Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 18
Vitamin D and Its Regulatory Role in Pancreatic β-Cell Function: Implications for Diabetes
Authors Meng X, Yang X, Lu H, Yang X, Hu D, Wu H
Received 18 July 2025
Accepted for publication 20 November 2025
Published 23 December 2025 Volume 2025:18 Pages 4741—4754
DOI https://doi.org/10.2147/DMSO.S551390
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Rebecca Baqiyyah Conway
Xingpei Meng,1,* Xue Yang,2,* Hua Lu,3 Xuehua Yang,4 Demei Hu,4 Hongping Wu4
1Department of General Practice, School of Clinical Medicine, Guizhou Medical University, Guiyang, Guizhou, 550000, People’s Republic of China; 2Department of General Practice, The People’s Hospital Of QianNan, Duyun, Qiannan Buyi and Miao Autonomous Prefecture, Guizhou Province, 558000, People’s Republic of China; 3Department of General Practice Medicine, The Third Affiliated Hospital of Guizhou Medical University, Duyun, Qiannan Buyi and Miao Autonomous Prefecture, Guizhou Province, 558000, People’s Republic of China; 4Department of Endocrinology, The Third Affiliated Hospital of Guizhou Medical University, Duyun, Qiannan Buyi and Miao Autonomous Prefecture, Guizhou Province, 558000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xue Yang, Department of General Practice, The People’s Hospital Of QianNan, No. 9, Wenfeng Road, Duyun, Qiannan Buyi and Miao Autonomous Prefecture, Guizhou Province, 558000, People’s Republic of China, Tel +86 15180775530, Email [email protected]
Abstract: Pancreatic β-cell dysfunction represents a key pathological feature in the development and progression of diabetes mellitus. Accumulating evidence has confirmed the widespread expression of vitamin D receptors in pancreatic tissue, suggesting a potential regulatory role in glucose metabolism. Epidemiological studies have consistently reported an association between vitamin D deficiency and increased diabetes incidence, impaired insulin secretion, and poor glycemic control. Vitamin D has been known to support pancreatic islet function by modulating β-cell proliferation, enhancing insulin synthesis and secretion, and mitigating inflammatory responses and oxidative stress. This review systematically discusses vitamin D metabolism and physiological functions, the role of pancreatic islet dysfunction in diabetes pathogenesis, vitamin D receptor expression and activity in pancreatic tissue, epidemiological correlations between vitamin D status and diabetes risk, and the molecular mechanisms through which vitamin D influences β-cell function. Furthermore, this review examines the therapeutic implications of vitamin D supplementation for the prevention and management of diabetes. It contributes to a growing body of knowledge that informs potential strategies to improve diabetes outcomes through vitamin D-related pathways.
Keywords: diabetes mellitus, pancreatic β cells, pancreatic islet function, vitamin D, vitamin D receptor
Introduction
The global prevalence of diabetes mellitus (DM) has continued to rise in recent years, posing a significant public health concern. According to the latest estimates from the International Diabetes Federation (IDF Diabetes Atlas, 11th Edition, 2025), approximately 588.7 million adults aged 20–79 years were living with diabetes worldwide in 2024, with the vast majority having type 2 diabetes mellitus (T2DM). Based on demographic trends, this number is projected to increase to 852.5 million by 2050.1,2 T2DM is a metabolic disorder characterized by insulin resistance (IR) and the progressive dysfunction of pancreatic β cells.3 The deterioration of β-cell function is a key contributor to the progression of T2DM and involves several interrelated pathogenic mechanisms, including glucotoxicity, lipotoxicity, endoplasmic reticulum (ER) stress, oxidative stress, and chronic low-grade inflammation.4,5
Historically, vitamin D has been primarily associated with the regulation of calcium and phosphate homeostasis and the maintenance of bone health. However, emerging evidence has indicated the widespread expression of vitamin D receptors (VDR) across various tissue types, indicating broader physiological roles beyond skeletal metabolism.6 Dysregulation of vitamin D metabolism has been implicated in the impairment of insulin secretion by β cells.7 Epidemiological studies have identified a significant association between vitamin D deficiency, defined as serum 25-hydroxyvitamin D [25(OH)D] concentrations below 20 ng/mL, and increased incidence of T2DM, reduced insulin secretory capacity, and poor glycemic control, indicating a potential regulatory function of vitamin D in glucose metabolism.8,9
Despite consistent epidemiological findings linking vitamin D deficiency to adverse metabolic outcomes, the extent to which vitamin D directly enhances pancreatic β-cell function remains a subject of ongoing investigation. This review provides a systematic examination of the regulatory mechanisms through which vitamin D influences pancreatic islet function and explores its potential therapeutic implications for the prevention and management of diabetes.
The scale of this epidemic is further underscored by its substantial human and economic costs. The same report estimates that 252 million adults are living with undiagnosed diabetes,2 and diabetes was responsible for over 3.4 million deaths in 2024. Furthermore, the global economic burden is staggering, with approximately USD 1 trillion spent on diabetes-related healthcare, accounting for 12% of total global health expenditure.2 Confronted with this immense and growing burden, identifying modifiable risk factors and novel therapeutic strategies is of paramount importance. The role of vitamin D in pancreatic β-cell function represents one such promising avenue of investigation.
Vitamin D Overview
Vitamin D, a fat-soluble vitamin, primarily exists in two forms: vitamin D2 and vitamin D3. Within the human body, vitamin D undergoes two sequential hydroxylation reactions, first in the liver and subsequently in the kidneys to produce its biologically active metabolite, 1,25-dihydroxyvitamin D3. This metabolite exerts its physiological effects by binding to the vitamin D receptor. The primary functions of vitamin D include facilitating the intestinal absorption of calcium and phosphorus, regulating bone metabolism, and maintaining calcium homeostasis.
Vitamin D is acquired via two primary pathways: endogenous synthesis through ultraviolet radiation-induced conversion of 7-dehydrocholesterol in the skin and dietary intake. Dietary sources of vitamin D include fatty fish, egg yolks, and fortified dairy products. Once in the body, vitamin D undergoes a series of metabolic transformations. Initially, 7-dehydrocholesterol in the skin is photochemically converted to vitamin D3, or vitamin D3 is absorbed directly from dietary sources. This is followed by hepatic conversion to 25-hydroxyvitamin D3 [25(OH)D3], the principal circulating form. In the kidneys, 25(OH)D3 is further hydroxylated to form the active metabolite 1,25-dihydroxyvitamin D3 [1,25-(OH)2D3], as well as 24R,25-dihydroxyvitamin D3 [24R,25(OH)2D3]. These metabolites are transported to target tissues via plasma vitamin D binding protein and are ultimately excreted through bile and urine.
The hepatic conversion of vitamin D3 to 25(OH)D3 occurs in microsomal and mitochondrial compartments and is catalyzed by vitamin D3-25-hydroxylase. The activity of this enzyme is only partially inhibited by its product, 25(OH)D3. As a result, increased intake of vitamin D3 leads to a proportional increase in 25(OH)D3 synthesis, establishing a positive correlation between serum 25(OH)D3 concentrations and vitamin D3 intake. Owing to its stability and prevalence in circulation, serum 25(OH)D3 concentration is widely used as a biomarker to assess vitamin D nutritional status.10
Advancements in research have significantly expanded the understanding of vitamin D. Initially classified solely as a nutrient involved in calcium and phosphate metabolism, vitamin D has since been recognized, particularly following the identification of vitamin D receptor, as a steroid hormone with regulatory roles in diverse physiological systems.11
The vitamin D receptor is a member of the nuclear receptor superfamily of transcription factors. Upon interaction with the active form of vitamin D (1,25-dihydroxyvitamin D3), the vitamin D receptor initiates transcription of target genes that govern key biological processes, including cellular proliferation, differentiation, and immune modulation.12 Vitamin D receptor expression and function are modulated by various factors such as vitamin D availability, hormonal signals, and environmental influences.13 Expression of vitamin D receptor has been observed in multiple pancreatic cell types, with particularly high levels noted in pancreatic β cells.14 As the principal endocrine cell type responsible for insulin synthesis and secretion, pancreatic β cells are directly relevant to the metabolic actions of vitamin D.
