Back to Archived Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 16
Ultrasonic-Assisted Synthesis of Heterocyclic Curcumin Analogs as Antidiabetic, Antibacterial, and Antioxidant Agents Combined with in vitro and in silico Studies
Authors Zelelew D ![]() , Endale M
, Endale M ![]() , Melaku Y, Geremew T
, Melaku Y, Geremew T ![]() , Eswaramoorthy R
, Eswaramoorthy R ![]() , Tufa LT, Choi Y
, Tufa LT, Choi Y ![]() , Lee J
, Lee J
Received 3 February 2023
Accepted for publication 11 July 2023
Published 28 July 2023 Volume 2023:16 Pages 61—91
DOI https://doi.org/10.2147/AABC.S403413
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Frank Boeckler
Demis Zelelew,1 Milkyas Endale,1 Yadessa Melaku,1 Teshome Geremew,2 Rajalakshmanan Eswaramoorthy,3 Lemma Teshome Tufa,1,4 Youngeun Choi,5 Jaebeom Lee5
1Department of Applied Chemistry, School of Applied Natural Science, Adama Science and Technology University, Adama, Ethiopia; 2Department of Applied Biology, School of Applied Natural Science, Adama Science and Technology University, Adama, Ethiopia; 3Department of Biomaterials, Saveetha Dental College and Hospital, Saveetha University, Chennai, 600 077, India; 4Research Institute of Materials Chemistry, Chungnam National University, Daejeon, 34134, Republic of Korea; 5Department of Chemistry, Department of Chemistry Engineering and Applied Chemistry, Chungnam National University, Daejeon, 34134, Republic of Korea
Correspondence: Milkyas Endale; Demis Zelelew, Department of Applied Chemistry, School of Applied Natural Science, Adama Science and Technology University, P.O. Box 1888, Adama, Ethiopia, Email [email protected]; [email protected]; [email protected]
Background: Heterocyclic analogs of curcumin have a wide range of therapeutic potential and the ability to control the activity of a variety of metabolic enzymes.
Methods: 1H-NMR and 13C-NMR spectroscopic techniques were used to determine the structures of synthesized compounds. The agar disc diffusion method and α-amylase inhibition assay were used to examine the antibacterial and anti-diabetic potential of the compounds against α-amylase enzyme inhibitory activity, respectively. DPPH-free radical scavenging and lipid peroxidation inhibition assays were used to assess the in vitro antioxidant potential.
Results and Discussion: In this work, nine heterocyclic analogs derived from curcumin precursors under ultrasonic irradiation were synthesized in excellent yields (81.4– 93.7%) with improved reaction time. Results of antibacterial activities revealed that compounds 8, and 11 displayed mean inhibition zone of 13.00± 0.57, and 19.66± 00 mm, respectively, compared to amoxicillin (12.87± 1.41 mm) at 500 μg/mL against E. coli, while compounds 8, 11 and 16 displayed mean inhibition zone of 17.67± 0.57, 14.33± 0.57 and 23.33± 00 mm, respectively, compared to amoxicillin (13.75± 1.83 mm) at 500 μg/mL against P. aeruginosa. Compound 11 displayed a mean inhibition zone of 11.33± 0.57 mm compared to amoxicillin (10.75± 1.83 mm) at 500 μg/mL against S. aureus. Compound 11 displayed higher binding affinities of − 7.5 and − 8.3 Kcal/mol with penicillin-binding proteins (PBPs) and β-lactamases producing bacterial strains, compared to amoxicillin (− 7.2 and − 7.9 Kcal/mol, respectively), these results are in good agreement with the in vitro antibacterial activities. In vitro antidiabetic potential on α-amylase enzyme revealed that compounds 11 (IC50=7.59 μg/mL) and 16 (IC50=4.08 μg/mL) have higher inhibitory activities than acarbose (IC50=8.0 μg/mL). Compound 8 showed promising antioxidant inhibition efficacy of DPPH (IC50 = 2.44 g/mL) compared to ascorbic acid (IC50=1.24 g/mL), while compound 16 revealed 89.9± 20.42% inhibition of peroxide generation showing its potential in reducing the development of lipid peroxides. In silico molecular docking analysis, results are in good agreement with in vitro biological activity. In silico ADMET profiles suggested the adequate oral drug-likeness potential of the compounds without adverse effects.
Conclusion: According to our findings, both biological activities and in silico computational studies results demonstrated that compounds 8, 11, and 16 are promising α-amylase inhibitors and antibacterial agents against E. coli, P. aeruginosa, and S. aureus, whereas compound 8 was found to be a promising antioxidant agent.
Keywords: ultrasonic-irradiation, α-amylase, antibacterial, antioxidant, heterocyclic curcumin analogs, molecular docking, ADMET prediction
Introduction
Globally, the ever-growing population has put the life of humankind on the edge of being targeted by various diseases, among which, diabetes and antibiotic-resistance infectious diseases are the most common.1 Over the past decades, the global incidence rate of the disease has risen at an alarming rate, hence making it a major health problem of the 21st century.2 Diabetes mellitus (DM) is a chronic metabolic and epidemic disease characterized by constant hyperglycemia. In 2019, the World Health Organization (WHO) estimated that over 4.22 billion people around the globe have been affected by diabetes and this number is predicted to be about 6.42 billion, by 2040.3 There are two types of DM; type 1 DM results from damage of β-cells of the pancreas so that insulin cannot be synthesized adequately, and type 2 DM results from insufficiency of insulin action or resistance. It is interesting to note that people with uncontrolled DM (regardless of type) have changes in healing latency and a higher risk of contracting several newly emerging infectious (mostly bacterial) infections. Due to the immune system’s inability to successfully defend against invasive microorganisms, infections are a major problem in diabetics.4 Bacterial infections and diabetes can have a mutually reinforcing relationship, with some infections, such as periodontitis exacerbating insulin resistance. Antimicrobial resistance (AMR) is a significant problem in diabetes patients as a result of their susceptibility to infection as well as their frequent use of antibiotics to treat illnesses. Bacteria can develop resistance by changing target sites, making specialized efflux pumps, mutating their DNA to create new metabolic pathways, or creating β -lactamases, an enzyme that breaks down β -lactams.5 As reported by the International Diabetes Federation (IDF) in 2015, T2DM comprises approximately 90–95% of all cases of diabetes.6 One solution to mitigate diabetes, especially type 2 DM, is the regulation of postprandial blood glucose by inhibiting the main carbohydrate hydrolyzing enzymes of α-glucosidase/amylase in the intestine.7 Currently, prescribed acarbose as both sugar mimics oral α-glucosidase and α-amylase inhibitors remain in parallel with voglibose, and miglitol the most common drugs of T2DM; however, their application is hindered by the side effects. Moreover, the associated disadvantages required tedious multi-steps for preparation, and low natural abundance prompted the exploration of new molecules as inhibitors for the targeted enzyme.8,9 Reactive oxygen species (ROS) decrease insulin levels in hyperglycemia, which in turn regulates gene transcription and ultimately results in death. ROS is produced under a hyperglycemic condition which further implicates various disorders important to the development of diabetic complications. ROS are thought to damage cells by forming free radicals and lipid peroxides in cell membranes.10,11 Antioxidants that scavenge ROS are of great value in preventing the development and spread of oxidative diseases.12 They play an important role in protecting against deadly diseases by turning free radicals into ineffective states.13 Thus, the use of antioxidants in combination with an anti-diabetic and antibiotic drug will greatly help the immune system to combat insulin and bacterial resistance, respectively.6,14
Currently, lead compounds derived from natural products are emerging as biotherapeutic sources for drug discovery and development.15,16 Curcumin, is a polyphenolic substance obtained from the rhizomes of Curcuma longa L, displaying a wide spectrum of biological activities.17,18 The pharmacokinetic studies revealed that the β-diketone functionality of curcumin is a substrate for liver aldo keto reductase, which may be one of the reasons for its rapid metabolism in vivo.19 Due to an unstable β-diketone moiety in the curcumin structure, its poor stability, low bioavailability, and pharmacokinetic limitations have hindered the therapeutic efficacy and largely contributed to curcumin instability under physiological conditions.20,21 To overcome such limitations, several approaches have been explored and the replacement of central β-diketo functionality with mono carbonyl has resulted in many compounds with improving pharmacokinetic properties and bioavailability of curcumin while increasing its biological activities.22 Monocarbonyl curcuminoids’ bioactive representations exhibit fascinating biological activities that are superior to curcumin itself and are chemically more stable than curcumin in vitro.23 Studies have identified the diketo moiety as the cause of curcumins’ instability and it has been hypothesized that adding a heterocyclic ring structure to the heart of the curcumin scaffold would increase the molecule’s stability as well as its activity by a wide margin.24,25 Curcuminoids incorporated with a heterocyclic structure at the diketo site tend to have reduced rotational freedom, absence of tautomerism, and minimized metal chelation properties.26 In continuation of our efforts in finding antidiabetic, antibacterial, and antioxidant drug candidates, this work focused on the preparation of nitrogen-containing compounds of N-pyrazoline, pyrimidine-2-ol, thiazolo[3,2-α] pyrimidine, and β-substituted imidazole analogs through heterocyclization of suitably functionalized substrates of curcumin derivatives. On the other hand, over the years, eco-friendly ultrasonic irradiation approaches have been established for the synthesis of various heterocycles with spectacular results, such as reduced environmental pollution, low cost, high yields with short reaction times, experimental simplicity, enhanced selectivity, energy saving and ease of processing and handling.27,28 These desirable qualities have established the advantages of sonication over conventional heating.
In light of the aforementioned facts, we hereby report the synthesis of a new series of heterocyclics derived from curcumin scaffolds by following a greener route of ultrasonic irradiation. The synthesized compounds were evaluated for their antihyperglycemic activity through α-amylase inhibition as well as their antioxidant and antibacterial potentials, with the final attempt of obtaining improved efficiency, reduced side effects, and relatively low-cost agents. Furthermore, the in silico ADMET prediction and molecular docking studies were investigated to explore their potential to become clinical drugs.
Materials and Methods
All of the chemicals and reagents used in the study were purchased from commercial suppliers, including Merck (Germany), Sigma Aldrich Sigma Chemical, Co. (USA), and Loba Chemie, Pvt Ltd. (India) were used without additional purification. Thin layer chromatography (TLC) analysis was performed on pre-coated aluminum plate silica gel (particle size 60–120 mm) (Merck) G60F254 plates (Germany) plates employing visualizing spots in UV light of long and short wavelengths (254 and 365 nm) to determine the compounds’ purity. Column chromatography was carried out using silica gel with a mobile phase of n-hexane, ethyl acetate, and dichloromethane. Acarbose, amoxicillin, and ascorbic acid were utilized as antidiabetic, antibacterial, and antioxidant standard drugs, respectively, obtained from Ethiopian Pharmaceutical Manufacturing Factory (EPHARM).
Melting points were determined using the open capillary method in a Gallen-Kamp MFB-595 electric melting point equipment and are uncorrected. The ultrasonic-assisted reactions are performed in a “Spectralab model UMC 20 Ultrasonic cleaner” (horn type, made in ACE, USA). The instrument had an operating frequency of 40 kHz, output (nominal) power of 250 W, 1.3×10−2 m diameter of stainless-steel tip with an intensity of 3.4×105 W/m2, and 1.32×10−4 m2 surface area of the ultrasound irradiating face at Department of Materials Science and Engineering Laboratory, School of Mechanical, Chemical and Materials Engineering, Adama Science and Technology University. A double-beam UV–Visible spectrophotometer (Model 2201, India) was used to obtain UV spectra. The 1H-NMR, 13C-and 13C-DEPT-135 NMR spectra were recorded on a Bruker Avance III 400 nano at 400 and 100 MHz, respectively, with Tetramethylsilane (TMS) as an internal standard. The spectra were measured in DMSO-d6 and MeOD-d4 relative to TMS. The chemical shifts are expressed as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublets), and br s (broad singlet), respectively, and are presented together with coupling constants (J) in Hz (Hertz) in the appropriate solvent. In silico ADMET and molecular docking simulation, investigations were carried out using the Swiss ADMET/AdmetSAR and AutoDock vina 4.2.6 version software packages, respectively.
Synthesis Procedures
Synthesis of Curcumin (1,5-Bis(4-Hydroxy-3-Methoxyphenyl)penta-1,4-Dien-3-One (3)
Vanillin (4-hydroxy-3-methoxybenzaldehyde, 1.35 g, 8.84 mmol) (1) was combined with 5-mL acetone (2) in a well of a closed round-bottom flask, and the mixture was stirred for 5 minutes in an ice bath. Then, 2 mL of 20% NaOH in ethanol-water (1:1) was dropped into the solution, and the mixture was stirred at room temperature for approximately 1 hr.29 During the reaction period, the reactor was covered with aluminum foil to block out the light. Monitoring the reaction’s course was done by TLC using methanol/dichloromethane (1:9) as a mobile phase. The reaction was then worked up by adding 10% HCl solution to provide a solid product. The resulting solids were crushed into smaller pieces and filtered through filter paper under a vacuum, washed with cold water, and dried in a vacuum oven. Curcumin (3) was finally purified by recrystallization in ethanol to obtain a light-yellow solid. All the synthesized compounds were analyzed by 1H and 13C–NMR spectra are presented in Supplementary Information in Appendix Ia–c and IIa–c.
1,5-Bis(4-Hydroxy-3-Methoxyphenyl)penta-1,4-Dien-3-One (3)
Yellow solid powder; yield: 86.7%; m.p.: 144–146 °C; Rf (30% EtOAc in n-hexane) = 0.80;1H-NMR (400 MHz, MeOD-d4): 8.28 (s, 1H), 8.26 (s, 1H), 7.92 (d, J = 15.6 Hz, 2H), 7.76 (dd, J = 7.8, 1.8 Hz, 2H), 7.45 (d, J = 15.9 Hz, 2H), 7.13 (d, J = 8.2 Hz,2H), 6.58 (dd, J = 15.8 Hz, 2H), 3.83 (s, 6H); 13C-NMR (100 MHz, MeOD-d4): δ 190.0, 153.1, 149.9, 143.2, 139.3, 130.1, 125.0, 118.4, 110.9, 55.7; 13C−DEPT-135 (100 MHz, MeOD-d4): δ 148.5, 137.9, 129.7, 128.7, 123.6, 54.4.
Synthesis of Curcumin Derivatives from Dehydrozingerone (5–7)
Vanillin (1, 5 g, 0.033 mol) was dissolved in 80 mL of acetone (2), and then 50 mL of NaOH (2.0 g, 0.25 mol) was added gradually while being continuously stirred for 2 to 3 hrs. Reduced pressure was used to eliminate extra acetone.30 A yellow precipitate was produced after 0.1 N HCl acidification; this precipitate was extracted with CHCl3, and the organic layer was dried over anhydrous sodium sulfate. The yellow solid of (E)-4-(4-hydroxy-3-methoxyphenyl) but-3-en-2-one, (4) obtained was recrystallized from ethanol. Then, an equimolar amount of dehydrozingerone, (4) (0.06 mol), and the appropriate aromatic aldehyde derivatives, 5–7 (0.06 mol) were dissolved in a minimum amount of ethanol (5–10 mL), aqueous KOH (3 mL, 30%) was added dropwise slight modification method.31 After 30 min in an ice bath, the solid that had formed in the mixture was stirred at room temperature for 3 hrs, until the precipitate had formed. The reaction was stirred until benzaldehyde consumption was detected using TLC with an n-hexane: ethyl acetate eluent (9:1). The precipitate was extensively rinsed with cold distilled water and cold methanol (2×20 mL) after being filtered off. Curcumin analogs (5–7) were recrystallized from absolute ethanol (Supplementary Information in Appendix III–Va–c).
1-(4-Hydroxy-3-Methoxyphenyl)-5-(4-Nitrophenyl)penta-1,4-Dien-3-One (5)
Brown solid powder; yield: 86.7%; m.p.: 171–173 °C; Rf (30% EtOAc in n-hexane) = 0.73; 1H-NMR (400 MHz, MeOD-d4): 8.54 (s, 1H), 7.70 (d, J = 15.4 Hz, 1H), 7.64 (d, J = 15.6 Hz, 1H), 7.54 (dd, J = 8.9, 2.1 Hz, 1H), 7.10 (d, J = 15.7 Hz, 1H), 7.07 (dd, J = 8.4, 2.2 Hz, 1H), 6.99 (s, 1H), 6.95 (d, J = 11.1 Hz, 1H), 6.80 (d, J = 15.4 Hz, 1H), 6.74 (d, J = 9.0 Hz, 1H), 6.59 (d, J = 8.2 Hz, 1H), 3.83 (s, 3H); 13C-NMR (100 MHz, MeOD-d4): δ 191.4, 165.0, 153.81, 152.9, 147.7, 145.5, 144.5, 131.5, 127.9, 124.2, 121.9, 119.6, 113.1, 111.2, 110.8, 55.8; 13C−DEPT-135 (100 MHz, MeOD-d4): δ 146.3, 143.1, 130.1, 126.5, 120.5, 118.2, 117.6, 111.7, 109.4, 54.4.
1-(4-(Dimethylamino)phenyl)-5-(4-Hydroxy-3-Methoxyphenyl)penta-1,4-Dien-3-One (6)
Reddish solid powder; yield: 86.7%; m.p.: 200–202 °C; Rf (30% EtOAc in n-hexane) = 0.71;1H-NMR (400 MHz, MeOD-d4): 8.55 (s, 1H), 7.93 (d, J = 6.9 Hz, 1H), 7.78 (d, J =15.4 Hz, 1H), 7.69 (dd, J = 6.1, 2.1 Hz, 2H), 7.66 (s, 1H), 7.40 (dd, J = 10.5, 7.1 Hz, 1H), 7.34 (dd, J = 16.2, 1.4 Hz, 1H), 7.26 (d, J = 15.9 Hz, 1H), 7.11 (dd, J = 15.8, 2.1 Hz, 1H), 6.81 (d, J = 15.4 Hz, 1H), 6.58 (d, J = 8.2 Hz, 1H), 3.84 (s, 3H), 3.31 (s, 6H); 13C-NMR (100 MHz, MeOD-d4): δ 191.1, 170.3, 153.3, 149.3, 142.7, 136.7, 131.2, 130.2, 129.9, 129.4, 128.7, 127.2, 120.9, 118.3, 110.9, 55.8, 24.2; 13C DEPT-135 (100 MHz, MeOD-d4): δ 147.9, 141.3, 129.8, 128.8, 127.9, 127.3, 125.8, 118.4, 116.9, 109.5, 47.9.
