Back to Journals » Journal of Inflammation Research » Volume 14
WNT16 is Robustly Increased by Oncostatin M in Mouse Calvarial Osteoblasts and Acts as a Negative Feedback Regulator of Osteoclast Formation Induced by Oncostatin M
Authors Henning P, Movérare-Skrtic S, Westerlund A, Souza PPC, Floriano-Marcelino T ![]() , Nilsson KH, El Shahawy M
, Nilsson KH, El Shahawy M ![]() , Ohlsson C, Lerner UH
, Ohlsson C, Lerner UH
Received 4 June 2021
Accepted for publication 2 September 2021
Published 18 September 2021 Volume 2021:14 Pages 4723—4741
DOI https://doi.org/10.2147/JIR.S323435
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Petra Henning,1 Sofia Movérare-Skrtic,1 Anna Westerlund,1 Pedro Paulo Chaves de Souza,2,3 Thais Floriano-Marcelino,3 Karin H Nilsson,1 Maha El Shahawy,1,4 Claes Ohlsson,1 Ulf H Lerner1
1Department of Internal Medicine and Clinical Nutrition, Institute of Medicine, Sahlgrenska Osteoporosis Centre and Centre for Bone and Arthritis Research at the Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden; 2The Innovation in Biomaterials Laboratory, School of Dentistry, Federal University of Goiás, Goiânia, Brazil; 3Department of Physiology and Pathology, São Paulo State University (UNESP), School of Dentistry, Araraquara, Brazil; 4Department of Oral Biology, Faculty of Dentistry, Minia University, Minia, 61511, Egypt
Correspondence: Ulf H Lerner
University of Gothenburg, Klin Farm Lab, Vita Straket 11, Gothenburg, 41345, Sweden
Tel +46 70 651 91 03
Email [email protected]
Background: Bone loss is often observed adjacent to inflammatory processes. The WNT signaling pathways have been implicated as novel regulators of both immune responses and bone metabolism. WNT16 is important for cortical bone mass by inhibiting osteoclast differentiation, and we have here investigated the regulation of WNT16 by several members of the pro-inflammatory gp130 cytokine family.
Methods: The expression and regulation of Wnt16 in primary murine cells were studied by qPCR, scRNAseq and in situ hybridization. Signaling pathways were studied by siRNA silencing. The importance of oncostatin M (OSM)-induced WNT16 expression for osteoclastogenesis was studied in cells from Wnt16-deficient and wild-type mice.
Results: We found that IL-6/sIL-6R and OSM induce the expression of Wnt16 in primary mouse calvarial osteoblasts, with OSM being the most robust stimulator. The induction of Wnt16 by OSM was dependent on gp130 and OSM receptor (OSMR), and downstream signaling by the SHC1/STAT3 pathway, but independent of ERK. Stimulation of the calvarial cells with OSM resulted in enhanced numbers of mature, oversized osteoclasts when cells were isolated from Wnt16 deficient mice compared to cells from wild-type mice. OSM did not affect Wnt16 mRNA expression in bone marrow cell cultures, explained by the finding that Wnt16 and Osmr are expressed in distinctly different cells in bone marrow, nor was osteoclast differentiation different in OSM-stimulated bone marrow cell cultures isolated from Wnt16−/- or wild-type mice. Furthermore, we found that Wnt16 expression is substantially lower in cells from bone marrow compared to calvarial osteoblasts.
Conclusion: These findings demonstrate that OSM is a robust stimulator of Wnt16 mRNA in calvarial osteoblasts and that WNT16 acts as a negative feedback regulator of OSM-induced osteoclast formation in the calvarial bone cells, but not in the bone marrow.
Keywords: oncostatin M, WNT16, osteoclast, osteoblast
Introduction
WNT signaling through the canonical pathway is a potent regulator of bone mass, initially demonstrated by the seminal observations that loss-of-function mutations in the low-density lipoprotein receptor-related protein-5 (LRP5) are the cause of low bone mass in the osteoporosis pseudoglioma syndrome1 and that gain-of-function mutations are important for a high bone mass phenotype inherited in certain families.2,3 WNT canonical signaling is dependent on the binding of the ligand WNT to the co-receptor LRP5/6 and the signaling transducing receptor Frizzled.4 To date, 19 different WNTs and 10 different Frizzleds have been described. Canonical WNT signaling is also controlled by several inhibitors such as sclerostin and Dickkopf 1–4 (DKK1-4), which bind to extracellular domains of LRP5, and by inhibitors such as secreted Frizzled-related proteins 1–5 (SFRP1-5) and WNT inhibitory factor 1 (WIF1), which bind to soluble WNTs. The crucial role of WNTs for bone mass in humans is further demonstrated by the findings that loss-of-function mutations in the SOST gene (encoding sclerostin) are the cause of high bone mass in van Buchem’s disease and sclerosteosis,5,6 that mutations in the WNT1 gene cause early-onset osteoporosis and rare variants of osteogenesis imperfecta,7 and that recessive mutations in the secreted frizzled-related protein-4 (SFRP4) gene is the reason for low cortical bone mass in Pyle’s disease.8 Using a translational approach involving human and mouse genetic studies, together with human and mouse cell culture experiments, we have demonstrated the important role of WNT16 for osteoclast differentiation and bone mass in vivo and in vitro.9–11
Although most attention has been paid to the role of WNTs for bone formation, studies have also shown the important role of WNT signaling in controlling bone resorption by interacting with osteoclastogenesis induced by RANKL (receptor activator of NF-kB ligand).12,13 Thus, in vitro and in vivo experiments have demonstrated that WNT5a potentiates RANKL-induced osteoclast formation through non-canonical signaling, mediated by the co-receptor receptor tyrosine-kinase-like orphan receptor-2 (ROR2) in osteoclast progenitor cells.14,15 Cell culture experiments have shown that WNT3a can inhibit RANKL-induced osteoclastogenesis,16,17 although this could not be observed by Maeda et al.14 WNT4 has been shown to decrease RANKL-induced osteoclast differentiation of mouse bone marrow macrophages.18 Recently, it was shown that WNT1, in addition to its anabolic effect on osteoblasts, can inhibit osteoclast differentiation.19
Human genome-wide association studies initially indicated that a locus in the WNT16 gene was highly associated with bone mass,20,21 preferentially with cortical bone mass, and with increased susceptibility to forearm fractures.21 This observation has been confirmed by several subsequent studies. In mouse genetic studies, we found that global deletion of Wnt16 resulted in decreased cortical bone mass and spontaneous fractures, with no effect on trabecular bone.9 Deletion of Wnt16 in Runx2-expressing osteoblasts copied the phenotype of global deletion, demonstrating that WNT16 expression in the osteoblastic lineage is regulating cortical bone mass. The phenotype was associated with increased numbers of cortical osteoclasts and mechanistic studies showed that WNT16 inhibited RANKL-induced osteoclast formation using both mouse bone marrow macrophages and human peripheral blood monocytes as osteoclast progenitor cells. WNT16 was also found to enhance OPG, which suggests a second mechanism by which WNT16 can inhibit osteoclast formation. In subsequent studies, we have demonstrated that induced deletion of Wnt16 in adult mice decreases cortical bone mass,11 that the bone sparing effect of estrogen is independent of Wnt16,22 and that glucocorticoid-induced bone loss can be prevented by osteoblast-specific overexpression of Wnt16.23
Local bone resorption is commonly observed adjacent to inflammatory processes in diseases such as rheumatoid arthritis and periodontitis due to activation of osteoclast formation by pro-inflammatory cytokines, including those in the gp130 family.24–27 This family is made up by cytokines with sequence homologies, although not to a large extent, which share receptor components heterodimerizing with the signaling, transmembrane co-receptor gp130 or a gp130-like protein.25–27 Interleukin-6 (IL-6), IL-11, oncostatin M (OSM) and leukemia inhibitory factor (LIF) are members of the gp130 family, which have been shown to stimulate osteoclast formation through induction of the osteoclastogenic cytokine RANKL.25,26,28 We recently reported that OSM is a uniquely strong stimulator of RANKL, due to activation of the adapter protein Src homology 2 domain-containing, transforming protein-1 (SHC1).29 OSM was discovered as an inhibitor of melanoma cell proliferation released from macrophage differentiated U-937 histiocytic lymphoma cells,30 but was later found to be expressed by several other cell types including osteoblasts and osteocytes.31 A pathogenetic role of OSM has been suggested in a variety of diseases including inflammatory diseases such as rheumatoid arthritis,32 periodontitis,33 inflammatory bowel disease,34 pulmonary fibrosis35 and neurogenic heterotopic ossifications.36
Several components of the WNT signaling system have been associated with inflammatory processes and found to exert immune-related functions.37–43 As regards WNT16, it has been reported that the WNT16 gene is upregulated in areas of human articular cartilage with injury or osteoarthritis damage44 and that global deletion, or deletion of Wnt16 in chondrocytes, results in more severe outcome of experimentally induced osteoarthritis in mice.45,46 Overexpression of Wnt16 using intra-articular injection of adenovirus expressing Wnt16 attenuates osteoarthritis in mice.46 Overexpression of Wnt16 in osteoblasts, however, does not affect experimentally induced osteoarthritis in mice, while subchondral bone mass was increased.47 It was recently reported that Wnt16 expression is upregulated early during experimental osteoarthritis in the temporomandibular joint and that WNT16 inhibited expression of cartilage degrading enzymes in chondrocytes induced by IL-1β.48 It seems that WNT16 can be upregulated by unknown mechanisms in inflammatory processes. We have investigated how pro-inflammatory cytokines in the gp130 family can regulate Wnt16 expression in osteoblasts. We identified OSM as the most potent inducer of Wnt16 expression among the gp130 cytokines and further investigated how WNT16 affects osteoclastogenesis induced by OSM.
Materials and Methods
Animals
The Wnt16−/- mice have been described previously.9 Wnt16−/-, control wild-type littermates, C57Bl/6N (Gothenburg University) and Swiss (School of Dentistry at Araraquara) mice were used for isolation of primary cells for cultures. Mouse experiments performed in Gothenburg were approved by the Ethics Committee in Gothenburg (Approval no 1849–2018) and animal care followed the regulations for animal research from the Swedish Board of Agriculture (SJVFS 2019:9) and the European Commission Directive on the protection of animals used for scientific purposes (Directive 2010/63/EU). Mouse experiments performed in Araraquara were approved by the Ethics Committee at the School of Dentistry, Araraquara (CEUA No. 11/2015) and animal care followed the “Brazilian Guidelines for The Production, Maintenance and Use of Animals for Teaching or Research” from the National Council of Control in Animal Experimentation (CONCEA, Resolucao Normativa No 25, de 29 de Setembro de 2015).