Pancreatic β Cell Function in Diabetes
Pancreatic β cells, the principal cell type responsible for insulin secretion, experience both a reduction in cell mass and a decline in functional capacity during the pathogenesis of diabetes, ultimately resulting in impaired glycemic regulation. Chronic hyperglycemia imposes glucotoxic stress on β cells.15 In the context of IR, β cells initially respond by augmenting insulin secretion to preserve glucose homeostasis. However, sustained metabolic stress eventually contributes to β-cell dysfunction and exhaustion.
Evidence indicates that prolonged hyperglycemia induces ER stress, thereby activating the unfolded protein response (UPR) and apoptotic signaling cascades, including those mediated by C/EBP homologous protein and c-Jun N-terminal kinase (JNK). These pathways accelerate β cell apoptosis and contribute to disease progression.16 Clinical studies have demonstrated that in patients recently diagnosed with T2DM, pancreatic β-cell function declines at an estimated rate of 2–3% annually. This deterioration likely begins several years prior to clinical diagnosis.17
IR represents another defining characteristic of diabetes and contributes to a self-perpetuating cycle with pancreatic β cell dysfunction. In response to diminished insulin sensitivity in peripheral tissues, β cells increase insulin secretion, resulting in compensatory hyperinsulinemia.18,19 However, prolonged compensatory secretion has been associated with β-cell dedifferentiation and ultimately, apoptosis.20,21
In addition, the excessive release of free fatty acids (FFAs) from adipose tissue, a phenomenon known as lipotoxicity, along with elevated levels of proinflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukin-6, can further contribute to β-cell dysfunction.22,23 This interaction establishes a positive feedback loop characterized by metabolic inflammation and progressive β-cell damage.
Findings from autopsy studies have demonstrated a reduction in pancreatic β-cell mass by approximately 40% to 60% in patients with T2DM compared to those without the disease. Furthermore, this degree of β-cell loss is considered irreversible by the time clinical diagnosis is established.21 These observations highlight the potential importance of early intervention in mitigating β-cell decline and improving glycemic control.
Epidemiological Association Between Vitamin D Deficiency and Diabetes
Recent studies have demonstrated that pancreatic β cells express vitamin D receptors, which contribute to the regulation of β-cell survival. Vitamin D facilitates insulin secretion, thereby supporting the maintenance of normoglycemia.8 According to evidence from research, vitamin D deficiency may impair islet cell function, and islet dysfunction constitutes a fundamental pathological mechanism in the development of diabetes, particularly T2DM.24,25 Accordingly, serum vitamin D levels appear to be closely associated with both the onset and progression of diabetes.
This potential link between vitamin D and diabetes must be considered within the context of a rapidly expanding global diabetes epidemic. Recent estimates indicate that hundreds of millions of adults are affected, a significant number remain undiagnosed, and the disease imposes a trillion-dollar burden on healthcare systems worldwide.4 Against this backdrop, elucidating the contribution of widespread vitamin D deficiency to diabetes risk becomes not only a biological question but also a significant public health priority.
Extensive epidemiological studies have consistently reported a higher risk of T2DM among individuals with vitamin D deficiency. Data from the International Osteoporosis Foundation indicate that approximately 30–50% of the global population is affected by vitamin D deficiency, with prevalence estimates reaching 60–80% in Southeast Asia and 30–50% in Europe and North America.26
In a 20-year prospective cohort study involving 83,779 female nurses in the United States, Pittas et al reported that each 400 IU increase in daily vitamin D intake was associated with a 13% reduction in T2DM risk (relative risk [RR] = 0.87, 95% confidence interval [CI]: 0.80–0.95, p < 0.05). Additionally, participants who consumed more than 800 IU per day demonstrated a 33% lower risk of developing T2DM compared with those consuming less than 200 IU per day (RR = 0.67, 95% CI: 0.49–0.90).27 In a cross-sectional study conducted by Kayaniyil et al, each 10 nmol/L increase in 25(OH)D concentration was associated with a 1.3% improvement in the Matsuda insulin sensitivity index and a reduction in IR. Patients with vitamin D deficiency exhibited more pronounced IR, and a significant positive correlation was observed between serum vitamin D concentrations and pancreatic β cell function.28
A prospective study demonstrated that patients with baseline serum vitamin D concentrations below 30 nmol/L exhibited significantly elevated fasting plasma glucose levels (p < 0.001) and worsened IR (p = 0.02) after a 10-year follow-up. In contrast, participants with sufficient vitamin D levels (≥ 75 nmol/L) displayed markedly improved glucose metabolic profiles.29
Findings from interventional studies further support this association. In one trial assessing the preventive effects of vitamin D supplementation on diabetes, 2423 individuals with prediabetes were enrolled. Participants who received 4000 IU of vitamin D daily demonstrated a 12% reduction in diabetes incidence, although this difference did not reach statistical significance in the overall cohort. Notably, subgroup analysis indicated that among patients with baseline 25(OH)D levels below 30 nmol/L, diabetes risk was significantly reduced by 30%.30
In another study involving South Asian women with vitamin D deficiency, daily supplementation with 4000 IU of vitamin D for a duration of six months resulted in a 19.6% reduction in the IR index (p = 0.04) and a 12.8% decrease in fasting insulin levels compared with the placebo group (p = 0.03). In this cohort, serum 25(OH)D concentrations increased from a baseline of 31.5 nmol/L to 80.5 nmol/L in the intervention group (p < 0.001).31 Additionally, the co-administration of calcium supplementation was associated with further enhancement of metabolic outcomes.32
Mechanisms by Which Vitamin D Regulates Pancreatic β-Cell Function
Calcium Signaling Pathway (Figure 1)
Calcium signaling plays a central role in pancreatic β-cell physiology by regulating insulin secretion, primarily through oscillatory dynamics of intracellular calcium ions (Ca2+).33 Glucose is transported into β cells via glucose transporter 2 and undergoes metabolic conversion to generate adenosine triphosphate (ATP). The resulting increase in ATP concentration leads to closure of ATP-sensitive potassium (K_ATP) channels, inducing membrane depolarization. This electrical change subsequently activates voltage-dependent calcium channels, facilitating Ca2+ influx. The elevation in intracellular Ca2+ concentration promotes the fusion of insulin-containing granules with the plasma membrane, initiating insulin exocytosis.34 However, prolonged Ca2+ influx may lead to calcium overload, which activates apoptotic signaling pathways and contributes to β cell dysfunction.35
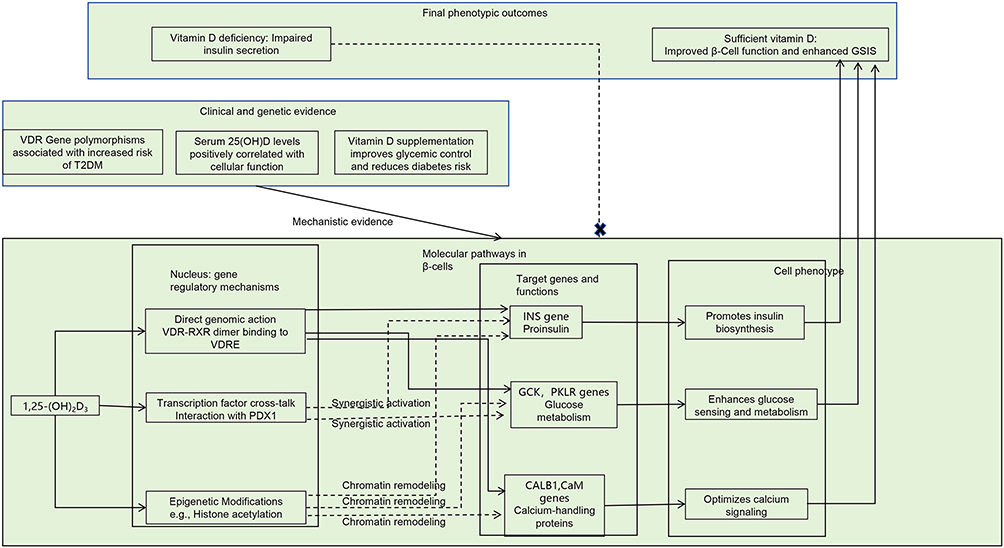 |
Figure 1 Vitamin D regulates β-cell calcium homeostasis and insulin secretion via genomic and non-genomic pathways. |
Vitamin D modulates this calcium-regulated process by supporting β cell function through a calcium signaling network. At the systemic level, 1,25-(OH)2D3 enhances intestinal calcium absorption by binding to vitamin D receptor and inducing the expression of the calcium-binding protein Calbindin-D9k. This action supports systemic calcium homeostasis and helps maintain a microenvironment conducive to optimal β cell function.36,37
At the molecular level, vitamin D regulates intracellular calcium dynamics through both genomic and non-genomic pathways. In genomic regulation, the 1,25-(OH)2D3–vitamin D receptor complex translocates into the nucleus and binds to vitamin D response elements (VDREs) within promoter regions of calcium-related genes. This interaction results in the upregulation of proteins such as Calbindin-D28k and the L-type voltage-dependent calcium channel Cav1.2, thereby enhancing glucose-stimulated calcium influx and supporting β-cell secretory activity.38,39
