Back to Journals » Journal of Pain Research » Volume 12
TRPV1 channel contributes to remifentanil-induced postoperative hyperalgesia via regulation of NMDA receptor trafficking in dorsal root ganglion
Authors Song C, Liu P, Zhao Q, Guo S, Wang G
Received 5 September 2018
Accepted for publication 22 November 2018
Published 15 February 2019 Volume 2019:12 Pages 667—677
DOI https://doi.org/10.2147/JPR.S186591
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Überall
Chengcheng Song,1–3 Peng Liu,3,4 Qi Zhao,1–3 Suqian Guo,1–3 Guolin Wang1–3
1Tianjin Research Institute of Anesthesiology, Tianjin, China; 2Department of Anesthesiology, Tianjin Medical University General Hospital, Tianjin, China; 3Tianjin Medical University, Tianjin, China; 4Department of General Surgery, Tianjin Medical University General Hospital, Tianjin, China
Background: Remifentanil is widely used in general anesthesia due to its reliability and rapid onset. However, remifentanil-induced postoperative hyperalgesia might be a challenge nowadays. Accumulating evidence suggests that the transient receptor potential vanilloid 1 (TRPV1) was involved in the development of neuropathic pain and hyperalgesia. However, the contribution of TRPV1 in modulating remifentanil-induced postoperative hyperalgesia is still unknown. The aim of this study is the contribution of TRPV1 to the surface expression of N-methyl-d-aspartate (NMDA) receptors in remifentanil-induced postoperative hyperalgesia.
Methods: The hot plate test and the Von Frey test were performed to evaluate thermal and mechanical hyperalgesia. Capsazepine (CPZ) was administrated intrathecally to confirm our results. TRPV1, NMDA receptors, CaMKII (calcium/calmodulin-dependent kinase II), and protein kinase C (PKC) in the dorsal root ganglion (DRG) were detected by Western blotting. Immunofluorescence assay was applied to analyze the distribution of TRPV1 and the relationship between TRPV1 and NMDA receptor subunit 1 (NR1).
Results: Remifentanil-induced both thermal and mechanical postoperative hyperalgesia. Here, we found the membrane trafficking of NR1, possibly due to the activation of TRPV1 in DRG neurons after remifentanil infusion. Furthermore, intrathecal injection of CPZ was able to relieve remifentanil-induced postoperative hyperalgesia according to a behavioral test and CPZ confirmed that TRPV1 is involved in NR1 trafficking. In addition, CaMKII/PKC but not protein kinase A (PKA) contributed to remifentanil-induced postoperative hyperalgesia.
Conclusion: Our study demonstrates that TRPV1 receptors are involved in remifentanil-induced postoperative hyperalgesia. TRPV1 contributes to the persistence of remifentanil-induced postoperative hyperalgesia through the trafficking of NMDA receptors via the activation of CaMKII-PKC signaling pathways in DRG neurons.
Keywords: opioid, pain, TRPV1, NMDA receptor, CaMKII
Introduction
Opioids are the cornerstone of acute and chronic pain therapy. A survey of opioid medication in pharmacological pain management, showed that the number of opioid prescriptions increased linearly. Their widespread use has risen over a 20-year period with a >10-fold increase in fentanyl-based opioid (fentanyl, remifentanil, and sufentanil) use throughout all clinical settings.1 Remifentanil is a potent short-acting opioid for general anesthesia and sedation in the intensive care unit.2 Although remifentanil offers several advantages in clinical practice, accumulation evidence from experimental and clinical research suggested that remifentanil could also generate and strengthen postoperative pain sensitization, known as remifentanil-induced postoperative hyperalgesia.3–6 Nonetheless, many studies have demonstrated that remifentanil could enhance postoperative analgesic consumption and cause pain hypersensitivity. However, its mechanism and effective therapy are still unclear.7,8 This study focused on the mechanism of remifentanil-induced postoperative hyperalgesia; we are hoping to provide new ideas for the treatment and prevention of remifentanil-induced postoperative hyperalgesia to relieve patients’ pain.
The transient receptor potential vanilloid 1 (TRPV1) is the first cloned mammalian TRP channel. TRPV1 is predominantly expressed on peripheral nociceptors. TRPV1 is localized mostly in small- to medium-sized neurons and the major cellular expression sites are unmyelinated C fibers and myelinated Aδ fibers.9–11 TRPV1 is a selective proton-sensitive cation channel, which can be blocked by capsazepine (CPZ), a TRPV1 channel antagonist. CPZ inhibits voltage-gated calcium channels and is also a ligand for TRPV1.10,12 TRPV1 acting as a transducer for peripheral noxious stimuli is reported to be an important mediator in various types of pain,13,14 including inflammatory pain, neuropathic pain, and cancer pain.15 However, the relationship between TRPV1 and remifentanil-induced postoperative hyperalgesia remains unknown.
Studies have shown that NMDA receptors, which are expressed in the central sensory neurons, are effective for central sensitization and pain hypersensitivity.16 Central N-methyl-D-aspartate (NMDA) receptors have been demonstrated to contribute to remifentanil-induced postoperative hyperalgesia.17 NMDA receptors synthesized in the peripheral dorsal root ganglion (DRG) neurons can transport the signal from the peripheral skin to the central spinal cord and brainstem trigeminal nucleus.18,19 NMDA receptors in primary sensory neurons play critical roles in pain conduction and contribute to the pain sensitization. NMDA receptors, including NMDA receptor subunit 1 (NR1) and NR2B (but not NR2A) subunits, are expressed in the DRG neurons.20 In addition, NR1 seems to be more closely related to nociception. It is reported that NR1 contributes to inflammatory pain sensitization increases during inflammation.21 NR1 has multiple amino acid residues in the C-terminals, and these domains are the substrates of calcium/calmodulin-dependent protein kinase II (CaMKII), protein kinase A (PKA), and protein kinase C (PKC). Some studies have demonstrated that CaMKII inhibitor KN93 could prevent allodynia and hyperalgesia by regulating NR1.22,23 Furthermore, the activation of TRPV1 causes the amount of Ca2+ influx into the neurons to increase, followed by the activation of calcium-mediated signal transduction,24,25 including activation of CaMKII, PKA, and PKC.26,27 Therefore, these findings raise the possibility that NMDA receptors contribute to remifentanil-induced postoperative hyperalgesia by modulating CaMKII signaling via TRPV1.