Cell Culture Media
The primary cell cultures were performed in complete α-MEM medium prepared by supplementing Minimum Essential Medium α (α-MEM, Gibco, Thermo Fisher Scientific, Waltham, MA, US) with 10% heat-inactivated fetal bovine serum (FBS, Sigma-Aldrich, Merck, Darmstadt, Germany), 2 mM GlutaMAX (Gibco), 50 µg/mL gentamicin (Gibco), 100 U/mL penicillin and 100 µg/mL streptomycin (Gibco). Osteogenic media were prepared by supplementing complete α-MEM medium with 2 mM β-glycerophospate (Sigma-Aldrich) and 0.2 mM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate (Asc-2P, Sigma-Aldrich).
Primary Calvarial Osteoblast Cultures
Primary calvarial bone cells from 3- to 5-day-old C57Bl/6N mice were isolated by collagenase digestion. For each cell isolation experiment, 8–14 dissected calvariae were initially washed in PBS and incubated in 5 mL 4 mM EDTA in PBS at 37°C, 600 rpm rotation table for two sequential 10 min digestions. Thereafter, the calvariae were incubated with 180 U/mL Collagenase type II (Worthington Biochemical Corporation, Lakewood, NJ, USA) in PBS for 10 sequential 10 min, 37°C, 600 rpm, 5 mL digestions. The last five collagenase fractions were pooled and centrifuged. Primary calvarial osteoblasts were cultured in complete α-MEM medium for 3–5 days in a T75 flask prior to re-seeding at the start of the experiments.
Isolated calvarial osteoblasts were seeded at a density of 20,000 cells/cm2 in osteogenic media and were treated with mRANKL (Lys158-Asp316), mOSM, hOSM, msIL6R, mIL6, mNP (CT-2), mIL31, mIL27, hCNTF, mCT-1, mIL-11 (R&D Systems, Minneapolis, MN, USA) or mLIF (Sigma-Aldrich) for 3, 6, 24 or 48h in 37°C, 5% CO2 and then harvested in RLT buffer (Qiagen, Hilden, Germany) containing 1% β-mercapto ethanol (Sigma-Aldrich) and stored at −80°C for gene expression analysis.
Osteoclast Formation in Calvarial Bone Cell Cultures
Calvarial bone cells isolated as described above contain mainly osteoblasts but also pre-osteoclasts, which after stimulation can differentiate to osteoclasts.49 The bone cells were seeded at a density of 20,000 cells/cm2 in osteogenic media and cultured in the presence of mOSM to induce RANKL production and osteoclast formation, and with or without hWNT16 (R&D Systems). Media were replenished every 3–4 days and cultures were stained for tartrate resistant acid phosphatase (TRAP) using a commercial kit (Sigma-Aldrich), or harvested for gene expression analyses at indicated days of culture. The numbers of TRAP-positive multinucleated osteoclasts (TRAP+MuOCL) containing either at least 3 or 10 nuclei, and the total surface per well covered with osteoclasts, were counted using the Osteomeasure (Osteometrics, Decatur, GA, USA) or the Bioquant Osteo software (Bioquant, Nashville, TN, USA).
Osteoclast Formation in Spleen Cell Cultures
Cells were obtained from spleens of 10–12-week-old wild-type C57BL/6N mice as previously described.50 The isolated cells were spot seeded (750,000 cells in 30 µL per 48-plate well) for 10 min, followed by addition of complete α-MEM and cultured for 24h. The cells were gently washed to remove non-adherent cells and then incubated in complete α-MEM with mM-CSF (30 ng/mL, R&D Systems) and mRANKL (4 ng/mL) with or without hWNT16. Media were replenished after 3 days and after 4 days the cells were stained with TRAP and numbers of osteoclasts with three or more nuclei counted. In separate experiments, cells were harvested after 4 days for RNA isolation and gene expression analyses.
Osteoclast Formation in Bone Marrow Cell Cultures
Mouse bone marrow cells were flushed from the femur and tibia of wild-type C57BL/6N mice. After centrifugation, the bone marrow cells were resuspended in complete α-MEM and 900,000 cells in 50 µL were spot-seeded in 48-well-plates. After cell attachment, complete α-MEM was added and the cells were cultured for 24 h before addition of mOSM in complete α-MEM. Media were replenished every 3–4 days. After 9 days, cultures were TRAP stained to visualize osteoclast formation and cells harvested for gene expression analyses. Osteoclasts were formed in foci in close contact with stromal cells. TRAP+ cells with at least 3 or 10 nuclei, as well as total surface per well covered with osteoclasts, were counted using the Osteomeasure or the Bioquant Osteo software.
Gene Silencing in Osteoblasts Using Small Interfering RNA
Calvarial osteoblasts from Swiss mice were isolated as described above and seeded at 1000 cells per well in 96-well plates in complete α-MEM. After overnight attachment, silencing of Osmr, Lifr, Il6st, Shc1, Stat3 or Mapk1/Mapk3 (Erk) was performed as previously described29 using Lipofectamine RNAiMAX and 30 nM of SilencerSelect siRNA (ThermoFisher Scientific) against Il6st (s68298), Lifr (s69223), Osmr (s71149), Shc1 (s73682) Stat3 (s74451), and a 1:1 mix for Mapk1 (s77104) and Mapk3 (s77116) (30 nM each). Cells treated with a scrambled siRNA (Scr, Assay ID AM4635) sequence served as controls. Forty-eight hours after the first silencing, the protocol was repeated. Twenty-four hours after the second silencing, the cells were incubated in medium containing either vehicle or mOSM. Twenty-four hours after addition of OSM, RNA was extracted, and gene expression analyzed. Gene expression is presented as % of expression in cells treated with Scr cultured in normal control media. The robust silencing efficacy of the oligonucleotides used for silencing of Osmr, Lifr, Il6st, Shc1 and Stat3 expression has been previously reported.29 We, here, show the efficacy of the oligonucleotides used to silence Mapk1 and Mapk3, which markedly reduce the mRNA expression of Mapk1 and Mapk3 (Supplementary Figure 1).
RNA Isolation and First-Strand cDNA Synthesis
Media were removed and cells were lysed with RLT lysis buffer (Qiagen) containing 1% β-mercapto ethanol (Sigma-Aldrich) and stored at −80°C. RNA was isolated using the RNAeasy micro kit with on-column DNase treatment (Qiagen). Single-strand cDNA was synthesized from total RNA using High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Thermo Fisher Scientific).
Quantitative Real-Time Polymerase Chain Reaction
Quantitative real-time PCR (qPCR) analyses were performed by using predesigned TaqMan Assays for Wnt16 (Mm00446420_m1), Tnfsf11 (Mm00441908_m1), Tnfrsf11b (Mm00435452_m1), Acp5 (Mm00475698_m1), Ctsk (Mm00484036_m1), Calcr (Mm00432282_m1), Il6st (Mm00439665_m1), Lifr (Mm00442942_m1), Osmr (Mm01307326_m1), Stat3 (Mm01219775_m1) and TaqMan Fast Advance Master Mix (Applied Biosystems, Thermo Fisher Scientific). Housekeeping gene 18S (Thermo Fisher Scientific) or Actb (Primer F: 5ʹ-GGACCTGACGGACTACCTCATG-3ʹ, Primer R: 5ʹ-TCTTTGATGTCACGCACGATTT-3ʹ, Probe: VIC-CCTGACCGAGCGTGGCTACAGCTTC-TAMRA) were used as endogenous control in all analyses. For Shc1, Mapk1 and Mapk3, amplification was performed using Fast SyBr Master Mix and gene-specific primers: Shc1 primer F: 5ʹ-GGTCCTGGGGTGAAAGTT-3ʹ and Shc1 Primer R: 5ʹ-TGTTCATGTCCAGGGTCTCA-3, Mapk1 primer F: ACCCAAGTGATGAGCCCATTG, Mapk1 primer R: TGCATTGAAAGTGCACACTGC, Mapk3 primer F: CACTGGCTTTCTGACGGAGT and Mapk3 primer R: GGATTTGGTGTAGCCCTTGGA. As endogenous control for the SyBr qPCR, the same Actb primers as above, excluding the probe, was used. Amplifications were performed on StepOnePlus Real-Time PCR System (Applied Biosystems). The relative quantity was analyzed using the delta-delta-Ct method and presented as % of control. To compare gene expression in bone marrow cell cultures and primary calvarial bone cell cultures, cDNA from three different experiments with the two cell culture types were analyzed in the same TaqMan assay and presented as % expression of the mean of the three bone marrow cell experiments. The relative expression of the endogenous PCR control 18S was analyzed vs amount of RNA reverse transcribed to cDNA and found to be similar between the two cell culture types (Supplementary Figure 2A).
In situ Hybridization (RNAscope)
To study the expression of Wnt16 mRNA in bone, chromogenic in situ hybridization was performed on lumbar vertebra 5 from 5-month-old wild-type C57Bl/6N mice. The bone samples were fixed overnight in 4% paraformaldehyde (PFA) in phosphate buffered saline. The specimens were demineralized for 3 weeks in 15% EDTA with 0.4% PFA, and thereafter processed for paraffin embedding. Consecutive 6-µm-thick sections were dewaxed and in situ hybridized using RNAscope technology (RNAscope 2.5 HD brown, Advanced Cell Diagnostics [ACD], Bio-Techne, Abingdon, UK). A modified RNAscope manual protocol was followed.51 The used probes were: Runt-related transcription factor 2 (Runx2, 414021, target sequence 3838–4821), collagen 1α1 (Col1a1, 319371, target 899 sequence 1686–4669) and Wnt16 (Wnt16, 401081, target sequence 453–1635). The bone sections were hybridized with the probe overnight followed by pretreatment and six amplification steps according to the instructions of the manufacturer. For further amplification of the signal, digoxigenin-labelled tyramide signal amplification (Akoya Biosciences, Marlborough, MA, USA) was performed. The signal was detected and visualized by alkaline phosphatase-conjugated anti-digoxigenin FAB fragment (Sigma-Aldrich) and liquid permanent red (DAKO, Agilent, Santa Clara, CA, USA), respectively. Subsequently, the sections were counterstained with Mayer´s Hematoxylin (Sigma-Aldrich) and Aquatex-mounted (Sigma-Aldrich).
