Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 12
Ursolic Acid Treatment Alleviates Diabetic Kidney Injury By Regulating The ARAP1/AT1R Signaling Pathway
Authors Ma TK, Xu L, Lu LX, Cao X, Li X, Li LL, Wang X, Fan QL ![]()
Received 8 July 2019
Accepted for publication 30 October 2019
Published 9 December 2019 Volume 2019:12 Pages 2597—2608
DOI https://doi.org/10.2147/DMSO.S222323
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Konstantinos Tziomalos
Tian-Kui Ma,1,* Li Xu,1,2,* Ling-Xu Lu,1,3,* Xu Cao,1 Xin Li,1 Lu-Lu Li,1 Xu Wang,4 Qiu-Ling Fan1
1Department of Nephrology, The First Hospital of China Medical University, Shenyang, Liaoning, People’s Republic of China; 2Department of Clinical Laboratories, The First Hospital of China Medical University, Shenyang, Liaoning, People’s Republic of China; 3The First Respiratory Department, General Hospital of Fushun Mining Bureau, Fushun, Liaoning, People’s Republic of China; 4Department of Gastroenterology, The First Hospital of China Medical University, Shenyang, Liaoning, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qiu-Ling Fan Email [email protected]
Purpose: This study aimed to investigate whether ursolic acid (UA) mitigates renal inflammation, oxidative stress and fibrosis by regulating the angiotensin II type 1 receptor-associated protein (ARAP1)/angiotensin II type 1 receptor (AT1R) signaling pathway and subsequently alleviating renal damage.
Methods: db/db mice were divided randomly into a diabetic nephropathy (DN) group and a UA treatment group. Light microscopy and electron microscopy were used to observe pathological changes in renal tissues. Immunohistochemistry (IHC) was employed to examine changes in the expression of ARAP1, AT1R, 8-hydroxydeoxyguanosine (8-OHdG), NADPH oxidase 2 (NOX2), the extracellular matrix protein fibronectin (FN), IL-1β and IL-18 in renal tissues. Western blotting and RT-qPCR were used to detect the respective changes in the protein and mRNA levels of ARAP1, AT1R, NOX4, NOX2, transforming growth factor-β1 (TGF-β1), FN, collagen IV, IL-1β and IL-18 in renal tissues and mesangial cells. In addition, immunofluorescence staining was employed to examine changes in FN and NOX2 expression in mesangial cells.
Results: UA treatment effectively reduced the body weights and blood glucose levels of db/db mice (p<0.05) as well as the urinary albumin/creatinine ratio (p<0.05). In addition, the renal tissue lesions and glomerulosclerosis index of the db/db mice were significantly improved after treatment (p<0.01). Histochemical analysis results showed significantly lower expression levels of ARAP1, AT1R, FN, NOX2, 8-OHdG, IL-1β and IL-18 in renal tissues in the UA treatment group than in the DN group. Western blotting and RT-qPCR data also revealed UA-induced decreases in the renal levels of the ARAP1, AT1, NOX4, NOX2, TGF-β1, FN, collagen IV, IL-1β and IL-18 proteins in vivo and/or in vitro (p<0.01). ARAP1 knockdown effectively reduced the expression of NOX2 and FN in vitro.
Conclusion: UA alleviated renal damage in type 2 diabetic db/db mice by downregulating proteins in the ARAP1/AT1R signaling pathway to inhibit extracellular matrix accumulation, renal inflammation, fibrosis and oxidative stress.
Keywords: ursolic acid, diabetic nephropathy, oxidative stress, renal fibrosis, ARAP1, AT1R
Introduction
Diabetes is a growing global health problem. According to the International Diabetes Federation (IDF), the global prevalence of diabetes in 2017 was approximately 8.3%, affecting 425 million adults. By 2045, this number will increase by 48%, thus affecting 700 million people.1 Diabetic nephropathy (DN), a major complication of advanced diabetes, affects approximately one-third of patients with diabetes and is the most common cause of end-stage renal disease (ESRD) and the leading cause of death among patients with diabetes.2 The exact mechanism underlying the pathogenesis of DN has not yet been elucidated, and no clinical drugs can effectively reverse the progression of DN. Therefore, new treatments for DN are urgently needed.
Angiotensin II (Ang II) is the most important component of the renin-angiotensin system (RAS), and its downstream receptor, the angiotensin II type 1 receptor (AT1R), plays key roles in promoting inflammation and fibrosis, stimulating growth, and generating oxygen free radicals.3,4 The AT1R-associated protein has been shown to locally regulate AT1R functions. Guo et al5 identified the AT1R-associated protein angiotensin II type 1 receptor-associated protein 1 (ARAP1). ARAP1, which is a member of the renin-angiotensin system (RAS), binds to the C-terminal region of the AT1R and promotes the recycling of the receptor to the cell membrane, the increasing the sensitivity of the AT1R to Ang II and activating the RAS.5 However, researchers have not clearly determined whether ARAP1 regulates DN through the AT1R.
Ursolic acid (UA) is a pentacyclic triterpenoid derived from the berries, fruits, leaves and flowers of many medicinal plants. It has been used for centuries in Asia as an antitumor, anti-inflammatory, antihyperglycemic and immunomodulatory drug.6,7 However, its exact molecular mechanism and potential beneficial effects remain unclear. UA likely reduces extracellular matrix accumulation and suppresses cellular hypertrophy and proliferation by inhibiting miRNA-21 overexpression in mesangial cells cultured under high glucose conditions; in this manner, UA upregulates phosphatase and tensin homolog deleted on chromosome ten (PTEN) expression, inhibits aberrant activation of the phosphoinositide 3-kinase (P13K)/Akt/mammalian target of rapamycin (mTOR) signaling pathway, and enhances autophagy.8,9 Currently, no published studies have examined whether UA reduces renal oxidative stress, inflammation and fibrosis or alleviates renal damage in diabetic db/db mice by regulating the ARAP1/AT1R signaling pathway.