In our study, using the incisional postoperative pain model, we investigated intracellular signaling pathways of remifentanil-induced postoperative hyperalgesia initiated by TRPV1 activation in DRG neurons. Since TRPV1 receptors are involved in pain hypersensitivity, we explored whether the activation of TRPV1 receptors is responsible for the regulation of NMDA receptors through CaMKII pathways in remifentanil-induced postoperative hyperalgesia. Our results will increase the understanding of the molecular mechanism of remifentanil-induced postoperative hyperalgesia.
Materials and methods
Animal preparation
Adult male Sprague-Dawley rats weighing 240–250 g were supplied by the Laboratory Animal Center of the Chinese People’s Liberation Army. Three to four rats were housed in a plastic cage with soft bedding and standard laboratory food and water. They were kept in a standard climate-controlled conditions and strict light-controlled conditions. According to the International Association for the Study of Pain guidelines for pain research in animals, all animal protocols were tested with the approval of the Animal Care and Use Committee at the Tianjin General Hospital affiliated to Tianjin Medical University. All treatment conditions were blinded from the experimenters. Six rats in each group were randomly divided into treatment groups before any assessment was performed.
Surgical procedure
The incisional surgery pain model was confirmed in our laboratory.28 First, the rats were anesthetized with 2% sevoflurane (Maruishi Pharmaceutical Co., Osaka, Japan) for 60 minutes via a nose mask. The right hind paw of rats were cut with a 1 cm longitudinal incision, starting 0.5 cm from the edge of the heel and extending to the toes, through the skin and fascia, then the plantaris muscle was exposed and incised longitudinally. After hemostasis with gentle pressure, the skin was sutured with 4–0 silk and covered with erythromycin ointment to prevent infection. Control rats had a sham procedure without incision of the right paw.
Intrathecal catheter placement
For intrathecal drug administration, 32G intrathecal catheters were inserted through the atlanto-occipital membrane into the lumbar enlargement. Animals who failed to show hind limb paralysis from the lidocaine (2%, 5 µL) were not induced in the experiments.29
Drugs
The rats were intravenously administrated remifentanil (Yichang Renfu Pharmaceutical Co., Yichang, China) for 60 minutes, the infusion rate was 1.0 µg/kg/min, and the remifentanil dosage was verified in our previous study according to the clinical dose.30 Saline was used as the control treatment, and the infusion rate was 0.1 mL kg/min and lasted for 60 minutes.
CPZ (Sigma-Aldrich Co., St. Louis, MO, USA) was dissolved at 100 mmol/L in 100% dimethyl sulfoxide and stored at –20°C until further processing. CPZ was diluted with saline to a final concentration and CPZ (12 µg, 10 mL) was intrathecally injected 30 minutes before intravenous infusion of saline/remifentanil.31 A schematic view of the experimental design is shown in Figure 1.
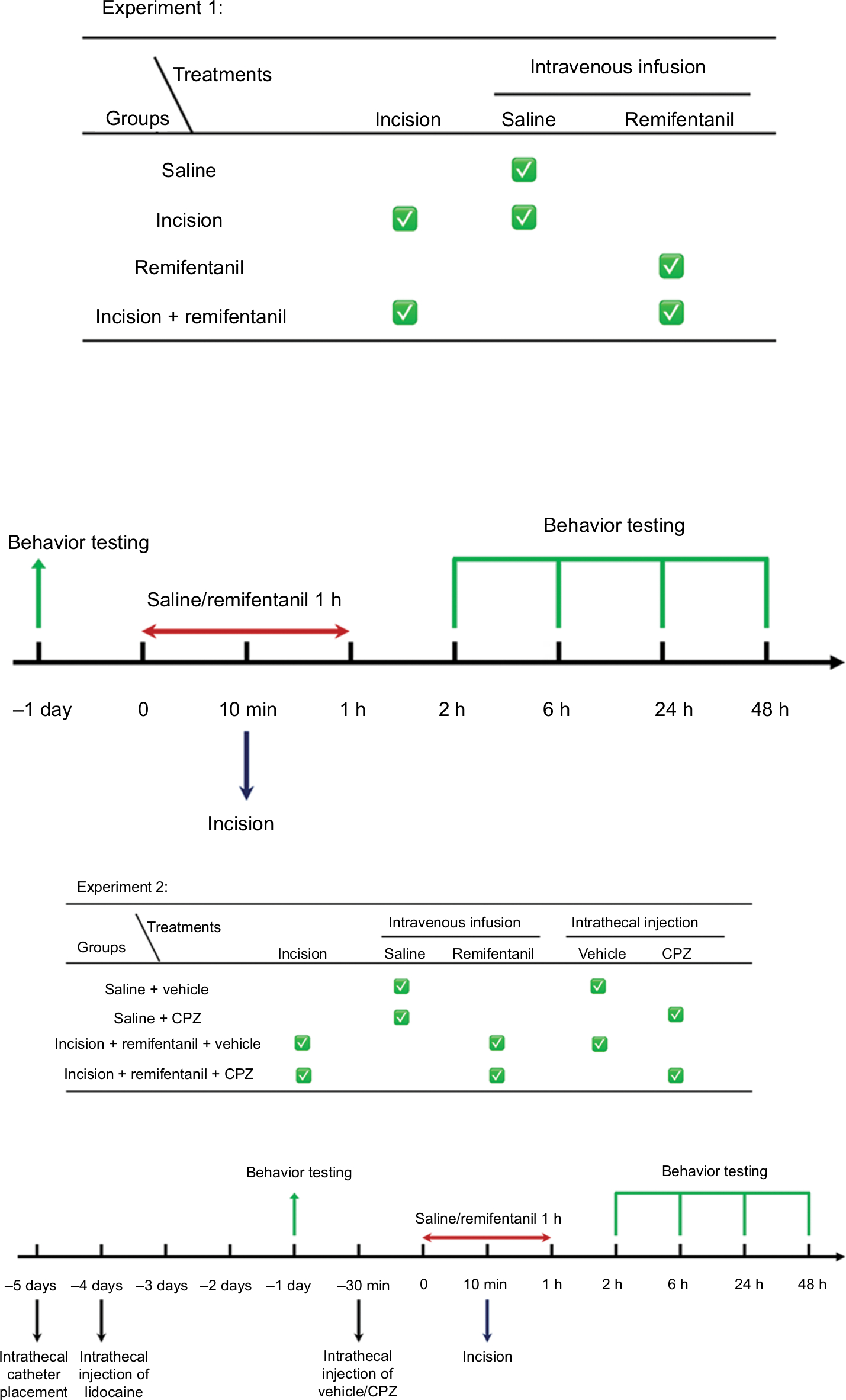 | Figure 1 Illustration of the experimental design. Notes: Experiment 1 is to establish a RIH pain model and to confirm whether TRPV1 and NR1 trafficking is involved in RIH. Experiment 2 is to examine the effect of CPZ on RIH and to further confirm the mechanism of TRPV1 involvement in RIH. Abbreviations: CPZ, capsazepine; NR1, N-methyl-D-aspartate receptor subunit 1; RIH, remifentanil-induced postoperative hyperalgesia; TRPV1, transient receptor potential vanilloid 1. |
Behavior testing
To test the mechanical sensitivity, rats were placed in 20 cm ×20 cm ×20 cm chambers with a mesh bottom and acclimated for 1 hour before any examination. Mechanical stimuli in the rat’s right hind paw were measured with Von Frey filaments (BSEVF3; Harvard Apparatus, Holliston, MA, USA), which exerts force in log increments. The pressure (g) when a positive response (withdrawal, shaking, or licking the paws) appeared was defined as paw withdrawal threshold (PWT). PWT was repeated three times with an interval of 5 minutes and, then, the average value of the three trials was evaluated. A cutoff threshold of 50 g was used to avoid tissue damage in the right hind paw.