Single Cell RNA Sequencing (scRNAseq)
To analyse the expression of Wnt16, Osmr and Tnfsf11 in bone marrow stromal cells, we used the publicly available scRNAseq analysis tool nicheExplorer made available by Iannis Aifantis laboratory (http://aifantislab.com/niche) in an interactive data browser as a supplement to their published scRNAseq data of mouse bone marrow vascular, perivascular, and osteoblast cell populations.52 In addition to scRNAseq analysis by nicheExplorer, the recently published scRNAseq data from Cxcl12-eGFP expressing bone marrow stromal cells53 was downloaded from GEO project GSE136970 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE136970). Both datasets (GSM4064136Cxcl12GFPCxcl12CE#1 and GSM4064137Cxcl12GFPCxcl12CE#2) were imported into R and analyzed using Seurat. Cells with less than 1000 genes per cell were filtered out and the two datasets were merged, resulting in 8026 cells from the two datasets. We further filtered out cells with more than 15% mitochondrial read content, resulting in 7332 cells remaining for analysis. Further analysis was performed after normalization of the data using the logNormalize method, and UMAP cluster analysis was done using the first 10 principal components (1:10 dimensions). We obtained 8 different cell clusters (Supplementary Figure 3A) Heat map and feature plots of selected genes identified the clusters as four osteogenic/adipogenic (cluster 0, 1, 4 and 7), one mitotic (cluster 6), one endothelial (cluster 5) and two haematopoetic (cluster 2 and 3) that were used for feature plots of Wnt16, Osmr and Tnfsf11 (Supplementary Figure 3B–D).
Statistical Analysis
Data are presented as absolute values or as % of untreated control cells ±SEM. Statistically significant effects were determined by Student’s t-test, one-way ANOVA followed by Dunnett’s multiple comparison test vs CTRL, or by two-way ANOVA for interaction followed by Sidak’s multiple comparison test for CTRL vs OSM-treated groups or for Scr vs siRNA-treated groups. All experiments contained at least four wells per genotype or treatment group and were repeated at least 3 times with similar results.
Results
OSM and IL-6 Induces Wnt16 Expression in Primary Calvarial Osteoblast Cultures
When mouse osteoblasts, isolated from the periosteum of newborn calvariae, were treated for 24 h with a variety of cytokines in the gp130 family (100 ng/mL), it was observed that mOSM and IL-6 (100 ng/mL) in the presence of sIL-6R (100 ng/mL), strongly induced Wnt16 mRNA expression, whereas IL-11, hOSM, LIF, CT-1 (cardiotrophin-1), CNTF (ciliary neurotrophic factor), NP (neuropoietin), IL-27 and IL-31 had no effect (Figure 1A). The expression of Tnfsf11 mRNA (encoding RANKL) was also induced by mOSM and IL-6+sIL-6R, whereas the other cytokines were without effect (Figure 1B). mOSM and IL-6+sIL-6R induced Wnt16 mRNA to the same degree, but IL-6+sIL-6R was considerably less effective as stimulator of Tnfsf11 mRNA than mOSM. The expression of Tnfrsf11b (encoding OPG) was slightly increased by IL-27, but unaffected by the other gp130 family cytokines (Figure 1C). IL-6 had no effect on Wnt16 or Tnfsf11 mRNA expression in the absence of sIL-6R (data not shown).
 |
Figure 1 Gene expression of Wnt16, Tnfsf11 and Tnfrsf11b following stimulation with gp130 cytokines. Primary calvarial osteoblast cultures were treated with cytokines in the gp130 family (100 ng/mL). The expression of Wnt16 (A), Tnfsf11 (B) and Tnfrsf11b (C) was analyzed after 24h. Individual values are presented in all graphs with the mean shown as horizontal lines and ±SEM as vertical lines. ***P<0.001, *P < 0.05 vs control (CTRL), one-way ANOVA followed by Dunnett’s multiple comparison test. |
The effect by mOSM on Wnt16 mRNA was time dependent with no effect at 3 and 6 h, but with a gradually increased response at 24 and 48 h (Figure 2A). Tnfsf11 mRNA was increased by mOSM after 3 h and gradually increased until 48 h (Figure 2B). The stimulatory effect by mOSM on Wnt16 mRNA was observed at and above 1 ng/mL with half maximal effect at 3 ng/mL (Figure 2C).
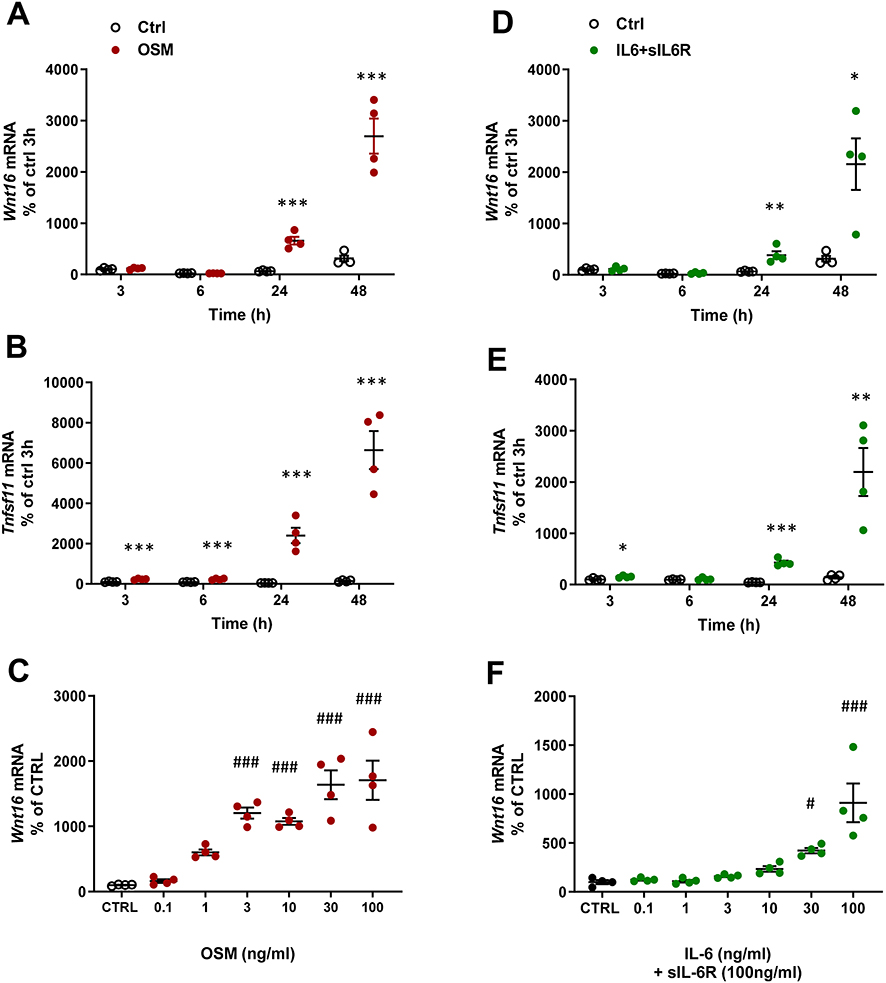 |
Figure 2 OSM and IL-6 time-and dose-dependently increase the expression of Wnt16 and Tnfsf11. Primary calvarial osteoblast cultures were treated with mOSM (A–C) or with IL-6 (D–F) together with sIL-6R. Expression of Wnt16 (A and D) and Tnfsf11 (B and E) was analyzed after stimulation with 100 ng/mL mOSM (A and B) or 100 ng/mL IL-6 together with 100 ng/mL sIL-6R (D and E) for 3, 6, 24 and 48 h. Expression of Wnt16 24h after addition of mOSM (0.1–100 ng/mL) (C) or IL-6 (0.1–100 ng/mL) together with sIL-6R (100 ng/mL) (F) for 24h. Individual values are presented in all graphs with the mean shown as horizontal lines and ±SEM as vertical lines. ***P<0.001, **P < 0.01, *P < 0.05 vs untreated control (CTRL) at each time point, Student’s t-test. ###P<0.001, #P < 0.05 vs control, one-way ANOVA followed by Dunnett’s multiple comparison test. |
In the presence of sIL-6R, IL-6 caused a time-dependent upregulation of Wnt16 and Tnfsf11 mRNA (Figure 2D and E). While the Wnt16 induction by IL-6+ sIL-6R was similar to the effect observed by mOSM, the Tnfsf11 response to IL-6 + sIL-6R was lower in magnitude compared to mOSM (Figures 1A and B; 2B and E). The increased mRNA expression of Wnt16 and Tnfsf11 caused by IL-6+sIL-6R was dependent on the concentration of IL-6, with half maximal effect observed at 30 ng/mL IL-6 + 100 ng/mL sIL-6R (Figure 2F).
These observations demonstrate that OSM and IL-6 (in the presence of sIL-6R) strongly upregulate Wnt16 mRNA and that OSM is a more robust inducer of Tnfsf11 mRNA in primary bone cells from mouse calvariae. The results further indicate that the effect is mediated via the OSMR since only mOSM and not hOSM and LIF, which both only bind the LIFR in mouse, increase Wnt16 expression.
Recombinant WNT16 Inhibits OSM-Induced Osteoclast Formation in Calvarial Bone Cell Cultures
Since our data show that OSM and IL-6+sIL-6R are equally effective in stimulating Wnt16 mRNA, but that OSM is a more robust stimulator of the osteoclastogenic cytokine RANKL, we focused further experiments on studies aiming to investigate the possible role of WNT16 in OSM-induced osteoclast formation. For this purpose, we added recombinant WNT16 to mOSM-stimulated mouse calvarial bone cell cultures. These cell cultures contain mainly osteoblasts, but also osteoclast progenitor cells, and therefore can be used as a co-culture of osteoclast progenitors and osteoblasts suitable for studies on the formation of mature osteoclasts.49
Recombinant WNT16 reduced osteoclast formation in mOSM-stimulated calvarial bone cell cultures (Figure 3A and B). This effect was associated with decreased mRNA expression of the osteoclastic genes Acp5 (encoding TRAP) and Calcr (encoding calcitonin receptor) by WNT16 (Figure 3C and D), without any significant effect on Tnfsf11 mRNA or Tnfrsf11b mRNA (Figure 3E and F).