Materials And Methods
Experimental Animals, Groups, And Sample Collection
Specific pathogen-free (SPF) grade 9-week-old male db/m (BKS. Cg-leprdb/+, n=10) and db/db (BKS. Cg-leprdb/leprdb, n=20) mice were purchased from the Model Animal Research Center of Nanjing University, China. UA for mice was purchased from Solarbio, Beijing, China. This study fully complies with the Ethical Guidelines of the International Association for the Study of Diabetes, and the experimental methods were reviewed and approved by the Institutional Animal Care and Use Committee of China Medical University. The animals were housed at the Laboratory Animal Center of China Medical University at 23±3°C under a 12 hr light/dark cycle. The animals were divided randomly into two groups: the diabetic group (db/db group, n=10) and the UA treatment group (db/db+0.3% UA, n=10). The UA treatment group was fed a diet containing 0.3% UA (0.3 g of UA per 100 g of standard feed) for 10 weeks, the therapeutic dose is determined by our previous studies8,9,38 and other references.34–37 The animal experiments were started when the mice reached ten weeks of age. We fasted the mice for 12 hrs without limiting water intake, then the body weights of the mice were examined, and blood glucose levels were analyzed at regular intervals from blood samples collected from the tail vein. Urine samples were collected from a random sample of mice at 20th week, urinary albumin levels were measured using an ELISA, and urinary creatinine levels were examined using the picric acid method. Specimens were collected after euthanasia at 20 weeks of age.
Cell Culture And ARAP1 Knockdown
The mouse mesangial cell line SV40 MES 13 was purchased from the American Type Culture Collection (ATCC) and cultured in DMEM/F12 (Gibco, Grand Island, NY, USA) containing 5% serum and 5 mM glucose for the normal glucose (NG) condition or 25 mM glucose for the high glucose (HG) condition. And we used the same molar concentration of mannitol as the osmotic control group (MA). The glucose concentration was chosen based on previous studies from our study group.8,9
UA for cell culture was purchased from Sigma-Aldrich (U6753), and the concentration (2.5μM) was chosen according to our previous studies.8,9,38
We constructed three siRNA sequences, which were used to transfect mesangial cells. After 24–48 hrs, the knockdown efficiency was detected by RT-qPCR. The knockdown group was transfected with jetPRIME® (New York, US) according to the manufacturer’s instructions.
Histopathological Examination
Renal tissues were fixed and embedded in paraffin, followed by sectioning and Hematoxylin-Eosin(H-E), Masson’s trichrome and periodic acid-Schiff (PAS) staining. Pathological changes in the glomerular basement membrane and mesangial matrix of the kidneys were observed in different visual fields under a light microscope, and the glomerulosclerosis index (GSI) was calculated. The extent of mesangial matrix hypertrophy was assessed as described in the literature: 20 glomeruli were selected randomly from each section, and the GSI and degree of mesangial matrix hypertrophy were calculated.10
Immunohistochemistry
Paraffin sections were dewaxed, rehydrated, and blocked with nonimmunized animal serum. The serum was then removed, and the sections were incubated with a primary antibody at 4°C overnight. The antibody dilutions were 1:100 for ARAP1 (Protein Tech 11559-1-AP), 1:100 for AT1R (Sigma, SAB3500209), 1:120 for NADPH oxidase 2 (NOX2) (Abcam, ab80508), 1:100 for NOX4 (Abcam, ab133303), 1:50 for 8-hydroxydeoxyguanosine (8-OHdG, Abcam, ab48508), 1:80 for Fibronectin (FN, Protein Tech, 15613-1-AP), 1:50 for IL-1β (absin, abs135771) and 1:50 for IL-18 (absin, abs135772). PBS was used as a negative control. The sections were washed and incubated with an anti-rabbit IgG antibody (Invitrogen, US) at room temperature and then stained with diaminobenzidine (DAB) for color development. Finally, they were observed and imaged under a Leica microscope.
Immunofluorescence Staining
Cells cultured on coverslips were fixed with 4% paraformaldehyde and treated with 0.1% Triton X to permeabilize the cell membrane. The cells were then blocked with nonimmunized animal serum and incubated with a primary antibody at 4°C overnight. The cells were rinsed with PBS three times and incubated with a fluorescently labeled secondary antibody at room temperature. DAPI was used to stain the nuclei. The cells on the coverslips were rinsed and mounted and finally observed and imaged under a Leica fluorescence microscope.
Real-Time Quantitative PCR (RT-qPCR)
Total RNA was extracted from the cells of each group and reverse transcribed into cDNA. PCR amplification was performed using Taq DNA polymerase and cDNA templates. The primer sequences are shown in Supplemental Table 1. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal standard. The cycle threshold (CT) values from RT-qPCR were analyzed using the 2−ΔΔCT method to calculate the fold change in expression. Each experimental group was analyzed in triplicate.
Western Blotting
Total protein was extracted with a RIPA lysis solution. The proteins were denatured and then separated using SDS-PAGE. Immunoblotting was performed using antibodies specific for ARAP1 (Protein Tech 11559-1-AP 1:500 for tissue sample; Absin abs134266 1:200 for cell sample), AT1R (Sigma, SAB3500209, 1:500 for tissue sample; Absin abs135212, 1:300 for cell sample), FN (Protein Tech, 15613-1-AP, 1:200), collagen IV (Col IV, Protein Tech, 55131-1-AP, 1:500), NOX4 (Abcam, ab133303, 1:200), NOX2 (Abcam, ab80508, 1:500), TGF-β1 (Abcam, ab92486, 1:500), IL-1β (absin, abs135771, 1:500), and IL-18 (absin, abs135772, 1:200). A semiquantitative analysis was conducted using ImageJ software.
Statistical Analysis
The statistical software SPSS17.0 was used for the analysis. Data with a normal distribution are presented as the means±standard deviation. The data from multiple groups were compared using a one-way ANOVA, differences between groups were subjected to Fisher’s Least Significant Difference post hoc test for multiple comparisons, and pairwise comparisons were conducted using the t-test. Differences were considered statistically significant at p<0.05.
Results
Effects Of UA On Body Weight, Blood Glucose Levels, And Serum Creatinine Levels In Diabetic Db/Db Mice
As shown in Figure 1A, the mice in each group showed increasing body weights with age, and the body weights were significantly higher in db/db mice than in db/m mice at various ages (p<0.05). After 6 weeks of UA treatment, the body weights of the db/db mice were lower than those of the untreated group (p<0.05), and the most significant treatment effect was observed after 8 weeks of treatment (p<0.01). Compared with those of the untreated group, the blood glucose levels of the db/db mice decreased after 6 weeks of treatment with UA (p<0.05), and the hypoglycemic effect was more pronounced after 10 weeks of treatment (p<0.01) (Figure 1B and C). The urinary albumin/creatinine ratio was significantly increased in the db/db mice (p<0.01), and UA treatment reduced the urinary albumin/creatinine ratio of the mice in the db/db group (p<0.05) (Figure 1D and E).