Along with genomic mechanisms, vitamin D also activates non-genomic signaling pathways. One such pathway involves the rapid stimulation of phospholipase C (PLC), which facilitates inositol trisphosphate-mediated calcium release from intracellular stores. This cascade increases the activity of calcium/calmodulin-dependent protein kinase (CAMK) and modulates the mitochondrial calcium uniporter, thereby supporting cellular energy metabolism through the PLC–CAMK–cAMP response element-binding protein signaling axis.40
The functional effects of vitamin D are evident in both insulin secretion and β-cell viability. During glucose metabolism, an elevated ATP/adenosine diphosphate ratio induces closure of K_ATP channels and membrane depolarization, leading to activation of L-type calcium channels, whose expression is upregulated by vitamin D. This process enhances calcium influx into β cells.34 The resulting increase in intracellular calcium activates CAMKII, which promotes the fusion of insulin granules with the plasma membrane and augments glucose-stimulated insulin secretion (GSIS).33
Concurrently, vitamin D modulates intracellular calcium homeostasis through the calcium-binding protein Calbindin-D28k, which acts as a buffer for cytosolic calcium, thereby reducing calcium peak amplitudes under pro-apoptotic conditions. Additionally, vitamin D upregulates the expression of the anti-apoptotic protein B-cell lymphoma 2 (Bcl-2) and suppresses the activation of caspase-3, contributing to a reduction in β-cell apoptosis.35,41 Vitamin D reverses insulin secretory impairments resulting from dysregulated calcium signaling in T2DM, attenuates calcium overload induced by chronic hyperglycemia, and inhibits calcium-dependent apoptosis triggered by oxidative stress, thereby providing protective effects on pancreatic β cells under pathological conditions.
Although some studies have reported no significant association between vitamin D deficiency and glucose tolerance, the preponderance of clinical evidence supports a positive correlation between vitamin D status and β-cell functional capacity.42,43 These findings propose that therapeutic strategies targeting the vitamin D–calcium signaling axis represent a promising approach for the management of diabetes.44
Regulation of Gene Expression (Figure 2)
Vitamin D enhances pancreatic β-cell function through a multilevel regulatory network involving gene expression. Initial evidence by Norman demonstrated that vitamin D deficiency suppresses insulin secretion.8 Subsequent molecular studies indicated that 1,25-(OH)2D3 modulates the expression of several key genes in pancreatic β cells through the vitamin D receptor. These regulatory effects include the upregulation of preproinsulin mRNA expression, modulation of calmodulin and other calcium-binding proteins, and increased expression of metabolic enzymes such as glucokinase and pyruvate kinase.45–48
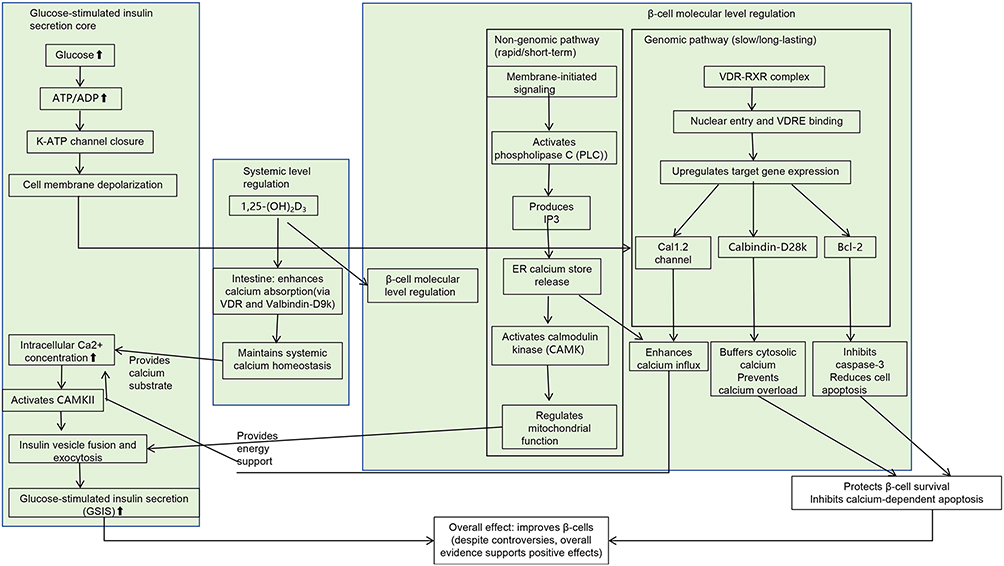 |
Figure 2 Vitamin D modulates β-cell gene expression through multiple pathways. |
Chuang et al reported high levels of vitamin D receptor expression in pancreatic β cells and demonstrated that functional deficiency of vitamin D receptor leads to impaired insulin secretion.49 Additionally, genetic polymorphisms in the vitamin D receptor gene have been associated with an elevated risk of T2DM.50 Clinical studies have consistently indicated a positive correlation between 25(OH)D levels and β-cell function.51 Vitamin D supplementation has been associated with improvements in glycemic control, and an inverse association has been observed between vitamin D intake and the risk of developing diabetes.27,32,52
At the molecular level, the vitamin D–vitamin D receptor complex exerts regulatory effects through multiple synergistic mechanisms. These include direct binding to VDREs, interaction with transcription factors such as pancreatic and duodenal homeobox 1, and the facilitation of epigenetic modifications. Collectively, these mechanisms contribute to the transcriptional regulation of genes crucial for maintaining β-cell function.53,54
Antioxidant Mechanisms
The antioxidant properties of vitamin D contribute to the preservation of pancreatic β-cell function. Pancreatic β cells generate excessive amounts of reactive oxygen species (ROS), leading to oxidative stress under hyperglycemic conditions. This elevated oxidative burden adversely affects β-cell viability and impairs insulin secretory function.55 Giacco and Brownlee identified three principal mechanisms by which ROS contribute to β-cell damage: (1) direct induction of mitochondrial dysfunction, thereby disrupting cellular energy homeostasis; (2) amplification of ER stress pathways via activation of the UPR; and (3) direct oxidative damage to β-cell DNA, resulting in genomic instability. These mechanisms collectively promote β-cell dysfunction and apoptosis.56
In obesity-associated diabetes, lipotoxicity caused by elevated levels of FFAs further exacerbates oxidative stress and contributes to β-cell injury.57 Experimental findings by Liu et al demonstrated that 1,25-(OH)2D3 upregulates the expression of antioxidant enzymes such as superoxide dismutase and glutathione peroxidase, while concurrently inhibiting the activity of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. In addition, vitamin D enhances the cellular antioxidant defense system by modulating the nuclear factor erythroid 2–related factor 2/antioxidant response element signaling pathway, thereby mitigating oxidative stress–induced cellular injury.58
Oxidative stress–mediated activation of the JNK pathway is recognized as a key mechanism in promoting β-cell apoptosis. Evidence indicates that vitamin D suppresses activation of the JNK pathway, reduces the accumulation of lipid peroxidation products, preserves mitochondrial function, and maintains ATP synthesis. Collectively, these actions contribute to the restoration of glucose-stimulated insulin secretion.59
Anti-Apoptotic Mechanisms
Apoptosis is a major mechanism contributing to the loss of pancreatic β-cell function, particularly under metabolic conditions such as diabetes, where inflammatory cytokines and chronic hyperglycemia promote β-cell death. Vitamin D inhibits β-cell apoptosis through multiple coordinated molecular pathways. Initial evidence provided by Riachy demonstrated that 1,25-(OH)2D3 enhances β-cell survival in the presence of cytokine stimulation by modulating the expression of Bcl-2 family proteins, specifically by upregulating the anti-apoptotic protein Bcl-2 and downregulating the pro-apoptotic protein Bax.60
Subsequent studies indicated that 1,25-(OH)2D3 also induces the expression of A20, an anti-apoptotic protein that inhibits the activation of caspase-8 and caspase-3, as well as the JNK signaling pathway. A20 acts as a negative regulator of the nuclear factor-kappa B (NF-κB) pathway and downregulates pro-apoptotic mediators, thereby enhancing cellular resistance to apoptosis.61 Furthermore, vitamin D downregulates the expression of Fas, a pro-apoptotic receptor involved in the extrinsic apoptosis pathway. Suppression of Fas expression and inhibition of caspase-8 activation collectively contribute to reduced apoptosis in β cells.62
Also, vitamin D inhibits the assembly of the NLRP3 inflammasome through activation of the AMP-activated protein kinase (AMPK) pathway, thereby reducing β-cell apoptosis via the AMPK–NLRP3 signaling axis.63 Vitamin D has also been reported to indirectly protect β cells by inducing the expression of cathepsin G (CatG) in CD4+ T cells, which facilitates the inactivation of autoreactive T cells and contributes to immune-mediated protection of β cells.64
Taken together, these findings demonstrate that vitamin D exerts protective effects on pancreatic β cells through the integrated regulation of Bcl-2 family proteins, A20, Fas/Fas ligand signaling, the AMPK–NLRP3 inflammasome axis, and immune-modulatory mechanisms.