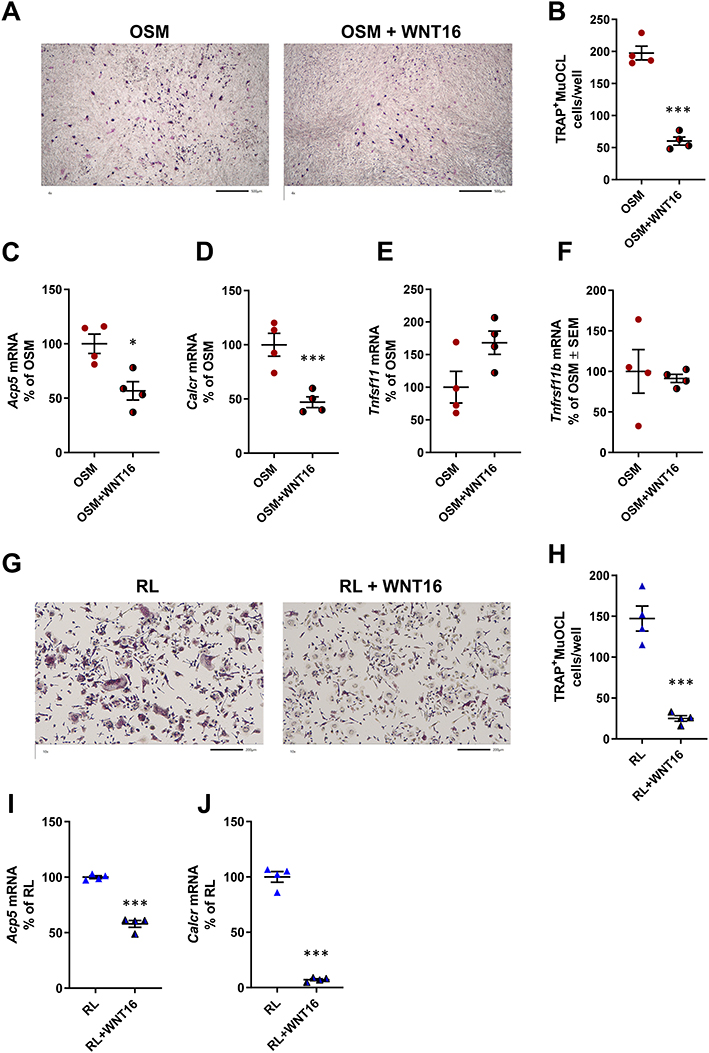 |
Figure 3 Recombinant exogenous WNT16 inhibits osteoclastogenesis in primary calvarial bone cell cultures and in spleen cell cultures. Primary calvarial bone cells were stimulated with mOSM (30 ng/mL) in the presence or absence of WNT16 (1 µg/mL) for 9 days. Cells were TRAP stained (A) and number of TRAP+MuOCL with more than 3 nuclei (B) was analyzed. Scale bars, 500 µm. Gene expression of Acp5 (C), Calcr (D), Tnfs11 (E) and Tnfrs11b (F). Primary spleen cell cultures were stimulated with 4 ng/mL RANKL in the presence or absence of 1 µg/mL WNT16 for 4 days. Cells were TRAP stained (G) and number of TRAP+MuOCL with more than 3 nuclei (H) was analyzed. Scale bars, 200 µm. Gene expression of Acp5 (I) and Calcr (J). Individual values are presented in all graphs with the mean shown as horizontal lines and ±SEM as vertical lines. ***P<0.001, *P < 0.05 vs mOSM or mRANKL-treated cells, Student’s t-test. |
These experiments demonstrate that exogenous WNT16 inhibits OSM-stimulated osteoclastogenesis, most likely by targeting osteoclast progenitors.
Recombinant WNT16 Inhibits Osteoclastogenesis in Spleen Cell Cultures
Having observed that osteoclast progenitors in mouse bone marrow and human peripheral blood,9 as well as in mouse calvariae (present study), are sensitive to WNT16-induced inhibition, we assessed if osteoclast progenitors in spleen are also sensitive to WNT16. This was prompted by the recent finding that spleen macrophages derived from Cx3cr1+ yolk-sac progenitors contribute to osteoclast formation during injury-induced inflammation and bone healing.54 Since spleen lacks osteoclastogenic stromal cells sensitive to RANKL-stimulating hormones and cytokines, we used recombinant RANKL to induce osteoclastogenesis. We found that WNT16 robustly inhibited the numbers of TRAP+MuOCL in RANKL-stimulated mouse spleen cell cultures (Figure 3G and H) and that this response was associated with decreased expression of osteoclastic genes (Acp5, Calcr; Figure 3I and J).
Increased Osteoclast Formation in OSM-Stimulated Mouse Calvarial Bone Cell Cultures from Wnt16−/- Mice
Since recombinant exogenous WNT16 was found to inhibit OSM-stimulated osteoclast formation in calvarial bone cell cultures, we next evaluated the role of endogenous WNT16. For this purpose, we stimulated calvarial bone cell cultures from wild-type mice and from Wnt16 gene deficient mice with mOSM and assessed osteoclast formation and expression of osteoclastogenic and osteoclastic genes.
No TRAP+MuOCL could be observed in unstimulated control cells (data not shown), but treatment with mOSM (100 ng/mL) for 11 days enhanced the differentiation of mono- and multinucleated TRAP+ cells (Figure 4A). The total number of TRAP+MuOCL (≥3 nuclei/cell) was not different in cell cultures from the two genotypes (Figure 4B). Interestingly, however, the numbers of large TRAP+MuOCL (≥10 nuclei/cell) was clearly enhanced in mOSM-stimulated cultures from Wnt16−/- mice (Figure 4C), resulting in a threefold increase of the area of all TRAP+MuOCL (Figure 4D). Expression of the osteoclastic genes Acp5 and Ctsk (encoding cathepsin K) was also more pronounced when calvarial cells from Wnt16−/- mice were used compared to the response in wild-type cells (Figure 4E and F). The difference in osteoclast formation between cells from wild-type and Wnt16−/- mice was not due to difference in mRNA expression of Tnfsf11 or Tnfrsf11b (encoding OPG) caused by mOSM treatment (Figure 4G and H).
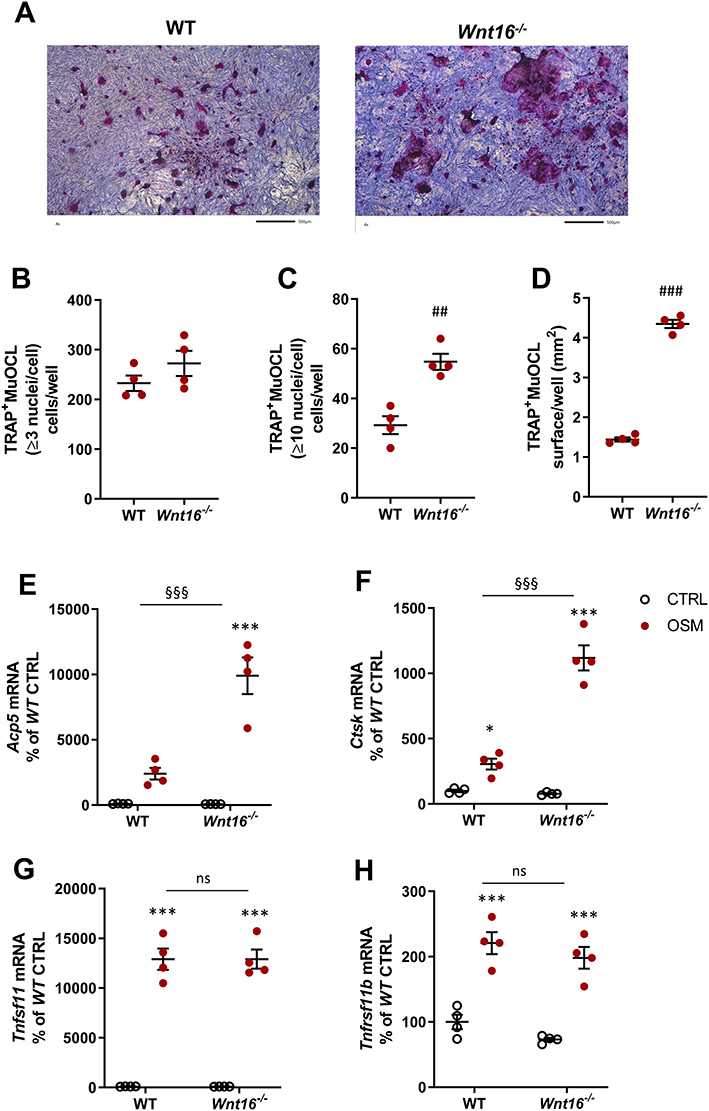 |
Figure 4 Osteoclastogenesis stimulated by OSM is enhanced in primary calvarial bone cell cultures from Wnt16−/- mice. Primary calvarial periosteal cells from wild-type (WT) and Wnt16−/- mice were stimulated with 100 ng/mL mOSM for 11 days. Cells were TRAP stained (A) and numbers of TRAP+MuOCL with more than 3 (B) and 10 nuclei (C) as well as the total surface (D) covered by TRAP+MuOCL were analyzed. Scale bars, 500 µm. Expression of Acp5 (E), Ctsk (F), Tnfs11 (G) and Tnfrs11b (H) after mOSM treatment for 11 days. Individual values are presented in all graphs with the mean shown as horizontal lines and ±SEM as vertical lines. ###P<0.001, ##P < 0.01 vs WT, Student’s t-test. ***P<0.001*P < 0.05 vs genotype specific untreated control, two-way ANOVA followed by Sidak’s multiple comparison test for the effect of mOSM treatment. §§§P<0.001, ns (not significant) P>0.05 for the effect by mOSM in WT vs the effect by mOSM in Wnt16−/- cells analyzed by interaction in two-way ANOVA. |
Osteoclast Formation in OSM-Stimulated Bone Marrow Cell Cultures is Similar in Cell from Wild-Type and Wnt16−/- Mice
We also assessed the role of endogenous WNT16 in mouse bone marrow cell (BMC) cultures. These cell cultures contain bone marrow stromal cells and hematopoietic cells, including osteoclast progenitors, and are frequently used to study osteoclast formation. In contrast to the calvarial bone cell cultures, no difference of total osteoclast numbers (>3 nuclei/cell), numbers of large osteoclasts (>10 nuclei/cell) or total area of osteoclasts was observed in mOSM-stimulated BMC cultures from wild-type and Wnt16−/- mice (Figure 5A–D). Nor could we observe any difference in the mRNA expression of Acp5 or Ctsk in mOSM-stimulated BMC from wild-type or Wnt16−/- mice (Figure 5E and F). The BMC cultures from wild-type but not from Wnt16−/- mice expressed Wnt16 mRNA, but OSM did not affect the expression in cells from wild-type mice (Figure 5G). The expression of Osmr mRNA was verified in the BMC cells, which, together with the finding showing that mOSM induced the expression of Tnfsf11 mRNA in the BMC cultures, indicates that the BMC cultures contain cells that are capable of responding to mOSM (Figure 5H and I).