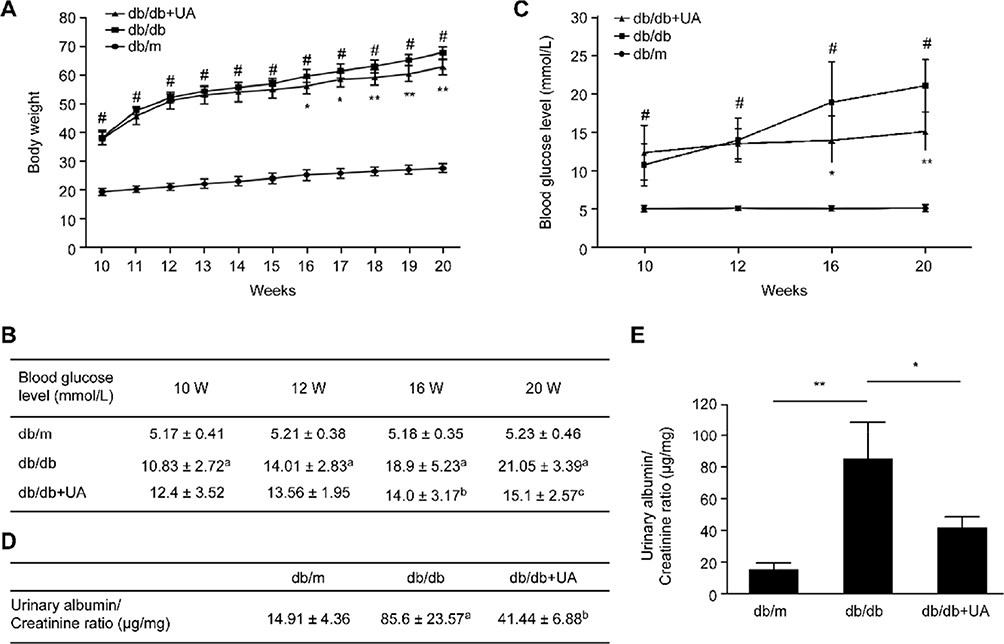 |
Figure 1 General characteristics of db/db mice after UA treatment. (A) Weekly changes in the body weights of mice. (B, C) Blood glucose levels were measured in blood samples collected from the tail vein. (D, E) Changes in the blood glucose levels and urinary albumin/creatinine ratio in 20-week-old mice. ap<0.05 compared with db/m mice, bp<0.05 compared with diabetic db/db mice, and cp<0.01 compared with diabetic db/db mice. #p<0.05 compared with db/m mice, *p<0.05 compared with diabetic db/db mice, and **p<0.01 compared with diabetic db/db mice. db/m group, n=10; db/db group, n=10; and db/db+UA group, n=10. |
Effects Of UA On Renal Histopathology
A histology analysis showed significant glomerular basement membrane thickening and mesangial matrix accumulation in db/db mice, along with significant increases in the GSI and glomerular mesangial proliferation (p<0.01). These pathological changes were significantly improved in the mice after UA treatment. Furthermore, the GSI and extent of hypertrophy were significantly reduced (p<0.05). The thickness of the basement membrane, the degree of podocyte foot process effacement, and changes in the fenestral structure of endothelial cells were observed under an electron microscope. The db/db mice exhibited significant basement membrane thickening and increased podocyte foot process effacement. These symptoms were significantly relieved in the UA group (Figure 2A–E).
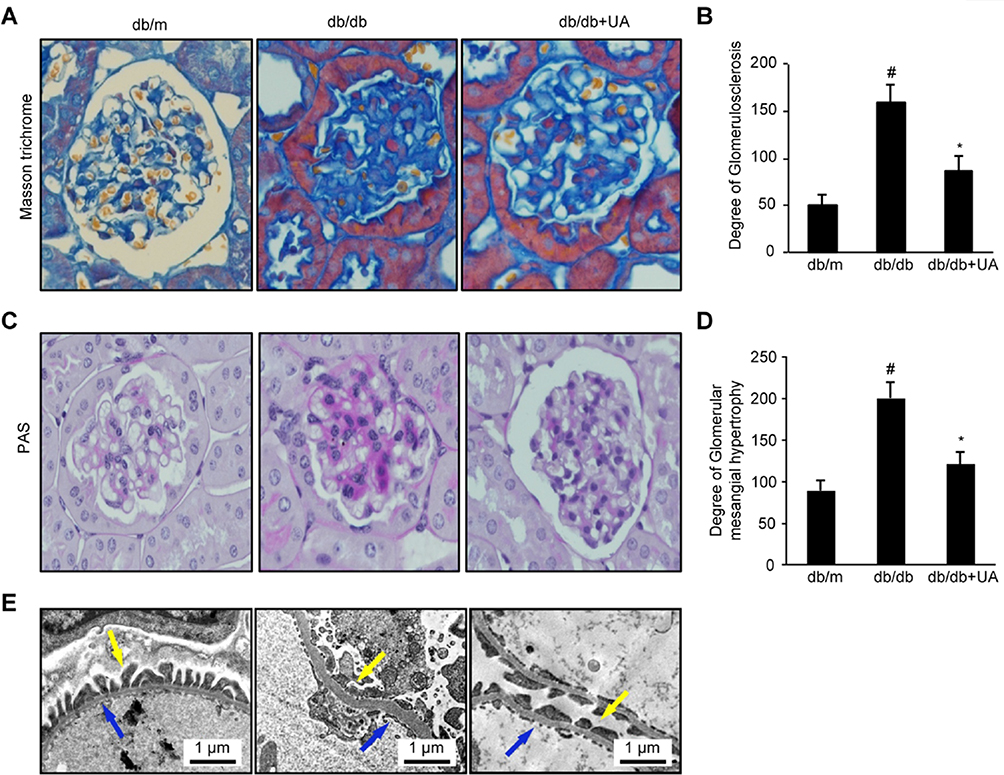 |
Figure 2 UA treatment reduces renal histological changes in db/db mice. (A) Images of Masson’s trichrome staining of kidney tissues from 20-week-old mice (×400). (B) Semiquantitative analysis of the GSI as determined by Masson’s trichrome staining. (C) Images of PAS staining of kidney tissues from 20-week-old mice (×400). (D) Semiquantitative analysis of glomerular mesangial matrix hypertrophy as determined by PAS staining. (E) Electron microscopy images of basement membrane thickness (blue arrow) and the degree of podocyte foot process effacement (yellow arrow). #p<0.01 compared with db/m mice and *p<0.05 compared with diabetic db/db mice. db/m group, n=10; db/db group, n=10; and db/db+UA group, n=10. |
Effects Of UA Treatment On The Changes In The Renal Expression Levels Of Molecules In The ARAP1 And AT1R Pathways In Mice
As shown in Figure 3, compared with the db/m group, the db/db group exhibited significantly increased expression levels of ARAP1 and AT1R in their kidney tissues (p<0.01), while the levels in the UA treatment group were markedly reduced (p<0.05). Western blotting and RT-qPCR results showed significantly higher protein and mRNA expression levels, respectively, of ARAP1 and AT1R in the renal tissues obtained from db/db mice than in the renal tissues from db/m mice (p<0.01). After UA treatment, the protein expression levels of ARAP1 (p<0.05) and AT1R (p<0.05) were significantly decreased (Figure 4).