Before the thermal sensitivity test, the rats were accustomed to a test environment for 1 hour to achieve immobility. Thermal sensitivity was measured with a hot plate instrument (YLS-6B; Zhenghua Biological Instrument Equipment Co., Hubei, China). Rats were placed on a 50°C hotplate. The time it took to receive a positive response (rat lifted its foot) was defined as the paw withdrawal latency (PWL). Each trial of PWL was repeated and the mechanical hyperalgesia was assessed. A cutoff time was set at 25 seconds to prevent paw tissue damage.
PWL was measured in the same animals 10 minutes after the determination of PWT. Hypersensitivity was measured at a baseline value 24 hours before surgery and subsequently a 2, 6, 24, and 48 hours after surgery. After the behavioral test, rats were deeply anesthetized and the L4–L6 segments of DRG tissues were taken for molecular biological analysis.
Western blotting analysis
The protein used for the Western blot was extracted from the L4–L6 segments of DRG. Then, the DRG was stored at –80°C until needed. For protein extraction, the ipsilateral portion of DRG was lysed in RIPA buffer (KeyGen Biotech Co., Ltd., Nanjing, China). The supernatant was collected as the total protein after centrifugation. The membrane fraction of DRG was extracted with the membrane compartment protein extraction kit (BioChain Institute, Inc., Newark, CA, USA). Protein concentrations were estimated by the BCA protein assay. Equal amount of protein extracts was separated by SDS-PAGE and transferred onto a polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA, USA). The Millipore membrane was blocked with 5% nonfat milk for 1 hour and incubated with the following primary antibodies: rabbit anti-TRPV1 polyclonal antibody (1:1,000; OriGene, Rockville, MD, USA), anti-NR1 monoclonal antibody (1:1,000; Abcam, Cambridge, UK), rabbit anti-CaMKII polyclonal antibody (1:1,000; GeneTex, Inc., Irvine, CA, USA), PKA catalytic subunit (1:2,000; Abcam), rabbit anti-PKC polyclonal antibody (1:2,000; Abcam), and mouse monoclonal anti-β-actin antibody (1:3,000; Abclonal, Wuhan, China) at 4°C overnight. Next, the primary antibodies were washed and followed by peroxidase-conjugated affinipure goat anti-mouse IgG antibody (1:5,000; Proteintech, Wuhan, China) or anti-rabbit IgG antibody (1:8,000; Proteintech). The density of each band was measured and analyzed by GeneTools Match software (Syngene, Cambridge, UK).
Immunofluorescence
After appropriate survival times, deeply anesthetized animals were perfused with PBS and 4% paraformaldehyde. L4–L6 DRG tissues were rapidly removed, post-fixed in 4% paraformaldehyde overnight, and embedded with OCT compound medium (CellPath Ltd, Newtown, UK). DRG tissues were cut using a cryomicrotome, with a thickness of 5 µm. The sections were first blocked with 5% goat serum for 1 hour at room temperature and then incubated with the following primary antibodies overnight at 4°C: anti-TRPV1 (rabbit, 1:40; OriGene) followed by goat anti-rabbit IgG H&L antibody (1:100; Proteintech).
For double immunofluorescence, the sections were first blocked with 5% horse serum for 1 hour at room temperature and then incubated with a mixture of anti-TRPV1 and anti-NR1 primary antibodies (mouse, 1:50; Abcam) overnight at 4°C, followed by a mixture of goat anti-rabbit IgG H&L antibody and Dylight 594-conjugated goat anti-mouse secondary antibodies (1:200; Abbkine Scientific Co., Ltd., Wuhan, China). Sections were examined under an Olympus fluorescence microscope. The data analysis was performed using ImageJ software.
Statistical analyses
Statistical analyses with SPSS Statistics version 22.0 (IBM Corporation, Armonk, NY, USA) were used for calculations. Data from behavior testing were assessed by two-way ANOVA. The results of Western blot and immunofluorescence were assessed by one-way ANOVA. P<0.05 was considered statistically significant. Data were presented as mean ± SD.
Results
Mechanical and thermal hyperalgesia in remifentanil-induced postoperative hyperalgesia rats
There were no significant differences between baseline PWLs to thermal stimulation and baseline PWTs to von Frey filaments in the treatment groups before the surgery (–24 hours) (P>0.05, Figure 2A and B). After surgery, PWT and PWL in the incision and + remifentanil group decreased at 2, 6, 24, and 48 hours, and remifentanil-induced postoperative hyperalgesia was noticeable and maintained from 2 to 48 hours, (P<0.05, Figure 2A and B). Moreover, it reached a peak at 24 hours and remifentanil appeared to facilitate hyperalgesia induced by incision. On the contrary, PWT and PWL did not demonstrate significant changes in saline rats (P>0.05, Figure 2A and B). These results reflect that the remifentanil-induced postoperative hyperalgesia rat model was successfully established.