1-(4-Hydroxy-3-Methoxyphenyl)-5-Phenylpenta-1,4-Dien-3-One (7)
Dark brown solid powder; yield: 86.7%; m.p.: 164–166 °C; Rf (30% EtOAc in n-hexane) =0.69; 1H-NMR (400 MHz, MeOD-d4): 8.49 (s, 1H), 7.49 (d, J = 15.8 Hz, 1H), 6.95 (s, 1H), 6.93 (dd, J = 8.3, 2.2 Hz, 2H), 6.51 (d, J = 7.9 Hz, 2H), 6.35 (d, J = 15.8 Hz, 1H), 3.73 (s, 3H); 13C-NMR (100 MHz, MeOD-d4): δ 180.4, 170.5, 165.1, 161.3, 152.9, 149.5, 127.7, 120.3, 119.9, 119.8, 110.8, 55.8; 13C−DEPT-135 (100 MHz, MeOD-d4): δ 148.2, 126.4, 118.3, 109.4, 54.5.
Synthesis Procedures for the Preparation of Heterocyclic Curcumin Analogs (8–16) via Ultrasonic Irradiation-Assisted Approach
The general strategy for the synthesis of title compounds under ultrasonic radiation promoted procedure:
A mixture of curcumin derivatives, 5–7, and different nucleophilic reagents, namely, hydrazine hydrate, urea, 2-amino-4-substituted phenyl-thiazole and imidazole in catalytic condition under ethanol (10 mL) was sealed in the Pyrex tube was sonicated in an ultrasonic bath at 45–60°C for the appropriate time. Cold/hot water was added or removed manually to maintain the temperature of the ultrasonic bath. The reaction mixture was cooled to room temperature after it had finished, as determined by TLC using n-hexane: ethyl acetate (9:1 v/v) as the eluent. The precipitated product was filtered, washed with water and acetone, and then vacuum-dried. The obtained solid (8–16) was recrystallized from ethanol. In most cases, no further purification was necessary.32 The structures of the synthesized compounds were fully confirmed by employing physicochemical parameters, and spectroscopic techniques (1H-NMR, and 13C−NMR). The substitution pattern at the aryl rings was carefully chosen to impart a variety of electronic environments, which would undoubtedly impact the target compounds’ pattern of anti-diabetic, anti-bacterial, and anti-oxidant activities.
Synthesis of N-Pyrazoline Analogs, 8 and 9
A mixture of curcumin derivatives, 5–7 (1 mmol), and hydrazine hydrate (10 mmol, 98%) in absolute ethanol (10 mL) and then subjected to ultrasonic irradiation. When the reaction was finished, as determined by TLC, the reaction mixture was cooled to room temperature and water was added. The precipitated product underwent filtering, acetone and water washing, and vacuum drying before being dried. The obtained solid products of curcumin-based N-pyrazoline analogs (8 and 9) were recrystallized from ethanol (Supplementary Information in Appendix Via–c and VIIa–c).
4-(-2-(3-(4-(Dimethylamino)phenyl)-4,5-Dihydro-1H-Pyrazol-5-yl)vinyl)-2-Methoxyphenol (8)
Pale yellow solid powder; yield: 87.8%; m.p.: 215–217 °C; Rf (30% EtOAc in n-hexane) = 0.67; 1H-NMR (400 MHz, DMSO-d6): 7.51 (s, 1H), 7.22 (s, 1H, NH), 7.14 (dd, J = 7.9, 2.1 Hz, 1H), 6.90 (d, J = 15.1 Hz, 1H), 6.72 (d, J = 10.6 Hz, 1H), 6.48 (dd, J = 6.9, 1.6 Hz, 2H), 6.38 (d, J = 8.2 Hz, 1H), 6.29 (d, J = 2.4 Hz, 1H, olefinic H), 6.29–6.23 (m, 1H), 5.90 (d, J = 15.8 Hz, 1H, olefinic H), 4.11 (dd, J =11.6, 4.0 Hz, 1H, pyrazoline HX), 3.39 (s, 1H, OCH3), 2.47 (dd, J =17.6, 11.6 Hz, 1H, pyrazoline HM), 2.07 (dd, J = 17.6, 4.0 Hz, 1H, pyrazoline HA); 13C-NMR (100 MHz, DMSO–d6): δ 155.1, 153.2, 150.9, 144.3, 136.2, 134.7, 128.6, 128.4, 126.6, 118.5, 114.3, 114.2, 59.4, 55.5, 44.4, 39.6; 13C DEPT-135 (100 MHz, DMSO-d6): δ 150.8, 140.6, 126.4, 118.8, 115.1, 112.1, 110.6, 63.2, 55.5, 44.7.
4-(-2-(4,5-Dihydro-3-Phenyl-1H-Pyrazol-5-yl)vinyl)-2-Methoxyphenol (9)
Yellow solid powder; yield: 91.2%; m.p.: 146–148 °C; Rf (30% EtOAc in n-hexane) = 0.68; 1H-NMR (400 MHz, DMSO-d6): 8.51 (s, 1H), 7.42 (s, 1H, NH), 7.91 (dd, J =7.8, 1.1 Hz, 1H), 7.82 (d, J = 7.6 Hz, 1H), 7.74 (dd, J = 15.6, 7.7 Hz, 1H), 7.53 (d, J = 7.8 Hz, 1H), 7.35 (s, 1H), 7.26 (dd, J = 8.1, 4.5 Hz, 1H), 7.12 (d, J = 2.1 Hz, 2H), 6.92 (d, J = 6.3 Hz, 1H, olefinic H), 6.82–6.70 (m, 1H), 6.64 (dd, J = 8.1, 2.1 Hz, 1H), 6.59 (d, J = 16.2 Hz, 1H, olefinic H), 4.78 (dd, J = 10.2, 4.0 Hz, 1H, pyrazoline HX), 3.29 (dd, J = 16.0, 10.6 Hz, 1H, pyrazoline HM), 2.69 (dd, J =10.9, 5.5 Hz, 1H, pyrazoline HA), 3.78 (s, 3H, OCH3); 13C-NMR (100 MHz, DMSO-d6): δ 153.3, 151.9, 150.9, 148.2, 147.2, 143.3, 132.6, 128.5, 126.6, 120.6, 119.1, 115.9, 110.0, 63.5, 55.7, 43.9; 13C DEPT-135 (100 MHz, DMSO-d6): δ 132.8, 128.7, 127.0, 120.5, 119.2, 116.1, 111.8, 110.2, 63.7, 55.9, 40.3.
Synthesis of 4-Phenyl-6-Styrylpyrimidine-2-ol Analogs (10 and 11)
Synthesis of 4-phenyl-6-styrylpyrimidine-2-olcompounds (10 and 11) involves the reaction of monocarbonyl curcumin derivatives, 5–7 (1 mmol) with urea (0.6g, 1 mmol) dissolved in an ethanolic sodium hydroxide (4 g NaOH and 10 mL ethanol) solution and then subjected to the ultrasonic irradiation. The reaction mixture was cooled to room temperature when it had finished the reaction as indicated by TLC. The precipitated product was filtered, washed with water and acetone, and then vacuum-dried. The 4-phenyl-6-styrylpyrimidine-2-ol analogs precipitate obtained (10 and 11) was filtered, washed, and recrystallized in ethanol (Supplementary Information in Appendix VIIIa–c and IXa–c).
4-(4-Hydroxy-3-Methoxystyryl)-6-(4-(Dimethylamino)phenyl)pyrimidin-2-ol (10)
Brown solid powder; yield: 90.3%; m.p.: 184–186 °C; Rf (30% EtOAc in n-hexane) = 0.59; 1H-NMR (400 MHz, DMSO-d6): 9.40 (s, 1H), 8.23 (s, 1H), 7.43 (d, J = 9.0 Hz, 2H), 7.66 (d, J = 15.6 Hz, 1H), 7.33 (dd, J = 8.8, 1.8 Hz, 1H), 6.75 (d, J = 15.8 Hz, 1H), 6.53 (d, J = 9.1 Hz, 2H), 6.48 (d, J = 9.0 Hz, 1H, olefinic H), 6.32 (d, J = 6.5 Hz, 1H, olefinic H), 5.32 (s, 1H, Ar-CH, pyrimidine H-4), 2.78 (s, 6H, –N(CH3)2); 13C-NMR (100 MHz, DMSO-d6): δ 166.6, 160.2, 154.4, 151.9, 151.1, 142.7, 131.8, 130.3, 128.1, 124.6, 122.3, 121.1, 119.1, 112.0, 111.2, 110.7, 55.58, 43.3; 13C DEPT-135 (100 MHz, DMSO-d6): δ 142.9, 130.7, 130.6, 121.4, 112.3, 111.5, 111.0, 55.7, 40.5.
4-(4-Hydroxy-3-Methoxystyryl)-6-Phenylpyrimidin-2-ol (11)
Brown solid powder; yield: 86.1%; m.p.: 166–168 °C; Rf (30% EtOAc in n-hexane) = 0.50; 1H-NMR (400 MHz, DMSO-d6): 8.91 (s, 1H), 8.19 (s, 1H), 7.56 (dd, J =7.2, 2.2 Hz, 1H), 7.18 (d, J = 7.9 Hz, 1H), 7.11–6.88 (m, 1H), 6.63 (d, J = 8.2 Hz, 1H), 6.10 (d, J = 18.6 Hz, 1H, olefinic H), 5.87 (d, J = 8.3 Hz, 1H, olefinic H), 5.29 (s, 1H, Ar-CH, pyrimidine H-4), 4.20 (s, 1H). 13C-NMR (100 MHz, DMSO-d6): δ 175.14, 171.0, 169.9, 166.7, 160.3, 140.5, 129.2, 128.9, 128.8, 128.3, 127.3, 127.2, 126.6, 117.7, 54.8; 13C DEPT-135 (100 MHz, DMSO-d6): δ 130.2, 129.4, 129.1, 128.5, 127.6, 127.4, 126.9, 55.0.
Synthesis of 5H-Thiazolo[3,2-α] Pyrimidines Analogs (12–14)
A solution of 2-amino-4-substituted phenylthiazole (1.5 mmol) in ethanol (10 mL) was mixed with the appropriate curcumin derivatives 5–7 (1.5 mmol) in presence of Na metal (0.5 g) and then subjected to the ultrasonic irradiation. The reaction mixture was cooled to room temperature when it had finished the reaction as determined by TLC. The precipitated product underwent filtering, acetone and water washing, and vacuum drying before being dried. The obtained solid thiazolo[3,2-α] pyrimidine analogs (12–14) was recrystallized from ethanol. In most cases, no further purification was necessary (Supplementary Information in Appendix X–XIIa–c).
2-(7-(4-Hydroxy-3-Methoxystyryl)-5-(4-Nitrophenyl)-5H-Thiazolo[3,2-α]pyrimidin-3-yl)phenol (12)
Brown solid powder; yield: 93.7%;m.p.: 177–179 °C; Rf (30% EtOAc in n-hexane) = 0.77; 1H-NMR (400 MHz, DMSO-d6): 9.12 (s, 1H), 8.35 (s, 1H), 7.94 (s, 1H, Ar-H of thiazole-H)7.54 (d, J = 9.4 Hz, 2H), 7.01 (dd, J = 15.2, 1.9 Hz, 2H), 6.95 (d, J = 8.3 Hz, 1H), 6.81 (d, J = 15.2 Hz, 1H, olefinic H), 6.56–6.43 (m, 6H), 6.06 (d, J = 8.3 Hz, 1H, olefinic H), 6.29 (s, 1H, Ar-CH, pyrimidine H), 5.41 (s, 1H, benzylic-H), 3.58 (s, 3H, OCH3); 13C-NMR (100 MHz, DMSO-d6): δ 186.2, 175.5, 171.9, 170.2, 166.9, 162.7, 159.9, 151.3, 131.9, 131.5, 130.1, 124.5, 120.4, 117.8, 117.2, 116.9, 116.1, 115.8, 54.6; 13C DEPT-135 (100 MHz, DMSO-d6): δ 131.9, 130.5, 117.6, 116.5, 116.2, 54.9.
2-(7-(4-Hydroxy-3-Methoxystyryl)-5-(4-(Dimethylamino)phenyl)-5H-Thiazolo[3,2-α]pyrimidin-3-yl)phenol (13)
Pale yellow solid powder; yield: 91.0%; m.p.: 196–198 °C; Rf (30% EtOAc in n-hexane) = 0.66; 1H-NMR (400 MHz, DMSO-d6): 9.14 (s, 1H), 8.41 (s, 1H), 7.60 (d, J = 9.1 Hz, 1H), 7.58 (d, J = 2.1 Hz, 0H), 7.05 (d, J = 7.3 Hz, 1H), 6.84 (s, 1H, Ar-H of thiazole-H), 6.70 (d, J = 9.0 Hz, 1H), 6.55–6.48 (m, 6H), 6.07 (d, J = 8.3 Hz, 1H), 5.40 (s, 1H, Ar-CH, pyrimidine H), 4.26 (s, 1H, benzylic-H), 3.56 (s, 3H), 2.16 (s, 6H,–N(CH3)2); 13C-NMR (100 MHz, DMSO-d6): δ 189.9, 185.9, 171.8, 170.7, 166.7, 162.8, 159.8, 154.3, 131.6, 131.4, 130.0, 124.5, 117.4, 116.0, 115.8, 111.1, 54.5, 40.1, 27.4; 13C DEPT-135 (100 MHz, DMSO-d6): δ 131.2, 129.8, 129.5, 127.6, 115.8, 115.5, 111.8, 111.6, 111.5, 110.8, 54.3.
2-(7-(4-Hydroxy-3-Methoxystyryl)-5-Phenyl-5H-Thiazolo[3,2-α]pyrimidin-3-yl)phenol (14)
Brown solid powder; yield: 89.6%; m.p.: 142–144 °C; Rf (30% EtOAc in n-hexane) = 0.71; 1H-NMR (400 MHz, DMSO-d6): 9.08 (s, 1H), 8.28 (s, 1H), 8.22 (s, 1H, Ar-CH, pyrimidine H-4), 7.71 (d, J = 7.4 Hz, 1H), 7.50 (dd, J = 7.6, 2.7 Hz, 1H), 7.11 (dd, J = 12.3, 6.9 Hz, 1H), 6.98 (d, J = 2.8 Hz, 1H), 6.96 (d, J = 2.8 Hz, 1H), 6.91 (d, J =8.4 Hz, 1H), 6.78 (d, J = 12.7 Hz, 1H, olefinic H), 6.48–6.37 (m, 6H), 6.04 (d, J = 8.3 Hz, 1H, olefinic H), 5.38 (s, 1H, Ar-CH, pyrimidine H), 4.22 (s, 1H, benzylic-H), 3.22 (s, 3H); 13C-NMR (100 MHz, DMSO-d6): δ 186.5, 175.8, 172.2, 170.1, 167.2, 163.2, 162.5, 160.1, 151.3, 131.6, 130.2, 129.1, 129.0, 127.3, 120.3, 118.1, 116.4, 121.8, 115.9, 54.6, 40.0; 13C DEPT-135 (100 MHz, DMSO-d6): δ 131.4, 129.9, 128.9, 128.8, 127.0, 116.1, 115.6, 54.4.
Synthesis of Michael addition products of N-Imidazole analogs (15 and 16)
A mixture of imidazole (1 mmol) and curcumin derivatives, 5–7 (1.2 mmol), were added to a 10-mL flask containing anhydrous K3PO4 (0.25 mmol) in CH3CN (10 mL), and the mixture was stirred at room temperature for some time and then subjected to the ultrasonic irradiation. The reaction mixture was cooled to room temperature when it had finished the reaction as determined by TLC. The precipitated product underwent filtering, acetone and water washing, and vacuum drying before being dried. The obtained corresponding products of Michael adducts (15 and 16) were recrystallized from ethanol (Supplementary Information in Appendix XIIIa–c and XIVa–c).
1-(4-Hydroxy-3-Methoxyphenyl)-5-(1H-Imidazol-1-yl)-5-(4-Nitrophenyl)pent-1-En-3-One (15)
Greenish solid powder; yield: 81.5%; m.p.: 177–179 °C; Rf (30% EtOAc in n-hexane) = 0.77; 1H-NMR (400 MHz, DMSO -d6): 8.53 (s, 1H), 8.22 (d, J = 13.8 Hz, 1H), 8.09 (d, J = 13.0 Hz, 1H), 7.96 (d, J = 6.8 Hz, 7H), 7.60 (d, J = 16.4 Hz, 4H), 7.56 (s, 1H, imidazole CH, -HC=CH-)), 7.51 (s, 1H, imidazole CH, -HC=CH-), 7.49 (s, 1H, -CH), 7.42 (d, J = 15.8 Hz, 1H), 6.86 (s, 1H, olefinic H), 6.41 (d, J = 14.8 Hz, 1H), 6.03 (m, 1H), 3.76 (dd, J = 21.1, 4.6 Hz, 1H), 3.73 (dd, J = 16.9, 8.8 Hz, 1H), 3.63 (s, 3H, OCH3); 13C-NMR (100 MHz, DMSO -d6): δ 195.5, 183.6, 147.6, 145.7, 140.8, 134.3, 132.1, 126.6, 124.4, 120.5, 119.1, 115.4, 112.3, 110.9, 63.4, 54.8, 44.9; 13C DEPT-135 (100 MHz, DMSO -d6): δ 150.2, 147.2, 146.6, 142.6, 128.8, 123.9, 110.6, 63.2, 55.5, 54.8.
5-(4-(Dimethylamino)phenyl)-1-(4-Hydroxy-3-Methoxyphenyl)-5-(1H-Imidazol-1-yl)pent-1-En-3-One (16)
Reddish solid powder; yield: 89.7%; m.p.: 184–186 °C; Rf (30% EtOAc in n-hexane) = 0.72; 1H-NMR (400 MHz, DMSO-d6): 8.55 (s, 1H), 8.50 (s, 1H, imidazole CH, -HC=CH-), 7.96 (s, 1H, -CH), 7.69 (d, J = 9.0 Hz, 1H), 7.66 −7.62 (m, 2H), 7.60–7.50 (m, 2H), 7.42 (d, J = 15.8 Hz, 1H), 7.11 (s, 1H, imidazole CH, -HC=CH-), 7.06–6.95 (m, 4H), 6.76 (dd, J = 16.1, 9.0 Hz, 3H), 6.53–6.34 (m, 1H), 3.77 (dd, J = 15.8, 3.6 Hz, 1H), 3.75 (dd, J = 14.2, 2.9 Hz, 1H), 3.72 (s, 3H), 3.04 (s, 6H); 13C-NMR (100 MHz, DMSO-d6): δ 189.9, 165.8, 159.8, 154.2, 151.5, 150.0, 145.3, 143.8, 141.0, 135.2, 130.1, 129.8, 124.5, 122.5, 121.7, 63.2, 55.2, 40.6, 27.5; 13C DEPT-135 (100 MHz, DMSO-d6): δ 145.1, 143.5, 142.1, 140.7, 134.9, 129.8, 129.6, 121.4, 120.8, 116.9, 116.7, 111.6, 111.4, 110.8, 110.1, 54.9.