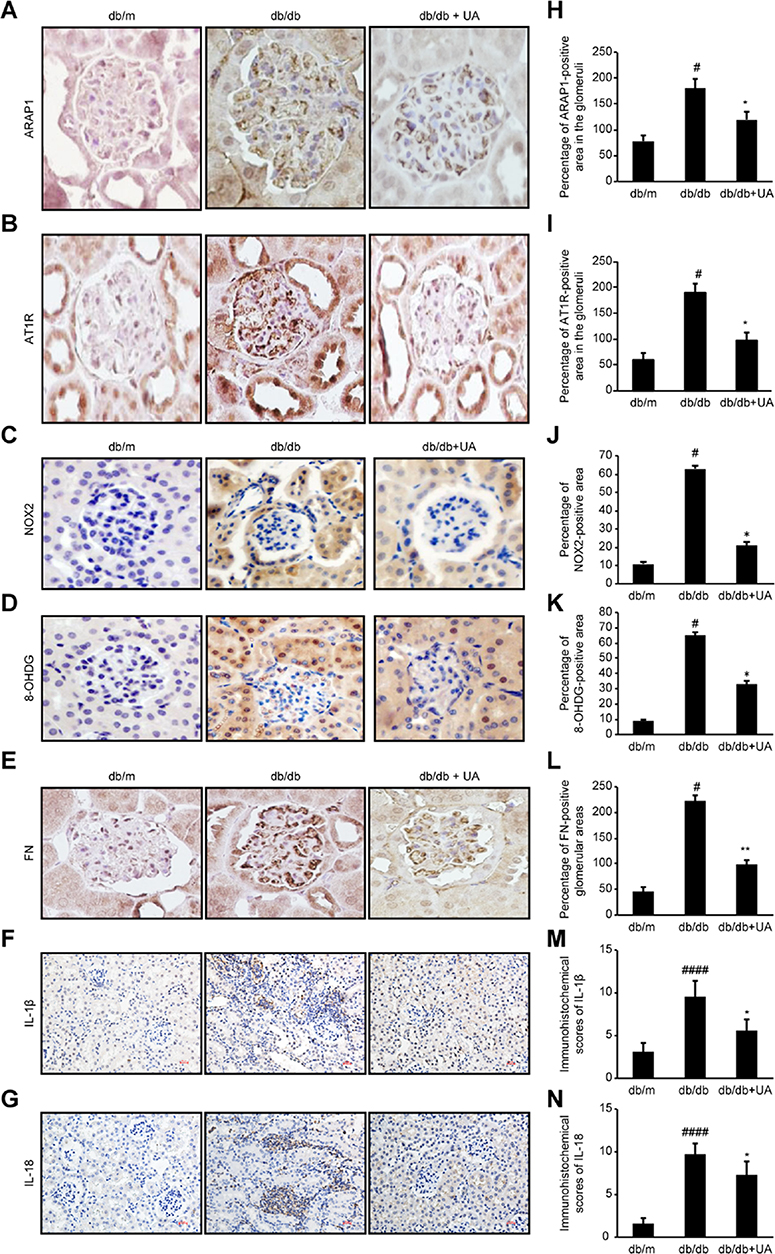 |
Figure 3 Changes in ARAP1, AT1R, NOX2, 8-OHdG, FN, IL-1β and IL-18 expression in the kidney tissues of 20-week-old mice were assessed by immunohistochemistry. (A-G) Immunohistochemistry for ARAP1, AT1R, NOX2, 8-OHdG, FN, IL-1β and IL-18 expression. (H-N) Semiquantitative analysis of the immunohistochemistry results. #p<0.05, ####p<0.0001 compared with db/m mice and *p<0.05, **p<0.01 compared with diabetic db/db mice. |
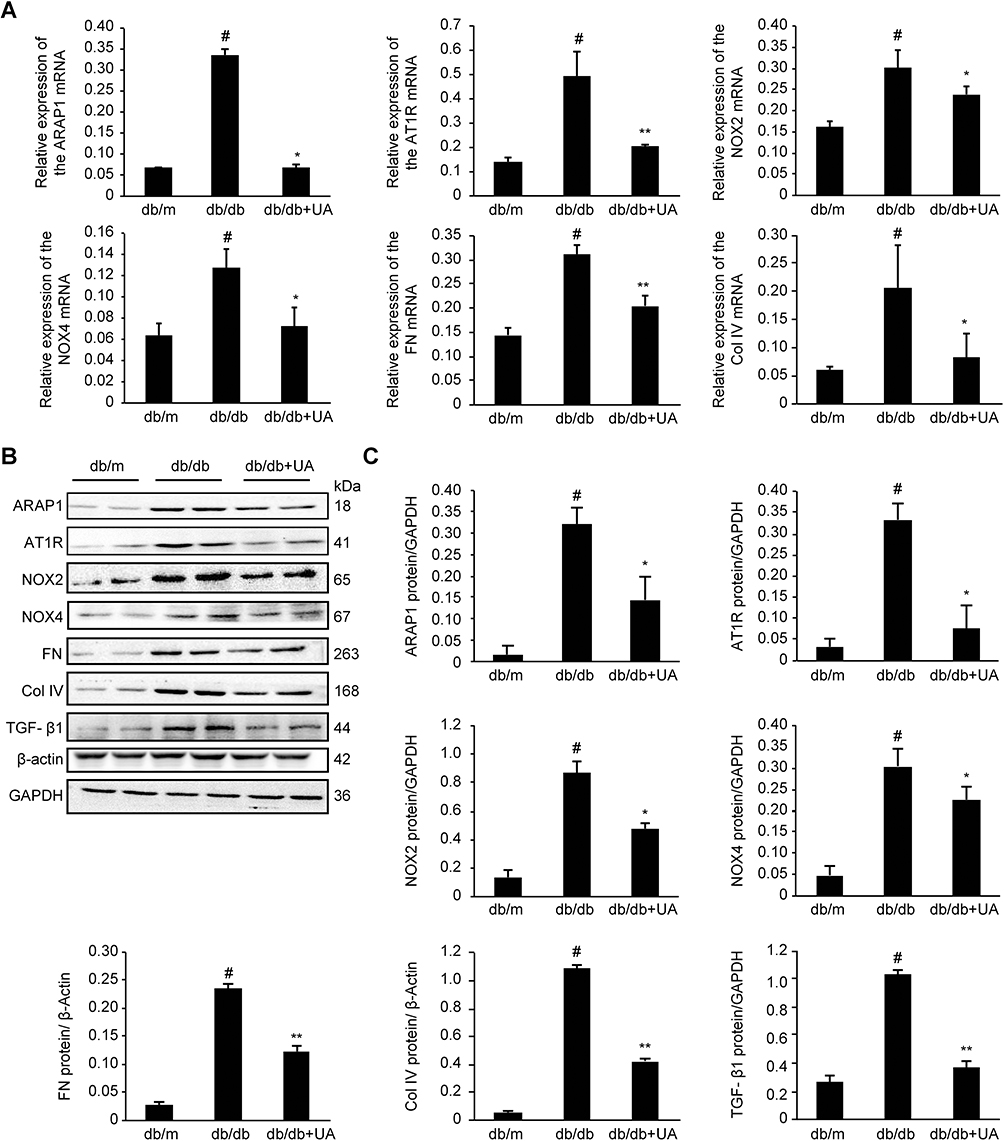 |
Figure 4 Protein and mRNA expression in the renal cortex of 20-week-old mice as detected by Western blotting and RT-qPCR, respectively. (A) Changes in ARAP1, AT1R, NOX2, NOX4, FN and Col IV mRNA expression as detected by RT-qPCR. (B) Changes in the levels of ARAP1, AT1R, NOX2, NOX4, FN, Col IV, and TGF-β1 proteins were detected using Western blotting. #p<0.01 compared with db/m mice; *p<0.05 and **p<0.01 compared with diabetic db/db mice. |
Effects Of UA On Oxidative Stress And Renal Inflammation
Inflammation is one of the main characteristics of diabetic kidney injury. The inflammation index indicators, IL-1β and IL-18, expression were significantly increased in the db/db group compared with db/m group (p<0.01), and also significantly decreased after UA therapy (p<0.01)(Figure 3). These changes indicate that UA may have anti-renal inflammatory functions, as shown in previous studies.11,12 The levels of o xidative stress markers, including NOX2, NOX4 and 8-OHdG, were assessed in renal tissues to determine whether UA treatment inhibits oxidative stress during diabetic kidney injury (Figure 3). The Western blotting and RT-qPCR results revealed significantly increased levels of NOX2 and NOX4 in the renal tissues obtained from db/db mice (p<0.01), but these levels were markedly reduced after UA treatment (p<0.05)(Figure 4B). Immunohistochemistry also confirmed significantly increased NOX2 and 8-OHdG expression levels in the renal cortex of the db/db group, and this expression was significantly reduced after UA treatment (p<0.05) (Figure 3).
UA Alleviated Extracellular Matrix Accumulation In db/db Mice
Western blotting was used to detect the levels of the TGF-β1, FN and Col IV proteins in the renal cortexes of each group. The levels of TGF-β1, FN and Col IV were significantly increased (p<0.01) in the db/db group, and treatment with UA mitigated extracellular matrix FN accumulation and reduced TGF-β1 and Col IV levels (p<0.01). Similar results were observed via RT-qPCR analysis (Figure 4). The immunohistochemical analysis results showed that the level of the extracellular matrix protein FN was significantly reduced after UA treatment (p<0.01)(Figure 3).
UA Can Reduce Cell Inflammation And The Expression Of Extracellular Matrix Proteins In Vitro