Immunomodulatory Mechanisms
Evidence indicates that vitamin D contributes to the improvement of pancreatic β-cell function by modulating immune responses. During the progression of diabetes, immune dysregulation is frequently observed, characterized by elevated levels of pro-inflammatory cytokines, including interleukin-1β (IL-1β), TNF-α, and interferon-gamma (IFN-γ), as well as impaired function of regulatory T cells (Tregs). This immune imbalance plays a significant role in mediating β-cell injury and dysfunction.
Under these pathological conditions, vitamin D regulates immune function by inhibiting the activation of T helper 1 and T helper 17 cells, while promoting the differentiation of Tregs. These immunomodulatory effects improve the immune microenvironment and mitigate autoimmune-mediated β-cell damage.65 Furthermore, vitamin D supplementation upregulates the expression of CatG in CD4⁺ T cells. CatG degrades key components of the T cell receptor signaling pathway, leading to the functional inactivation of autoreactive T cells. This mechanism reduces immune-mediated β-cell destruction and contributes to the preservation of β-cell function. Notably, vitamin D regulates CatG expression through vitamin D receptor-dependent transcriptional mechanisms.64
In preclinical studies, Gysemans et al demonstrated that the combined administration of a vitamin D analog and interferon-β effectively suppressed insulitis in non-obese diabetic mice, thereby preserving pancreatic β-cell function. This synergistic effect was attributed to the vitamin D-induced modulation of dendritic cell phenotypes, along with immunoregulatory enhancement mediated by interferon-β.66
Anti-Inflammatory Mechanisms
Evidence indicates that vitamin D exerts anti-inflammatory activity that contributes to the preservation of islet cell function and attenuation of pancreatic injury associated with diabetes. T2DM is primarily recognized as a chronic inflammatory condition, wherein low-grade systemic inflammation plays a key role in disease pathogenesis. The anti-inflammatory properties of vitamin D position it as a key modulator of such chronic inflammatory responses. By downregulating the expression of pro-inflammatory cytokines, vitamin D reduces pancreatic inflammation and facilitates the restoration of a microenvironment conducive to β-cell functional recovery.67
Studies have indicated that vitamin D decreases the expression of chemokines produced by pancreatic islet cells, including CCL2, CCL3, and CXCL10. This reduction in chemokine expression limits the infiltration of immune cells into the islets and modulates the local inflammatory environment within pancreatic tissue.68 The protective role of vitamin D was first demonstrated by Riachy, who presented that 1,25-(OH)2D3 exerts beneficial effects on human islets exposed to pro-inflammatory cytokines.
The study by Riachy et al demonstrated that 1,25-(OH)2D3 significantly attenuates the cytotoxic effects of pro-inflammatory cytokines, including IL-1β, TNF-α, and IFN-γ, on human islet cells.61 This action contributes to the protection of pancreatic β cells against inflammatory injury. The underlying mechanism involves downregulation of the NF-κB signaling pathway and modulation of ER stress responses.60 Further supporting evidence was provided by Wolden-Kirk, who confirmed that vitamin D alleviates β-cell dysfunction induced by inflammatory cytokines through regulation of ER stress pathways.69
Additionally, a study conducted by Wu demonstrated that vitamin D reduced the assembly of the NLRP3 inflammasome through phosphorylation of AMPK, thereby decreasing the production of inflammatory cytokines such as IL-1β and interleukin-18. This reduction mitigates inflammation-induced β-cell injury and contributes to improved β-cell function.63
Furthermore, recent findings by Jia reported a synergistic effect between vitamin D and glibenclamide, a sulfonylurea compound. The combined therapy enhanced the protective effect of glibenclamide on β cells by suppressing the inflammatory response through inhibition of the NF-κB pathway. This combination promoted the restoration and functional recovery of pancreatic islet cells.70
Inhibition of Glucagon Secretion
Studies have indicated that vitamin D enhances pancreatic islet function not only by promoting insulin secretion but also by inhibiting glucagon release. Glucagon, a hormone secreted by pancreatic α cells, plays a key role in elevating blood glucose levels.71 Vitamin D regulates glucagon secretion through modulation of calcium signaling pathways within the pancreas. Specifically, vitamin D enhances calcium uptake and storage, increases calcineurin activity, and subsequently suppresses glucagon release. This suppression contributes to reductions in blood glucose levels and lessens the functional burden on α cells, thereby supporting their viability. Through inhibition of glucagon secretion, vitamin D indirectly contributes to improved β-cell function. Given the antagonistic relationship between insulin and glucagon in the regulation of glucose homeostasis, vitamin D facilitates hormonal balance within the islets and enhances overall pancreatic endocrine function.