Biological Activity Evaluations
In vitro Antibacterial Susceptibility Assay
The agar disc diffusion method was used to evaluate each bacterial strain’s antibacterial susceptibility to all synthetic compounds according to the Clinical and Laboratory Standards Institute’s standard protocols (CLSI).33 Each of these synthesized compounds was tested individually against the Gram-positive bacteria Staphylococcus aureus (ATCC 29213) and Streptococcus pyogenes (ATCC 27853), and Gram-negative bacteria Escherichia coli (ATCC 25922) and Pseudomonas aeruginosa (ATCC 27853) gained from the Adama Public Health Research & Referral Laboratory Center. According to Bergey’s Manual of Determinative Bacteriology, the colony morphology, Gram staining, and common biochemical assays were used to identify and authenticate the bacterial strains.34 The antibacterial activity was assessed by measuring the zone of inhibition against the test organisms and comparing it to amoxicillin as a reference drug at 250 and 500 µg/mL, respectively, while DMSO served as the negative control. The experiments were performed in triplicate, and the average clear zone (inhibition zone diameter) around each compound was measured with a caliper in mm.35
In vitro Antidiabetic Activity Using the α-Amylase Inhibitory Assay
The pancreatic porcine α-amylase inhibitory activity was used to assess the antidiabetic activity of test samples adapted with modifications.36 Briefly, the test compounds, enzyme, and soluble starch were dissolved in 20 mM sodium phosphate buffer containing 6 mM NaCl (pH 6.9). The synthesized compounds at different concentrations were dissolved in the buffer solution. To a test tube, 250 μL of pancreatic porcine α-amylase (1 U/mL, dissolved in the buffer (pH 6.9) and 100 μL of synthesized compounds at a concentration ranging from 1.25 to 10 µg/mL were added. Before adding 250 mL of 0.5% starch, the mixture was pre-incubated at 37 °C for 15 min. The mixture was then vortexed and incubated again at 37 °C for 15 min followed by the reaction termination using 1 mL of dinitrosalicylic acid (DNS) color reagent. The tubes were placed in a boiling water bath for 5 min, cooled to room temperature, and diluted. Two hundred microliters of the reaction mixture were taken into a 96-well clear plate, and the absorbance was read at 540 nm by Thermo Scientific TM GO Microplate Spectrophotometer. The control α-amylase at 1 U/mL without any inhibitor represented 100% enzyme activity. Appropriate synthesized compounds and control containing the reaction mixture except the enzyme were used to correct for the color interference. A known α-amylase inhibitor, acarbose was used for comparison studies. The percentage inhibition of the test sample on α-amylase was calculated as:
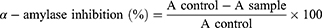
where A sample and A control represent the absorbance of the sample and control, respectively. By graphing inhibition percentages versus sample concentrations, the concentration of the drug that provides 50% inhibition (IC50) was determined. Each assay was performed in triplicate.
In vitro Antioxidant Activities
DPPH Free Radical Scavenging Test
The ability of the synthesized compounds to transfer hydrogen atoms or electrons was measured by bleaching the purple color of a methanolic solution of DPPH with minor adjustments. In vitro antioxidant efficacy screening of the synthesized compounds was determined via DPPH (2,2-diphenyl-1-picrylhydrazyl) radical scavenging assay.37 Various concentrations of synthesis analogs were prepared in methanol. DPPH (0.012%, 1 mL) in methanol was added to each concentration. The prepared solution was left to stand in dark at room temperature for 30 min. The absorbance was measured at λ= 517 nm for each solution against blank was recorded. Ascorbic acid was used as the standard solution and methanol was the blank reading solution, which was calculated by the following equation:
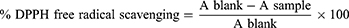
Where A sample and A blank represent the absorbance of the test sample and control reaction (containing all reagents except the test compound), respectively. Inhibitory concentrations were obtained using inhibition curves by correlating the percentage of DPPH inhibition with the concentration of each sample tested.38 Statistical analysis of the mean and standard deviation of each experiment was performed in triplicates.
Anti-Lipid Peroxidation Inhibition Assay
Lipid peroxidation is extremely harmful and occurs as a self-sustaining chain reaction. Reactive oxygen species are thought to cause cell damage through the production of free radicals and lipid peroxide radicals in cell membranes.39 With a few minor adjustments, the lipid peroxidation inhibition experiment was carried out as specified in method.40 The synthesized compounds were homogenized using the homogenizer, and the supernatant was used for the assay. The Thiobarbituric acid-Trichloroacetic acid (TBA-TCA) mixture was prepared by dissolving 0.392 g TBA in 75 mL of 0.25 M HCl, followed by the addition of 5 g TCA, and made up to 100 mL of 0.25 M HCl. In this assay, 500 mL of the TBA-TCA mixture was added to a mixture containing 250 mL of homogenate and 200 mL PBS buffer, in a clean test tube. These test tubes were covered with aluminum foil and boiled in a water bath at 100 °C for 30 min. The mixture was allowed to cool at room temperature, centrifuged at 2000 rpm for 10 min and the supernatant was transferred into a 96-microwell plate. Absorbance was read on a microplate reader at 540 nm. Ascorbic acid was used as a positive control. The percentage of lipid peroxidation inhibition was calculated from the following equation:
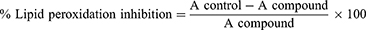
Where A compound and A control are the absorbances of the sample and control, respectively. The concentration needed to accomplish 50% inhibition of phospholipid oxidation (IC50) in linoleic acid was calculated by plotting the proportion of lipid peroxidation inhibition against the synthetic compound concentration.
In silico Computational Studies
Prediction of Drug-Likeness, Pharmacokinetics, and Pharmacodynamics (ADMET) Properties
Computer-assisted drug discovery technology minimizes the time and financial burden associated with the drug discovery process. In silico ADMET screening of drugs could avoid the tremendous cost and time associated with the in vivo experiments, and attracted more attention recently.41 Thus, pharmacodynamics (potency, affinity, efficacy, and selectivity), and pharmacokinetic (ADME: absorption, distribution, metabolism, excretion, and toxicity) data of molecules were evaluated and analyzed by that technology.42 This prediction is based on a concept that has been established by the Lipinski rule of five43 and Veber parameters.44 SwissADME (http://www.swissadme.ch/) and AdmetSAR (http://lmmd.ecust.edu.cn/admetsar1) online servers were used to assess the compounds’ ADME descriptors of pharmacokinetics and drug-likeness features. To estimate in silico pharmacokinetic parameters and other molecular characteristics, SwissADME tools were used to convert compound structures to their canonical simplified molecular-input line-entry system (SMILEs) based on the reported method.45 Pro Tox II web server (https://tox-new.charite.de/) was used to anticipate the organ toxicity profiles and toxicological endpoints of the ligands and toxicity class.
Molecular Docking Study
The most potent compounds’ modes of binding to target proteins were examined using molecular docking experiments. Using the ChemOffice tool (Chem Draw 16.0) and the appropriate 2D orientation, the chemical structures of the compounds were represented, and the energy of each molecule was minimized using ChemBio3D. The ligand molecules that had their energy minimized were then sent into AutoDock Vina to perform the docking simulation. The protein preparation was done using the reported standard protocol.46 The crystal structures were obtained from the most potent compounds in the active site of DNA gyrase B (PDB ID: 6F86), the N-terminal domain of PqsA (PDB ID:5OE3), Pyruvate Kinase (PDB ID:3T07), LuxS of S. pyogenes (PDB ID: 4XCH), Penicillin-binding proteins (PBPs) (PDB ID: 1VQQ), β-lactamases (PDB ID: 1IYS), human peroxiredoxin 5 (PDB ID: 1HD2), and α-amylase enzyme (PDB ID: 4W93) were downloaded from the protein data bank.47 The dimensions of the grid box were fitted to enclose the pocket site established in (PDB ID: 6F86) [62x30x64], [15x14x19], (PDB ID:5OE3) [38x-2x17], [19x21x14], (PDB ID:3T07) [−15x-3x2], [18x14x16], (PDB ID: 4XCH) [46x45x39], [14x12x17], (PDB ID: 1VQQ) [18x32x38], [16x18x17], (PDB ID: 1IYS) [−5x40x29], [21x22x14], (PDB ID:1HD2) [6x44x33], [15x18x17], (PDB ID: 4W93) [−11.95x10.64x-17.96] and [26.5x15.8x16.39] Å (x, y, and z). Meanwhile, possible interactions, ligand orientations, and image preparation were investigated using Discovery studio Visualizer 2021 (Biovia).48
Statistical Data Analysis
All experiments were performed in triplicate (n =3). The in vitro antidiabetic and antioxidant activities of synthesized compounds were expressed as 50% inhibitory concentration (IC50) values. All data were presented as the mean ± standard error of the mean (95% confidence intervals) of three independent experiments. Office Excel 2016 software (Microsoft, Redmond, WA, USA) was used for the statistical and graphical analysis. Statistical significance was assessed using a one-way analysis of variance ANOVA followed by a t-test to determine the degree of significance with the single normal and experimental control group. When the P-value <0.05, differences between data sets were judged statistically significant.
Result and Discussions
Chemistry
The chemical susceptibility of the β-diketone linker in the structure of curcumin to hydrolysis and metabolism has made it crucial to explore structurally modified analogs of curcumin without such shortcomings.49 Hence, extensive research has been done on synthetic modifications of the curcumin molecular framework, mostly targeted at enhancing its bioactivities.26,50 In this paper, twelve heterocyclic analogs derived from curcumin derivatives were synthesized under the ultrasonic-assisted method, and their antidiabetic, antibacterial, and antioxidant activities were evaluated. Thus, in this work, the synthesis of title compounds of heterocyclic curcumin analogs involves the reaction of monocarbonyl curcumin derivatives, 5–7 (1 mmol), in the presence of different nucleophilic reagents, namely; hydrazine hydrate (10 mmol), urea (1 mmol), 2-amino-4-substituted phenylthiazole (1.5 mmol), and imidazole (1 mmol) were dissolved in ethanolic sodium hydroxide resulted in the formation of the corresponding products of pyrazolines (8 and 9), 4-phenyl-6-styryl pyrimidine-2-ol (10 and 11), thiazolo[3,2-α]pyrimidines (12–14) and β-substituted imidazole compounds (15 and 16). The corresponding asymmetrical heterocyclic C5-curcumin analogs, 8–16 were synthesized regioselectivity at one side of curcumin, due to the effect of two electron donating groups of –OH and –OCH3 that stabilize the α,β-unsaturated group of curcumin, while the less stabilized part undergoes cyclization easily, and all the synthesized compounds were then purified and characterized by their 1H-NMR, and 13C-NMR spectroscopy techniques.
In terms of the environment, reaction time, high yield, simplicity of workup, and isolation of products, ultrasound-promoted technology is more effective.51 Additionally, ultrasonic irradiation has a number of benefits due to solvents being costly, hazardous, and challenging to remove especially when they are aprotic dipolar solvents with a high boiling point and harmful to the environment.52 The reaction time and yield data of newly synthesized compounds by ultrasonic methods are presented in Table 1. An efficient and greener synthesis of heterocyclic curcumin analogs via ultrasonic-aided approach is preferable due to increased yields, time savings, and achievement of environmentally friendly reactions, which makes it a useful and eco-friendly strategy compared to the conventional method. It was observed that the reactions which required 6–8 h by a conventional method were completed within 45–60 min by the ultrasonic irradiation method at 45–60 °C, and the yields of corresponding products have been improved in excellent yields (81.4 to 93.7%). It has also been observed that reactions under ultrasonic irradiation are generally easier to workup and have less energy consumption than conventional heating processes.26,53 This finding is due to the cavitation phenomenon, where high pressure and energy are generated within seconds, accelerating reaction rates compared to traditional methods, resulting in turbulence in the liquid and enhanced mass transfer in that region. This method has riddled the limitations such as high temperature, extended reaction time, expensive reagents, and hazardous reaction conditions. Additionally, reactions under ultrasound irradiation represent environmentally friendly processes, using small amounts of solvents and consuming less energy.54
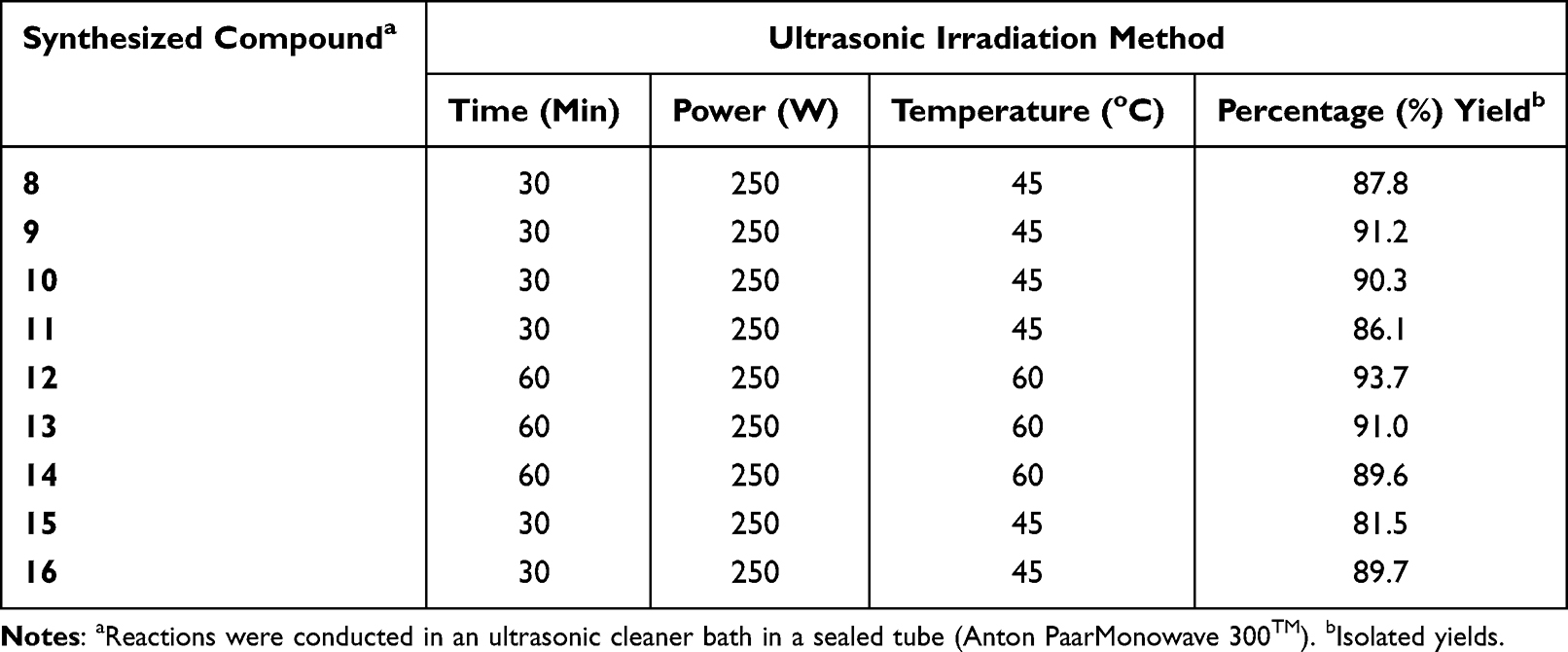 |
Table 1 Time and Yield Data of Synthesized Compounds (8–16) Using Ultrasonic Irradiation Techniques |
General Synthesis
General Procedure for the Synthesis of Curcumin and Its Derivatives (3 and 5–7)
Synthesis of curcumin and its derivatives were done at room temperature by combining substituted acetophenone and dehydrozingerone with various substituted aryl aldehydes using a base-catalyzed Claisen-Schmidt condensation reaction for symmetrical curcumin derivatives, respectively (Schemes 1 and 2). In the 1H–NMR spectra of compounds, 3 and 5–7 the H-β and H-α protons appeared each as a doublet at δ 7.11–7.71 and δ 7.33–7.40, respectively, with a coupling constant between them J=15.6 Hz, which is consistent with a trans configuration. The 1H-NMR spectrum of synthesized compounds displayed multiplet at δ 7.91–8.54, indicating the aromatic C-H linkage and the methoxy and hydroxyl groups appeared at δ 3.97 and 8.28 ranges, respectively. Different chemical shift positions consequently resonated with these protons. A variation in the substituent position of curcumin phenyl groups, which results in various chemical shift values, is the reason for this alteration in the scaffold’s structure. In the 13C-NMR- spectrum for the compounds the C-β and C-α signals appeared at δ 148.2 and δ 146.6 ranges, respectively, while the C=O appeared at δ 188.8–190.7. However, all the aromatic carbon peaks appeared in the expected regions (Supplementary Information in Appendix XV).
 |
Scheme 1 Synthesis of monocarbonyl curcumin compounds. Reagent and conditions: (a) NaOH (20%), ethanol, rt, stir for 1 h. |
 |
Scheme 2 Synthesis of dehydrozingerone-based monocarbonyl curcumin derivatives. Reagent and conditions: (a) NaOH (20%), rt, stir for 1 h; (b) substituted benzaldehyde, KOH (30%), ethanol, rt, stir for 2–3 h, overnight. |
General Procedure for the Synthesis of N-Pyrazoline Analogs Derived from Curcumin Derivatives (8 and 9)
Cyclization of curcumin precursors (5–7) with a 1.4-Michael addition of hydrazine derivatives in 10.0 mL of ethanol gives pyrazoline derivatives. Monitoring of reaction progress was done using TLC and the precipitate formed was filtered-off, washed with cold water, and dried. The solid product was recrystallized from ethanol. The synthesis of new monocarbonyl curcumin-based pyrazoline derivatives, 8 and 9 followed the general pathway outlined in Scheme 3. The 1H-NMR spectrum of the pyrazoline ring showed three different signals as a doublet of doublet attributed to diastereotopic protons (HA, HM, and HX) protons due to the AMX spin system in the structures. The HA, HM, and Hx protons appeared at 3.07–3.38 ppm (JAM=17.24 Hz), 3.77–3.98 ppm (JAX= 5.98 Hz), and 5.41–5.68 ppm (JMX= 11.90 Hz), suggesting vicinal coupling of methane with magnetically non-equivalents methylene protons, respectively. NH-protons of pyrazoline groups appeared as a singlet peak at 6.09 (s, NH, 1H). The α,β- unsaturated double bond appeared at δ 6.89–6.00, with a coupling constant between them of J= 15.6 Hz, which agrees with a trans-configuration. The 13C-NMR spectrum shows that the methylene carbon, sp3 methine, and sp2 quaternary carbons of the pyrazoline ring appeared at C3 (147.5–148.7), C4 (41.5–43.5) and C5 (57.1–63.5) ppm, respectively. In the 13C-NMR spectrum for the compounds, the C-β and C-δ signals appeared around δ 148.2 and δ 146.6 ranges, respectively. All the other protons and peaks of aromatics were observed in the expected regions (Supplementary Information in Appendix XVI).
 |
Scheme 3 Ultrasonic irradiation-assisted synthesis of heterocyclic curcumin analogs (8–16). Reagents and conditions: (a) hydrazine hydrate, EtOH; (b) NH2CONH2, 40% NaOH, EtOH; (c) Na metal, ethanol; (d) Imidazole, 25 mol% K3PO4, CH3CN,))), 45–60 °C, 250 W for 30–60 min. |
General Procedure for the Synthesis of 4-Phenyl-6-Styrylpyrimidine-2-Ol analogs Derived from Curcumin Derivatives (10 and 11)
The preparation of 4-phenyl-6-styrylpyrimidine-2-olderivatives involves starting from equimolar quantities of curcumin derivatives (5–7) by cyclization with the nucleophilic regent of urea separately, in NaOH, and 10.0 mL ethanol. TLC was used to monitor the reaction’s progress, and the precipitate that had developed was filtered out, rinsed in cold water, and dried. The solid product of 4-phenyl-6-styrylpyrimidine-2-ol (10 and 11) was recrystallized from ethanol (Scheme 3). The 1H-NMR spectrum displayed singlet at δ 3.79 and δ 5.04 revealing the presence of methoxy, and hydroxyl functionality of pyrimidin-2-ol, respectively. The methane proton of the pyrimidine ring appears at δ 5.35. The H-β and H-α protons appeared each as a doublet at δ 7.1–7.7 and 7.3–7.4, respectively, with a coupling constant between them of J= 15.6 Hz, which agrees with a trans-configuration. The 13C-NMR spectrum revealed that δ 88.16 (pyrimidine-CH), 165.02, 150.52, 157.21 (Pyrimidine-C 2/4/6), respectively. The peaks of C-β and C-α signals appeared at δ 148.2 and δ 146.6 ranges, respectively (Supplementary Information in Appendix XVI).