UA has previously been reported to have anti-inflammatory effects.11,12 To investigate the anti-inflammatory function of UA in DN conditions in vitro, we treated HG condition cultured mesangial cells with UA. We found that the levels of the inflammatory markers IL-1β and IL-18 were significantly decreased after UA treatment (p<0.0001) (Figure 5 A–F), as well as the expression of the extracellular matrix protein Col IV (p<0.0001).
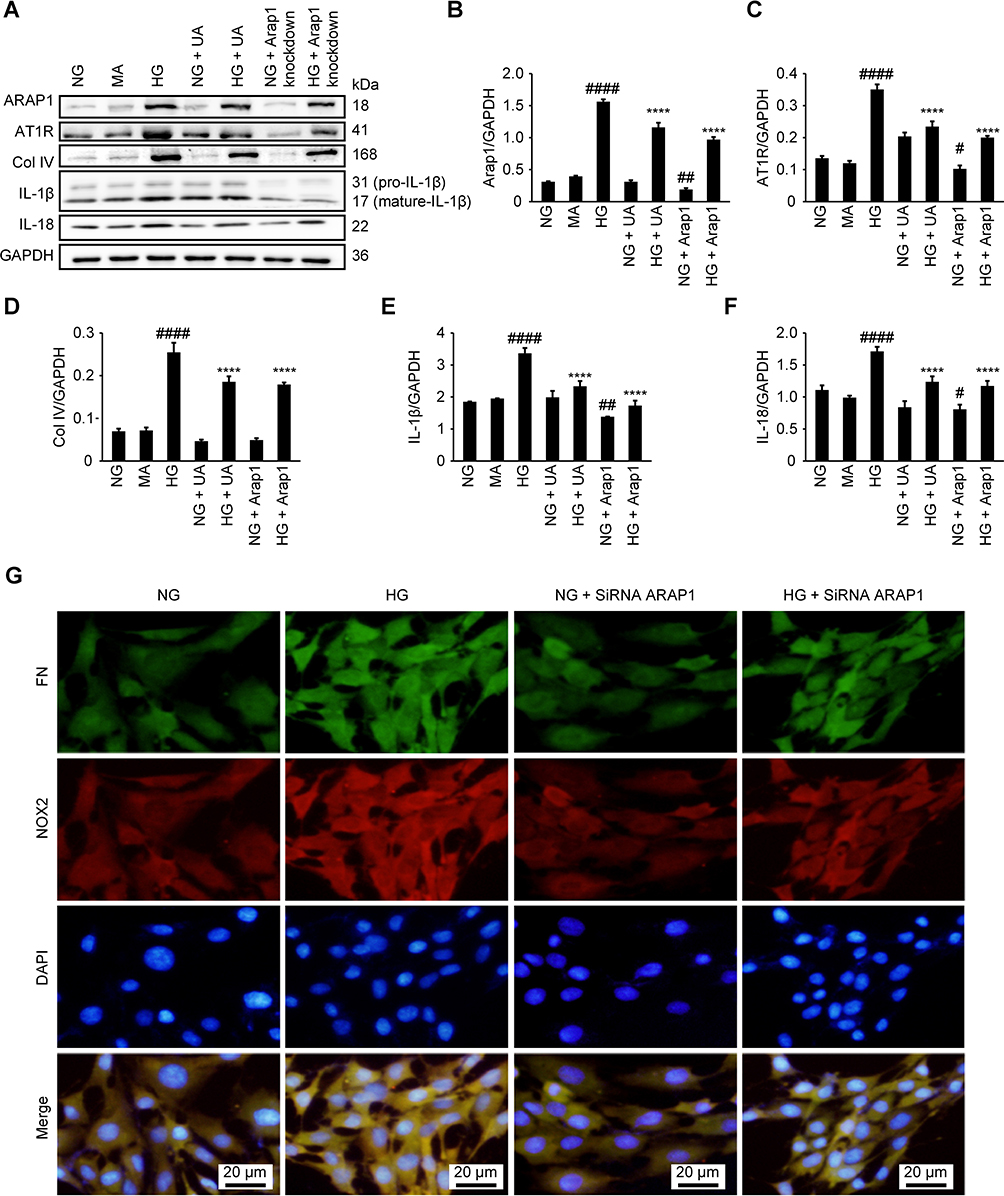 |
Figure 5 UA treatment and ARAP1 knockdown reduced mesangial cell inflammation, extracellular matrix protein expression, fibrosis and oxidative stress. (A) Changes in ARAP1, AT1R, Col IV, IL-1β and IL-18 protein levels detected by Western blotting. (B–F) Semiquantitative analysis of Western blotting results. (G) Changes in the expression of FN and NOX2 in each group as assessed using fluorescence microscopy (x200). #P<0.05, ##P<0.01, ####P<0.0001 compared with the NG group, and *p<0.05 ****p<0.0001 compared with the HG group. |
The ARAP1 Protein Increased The Expression Of AT1R
We constructed three siRNA sequences, which were used to transfect mesangial cells. After 24–48 hrs, the knockdown efficiency was detected by RT-qPCR, and the most efficient sequence (# 3) was used for subsequent experiments (Supplemental Figure 1). We used ARAP1 siRNA-3, which specifically inhibits ARAP1 expression, in glomerular mesangial cells to further examine the regulatory effects of ARAP1 and AT1R. Western blotting was employed to examine the ARAP1 and AT1R protein levels in each group, and we found AT1R expression was significantly decreased after ARAP1 knockdown (p<0.0001). These results indicated that ARAP1 may regulate the expression of AT1R (Figure 5A–C).
ARAP1 Inhibition Reduced The Levels Of Oxidative Stress Markers And The Expression Of Extracellular Matrix Proteins
ARAP1 expression was specifically inhibited by the ARAP1-knockdown siRNA (# 3). Col IV protein levels were examined in each group by Western blotting, and ARAP1 knockdown decreased the secretion of this extracellular matrix protein (p<0.0001) (Figure 5A and D). The expression of FN and NOX2 in each group was examined by immunofluorescence staining to further verify the role of ARAP1 in glomerular mesangial cells cultured in a HG environment in vitro. ARAP1 knockdown suppressed FN and NOX2 expression in the glomerular mesangial cells cultured in a HG environment and alleviated the increases in extracellular matrix FN secretion and oxidative stress (Figure 5G).
Discussion