 |
Figure 5 Osteoclastogenesis stimulated by OSM is unchanged in primary bone marrow cell cultures from Wnt16−/- mice. Primary bone marrow cell cultures from WT and Wnt16−/- mice were stimulated with 100 ng/mL mOSM for 9 days. Photos of TRAP stained cells (A), and numbers of TRAP+MuOCL with more than 3 (B) and 10 nuclei (C) as well as the total surface covered by TRAP+MuOCL (D) were analyzed. Scale bars, 200 µm. Expression of Acp5 (E), Ctsk (F), Wnt16 (G), Osmr (H) and Tnfs11 (I) after mOSM treatment for 9 days. Individual values are presented in all graphs with the mean shown as horizontal lines and ±SEM as vertical lines. ***P<0.001*P < 0.05 vs genotype specific untreated control; two-way ANOVA followed by Sidak’s multiple comparison test. ns (not significant) P>0.05 for the effect by mOSM in WT vs the effect by mOSM in Wnt16−/- cells analyzed by interaction in two-way ANOVA. |
Wnt16 Expression is Lower in Mouse Bone Marrow Cell Cultures Than in Calvarial Bone Cell Cultures
We were intrigued by the fact that OSM-stimulated osteoclastogenesis was increased in Wnt16−/-calvarial bone cell cultures, but not in Wnt16−/- bone marrow cell cultures, compared to cultures from wild-type mice. Therefore, we compared the expression levels of Wnt16 in the two cell culture systems. The expression of Wnt16 was 200–600 times higher in calvarial bone cell cultures than in bone marrow cell cultures (Figure 6A). The expression of Osmr and Runx2, however, was only slightly higher in the calvarial bone cell cultures, the difference being only two- to four- and three- to sixfold, respectively (Supplementary Figure 2B and C). This indicates that although cells in the BMC cultures and the calvarial osteoblasts cultures express a functional OSMR and both express the osteoblastic transcription factor Runx2, these cell cultures are fundamentally different in the constitutive and OSM-induced expression and regulation of Wnt16.
 |
Figure 6 Expression of Wnt16 in calvarial bone cells and bone marrow cells analyzed by PCR, in situ hybridization and scRNAseq. (A) Expression of Wnt16 in three bone marrow cell (BMC) cultures and three calvarial bone cell (cOBL) cultures cultured in osteogenic media for 7 and 6–8 days, respectively. Individual values are presented with the mean shown as horizontal lines and ±SEM as vertical lines. ***P<0.001 BMC vs cOBL, two-way ANOVA. (B) RNAscope of Wnt16 expression (red) in L5 vertebrae. Scale bar, 50 µm. (C–F) Single cell RNA sequencing analysis of bone marrow stromal cells using nicheExplorer.52 Reprinted with permission from Springer Nature Customer Service Centre GmbH; Springer Nature; Nature; The bone marrow microenvironment at single-cell resolution, Anastasia N. Tikhonova et al, © 2019, www.aifantislab.com/niche. (C) t-SNE visualization of endothelial (V1-2), perivascular (P1-4), osteo-lineage (O1-3) and one mitotic cell cluster (cycling) identified by Tikhovona et al.52 Feature plots of Wnt16 (D), Osmr (E) and Tnfsf11 (F) expression. |
Wnt16 is Expressed in Cortical Bone but Not in Bone Marrow Cells
To further investigate the difference between bone marrow cells and osteoblasts on bone surfaces we performed RNAScope. Using this technique, we found that Wnt16 mRNA is expressed in Runx2 and Col1a1 expressing cells on cortical bone, but not in bone marrow cells (Figure 6B, Supplementary Figure 2).
Wnt16 and Osmr are Expressed in Distinctly Different Bone Marrow Stromal Cells
While both Osmr and Wnt16 were detected in bone marrow cell cultures using qPCR, the expression of Wnt16 was not increased by addition of recombinant OSM, in contrast to calvarial cell cultures. We hypothesized that Osmr and Wnt16 could be expressed in different cell types in the bone marrow cell cultures. To investigate this, we used two different, recently published and publicly available scRNAseq data sets of bone marrow stromal cells.52,53 Tikhonova et al performed scRNAseq analysis of bone marrow stromal cells expressing Cdh5 (found in vasculature), Lepr (perivascular stromal stem cells) and Col1a1 (osteoblastic cells) and clustered them into two vascular (V1-2), four perivascular stromal cells (P1-4) and three osteo-lineage cell clusters (O1-3) (Figure 6C).52 Using this dataset, we found that while Wnt16 was expressed almost exclusively in two of the osteo-lineage cell clusters (O1 and O2), these clusters expressed almost no Osmr (Figure 6D and E). Osmr was expressed in both Lepr expressing stromal cells and Chd5 expressing vasculature cells, but only the Lepr expressing stromal cells expressed Tnfsf11 (Figure 6E and F). This suggests that it is the Lepr expressing stromal cells that increase the expression of Tnfsf11 after addition of mOSM to bone marrow cell cultures and that the osteo-lineage cells that express Wnt16 are unresponsive to mOSM due to lack of Osmr expression. The lack of Wnt16 expression in bone marrow stromal cells that express Osmr and Tnfsf11 was verified in a second scRNA data set of Cxcl12 expressing bone marrow stromal stem cells53 (Supplementary Figure 3).
Signaling Pathways Involved in OSM-Induced Wnt16 Expression Through the OSM Receptor
We next evaluated the receptor and signaling pathways involved in OSM-induced Wnt16 mRNA expression. OSM binds to a heterodimeric complex between the OSMR and gp130.55 Mouse OSM acts mainly through the OSMR:gp130 heterodimer,55,56 but interestingly it has been shown that mouse OSM can decrease sclerostin expression in osteoblasts through the LIFR.31 We, therefore, assessed the importance of OSMR and LIFR for the effect of mOSM on Wnt16 mRNA induction. By silencing Osmr and Lifr in calvarial osteoblasts, we observed that mOSM acts through the OSMR, but not through the LIFR, to induce Wnt16 mRNA (Figure 7A and B). The importance of gp130-mediated signaling was demonstrated by the observation that silencing of Il6st (encoding gp130) decreased mOSM-induced Wnt16 mRNA expression (Figure 7C).
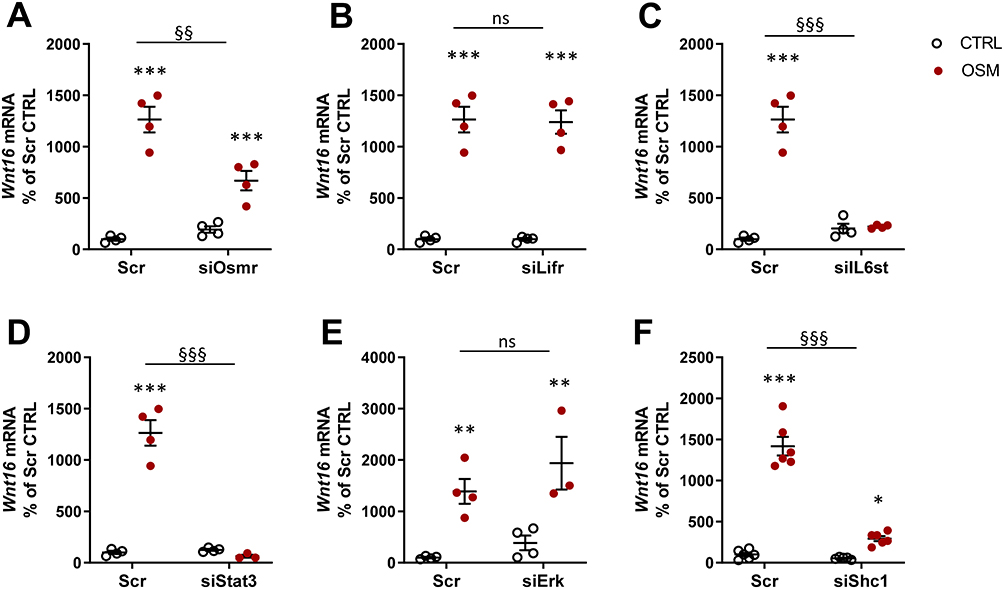 |
Figure 7 OSM induces Wnt16 expression through the OSMR/gp130/SHC1/STAT3 axis. The expression of Osmr (A), Lifr (B), IL6st (encoding gp130) (C), Stat3 (D), Erk (Mapk1/Mapk3) (E), and Shc1 (F) was silenced in primary calvarial osteoblast cultures using siRNA. Cells were treated with 100 ng/mL mOSM for 24 h and thereafter Wnt16 expression was analyzed. Scrambled siRNA (Scr) was used as control. Individual values are presented in all graphs with the mean shown as horizontal lines and ±SEM as vertical lines. ***P<0.001, **P < 0.01, *P < 0.05 vs respective untreated control, two-way ANOVA followed by Sidak’s multiple comparison test. §§P<0.01, §§§P<0.001, ns (not significant) P>0.05 for the effect by mOSM in siRNA-treated cells vs the effect by mOSM in Scr-treated cells analyzed by interaction in two-way ANOVA. |
OSMR:gp130 activates the JAK-STAT pathway initiated by JAK1- and JAK2-mediated phosphorylations of several Tyr residues at the membrane-proximal domains of gp130 and OSMR, followed by recruitment and phosphorylation of the transcription factor STAT3 and subsequent translocation of dimeric STAT3 to the nucleus.57–59 We have recently shown that OSM robustly stimulates the phosphorylation of STAT3 in mouse calvarial osteoblasts.29 Here, we demonstrate that silencing of Stat3 resulted in decreased Wnt16 mRNA expression induced by mOSM (Figure 7D).