Moreover, reduced glucagon secretion helps mitigate pancreatic injury associated with hyperglycemia, providing additional support for pancreatic tissue health. Bourlon et al reported that vitamin D3 deficiency decreases the expression of the calcium-binding protein calbindin-D28k in both α and β cells within rat islets, resulting in impaired insulin and glucagon secretion. Conversely, vitamin D upregulates calbindin-D28k expression, which leads to a reduction in intracellular free calcium concentrations and suppression of calcium-dependent glucagon secretion. This regulatory effect results in decreased basal and stimulated glucagon release.72
Inhibition of glucagon secretion has also been proposed to attenuate glucotoxic effects of hyperglucagonemia on β cells, improve the islet microenvironment, reduce the metabolic burden on β cells, and enhance glucose-stimulated insulin secretion, thereby supporting β-cell function. Immunohistochemical studies conducted by Redecker and Cetin identified expression of calcium-binding proteins such as calcineurin and calretinin in rodent islet cells. These proteins play key roles in pancreatic endocrine regulation. Vitamin D influences the expression of these calcium-binding proteins and contributes to glucagon suppression by maintaining intracellular calcium homeostasis, ultimately aiding in the stabilization of blood glucose levels.73
Inhibition of the Renin–Angiotensin System
In T2DM, overactivation of the renin–angiotensin system (RAS) within pancreatic islets has been implicated in the pathogenesis of β-cell dysfunction. Angiotensin II (Ang II), acting through the angiotensin II type 1 receptor (AT1 receptor), promotes oxidative stress and apoptosis in β cells, inhibits glucose-stimulated insulin secretion, and contributes to islet fibrosis. These findings underscore the detrimental role of RAS activation in β-cell impairment during T2DM progression.74
Yuan et al elucidated the molecular mechanism by which 1,25-(OH)2D3 inhibits RAS activity. Specifically, 1,25-(OH)2D3 binds to the negative vitamin D response element in the promoter region of the renin gene through the vitamin D receptor, thereby blocking the transcriptional activity of the cyclic AMP response element. This interaction suppresses renin gene transcription.75 Complementary findings by Cheng demonstrated that treatment with 1,25-(OH)2D3 reduces Ang II production in pancreatic islets, downregulates AT1 receptor expression, and upregulates expression of the angiotensin II type 2 receptor. These molecular changes contribute to β cell protection through multiple synergistic mechanisms: (1) Reduced Ang II levels lead to reduced NADPH oxidase activity and reduced ROS generation, thereby alleviating oxidative stress; (2) Downregulation of the pro-apoptotic protein Bax and upregulation of the anti-apoptotic protein Bcl-2 promote β-cell survival; (3) Glucose-stimulated insulin secretion responsiveness is enhanced; and (4) Suppression of transforming growth factor-beta 1 signaling reduces collagen deposition, thereby attenuating islet fibrosis.76
Furthermore, studies by Leung and Cheng highlighted a regulatory interaction between glucagon-like peptide-1 (GLP-1) and Ang II in maintaining islet cell function. Vitamin D may contribute to islet preservation by modulating crosstalk between GLP-1 and the RAS. A synergistic effect has also been observed between vitamin D–mediated RAS inhibition and GLP-1–induced β-cell proliferation. These complementary mechanisms help preserve the islet microenvironment and support β-cell functional integrity.77
From these observations, Leung proposed that therapeutic strategies targeting the vitamin D-vitamin D receptor-RAS signaling axis may offer clinical benefits for individuals with diabetes characterized by RAS overactivation. However, further research is warranted to optimize treatment regimens and establish standardized outcome measures to fully realize the therapeutic potential of this pathway.78
Application of Vitamin D Supplementation in Improving Islet Cell Function
Pancreatic β-cell dysfunction represents a central pathogenic mechanism in the development of DM (Table 1). Although numerous studies have proposed that vitamin D may exert regulatory effects on pancreatic islet cells, ongoing debate persists regarding the extent to which vitamin D supplementation can improve β-cell dysfunction, delay diabetes onset, or enhance glycemic control.
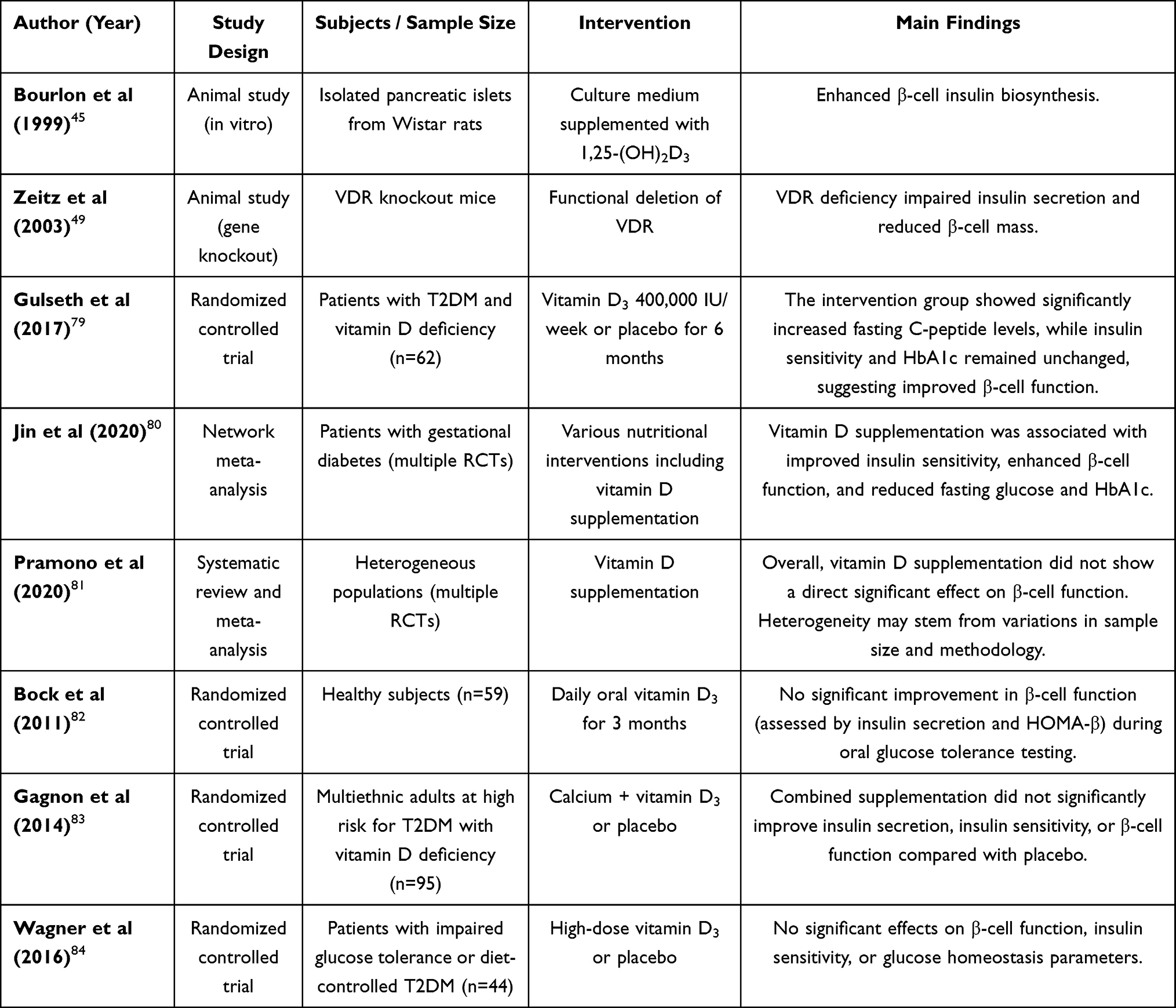 |
Table 1 Summary of Studies on the Effects of Vitamin D Supplementation on Pancreatic β-Cell Function and Glucose Metabolism |
Given the immense global burden of diabetes, which costs healthcare systems approximately USD 1 trillion annually and affects nearly 600 million adults, there is an urgent need for accessible and cost-effective adjuvant therapies.1 Vitamin D supplementation, if proven effective, could represent such a strategy, particularly for high-risk populations with identified deficiency.2
In an in vitro study, Bourlon demonstrated that the addition of 1,25-(OH)2D3 to the culture medium of isolated Wistar rat islets enhanced the biosynthetic capacity of β cells.45 Similarly, Zeitz conducted experiments using vitamin D receptor gene knockout mice and observed reduced insulin secretory capacity and a decreased number of β cells compared to wild-type controls, indicating a key role for vitamin D receptor in maintaining β cell function.49 In a clinical study involving 71 patients with T2DM and vitamin D deficiency, participants were randomized to receive either weekly high-dose vitamin D supplementation (20,000 IU/week) or placebo for 12 weeks. The intervention group demonstrated increased 25(OH)D and fasting C-peptide levels; however, no significant changes were observed in insulin sensitivity or glycated hemoglobin. These findings indicate that short-term high-dose vitamin D supplementation may improve β-cell function through mechanisms that are independent of enhanced insulin sensitivity.79
A network meta-analysis assessing nutritional interventions for glucose homeostasis in individuals with gestational DM included randomized controlled trials published up to 2019. Among various interventions assessed such as vitamin D supplementation, dietary fiber, probiotics, and adherence to a Mediterranean diet, vitamin D supplementation was associated with improvements in insulin sensitivity and β-cell function, along with reductions in fasting plasma glucose and glycated hemoglobin levels.80
However, conflicting evidence exists. A meta-analysis conducted in a heterogeneous population concluded that vitamin D supplementation did not exert a direct effect on β-cell function overall. This conclusion may be attributable to the limited sample sizes of the included trials and the invasive nature of β-cell function assessment methodologies.81 In a separate randomized controlled trial involving 40 healthy participants, participants were randomized to receive daily oral vitamin D3 for six weeks. β-cell function, assessed using the oral glucose tolerance test, presented no significant improvement in indices such as insulin secretion or HOMA-β.82 Other studies have similarly failed to demonstrate beneficial effects of vitamin D on β-cell function.83,84 The inconsistencies between preclinical and clinical findings may be due to the greater complexity of β-cell repair mechanisms in humans. Additionally, heterogeneity across clinical studies may reflect variations in diabetes stage, sample size, supplementation dose and duration, as well as methodological differences in outcome assessment.
