Back to Journals » Journal of Hepatocellular Carcinoma » Volume 13
Tumor-Associated Macrophages in Hepatocellular Carcinoma: From Ontogeny and Heterogeneity to Immune Evasion and Therapeutic Targeting
Authors Su Y, Liang Y ![]() , Zhong D, Chen Y, Ma SS, Huang X
, Zhong D, Chen Y, Ma SS, Huang X ![]()
Received 11 April 2026
Accepted for publication 29 May 2026
Published 16 June 2026 Volume 2026:13 616101
DOI https://doi.org/10.2147/JHC.S616101
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr David Gerber
Yuhao Su, Yuxin Liang, Deyuan Zhong, Yahui Chen, Shuo Shuo Ma, Xiaolun Huang
Department of Liver Transplantation Center and HBP Surgery, Sichuan Cancer Hospital & Institute, Sichuan Cancer Center, School of Medicine, University of Electronic Science and Technology of China, Chengdu, People’s Republic of China
Correspondence: Xiaolun Huang, Department of Liver Transplantation Center and HBP Surgery, Sichuan Cancer Hospital & Institute, Sichuan Cancer Center, School of Medicine, University of Electronic Science and Technology of China, Chengdu, People’s Republic of China, Email [email protected]
Abstract: Hepatocellular carcinoma (HCC) remains one of the leading causes of cancer morbidity and mortality worldwide and typically arises within a profoundly immunosuppressive tumor microenvironment (TME). Among the myeloid populations that infiltrate the HCC microenvironment, tumor-associated macrophages (TAMs) are numerically dominant and function as central orchestrators of tumor initiation, progression, and immune evasion. TAMs originate from both tissue-resident Kupffer cells and bone marrow–derived monocytes, and are shaped by TME-derived cues such as hypoxia, metabolic stress, and inflammatory mediators that polarize them toward a tumor-promoting, M2-like phenotype. Once activated, TAMs release pro-angiogenic and trophic factors that sustain vascular remodeling and tumor proliferation, and they induce epithelial–mesenchymal transition (EMT) and stemness-associated programs that facilitate invasion and metastasis. Concomitantly, TAMs suppress antitumor immunity through the secretion of immunosuppressive cytokines such as IL-10 and TGF-β, the expression of PD-L1, and the recruitment of regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs). Clinically, a high density of TAM infiltration is strongly associated with aggressive disease features, early recurrence, and poor prognosis. Given their dual roles in promoting immune escape and therapeutic resistance, TAMs have emerged as critical nodes within the HCC ecosystem and as promising targets for translational intervention. This review summarizes current understanding of TAM ontogeny, phenotypic diversity, and functional heterogeneity in HCC, delineates the molecular and immunological mechanisms through which they drive tumor progression, and discusses emerging TAM-targeted therapeutic strategies with clinical potential including CSF1R inhibition, CD47–SIRPα blockade, and chimeric antigen receptor–macrophage (CAR-M) therapies.
Keywords: hepatocellular carcinoma, tumor-associated macrophages, immune evasion, progression, therapeutic strategies
Introduction
Liver cancer ranks seventh in global incidence and third in cancer-related mortality, with hepatocellular carcinoma (HCC) accounting for approximately 80% of all cases.1 Its rising prevalence is strongly linked to the increasing burden of risk factors such as chronic hepatitis B virus (HBV) infection, non-alcoholic fatty liver disease, and unhealthy lifestyle behaviors.2 Although surgical resection and locoregional therapies are effective for early-stage disease, the majority of HCC patients are diagnosed at advanced stages, where curative options are limited. Historically, the lack of effective systemic therapies has contributed to persistently high mortality rates. The advent of immunotherapies, particularly immune checkpoint inhibitors (ICIs), has brought new therapeutic opportunities for patients with advanced HCC.3,4 However, their clinical efficacy remains constrained by the profoundly immunosuppressive TME.
The HCC TME is composed of diverse non-tumor elements, including the extracellular matrix (ECM), fibroblasts, and a complex array of immune cell populations.5 Interactions among these components establish a protective niche that shields tumor cells from immune elimination and promotes resistance to conventional therapies.6 Among the immune infiltrates, TAMs constitute a predominant population, accounting for approximately 30–50% of all immune cells in the TME, and play a pivotal role in orchestrating tumor progression and immune modulation.7 More importantly, TAMs function as central orchestrators of tumor progression and immune suppression by integrating inflammatory signaling, angiogenesis, fibrosis, metabolic adaptation, and T-cell dysfunction.8 Emerging evidence further indicates that TAMs represent a mechanistically distinct barrier to ICI efficacy through their ability to suppress cytotoxic T-cell activity, recruit immunosuppressive cell populations, and sustain an immune-excluded TME.9 Consequently, TAMs have emerged as particularly attractive therapeutic targets among the diverse cellular components of the HCC microenvironment. Accumulating clinical evidence indicates that elevated TAM infiltration in HCC tissues correlates with increased tumor size, vascular invasion, metastasis, and poor prognosis. By promoting angiogenesis, enhancing tumor invasion and metastasis, and mediating immune evasion, TAMs exert multifaceted protumor functions throughout the course of HCC development.10 Notably, accumulating studies suggest that the composition, polarization state, and functional properties of TAMs may vary across different etiological backgrounds of HCC, including HBV-, HCV-, and NAFLD-associated disease, potentially contributing to distinct immune landscapes and therapeutic responses.11,12
Given their pivotal roles in tumor progression and the establishment of immune tolerance, TAMs have garnered growing interest as promising therapeutic targets in cancer immunology. Building upon a comprehensive overview of their biological origins, phenotypic plasticity, and functional diversity, this review focuses on the mechanisms through which TAMs mediate immune evasion in HCC. Furthermore, it summarizes emerging therapeutic strategies aimed at reprogramming or depleting TAMs and discusses the current challenges and future opportunities associated with translating TAM-targeted interventions into clinical practice.
Ontogeny and Characteristics of TAMs in HCC
Ontogeny and Developmental Lineages of TAMs
Macrophages in the liver originate from two ontogenetically distinct lineages that jointly shape the hepatic immune landscape. The first lineage consists of tissue-resident macrophages, primarily Kupffer cells (KCs), which arise from yolk sac or fetal liver erythromyeloid progenitors (EMPs) that express the macrophage colony-stimulating factor receptor (CSF1R). These embryonically derived macrophages seed the liver early in development and maintain their population through self-renewal under homeostatic conditions.13 Increasing evidence from fate-mapping and single-cell transcriptomic studies has revealed that resident macrophages in the healthy liver are transcriptionally and functionally distinct from infiltrating monocyte-derived counterparts, exhibiting a tolerogenic phenotype that contributes to hepatic immune homeostasis.14 The second lineage comprises bone marrow–derived monocytes, which are recruited into the liver through the bloodstream in response to inflammatory or tumor-derived signals. During chronic liver injury and hepatocarcinogenesis, persistent inflammation, fibrogenesis, and hepatocyte death stimulate the production of chemokines such as CCL2, CCL5, and CXCL10, which drive the recruitment of CCR2⁺ inflammatory monocytes.15 Upon entering the TME, these monocytes differentiate into TAMs and gradually replace or complement the resident KC population.16 In advanced HCC, this process results in the enrichment of a highly plastic CCR2⁺ TAM subset characterized by elevated expression of CSF1R, PD-L1, and CD206, reflecting their immunosuppressive and pro-tumorigenic potential.17
Beyond bone marrow–derived monocytes, splenic reservoirs have been identified as additional sources of macrophage precursors, particularly under conditions of systemic inflammation or hepatic injury.15 These splenic monocytes migrate to the liver through CCL2-dependent chemotaxis, where they further contribute to TAM expansion. Collectively, both embryonic and peripheral macrophage lineages undergo continuous phenotypic remodeling under the influence of tumor-derived cytokines (eg., IL-4, IL-10, CSF1) and metabolic cues (eg., hypoxia, lactic acidosis), which skew them toward an M2-like, tumor-supportive state.
This dual ontogeny underlies the remarkable heterogeneity of TAMs in HCC, wherein resident KCs and infiltrating monocyte-derived macrophages coexist and cooperatively establish an immunosuppressive niche. Dissecting the developmental hierarchy and lineage plasticity of these macrophage subsets is therefore crucial for designing therapeutic strategies aimed at selectively reprogramming or depleting tumor-promoting macrophage populations.
Etiology-Specific Heterogeneity of TAMs in the HCC
Emerging evidence suggests that TAM composition and functional polarization in HCC are strongly influenced by the underlying disease etiology.12 Chronic HBV- and HCV-associated inflammation, as well as metabolic dysfunction in NAFLD-related HCC, generate distinct hepatic immune microenvironments that may shape macrophage recruitment, activation, and immunosuppressive activity in different ways.18
In HBV-and HCV-related HCC, persistent viral antigen exposure and chronic inflammatory signaling promote the accumulation of immunosuppressive TAMs characterized by increased PD-L1 expression, IL-10 secretion, and T-cell exhaustion. HBV-associated cytokine networks may further enhance macrophage-mediated immune tolerance and facilitate tumor progression.15
By contrast, NAFLD-related HCC is closely associated with metabolic inflammation and lipid dysregulation. Recent studies have identified lipid-associated macrophage subsets enriched in steatotic livers, which exhibit altered metabolic programming, enhanced inflammatory signaling, and distinct immunoregulatory properties.11 These TAM populations may contribute to the reduced responsiveness to immune checkpoint blockade observed in some patients with NAFLD-associated HCC.19
Phenotypic and Functional Polarization of TAMs in the HCC
Macrophages exhibit remarkable functional plasticity shaped by local microenvironmental cues, which allows them to dynamically transition between proinflammatory and immunoregulatory states. This polarization is traditionally conceptualized along an M1–M2 spectrum. Classically activated M1 macrophages arise in response to interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), or lipopolysaccharide (LPS), and are characterized by potent proinflammatory and antitumor activities. They secrete cytokines such as interleukin-12 (IL-12) and interleukin-1β (IL-1β), produce reactive oxygen and nitrogen species, and reinforce Th1-type immune responses, thereby promoting tumoricidal activity and antigen presentation.20,21 Conversely, alternatively activated M2 macrophages are induced by interleukin-4 (IL-4), interleukin-13 (IL-13), interleukin-10 (IL-10), or macrophage colony-stimulating factor (M-CSF). These macrophages exhibit high expression of immunoregulatory and tissue-repair markers, including arginase-1 (Arg1), CD163, and CD206, and secrete IL-10 and transforming growth factor-β (TGF-β), which collectively suppress Th1 and cytotoxic T-cell activity while promoting angiogenesis, matrix remodeling, and tumor progression.16 Functionally, these M2-like programs foster an immunosuppressive milieu that favors tumor cell survival and metastasis.22 These features suppress Th1 and cytotoxic T-cell activity, promote angiogenesis, and facilitate tumor growth.