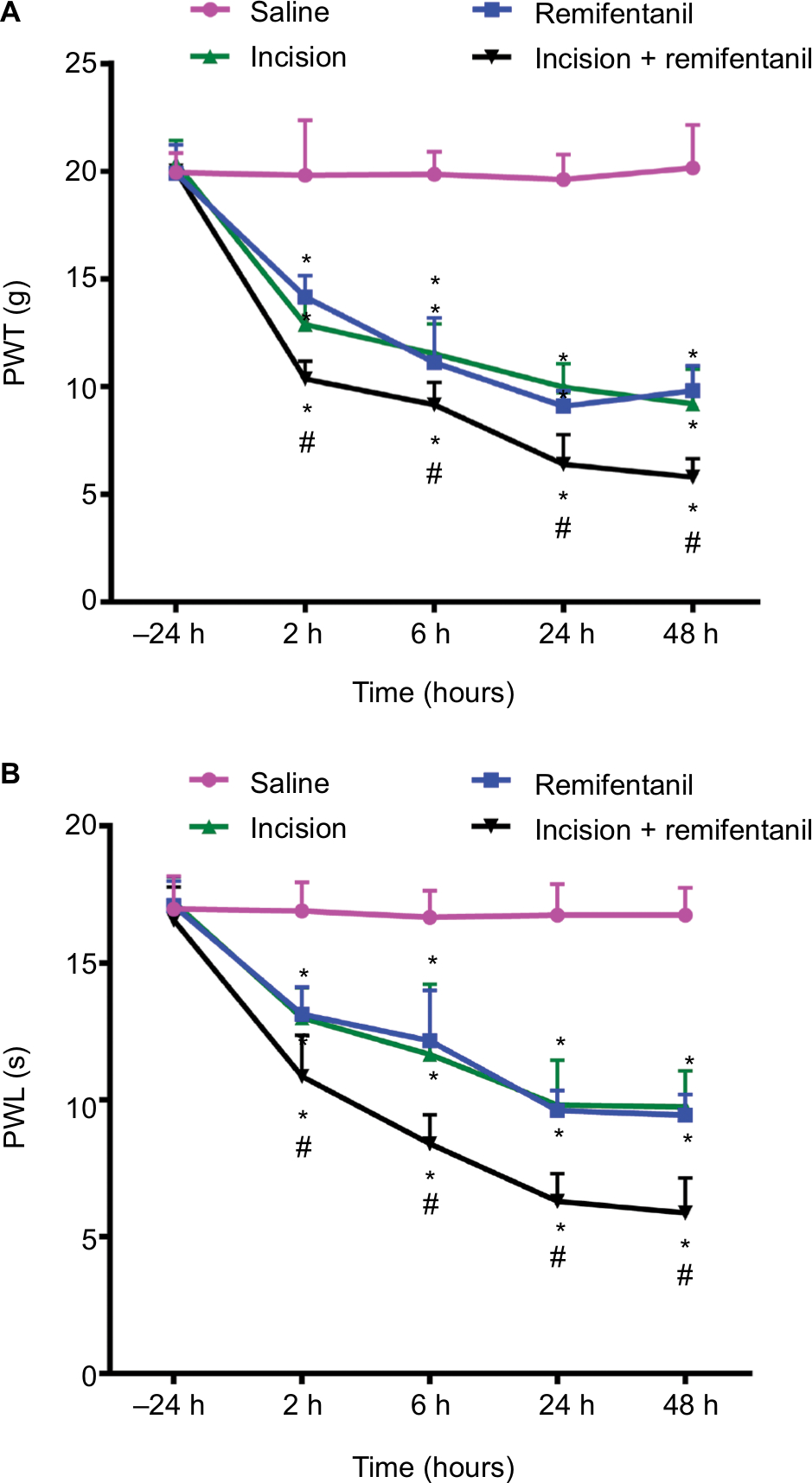 | Figure 2 The effect of remifentanil-induced postoperative hyperalgesia. Notes: (A) PWL and (B) PWT in the ipsilateral hind-paws were evaluated at different times. The baseline numbers of PWL and PWT were similar in all groups. When compared with rats receiving saline, incision and remifentanil significantly decreased PWL and PWT and maintained from 2 to 48 hours. Moreover, incision and remifentanil treatment significantly enhanced thermal and mechanical hyperalgesia induced by incision. Data represent mean ± SEM, n=6, analyzed by the two-way ANOVA; *P<0.05 vs saline group; #P<0.05 vs incision group. Abbreviations: PWL, paw withdrawal latency; PWT, paw withdrawal threshold; SEM, standard error of measurement. |
Expression of TRPV1 and NR1 trafficking in DRG contributes to remifentanil-induced postoperative hyperalgesia
To determine whether and how TRPV1 regulates remifentanil-induced postoperative hyperalgesia, we first examined the alteration in TRPV1 levels in DRG using the Western blot assay. When compared with the saline group, the expression of TRPV1 in DRG dramatically increased in rats receiving incision and, remifentanil, and especially in the incision + remifentanil group (P<0.05, Figure 3A and B).
