neulet.2019.134649
264. Xing F, Liu Y, Wu SY, et al. Loss of XIST in Breast Cancer Activates MSN-c-Met and Reprograms Microglia via Exosomal miRNA to Promote Brain Metastasis. Cancer Res. 2018;78(15):4316–4330. doi:10.1158/0008-5472.CAN-18-1102
265. Yan XW, Liu HJ, Hong YX, Meng T, Du J, Chang C. lncRNA XIST induces Aβ accumulation and neuroinflammation by the epigenetic repression of NEP in Alzheimer’s disease. J Neurogenet. 2022;36(1):11–20. doi:10.1080/01677063.2022.2028784
266. Ye Y, He X, Lu F, et al. A lincRNA-p21/miR-181 family feedback loop regulates microglial activation during systemic LPS- and MPTP- induced neuroinflammation. Cell Death Dis. 2018;9(8):803. doi:10.1038/s41419-018-0821-5
267. Wang Q, Ge X, Zhang J, Chen L. Effect of lncRNA WT1-AS regulating WT1 on oxidative stress injury and apoptosis of neurons in Alzheimer’s disease via inhibition of the miR-375/SIX4 axis. Aging. 2020;12(23):23974–23995. doi:10.18632/aging.104079
268. Wen Y, Yu Y, Fu X. LncRNA Gm4419 contributes to OGD/R injury of cerebral microglial cells via IκB phosphorylation and NF-κB activation. Biochem Biophys Res Commun. 2017;487(4):923–929. doi:10.1016/j.bbrc.2017.05.005
269. Pang H, Ren Y, Li H, Chen C, Zheng X. LncRNAs linc00311 and AK141205 are identified as new regulators in STAT3-mediated neuropathic pain in bCCI rats. Eur J Pharmacol. 2020;868:172880. doi:10.1016/j.ejphar.2019.172880
270. Fu CH, Zhang BH, Fang CZ, et al. Long non-coding RNA CRNDE deteriorates intrauterine infection-induced neonatal brain injury. Mol Cell Probes. 2020;52:101565. doi:10.1016/j.mcp.2020.101565
271. Yang X, Chen Y, Li J, et al. Hypertonic saline maintains coagulofibrinolytic homeostasis following moderate‑to‑severe traumatic brain injury by regulating monocyte phenotype via expression of lncRNAs. Mol Med Rep. 2019;19(2):1083–1091. doi:10.3892/mmr.2018.9748
272. Zhang X, Zhu XL, Ji BY, et al. LncRNA-1810034E14Rik reduces microglia activation in experimental ischemic stroke. J Neuroinflammation. 2019;16(1):75. doi:10.1186/s12974-019-1464-x
273. Xiang W, Jiang L, Zhou Y, et al. The lncRNA Ftx/miR-382-5p/Nrg1 axis improves the inflammation response of microglia and spinal cord injury repair. Neurochem Int. 2021;143:104929. doi:10.1016/j.neuint.2020.104929
274. Shao M, Jin M, Xu S, et al. Exosomes from Long Noncoding RNA-Gm37494-ADSCs Repair Spinal Cord Injury via Shifting Microglial M1/M2 Polarization. Inflammation. 2020;43(4):1536–1547. doi:10.1007/s10753-020-01230-z
275. Cui SY, Zhang W, Cui ZM, et al. Knockdown of long non-coding RNA LEF1-AS1 attenuates apoptosis and inflammatory injury of microglia cells following spinal cord injury. J Orthop Surg Res. 2021;16(1):6. doi:10.1186/s13018-020-02041-6
276. Xu S, Wang J, Jiang J, et al. TLR4 promotes microglial pyroptosis via lncRNA-F630028O10Rik by activating PI3K/AKT pathway after spinal cord injury. Cell Death Dis. 2020;11(8):693. doi:10.1038/s41419-020-02824-z
277. Wang H, Liao S, Li H, Chen Y, Yu J. Long Non-coding RNA TUG1 Sponges Mir-145a-5p to Regulate Microglial Polarization After Oxygen-Glucose Deprivation. Front Mol Neurosci. 2019;12:215. doi:10.3389/fnmol.2019.00215
278. Li Z, Zhang J, Zheng H, et al. Modulating lncRNA SNHG15/CDK6/miR-627 circuit by palbociclib, overcomes temozolomide resistance and reduces M2-polarization of glioma associated microglia in glioblastoma multiforme. J Exp Clin Cancer Res. 2019;38(1):380. doi:10.1186/s13046-019-1371-0
279. Herman AB, Tsitsipatis D, Gorospe M. Integrated lncRNA function upon genomic and epigenomic regulation. Mol Cell. 2022;82(12):2252–2266. doi:10.1016/j.molcel.2022.05.027
280. Bridges MC, Daulagala AC, Kourtidis A. LNCcation: lncRNA localization and function. J Cell Biol. 2021;220(2). doi:10.1083/jcb.202009045
281. Wang SH, Zhu XL, Wang F, et al. LncRNA H19 governs mitophagy and restores mitochondrial respiration in the heart through Pink1/Parkin signaling during obesity. Cell Death Dis. 2021;12(6):557. doi:10.1038/s41419-021-03821-6
282. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist. Nat Neurosci. 2016;19(8):987–991. doi:10.1038/nn.4338
283. Wendimu MY, Hooks SB. Microglia Phenotypes in Aging and Neurodegenerative Diseases. Cells. 2022;11(13). doi:10.3390/cells11132091
284. Paolicelli RC, Sierra A, Stevens B, et al. Microglia states and nomenclature: a field at its crossroads. Neuron. 2022;110(21):3458–3483. doi:10.1016/j.neuron.2022.10.020
285. Treiber T, Treiber N, Meister G. Publisher correction: regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cell Biol. 2019;20(5):321. doi:10.1038/s41580-019-0106-6
286. Khorkova O, Stahl J, Joji A, Volmar CH, Wahlestedt C. Amplifying gene expression with RNA-targeted therapeutics. Nat Rev Drug Discov. 2023;22(7):539–561. doi:10.1038/s41573-023-00704-7
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Protective Effects of Pioglitazone on Cognitive Impairment and the Underlying Mechanisms: A Review of Literature
Alhowail A, Alsikhan R, Alsaud M, Aldubayan M, Rabbani SI
Drug Design, Development and Therapy 2022, 16:2919-2931
Published Date: 31 August 2022
Advances in the Understanding of the Correlation Between Neuroinflammation and Microglia in Alzheimer’s Disease
Yan H, Wang W, Cui T, Shao Y, Li M, Fang L, Feng L
ImmunoTargets and Therapy 2024, 13:287-304
Published Date: 12 June 2024
Prevention and Treatment Strategies for Alzheimer’s Disease: Focusing on Microglia and Astrocytes in Neuroinflammation
Zhang S, Gao Z, Feng L, Li M
Journal of Inflammation Research 2024, 17:7235-7259
Published Date: 13 October 2024
Jiawei Qifuyin Enhances Immunity and Improves Cognitive Impairment in APP/PS1 Mice Through Modulation of Neuroinflammatory Pathways
Cheng J, Li W, Wang L, Gao Y, Ma Y, Zhou M, Yang T, Yue C, Yan L, Lyu Y
Journal of Inflammation Research 2024, 17:9021-9040
Published Date: 18 November 2024
Advances in the Understanding of Mitochondrial Inflammatory Regulation in the Pathogenesis of Alzheimer’s Disease
Zhang RZ, Li L, Ma C, Dou TT, Wei YT, Yan XK
Journal of Inflammation Research 2025, 18:14475-14491
Published Date: 23 October 2025