It is important to emphasize that TAMs are not identical to conventional M2 macrophages. Within the HCC tumor microenvironment, a variety of signaling cues, including hypoxia, lactic acidosis, and tumor-derived cytokines such as CSF1, IL-6, and VEGF, collectively reprogram macrophages toward an M2-like phenotype that supports tumor growth and immune suppression. In HCC tissues, most TAMs display high expression of M2-associated markers (eg., CD163, CD206, and MARCO) and low expression of M1-associated markers (eg., IL-12, MHC-II, CD80), indicating a profound shift from immune activation to immune suppression.23 This phenotypic polarization diminishes their antigen-presenting and cytotoxic capacities while enhancing their proangiogenic, profibrotic, and immunosuppressive functions.24 Clinical studies have confirmed that the majority of macrophages in HCC are CD68⁺CD163⁺ M2-type cells, with infiltration density positively correlated with tumor grade, vascular invasion, and early recurrence.25
At the molecular level, TAM polarization in HCC is orchestrated by diverse intracellular signaling pathways. Activation of STAT3, STAT6, and PI3K–AKT–mTOR cascades reinforces M2-like transcriptional programs, while suppression of NF-κB and IRF5 signaling dampens M1-associated proinflammatory responses. Moreover, transcription factors such as c-Maf, KLF4, and PPARγ act as master regulators that sustain M2 polarization and immunosuppressive gene expression. In parallel, metabolic reprogramming plays a crucial role: M1 macrophages rely predominantly on glycolysis and pentose phosphate pathway activity to sustain inflammatory responses, whereas M2-like TAMs depend on oxidative phosphorylation and fatty acid oxidation, providing metabolic flexibility that enhances survival within hypoxic and nutrient-depleted tumor niches.
Recent single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics analyses have revealed extensive intra-tumoral heterogeneity among TAMs in HCC. Distinct subsets have been identified, including angiogenic TAMs (VEGFA⁺, SPP1⁺), immunoregulatory TAMs (CD163⁺MARCO⁺), and inflammatory TAMs (IL1B⁺TNF⁺), reflecting spatial adaptation to different microenvironmental regions. In particular, SPP1⁺ (osteopontin-positive) TAMs localize preferentially to the invasive front and perivascular regions, where they promote extracellular matrix remodeling and EMT.
Collectively, these findings illustrate that TAMs in HCC are a highly heterogeneous and plastic population, dynamically shaped by tumor-derived metabolic, inflammatory, and stromal cues. Their skewing toward an immunosuppressive M2-like phenotype constitutes a central mechanism of immune evasion and therapeutic resistance. Thus, decoding the molecular circuitry governing TAM polarization and spatial organization is pivotal for developing strategies to reprogram or selectively deplete tumor-promoting macrophage subsets in HCC.
TAM-Mediated Mechanisms Driving HCC Progression
TAMs drive the progression of HCC through multiple interconnected mechanisms, including the promotion of angiogenesis, facilitation of tumor invasion and metastasis, and induction of therapeutic resistance. These processes establish a pro-tumorigenic microenvironment that supports cancer cell survival, proliferation, and dissemination. (Table 1).
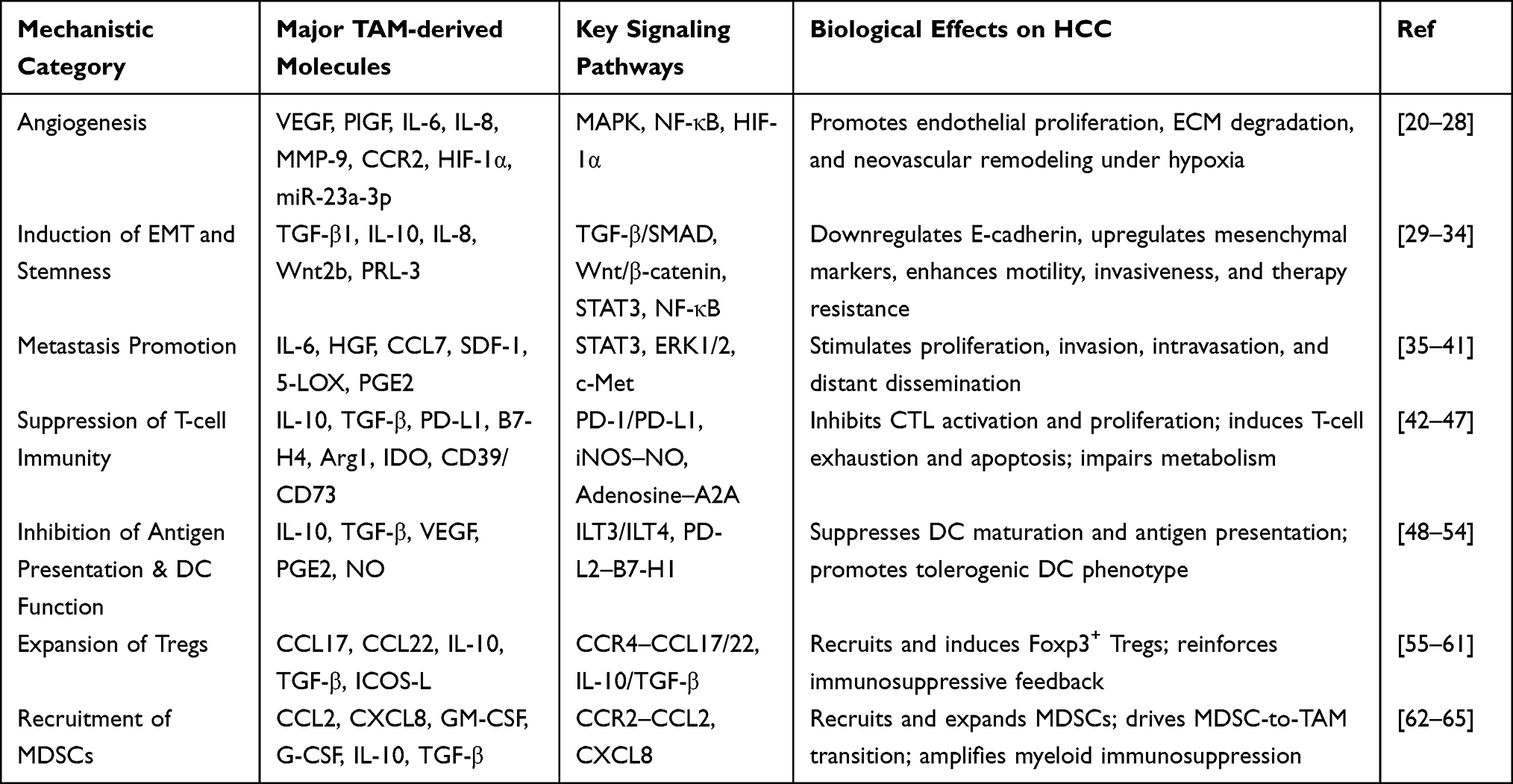 |
Table 1 Major Mechanistic Pathways by Which TAMs Promote HCC Progression |
TAMs Promote Angiogenesis
Neovascularization is a critical process for tumor growth and expansion. HCC is widely recognized as a “hypervascular” tumor, and microvessel density within tumor tissues is closely associated with prognosis. Studies have shown a positive correlation between TAM infiltration and microvessel density in HCC, underscoring their pivotal role in tumor angiogenesis.26 TAMs promote angiogenesis and vascular remodeling through several mechanisms.
M2-polarized TAMs secrete abundant pro-angiogenic factors such as VEGF, which directly stimulate endothelial cell proliferation and neovascular formation.27 In HCC, interactions between tumor cells with elevated PRL-3 expression and TAMs activate the MAPK pathway in macrophages, inducing IL-6/IL-8–mediated EMT and invasiveness.28 Simultaneously, PRL-3 enhances NF-κB signaling in tumor cells and upregulates VEGFA, thereby promoting angiogenesis.