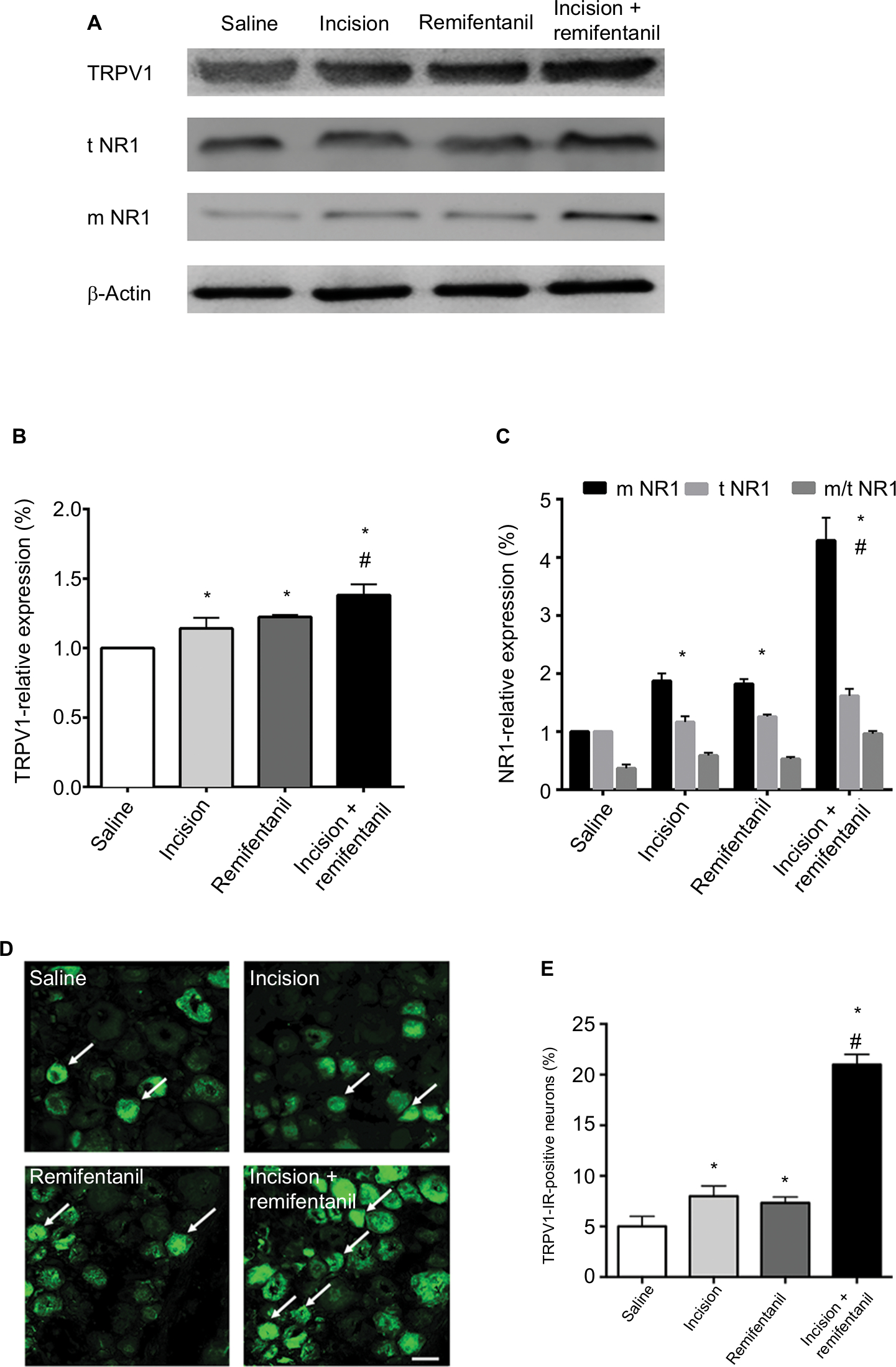 | Figure 3 Expression of TRPV1 and NR1 receptor trafficking in rat DRG after remifentanil-induced postoperative hyperalgesia. Notes: (A) Western blot of TRPV1 and t and m NR1 expressions in ipsilateral L4 and L5 DRG. (B and C) Quantification of bands of Western blot of TRPV1 and t and m NR1 in DRG. β-Actin is used as an internal control. Results are expressed as mean ± SD for n=6 rats. Data are analyzed using the one-way ANOVA with Dunnett’s test post hoc comparisons. *P<0.01 vs saline group; #P<0.01 vs incision group. (D) Immunohistochemical analyses of TRPV1 (arrows) in DRgs. (E) Statistical histogram of the immunofluorescence of TRPV1 immunoreactive positive neurons. n=6 rats per group, scale bar =50 μm. *P<0.01 vs saline group; #P<0.01 vs incision group. Abbreviations: DRG, dorsal root ganglion; m, membrane; IR, immunoreactive; NR1, N-methyl-D-aspartate receptor subunit 1; t, total; TRPV1, transient receptor potential vanilloid 1. |
Immunofluorescence assay was used to detect the expression and activation of TRPV1 in DRG. Consistent with the results of the Western blot analysis, TRPV1 was inconspicuous in the saline group, whereas there was an accumulation of TRPV1 in the incision and remifentanil groups, especially in the incision + remifentanil group (P<0.05, Figure 3D and E). This data showed that remifentanil infusion and plantar incision might induce hyperalgesia by the upregulation of TRPV1.
In addition, the expression of NR1 in both total and membrane proteins was considerably elevated (P<0.05, Figure 3A and C) after incision with remifentanil infusion. It is also showed that the trafficking of NR1 from the cytosol to the membrane was increased, which was represented by an increase in the ratio of membrane: total NR1 (P<0.05, Figure 3A and C).
TRPV1 antagonist CPZ can partly improve remifentanil-induced postoperative hyperalgesia
It is virtually unknown whether blocking TRPV1 reversed remifentanil-induced thermal and mechanical hypersensitivity in rats; we used intrathecal delivery of a TRPV1 antagonist, CPZ, to determine this.32 The baseline of PWL and PWT was conducted in separate groups of rats prior to the infusion of remifentanil or saline. When compared with corresponding nonCPZ-treated groups, the PWT and PWL were markedly increased at 2, 6, 24, and 48 hours after intrathecal administration of CPZ (P<0.05). These results suggest that CPZ significantly prevented remifentanil-induced thermal and mechanical hyperalgesia (P<0.01, Figure 4A and B). Of note, this dose of CPZ did not induce antinociception in saline-treated mice (P>0.05, Figure 4A and B).