Activation of the RAS in renal tissues plays an important role in the progression of DN.13 Ang II is the main active peptide of the RAS and exerts its primary pathophysiological functions by binding to AT1R, which is a protein that plays a crucial role in the pathogenesis of DN. Angiotensin-converting enzyme inhibitors (ACEIs) and Ang II receptor blockers (ARBs) are currently the most effective treatments for DN; these treatments reduce the occurrence of albuminuria, delay renal dysfunction and alleviate structural damage in renal tissues.14–16
ARAP1, which is a member of the RAS, binds to the C-terminal region of the AT1R and promotes receptor recycling to the cell membrane, increasing the expression of AT1R and its sensitivity to Ang II.5 ARAP1 is expressed in various organs in mice, with the highest levels observed in the heart, kidney, aorta and adrenal gland. ARAP1 is expressed primarily in the vasculature and glomerular mesangial cells in the kidneys. ARAP1 is also expressed in mouse glomerular mesangial cells and participates in mesangial cell growth and the secretion of extracellular matrix proteins such as Col IV and FN by regulating AT1R-mediated Ang II signaling. ARAP1 is involved in the regulation of its downstream targets, such as phospholipase C, protein kinase, tyrosine kinase/phosphatase, and Rho kinase, and in the generation of reactive oxygen species (ROS) by regulating Ang II type 1 and type 2 receptor expression; in this manner, ARAP1 participates in the mechanisms regulating endothelial dysfunction and hyperresponsiveness, thus altering vascular smooth muscle cell growth, apoptosis, fibrosis, contractibility, calcification and inflammation-related macrophage infiltration and increasing redox sensitivity.17 Transgenic mice overexpressing ARAP1 in the proximal tubules exhibit salt sensitivity and Ang II-dependent hypertension with apparent renal hypertrophy.18 Based on these data, ARAP1 enhances AT1R-dependent renal signal transduction. In addition, the blood pressure of mice with endotoxemia is significantly decreased. Although the concentration of Ang II in the body was increased, ARAP1 and AT1R expression was significantly reduced.19 The expression of ARAP1 and AT1R in mesangial cells was significantly reduced after treatment in vitro with proinflammatory factors, whereas ARAP1 and AT1R expression was increased after the administration of ACEIs.19 Under the conditions of DN, the kidney produces large quantities of proinflammatory factors, and the Ang II concentration in the body is increased.20–22 However, the mutual regulation between ARAP1 and AT1R in DN and their effects on diabetic kidney injury have not been reported in the literature. In the present study, the expression levels of ARAP1, AT1R, TGF-β1, NOX2, NOX4, 8-OHdG, FN, Col IV, IL-1β and IL-18 were significantly higher in the renal cortexes of 20-week-old db/db mice than in those in the control group, suggesting that ARAP1 and AT1R may be involved in kidney fibrosis and the oxidative stress response, which cause kidney damage in DN.