TAMs also indirectly support angiogenesis through ECM remodeling. They secrete proteolytic enzymes such as matrix metalloproteinases (MMP-9 and MMP-2), which degrade tumor stroma and vascular basement membranes.29 This not only facilitates tumor invasion but also releases growth factors such as VEGF previously sequestered in the matrix, accelerating angiogenic processes.30 TAMs have been identified as the major source of MMP-9 at the invasive front of HCC, highlighting their central role in matrix degradation and angiogenesis. In addition, TAMs secrete placental growth factor (PlGF), a VEGF family member that further amplifies pro-angiogenic signaling.31
Hypoxia within the TME further enhances TAM-driven angiogenesis. Under hypoxic conditions, TAMs upregulate chemokine receptors such as CCR2. Bartneck et al reported that in HCC developing on a fibrotic background, highly vascularized tumor regions were enriched with CCR2+ TAM subsets, which not only accumulated in response to hypoxic–ischemic signals but also promoted vascular growth and remodeling.32 Hypoxia also induces upregulation of HIF-1α in TAMs, which in turn increases production of VEGF and other angiogenic mediators.33 Moreover, microRNAs contribute to TAM-mediated angiogenic regulation. Exosomes released by M2-type TAMs deliver miR-23a-3p to HCC cells and endothelial cells, targeting PTEN and TJP1, respectively.34 This promotes EMT in tumor cells while downregulating tight junction proteins such as occludin and claudin-5 in endothelial cells, thereby enhancing vascular permeability and angiogenesis.
TAMs and EMT
TAMs secrete a broad spectrum of cytokines, chemokines, and growth factors that profoundly influence tumor cell plasticity and behavior. One of their key tumor-promoting functions is the induction of epithelial–mesenchymal transition (EMT), a process in which epithelial tumor cells lose their polarity and intercellular adhesion while acquiring mesenchymal characteristics, thereby gaining enhanced motility, invasiveness, and resistance to apoptosis. EMT is not only a critical driver of tumor dissemination and metastasis but also contributes to stemness acquisition and therapeutic resistance in hepatocellular carcinoma. Through sustained paracrine interactions, TAM-derived mediators remodel the extracellular matrix, activate fibroblasts, and engage multiple signaling pathways such as TGF-β/SMAD, Wnt/β-catenin, and NF-κB, which cooperatively initiate EMT programs in tumor cells. Among these mediators, TGF-β secreted by TAMs acts as a principal inducer of EMT by upregulating transcriptional repressors including Snail, Slug, and Twist, thereby downregulating E-cadherin expression and promoting cytoskeletal reorganization. This cytokine-driven reprogramming ultimately confers tumor cells with greater migratory and invasive capacity, facilitating intrahepatic and distant metastasis.35
Studies have shown that TAMs in HCC tissues secrete high levels of TGF-β1, which endows HCC with stem cell–like properties and an EMT phenotype, characterized by upregulation of mesenchymal markers, ultimately increasing metastatic potential.36 In intrahepatic cholangiocarcinoma (ICC), tumor cells induce monocytes to differentiate into M2-polarized TAMs, which secrete IL-10 to activate STAT3 signaling in cancer cells, thereby promoting EMT and invasiveness.37 Similarly, Jiang et al reported that HCC-educated macrophages upregulate Wnt2b and activate the β-catenin/c-Myc pathway, driving polarization toward an M2-like phenotype accompanied by enhanced glycolysis, which in turn facilitates EMT and migration of HCC cells.38 Another study found that neurotensin (NTS) stimulates tumor cells to secrete IL-8 via MAPK and NF-κB signaling, rather than PKC/PI3K pathways.39 This IL-8 promotes M2 polarization of TAMs and induces EMT and invasiveness in HCC cells. Clinically, elevated intratumoral IL-8 levels correlate with increased metastasis and poor prognosis in HCC patients.40
TAMs Promote HCC Metastasis
TAMs also facilitate metastasis by secreting chemokines and growth factors that directly attract or stimulate tumor cells to migrate toward blood vessels or lymphatic channels. CD163+ M2-polarized TAMs are associated with poor outcomes in HCC, although the role of their lipoxygenase pathway had remained unclear.41 Recent evidence demonstrates that CD163+ TAMs express 5-lipoxygenase (5-LOX) and produce leukotrienes such as LTB4 and LTC/D/E4, which activate ERK1/2 signaling in tumor cells and enhance both proliferation and stemness.42 Inhibition of 5-LOX with zileuton suppressed tumor progression in mouse models, while patient cohorts and TCGA analyses further linked high 5-LOX expression to worse survival.
TREM1, a receptor that amplifies myeloid inflammatory signaling, has gained increasing attention in the context of HCC metastasis. Studies show that TREM1high TAMs secrete CCL7, inducing a metastasis-associated phenotype in tumor cells characterized by EMT.43 Knockdown of TREM1 reduces the pro-metastatic effects of TAMs, and high TREM1 expression correlates significantly with metastasis and poor prognosis. In HCC, infiltration of TAMs coupled with activation of the STAT3 pathway also predicts more aggressive biological behavior.44 Tumors positive for phosphorylated STAT3 (pSTAT3) are frequently associated with elevated alpha-fetoprotein (AFP), larger tumor size, and increased intrahepatic metastases. Mechanistically, TAMs secrete IL-6, which induces STAT3 phosphorylation, thereby promoting HCC cell proliferation and migration.45
Additional studies by Mano et al revealed that TAMs induce HGF expression and activate the c-Met signaling pathway, thereby enhancing the invasiveness of HCC cells.46 Moreover, TAM-derived stromal cell–derived factor-1 (SDF-1/CXCL12) recruits tumor cells along chemotactic gradients and promotes endothelial expression of adhesion molecules, facilitating tumor cell intravasation and hematogenous dissemination. In highly aggressive HCC, macrophage-derived prostaglandin E2 (PGE2) has also been observed to upregulate epigenetic regulators such as UHRF1 in tumor cells, forming a self-reinforcing loop that drives tumor progression.47
TAMs Promote Immune Evasion in HCC
The ability of tumors to survive and grow under immune surveillance reflects the development of multiple immune evasion strategies. In HCC, immune escape is closely linked to an immunosuppressive cellular network within the TME, among which TAMs are considered a central driver. TAMs orchestrate immune evasion by suppressing effector T-cell function, inducing tolerogenic DCs, and recruiting Tregs and MDSCs, collectively shaping an immune “privilege” niche that allows tumor persistence.48
Suppression of Antitumor T-Cell Immunity
Effective antitumor immunity primarily depends on the coordinated activity of cytotoxic T lymphocytes (CTLs) and CD8⁺ T cells, which recognize tumor-associated antigens and mediate tumor elimination through direct cytolysis and cytokine-driven immune amplification. In HCC, however, the intratumoral T-cell compartment is profoundly dysfunctional, characterized by functional exhaustion, impaired proliferation, and diminished production of effector cytokines. This immune dysfunction results from chronic antigenic stimulation and the persistent inflammatory milieu that define the HCC TME.
Within this immunosuppressive context, tumor-associated macrophages (TAMs) serve as critical mediators that orchestrate T-cell suppression through multiple converging mechanisms. By secreting immunoregulatory cytokines such as interleukin-10 (IL-10) and transforming growth factor-β (TGF-β), and by expressing immune checkpoint ligands including PD-L1, B7-H4, and VISTA, TAMs directly inhibit CD8⁺ T-cell activation and proliferation. Furthermore, TAMs modulate T-cell metabolism through arginase-1 (Arg1)–mediated depletion of L-arginine and inducible nitric oxide synthase (iNOS)–driven nitric oxide production, both of which impair T-cell receptor (TCR) signaling and effector function. In addition, TAMs upregulate inhibitory receptors and ligands, most notably PD-L1 and members of the B7 family.49 PD-L1, commonly elevated in M2-polarized TAMs, binds to PD-1 on T cells and delivers inhibitory signals that induce T-cell exhaustion and apoptosis. In HCC, Ruf et al observed that CD163+ TAMs with high PD-L1 expression directly contact MAIT cells, leading to their functional impairment and apoptosis.50 Sustained activation of the PD-1/PD-L1 axis is also a major mechanism underlying CTL dysfunction. Through these pathways, TAMs effectively paralyze antitumor T-cell responses.
Metabolic reprogramming further contributes to suppression of T-cell activity. M2-TAMs express high levels of arginase I (Arg1), which depletes extracellular L-arginine, an amino acid essential for T-cell proliferation and cytotoxic molecule synthesis. Similarly, indoleamine 2,3-dioxygenase (IDO) expressed by macrophages depletes tryptophan, impairing T-cell function.51 Another important mechanism is the accumulation of adenosine. TAMs frequently overexpress CD39 and CD73, ectoenzymes that hydrolyze ATP and other nucleotides into adenosine. Adenosine strongly suppresses T-cell cytotoxicity and induces production of inhibitory cytokines via the A2A receptor.52 In HCC mouse models, TAM-mediated upregulation of CD39/CD73 increased intratumoral adenosine concentrations, directly correlating with CTL depletion and functional impairment.53
Impairment of Dendritic Cell Function and Antigen Presentation
DCs are key antigen-presenting cells required to initiate T-cell priming. Effective antitumor immunity depends on DC uptake and presentation of tumor antigens to activate T cells. In HCC, however, DCs often display impaired or tolerogenic phenotypes, a process strongly influenced by TAMs.54
IL-10 and TGF-β secreted by TAMs suppress DC maturation and function. In HCC, TAM-derived IL-10 drives infiltrating DCs toward a tolerogenic phenotype characterized by low IL-12 and high IL-10 production, rendering them incapable of effectively activating CD8+ T cells.55 TGF-β induces tolerogenic DCs by upregulating inhibitory molecules such as ILT3 and ILT4, further impairing antigen presentation.56 VEGF, produced by TAMs and tumor cells, inhibits DC precursor differentiation and maturation, reducing the pool of functional DCs. Guenther et al reported that VEGF-rich environments decrease both DC abundance and function, explaining the association of high VEGF expression with immunosuppressive TME.57
Even when DCs are present within tumors, TAMs can directly interfere with their antigen-presenting efficiency. First, TAMs may competitively engulf tumor antigens: while macrophages phagocytose large amounts of tumor material, their M2-skewed phenotype results in weak antigen processing and presentation, thereby reducing the antigen load available to DCs.58 Second, direct cell–cell contact between TAMs and DCs can silence DC function. TAM surface molecules such as PD-L2 and B7-H1 interact with DC receptors to induce suppressive phenotypes or apoptosis.59 Third, TAM-derived factors including PGE2 and nitric oxide (NO) paracrinally inhibit antigen processing in DCs.60
Collectively, through cytokine secretion, competitive antigen uptake, and direct cell contact, TAMs maintain DCs in immature, tolerogenic, or apoptotic states. As a result, antigen presentation is impaired, T cells remain insufficiently activated, and antitumor immunity is blunted. By disabling DC function, TAMs disrupt the bridge between innate and adaptive immunity, thereby enabling tumors to successfully evade immune surveillance.