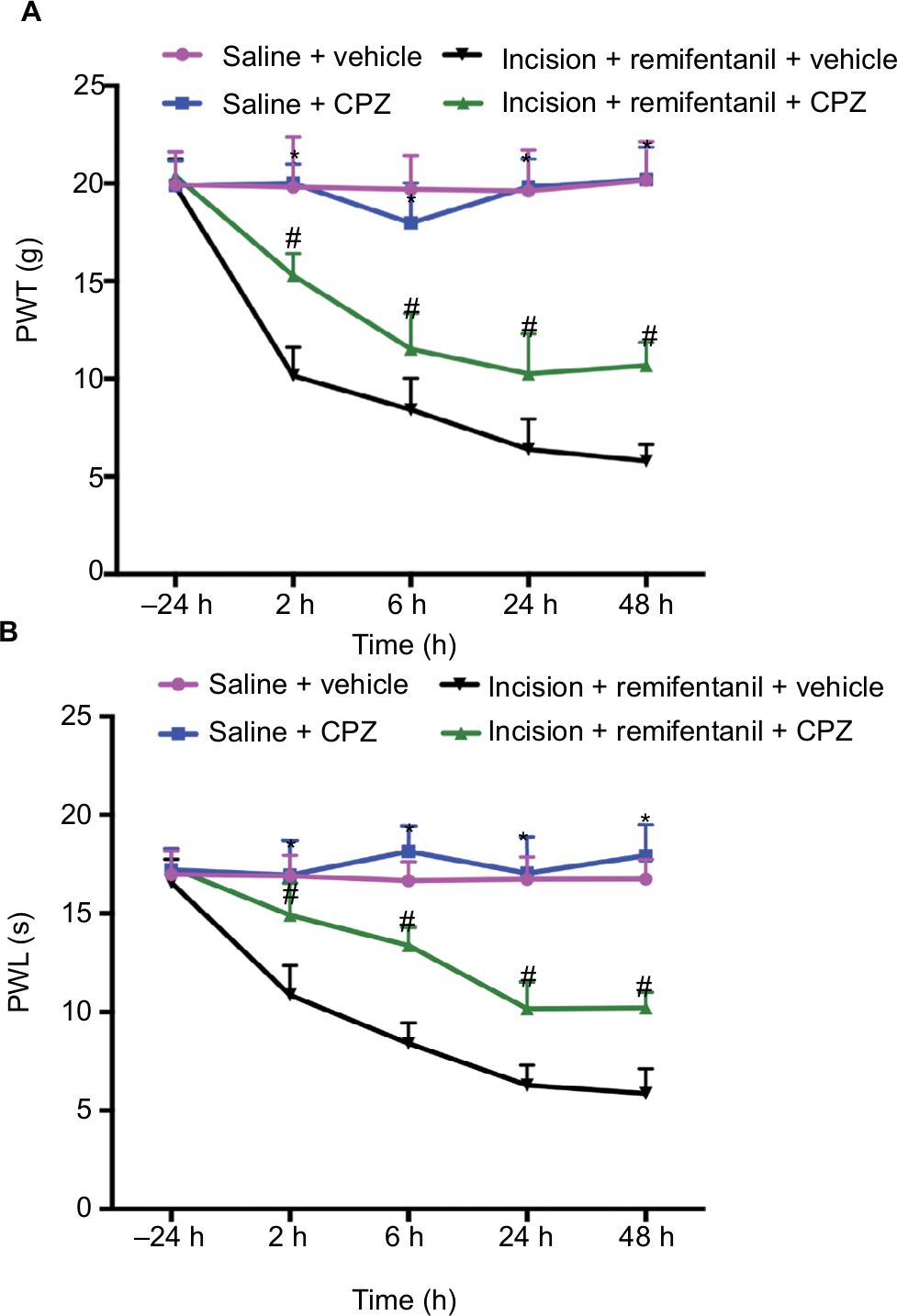 | Figure 4 The effects of TRPV1 antagonist CPZ on remifentanil-induced postoperative hyperalgesia. Notes: CPZ was intrathecally injected in the saline + CPZ group and incision + remifentanil + CPZ group. (A) PWL and (B) PWT were evaluated at –24, 2, 6, 24, and 48 hours after infusion. Results are expressed as mean ± SD for n=6 rats. Data are analyzed using the one-way ANOVA with Dunnett’s test post hoc comparisons. *P<0.01 vs saline group; #P<0.01 vs incision + remifentanil group. Abbreviations: CPZ, capsazepine; PWL, paw withdrawal latency; PWT, paw withdrawal threshold; TRPV1, transient receptor potential vanilloid 1. |
Effect of CPZ on NR1 membrane trafficking in remifentanil-induced postoperative hyperalgesia
Next, Western blot data indicated that intrathecal pretreatment with TRPV1 antagonist, CPZ, blocked the increase in TRPV1 in remifentanil-induced postoperative hyperalgesia (P<0.01, Figure 5A and B). The study has shown that the function of NMDA receptors plays an essential role in remifentanil-induced postoperative hyperalgesia.17 Thus, we determined that NMDA receptor were involved in remifentanil-induced postoperative hyperalgesia via increased TRPV1. Compared with the control group, NR1 levels were significantly increased after surgery and remifentanil infusion (P<0.01, Figure 3A and C). Moreover, CPZ suppressed the level of NR1, especially the trafficking of NR1, compared with the incision + remifentanil group (P<0.01, Figure 5A and C). To further verify the relationship between TRPV1 and the NR1 receptor in remifentanil-induced postoperative hyperalgesia, immunofluorescence was used and it showed that CPZ significantly reduced the interaction between TRPV1 and NR1 (P<0.01, Figure 5D and E). Taken together, these findings indicate that TRPV1 potentiates NMDA receptor, which can be blocked by TRPV1 antagonists.