Activation of JAKs by OSM results in phosphorylation also of the Tyr759 residue in gp130 and subsequent recruitment and activation of the tyrosine phosphatase SHP-2, which forms a complex with GRB2 and SON.60 This leads to activation of the RAS/RAF/MAPK pathway.61 In accordance with these observations, OSM causes a rapid activation of the MAPK ERK in mouse calvarial osteoblasts.29 Silencing of Erk (Mapk1 and Mapk3), however, did not affect Wnt16 mRNA expression induced by stimulation with mOSM (Figure 7E).
A unique feature of the OSMR is the recruitment of the adapter protein SHC1 to Tyr861.62,63 SHC1 contains both phosphotyrosine binding (PTB) and SH2 domains, allowing the recruitment of the adapter GRB2, which by forming a complex with SOS activates the RAS/RAF/MAPK pathway.62,63 We have shown that OSM induces the phosphorylation of SHC1 in mouse calvarial osteoblasts.29 The importance of this adapter protein for the induction of Wnt16 is demonstrated by the finding in the present study showing that silencing of Shc1 decreased mOSM-induced Wnt16 mRNA expression (Figure 7F).
Discussion
In our efforts to elucidate how WNT16 is regulated in osteoblasts by cytokines in the gp130 family we observed that the cytokine OSM is a potent and robust inducer of Wnt16 mRNA expression in mouse calvarial osteoblasts. OSM plays a role in physiological bone remodeling as demonstrated by the findings that global deletion of the Osmr gene in mice results in increased bone mass and decreased numbers of osteoclasts.31 The latter observation is in agreement with the observation showing that OSM stimulates osteoclast formation in organ cultured mouse calvarial bones28 and Tnfsf11 mRNA expression in calvarial osteoblasts (present investigation) as well as in several human and murine osteoblastic cell lines.31,64 We, here, report that OSM, through an OSMR/gp130/SHC1/STAT3-mediated pathway induces Wnt16 mRNA expression in mouse calvarial osteoblasts, but not in bone marrow stromal cells, and that WNT16 acts as a negative feedback regulator of OSM-induced osteoclastogenesis.
By comparing all cytokines in the gp130 family, we found that only OSM and IL-6 (in the presence of soluble IL-6 receptor), time- and concentration-dependently, induced the mRNA expression of Wnt16 in calvarial osteoblasts. IL-11, LIF, CT-1, CNTF, NP, IL-27 and IL-31 were all without effect. Similarly, OSM and IL-6/sIL-6R, but not the other cytokines, induced the mRNA expression of Tnfsf11. The requirement of soluble IL-6 receptor to obtain an effect by IL-6 is due to the low expression of IL-6 receptors in osteoblasts65 and, therefore, the activation by the IL-6/sIL-6R complex is due to binding to the extracellular domain of gp130, so-called IL-6 trans-signaling.66 Since OSM was a more robust stimulator of Wnt16 and Tnfsf11 mRNA than IL-6/sIL-6R in the primary calvarial osteoblasts, we focused on OSM for our subsequent studies on osteoclast formation and signaling pathways.
We used cells from wild-type and Wnt16−/- mice to assess the role of WNT16 in OSM-induced osteoclastogenesis. Since osteoclasts can be differentiated from progenitor cells present at the surfaces of cortical calvarial bone, we utilized a cell culture system consisting of isolated calvarial bone cells that we and others have previously reported can be used for studies on osteoclastogenesis when stimulated by either parathyroid hormone or RANKL.49,67 In these cultures, osteoclast differentiation was enhanced by OSM as evidenced by upregulated mRNA expression of Acp5 and Ctsk and the responses were significantly enhanced in cultures from mice with Wnt16 deletion. The numbers of osteoclasts with three nuclei or more were not changed, but the osteoclasts were considerably larger in cultures lacking Wnt16 expression and counting of oversized osteoclasts with more than ten nuclei showed a two-fold increase of these cells. The difference in osteoclastogenesis was not likely due to changes in RANKL or OPG since OSM increased the mRNA expression of Tnfsf11 and Tnfrsf11b to the same extent in cell cultures from wild-type and Wnt16−/- mice. These data are in agreement with our previous findings showing that WNT16 interferes with signaling downstream RANK in osteoclast progenitor cells.9 Although a wide variety of osteoclast progenitor cells, including those in bone marrow and blood,9 as well as those in periosteum and spleen (present study), are responsive to WNT16-induced inhibition, WNT16 regulates bone mass and osteoclastogenesis exclusively in cortical bone, with no effect on these parameters in trabecular bone.9 This could possibly be due to that expression of Wnt16 is regulated preferentially in cortical osteoblasts. In agreement with this view, we here show, using in situ hybridization, that Wnt16 mRNA could be detected in cortical bone, but not in bone marrow cells. While no Wnt16 expression could be detected in bone marrow cells by in situ hybridization, Wnt16 mRNA expression could be detected in bone marrow osteoprogenitor cells by scRNAseq and in cultured bone marrow cells by qPCR. The relative difference in Wnt16 expression was shown by the finding that Wnt16 is markedly less expressed in cultured bone marrow cells vs calvarial osteoblasts when assessed by qPCR analysis. Moreover, we found that OSM induced Wnt16 expression in calvarial osteoblasts, but not in bone marrow cells, and that OSM induced osteoclastogenesis is similar in bone marrow cell cultures from wild-type and Wnt16−/- mice, in contrast to the inhibitory role of WNT16 in the calvarial cells. The reason why OSM does not affect Wnt16 expression in bone marrow cells is explained by observations using two different sets of scRNASeq analyses showing that Osmr and Wnt16 are expressed in distinctly different osteogenic cell types. It is likely that the WNT16 levels are too low to affect osteoclast differentiation in the bone marrow cells cultures.
Having established that OSM robustly upregulates Wnt16 mRNA expression in calvarial osteoblasts and that deletion of Wnt16 results in increased osteoclast formation in such cultures, we next sought to investigate which receptors were used and which downstream signaling pathways were involved in the OSM-induced expression of Wnt16. Mouse OSM, which has been used in the present study, is known to utilize the mouse OSMR, whereas human OSM utilizes the LIFR in mouse cells.55,56 It has been found, however, that mouse OSM activates the mouse LIFR when decreasing Sost expression in mouse calvarial osteocytes.31 Furthermore, 172 identical genes are regulated by mouse OSM and LIF in osteoblasts from Osmr−/- mice, which demonstrates that OSM can utilize the LIFR:gp130 complex to regulate several genes.68 We, therefore, silenced Il6st (encoding gp130), Lifr and Osmr and found that expression of Il6st and Osmr, but not that of Lifr, is important for efficient OSM induction of Wnt16. These findings, together with the fact that cytokines utilizing the LIFR, such as hOSM, LIF, CT-1 and CNTF, did not regulate Wnt16 mRNA expression, demonstrate that OSM is dependent on activation of the OSMR:gp130 complex to induce Wnt16 mRNA expression.
The OSMR has no intrinsic tyrosine kinase activity, but activation of the OSMR:gp130 complex results in phosphorylation of several Tyr residues in the intracellular domains of gp130 and OSMR by constitutively connected JAKs.57,58 This leads to recruitment of STAT3, which after homo-dimerization is translocated to the nuclei to regulate genes with STAT-responsive elements. Silencing of Stat3 abolished OSM-induced Wnt16 mRNA expression, demonstrating the crucial role of this transcription factor. Ligation of the OSMR results also in activation of MAPKs, including ERK, in several cell types61 as well as in the calvarial osteoblasts.29 Silencing of Erk, however, showed that ERK is not involved in induction of Wnt16 mRNA by OSM in calvarial osteoblasts.
In contrast to other receptors for cytokines in the gp130 family, the OSMR activates a non-redundant signaling pathway by recruiting the adapter protein SHC1 through a SH2 domain to Tyr861 phosphorylated by JAK. SHC proteins are phosphotyrosine adapters important for downstream signaling of certain receptors and four members of the SHC family have been described.69 We have recently shown that calvarial osteoblasts express all three isoforms (p46, p52 and p66) of the adapter protein SHC1, but not SHC2, SHC3 and SHC4.29 We found that SHC1 is an important part of signaling downstream the OSMR since silencing of Shc1 inhibited OSM-induced Wnt16 mRNA expression, which might explain why OSM is a more robust stimulator of Wnt16 mRNA in calvarial osteoblasts than other cytokines in the gp130 family.
Although the observation that mice with Osmr deletion have increased trabecular and cortical bone mass and decreased numbers of osteoclasts, indicating that OSM might have a role in physiological bone resorption,31 our present findings should not be interpreted as OSM-induced Wnt16 having a role in cortical bone remodeling explaining the low cortical bone mass in the appendicular skeleton of Wnt16−/- mice since a limitation of the present study is that we have focused our studies on the effect of OSM on in vitro cultured primary calvarial osteoblast. Further studies are needed to investigate the effect of OSM on WNT16 expression in cortical osteoblasts from long bones and its importance for osteoclastogenesis in vivo.