Future investigations should prioritize large-scale, well-designed randomized controlled trials to more definitively elucidate the role of vitamin D in diabetes prevention and management. Consideration should also be given to potential synergistic effects between vitamin D and other nutrients or pharmacological agents. The development of individualized intervention strategies and novel vitamin D analogs or selective vitamin D receptor modulators may provide more targeted and effective approaches for diabetes therapy.
Conclusion and Perspectives
The relationship between vitamin D and pancreatic β-cell dysfunction has emerged as a significant focus in contemporary medical research. Beyond its well-established role in bone metabolism and the prevention of osteoporosis, vitamin D regulates β-cell function through various mechanisms, including the promotion of insulin secretion, inhibition of apoptosis, and attenuation of inflammation and oxidative stress. Advancements in experimental methodologies are expected to further elucidate the multifaceted roles of vitamin D in metabolic regulation, thereby providing a theoretical basis for the treatment of endocrine disorders.
Although existing evidence supports the potential of vitamin D in the prevention and management of DM, key parameters such as optimal supplementation dosage, timing of intervention, and the development of individualized therapeutic strategies remain to be fully defined. Future research should incorporate integrative approaches derived from genomics and metabolomics to examine the regulatory role of the vitamin D–vitamin D receptor signaling pathway in pancreatic islet function. Such investigations may contribute to the advancement of precision medicine strategies for the treatment of diabetes.
Future research should prioritize large-scale trials to translate these mechanistic insights into practical, cost-effective interventions. Success in this endeavor could offer a valuable strategy to mitigate the immense personal and societal costs of diabetes, embodied by the hundreds of millions affected and the trillion-dollar economic burden.
Abbreviations
VDR, Vitamin D receptor; IDF, International Diabetes Federation; 25(OH)D3, 25-Hydroxyvitamin D3; T2DM, Type 2 Diabetes Mellitus; 1,25-(OH)2D3, 1, 25-dihydroxyvitamin D3; 24R,25(OH)2D3, 24R,25-dihydroxyvitamin D3; ER stress, Endoplasmic reticulum stress; UPR, Unfolded protein reaction; IR, Insulin Resistance; FPG, Fasting Plasma Glucose; Ca2+, Calcium ion; GLUT2, Glucose Transporter 2; ATP, Adenosine Triphosphate; KATP, ATP-sensitive potassium channel; VDCC, Voltage-dependent calcium channel; VDRE, Vitamin D reaction element; PDX-1, Pancreatic and Duodenal Homeobox 1; Cav1.2, L-type voltage-dependent calcium channel; PLC, Phospholipase C; CAMK, Calmodulin kinase; MCU, Mitochondrial calcium unidirectional transporter; CAMKII, Ca2⁺/Calcium/calmodulin-dependent protein kinase II; GSIS, Glucose-Stimulated Insulin Secretion; ROS, Reactive oxygen species; SOD, Superoxide dismutase; GPx, Glutathione peroxidase; NOX, NADPH Oxidase; JNK, c-Jun N-terminal Kinase; Treg, Regulatory T cells; CatG, Cathepsin G; NOD, Non-obese diabetes; RAS, Renin-angiotensin system; AngII, Angiotensin II; nVDRE, Negative Vitamin D Reaction element; TGF-β1, Transforming growth factor -β1; GLP-1, Glucagon-like peptide-1; GDM, Gestational Diabetes Mellitus; OGTT, Oral Glucose Tolerance Test.
Data Sharing Statement
The datasets used or analysed during the current study are available from the corresponding author on reasonable request.
Author Contributions
Conceptualization and Methodology:: Xue Yang, Xingpei Meng, Hua Lu, Xuehua Yang, Demei Hu, Hongping Wu
Investigation: Demei Hu
Formal analysis: Xue Yang, Hua Lu, Demei Hu
Funding acquisition: Xue Yang
Writing – original draft: Xue Yang, Xingpei Meng, Hua Lu, Xuehua Yang, Demei Hu, Hongping Wu
Writing – review and editing: Xue Yang, Xingpei Meng
All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was supported by Science and Technology Fund Project of the Health Commission of Guizhou Province (No: gzwkj2023-272).
Disclosure
The authors declare that they have no conflict of interest regarding this work.
References
1. Hu FB. Globalization of diabetes: the role of diet, lifestyle, and genes. Diabetes Care. 2011;34(6):1249–1257. doi:10.2337/dc11-0442
2. Duncan BB, Magliano DJ, Boyko EJ. IDF diabetes atlas 11th edition 2025: global prevalence and projections for 2050. Nephrol Dial Transplant. 2025;gfaf177. doi:10.1093/ndt/gfaf177
3. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840–846. doi:10.1038/nature05482
4. Defronzo RA, Ferrannini E, Groop L, et al. Type 2 diabetes mellitus. Nat Rev Dis Prim. 2015;1(1):1–22.
5. Weir GC, Gaglia J, Bonner-Weir S. Inadequate β-cell mass is essential for the pathogenesis of type 2 diabetes. Lancet Diabetes Endocrinol. 2020;8(3):249–256. doi:10.1016/S2213-8587(20)30022-X
6. Carlberg C, Campbell MJ. Vitamin D receptor signaling mechanisms: integrated actions of a well-defined transcription factor. Steroids. 2013;78(2):127–136. doi:10.1016/j.steroids.2012.10.019
7. Wang Y, He D, Ni C, et al. Vitamin D induces autophagy of pancreatic β-cells and enhances insulin secretion. Mol Med Rep. 2016;14(3):2644–2650. doi:10.3892/mmr.2016.5531
8. Norman AW, Frankel JB, Heldt AM, et al. Vitamin D deficiency inhibits pancreatic secretion of insulin. Science. 1980;209(4458):823–825. doi:10.1126/science.6250216
9. Li X, Liu Y, Zheng Y, et al. The effect of vitamin D supplementation on glycemic control in type 2 diabetes patients: a systematic review and meta-analysis. Nutrients. 2018;10(3):375. doi:10.3390/nu10030375
10. Christakos S, Dhawan P, Verstuyf A, et al. Vitamin D: metabolism, molecular mechanism of action, and pleiotropic effects. Physiol Rev. 2016;96(1):365–408. doi:10.1152/physrev.00014.2015
11. Norman AW. From vitamin D to hormone D: fundamentals of the vitamin D endocrine system essential for good health1. Am J Clin Nutr. 2008;88(2):491S–9S. doi:10.1093/ajcn/88.2.491S
12. Aranow C. Vitamin D and the immune system. J Investig Med. 2011;59(6):881–886. doi:10.2310/JIM.0b013e31821b8755
13. Pike JW, Meyer MB. The vitamin D receptor: new paradigms for the regulation of gene expression by 1,25-dihydroxyvitamin D3. Rheum Dis Clin North Am. 2012;38(1):13–27. doi:10.1016/j.rdc.2012.03.004
14. Norman AW. Vitamin D receptor: new assignments for an already busy receptor. Endocrinology. 2006;147(12):5542–5548. doi:10.1210/en.2006-0946
15. Del Prato S. Role of glucotoxicity and lipotoxicity in the pathophysiology of type 2 diabetes mellitus and emerging treatment strategies. Diabet Med. 2009;26(12):1185–1192. doi:10.1111/j.1464-5491.2009.02847.x
16. Lee JH, Lee J. Endoplasmic Reticulum (ER) stress and its role in pancreatic β-cell dysfunction and senescence in type 2 diabetes. Int J Mol Sci. 2022;23(9):4843.