Promotion of Regulatory T-Cell Expansion and Function
Tregs are key suppressors of antitumor immunity in the TME. They inhibit effector T-cell and NK-cell activity through secretion of inhibitory cytokines and direct cell–cell contact. In HCC, high levels of Treg infiltration are frequently observed, and TAMs are major drivers of their accumulation and function.61
TAMs secrete chemokines that specifically attract Tregs, most notably CCL22 and CCL17, both ligands for CCR4. In HCC, elevated CCL22 secretion by macrophages is directly correlated with increased intratumoral Foxp3+ Tregs.62 Tregs expressing CCR4 migrate along the CCL22 gradient and preferentially accumulate in TAM-rich regions. Similarly, M2-polarized macrophages produce CCL17 and CCL22, reinforcing Treg recruitment into tumors.63
Beyond recruitment, TAMs also promote the differentiation of naïve T cells into Tregs. IL-10 and TGF-β, abundant products of M2-TAMs, are key drivers of peripheral Treg (pTreg) induction. Under chronic antigen stimulation and exposure to IL-10/TGF-β, conventional CD4+ T cells progressively acquire Foxp3 expression and convert into induced Tregs. The close spatial association between TAMs and Tregs ensures a local cytokine-rich milieu that favors this conversion.64 Moreover, TAM surface molecules such as ICOS-L can interact with naïve T cells to bias their differentiation toward IL-10–secreting regulatory phenotypes. Thus, through both soluble factors and surface ligands, TAMs promote expansion of the Treg pool in tumors.65
Interestingly, TAMs and Tregs engage in a reciprocal “conspiracy.” Treg-derived IL-10 reinforces M2 polarization in macrophages, while TAM-derived IDO promotes Treg survival and suppressive function.66 This feedback loop stabilizes a profoundly immunosuppressive microenvironment. Wu et al further observed that under hypoxic conditions, TREM-1+ TAMs not only impair CD8+ T cells but also act synergistically with Tregs to amplify immunosuppression.67
In HCC, high Treg infiltration is associated with poor prognosis and diminished response to immunotherapy, with TAMs being a principal orchestrator of this process. Accordingly, simultaneous targeting of TAMs and Tregs is considered a promising approach to dismantle tumor immune evasion.
Recruitment and Activation of Myeloid-Derived Suppressor Cells
MDSCs are immature myeloid cells that strongly suppress T-cell and NK-cell function in the TME. In HCC, MDSC accumulation is frequently observed, and together with TAMs they establish a cooperative immunosuppressive network.68
TAMs secrete chemokines that attract MDSC precursors to tumors. CCL2 and CXCL8 are notable examples, as they recruit not only monocytes but also mobilize MDSCs from the bone marrow into circulation and subsequently to tumor sites.69 Blocking the CCL2/CCR2 axis reduces both TAM and MDSC infiltration, thereby improving the immune microenvironment. In addition, TAM-derived growth factors such as GM-CSF and G-CSF facilitate expansion and mobilization of MDSCs in the bone marrow, increasing their availability in peripheral blood.70
Within tumors, MDSCs can further differentiate into macrophage-like cells. Monocytic MDSCs, upon entering the TME, may acquire TAM phenotypes. Signals from TAMs, including IL-10 and TGF-β, likely drive this differentiation, expanding and stabilizing the immunosuppressive myeloid compartment. In turn, MDSC-derived TAMs typically adopt M2 phenotypes, perpetuating immunosuppression.
TAMs and MDSCs share functional overlap in expressing metabolic enzymes such as Arg1, IDO, and NOS2, as well as secreting immunosuppressive factors including IL-10 and PGE2. They mutually reinforce each other: MDSC-derived IL-10 promotes M2 polarization of macrophages, while TAM-derived CCL2 recruits additional MDSCs.71 Together, they deplete essential metabolites such as arginine and generate reactive oxygen/nitrogen species, imposing a dual blockade on effector T cells. In HCC, high expression of Arg1 by both MDSCs and TAMs profoundly lowers arginine availability, virtually abolishing T-cell proliferation and cytotoxic function.72
This synergistic suppression explains why eliminating either TAMs or MDSCs alone is insufficient to reverse immunosuppression; combined strategies are often necessary to restore effective antitumor immunity. TAMs promotes the progression of HCC through multiple mechanisms. (Figure 1).
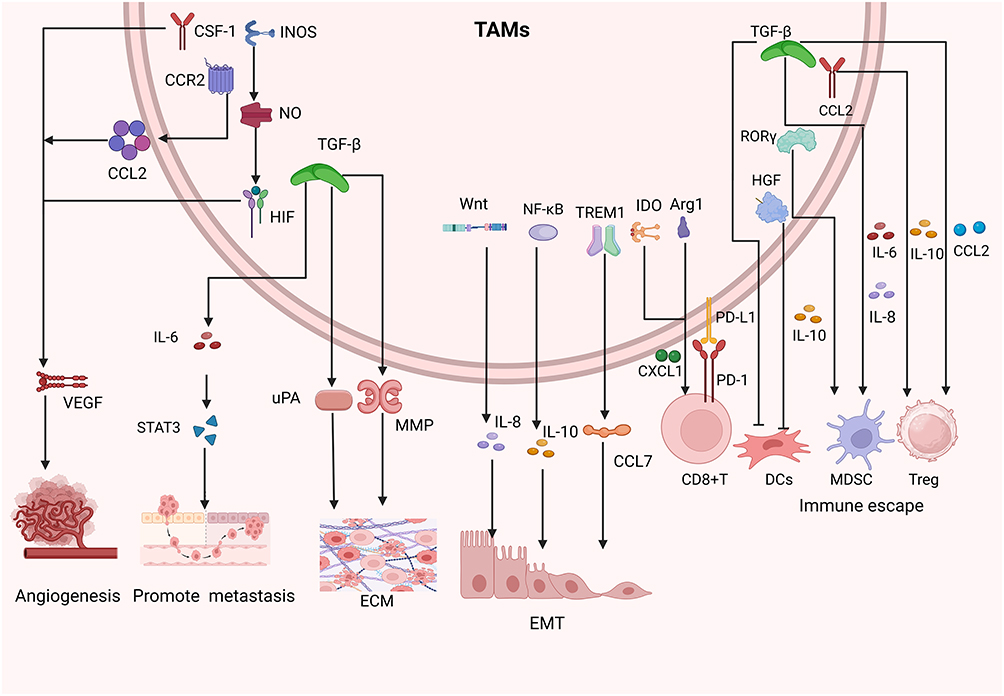 |
Figure 1 Mechanisms by which TAMs promote the progression and immune evasion of HCC. TAMs enhance angiogenesis through pro-angiogenic factors, facilitate invasion and metastasis by remodeling the ECM and activating STAT3 signaling, and suppress antitumor immunity via PD-L1/PD-1 interactions, Arg1- and IDO-mediated metabolic regulation, and secretion of IL-10 and TGF-β. They also impair dendritic cell function and recruit Tregs and MDSCs, collectively shaping an immunosuppressive TME that favors tumor growth and dissemination. |
Therapeutic Strategies Targeting TAMs
Given the critical role of TAMs in HCC progression and immune evasion, multiple therapeutic strategies have recently emerged that target these cells. Broadly, these approaches can be divided into three categories: (1) inhibiting or depleting protumor TAMs by reducing their abundance or suppressing their tumor-supportive functions. (2) activating or enhancing antitumor TAMs by promoting polarization toward an M1 phenotype with enhanced cytotoxicity and antigen-presenting capacity.
Inhibition of Protumor TAMs
Direct depletion of TAMs is one of the most straightforward strategies and can be achieved by blocking survival signals or recruitment pathways. Macrophage differentiation and survival are strongly dependent on colony-stimulating factor-1 (CSF-1, also known as M-CSF) and its receptor CSF1R, making this pathway a prime target.73 In HCC and other tumors, infiltration of CSF1R+ TAMs is increased and correlates with poor prognosis. Pharmacologic inhibition of CSF1R reduces TAM density within tumors and enhances antitumor immunity. Several CSF1R inhibitors are currently under clinical investigation in multiple solid tumors, including monoclonal antibodies such as Emactuzumab (RG7155) and small-molecule tyrosine kinase inhibitors such as Pexidartinib,74 however, robust HCC-specific clinical evidence remains limited. Early studies demonstrated decreased macrophage numbers and increased T-cell activity in some solid tumors, although efficacy in HCC remains to be validated.75
Targeting monocyte recruitment is another approach. CCR2 antagonists have been shown to reduce TAMs and MDSCs and enhance CD8+ T-cell activity in murine HCC. However, clinical efficacy of this axis has been modest. For example, a Phase II trial of the CCL2-neutralizing antibody Carlumab failed due to compensatory upregulation of CCL2 by tumor cells, which offset drug activity.76
Beyond depletion, strategies aim to attenuate protumor functions of TAMs. Since TAMs secrete VEGF and other angiogenic mediators, anti-angiogenic therapies can counteract their effects. The first-line HCC regimen combining atezolizumab with bevacizumab exemplifies this dual blockade of PD-L1 and VEGF, simultaneously relieving immune suppression and angiogenesis. Bevacizumab, in particular, neutralizes VEGF produced by TAMs and other cells.77 Another noteworthy target is CD163, a classical M2 marker. Strategies include conjugating drugs to CD163 for selective delivery to TAMs, or employing anti-CD163 antibodies to trigger antibody-dependent cellular cytotoxicity (ADCC).78
Overall, therapies targeting key protumor molecules or markers in TAMs represent promising means of weakening their support for tumor progression.