General Procedure for the Synthesis of Thiazolo[3,2-α] Pyrimidine Analogs Derived from Curcumin Derivatives(12–14)
The synthesis procedure used to prepare thiazolo[3,2-α] pyrimidine analogs derived from curcumin analogs is illustrated in Scheme 3. The target compounds were obtained by a 1.4-nucleophilic addition of 2-amino-4-substituted phenyl-thiazole (4-(2-aminothiazol-4-yl) phenol) with curcumin derivatives with sodium ethoxide as solvent. The 1H-NMR spectrum of title compounds (12–14) revealed the presence of singlet peak (1H, br) at δ 6.05 and (3H, s) at δ 3.90 of hydroxyl and methoxy protons, respectively. The spectrum shows a signal at δ 5.4–6.9 assigned for the C-H thiazolopyrimidine proton (pyrimidine H-4), δ 7.41 for methane proton, and the thiazole proton shows singlet around δ 6.90–8.52. The H-β and H-α protons appeared each one as a doublet at δ = 7.75–7.73 (1H, d, J= 8.16 Hz), and δ = 7.32–7.39 (1H, d, J=16.20 Hz), respectively. The 13C-NMR shows a characteristic peak at δ 167.0–168.0 corresponding to C-2 of the thiazole ring. The peak appeared around δ 195.7–168.0 corresponding to (-S-C(N)=N- of thiazolo-pyrimidine, the appearance of this downfield shift around δ 43.9 for the pyrimidine H-4 indicates the effect of the environment around carbon-4. The peaks of the C-β and C-α signals appeared at δ 148.2 and δ 146.6 ranges, respectively (Supplementary Information in Appendix XVI).
General Procedure for the Synthesis of the Aza-Michael Addition Product of β-Substituted Imidazole Analogs Derived from Curcumin Derivatives (15 and 16)
The curcumin derivatives and imidazole were reacted via an aza-Michael addition type reaction in the presence of anhydrous K3PO4 in CH3CN to produce β-substituted imidazole analogs (Scheme 3). 1H-NMR spectra of target compounds, (15 and 16) revealed the presence of singlet peak (1H, br) at δ 6.05 and (3H, s) at δ.3.90 of hydroxyl and methoxy protons and the spectrum shows δ 3.2 (dd, JAM = 4.6 Hz, JAX= 16.9 Hz; 1H), 3.6 (dd, JAM= 8.8 Hz, JAX= 16.9 Hz, 1H), 4.98–5.03 (m, 1H), due to the vicinal coupling with the non-equivalent protons of the methylene and methine group. The spectrum H-β and H-α protons appeared each as a doublet at δ = 7.75–7.73 (1H, d, J= 2.16 Hz), and δ = 7.32–7.39 (1H, d, J= 16.20 Hz), respectively, with coupling constant between them of J= 15.6 Hz, which agrees with a trans-configuration. The imidazole C2 and vinylic protons appear singlet at 7.96 (s, 1H, -CH), and doublet at 7.76–8.50 (1H, d, imidazole CH, J= 15.6 Hz, -HC=CH-), respectively. Besides, the 13C NMR chemical shift values of carbonyl carbon peak appear atδ 200.4, the methine carbon around δ 49.9, and methylene carbon at δ 40.3–47.3, while C-β and C-α signals appeared around δ 148.2 and δ 146.6 ranges, respectively (Supplementary Information in Appendix XVI).
Biological Activities
In vitro Antibacterial Activity
Using a disk diffusion assay, the synthesized compounds were tested for in vitro antibacterial activity against two species of Gram-positive and Gram-negative bacteria (Figure 1). The antibacterial activity of the nine series of compounds (8–16) was evaluated by measuring the zone of inhibition against the test organisms. Among these, compounds 8, and 11 exhibited the highest antibacterial activities against E. coli with a mean inhibition zone of 13.00± 0.57, and 19.66±00 mm diameter, respectively, at 500 μg/mL compared to the results of standard drug amoxicillin (IZD = 12.87±1.41 mm) at the same concentration, while compound 10 (11.00±0.57 mm) demonstrated comparable activities at the same concentration. In addition, Compounds 8, 11, and 16 showed excellent potency activities against P. aeruginosa with a mean inhibition zone of 17.67±0.57, 14.33±0.57, and 23.33±00 mm diameter, respectively, at 500 μg/mL compared to amoxicillin (IZD = 13.75±1.83 mm) at the same concentration. Moreover, Compound 12 showed the most potent activities against S. aureus with a mean inhibition zone of 11.33±0.57 mm diameter at 500 μg/mL, whereas compound 11 showed comparable activities with a mean inhibition zone of 17.00± 0.57mm at 250 μg/mL compared to amoxicillin (IZD = 10.75±1.83 and 9.37±1.50 mm, respectively) at the same concentration. On the other hand, compounds 9 and 11 displayed comparable activities against S. pyogenes with a mean inhibition zone of 11.33±0.57 mm and 15.67±0.57 mm at 500 μg/mL, respectively, compared to amoxicillin (IZD = 14.75±2.00 mm) at the same concentration. The remaining synthesized compounds exhibited moderate to weak antibacterial activities in the tested bacterial strains, while compound 11 (19.66±00, 14.33±0.57, 17.33±0.57 and 15.67±0.57 mm) exhibited excellent antibacterial potency against Penicillin-binding proteins (PBPs) and β-lactamases producing bacterial strains of E. coli, P. aeruginosa, S. aureus, and S. pyogenes at 500 μg/mL compared to the reference drug, amoxicillin (IZD = 12.875±1.41, 13.75±1.83, 10.75±1.83 and 14.75±2.00 mm, respectively), this is due to involvement of 4-phenyl-6-styrylpyrimidine-2-ol and curcumin moiety of –OH groups are responsible for the activation of the β-lactam ring, this provides an anionic site (hydrophobicity), which appears to be necessary for interacting with the target enzymes, and encourages the acylation of the PBPs’ active site serine (Supplementary Information Appendix XVII). Thus, these powerful antibacterial compounds might be thought of as viable lead compounds for the development of new antibacterial drugs.
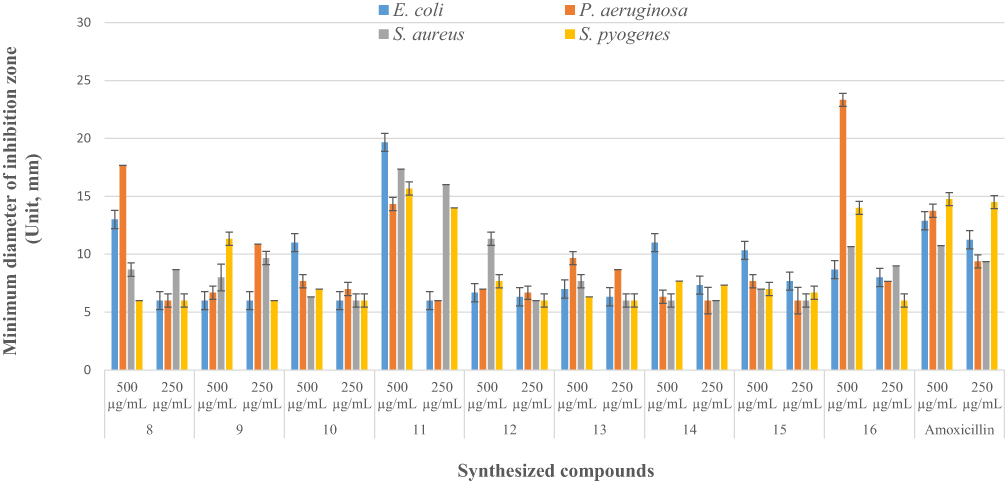 |
Figure 1 Diameter of inhibition zone of the synthesized compounds (8–16) in mm (mean ± SD) at 250 and 500 μg/mL concentrations. |
The design and discovery of new antibacterial drugs depend heavily on structure-activity-based mechanisms of antibacterial action. By comparing the obtained results (Table 2) with the compound structures, the following structural activity relationships (SARs) may be concluded: Compared to compounds without any substituted systems, compounds with phenyl rings that have electron-donating and electron-withdrawing substituents are more powerful. The presence of electron-withdrawing groups (p-NO2) at the para position of compounds 8, 11, and 16 on pyrazoline, 4-phenyl-6-styrylpyrimidine-2-ol and β-substituted imidazoles analogs derived from curcuminoid moieties may enhance (increase) the antibacterial activity against E. coli and P. aeruginosa. Moreover, the presence of electron donating groups –OH and –N(CH3)2 at the para position of compound 11 on 4-phenyl-6-styrylpyrimidine-2-ol analogs and curcumin moiety of –OH groups could increase the antibacterial activity against E. coli, P. aeruginosa, S. aureus, and S. pyogenes. Structural activity relationship revealed that the nature of the phenyl moiety of aromatic rings considerably influences the activity.
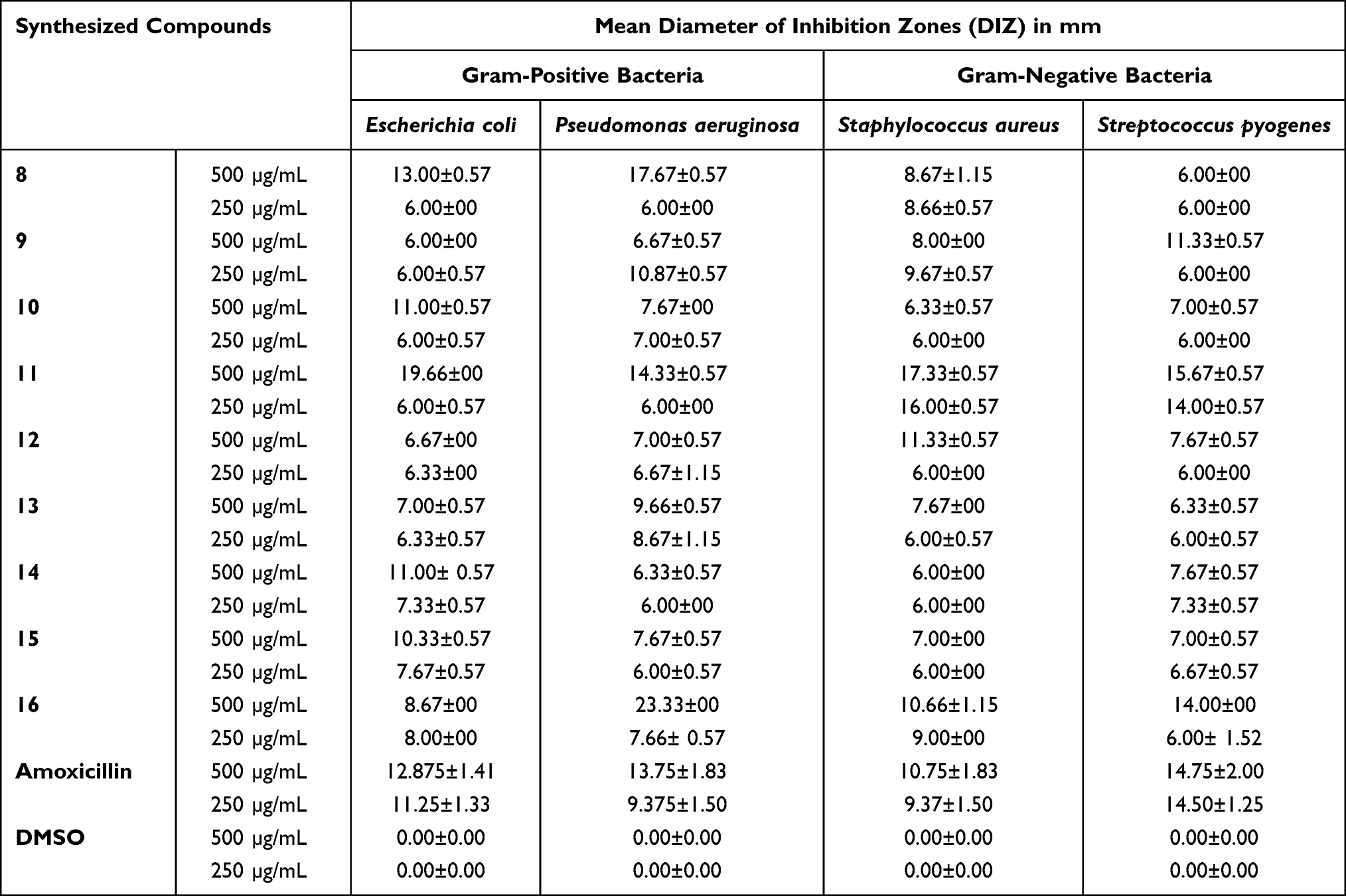 |
Table 2 Antibacterial Activity of Heterocyclics Curcuminoid Analogs (8–16) Against Selected Bacterial Strains |
In vitro α-Amylase Inhibitory Activity
Inhibition of the digestive enzyme (α-amylase) decreases the rate of glucose absorption by extending the digestion and absorption time of carbohydrates and is one of the therapeutic approaches for controlling the level of glucose in T2DM.36,55 The synthesized compounds were evaluated in vitro inhibitory potencies against α-amylase enzyme in a dose-dependent manner (1.25–10 μg/mL) and the results are displayed in Table 3. Based on the obtained results of % inhibition of alpha-amylase enzyme and IC50 values are illustrated in Figure 2, Interestingly, the tested heterocyclic curcuminoid analogs (8–16) showed excellent to potent α-amylase inhibitory activity with IC50 ranging from 4.08±0.23 to 102.65±0.23 μg/mL, in comparison to the standard drug, acarbose (IC50 =8.0 μg/mL). Among these, compounds 8, 11, and 16 showed the highest activity against α-amylase with IC50 = 22.03, 7.59, and 4.08 μg/mL, respectively, compared to acarbose (IC50 =8.0 μg/mL). These compounds inhibited the α-amylase activity of85.32±0.36%, 85.17±0.55%, and 87.15±0.46 at 10 µg/mL concentration, respectively. It was observed that compound 16 was found to be the most active inhibitor with an inhibitory effect up to 2-fold better than acarbose. This enhanced activity of compounds 8 and 16 was found to be most potent which might be due to the presence of electron-donating a group of dimethyl amino (–N(CH3)2) moiety on the para-position of the aryl ring of N-pyrazolines and β-substituted imidazole analogs, while compound 11 exhibited better potentials with unsubstituted phenyl ring of 4-phenyl-6-styrylpyrimidine-2-ol group that might be attributed to better binding with the active site of the α-amylase. As the concentration of tested compounds increases, the percentage of inhibition is also increased. The structural activity relationship was established for all compounds which depend upon the different substituents EWG or EDG on the phenyl ring of curcumin and its heterocyclic part. Based on the results, the constituent that is responsible for an α-amylase inhibitor is possibly the phenols at curcumin since their antioxidant properties can prevent damage from reactive oxygen species and also bind and inhibit an α-amylase.56 The introduction of strong electron-withdrawing group p-NO2 substitution (12, IC50=102.65±0.23 μg/mL) into the thiazolo[3,2-α] pyrimidines moiety can reduce the inhibitory activity. Thus, the most active compounds diminished the levels of enzymes, which are in charge of catalyzing the hydrolysis of complex carbohydrates, and improved the utilization rate of glucose by lowering the postprandial hyperglycemia.36
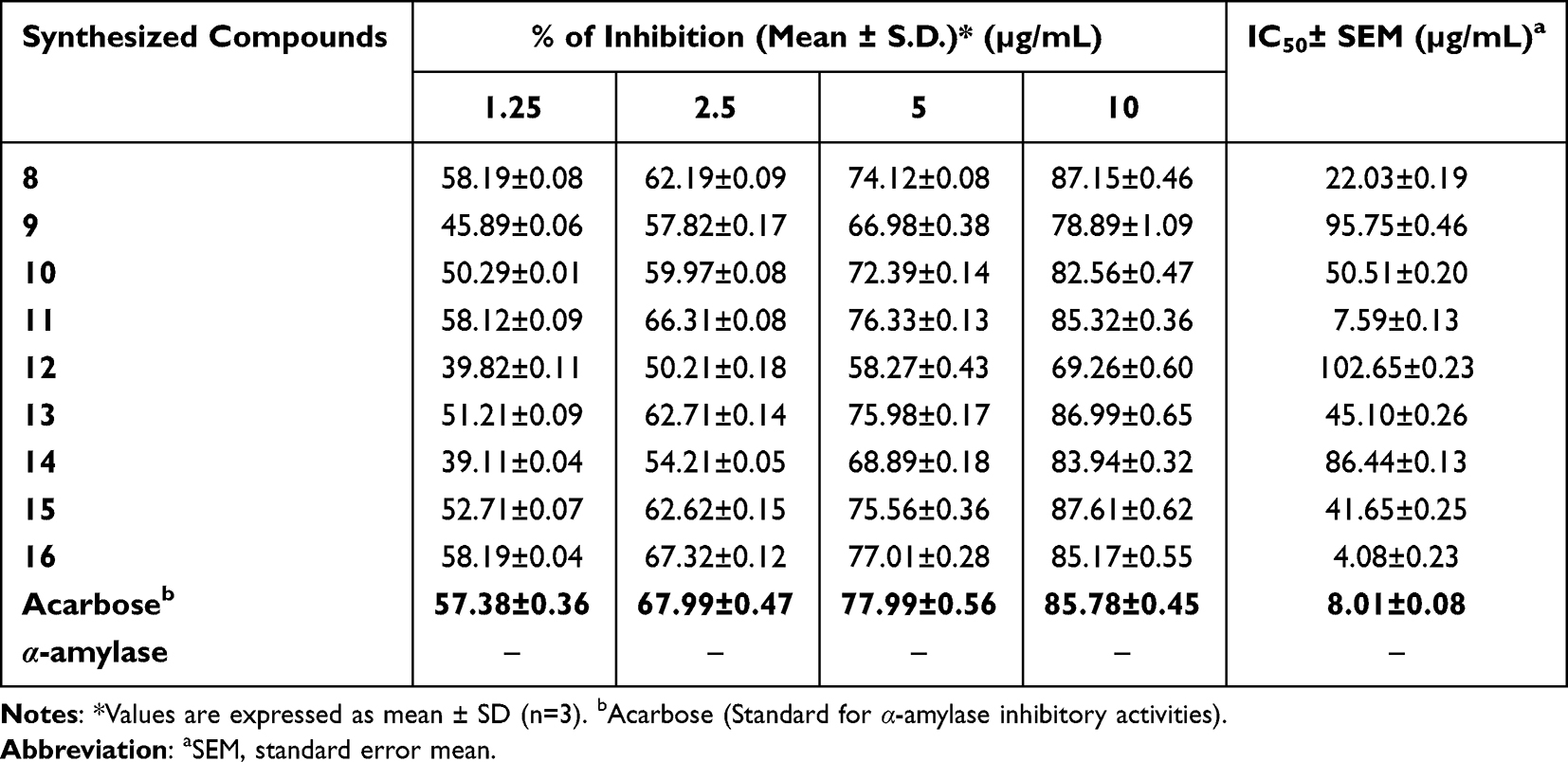 |
Table 3 % Inhibition of α-Amylase Enzyme and IC50 Values of Heterocyclics Curcuminoid Analogs (8–16) and Standard Acarbose at Different Concentrations |
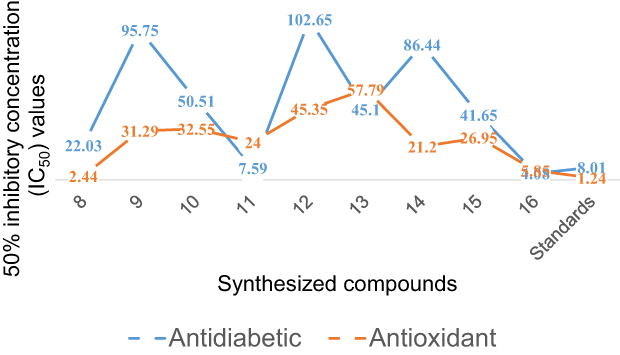 |
Figure 2 IC50 values of antidiabetic and antioxidant activities of synthesized compounds, 8–16 compared to that of standard drugs, acarbose, and ascorbic acid, respectively. |
In vitro Antioxidant Activities
DPPH Scavenging Activity