In summary, we show that OSM is a strong stimulator of anti-osteoclastogenic WNT16 in calvarial osteoblasts through the OSMR/gp130/SHC1/STAT3 pathway (Figure 8). OSM is not only known as a stimulator of RANKL and osteoclast formation through the OSMR but also as an inducer of bone formation by decreasing Sost expression through the LIFR.31 Although stimulation of osteoclast formation and bone resorption by OSM have been observed both in vitro and in vivo, some reports indicate that the dominant role of OSM in vivo is to enhance bone formation.27,31 The robust upregulation of WNT16 by OSM could be a mechanism to decrease osteoclastogenesis, thereby contributing to the OSM-induced increase in bone mass. Our findings may also suggest that OSM-induced WNT16 may be a mechanism participating in the resolution of inflammation, where increased bone resorption is initially caused by a variety of osteoclastogenic proinflammatory cytokines and then followed by enhanced bone formation. The proinflammatory cytokine OSM may be more important for the bone formation than the resorptive response both by increasing WNT16 to decrease resorption and by directly stimulating osteoblastic bone formation.31 In addition, WNT16 may also contribute to the bone formation response since it has been shown that global and inducible Wnt16 deletion is associated not only with enhanced numbers of osteoclasts but also with decreased bone formation.11,70 In favor of this bone anabolic hypothesis is the importance of OSM in the healing of experimental fractures in mice.71
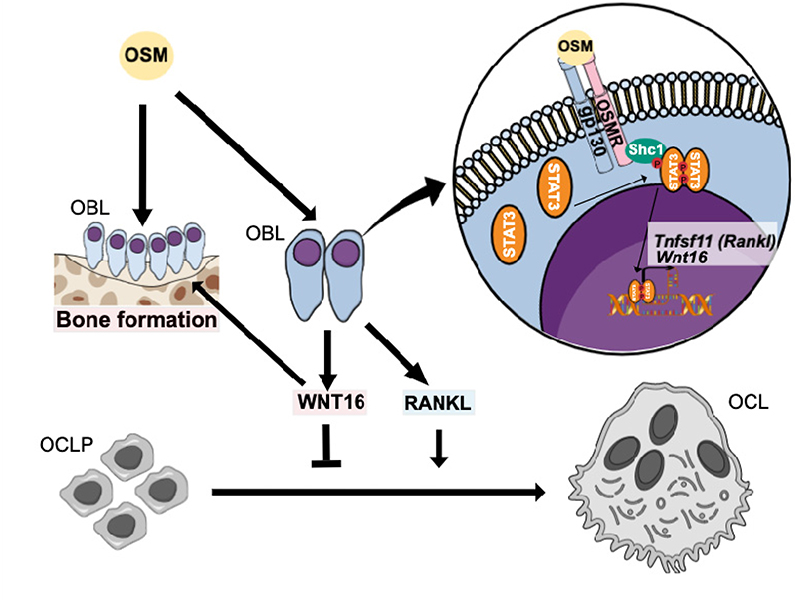 |
Figure 8 Proposed effects and signaling pathways by OSM in bone cells. OSM increases bone resorption by inducing the expression of osteoclastogenic RANKL by osteoblasts. We, here, show that OSM also induces the expression of WNT16 that act as a negative feedback regulator of osteoclast formation in calvarial periosteal cells. The induction of Wnt16 by OSM is dependent on gp130 and OSM receptor (OSMR), and downstream signaling by the SHC1/STAT3 pathway. The inhibition of osteoclastogenesis by WNT16, together with a stimulatory effect on bone formation by OSM27,31 and WNT16,11,70 will cause an OSM-dependent increase of bone mass. Abbreviations: OBL, osteoblast; OCL, osteoclast. |
Acknowledgments
This study was supported by the Swedish Research Council, the Swedish state under the agreement between the Swedish government and the county councils, the ALF-agreement in Gothenburg (grant numbers 237551, 238261 and 226481), the Swedish Foundation of Strategic Research, the IngaBritt and Arne Lundberg Foundation, the Torsten and Ragnar Söderberg’s Foundation, the Knut and Alice Wallenberg Foundation, the Novo Nordisk Foundation, the Swedish Rheumatism Association, the Royal 80 Year Fund of King Gustav V, and the County Council of Västerbotten, grants #2014/05283-3 and 2015/00410-0, São Paulo Research Foundation (FAPESP) and by a scholarship to TF-M from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior–Brasil (CAPES)–Finance code 001 and grant #061/2013 (PVE080/2012).
Disclosure
Dr Sofia Movérare-Skrtic reports grants from the Swedish Research Council, grants from the Swedish state under the agreement between the Swedish government and the county councils, the ALF-agreement in Gothenburg, grants from the Novo Nordisk Foundation, during the conduct of the study. Dr Pedro Paulo Chaves de Souza reports grants from São Paulo Research Foundation (FAPESP), grants from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior–Brasil (CAPES), during the conduct of the study. Prof. Dr Claes Ohlsson reports grants from The Swedish Research Council, grants from the Swedish state under the agreement between the Swedish government and the county councils, the ALF-agreement, the Torsten Söderberg Foundation, the Novo Nordisk Foundation, the IngaBritt and Arne Lundberg Foundation, the Knut and Alice Wallenberg Foundation, during the conduct of the study; In addition, Prof. Dr Claes Ohlsson has two Patents/patent applications within the field of probiotic treatment and osteoporosis pending. Professor Ulf Lerner reports grants from the Swedish Research Council, the Swedish state under the agreement between the Swedish government and the county councils, the ALF-agreement in Gothenburg (grant numbers 237551), IngaBritt and Arne Lundberg Foundation, the Swedish Rheumatism Association, the Royal 80 Year Fund of King Gustav V, and the County Council of Västerbotten, grants #2014/05283-3 and 2015/00410-0, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Gong Y, Slee RB, Fukai N, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107(4):513–523. doi:10.1016/S0092-8674(01)00571-2
2. Boyden LM, Mao J, Belsky J, et al. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346(20):1513–1521. doi:10.1056/NEJMoa013444
3. Little RD, Carulli JP, Del Mastro RG, et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. 2002;70(1):11–19. doi:10.1086/338450
4. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–1205. doi:10.1016/j.cell.2012.05.012
5. Balemans W, Ebeling M, Patel N, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. 2001;10(5):537–543. doi:10.1093/hmg/10.5.537
6. Balemans W, Patel N, Ebeling M, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39(2):91–97. doi:10.1136/jmg.39.2.91
7. Laine CM, Joeng KS, Campeau PM, et al. WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N Engl J Med. 2013;368(19):1809–1816. doi:10.1056/NEJMoa1215458
8. Kiper POS, Saito H, Gori F, et al. Cortical-bone fragility–insights from sfrp4 deficiency in pyle’s disease. N Engl J Med. 2016;374(26):2553–2562. doi:10.1056/NEJMoa1509342
9. Moverare-Skrtic S, Henning P, Liu X, et al. Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat Med. 2014;20(11):1279–1288. doi:10.1038/nm.3654
10. Gori F, Lerner U, Ohlsson C, Baron R. A new WNT on the bone: WNT16, cortical bone thickness, porosity and fractures. Bonekey Rep. 2015;4:669. doi:10.1038/bonekey.2015.36
11. Ohlsson C, Henning P, Nilsson KH, et al. Inducible Wnt16 inactivation: WNT16 regulates cortical bone thickness in adult mice. J Endocrinol. 2018;237(2):113–122. doi:10.1530/JOE-18-0020
12. Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19(2):179–192. doi:10.1038/nm.3074
13. Lerner UH, The OC. WNT system: background and its role in bone. J Intern Med. 2015;277(6):630–649. doi:10.1111/joim.12368
14. Maeda K, Kobayashi Y, Udagawa N, et al. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat Med. 2012;18(3):405–412. doi:10.1038/nm.2653
15. Chen K, Ng PY, Chen R, et al. Sfrp4 repression of the Ror2/Jnk cascade in osteoclasts protects cortical bone from excessive endosteal resorption. Proc Natl Acad Sci U S A. 2019;116(28):14138–14143. doi:10.1073/pnas.1900881116
16. Albers J, Keller J, Baranowsky A, et al. Canonical Wnt signaling inhibits osteoclastogenesis independent of osteoprotegerin. J Cell Biol. 2013;200(4):537–549. doi:10.1083/jcb.201207142
17. Weivoda MM, Ruan M, Hachfeld CM, et al. Wnt signaling inhibits osteoclast differentiation by activating canonical and noncanonical cAMP/PKA pathways. J Bone Miner Res. 2016;31(1):65–75. doi:10.1002/jbmr.2599
18. Yu B, Chang J, Liu Y, et al. Wnt4 signaling prevents skeletal aging and inflammation by inhibiting nuclear factor-kappaB. Nat Med. 2014;20(9):1009–1017. doi:10.1038/nm.3586
19. Wang F, Tarkkonen K, Nieminen-Pihala V, et al. Mesenchymal cell-derived juxtacrine wnt1 signaling regulates osteoblast activity and osteoclast differentiation. J Bone Miner Res. 201934(6):1129–1142. doi:10.1002/jbmr.3680.
20. Estrada K, Styrkarsdottir U, Evangelou E, et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet. 2012;44(5):491–501. doi:10.1038/ng.2249
21. Zheng HF, Tobias JH, Duncan E, et al. WNT16 influences bone mineral density, cortical bone thickness, bone strength, and osteoporotic fracture risk. PLoS Genet. 2012;8(7):e1002745. doi:10.1371/journal.pgen.1002745
22. Moverare-Skrtic S, Wu J, Henning P, et al. The bone-sparing effects of estrogen and WNT16 are independent of each other. Proc Natl Acad Sci U S A. 2015;112(48):14972–14977. doi:10.1073/pnas.1520408112
23. Ohlsson C, Nilsson KH, Henning P, et al. WNT16 overexpression partly protects against glucocorticoid-induced bone loss. Am J Physiol Endocrinol Metab. 2018;314(6):E597–E604. doi:10.1152/ajpendo.00292.2017
24. Schett G, Gravallese E. Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nat Rev Rheumatol. 2012;8(11):656–664. doi:10.1038/nrrheum.2012.153
25. Souza PP, Lerner UH. The role of cytokines in inflammatory bone loss. Immunol Invest. 2013;42(7):555–622.
26. Sims NA, Walsh NC. GP130 cytokines and bone remodelling in health and disease. BMB Rep. 2010;43(8):513–523. doi:10.5483/BMBRep.2010.43.8.513
27. Lerner UH. Role of interleukins on physiological and pathological bone resorption and bone formation. II. Effects by cytokines in the IL-6 and IL-10 families. In: Encyclopedia of Bone. Vol. 2. Oxford: Academic Press; 2020:67–87.