Activation of Antitumor TAMs
Reinvigorating the antitumor potential of TAMs can enhance phagocytosis and tumor killing while promoting adaptive immune activation. Tumor cells frequently escape clearance by expressing “don’t eat me” signals, the best-known being CD47. By binding to SIRPα on macrophages, CD47 delivers a strong inhibitory phagocytosis signal.79 HCC and other cancers commonly upregulate CD47 to evade macrophage-mediated killing. This has motivated the development of CD47–SIRPα blockade strategies, including anti-CD47 monoclonal antibodies and SIRPα–Fc fusion proteins.80 These approaches have shown promise in hematologic malignancies and are now being tested in solid tumors including HCC.
Macrophage activation can also be achieved through costimulatory receptors such as CD40. CD40 is expressed on macrophages and DCs, and its engagement induces robust activation. Agonistic anti-CD40 antibodies (eg., Selicrelumab) mimic T-cell help signals, driving TAMs to release pro-inflammatory cytokines and cytotoxic mediators, effectively reprogramming them toward an M1 phenotype.81
As innate immune cells, macrophages can be stimulated via pattern recognition receptors (PRRs) to release pro-inflammatory mediators. This principle has been leveraged by using pathogen-associated molecular pattern (PAMP) mimetics in cancer immunotherapy. TLR agonists such as Poly I:C (TLR3), imiquimod or R848 (TLR7/8) can activate macrophages to produce IL-12 and TNF-α, thereby enhancing tumoricidal capacity.82,83 Similarly, STING agonists and NOD-like receptor ligands represent additional approaches. While effective, systemic inflammation remains a concern, suggesting that local delivery or transient stimulation may be optimal.
A novel strategy involves genetic engineering of macrophages, termed CAR-M (chimeric antigen receptor macrophages).84 By equipping macrophages or monocytes with tumor-specific receptors, CAR-Ms can selectively phagocytose tumor cells and sustain immune activity within the TME.85 In HCC, antigens such as GPC3 represent promising targets, and CAR-M therapy may help overcome the immunosuppressive barriers that limit CAR-T efficacy in solid tumors. We have also made a detailed summary of the relevant clinical trials targeting TAMs. (Table 2).
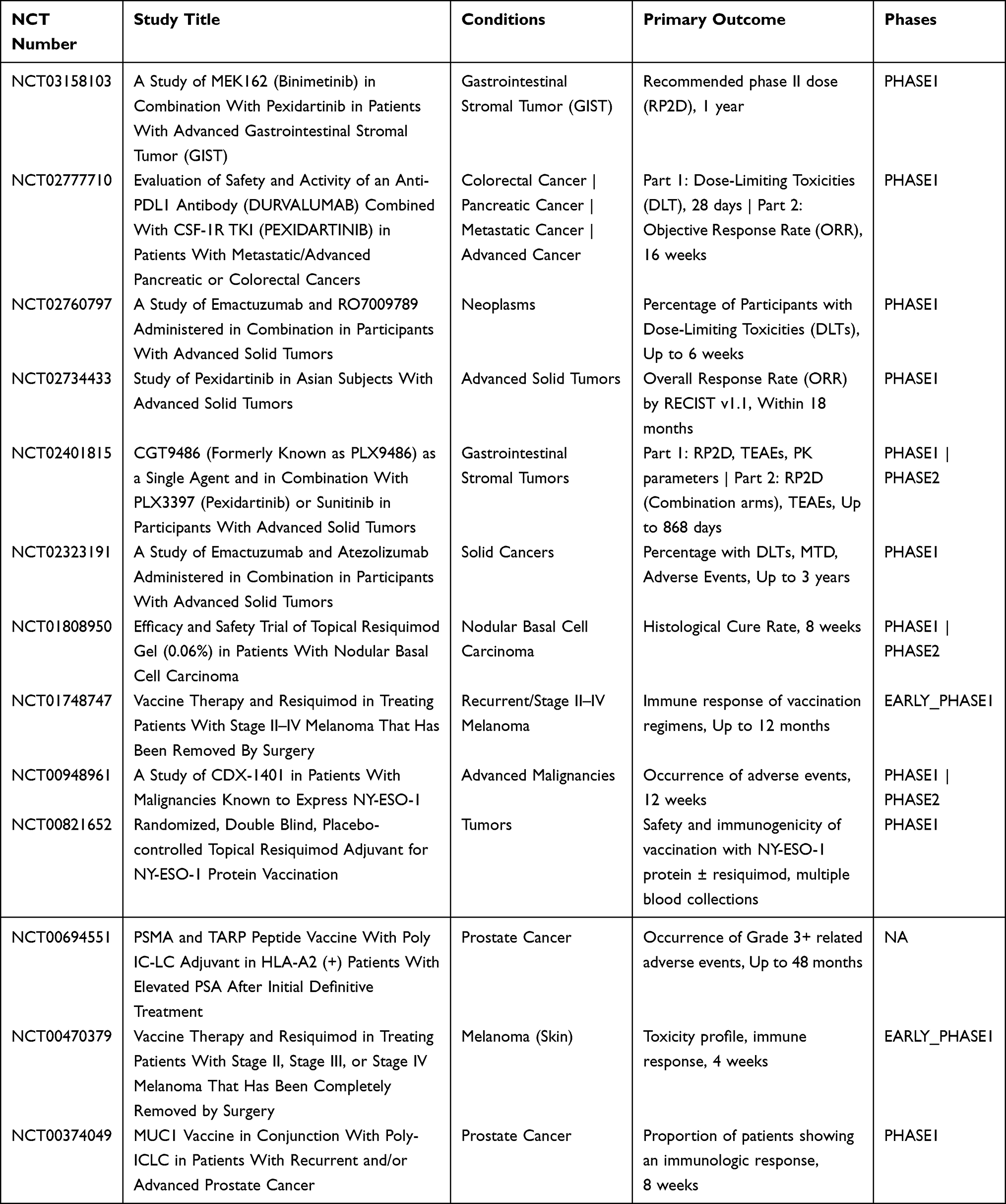 |
Table 2 Progress of TAM-Targeted Therapeutic Strategies in Solid Tumor Clinical Trials |
Challenges and Translational Barriers of TAM-Targeted Therapy in HCC
Despite encouraging preclinical findings and early clinical evidence from multiple solid tumors, the translational applicability of TAM-targeted therapies to HCC remains incompletely understood. Most currently available evidence regarding CSF1R inhibitors, CCR2 blockade, CD47–SIRPα inhibition, and CAR-M therapy has been generated in non-hepatic malignancies, including melanoma, colorectal cancer, pancreatic cancer, and sarcoma.86–88 Whether these findings can be directly extrapolated to HCC requires careful consideration, given the unique immunobiology of the liver.
Unlike many other organs, the liver is characterized by a highly tolerogenic immune environment due to its continuous exposure to gut-derived antigens through the portal circulation.12 This physiological immune tolerance contributes to a suppressive hepatic microenvironment that may fundamentally alter macrophage activation, antigen presentation, and responses to immunotherapy. In addition, hepatic macrophage populations are highly heterogeneous and include both infiltrating monocyte-derived macrophages and tissue-resident Kupffer cells, which possess distinct ontogeny, transcriptional programs, and immunological functions.89 Evidence suggests that Kupffer cells may exhibit context-dependent protumor or antitumor properties during hepatocarcinogenesis, further complicating therapeutic targeting strategies.90 Moreover, the dual blood supply of the liver and the extensive crosstalk among hepatocytes, hepatic stellate cells, sinusoidal endothelial cells, and immune populations create a uniquely complex tumor microenvironment in HCC. These liver-specific features may influence drug delivery, macrophage polarization, and therapeutic responsiveness in ways that differ substantially from other solid tumors.90
Importantly, clinical evidence supporting TAM-targeted therapy specifically in HCC remains limited. Most published clinical studies are early-phase trials involving heterogeneous tumor types, and HCC-specific efficacy data are still lacking. Therefore, although TAM-directed approaches represent promising therapeutic avenues, further investigation is required to clarify the functional heterogeneity of hepatic TAM subsets, identify HCC-specific biomarkers, and determine which patient populations are most likely to benefit from macrophage-targeted interventions. Future studies integrating single-cell transcriptomics, spatial immunophenotyping, and etiology-specific immune profiling may help overcome these translational barriers and facilitate the development of more precise TAM-centered therapeutic strategies for HCC.
Conclusion
TAMs are integral orchestrators of the initiation, progression, and immune evasion of HCC. They not only promote tumor growth, angiogenesis, invasion, and metastasis but also establish a profoundly immunosuppressive microenvironment that enables tumor cells to evade immune surveillance. Both clinical and experimental studies consistently demonstrate that elevated TAM infiltration correlates with advanced disease stage, poor patient prognosis, and diminished responsiveness to immunotherapy, underscoring their potential as pivotal therapeutic targets.