The synthesized compounds were tested as antioxidant agents using a 2.2-diphenyl-1-picrylhydrazyl (DPPH) method at λ = 517 nm. The purple color of DPPH fades or disappears when it reacts with a hydrogen donor, resulting in its conversion to 2.2-diphenyl-1-picrylhydrazine and a lowering in absorbance.57 In the presence of scavengers in the fractions, the lower the absorption, the more effective the antioxidant activity is. Based on the experimental results, the synthesized compounds (8–16) series exhibit good in vitro DPPH radical scavenging abilities, and the results are presented in Table 4 and Supplementary Information in Appendix XVIII. The results of antioxidant by DPPH assay revealed that the percentage of inhibition was increased with the increase in concentration test samples. At a concentration of 200 µg/mL, the majority of the compounds showed greater than 80–94% DPPH-free radical scavenging activity. Of the tested compounds, the IC50 of antioxidant activity data of compound 8 showed the highest percent inhibition of the DPPH with an IC50 value of 2.44μg/mL compared with standard ascorbic acid drug (IC50 = 1.24 μg/mL), indicating that heterocyclics derived curcumin, 8 exhibited best antioxidant activity compared to ascorbic acid. In other words, these compounds have the ability of electron donors to scavenge free radicals. This could be due to the presence of hydroxyl groups at the para positions (p-OH) of curcumin phenyl rings or pyrazoline moiety itself that stabilize the radicals for their strong electron-donating properties, respectively. The antioxidant capacity of all the tested compounds is explained by the redox potential of phenolic compounds, which allows them to function as reducing agents, hydrogen donors, and singlet oxygen scavengers. Comparison between the inhibition percentages of the tested compounds at concentration ranges of 12.5 to 200 µg/Ml (Figure 3). Comparison between the values of % RSA, as well as the IC50 of the synthesized compounds, compared to ascorbic acid is illustrated in Figure 2.
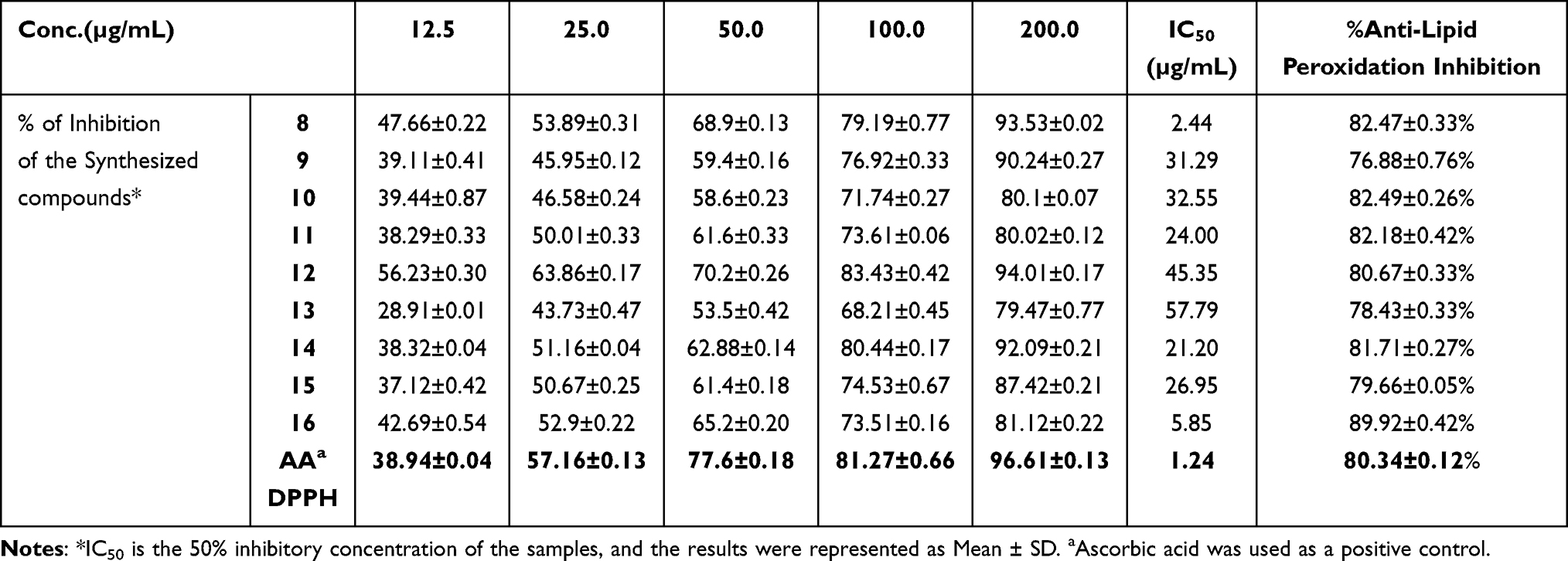 |
Table 4 In vitro Antioxidant and Anti-Lipid Peroxidation Inhibition Test with Various Concentrations of 8–16 |
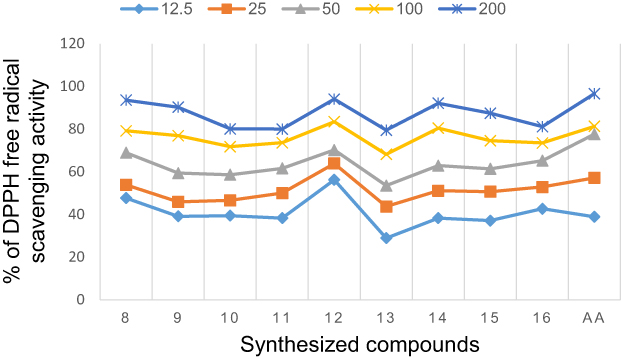 |
Figure 3 Percentage inhibition of the synthesized compounds (8–16) and the standard drug at the different concentration range. |
In vitro Anti-Lipid Peroxidation
Oxidative degradation of polyunsaturated fatty acids in membrane phospholipids is known as lipid peroxidation.58 The lipid peroxide removal effects of heterocyclic curcumin analogs reflect their antioxidant activities of them. The synthesized compounds (8–16) were also investigated for their anti-lipid peroxidation potential with the result summarized in Table 4. According to data on lipid peroxidation, compound 16 inhibits the production of peroxides by 89.92±0.42%, indicating that it can prevent the development of lipid peroxides. However, compared to ascorbic acid, a naturally occurring antioxidant, the synthesized compounds showed only a modest ability to suppress peroxide generation. The results showed that the investigated heterocyclic curcumin analogs eliminate free radicals, particularly lipid peroxide radicals, by adding a hydrogen atom to them, which inhibits the lipid peroxidation reaction. The activity values related to the antioxidant effects of the title compounds on % lipid peroxidation (Figure 4).
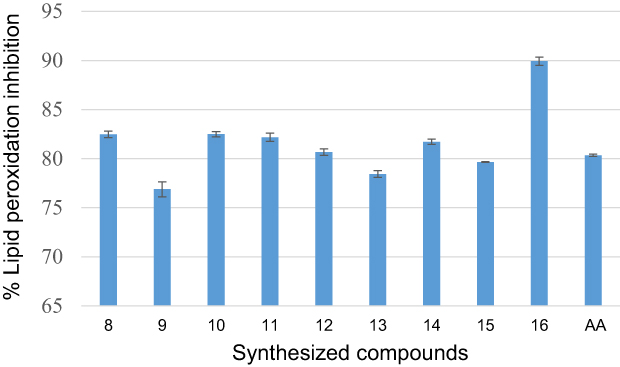 |
Figure 4 Percentage lipid peroxidation inhibitory effect in µg/mL of synthesized compounds (8–16) and ascorbic acid. |
In silico Computational Studies
Prediction of Drug-Likeness and ADMET Profiles
Prediction of the physicochemical characters, pharmacokinetics, and toxicity is an important tool in the drug discovery of biologically active agents.59 Many potential therapeutic candidates fail to reach the clinic due to their unfavorable absorption, distribution, metabolism, elimination, and toxicity (ADMET) factors.60 In this study, drug-likeness, pharmacokinetic, and toxicity parameters of synthesized compounds (8–16) for prediction of ADMET descriptors by using SwissADME/ AdmetSAR and Pro Tox II44 online servers. In the filter that combines the drug-likeness criteria of Lipinski’s “rule of five” (MW ≤ 500, ilog P ≤5, HBD ≤ 5, and HBA ≤ 10)61 and Veber’s parameters (TPSA ≤ 140Å2 and rotatable bonds ≤10).43 The percent absorption of the synthesized compounds was calculated using the formula %Abs = 109–0.345TPSA.62 Drug candidates showing violation of more than one of these rules will lead to difficulty in oral absorption.63 Using the AdmetSAR/Pro Tox II Web server tools, the synthesized compounds’ organ toxicity profiles, ligand toxicological endpoints, and LD50 were predicted.64
Oral bioavailability is accounted as a crucial characteristic for the discovery of efficacious medicinal drugs. The obtained results as illustrated in Table 5 and Supplementary Information Appendix XIX showed all compounds obey Lipinski’s rule of five, confirming their easy binding to receptors. Molecular weights of all the synthesized analogs were found to be ˂500 for most of the derivatives suggesting easy absorption, diffusion, and transportation. Molecular aquaphobic (iLog P) values of all synthesized compounds were found to be ˂5, demonstrating good membrane permeability of the compounds. The number of HBD (nOHNH) and the number of HBA (nON) values in all the synthesized analogs were determined to be ˂5, which is in the range of 1–2, and ˂10, which is in the range of 3–7, respectively. Topological polar surface area (TPSA) is a very useful parameter for the prediction of the transport of drug molecules. All the tested compounds had TPSA values <140 Å, in the range of 57.09–121.29 Å, with only one violation of 12, suggesting promising oral availability and were thus, they are expected to have good solubility, capacity for penetrating cell membranes, and intestinal absorption. It can be noticed that all the titled compounds displayed %Abs ranging from 58.16% to 81.43% indicating significant oral bioavailability and the numbers of rotational bonds are less than 10, confirming their good flexibility. They exhibited good lipophilicity (a major factor in the quality of drug candidate) given by the values of consensus Log Po/w in the range of 2.04–4.23 with better bioavailability score (0.55) meaning acceptable drug-likeness criteria. The greater lipophilicity of these compounds 8, 9, 11, 12, 14, and 16 may protect against ROS damage and is due in part to their smaller polar surface area which is another useful descriptor of the oral bioavailability and drug transport properties. The value of milogP (octanol-water partition coefficient) of all compounds is less than 5.0 (2.04–4.23) showing their high affinity and excellent permeability across the lipid bilayers of biological membranes. Results revealed that compounds, 8, 9, and 12–14 have the least lipophilicity (mlogP) and are biologically more active, but the remaining compounds (greater lipophilicity) are less active against the tested bacteria. Our results exhibited that there is a clear relationship between lipophilicity and antibacterial activity.65 For examined compounds, the MR values vary from 93.38 to 149.63, indicating the demand for preclinical development and the likelihood that they will become drugs. In light of the above results, we achieved that all tested compounds satisfy the “rule of five” and meet all criteria for good permeability and bioavailability.
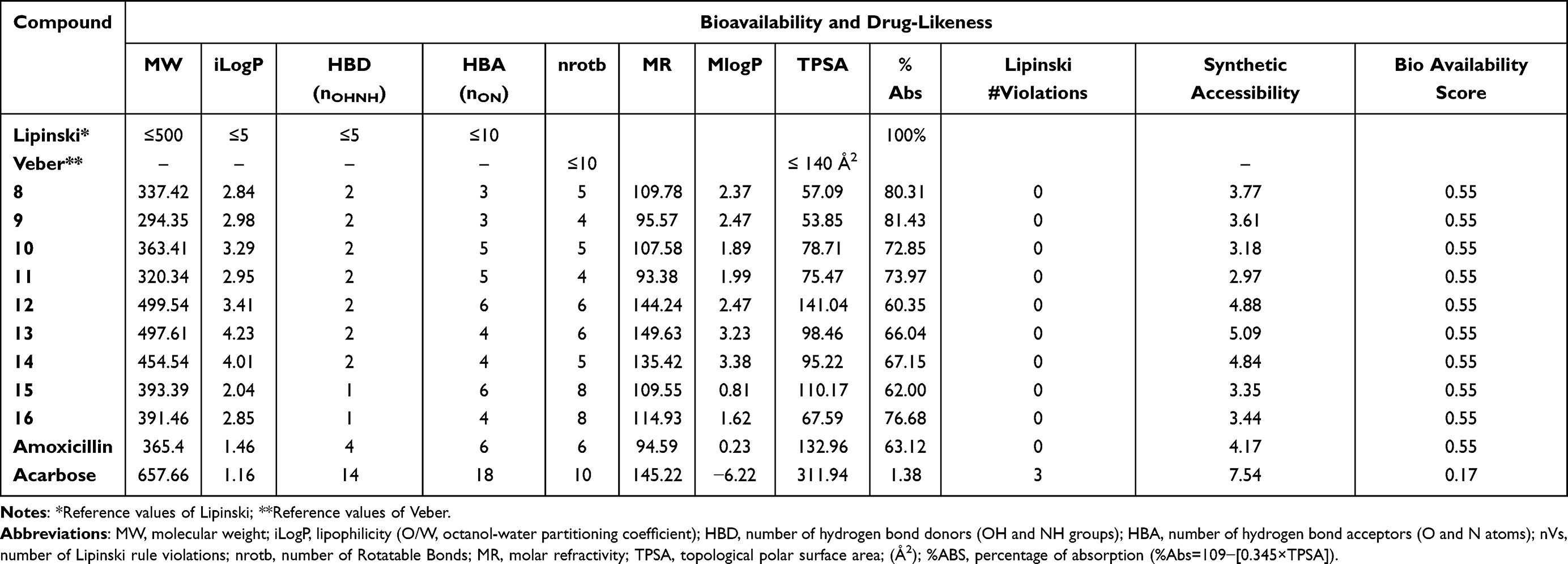 |
Table 5 Bioavailability, and Drug-Likeness Assessment of Synthesized Compounds, 8–16 Using SwissADME Online Server |
AdmetSAR online database was utilized to predict the absorption, distribution, metabolism, excretion, and toxicity (ADMET) of the target compounds as illustrated in Table 6. In silico pharmacokinetic characteristics, results revealed that compounds most active compounds had superior intestinal absorption to amoxicillin and acarbose with HIA and PPB values near (1) 100%, indicating that the synthesized compounds are showing good absorption and brain penetration properties to cross the gut wall by passive diffusion mechanism to reach the target but are impermeable to Caco-2. All of the substances have a high PPB%, with values of more than 75%, indicating prolonged half-lives more than amoxicillin and acarbose (0.348 and 0.302, respectively). The results revealed that compounds 8 (0.888), 11 (0.946), and 16 (0.757) had high PPB values, indicating longer half-lives than amoxicillin and acarbose.66 Skin permeability (SP, LogKp) was found to be somewhat below the permitted range of −5.25 to −6.69 cm/sec and a substance with a low negative log Kp value has higher absorption into human skin. Furthermore, glycoprotein (P-gp) inhibition measurements are used to predict the excretion characteristics of the tested compounds. P-gp and cytochrome P450 (CYP) are known to help biological membranes, such as those in the gastrointestinal tract or the brain, defend themselves against xenobiotics by biotransforming them.67 The actions of cytochrome P450 and its isoform enzymes were used to predict the metabolic characteristics of the compounds. In this investigation, almost all compounds showed inhibitory activity against CYP3A4, CYP2C9, and CYP2C19, but they did not show inhibitory behavior toward CYP2D6. In our study, none of the synthesized compounds inhibited P-gp substrates, except 8, and 9. Computationally toxicity risks such as AMES mutagenicity, hepatotoxicity, carcinogenicity, and skin sensitization were investigated. In silico pharmacodynamics studies indicated that compounds 8, 11, and 16 were predicted to be non-mutagenic, non-tumorigenic, non-hepatotoxic, non-irritant, AMES nontoxic and without any skin-sensitive effects, which gives them the property to be acceptable lead-like potential for future development of safe and efficient drugs. For further investigation of the in vivo antibacterial and antidiabetic activity, the computed LD50 in rats from the safer category III acute oral toxicity model seems to be sufficiently safe (2.29–2.41 mol/kg) as shown in Table 7.