According to Sonta et al,23 treatment with the ARB olmesartan reduced NOX4 expression and oxidative stress levels in streptozotocin (STZ)-induced diabetic mice. NOX4 is a molecule that plays a key role in TGF-β1-driven fibrosis;24 it is the main isoform expressed in the kidney and participates in renal ROS production under basic and pathological conditions such as DN and chronic kidney disease.25–27 NOX4 also plays an important role in the pathogenesis and progression of DN. In addition, 8-OHdG is used as an indicator of the degree of oxidative damage to DNA. The DNA helix is composed of four different nucleobases: adenine, thymine, cytosine and guanine. Because guanine has the lowest ionization potential, it is the most susceptible to oxidation.28 A guanine base with a double bond hydroxyl radical at the C-8 position results in the production of 8-OHdG. Increased expression of 8-OHdG is associated with the pathogenesis and progression of cancer29 and cardiovascular disease, and it is considered a sensitive biomarker of oxidative DNA damage in patients with diabetes.21 Significantly higher urinary excretion of 8-OHdG has been observed in patients with type 2 diabetes than in healthy control subjects. As shown in the study by Kanauchi et al,21 urinary 8-OHdG excretion increases with tubulointerstitial lesion severity in subjects with DN. Both short- and long-term blood glucose fluctuations and oxidative stress are correlated with 8-OHdG levels. Ang II produces ROS through the AT1R, thereby activating Jun N-terminal kinase (JNK), TGF-β1, nuclear factor-κB (NF-κB) and 5ʹ adenosine monophosphate-activated protein kinase (AMPK) and inducing the inflammatory response.22 In the pathophysiological environment, AT1R activation is closely related to the degree of fibrosis progression in various tissues. Ang II-mediated regulation of blood pressure is modulated by factors regulating AT1R activity, including the local regulation of AT1R expression, receptor desensitization, and receptor heterogenicity.22
UA and its analogs are widely used treatments for various cancers, inflammatory diseases, diabetes, Parkinson’s disease, Alzheimer’s disease, hepatitis B and other diseases because of their anti-inflammatory, anticancer, antioxidant, hypoglycemic and blood lipid-lowering activities.30 According to Zhou et al,31 low-dose oral UA significantly improved glomerular hypertrophy and extracellular matrix accumulation in STZ-induced diabetic mice by inhibiting activation of the signal transducer and activator of transcription-3 (STAT-3), extracellular signal-regulated kinase 1/2 (ERK1/2) and JNK signaling pathways and suppressing the overexpression of inducible nitric oxide synthase (iNOS). In a study by Qi et al,32 UA significantly reduced blood glucose, blood urea nitrogen (BUN), serum creatinine (Cr), superoxide dismutase (SOD), malondialdehyde (MDA), tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) levels and improved diabetic kidney injury in diabetic mice. Dietary UA supplementation improves blood sugar and blood lipid levels, lowers blood lipid accumulation in the liver, and increases antioxidant enzyme activity in rodent models of metabolic diseases.33 In the present study, the renal expression of ARAP1, AT1R, NOX2, NOX4, 8-OHdG, TGF-β1, FN, Col IV, IL-1β and IL-18 was substantially increased in 20-week-old db/db mice, while treatment with UA decreased the expression of these signaling proteins, indicating that UA may reduce the level of kidney injury in subjects with DN by inhibiting the ARAP1/AT1R signaling pathway and reducing the expression of NOX2, NOX4, 8-OHdG, TGF-β1, FN, Col IV, IL-1β and IL-18. Our siRNA knockdown study further confirmed the interaction between the ARAP1/AT1R pathways, revealing that decreased ARAP1 expression reduced the levels of the oxidative stress-related product NOX2 and the extracellular matrix protein FN. Further investigations into the mechanism underlying the effect of UA on relieving DN will be very important for guiding the clinical treatment of patients with DN.
Author Contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. International Diabetes Federation. IDF Diabetes. Brussels, Belgium: International Diabetes Federation; 2017.
2. Dounousi E, Duni A, Leivaditis K, Vaios V, Eleftheriadis T, Liakopoulos V. Improvements in the management of diabetic nephropathy. Rev Diabet Stud. 2015;12(1–2):119–133. doi:10.1900/RDS.2015.12.119
3. Colafella KM, Hilliard LM, Denton KM. Epochs in the depressor/pressor balance of the renin-angiotensin system. Clin Sci. 2016;130(10):761–771. doi:10.1042/CS20150939
4. Fukuda A, Wickman LT, Venkatareddy MP, et al. Angiotensin II-dependent persistent podocyte loss from destabilized glomeruli causes progression of end stage kidney disease. Kidney Int. 2012;81(1):40–55. doi:10.1038/ki.2011.306
5. Guo DF, Chenier I, Tardif V, Orlov SN, Inagami T. Type 1 angiotensin II receptor-associated protein ARAP1 binds and recycles the receptor to the plasma membrane. Biochem Biophys Res Commun. 2003;310(4):1254–1265. doi:10.1016/j.bbrc.2003.09.154
6. Lee CH, Wu SL, Chen JC, et al. Eriobotrya japonica leaf and its triterpenes inhibited lipopolysaccharide-induced cytokines and inducible enzyme production via the nuclear factor-kappaB signaling pathway in lung epithelial cells. Am J Chin Med. 2008;36(6):1185–1198. doi:10.1142/S0192415X0800651X
7. Liu J. Pharmacology of oleanolic acid and ursolic acid. J Ethnopharmacol. 1995;49(2):57–68. doi:10.1016/0378-8741(95)90032-2
8. Lu X, Fan Q, Xu L, et al. Ursolic acid attenuates diabetic mesangial cell injury through the up-regulation of autophagy via miRNA-21/PTEN/Akt/mTOR suppression. PLoS One. 2015;10(2):e0117400. doi:10.1371/journal.pone.0117400
9. Wang EM, Fan QL, Yue Y, Xu L. Ursolic acid attenuates high glucose-mediated mesangial cell injury by inhibiting the phosphatidylinositol 3-kinase/akt/mammalian target of rapamycin (PI3K/Akt/mTOR) signaling pathway. Med Sci Monit. 2018;24846–24854.