Therapeutic strategies targeting TAMs, including the inhibition of their recruitment and survival, reprogramming toward tumoricidal phenotypes, and enhancement of their phagocytic or antigen-presenting functions, have demonstrated encouraging results in preclinical models, with several approaches currently progressing into clinical evaluation. In particular, combinatorial regimens that integrate TAM-targeted agents with immune checkpoint blockade may offer a means to overcome immune tolerance and reinvigorate antitumor immunity in HCC.
Nevertheless, significant challenges persist, including the functional heterogeneity of TAM subsets, tumor-derived compensatory mechanisms, and potential safety concerns associated with macrophage modulation. Importantly, emerging evidence suggests that TAM biology is not uniform across HCC etiologies. HBV-, HCV-, and NAFLD-associated HCC exhibit distinct inflammatory and metabolic milieus that shape divergent immune microenvironments and may differentially influence TAM recruitment, polarization, and functional states. This etiology-dependent heterogeneity may also contribute to variable therapeutic responses, highlighting a critical gap in current translational research. Future studies should therefore prioritize etiology-stratified investigation of TAM-targeted interventions, alongside the identification of reliable molecular markers defining TAM subpopulations, delineation of their cross-talk networks with other immunoregulatory cells, and the development of optimized delivery platforms and rational combination strategies. With continued mechanistic insight and rigorous clinical validation, TAM-centered therapeutic interventions hold substantial promise to transition from conceptual frameworks to clinical reality. Ultimately, precise modulation of TAMs represents a transformative frontier in HCC therapy, offering the potential to improve patient outcomes when incorporated into comprehensive and personalized treatment paradigms.
Abbreviations
ADCC, antibody-dependent cellular cytotoxicity; AFP, alpha-fetoprotein; Arg1, arginase-1; CAR-M, chimeric antigen receptor macrophage; CCR2, C–C chemokine receptor type 2; CCL, C–C motif chemokine ligand; CD, cluster of differentiation; CSF1 (M-CSF), macrophage colony-stimulating factor; CSF1R, colony-stimulating factor 1 receptor; CTL, cytotoxic T lymphocyte; CXCL8, C–X–C motif chemokine ligand 8 (IL-8); DC, dendritic cell; ECM, extracellular matrix; EMT, epithelial–mesenchymal transition; EMP, erythromyeloid progenitor; ERK1/2, extracellular signal-regulated kinases 1/2; GM-CSF, granulocyte–macrophage colony-stimulating factor; G-CSF, granulocyte colony-stimulating factor; HCC, hepatocellular carcinoma; HGF, hepatocyte growth factor; HIF-1α, hypoxia-inducible factor 1 alpha; HBV, hepatitis B virus; ICIs, immune checkpoint inhibitors; ICOS-L, inducible T-cell costimulator ligand; IDO, indoleamine 2,3-dioxygenase; IFN-γ, interferon-gamma; IL, interleukin; ILT3/ILT4, immunoglobulin-like transcript 3/4; iNOS, inducible nitric oxide synthase; KC, Kupffer cell; KLF4, Krüppel-like factor 4; LPS, lipopolysaccharide; LTB4, leukotriene B4; M1/M2, classically/alternatively activated macrophage phenotype; MAPK, mitogen-activated protein kinase; MARCO, macrophage receptor with collagenous structure; MDSC, myeloid-derived suppressor cell; MHC-II, major histocompatibility complex class II; MMP, matrix metalloproteinase; mTOR, mechanistic target of rapamycin; NF-κB, nuclear factor kappa B; NK cell, natural killer cell; NOD, nucleotide-binding oligomerization domain; NO, nitric oxide; NOS2, nitric oxide synthase 2; NTS, neurotensin; PAMP, pathogen-associated molecular pattern; PD-1, programmed cell death protein 1; PD-L1/PD-L2, programmed death-ligand 1/2; PGE2, prostaglandin E2; PI3K, phosphatidylinositol 3-kinase; PlGF, placental growth factor; PRL-3, phosphatase of regenerating liver 3; PPARγ, peroxisome proliferator-activated receptor gamma; SDF-1 (CXCL12), stromal cell–derived factor 1; SIRPα, signal regulatory protein alpha; SPP1, secreted phosphoprotein 1 (osteopontin); STAT3/STAT6, signal transducer and activator of transcription 3/6; STING, stimulator of interferon genes; TAM, tumor-associated macrophage; TCR, T-cell receptor; TGF-β, transforming growth factor beta; Th1, type 1 helper T cell; TLR, toll-like receptor; TME, tumor microenvironment; TNF-α, tumor necrosis factor alpha; TREM1, triggering receptor expressed on myeloid cells 1; Treg, regulatory T cell; UHRF1, ubiquitin-like with PHD and ring finger domains 1; VEGF/VEGFA, vascular endothelial growth factor A; VISTA, V-domain immunoglobulin suppressor of T cell activation; Wnt, wingless/integrated signaling pathway.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Vogel A, Meyer T, Sapisochin G, Salem R, Saborowski A. Hepatocellular carcinoma. Lancet. 2022;400(10360):1345–15. doi:10.1016/S0140-6736(22)01200-4
2. Rajbhandari R, Nguyen VH, Knoble A, Fricker G, Chung RT. Advances in the management of hepatitis B. BMJ. 2025;389:e079579.
3. Affo S, Filliol A, Gores GJ, Schwabe RF. Fibroblasts in liver cancer: functions and therapeutic translation. Lancet Gastroenterol Hepatol. 2023;8(8):748–759. doi:10.1016/S2468-1253(23)00111-5
4. Thomas JA, Kendall BJ, El-Serag HB, Thrift AP, Macdonald GA. Hepatocellular and extrahepatic cancer risk in people with non-alcoholic fatty liver disease. Lancet Gastroenterol Hepatol. 2024;9(2):159–169. doi:10.1016/S2468-1253(23)00275-3
5. Cheng K, Cai N, Zhu J, Yang X, Liang H, Zhang W. Tumor-associated macrophages in liver cancer: from mechanisms to therapy. Cancer Commun. 2022;42(11):1112–1140. doi:10.1002/cac2.12345
6. Chen C, Wang Z, Ding Y, Qin Y. Tumor microenvironment-mediated immune evasion in hepatocellular carcinoma. Front Immunol. 2023;14:1133308. doi:10.3389/fimmu.2023.1133308
7. Wang Z, Wang Y, Gao P, Ding J. Immune checkpoint inhibitor resistance in hepatocellular carcinoma. Cancer Lett. 2023;555:216038. doi:10.1016/j.canlet.2022.216038
8. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 2019;30(1):36–50. doi:10.1016/j.cmet.2019.06.001
9. Peranzoni E, Lemoine J, Vimeux L, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci U S A. 2018;115(17):E4041–e4050.
10. Ezhilarasan D, Najimi M. Deciphering the possible reciprocal loop between hepatic stellate cells and cancer cells in the tumor microenvironment of the liver. Crit rev oncol/hematol. 2023;182:103902. doi:10.1016/j.critrevonc.2022.103902
11. Pfister D, Núñez NG, Pinyol R, et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. 2021;592(7854):450–456. doi:10.1038/s41586-021-03362-0
12. Ringelhan M, Pfister D, O’Connor T, Pikarsky E, Heikenwalder M. The immunology of hepatocellular carcinoma. Nat Immunol. 2018;19(3):222–232. doi:10.1038/s41590-018-0044-z
13. Buonomo EL, Mei S, Guinn SR, et al. Liver stromal cells restrict macrophage maturation and stromal IL-6 limits the differentiation of cirrhosis-linked macrophages. J Hepatol. 2022;76(5):1127–1137. doi:10.1016/j.jhep.2021.12.036
14. Liu J, Chen S, Wang W, et al. Cancer-associated fibroblasts promote hepatocellular carcinoma metastasis through chemokine-activated hedgehog and TGF-β pathways. Cancer Lett. 2016;379(1):49–59. doi:10.1016/j.canlet.2016.05.022
15. Li X, Yao W, Yuan Y, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66(1):157–167. doi:10.1136/gutjnl-2015-310514
16. Sathe A, Mason K, Grimes SM, et al. Colorectal cancer metastases in the liver establish immunosuppressive spatial networking between tumor-associated spp1+ macrophages and fibroblasts. Clin Cancer Res. 2023;29(1):244–260. doi:10.1158/1078-0432.CCR-22-2041
17. Cortés M, Sanchez-Moral L, de Barrios O, et al. Tumor-associated macrophages (TAMs) depend on ZEB1 for their cancer-promoting roles. EMBO J. 2017;36(22):3336–3355. doi:10.15252/embj.201797345
18. Fu Y, Liu S, Zeng S, Shen H. From bench to bed: the tumor immune microenvironment and current immunotherapeutic strategies for hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38(1):396. doi:10.1186/s13046-019-1396-4
19. Lurje I, Uluk D, Hammerich L, Pratschke J, Tacke F, Lurje G. Comparing hypothermic oxygenated and normothermic liver machine perfusion: translation matters. J Hepatol. 2024;80(4):e163–e165. doi:10.1016/j.jhep.2023.09.027
20. Chu X, Tian Y, Lv C. Decoding the spatiotemporal heterogeneity of tumor-associated macrophages. Mol Cancer. 2024;23(1):150. doi:10.1186/s12943-024-02064-1
21. Mulder K, Patel AA, Kong WT, et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity. 2021;54(8):1883–1900.e1885. doi:10.1016/j.immuni.2021.07.007
22. Helal IM, Kamal MA, Abd El-Aziz MK. El Tayebi HM: epigenetic tuning of tumour-associated macrophages (TAMs): a potential approach in hepatocellular carcinoma (HCC) immunotherapy. Expert Rev. Mol. Med. 2024;26:e18.