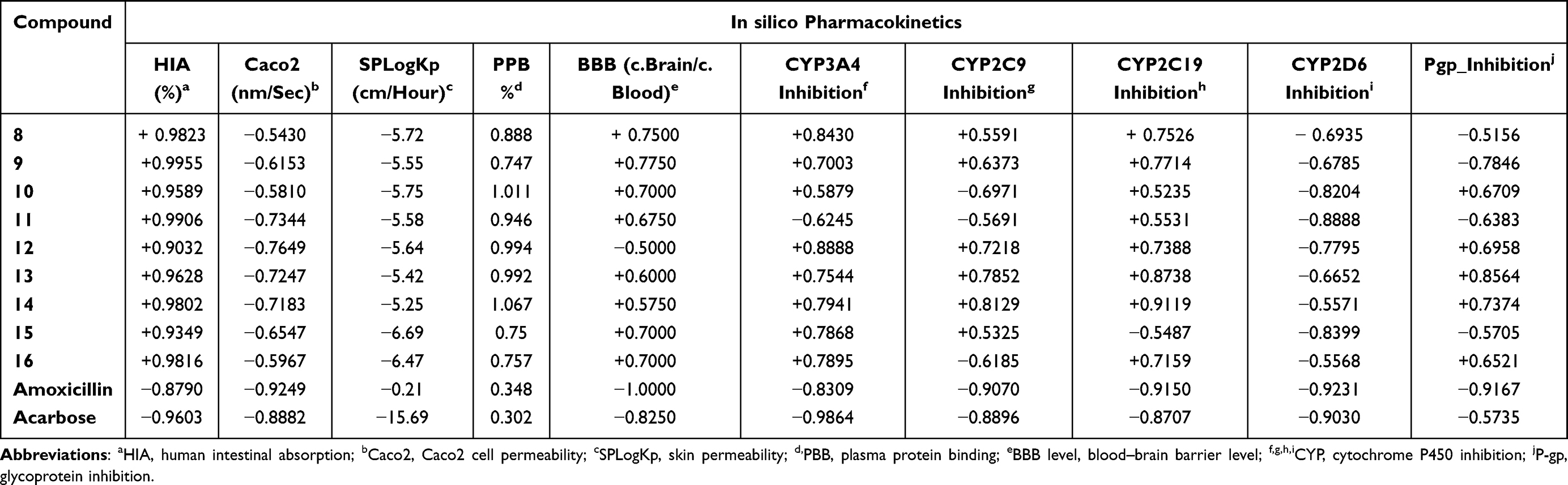 |
Table 6 Pharmacokinetic (ADME) Properties Prediction of Synthesized Compounds (8–16) Using AdmetSAR Online Platform |
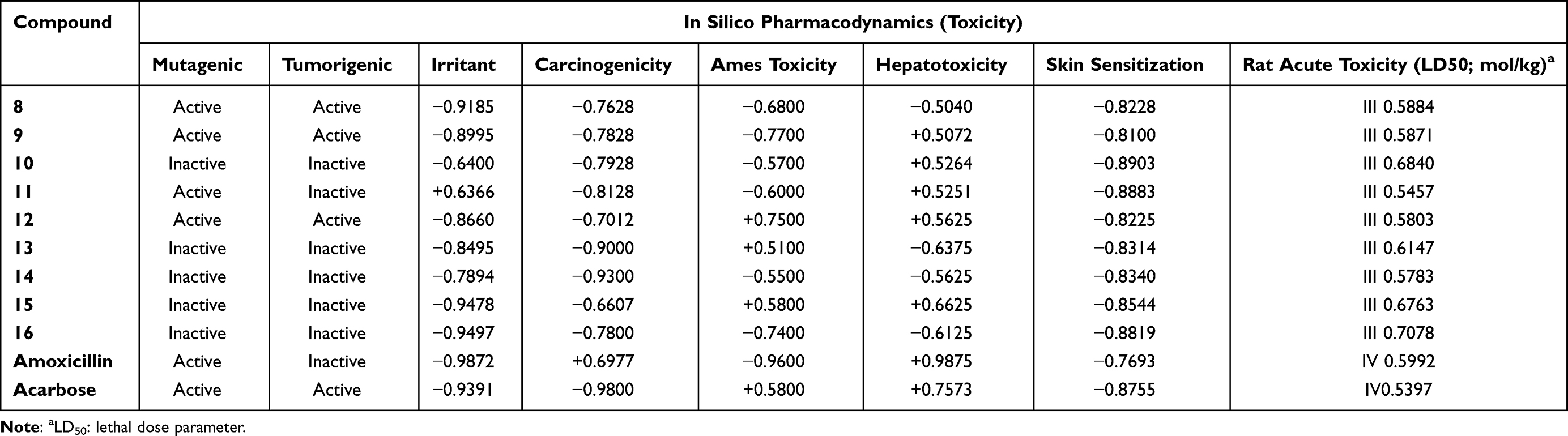 |
Table 7 In silico Toxicity Prediction of Compounds (8–16) and Amoxicillin Using AdmetSAR and Pro Tox-II Online Servers |
The bioavailability radar plot indicates that 8, 11, and 16 fall entirely in the pink area confirming their potent drug-likeness and their better bioavailability profiles.68 The compound does not violate any of Lipinski and Veber’s parameters, even though it violates type Fsp3 0.25, unsaturation score (INSATU), and it satisfies the requirements within the ideal spectrum that combines drug-likeness criteria. The BOILED-Egg server forecasts these pharmacokinetics using wLogP and TPSA.69 Compounds 8, 9, 11, and 15 appeared in the white ellipse (HIA), justifying their high likelihood of being passively absorbed by the gastro-intestinal tract and bioavailability, whereas compounds 14, 12–14, and 16 appeared in the yellow ellipse (BBB), justifying their high likelihood of passing through the blood–brain barrier and demonstrating their ability to cross the blood–brain barrier. All the above in silico ADME parameters of the most active compounds (8, 11, and 18) may help in the further development of new drug candidates with good and acceptable oral bioavailability.
Molecular Docking Simulation Study
Over the years, computational simulation approaches are gaining popularity due to the speed and accuracy of docking methods having improved, and these methods now play a significant role in structure-based drug design.70 The docking scores, binding energies, and interaction with amino acids of the prepared hits are presented in Supplementary Information in Appendix XX–XXVII and (2D and 3D), the results of which are shown in Supplementary Information in Appendix XXVIIIa–XXXVd. The interactions of ligands with H-bonds, hydrophobic molecules, π-cations/anion, and alkyl groups, as well as van der Waals interactions, were compiled.
Molecular Docking Studies of Synthesized Compounds Against E. coli DNA Gyrase B
DNA gyrase B is a member of bacterial type IIA topoisomerase enzymes that control the topology of DNA during processes of transcription, replication, and recombination by introducing transient breaks (negative supercoils) to both DNA strands, which provide the energy necessary for the catalytic function of the enzyme through ATP hydrolysis. Inhibiting the ATPase activity of DNA Gyrase B blocks the introduction of negative supercoils in DNA and traps the chromosome in a positive impact on cell physiology and division.71 It was found that the synthesized compounds (8–16) have minimum binding energy ranging from −6.4 to −7.9 kcal/mol, signifying the inhibitory effect of all compounds against E. coli DNA gyrase B, as shown in Supplementary Information in Appendix XX and Appendix XXVIIIa–j, respectively. The docking results (high negative binding affinity) agreed with the in vitro biological activity effects of 11(−7.9 kcal/mol) against E. coli as compared to standard amoxicillin (−6.1 kcal/mol as shown in Figure 5). Gly-77 and Arg-76 were involved in hydrogen bonding with –OH of styrylpyrimidine-2-ol group. Hydrophobic/π-cation interactions were observed for Thr-165, Ala-47, Ile-94, Pro-79, Ile-78, and Asn-46 with methoxy and pyrimidine rings, respectively. On the other hand, Asp-73, Gly-75, and Glu-50 exhibited Van der Waals residual interactions, respectively.
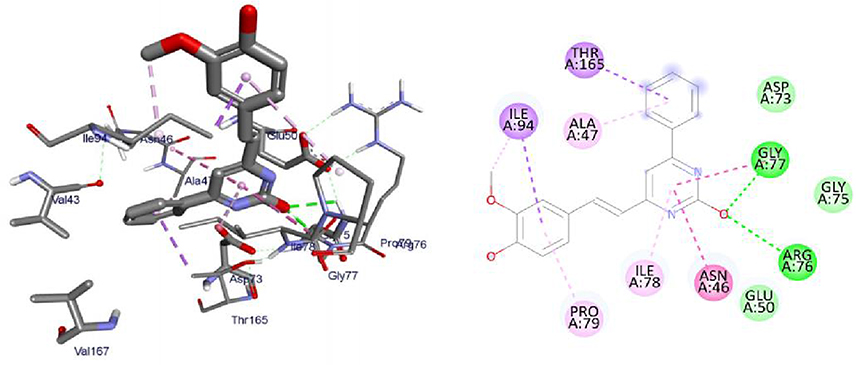 |
Figure 5 3D (right) and 2D (left) representations of the binding interactions of 11 against E. coli DNA Gyrase B (PDB ID: 6F86). |
Molecular Docking Studies of Synthesized Compounds Against PqsA
PqsA protein is responsible for priming anthranilate for entry in to the 2-alkyl-4(1H)-quinolones (Pseudomonas quinolone signal, PQS) biosynthesis pathway, and this enzyme may serve as a useful in vitro indicator for potential agents to disrupt quinolone signaling in P. aeruginosa. Inhibition of the PQS quorum sensing system is an attractive target for the development of alternative therapies against multidrug-resistant P. aeruginosa. Anthraniloyl-AMP mimics are competitive antagonists of the enzyme PqsA, and block the production (secretion) of virulence factors PQS, and the biofilm formation ability of P. aeruginosa is significantly reduced without affecting bacterial growth.72 It was found that the synthesized compounds (8–16) have minimum binding energy ranging from −4.6 to −9.1 kcal/mol in the binding pocket of the N-terminal domain of PqsA, with the best result achieved for compound 16 (−9.1 kcal/mol) as compared to amoxicillin (−7.9 kcal/mol), indicating that the compounds are promising antibacterial agents against P. aeruginosa, as depicted in Supplementary Information in Appendix XXI and Appendix XXIXa–j. Thus, the results of molecular docking are in good relationship with the results of compound 16 in vitro biological activity against P. aeruginosa (Figure 6). Val-309 was involved in hydrogen bonding with -OH of curcumin moiety. Hydrophobic/π-cation interactions were observed for His-308, Gly-302, Asp-382 (at imidazole group), Ala-303, Ile-301, Gly-279, Asp-299, Ala-278, Gly-300, His-394, and Phe-209 with nitrogen atom of –N(CH3)2 group, while Thr-304, Thr-164, Arg-397, Thr-380, Ser-280, Asp-299, Ala-278, Gly-300, His-394, and Phe-209 showed Van der Waals residual interactions, respectively.
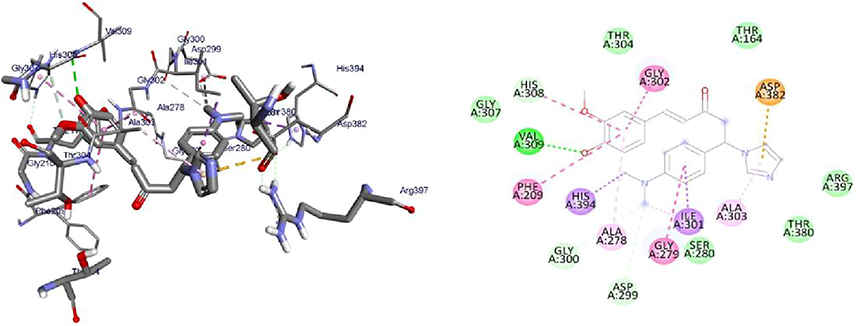 |
Figure 6 3D (right) and 2D (left) representations of the binding interactions of 16 against PqsA (PDB ID: 5OE3). |
Molecular Docking Studies of Synthesized Compounds Against S. aureus Pyruvate Kinase
Pyruvate Kinase (PK) enzyme catalyzes the rate-limiting, final step in glycolysis (carbohydrate metabolism) involving the irreversible conversion of phosphoenolpyruvate (PEP) into pyruvate with the subsequent phosphorylation of ADP into ATP. Both the products and substrates of PK are involved in a number of additional biological pathways, therefore providing a critical intervention point to disrupt whole-cell bacterial metabolism. Its inhibition would lead to reduced ATP levels, causing bacterial metabolism to be severely disrupted, leading to a lack of bacterial growth.73 It was found that the synthesized compounds (8–16) have minimum binding energy ranging from −5.1 to −5.8 kcal/mol in the pyruvate kinase binding pocket, while the best result achieved with compound 12 (−5.8 kcal/mol) indicates that the compounds are promising antibacterial agents against S. aureus, as presented in Supplementary Information in Appendix XXII and Appendix XXXa–j. The docking results are consistent with the results of 12 in vitro bioactivity against S. aureus (Figure 7). Compound 12 (Asn-465, Val-456, Gly-462, Thr-463) was involved in hydrogen bonding with –OH and methoxy groups of curcumin and –OH thiazole moiety, respectively. Hydrophobic/π-cation interactions were observed for Val-456, Leu-355, Ala-358, Met-467, Ile-359 and Ile-359, Ala-358, Pro-457, Leu-355 with nitrogen atom of –N(CH3)2 and pyrimidine-2-ol nitrogen atom, whereas Thr-461, Asn-465, and Ser-362 displayed Van der Waals residual interactions, respectively.
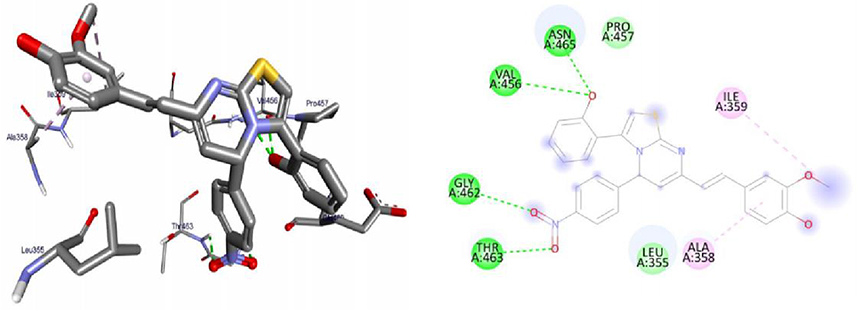 |
Figure 7 3D (right) and 2D (left) representations of the binding interactions of 12 against S. aureus Pyruvate Kinase (PDB ID: 3T07). |
Molecular Docking Studies of Synthesized Compounds Against LuxS of S. Pyogenes
LuxS protein is required for the biosynthesis of the type 2 autoinducer, AI-2, which is involved in quorum sensing in a wide range of bacterial species including S. pyogenes. Bacteria sense the density of the surrounding population via AI-2 molecules, and this regulates gene expression and biofilm formation.74 The synthesized compounds, 8–16 were found to have minimum binding energies of −1.6 to −5.6 kcal/mol within the binding pocket of the LuxS protein, as shown in Supplementary Information in Appendix XXIII and Appendix XXXIa–j. The best results were attained with compound 11 (−5.6 kcal/mol) suggesting that these compounds are promising antibacterial agents against S. pyogenes (Figure 8). The results of in silico docking are in good correlation with the results of bioactivity against S. pyogenes. Hydrophobic/π-cation interactions were observed for Ser-126, His-61, Pro-79, Leu-56, and His-57 with -OH and methoxy groups of curcumin moiety, while, Gly-128, Asn-129, and Thr-53 showed Van der Waals residual interactions, respectively.
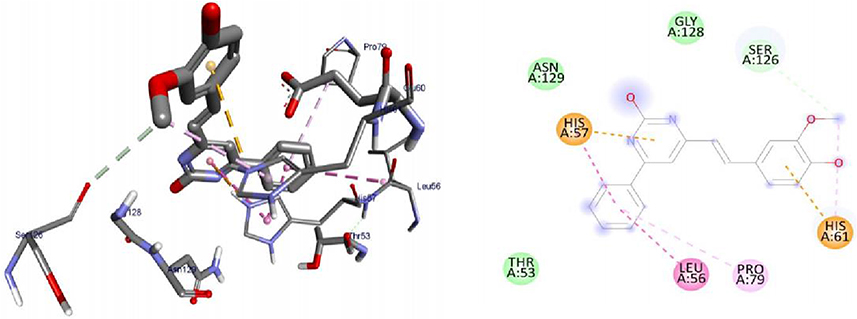 |
Figure 8 3D (right) and 2D (left) representations of the binding interactions of 11 against LuxS of S. pyogenes (PDB ID: 4XCH). |
Molecular Docking Studies of Synthesized Compounds Against Penicillin Binding Proteins (PBPs)
Penicillin-binding proteins (PBPs) remain attractive targets for developing new antibiotic agents due to their involvement in the end stages of the synthesis of bacterial cell wall biosynthesis of peptidoglycan.75 The peptidoglycan layer is important for cell wall structural integrity, especially in Gram-positive and negative organisms. The inhibition of PBPs leads to irregularity in the form of bacterial cell wall structure such as elongation, lesions, the loss of permeability, and cell lysis. The β-lactam antibiotics inhibit both transpeptidase and DD-carboxypeptidase activities by acylating the active-site serine of PBPs.76,77 The synthesized compounds, 8, 11, and 16 were found to have minimum binding energies of −6.7 to −7.5 kcal/mol within the binding pocket of the penicillin-binding proteins (PBPs), as shown in Supplementary Information in Appendix XXIV and Appendix XXXIIa–d. The highest results were attained with compound 11 (−7.5 kcal/mol) compared to reference drug amoxicillin (−7.2 kcal/mol), suggesting that these compounds are promising antibacterial agents against E. coli, P. aeruginosa, S. aureus, and S. pyogenes (Figure 9). The results of in silico docking are in good correlation with the results of bioactivity against penicillin binding proteins (PBPs) producing bacterial strains. Compound 11 was involved in hydrogen bonding of Ser-240 with –OH groups of curcumin. Hydrophobic/π-cation interactions were observed for His-293, Val-277, Asp-295, Ala-276, and Lys-273 aromatic rings of curcumin moiety, while Met-372, Glu-239, Ser-149, Lys-148, and Glu-294 showed Van der Waals residual interactions, respectively.