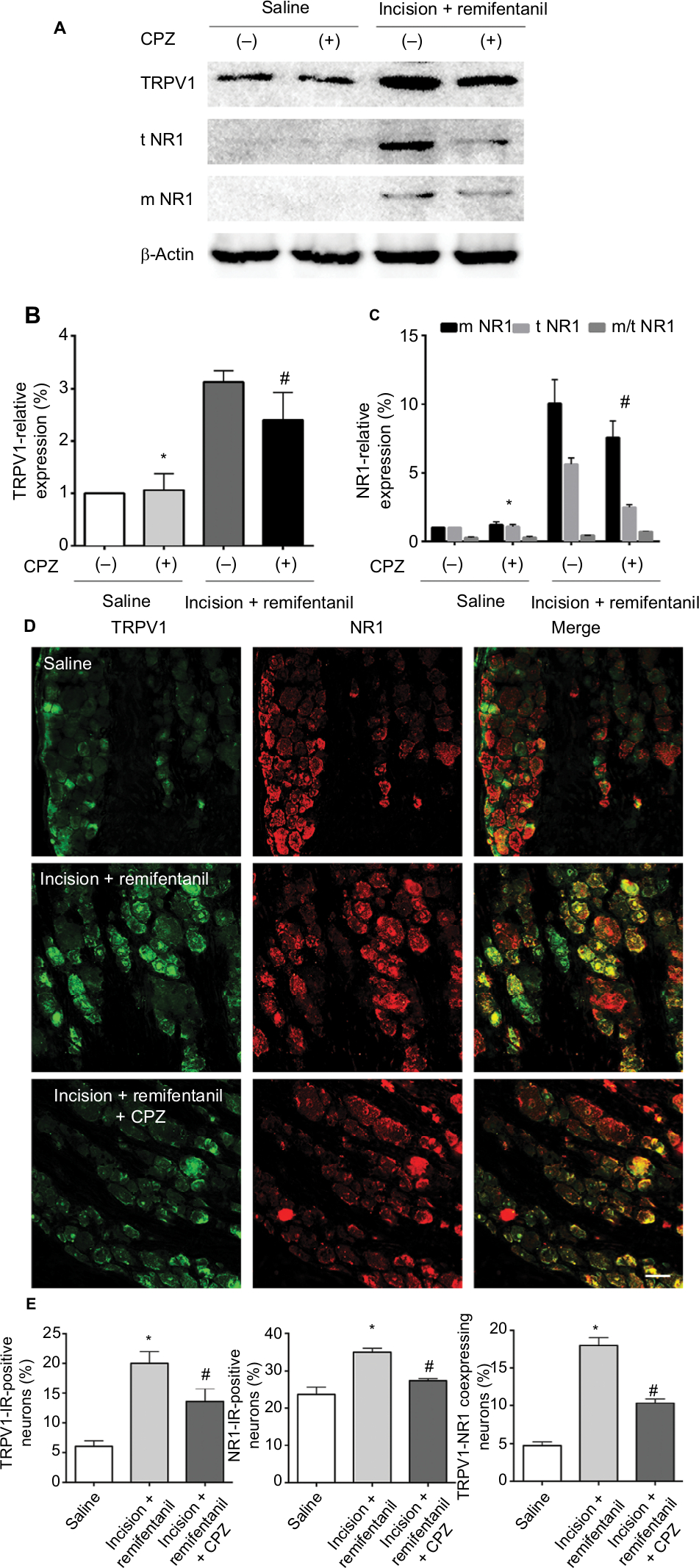 | Figure 5 The effects of CPZ on NR1 trafficking in remifentanil-induced postoperative hyperalgesia. Notes: (A) Western blot of TRPV1 and NR1 expression in DRG. (B and C) Quantification of bands of Western blot for TRPV1 and NR1 in DRG. β-Actin is used as an internal control. (D) Double immunofluorescence of TRPV1 and NR1 in DRG. (E) Statistical histogram of the double immunofluorescence of TRPV1 and NR1 immunoreactive positive neurons. Results are expressed as mean ± SD for n=6 rats, scale bar =50 µm. Data are analyzed using the one-way ANOVA with Dunnett’s test post hoc comparisons. *P<0.01 vs saline group; #P<0.01 vs incision + remifentanil group. Abbreviations: CPZ, capsazepine; DRG, dorsal root ganglion; IR, immunoreactive; m, membrane; NR1, N-methyl-D-aspartate receptor subunit 1; t, total; TRPV1, transient receptor potential vanilloid 1. |
Blockage of TRPV1 attenuates the expression of CaMKII/PKC in remifentanil-induced postoperative hyperalgesia
To examine whether CaMKII is associated with TRPV1-remifentanil-induced postoperative hyperalgesia, we tested the protein levels of CaMKII. The results showed a significant increase in the expression of CaMKII in DRG after incision + remifentanil treatment. Furthermore, the level of CaMKII was decreased by the blockade of TRPV1. These results suggested that CaMKII was involved in TRPV1-induced remifentanil postoperative hyperalgesia (P<0.01, Figure 6A and B). We further checked PKC, which was activated via TRPV1 in remifentanil-induced postoperative hyperalgesia (P<0.01, Figure 6A and D), but PKA was not involved in TRPV1-induced remifentanil postoperative hyperalgesia (P>0.05, Figure 6A and C). These results suggest that CaMKII/PKC pathways play an important role in the mechanisms of remifentanil postoperative hyperalgesia.