23. Chen S, Morine Y, Tokuda K, et al. Cancer‑associated fibroblast‑induced M2‑polarized macrophages promote hepatocellular carcinoma progression via the plasminogen activator inhibitor‑1 pathway. Int J Oncol. 2021;59(2). doi:10.3892/ijo.2021.5239
24. Liu C, Xie J, Lin B, et al. Pan-cancer single-cell and spatial-resolved profiling reveals the immunosuppressive role of apoe+ macrophages in immune checkpoint inhibitor therapy. Adv. Sci. 2024;11(23):e2401061. doi:10.1002/advs.202401061
25. Svensson MC, Svensson M, Nodin B, et al. High infiltration of cd68+/cd163- macrophages is an adverse prognostic factor after neoadjuvant chemotherapy in esophageal and gastric adenocarcinoma. J. Innate Immun. 2022;14(6):615–628. doi:10.1159/000524434
26. Dallavalasa S, Beeraka NM, Basavaraju CG, et al. The Role of Tumor Associated Macrophages (TAMs) in cancer progression, chemoresistance, angiogenesis and metastasis - current status. Curr. Med. Chem. 2021;28(39):8203–8236. doi:10.2174/0929867328666210720143721
27. Do MH, Shi W, Ji L, et al. Reprogramming tumor-associated macrophages to outcompete endovascular endothelial progenitor cells and suppress tumor neoangiogenesis. Immunity. 2023;56(11):2555–2569.e2555. doi:10.1016/j.immuni.2023.10.010
28. Zhang T, Liu L, Lai W, et al. Interaction with tumor‑associated macrophages promotes PRL‑3‑induced invasion of colorectal cancer cells via MAPK pathway‑induced EMT and NF‑κB signaling‑induced angiogenesis. Oncol Rep. 2019;41(5):2790–2802. doi:10.3892/or.2019.7049
29. Toledo B, Zhu Chen L, Paniagua-Sancho M, Marchal JA, Perán M, Giovannetti E. Deciphering the performance of macrophages in tumour microenvironment: a call for precision immunotherapy. J hematol oncol. 2024;17(1):44.
30. Yang C-L, Song R, Hu J-W, et al. Integrating single-cell and bulk RNA sequencing reveals CK19 + cancer stem cells and their specific SPP1 + tumor-associated macrophage niche in HBV-related hepatocellular carcinoma. Hepatol Internat. 2024;18(1):73–90. doi:10.1007/s12072-023-10615-9
31. Tjiu JW, Chen JS, Shun CT, et al. Inoue H et al: tumor-associated macrophage-induced invasion and angiogenesis of human basal cell carcinoma cells by cyclooxygenase-2 induction. J Invest Dermatol. 2009;129(4):1016–1025. doi:10.1038/jid.2008.310
32. Bartneck M, Schrammen PL, Möckel D, et al. The CCR2(+) macrophage subset promotes pathogenic angiogenesis for tumor vascularization in fibrotic livers. CMGH. 2019;7(2):371–390. doi:10.1016/j.jcmgh.2018.10.007
33. Xie P, Guo L, Yu Q, et al. ACE2 Enhances Sensitivity to PD-L1 blockade by inhibiting macrophage-induced immunosuppression and angiogenesis. Cancer Res. 2025;85(2):299–313. doi:10.1158/0008-5472.CAN-24-0954
34. Lu Y, Han G, Zhang Y, et al. M2 macrophage-secreted exosomes promote metastasis and increase vascular permeability in hepatocellular carcinoma. Cell Commun. Signal. 2023;21(1):299. doi:10.1186/s12964-022-00872-w
35. Han S, Bao X, Zou Y, et al. d-lactate modulates M2 tumor-associated macrophages and remodels immunosuppressive tumor microenvironment for hepatocellular carcinoma. Sci Adv. 2023;9(29):eadg2697. doi:10.1126/sciadv.adg2697
36. Zhu F, Li X, Chen S, Zeng Q, Zhao Y, Luo F. Tumor-associated macrophage or chemokine ligand CCL17 positively regulates the tumorigenesis of hepatocellular carcinoma. Med Oncol. 2016;33(2):17. doi:10.1007/s12032-016-0729-9
37. Yang LF, Zhang ZB, Wang L. S100A9 promotes tumor-associated macrophage for M2 macrophage polarization to drive human liver cancer progression: an in vitro study. Kaohsiung J. Med. Sci. 2023;39(4):345–353. doi:10.1002/kjm2.12651
38. Cai J, Song L, Zhang F, et al. Targeting SRSF10 might inhibit M2 macrophage polarization and potentiate anti-PD-1 therapy in hepatocellular carcinoma. Cancer Commun. 2024;44(11):1231–1260. doi:10.1002/cac2.12607
39. Jiang Y, Han Q, Zhao H, Zhang J. Promotion of epithelial-mesenchymal transformation by hepatocellular carcinoma-educated macrophages through Wnt2b/β-catenin/c-Myc signaling and reprogramming glycolysis. J Exp Clin Cancer. 2021;40(1):13. doi:10.1186/s13046-020-01808-3
40. Liu M, Zhong YB, Shao J, Zhang C, Shi C. Tumor-associated macrophages promote human hepatoma Huh-7 cell migration and invasion through the Gli2/IGF-II/ERK1/2 axis by secreting TGF-β1. Cancer Biol Ther. 2020;21(11):1041–1050. doi:10.1080/15384047.2020.1824478
41. Bagchi S, Yuan R, Huang HL, et al. The acid-sensing receptor GPR65 on tumor macrophages drives tumor growth in obesity. Sci immunol. 2024;9(100):eadg6453. doi:10.1126/sciimmunol.adg6453
42. Nosaka T, Murata Y, Takahashi K, et al. Hepatocellular carcinoma progression promoted by 5-lipoxygenase activity in CD163(+) tumor-associated macrophages. Biomed Pharmacothe. 2023;162:114592. doi:10.1016/j.biopha.2023.114592
43. Huang S, He L, Zhao Y, et al. TREM1(+) tumor-associated macrophages secrete CCL7 to promote hepatocellular carcinoma metastasis. J Cancer Res Clin Oncol. 2024;150(6):320. doi:10.1007/s00432-024-05831-1
44. Yuan H, Lin Z, Liu Y, et al. Intrahepatic cholangiocarcinoma induced M2-polarized tumor-associated macrophages facilitate tumor growth and invasiveness. Can Cell Inter. 2020;20(1):586. doi:10.1186/s12935-020-01687-w
45. Yao RR, Li JH, Zhang R, Chen RX, Wang YH. M2-polarized tumor-associated macrophages facilitated migration and epithelial-mesenchymal transition of HCC cells via the TLR4/STAT3 signaling pathway. World J. Surg. Oncol. 2018;16(1):9. doi:10.1186/s12957-018-1312-y
46. Mano Y, Aishima S, Fujita N, et al. Tumor-associated macrophage promotes tumor progression via STAT3 signaling in hepatocellular carcinoma. Pathobiology. 2013;80(3):146–154. doi:10.1159/000346196
47. Che D, Zhang S, Jing Z, et al. Macrophages induce EMT to promote invasion of lung cancer cells through the IL-6-mediated COX-2/PGE(2)/β-catenin signalling pathway. Mol Immunol. 2017;90:197–210. doi:10.1016/j.molimm.2017.06.018
48. Cai J, Zhang P, Cai Y, et al. Lactylation-driven nupr1 promotes immunosuppression of tumor-infiltrating macrophages in hepatocellular carcinoma. Adv. Sci. 2025;12(20):e2413095. doi:10.1002/advs.202413095
49. Xie M, Lin Z, Ji X, et al. FGF19/FGFR4-mediated elevation of ETV4 facilitates hepatocellular carcinoma metastasis by upregulating PD-L1 and CCL2. J Hepatol. 2023;79(1):109–125. doi:10.1016/j.jhep.2023.02.036
50. Ruf B, Bruhns M, Babaei S, et al. Tumor-associated macrophages trigger MAIT cell dysfunction at the HCC invasive margin. Cell. 2023;186(17):3686–3705.e3632. doi:10.1016/j.cell.2023.07.026
51. Chen J, Lin Z, Liu L, et al. GOLM1 exacerbates CD8(+) T cell suppression in hepatocellular carcinoma by promoting exosomal PD-L1 transport into tumor-associated macrophages. Signal Transduct Target Ther. 2021;6(1):397. doi:10.1038/s41392-021-00784-0
52. Zhang F, Jiang Q, Cai J, et al. Activation of NOD1 on tumor-associated macrophages augments CD8(+) T cell-mediated antitumor immunity in hepatocellular carcinoma. Sci Adv. 2024;10(40):eadp8266. doi:10.1126/sciadv.adp8266
53. Lima CF, Tamegnon A, Rodriguez S, et al. Exploring the expression of adenosine pathway-related markers cd73 and cd39 in colorectal and pancreatic carcinomas characterized by multiplex immunofluorescence: a pilot study. Pathobiology. 2024;91(3):205–218. doi:10.1159/000534677
54. Zhang Q, He Y, Luo N, et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell. 2019;179(4):829–845.e820. doi:10.1016/j.cell.2019.10.003
55. Kang S, Mansurov A, Kurtanich T, et al. Engineered GM-CSF polarizes protumorigenic tumor-associated macrophages to an antitumorigenic phenotype and potently synergizes with IL-12 immunotherapy. J Immunother Cancer. 2024;12(12):e009541. doi:10.1136/jitc-2024-009541
56. Modak M, Mattes AK, Reiss D, et al. CD206+ tumor-associated macrophages cross-present tumor antigen and drive antitumor immunity. JCI Insight. 2022;7(11). doi:10.1172/jci.insight.155022.