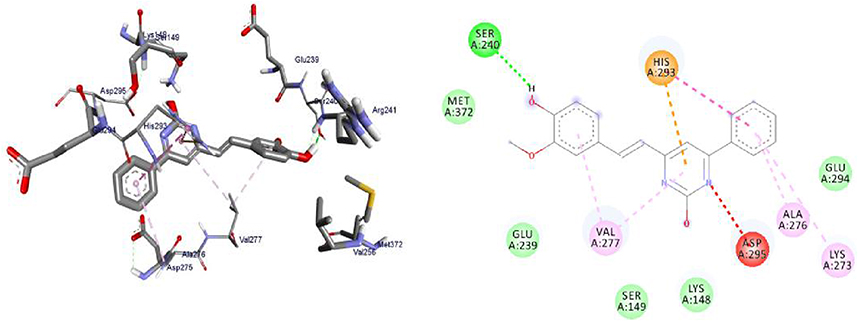 |
Figure 9 3D (right) and 2D (left) representations of the binding interactions of 11 against Penicillin binding proteins (PBPs) (PDB ID: 1VQQ). |
Molecular Docking Studies of Synthesized Compounds Against β-Lactamases
The production of β-lactamases is the major mechanism of bacterial resistance to β-lactam antibiotics (penicillins, cephalosporins, and carbapenems) and has been used effectively over several decades against different types of bacterial infections. This irreversible inhibition of the PBPs prevents the final cross-linking (transpeptidation) of the nascent peptidoglycan layer, disrupting cell wall synthesis. In the presence of these antibiotics, the PBPs form a lethal covalent penicilloyl-enzyme complex that blocks the normal transpeptidation reaction; this finally results in bacterial death. These enzymes hydrolyze the amide bond of the four-membered β-lactam ring and hence inactivate the antibiotics before they reach their target.78,79 The synthesized compounds, 8, 11, and 16 were found to have minimum binding energies of −6.9 to −7.7 kcal/mol within the binding pocket of the β-lactamases protein, as shown in Supplementary Information in Appendix XXV and Appendix XXXIIIa–d. The best highest results were obtained with compound 11 (−8.3 kcal/mol) compared to reference drug amoxicillin (−7.9 kcal/mol), demonstrating that these compounds are promising antibacterial agents against E. coli, P. aeruginosa, S. aureus, and S. pyogenes (Figure 10). The results of in silico docking are in good correlation with the results of bioactivity against β-lactamases producing bacterial strains. Compound 11 was involved in hydrogen bonding with Ser-70 at pyrimidine-2-ol of –OH. Hydrophobic/π-cation interactions were observed for Tyr-105, Asp-240, and Pro-167 with aromatic groups of curcumin moiety, while Asn-132, Ser-237, Thy-168, Thr-171, Asn-104, Asn-170, Glu-166, and Ser-130 showed Van der Waal's residual interactions, respectively.
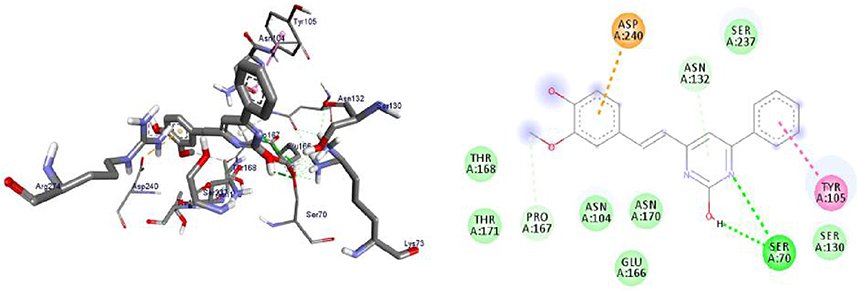 |
Figure 10 3D (right) and 2D (left) representations of the binding interactions of 11 against β-lactamases (PDB ID: 1IYS). |
Molecular Docking Studies of Synthesized Compounds Against Human Peroxiredoxin 5
Peroxiredoxin 5 (PRDX5) is a novel thioredoxin peroxidase recently identified in mammals, participating directly in eliminating hydrogen peroxide and neutralizing other reactive oxygen species.80,81 Functionally, PRDX5 has been implicated in antioxidant protective mechanisms as well as in signal transduction in cells.82 The synthesized compounds (8–16) have minimum binding energy ranging from (−4.6 to −5.6 kcal/mol) with a moderate result achieved using compound 9 (−5.6 kcal/mol), which revealed that the compounds are encouraging antioxidant effects within the target (Supplementary Information in Appendix XXVI and Appendix XXXIVa–j). The synthesized compounds’ antioxidant analysis revealed moderate to good activity. All substances additionally demonstrated a pertinent binding affinity and residual interaction for vitamin C of natural antioxidants. Compound 8 exhibited Thr-147 was involved in hydrogen bonding with –OH and methoxy groups of curcumin moiety. Hydrophobic/π-cation interactions were observed for Leu-116 with the aromatic group, while Arg-127, Asn-76, Arg-124, Phe-120, Thr-44, and Ile-119 showed Van der Waals residual interactions, respectively (Figure 11).
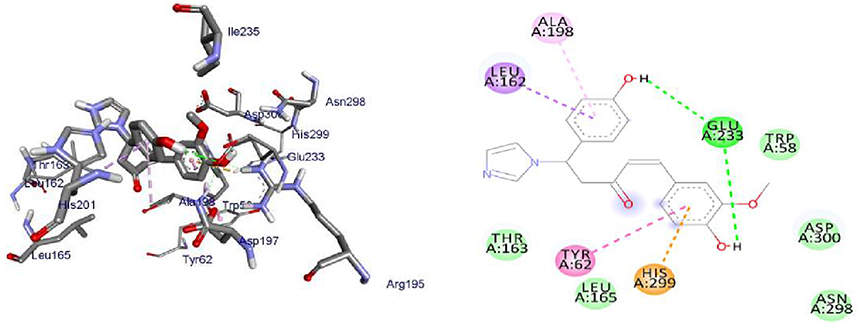 |
Figure 11 3D (right) and 2D (left) representations of the binding interactions of 8 against Human peroxiredoxin 5 (PDB ID: 1hd2). |
Molecular Docking Studies of Synthesized Compounds Against α-Amylase Enzyme
Alpha-amylase is a metalloenzyme containing Ca2+ ions in its active site and catalyzed the hydrolysis of starch into simple sugars. This enzyme fascinated extensive attention due to its capability of hydrolyzing the α-1,4-glycosidic connection of starch. Inhibition of these enzymes leads to minimization of absorption of carbohydrates from the small intestine, can control postprandial hyperglycemia, and reduce the risk of developing diabetics.36 It was found that the synthesized compounds (8–16) have minimum binding energy ranging from −7.4 to −8.4 kcal/mol in the binding pocket of the α-amylase enzyme, with the best result achieved for compounds 8 and 16 (−8.4 and −8.2 kcal/mol, respectively) and while comparable docking score exhibited for compound 11 (−7.4 kcal/mol) as compared to acarbose (−7.5 kcal/mol), signifying that the compounds are promising antidiabetic agents against the α-amylase enzyme, as depicted in Supplementary Information in Appendix XXVII and Appendix XXXVa–d. Thus, the results of molecular docking are in good correlation with the results of 8 and 16 in vitro α-amylase enzyme inhibition activities (Figures 12 and 13). Glu-233 was involved in hydrogen bonding with N-pyrazoline and –OH of curcumin moiety, respectively. The oxygen lone pair is strategically positioned within the curcumin-conjugated system to delocalize and increase electron density, which may account for the remarkable potency of these molecules. The delocalization is better able to stabilize (+ve)-charges over the benzene, which in turn improves the inhibitory action of the corresponding enzyme.83 Hydrophobic/π-cation interactions were observed for His-201, Trp-59, Trp-58, Ile-235 and His-299, Typ-62, Ala-198, Leu-162 with phenyl groups of curcumin, while Arg-195, Ala-198, Lys-200, Tyr-151, Tyr-62, Gln-63, and Tyr-58, Asp-300, Asn-298, Leu-165, Thr-163 showed Van der Waals residual interactions, respectively. Thus, the docking calculation was found in good correlations with the experimental results, which show the most potent anti-α-amylase activity of compounds 8, 11, and 16 compared to its analogs.
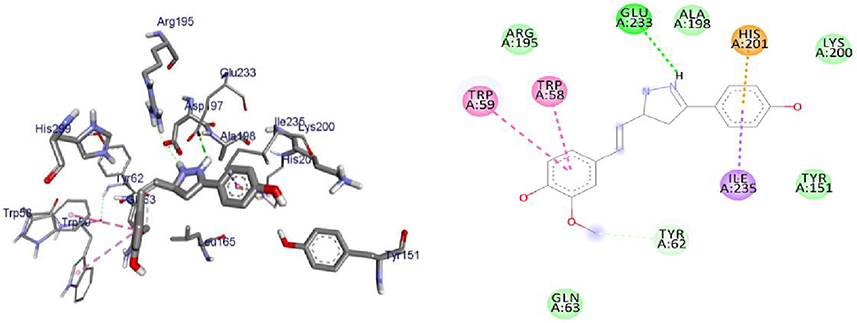 |
Figure 12 3D (right) and 2D (left) representations of the binding interactions of 8 against α-amylase enzyme (PDB ID: 4w93). |
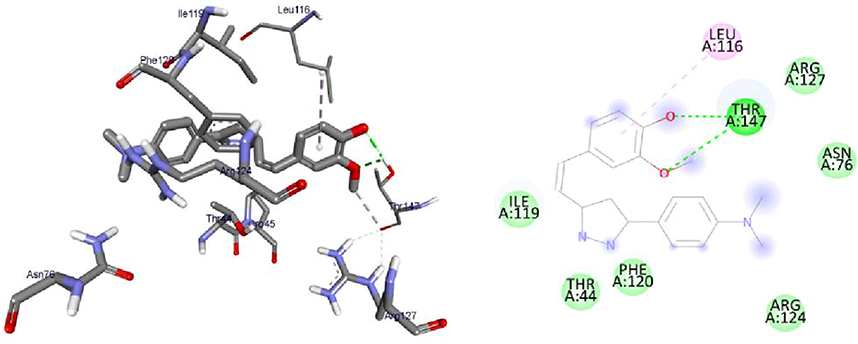 |
Figure 13 3D (right) and 2D (left) representations of the binding interactions of 16 against α-amylase enzyme (PDB ID: 4w93). |
Conclusion and Future Remarks
In the present study, nine series of heterocyclic C5-curcumin analogs were synthesized using an eco-friendly and effective ultrasonic irradiation-assisted protocol. Results of antibacterial activities revealed that compounds 8, and 11 displayed mean inhibition zone of 13.00±0.57, and 19.66±00 mm, respectively, compared to amoxicillin (12.87±1.41 mm) at 500 μg/mL against E. coli. Compounds 8, 11, and 16 displayed mean inhibition zone of 17.67±0.57, 14.33±0.57 and 23.33±00 mm, respectively, compared to amoxicillin (13.75±1.83 mm) at 500 μg/mL against P. aeruginosa, whereas compound 12 displayed mean inhibition zone of 11.33±0.57 mm compared to amoxicillin (10.75±1.83 mm) at 500 μg/mL against S. aureus. In vitro results for α-amylase activity revealed that compounds 11 and 16 had exhibited higher inhibitory activity potentials against α-amylase enzyme with IC50=7.59 µg/mL and 4.08 µg/mL, respectively, compared to the standard drug, acarbose (IC50= 8.0 µg/mL). Additionally, compound, 8 showed the highest antioxidant activities in the DPPH assay with an IC50 value of 2.44 μg/mL compared to natural antioxidant Vitamin C (1.24 μg/mL), while compound 16 showed potential in reducing the development of lipid peroxides by showing a promising suppression of peroxide formation by 89.92±0.42%. In silico analysis predicted that the synthesized compounds possessed good drug-likeness and drug-score properties to become orally active molecules.
Molecular docking study of compounds 9, 11, and 14 revealed higher DNA Gyrase B inhibitory effect with the binding affinity of −7.6, −7.9, and −7.6 Kcal/mol, respectively, and compounds 16, 15, and 11 displayed promising binding affinities of −9.1, −8.8, and −8.6 Kcal/mol, respectively, against PqsA compared to amoxicillin (−6.1 and −7.9 Kcal/mol, respectively). Compounds 9 and 12 displayed a higher binding affinity of −5.8 Kcal/mol with S. aureus Pyruvate Kinase, compared to amoxicillin (−4.9 Kcal/mol). Compound 11 displayed a binding affinity of −5.6 Kcal/mol with LuxS, compared to amoxicillin (−2.9 Kcal/mol), and Compound 11 displayed higher binding affinities of −7.5 and 8.3 Kcal/mol with penicillin-binding proteins (PBPs) and β-lactamases producing bacterial strains, compared to amoxicillin (−7.2 and −7.9 Kcal/mol, respectively), these results are in good agreement with the in vitro antibacterial activities exhibited by these compounds against E. coli, P. aeruginosa, S. aureus, and S. pyogenes. This is due to its involvement in hydrogen bonding with Serine-residue at pyrimidine-2-ol and curcumin moiety of the –OH group, which is able to acylate the active Serine residue. Compounds 8 and 14 displayed a binding affinity of −5.6 Kcal/mol against Human peroxiredoxin 5 compared to ascorbic acid (−5.4 Kcal/mol). While compounds 8 and 16 (−8.4 and −8.2 kcal/mol, respectively) exhibited better α-amylase inhibitory activity as compared to acarbose (−7.5 kcal/mol), indicating that these compounds are promising antidiabetic agents against the α-amylase enzyme. The in vitro antibacterial activity and molecular docking analysis revealed that compounds, 11, 16, and 12 are more potent antibacterial therapeutics agents against P. aeruginosa, E. coli, and S. aureus, whereas compound 14 was found to be promising antibacterial, and antioxidant agents, respectively. Thus, it could be concluded that synthesized compounds 8, 11, 16, 12, and 14 are entitled to be used as a future lead template for identifying more potent antidiabetic, antibacterial, and antioxidant candidates. Therefore, the result suggests that molecular docking studies of the most active compounds showed stronger binding interactions, and they are in good agreement with their in vitro biological activities as well as with in silico ADMET profiles demonstrating promising anti-diabetic properties, as well as moderate to strong antibacterial and free radical scavenging activities that may also assist decrease lipid profiles.
Abbreviations
DM, Diabetes mellitus; T2DM, type 2 diabetes mellitus; TLC, thin layer chromatography; AA, ascorbic acid; AMR, antimicrobial resistance; ROS, reactive oxygen species; ADMET, absorption, distribution, metabolism, excretion, and toxicity; ANOVA, analysis of variance; DPPH, 1.1-diphenyl-2-picrylhydrazyl; IC50, inhibitory concentration inhibiting 50% of growth; NMR, nuclear magnetic resonance; DIZ, diameter of inhibition zone; RSA, radical scavenging activity; UV, ultraviolet; TMS, tetramethylsilane; MeOD-d4, deuterated methanol; CLSI, Clinical and Laboratory Standards Institutes protocols; SMILEs, simplified molecular-input line-entry system; PDB ID, protein data bank identity; SAR, structural activity relationships; DMSO-d6, deuterated dimethyl sulfoxide; E. coli, Escherichia coli; S. aureus, Staphylococcus aureus; P. aeruginosa, Pseudomonas aeruginosa; S. pyogenes, Streptococcus pyogenes.
Data Sharing Statement
The data used to support the findings of this study are included within the manuscript and also submitted as supporting information.
Acknowledgments
The authors would like to acknowledge the Department of Applied Chemistry, School of Applied Natural Science, Adama Science and Technology University (ASTU), and Wachemo University for Ph.D. opportunity study and leave of absence, respectively, provided for DZ. Adama Public Health Research and Referral Laboratory Center, Ethiopia, has been greatly acknowledged for generous support during the antibacterial assay.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
National Research Foundation of Korea (Grant No. NRF-2021K1A3A1A16096990) and Adama Science and Technology University (ASTU) funded part of the work.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Bansal G, Thanikachalam PV, Maurya RK, Chawla P, Ramamurthy S. An overview on medicinal perspective of thiazolidine-2, 4-dione: A remarkable scaffold in the treatment of type 2 diabetes. J Adv Res. 2020;23:163–205.
2. Gomes MB, Rathmann W, Charbonnel B, et al. Treatment of type 2 diabetes mellitus worldwide: baseline patient characteristics in the global DISCOVER study. Diabetes Res Clin Pract. 2019;151:20–32. doi:10.1016/j.diabres.2019.03.024
3. World Health Organization. Noncommunicable diseases country profiles, 2018; 2018.
4. Huang C-H, Chiu C-H, Chen I-W, et al. Antimicrobial resistance and outcomes of community-onset bacterial bloodstream infections in patients with type 2 diabetes. J Glob Antimicrob Resist. 2018;15:271–276. doi:10.1016/j.jgar.2018.08.008
5. Chávez-Reyes J, Escárcega-González CE, Chavira-Suárez E, et al. Susceptibility for some infectious diseases in patients with diabetes: the key role of glycemia. Front Public Health. 2021;9:559595. doi:10.3389/fpubh.2021.559595
6. Hussain F, Khan Z, Jan MS, et al. Synthesis, in-vitro α-glucosidase inhibition, antioxidant, in-vivo antidiabetic and molecular docking studies of pyrrolidine-2,5-dione and thiazolidine-2,4-dione derivatives. Bioorg Chem. 2019;91:103128. doi:10.1016/j.bioorg.2019.103128
7. Saeedi M, Mohammadi-Khanaposhtani M, Asgari S, et al. Design, synthesis, in vitro, and in silico studies of novel diarylimidazole-1,2,3-triazole hybrids as potent α-glucosidase inhibitors. Bioorg Med Chem. 2019;27(23):115148. doi:10.1016/j.bmc.2019.115148
8. Rahim F, Ullah H, Javid MT, et al. Synthesis, in vitro evaluation and molecular docking studies of thiazole derivatives as new inhibitors of α-glucosidase. Bioorg Chem. 2015;62:15–21. doi:10.1016/j.bioorg.2015.06.006
9. Dhingra S, Sartelli M, Islam T, Haque M. Microbial resistance movements: an overview of global public health threats posed by antimicrobial resistance, and how best to counter. Front Public Health. 2020;8:535668. doi:10.3389/fpubh.2020.535668
10. Rao PS, Kalva S, Yerramilli A, Mamidi S. Free radicals and tissue damage: Role of antioxidants. Free Radic Antioxid. 2011;4:2–7.