17. U.K. prospective diabetes study 16. Overview of 6 years’ therapy of type II diabetes: a progressive disease. U.K. prospective diabetes study group. Diabetes. 1995;44(11):1249–1258. doi:10.2337/diab.44.11.1249
18. Gastaldelli A, Ferrannini E, Miyazaki Y, et al. Beta-cell dysfunction and glucose intolerance: results from the San Antonio metabolism (SAM) study. Diabetologia. 2004;47(1):31–39. doi:10.1007/s00125-003-1263-9
19. Stumvoll M, Goldstein BJ, Van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365(9467):1333–1346. doi:10.1016/S0140-6736(05)61032-X
20. Talchai C, Xuan S, Lin HV, et al. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell. 2012;150(6):1223–1234. doi:10.1016/j.cell.2012.07.029
21. Butler AE, Janson J, Bonner-Weir S, et al. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–110. doi:10.2337/diabetes.52.1.102
22. Unger RH, Scherer PE. Gluttony, sloth and the metabolic syndrome: a roadmap to lipotoxicity. Trends Endocrinol Metab. 2010;21(6):345–352. doi:10.1016/j.tem.2010.01.009
23. Donath MY. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat Rev Drug Discov. 2014;13(6):465–476. doi:10.1038/nrd4275
24. Berridge MJ. Vitamin D deficiency and diabetes. Biochem J. 2017;474(8):1321–1332. doi:10.1042/BCJ20170042
25. Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia. 2003;46(1):3–19. doi:10.1007/s00125-002-1009-0
26. Amrein K, Scherkl M, Hoffmann M, et al. Vitamin D deficiency 2.0: an update on the current status worldwide. Eur J Clin Nutr. 2020;74(11):1498–1513. doi:10.1038/s41430-020-0558-y
27. Pittas AG, Dawson-Hughes B, Li T, et al. Vitamin D and calcium intake in relation to type 2 diabetes in women. Diabetes Care. 2006;29(3):650–656. doi:10.2337/diacare.29.03.06.dc05-1961
28. Kayaniyil S, Vieth R, Retnakaran R, et al. Association of vitamin D with insulin resistance and beta-cell dysfunction in subjects at risk for type 2 diabetes. Diabetes Care. 2010;33(6):1379–1381. doi:10.2337/dc09-2321
29. Forouhi NG, Luan J, Cooper A, et al. Baseline serum 25-hydroxy vitamin d is predictive of future glycemic status and insulin resistance: the medical research council ely prospective study 1990-2000. Diabetes. 2008;57(10):2619–2625. doi:10.2337/db08-0593
30. Pittas AG, Dawson-Hughes B, Sheehan P, et al. Vitamin D supplementation and prevention of type 2 diabetes. N Engl J Med. 2019;381(6):520–530. doi:10.1056/NEJMoa1900906
31. Von Hurst PR, Stonehouse W, Coad J. Vitamin D supplementation reduces insulin resistance in South Asian women living in New Zealand who are insulin resistant and vitamin D deficient - a randomised, placebo-controlled trial. Br J Nutr. 2010;103(4):549–555. doi:10.1017/S0007114509992017
32. Pittas AG, Lau J, Hu FB, et al. The role of vitamin D and calcium in type 2 diabetes. A systematic review and meta-analysis. J Clin Endocrinol Metab. 2007;92(6):2017–2029. doi:10.1210/jc.2007-0298
33. Gilon P, Chae HY, Rutter GA, et al. Calcium signaling in pancreatic β-cells in health and in Type 2 diabetes. Cell Calcium. 2014;56(5):340–361. doi:10.1016/j.ceca.2014.09.001
34. Ashcroft FM, Rorsman P. Diabetes mellitus and the β cell: the last ten years. Cell. 2012;148(6):1160–1171. doi:10.1016/j.cell.2012.02.010
35. Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4(7):552–565. doi:10.1038/nrm1150
36. Christakos S, Dhawan P, Porta A, et al. Vitamin D and intestinal calcium absorption. Mol Cell Endocrinol. 2011;347(1–2):25–29. doi:10.1016/j.mce.2011.05.038
37. Hagström E, Hellman P, Lundgren E, et al. Serum calcium is independently associated with insulin sensitivity measured with euglycaemic-hyperinsulinaemic clamp in a community-based cohort. Diabetologia. 2007;50(2):317–324. doi:10.1007/s00125-006-0532-9
38. Berggård T, Miron S, Onnerfjord P, et al. Calbindin D28k exhibits properties characteristic of a Ca2+ sensor. J Biol Chem. 2002;277(19):16662–16672. doi:10.1074/jbc.M200415200
39. Pike JW. Genome-wide principles of gene regulation by the vitamin D receptor and its activating ligand. Mol Cell Endocrinol. 2011;347(1–2):3–10. doi:10.1016/j.mce.2011.05.012
40. Dalle S, Quoyer J, Varin E, et al. Roles and regulation of the transcription factor CREB in pancreatic β -cells. Curr Mol Pharmacol. 2011;4(3):187–195. doi:10.2174/1874467211104030187
41. Takiishi T, Gysemans C, Bouillon R, et al. Vitamin D and diabetes. Endocrinol Metab Clin North Am. 2010;39(2):419–446. doi:10.1016/j.ecl.2010.02.013
42. Ayesha I, Raghuramulu N. Oral glucose tolerance is unaltered in vitamin D-deficient rat. J Nutr Sci Vitaminol. 2000;46(3):115–118. doi:10.3177/jnsv.46.115
43. Szymczak-PAJOR I, Śliwińska A. Analysis of association between vitamin D deficiency and insulin resistance. Nutrients. 2019;11(4):794. doi:10.3390/nu11040794
44. Doyle ME, Egan JM. Pharmacological agents that directly modulate insulin secretion. Pharmacol Rev. 2003;55(1):105–131. doi:10.1124/pr.55.1.7
45. Bourlon PM, Billaudel B, Faure-Dussert A. Influence of vitamin D3 deficiency and 1,25 dihydroxyvitamin D3 on de novo insulin biosynthesis in the islets of the rat endocrine pancreas. J Endocrinol. 1999;160(1):87–95. doi:10.1677/joe.0.1600087
46. Kadowaki S, Norman AW. Dietary vitamin D is essential for normal insulin secretion from the perfused rat pancreas. J Clin Invest. 1984;73(3):759–766. doi:10.1172/JCI111269
47. Cade C, Norman AW. Vitamin D3 improves impaired glucose tolerance and insulin secretion in the vitamin D-deficient rat in vivo. Endocrinology. 1986;119(1):84–90. doi:10.1210/endo-119-1-84
48. Cade C, Norman AW. Rapid normalization/stimulation by 1,25-dihydroxyvitamin D3 of insulin secretion and glucose tolerance in the vitamin D-deficient rat. Endocrinology. 1987;120(4):1490–1497. doi:10.1210/endo-120-4-1490
49. Zeitz U, Weber K, Soegiarto DW, et al. Impaired insulin secretory capacity in mice lacking a functional vitamin D receptor. FASEB j. 2003;17(3):509–511. doi:10.1096/fj.02-0424fje
50. Zhang D, Cheng C, Wang Y, et al. The influence of VDR polymorphisms on the type 2 diabetes susceptibility in Chinese: an interaction with hypertriglyceridemia. Mol Genet Genomics. 2021;296(4):837–844. doi:10.1007/s00438-021-01784-z
51. Kayaniyil S, Retnakaran R, Harris SB, et al. Prospective associations of vitamin D with β-cell function and glycemia: the PROspective Metabolism and ISlet cell Evaluation (PROMISE) cohort study. Diabetes. 2011;60(11):2947–2953. doi:10.2337/db11-0465
52. Eftekhari MH, Akbarzadeh M, Dabbaghmanesh MH, et al. Impact of treatment with oral calcitriol on glucose indices in type 2 diabetes mellitus patients. Asia Pac J Clin Nutr. 2011;20(4):521–526.