28. Palmqvist P, Persson E, Conaway HH, Lerner UH. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae. J Immunol. 2002;169(6):3353–3362. doi:10.4049/jimmunol.169.6.3353
29. Persson E, Souza PPC, Floriano-Marcelino T, Conaway HH, Henning P, Lerner UH. Activation of Shc1 allows oncostatin M to induce RANKL and osteoclast formation more effectively than leukemia inhibitory factor. Front Immunol. 2019;10:1164. doi:10.3389/fimmu.2019.01164
30. Zarling JM, Shoyab M, Marquardt H, et al. Oncostatin M: a growth regulator produced by differentiated histiocytic lymphoma cells. Proc Natl Acad Sci U S A. 1986;83(24):9739–9743. doi:10.1073/pnas.83.24.9739
31. Walker EC, McGregor NE, Poulton IJ, et al. Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. J Clin Invest. 2010;120(2):582–592. doi:10.1172/JCI40568
32. Lisignoli G, Piacentini A, Toneguzzi S, et al. Osteoblasts and stromal cells isolated from femora in rheumatoid arthritis (RA) and osteoarthritis (OA) patients express IL-11, leukaemia inhibitory factor and oncostatin M. Clin Exp Immunol. 2000;119(2):346–353. doi:10.1046/j.1365-2249.2000.01114.x
33. Pradeep AR, Garima STM, Garima G, Raju A. Serum levels of oncostatin M (a gp 130 cytokine): an inflammatory biomarker in periodontal disease. Biomarkers. 2010;15(3):277–282. doi:10.3109/13547500903573209
34. West NR, Hegazy AN, Owens BMJ, et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat Med. 2017;23(5):579–589. doi:10.1038/nm.4307
35. Mozaffarian A, Brewer AW, Trueblood ES, et al. Mechanisms of oncostatin M-induced pulmonary inflammation and fibrosis. J Immunol. 2008;181(10):7243–7253. doi:10.4049/jimmunol.181.10.7243
36. Torossian F, Guerton B, Anginot A, et al. Macrophage-derived oncostatin M contributes to human and mouse neurogenic heterotopic ossifications. JCI Insight. 2017;2(21). doi:10.1172/jci.insight.96034
37. Blumenthal A, Ehlers S, Lauber J, et al. The Wingless homolog WNT5A and its receptor Frizzled-5 regulate inflammatory responses of human mononuclear cells induced by microbial stimulation. Blood. 2006;108(3):965–973. doi:10.1182/blood-2005-12-5046
38. Pereira C, Schaer DJ, Bachli EB, Kurrer MO, Schoedon G. Wnt5A/CaMKII signaling contributes to the inflammatory response of macrophages and is a target for the antiinflammatory action of activated protein C and interleukin-10. Arterioscler Thromb Vasc Biol. 2008;28(3):504–510. doi:10.1161/ATVBAHA.107.157438
39. Manicassamy S, Reizis B, Ravindran R, et al. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science. 2010;329(5993):849–853. doi:10.1126/science.1188510
40. Neumann J, Schaale K, Farhat K, et al. Frizzled1 is a marker of inflammatory macrophages, and its ligand Wnt3a is involved in reprogramming Mycobacterium tuberculosis-infected macrophages. FASEB J. 2010;24(11):4599–4612. doi:10.1096/fj.10-160994
41. Ouchi N, Higuchi A, Ohashi K, et al. Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science. 2010;329(5990):454–457. doi:10.1126/science.1188280
42. Irvine KM, Clouston AD, Gadd VL, et al. Deletion of Wntless in myeloid cells exacerbates liver fibrosis and the ductular reaction in chronic liver injury. Fibrogenesis Tissue Repair. 2015;8(1):19. doi:10.1186/s13069-015-0036-7
43. Gatica-Andrades M, Vagenas D, Kling J, et al. WNT ligands contribute to the immune response during septic shock and amplify endotoxemia-driven inflammation in mice. Blood Adv. 2017;1(16):1274–1286. doi:10.1182/bloodadvances.2017006163
44. Dell’accio F, De Bari C, Eltawil NM, Vanhummelen P, Pitzalis C. Identification of the molecular response of articular cartilage to injury, by microarray screening: wnt-16 expression and signaling after injury and in osteoarthritis. Arthritis Rheum. 2008;58(5):1410–1421. doi:10.1002/art.23444
45. Nalesso G, Thomas BL, Sherwood JC, et al. WNT16 antagonises excessive canonical WNT activation and protects cartilage in osteoarthritis. Ann Rheum Dis. 2017;76(1):218–226. doi:10.1136/annrheumdis-2015-208577
46. Tong W, Zeng Y, Chow DHK, et al. Wnt16 attenuates osteoarthritis progression through a PCP/JNK-mTORC1-PTHrP cascade. Ann Rheum Dis. 2019;78(4):551–561. doi:10.1136/annrheumdis-2018-214200
47. Tornqvist AE, Grahnemo L, Nilsson KH, Funck-Brentano T, Ohlsson C, Moverare-Skrtic S. Wnt16 overexpression in osteoblasts increases the subchondral bone mass but has no impact on osteoarthritis in young adult female mice. Calcif Tissue Int. 2020;107(1):31–40. doi:10.1007/s00223-020-00682-7
48. Liu X, Li X, Hua B, Yang X, Zheng J, Liu S. WNT16 is upregulated early in mouse TMJ osteoarthritis and protects fibrochondrocytes against IL-1beta induced inflammatory response by regulation of RUNX2/MMP13 cascade. Bone. 2021;143:115793. doi:10.1016/j.bone.2020.115793
49. Granholm S, Henning P, Lindholm C, Lerner UH. Osteoclast progenitor cells present in significant amounts in mouse calvarial osteoblast isolations and osteoclastogenesis increased by BMP-2. Bone. 2013;52(1):83–92. doi:10.1016/j.bone.2012.09.019
50. Granholm S, Lundberg P, Lerner UH. Calcitonin inhibits osteoclast formation in mouse haematopoetic cells independently of transcriptional regulation by receptor activator of NF-{kappa}B and c-Fms. J Endocrinol. 2007;195(3):415–427. doi:10.1677/JOE-07-0338
51. Lassen NE, Andersen TL, Ploen GG, et al. Coupling of bone resorption and formation in real time: new knowledge gained from human haversian BMUs. J Bone Miner Res. 2017;32(7):1395–1405. doi:10.1002/jbmr.3091
52. Tikhonova AN, Dolgalev I, Hu H, et al. The bone marrow microenvironment at single-cell resolution. Nature. 2019;569(7755):222–228. doi:10.1038/s41586-019-1104-8
53. Matsushita Y, Nagata M, Kozloff KM, et al. A Wnt-mediated transformation of the bone marrow stromal cell identity orchestrates skeletal regeneration. Nat Commun. 2020;11(1):332. doi:10.1038/s41467-019-14029-w
54. Yahara Y, Barrientos T, Tang YJ, et al. Erythromyeloid progenitors give rise to a population of osteoclasts that contribute to bone homeostasis and repair. Nat Cell Biol. 2020;22(1):49–59. doi:10.1038/s41556-019-0437-8
55. Hermanns HM. Oncostatin M and interleukin-31: cytokines, receptors, signal transduction and physiology. Cytokine Growth Factor Rev. 2015;26(5):545–558. doi:10.1016/j.cytogfr.2015.07.006
56. Richards CD. The enigmatic cytokine oncostatin m and roles in disease. ISRN Inflamm. 2013;2013:512103. doi:10.1155/2013/512103
57. Hermanns HM, Radtke S, Haan C, et al. Contributions of leukemia inhibitory factor receptor and oncostatin M receptor to signal transduction in heterodimeric complexes with glycoprotein 130. J Immunol. 1999;163(12):6651–6658.
58. Schmitz J, Dahmen H, Grimm C, et al. The cytoplasmic tyrosine motifs in full-length glycoprotein 130 have different roles in IL-6 signal transduction. J Immunol. 2000;164(2):848–854. doi:10.4049/jimmunol.164.2.848
59. Hintzen C, Evers C, Lippok BE, et al. Box 2 region of the oncostatin M receptor determines specificity for recruitment of Janus kinases and STAT5 activation. J Biol Chem. 2008;283(28):19465–19477. doi:10.1074/jbc.M710157200
60. Anhuf D, Weissenbach M, Schmitz J, et al. Signal transduction of IL-6, leukemia-inhibitory factor, and oncostatin M: structural receptor requirements for signal attenuation. J Immunol. 2000;165(5):2535–2543. doi:10.4049/jimmunol.165.5.2535
61. Neel BG, Gu H, The PL. ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28(6):284–293. doi:10.1016/S0968-0004(03)00091-4
62. Hermanns HM, Radtke S, Schaper F, Heinrich PC, Behrmann I. Non-redundant signal transduction of interleukin-6-type cytokines. The adapter protein Shc is specifically recruited to rhe oncostatin M receptor. J Biol Chem. 2000;275(52):40742–40748. doi:10.1074/jbc.M005408200
63. Wang Y, Robledo O, Kinzie E, et al. Receptor subunit-specific action of oncostatin M in hepatic cells and its modulation by leukemia inhibitory factor. J Biol Chem. 2000;275(33):25273–25285. doi:10.1074/jbc.M002296200
64. O’Brien CA, Gubrij I, Lin SC, Saylors RL, Manolagas SC. STAT3 activation in stromal/osteoblastic cells is required for induction of the receptor activator of NF-kappaB ligand and stimulation of osteoclastogenesis by gp130-utilizing cytokines or interleukin-1 but not 1,25-dihydroxyvitamin D3 or parathyroid hormone. J Biol Chem. 1999;274(27):19301–19308.
65. Udagawa N, Takahashi N, Katagiri T, et al. Interleukin (IL)-6 induction of osteoclast differentiation depends on IL-6 receptors expressed on osteoblastic cells but not on osteoclast progenitors. J Exp Med. 1995;182(5):1461–1468. doi:10.1084/jem.182.5.1461
66. McGregor NE, Murat M, Elango J, et al. IL-6 exhibits both cis- and trans-signaling in osteocytes and osteoblasts, but only trans-signaling promotes bone formation and osteoclastogenesis. J Biol Chem. 2019;294(19):7850–7863. doi:10.1074/jbc.RA119.008074
67. Jilka RL. Parathyroid hormone-stimulated development of osteoclasts in cultures of cells from neonatal murine calvaria. Bone. 1986;7(1):29–40. doi:10.1016/8756-3282(86)90149-3
68. Walker EC, Johnson RW, Hu Y, et al. Murine oncostatin M acts via leukemia inhibitory factor receptor to phosphorylate signal transducer and activator of transcription 3 (STAT3) but not STAT1, an effect that protects bone mass. J Biol Chem. 2016;291(41):21703–21716. doi:10.1074/jbc.M116.748483
69. Wills MK, Jones N. Teaching an old dogma new tricks: twenty years of Shc adaptor signalling. Biochem J. 2012;447(1):1–16. doi:10.1042/BJ20120769
70. Wergedal JE, Kesavan C, Brommage R, Das S, Mohan S. Role of WNT16 in the regulation of periosteal bone formation in female mice. Endocrinology. 2015;156(3):1023–1032. doi:10.1210/en.2014-1702
71. Guihard P, Boutet MA, Brounais-le Royer B, et al. Oncostatin m, an inflammatory cytokine produced by macrophages, supports intramembranous bone healing in a mouse model of tibia injury. Am J Pathol. 2015;185(3):765–775. doi:10.1016/j.ajpath.2014.11.008
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.