11. Su L-J, Zhang J-H, Gomez H, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev. 2019;2019:1.
12. Mady MF, Awad GE, Jørgensen KB. Ultrasound-assisted synthesis of novel 1,2,3-triazoles coupled diaryl sulfone moieties by the CuAAC reaction, and biological evaluation of them as antioxidant and antimicrobial agents. Eur J Med Chem. 2014;84:433–443. doi:10.1016/j.ejmech.2014.07.042
13. Shah MS, Rahman MM, Islam MD, et al. Synthesis, antimicrobial and antioxidant evaluation with in silico studies of new thiazole Schiff base derivatives. J Mol Struct. 2022;1248:131465. doi:10.1016/j.molstruc.2021.131465
14. Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, Nogueira-Machado JA. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018;9(2):1–9. doi:10.1038/s41419-017-0135-z
15. Atanasov AG, Zotchev SB, Dirsch VM, Supuran CT. Natural products in drug discovery: advances and opportunities. Nat Rev Drug Discov. 2021;20(3):200–216. doi:10.1038/s41573-020-00114-z
16. Thomford NE, Senthebane DA, Rowe A, et al. Natural products for drug discovery in the 21st century: innovations for novel drug discovery. Int J Mol Sci. 2018;19(6):1578. doi:10.3390/ijms19061578
17. Amalraj A, Pius A, Gopi S, Gopi S. Biological activities of curcuminoids, other biomolecules from turmeric and their derivatives – a review. J Tradit Complement Med. 2017;7(2):205–233. doi:10.1016/j.jtcme.2016.05.005
18. Oglah MK, Mustafa YF, Bashir MK, Jasim MH, Mustafa YF. Curcumin and its derivatives: A review of their biological activities. Syst Rev Pharm. 2020;11(3):472–481.
19. Rosemond MJC, John-Williams LS, Yamaguchi T, Fujishita T, Walsh JS. Enzymology of a carbonyl reduction clearance pathway for the HIV integrase inhibitor, S-1360: role of human liver cytosolic aldo-keto reductases. Chem Biol Interact. 2004;147(2):129–139. doi:10.1016/j.cbi.2003.12.001
20. da Silva CC, de Pereira CMP, Das Neves RN. Antiparasitic activity of synthetic curcumin monocarbonyl analogues against Trichomonas vaginalis. Biomed Pharmacother. 2019;111:367–377. doi:10.1016/j.biopha.2018.12.058
21. Azzi E, Alberti D, Parisotto S, Oppedisano A, Deagostino A. Design, synthesis and preliminary in-vitro studies of novel boronated monocarbonyl analogues of Curcumin (BMAC) for antitumor and β-amiloyd disaggregation activity. Bioorg Chem. 2019;93:103324. doi:10.1016/j.bioorg.2019.103324
22. Singh A, Singh JV, Rana A. Monocarbonyl curcumin-based molecular hybrids as potent antibacterial agents. ACS Omega. 2019;4(7):11673–11684. doi:10.1021/acsomega.9b01109
23. Sharma S, Gupta MK, Saxena AK, Bedi PMS. Triazole linked mono carbonyl curcumin-isatin bifunctional hybrids as novel anti tubulin agents: design, synthesis, biological evaluation and molecular modeling studies. Bioorg Med Chem. 2015;23(22):7165–7180. doi:10.1016/j.bmc.2015.10.013
24. Fadda AA, Badria FA, El-Attar KM. Synthesis and evaluation of curcumin analogues as cytotoxic agents. Med Chem Res. 2010;19:413–430. doi:10.1007/s00044-009-9199-3
25. Emam DR, Alhajoj AM, Elattar KM, Kheder NA, Fadda AA. Synthesis and evaluation of curcuminoid analogues as antioxidant and antibacterial agents. Molecules. 2017;22(6):971. doi:10.3390/molecules22060971
26. Rodrigues FC, Kumar NA, Thakur G. The potency of heterocyclic curcumin analogues: an evidence-based review. Pharm Res. 2021;166:105489. doi:10.1016/j.phrs.2021.105489
27. Mohammadi Ziarani G, Kheilkordi Z, Gholamzadeh P. Ultrasound-assisted synthesis of heterocyclic compounds. Mol Divers. 2020;24:771–820. doi:10.1007/s11030-019-09964-1
28. Kaur N. Ultrasound-assisted synthesis of six-membered n-heterocycles. Mini Rev Org Chem. 2018;15(6):520–536. doi:10.2174/1570193x15666180221152535
29. Ahmad MR, Sastry VG, Bano N, Anwar S. Synthesis of novel chalcone derivatives by conventional and microwave irradiation methods and their pharmacological activities. Arab J Chem. 2016;9:S931–S935. doi:10.1016/j.arabjc.2011.09.002
30. Mapoung S, Suzuki S, Limtrakul P, et al. Dehydrozingerone, a curcumin analog, as a potential anti-prostate cancer inhibitor in vitro and in vivo. Molecules. 2020;25(12):2737. doi:10.3390/molecules25122737
31. Yadav P, Lal K, Kumar A, Guru SK, Jaglan S, Bhushan S. Green synthesis and anticancer potential of chalcone linked-1,2,3-triazoles. Eur J Med Chem. 2017;126:944–953. doi:10.1016/j.ejmech.2016.11.030
32. Shabalala NG, Pagadala R, Jonnalagadda SB. Ultrasonic-accelerated rapid protocol for the improved synthesis of pyrazoles. Ultrason Sonochem. 2015;27:423–429. doi:10.1016/j.ultsonch.2015.06.005
33. Hudzicki J. Antibacterial activity of legundi leaf (vitex trifolia) essential oil using in-vitro and in-silico methods. Am Soc Microbiol. 2009;15:55–63.
34. Bergey DH. Bergey’s Manual of Determinative Bacteriology. Lippincott Williams & Wilkins; 1994.
35. Khaldi-Khellafi N, Makhloufi-Chebli M, Oukacha-Hikem D, et al. Green synthesis, antioxidant and antibacterial activities of 4-aryl-3,4-dihydropyrimidinones/thiones derivatives of curcumin. Theoretical calculations and mechanism study. J Mol Struct. 2019;1181:261–269. doi:10.1016/j.molstruc.2018.12.104
36. Chortani S, Horchani M, Romdhane A, Issaoui N, Jannet HB, Romdhane A. Design and synthesis of new benzopyrimidinone derivatives: α-amylase inhibitory activity, molecular docking and DFT studies. J Mol Struct. 2021;1230:129920. doi:10.1016/j.molstruc.2021.129920
37. Kenchappa R, Bodke YD, Chandrashekar A, Sindhe MA, Peethambar S. Synthesis of coumarin derivatives containing pyrazole and indenone rings as potent antioxidant and antihyperglycemic agents. Arab J Chem. 2017;10:S3895–S3906. doi:10.1016/j.arabjc.2014.05.029
38. Mehrabi M, Esmaeili S, Adibi H, Khodarahmi R, Adibi H, Khodarahmi R. Antioxidant and glycohydrolase inhibitory behavior of curcumin-based compounds: synthesis and evaluation of anti-diabetic properties in vitro. Bioorg Chem. 2021;110:104720. doi:10.1016/j.bioorg.2021.104720
39. Varma RR, Pandya JG, Vaidya FU, Pathak C, Bhatt BS, Patel MN. Biological activities of pyrazoline-indole based Re(I) carbonyls: DNA interaction, antibacterial, anticancer, ROS production, lipid peroxidation, in vivo and in vitro cytotoxicity studies. Chem Biol Interact. 2020;330:109231. doi:10.1016/j.cbi.2020.109231
40. Bajpai VK, Sharma A, Kang SC, Baek K-H. Antioxidant, lipid peroxidation inhibition and free radical scavenging efficacy of a diterpenoid compound sugiol isolated from Metasequoia glyptostroboides. Asian Pac J Trop Med. 2014;7(1):9–15. doi:10.1016/S1995-7645(13)60183-2
41. Ooms F. Molecular modeling and computer aided drug design. Examples of their applications in medicinal chemistry. Curr Med Chem. 2000;7(2):141–158. doi:10.2174/0929867003375317
42. Zhang J, Shan Y, Pan X, Wang C, Xu W, He L. Molecular docking, 3D-QSAR studies, and in silico ADME prediction of p-aminosalicylic acid derivatives as neuraminidase inhibitors. Chem Biol Drug Des. 2011;78(4):709–717. doi:10.1111/j.1747-0285.2011.01179.x
43. Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–2623. doi:10.1021/jm020017n
44. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(1):1–13. doi:10.1038/srep42717
45. Ahmed AH, Alkali YI. In silico pharmacokinetics and molecular docking studies of lead compounds derived from diospyros mespiliformis. PharmaTutor. 2019;7(3):31–37. doi:10.29161/PT.v7.i3.2019.31
46. Seeliger D, de Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des. 2010;24(5):417–422. doi:10.1007/s10822-010-9352-6
47. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi:10.1002/jcc.21334
48. Biovia DS. Discovery Studio Visualizer, San Diego. San Diego, CA: Dassault Systems; 2021.
49. Fu Y-S, Chen T-H, Weng C-F, Huang L, Lai D, Weng C-F. Pharmacological properties and underlying mechanisms of curcumin and prospects in medicinal potential. Biomed Pharmacother. 2021;141:111888. doi:10.1016/j.biopha.2021.111888
50. Borik RM, Fawzy NM, Abu-Bakr SM, Aly MS. Design, synthesis, anticancer evaluation and docking studies of novel heterocyclic derivatives obtained via reactions involving curcumin. Molecules. 2018;23(6):1398. doi:10.3390/molecules23061398
51. Nagasundaram N, Padmasree K, Santhosh S, Lalitha A, Sedhu N, Lalitha A. Ultrasound promoted synthesis of new azo fused dihydropyrano[2,3-c]pyrazole derivatives: in vitro antimicrobial, anticancer, DFT, in silico ADMET and Molecular docking studies. J Mol Struct. 2022;1263:133091. doi:10.1016/j.molstruc.2022.133091
52. Lahyani A, Trabelsi M. Ultrasonic-assisted synthesis of flavones by oxidative cyclization of 2′-hydroxychalcones using iodine monochloride. Ultrason Sonochem. 2016;31:626–630. doi:10.1016/j.ultsonch.2016.02.018
53. Eid NM, George RF. Facile synthesis of some pyrazoline-based compounds with promising anti-inflammatory activity. Future Med Chem. 2018;10(2):183–199. doi:10.4155/fmc-2017-0144
54. Zhang D-W, Zhao T-Q, Zhao T-Q, Lin F, Lin F, Lin F. Ultrasonic-assisted synthesis of 1,4-disubstituted 1,2,3-triazoles via various terminal acetylenes and azide and their quorum sensing inhibition. Ultrason Sonochem. 2017;36:343–353. doi:10.1016/j.ultsonch.2016.12.011
55. Khan S, Ullah H, Rahim F, Taha M, Hussain R. New thiazole-based thiazolidinone derivatives: synthesis, in vitro α-amylase, α-glucosidase activities and silico molecular docking study. Chem Data Collect. 2022;42:100967. doi:10.1016/j.cdc.2022.100967
56. Stagos D. Antioxidant activity of polyphenolic plant extracts. Antioxidants. 2019;9:19. doi:10.3390/antiox9010019
57. Idhayadhulla A, Xia L, Lee YR, Kim SH, Wee Y-J, Lee C-S. Synthesis of novel and diverse mollugin analogues and their antibacterial and antioxidant activities. Bioorg Chem. 2014;52:77–82. doi:10.1016/j.bioorg.2013.11.008
58. Halliwell B, Chirico S. Lipid peroxidation: its mechanism, measurement, and significance. Am J Clin Nutr. 1993;57(5):715S–725S. doi:10.1093/ajcn/57.5.715S
59. Ibrahim TS, Almalki AJ, Moustafa AH, et al. Novel 1,2,4-oxadiazole-chalcone/oxime hybrids as potential antibacterial DNA gyrase inhibitors: design, synthesis, ADMET prediction and molecular docking study. Bioorg Chem. 2021;111:104885. doi:10.1016/j.bioorg.2021.104885
60. Abishad P, Kurkure NV, Kurkure N, et al. In silico molecular docking and in vitro antimicrobial efficacy of phytochemicals against multi-drug-resistant enteroaggregative Escherichia coli and non-typhoidal Salmonella spp. Gut Pathog. 2021;13(1):1–11. doi:10.1186/s13099-021-00443-3
61. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23(1–3):3–25. doi:10.1016/S0169-409X(96)00423-1
62. Remko M, Remková A, Broer R. A comparative study of molecular structure, pKa, lipophilicity, solubility, absorption and polar surface area of some antiplatelet drugs. Int J Mol Sci. 2016;17(3):388. doi:10.3390/ijms17030388
63. Faizi M, Jahani R, Almasirad A. Novel 4-thiazolidinone derivatives as agonists of benzodiazepine receptors: design, synthesis and pharmacological evaluation. EXCLI J. 2017;16:52. doi:10.17179/excli2016-692
64. Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46(W1):W257–W263. doi:10.1093/nar/gky318
65. Brown P, Abdulle O, Boakes S, et al. Influence of lipophilicity on the antibacterial activity of polymyxin derivatives and on their ability to act as potentiators of rifampicin. ACS Infect Dis. 2021;7(4):894–905. doi:10.1021/acsinfecdis.0c00917
66. Alonso C, Carrer V, Espinosa S, et al. Prediction of the skin permeability of topical drugs using in silico and in vitro models. Eur J Pharm Sci. 2019;136:104945. doi:10.1016/j.ejps.2019.05.023
67. Finch A, Pillans P. P-glycoprotein and its role in drug-drug interactions. Aust Prescr. 2014;37(4):137–139. doi:10.18773/austprescr.2014.050
68. Islam MA, Pillay TS. Identification of promising anti-DNA gyrase antibacterial compounds using de novo design, molecular docking and molecular dynamics studies. J Biomol Struct Dyn. 2020;38(6):1798–1809. doi:10.1080/07391102.2019.1617785
69. Daina A, Zoete V. A BOILED-egg to predict gastrointestinal absorption and brain penetration of small molecules. Chem Med Chem. 2016;11(11):1117–1121. doi:10.1002/cmdc.201600182
70. Agnihotry S, Pathak RK, Srivastav A, Shukla PK, Gautam B. Molecular docking and structure-based drug design. In: Computer-Aided Drug Design. Springer; 2020:115–131.
71. Collin F, Karkare S, Maxwell A. Exploiting bacterial DNA gyrase as a drug target: current state and perspectives. Appl Microbiol Biotechnol. 2011;92(3):479–497. doi:10.1007/s00253-011-3557-z
72. Shaker B, Ahmad S, Thai TD, Eyun S-I, Na D. Rational drug design for pseudomonas aeruginosa pqsA enzyme: an in silico guided study to block biofilm formation. Front Mol Biosci. 2020;7:577316. doi:10.3389/fmolb.2020.577316
73. Kumar NS, Dullaghan EM, Finlay BB, et al. Discovery and optimization of a new class of pyruvate kinase inhibitors as potential therapeutics for the treatment of methicillin-resistant Staphylococcus aureus infections. Bioorg Med Chem. 2014;22(5):1708–1725. doi:10.1016/j.bmc.2014.01.020
74. Zhou L, Zhang Y, Zhu X, Pan J. Effects of gut microbiome and short-chain fatty acids (SCFAs) on finishing weight of meat rabbits. Front Microbiol. 2020;2020:2558.
75. Kumar K, Anbarasu A, Ramaiah S. Molecular docking and molecular dynamics studies on β-lactamases and penicillin binding proteins. Mol Biosyst. 2014;10(4):891–900. doi:10.1039/C3MB70537D
76. Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev. 2008;32(2):234–258. doi:10.1111/j.1574-6976.2008.00105.x
77. Martin JF, Alvarez-Alvarez R, Liras P. Penicillin-binding proteins, β-lactamases, and β-lactamase inhibitors in β-lactam-producing actinobacteria: self-resistance mechanisms. Int J Mol Sci. 2022;23(10):5662. doi:10.3390/ijms23105662
78. Tooke CL, Hinchliffe P, Bragginton EC, et al. β-lactamases and β-lactamase inhibitors in the 21st century. J Mol Biol. 2019;431(18):3472–3500. doi:10.1016/j.jmb.2019.04.002
79. N. González-Bello C, Rodríguez D, Pernas M, Rodriguez A, Colchón E. β-Lactamase inhibitors to restore the efficacy of antibiotics against superbugs. J Med Chem. 2019;63(5):1859–1881. doi:10.1021/acs.jmedchem.9b01279
80. Yuan J, Murrell GA, Trickett A, Landtmeters M, Knoops B, Wang M-X. Overexpression of antioxidant enzyme peroxiredoxin 5 protects human tendon cells against apoptosis and loss of cellular function during oxidative stress. Biochim Biophys Acta. 2004;1693(1):37–45. doi:10.1016/j.bbamcr.2004.04.006
81. Declercq J-P, Evrard C, Clippe A, Vander Stricht D, Bernard A, Knoops B. Crystal structure of human peroxiredoxin 5, a novel type of mammalian peroxiredoxin at 1.5 Å resolution. J Mol Biol. 2001;311(4):751–759. doi:10.1006/jmbi.2001.4853
82. Plaisant F, Clippe A, Vander Stricht D, Knoops B, Gressens P. Recombinant peroxiredoxin 5 protects against excitotoxic brain lesions in newborn mice. Free Radic Biol Med. 2003;34(7):862–872. doi:10.1016/S0891-5849(02)01440-5
83. Rahim F, Tariq S, Taha M, et al. New triazinoindole bearing thiazole/oxazole analogues: synthesis, α-amylase inhibitory potential and molecular docking study. Bioorg Chem. 2019;92:103284. doi:10.1016/j.bioorg.2019.103284
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.