53. Zhang L, Yuan L. Clinical research progress of vitamin d and type 2 diabetes. Int J Int Med. 2009;36(02):82–3+92.
54. Yang L, Ma J, Zhang X, et al. Protective role of the vitamin D receptor. Cell Immunol. 2012;279(2):160–166. doi:10.1016/j.cellimm.2012.10.002
55. Fiorentino TV, Prioletta A, Zuo P, et al. Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr Pharm Des. 2013;19(32):5695–5703. doi:10.2174/1381612811319320005
56. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107(9):1058–1070. doi:10.1161/CIRCRESAHA.110.223545
57. Unger RH. Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes. 1995;44(8):863–870. doi:10.2337/diab.44.8.863
58. Liu J, Zhang Y, Shi D, et al. Vitamin D alleviates type 2 diabetes mellitus by mitigating oxidative stress-induced pancreatic β-cell impairment. Exp Clin Endocrinol Diabetes. 2023;131(12):656–666. doi:10.1055/a-2191-9969
59. Kaneto H, Matsuoka TA, Nakatani Y, et al. Oxidative stress and the JNK pathway in diabetes. Curr Diabetes Rev. 2005;1(1):65–72. doi:10.2174/1573399052952613
60. Riachy R, Vandewalle B, Belaich S, et al. Beneficial effect of 1,25 dihydroxyvitamin D3 on cytokine-treated human pancreatic islets. J Endocrinol. 2001;169(1):161–168. doi:10.1677/joe.0.1690161
61. Riachy R, Vandewalle B, Kerr Conte J, et al. 1,25-dihydroxyvitamin D3 protects RINm5F and human islet cells against cytokine-induced apoptosis: implication of the antiapoptotic protein A20. Endocrinology. 2002;143(12):4809–4819. doi:10.1210/en.2002-220449
62. Riachy R, Vandewalle B, Moerman E, et al. 1,25-dihydroxyvitamin D3 protects human pancreatic islets against cytokine-induced apoptosis via down-regulation of the fas receptor. Apoptosis. 2006;11(2):151–159. doi:10.1007/s10495-006-3558-z
63. Wu M, Lu L, Guo K, et al. Vitamin D protects against high glucose-induced pancreatic β-cell dysfunction via AMPK-NLRP3 inflammasome pathway. Mol Cell Endocrinol. 2022;547:111596. doi:10.1016/j.mce.2022.111596
64. Lai X, Liu X, Cai X, et al. Vitamin D supplementation induces CatG-mediated CD4 + T cell inactivation and restores pancreatic β-cell function in mice with type 1 diabetes. Am J Physiol Endocrinol Metab. 2022;322(1):E74–e84. doi:10.1152/ajpendo.00066.2021
65. Alexander M, Cho E, Gliozheni E, et al. Pathology of diabetes-induced immune dysfunction. Int J Mol Sci. 2024;25(13):7105. doi:10.3390/ijms25137105
66. Gysemans C, Van Etten E, Overbergh L, et al. Treatment of autoimmune diabetes recurrence in non-obese diabetic mice by mouse interferon-beta in combination with an analogue of 1alpha,25-dihydroxyvitamin-D3. Clin Exp Immunol. 2002;128(2):213–220. doi:10.1046/j.1365-2249.2002.01825.x
67. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11(2):98–107. doi:10.1038/nri2925
68. Gysemans CA, Cardozo AK, Callewaert H, et al. 1,25-dihydroxyvitamin D3 modulates expression of chemokines and cytokines in pancreatic islets: implications for prevention of diabetes in nonobese diabetic mice. Endocrinology. 2005;146(4):1956–1964. doi:10.1210/en.2004-1322
69. Wolden-Kirk H, Rondas D, Bugliani M, et al. Discovery of molecular pathways mediating 1, 25-dihydroxyvitamin D3 protection against cytokine-induced inflammation and damage of human and male mouse islets of Langerhans. Endocrinology. 2014;155(3):736–747. doi:10.1210/en.2013-1409
70. Jia R, Liang L, Yin Y, et al. Vitamin D supplementation could enhance the effectiveness of glibenclamide in treating type 2 diabetes by improving the function of pancreatic β-cells through the NF-κB pathway. Biochem Biophys Res Commun. 2024;733:150596. doi:10.1016/j.bbrc.2024.150596
71. Quesada I, Tudurí E, Ripoll C, et al. Physiology of the pancreatic α-cell and glucagon secretion: role in glucose homeostasis and diabetes. J Endocrinol. 2008;199(1):5–19. doi:10.1677/JOE-08-0290
72. Bourlon PM, Faure-Dussert A, Billaudel B, et al. Relationship between calbindin-D28K levels in the A and B cells of the rat endocrine pancreas and the secretion of insulin and glucagon: influence of vitamin D3 deficiency and 1,25-dihydroxyvitamin D3. J Endocrinol. 1996;148(2):223–232. doi:10.1677/joe.0.1480223
73. Redecker P, Cetin Y. Rodent pancreatic islet cells contain the calcium-binding proteins calcineurin and calretinin. Histochem Cell Biol. 1997;108(2):133–139. doi:10.1007/s004180050154
74. Shao J. The role of the local renin-angiotensin system in type 2 diabetes. J Med Postgraduates. 2011;24(05):449–452.
75. Yuan W, Pan W, Kong J, et al. 1,25-dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. J Biol Chem. 2007;282(41):29821–29830. doi:10.1074/jbc.M705495200
76. Cheng Q, Li YC, Boucher BJ, et al. A novel role for vitamin D: modulation of expression and function of the local renin-angiotensin system in mouse pancreatic islets. Diabetologia. 2011;54(8):2077–2081. doi:10.1007/s00125-011-2100-1
77. Leung PS, Cheng Q. The novel roles of glucagon-like peptide-1, angiotensin II, and vitamin D in islet function. Adv Exp Med Biol. 2010;654:339–361.
78. Leung PS. The Potential Protective Action Of Vitamin D In Hepatic Insulin Resistance And Pancreatic Islet Dysfunction In Type 2 Diabetes Mellitus. Nutrients. 2016;8(3):147. doi:10.3390/nu8030147
79. Gulseth HL, Wium C, Angel K, et al. Effects of vitamin D supplementation on insulin sensitivity and insulin secretion in subjects with type 2 diabetes and vitamin D deficiency: a randomized controlled trial. Diabetes Care. 2017;40(7):872–878. doi:10.2337/dc16-2302
80. Jin S, Sha L, Dong J, et al. Effects of nutritional strategies on glucose homeostasis in gestational diabetes mellitus: a systematic review and network meta-analysis. J Diabetes Res. 2020;2020:6062478. doi:10.1155/2020/6062478
81. Pramono A, Jocken JWE, Blaak EE, et al. The effect of vitamin D supplementation on insulin sensitivity: a systematic review and meta-analysis. Diabetes Care. 2020;43(7):1659–1669. doi:10.2337/dc19-2265
82. Bock G, Prietl B, Mader JK, et al. The effect of vitamin D supplementation on peripheral regulatory T cells and β cell function in healthy humans: a randomized controlled trial. Diabetes Metab Res Rev. 2011;27(8):942–945. doi:10.1002/dmrr.1276
83. Gagnon C, Daly RM, Carpentier A, et al. Effects of combined calcium and vitamin D supplementation on insulin secretion, insulin sensitivity and β-cell function in multi-ethnic vitamin D-deficient adults at risk for type 2 diabetes: a pilot randomized, placebo-controlled trial. PLoS One. 2014;9(10):e109607. doi:10.1371/journal.pone.0109607
84. Wagner H, Alvarsson M, Mannheimer B, et al. No effect of high-dose vitamin D treatment on β-cell function, insulin sensitivity, or glucose homeostasis in subjects with abnormal glucose tolerance: a randomized clinical trial. Diabetes Care. 2016;39(3):345–352. doi:10.2337/dc15-1057
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Exploration and Identification of Vitamin D and Related Genes as Potential Biomarkers for Colorectal Tumors

Wang L, Xu R, Wang M, Wang M, Su S, Nian Y, Chen X
OncoTargets and Therapy 2025, 18:129-145
Published Date: 22 January 2025
Association Between Vitamin D and Microvascular Changes in Early Diabetic Retinopathy in Patients with Type 2 Diabetes
Wei Z, Wang K, Liu Y, Liu P, Tang Y, Chen L, Hou X, Yan F
Diabetes, Metabolic Syndrome and Obesity 2025, 18:4085-4095
Published Date: 8 November 2025