57. Guenther C. Stiffness regulates dendritic cell and macrophage subtype development and increased stiffness induces a tumor-associated macrophage phenotype in cancer co-cultures. Front Immunol. 2024;15:1434030. doi:10.3389/fimmu.2024.1434030
58. Sung PS. Crosstalk between tumor-associated macrophages and neighboring cells in hepatocellular carcinoma. Clinical and molecular hepatology. Clinical and Molecular Hepatology. 2022;28(3):333–350. doi:10.3350/cmh.2021.0308
59. Matsubara E, Shinchi Y, Komohara Y, et al. PD-L2 overexpression on tumor-associated macrophages is one of the predictors for better prognosis in lung adenocarcinoma. Med. Mol. Morphol. 2023;56(4):250–256. doi:10.1007/s00795-023-00361-0
60. Ma X, Guo Z, Wei X, et al. Spatial distribution and predictive significance of dendritic cells and macrophages in esophageal cancer treated with combined chemoradiotherapy and pd-1 blockade. Front Immunol. 2021;12:786429. doi:10.3389/fimmu.2021.786429
61. Chen J, Feng W, Sun M, et al. TGF-β1-induced sox18 elevation promotes hepatocellular carcinoma progression and metastasis through transcriptionally upregulating PD-L1 and CXCL12. Gastroenterology. 2024;167(2):264–280. doi:10.1053/j.gastro.2024.02.025
62. Yu X, Qian J, Ding L, et al. Galectin-1-induced tumor associated macrophages repress antitumor immunity in hepatocellular carcinoma through recruitment of Tregs. Adv. Sci. 2025;12(11):e2408788. doi:10.1002/advs.202408788
63. Wang Y, Chen W, Qiao S, et al. Lipid droplet accumulation mediates macrophage survival and Treg recruitment via the CCL20/CCR6 axis in human hepatocellular carcinoma. Cell. Mol. Immunol. 2024;21(10):1120–1130. doi:10.1038/s41423-024-01199-x
64. Zhou SL, Zhou ZJ, Hu ZQ, et al. Tumor-associated neutrophils recruit macrophages and t-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology. 2016;150(7):1646–1658.e1617. doi:10.1053/j.gastro.2016.02.040
65. Liu C, Chikina M, Deshpande R, et al. Treg cells promote the srebp1-dependent metabolic fitness of tumor-promoting macrophages via repression of cd8(+) t cell-derived interferon-γ. Immunity. 2019;51(2):381–397.e386. doi:10.1016/j.immuni.2019.06.017
66. Oh MH, Sun IH, Zhao L, et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J Clin Invest. 2020;130(7):3865–3884. doi:10.1172/JCI131859
67. Zheng H, Peng X, Yang S, et al. Targeting tumor-associated macrophages in hepatocellular carcinoma: biology, strategy, and immunotherapy. Cell Death Discov. 2023;9(1):65. doi:10.1038/s41420-023-01356-7
68. Chen XM, Liang YB, Zuo JX, et al. ZG16B: a key regulator of tumor progression and immune microenvironment modulation in cancer (Review). Int J Mol Med. 2026;57(3). doi:10.3892/ijmm.2026.5729
69. Feng PH, Yu CT, Chen KY, et al. S100A9(+) MDSC and TAM-mediated EGFR-TKI resistance in lung adenocarcinoma: the role of RELB. Oncotarget. 2018;9(7):7631–7643. doi:10.18632/oncotarget.24146
70. Loeuillard E, Yang J, Buckarma E, et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J Clin Invest. 2020;130(10):5380–5396. doi:10.1172/JCI137110
71. Chen S, Wang M, Lu T, et al. JMJD6 in tumor-associated macrophage regulates macrophage polarization and cancer progression via STAT3/IL-10 axis. Oncogene. 2023;42(37):2737–2750. doi:10.1038/s41388-023-02781-9
72. Xiao J, Wang S, Chen L, et al. 25-Hydroxycholesterol regulates lysosome AMP kinase activation and metabolic reprogramming to educate immunosuppressive macrophages. Immunity. 2024;57(5):1087–1104.e1087. doi:10.1016/j.immuni.2024.03.021
73. Fujiwara T, Yakoub MA, Chandler A, et al. CSF1/CSF1R Signaling Inhibitor Pexidartinib (PLX3397) reprograms tumor-associated macrophages and stimulates t-cell infiltration in the sarcoma microenvironment. Mol Cancer Ther. 2021;20(8):1388–1399. doi:10.1158/1535-7163.MCT-20-0591
74. Zhu Y, Yang J, Xu D, et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut. 2019;68(9):1653–1666. doi:10.1136/gutjnl-2019-318419
75. Ries CH, Cannarile MA, Hoves S, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25(6):846–859. doi:10.1016/j.ccr.2014.05.016
76. Obmolova G, Teplyakov A, Malia TJ, et al. Structural basis for high selectivity of anti-CCL2 neutralizing antibody CNTO 888. Mol Immunol. 2012;51(2):227–233. doi:10.1016/j.molimm.2012.03.022
77. Cappuyns S, Philips G, Vandecaveye V, et al. PD-1(-) CD45RA(+) effector-memory CD8 T cells and CXCL10(+) macrophages are associated with response to atezolizumab plus bevacizumab in advanced hepatocellular carcinoma. Nat Commun. 2023;14(1):7825. doi:10.1038/s41467-023-43381-1
78. Heemskerk N, Gruijs M, Temming AR, et al. Augmented antibody-based anticancer therapeutics boost neutrophil cytotoxicity. J Clin Invest. 2021;131(6). doi:10.1172/JCI134680.
79. Logtenberg MEW, Scheeren FA, Schumacher TN. The CD47-SIRPα Immune Checkpoint. Immunity. 2020;52(5):742–752. doi:10.1016/j.immuni.2020.04.011
80. van Duijn A, Van der Burg SH, Scheeren FA. CD47/SIRPα axis: bridging innate and adaptive immunity. J Immunother Cancer. 2022;10(7):e004589. doi:10.1136/jitc-2022-004589
81. Machiels JP, Gomez-Roca C, Michot JM, et al. Phase Ib study of anti-CSF-1R antibody emactuzumab in combination with CD40 agonist selicrelumab in advanced solid tumor patients. J Immunother Cancer. 2020;8(2):e001153. doi:10.1136/jitc-2020-001153
82. Chen E, Chen C, Niu Z, et al. Poly(I:C) preconditioning protects the heart against myocardial ischemia/reperfusion injury through TLR3/PI3K/Akt-dependent pathway. Signal Transduct Target Ther. 2020;5(1):216. doi:10.1038/s41392-020-00257-w
83. Wen F, Liu W, Li Y, et al. TLR7/8 agonist (R848) inhibit bovine X sperm motility via PI3K/GSK3α/β and PI3K/NFκB pathways. Int J Biol Macromol. 2023;232:123485. doi:10.1016/j.ijbiomac.2023.123485
84. Pierini S, Gabbasov R, Oliveira-Nunes MC, et al. Chimeric antigen receptor macrophages (CAR-M) sensitize HER2+ solid tumors to PD1 blockade in pre-clinical models. Nat Commun. 2025;16(1):706. doi:10.1038/s41467-024-55770-1
85. Wang M, Qin Z, Bian XW, Shi Y. Harnessing chimeric antigen receptor macrophages against solid tumors. Cancer Commun. 2025.
86. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. 2017;5(1):53. doi:10.1186/s40425-017-0257-y
87. Kzhyshkowska J, Shen J, Larionova I. Targeting of TAMs: can we be more clever than cancer cells? Cell Mol Immunol. 2024;21(12):1376–1409.
88. Qu T, Li B, Wang Y. Targeting CD47/SIRPα as a therapeutic strategy, where we are and where we are headed. Biomark Res. 2022;10(1):20. doi:10.1186/s40364-022-00373-5
89. Guilliams M, Scott CL. Does niche competition determine the origin of tissue-resident macrophages? Nat Rev Immunol. 2017;17(7):451–460.
90. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17(5):306–321.
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
CTB-193M12.5 Promotes Hepatocellular Carcinoma Progression via Enhancing NSD1-Mediated WNT10B/Wnt/β-Catenin Signaling Activation
Zhang S, Jiang M, Cao H, Xiong J, Xu J
Journal of Hepatocellular Carcinoma 2022, 9:553-569
Published Date: 7 June 2022
Clinical Results, Risk Factors, and Future Directions of Ultrasound-Guided Percutaneous Microwave Ablation for Hepatocellular Carcinoma
Dong TT, Wang L, Li M, Yin C, Li YY, Nie F
Journal of Hepatocellular Carcinoma 2023, 10:733-743
Published Date: 15 May 2023
Hsa_circ_0007991 Promotes Immune Evasion in Hepatocellular Carcinoma via Regulation of the miR-505-3p/CANX Axis
Wang L, Liang L, Qian J, Yu C, Shi Y, Yan X, Chen X
Journal of Hepatocellular Carcinoma 2025, 12:1337-1351
Published Date: 7 July 2025
A Critical Role of DC-SIGN+ Tumor-Associated Macrophages in Colorectal Cancer Immune Evasion and Progression via BCL-3-Mediated PD-L1 Expression
Zhang J, Zhao Y, Wang X, Miao C, Xu W, Wan C, Hu B, Qian F
ImmunoTargets and Therapy 2025, 14:1395-1410
Published Date: 10 December 2025
BRAP Promotes the Tumorigenesis of Hepatocellular Carcinoma by Corrupting Cancer Cell Cycle Regulation and Enhancing Immune Evasion
Guo Y, Gu R, Gao F, Liu L, Deng D, Zhang Q, Wang L, Liu Q, Lan L, Cang S
Journal of Hepatocellular Carcinoma 2025, 12:2771-2793
Published Date: 16 December 2025