10. Ninichuk V, Clauss S, Kulkarni O, et al. Late onset of Ccl2 blockade with the spiegelmer mNOX-E36-3ʹPEG prevents glomerulosclerosis and improves glomerular filtration rate in db/db mice. Am J Pathol. 2008;172(3):628–637. doi:10.2353/ajpath.2008.070601
11. Xu HL, Wang XT, Cheng Y, et al. Ursolic acid improves diabetic nephropathy via suppression of oxidative stress and inflammation in streptozotocin-induced rats. Biomed Pharmacother. 2018;105:915–921. doi:10.1016/j.biopha.2018.06.055
12. Wang XT, Gong Y, Zhou B, et al. Ursolic acid ameliorates oxidative stress, inflammation and fibrosis in diabetic cardiomyopathy rats. Biomed Pharmacother. 2018;97:1461–1467. doi:10.1016/j.biopha.2017.11.032
13. Rahimi Z. The role of renin angiotensin aldosterone system genes in diabetic nephropathy. Can J Diabetes. 2016;40(2):178–183. doi:10.1016/j.jcjd.2015.08.016
14. Bakris GL, Williams M, Dworkin L, et al. Preserving renal function in adults with hypertension and diabetes: a consensus approach. National kidney foundation hypertension and diabetes executive committees working group. Am J Kidney Dis. 2000;36(3):646–661. doi:10.1053/ajkd.2000.16225
15. Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345(12):861–869. doi:10.1056/NEJMoa011161
16. Lewis EJ, Hunsicker LG, Clarke WR, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001;345(12):851–860. doi:10.1056/NEJMoa011303
17. Savoia C, Burger D, Nishigaki N, Montezano A, Touyz RM. Angiotensin II and the vascular phenotype in hypertension. Expert Rev Mol Med. 2011;13:e11. doi:10.1017/S1462399411001815
18. Guo DF, Chenier I, Lavoie JL, et al. Development of hypertension and kidney hypertrophy in transgenic mice overexpressing ARAP1 gene in the kidney. Hypertension. 2006;48(3):453–459. doi:10.1161/01.HYP.0000230664.32874.52
19. Mederle K, Schweda F, Kattler V, et al. The angiotensin II AT1 receptor-associated protein Arap1 is involved in sepsis-induced hypotension. Crit Care. 2013;17(4):R130. doi:10.1186/cc12809
20. Castrop H, Hocherl K, Kurtz A, Schweda F, Todorov V, Wagner C. Physiology of kidney renin. Physiol Rev. 2010;90(2):607–673.
21. Kanauchi M, Nishioka H, Hashimoto T. Oxidative DNA damage and tubulointerstitial injury in diabetic nephropathy. Nephron. 2002;91(2):327–329. doi:10.1159/000058412
22. Kim N, Jung Y, Nam M, et al. Angiotensin II affects inflammation mechanisms via AMPK-related signalling pathways in HL-1 atrial myocytes. Sci Rep. 2017;7(1):10328. doi:10.1038/s41598-017-09675-3
23. Sonta T, Inoguchi T, Matsumoto S, et al. In vivo imaging of oxidative stress in the kidney of diabetic mice and its normalization by angiotensin II type 1 receptor blocker. Biochem Biophys Res Commun. 2005;330(2):415–422. doi:10.1016/j.bbrc.2005.02.174
24. Hecker L, Vittal R, Jones T, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. 2009;15(9):1077–1081. doi:10.1038/nm.2005
25. Gorin Y. Nox4 as a potential therapeutic target for treatment of uremic toxicity associated to chronic kidney disease. Kidney Int. 2013;83(4):541–543. doi:10.1038/ki.2012.434
26. Gorin Y, Wauquier F. Upstream regulators and downstream effectors of NADPH oxidases as novel therapeutic targets for diabetic kidney disease. Mol Cells. 2015;38(4):285–296. doi:10.14348/molcells.2015.0010
27. Jha JC, Gray SP, Barit D, et al. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol. 2014;25(6):1237–1254. doi:10.1681/ASN.2013070810
28. Yin B, Whyatt RM, Perera FP, Randall MC, Cooper TB, Santella RM. Determination of 8-hydroxydeoxyguanosine by an immunoaffinity chromatography-monoclonal antibody-based ELISA. Free Radic Biol Med. 1995;18(6):1023–1032. doi:10.1016/0891-5849(95)00003-G
29. Loft S, Poulsen HE. Cancer risk and oxidative DNA damage in man. J Mol Med (Berl). 1996;74(6):297–312. doi:10.1007/BF00207507
30. Hussain H, Green IR, Ali I, et al. Ursolic acid derivatives for pharmaceutical use: a patent review (2012–2016). Expert Opin Ther Pat. 2017;27(9):1061–1072. doi:10.1080/13543776.2017.1344219
31. Zhou Y, Li JS, Zhang X, Wu YJ, Huang K, Zheng L. Ursolic acid inhibits early lesions of diabetic nephropathy. Int J Mol Med. 2010;26(4):565–570. doi:10.3892/ijmm_00000500
32. Qi MY, Yang JJ, Zhou B, Pan DY, Sun X. Study on the protective effect of ursolic acid on alloxan-induced diabetic renal injury and its underlying mechanisms. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2014;30(5):445–448.
33. Jayaprakasam B, Olson LK, Schutzki RE, Tai MH, Nair MG. Amelioration of obesity and glucose intolerance in high-fat-fed C57BL/6 mice by anthocyanins and ursolic acid in Cornelian cherry (Cornus mas). J Agric Food Chem. 2006;54(1):243–248. doi:10.1021/jf0520342
34. Li D, Ren D, Luo Y, et al. Protective effects of ursolic acid against hepatotoxicity and endothelial dysfunction in mice with chronic high choline diet consumption. Chem Biol Interact. 2016;258:102–107. doi:10.1016/j.cbi.2016.08.019
35. Chu X, He X, Shi Z, et al. Ursolic acid increases energy expenditure through enhancing free fatty acid uptake and β-oxidation via an UCP3/AMPK-dependent pathway in skeletal muscle. Mol Nutr Food Res. 2015;59(8):1491–1503. doi:10.1002/mnfr.v59.8
36. Ullevig SL, Zhao Q, Zamora D, et al. Ursolic acid protects diabetic mice against monocyte dysfunction and accelerated atherosclerosis. Atherosclerosis. 2011;219(2):409–416. doi:10.1016/j.atherosclerosis.2011.06.013
37. Jia Y, Kim S, Kim J, et al. Ursolic acid improves lipid and glucose metabolism in high-fat-fed C57BL/6J mice by activating peroxisome proliferator-activated receptor alpha and hepatic autophagy. Mol Nutr Food Res. 2015;59(2):344–354. doi:10.1002/mnfr.201400399
38. Xu L, Fan Q, Wang X, et al. Ursolic acid improves podocyte injury caused by high glucose. Nephrol Dialysis Transplant. 2015;32:8.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.