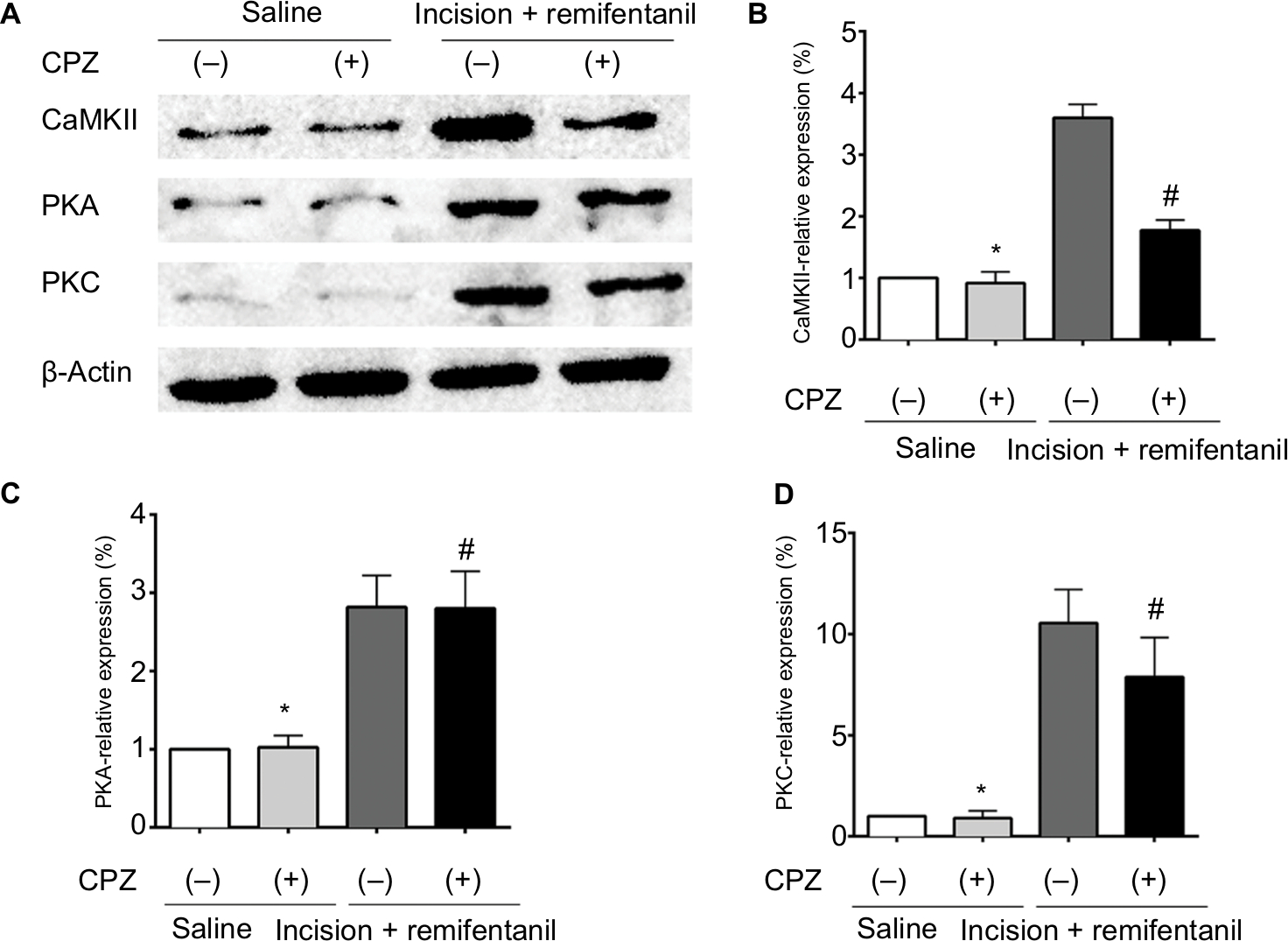 | Figure 6 CaMKII/PKC signaling is involved in remifentanil-induced postoperative hyperalgesia. Notes: (A) Western blot of CaMKII, PKA, and PKC expressions in DRG. (B–D) Quantification of bands of Western blot for CaMKII, PKA, and PKC expressions in DRG. β-Actin is used as an internal control. Results are expressed as mean ± SD for n=6 rats. Data are analyzed using the one-way ANOVA with Dunnett’s test post hoc comparisons. *P<0.01 vs saline group; #P<0.01 vs incision + remifentanil group. Abbreviations: CaMKII, calcium/calmodulin-dependent kinase II; CPZ, capsazepine; DRG, dorsal root ganglion; PKA, protein kinase A; PKC, protein kinase C. |
Discussion
Remifentanil is widely used for the management of operative pain as it maintains hemodynamic stability and controls intraoperative painful events.33 Evidence accumulated from both animal research and human clinical trials have reported that remifentanil infusion is associated with postoperative hyperalgesia and increases the requirements of postoperative analgesics.33,34 Conversely, there are some studies in which remifentanil did not induce hyperalgesia.35,36 Although remifentanil-induced postoperative hyperalgesia is still debated, the presence of remifentanil-induced postoperative hyperalgesia would be a clinical challenge in perioperative pain and an urgent problem to solve. Our results observed remifentanil-induced mechanical and thermal hyperalgesia and it reached a peak at 24 h after remifentanil infusion. Moreover, the pain threshold decreased from 2 to 48 hours after the stop of remifentanil infusion in our current study.
Remifentanil is a kind of μ-opioid receptor (MOR) agonist. It is known that MORs are kinds of GPCRs that are closely related to TRP channels. TRP channels and MORs are cell surface proteins in the sensory nerves, and they are the “front line” of the body. This GPCR–TRP channels axis is vitally essential for pathological changes functioning at sensing noxious, irritating, and inflammatory stimulants.24,37 The pain signals that are received by the GPCRs converge on TRP channels and alter TRP channels’ activity or expression, leading to amplification of the effects of the GPCRs. Based on the literature, we hypothesized that TRPV1 contributes to remifentanil-induced postoperative hyperalgesia and the sensitization of the sensory DRG neurons.
Furthermore, TRPV1 is a cation channel that can alter calcium permeability and pore diameter to allow an influx of calcium. Mounting evidence suggests that TRPV1 activation in the peripheral nervous system is partly responsible for some severe painful disorders, such as inflammation, nerve injury, diabetic neuropathic pain, and primary bone cancer pain.38–44 Our results showed that sustained remifentanil administration increases the expression and function of TRPV1 in the peripheral sensory neurons, which has an essential effect on sustained remifentanil-induced thermal and mechanical hypersensitivities. Our behavioral experiments also confirmed that intrathecal administration of CPZ blocked the thermal hyperalgesia as well as the mechanical hyperalgesia following sustained remifentanil administration in SD rats and, thus, suggested an action of TRPV1 in regulating remifentanil-induced postoperative hyperalgesia.
Although it has been shown that the excitatory neurotransmitter NMDA receptors play a central role in the development of remifentanil-induced postoperative hyperalgesia, particularly in the generation of central sensitization,6 whether similar mechanisms exist in the peripheral sensory neurons are unknown. NMDA receptors in primary afferents also play crucial roles not only in the peripheral sensitization but also in the central sensitization. Previous studies have indicated that NR1, a type of NMDAR, is localized in essentially every DRG cell.19,45 It is particularly expressed in C fibers. Moreover, TRPV1 is predominantly expressed in C fibers. Our results showed that NMDA receptors become activated after the administration of remifentaniland that this increase in NMDA receptors was significantly inhibited by CPZ. Together, these results indicate that NMDA receptors are essential for the development of remifentanil-induced postoperative hyperalgesia and are activated downstream to TRPV1 (Figure 6).
The accumulated evidence has shown that CaMKII is a serine/threonine protein kinase that is involved in Ca2+ signaling and hyperalgesia.46,47 Indeed, many studies suggest that CaMKII plays an important role in pain control.46,48,49 Meanwhile, TRPV1 is a selective Ca2+-permeable channel and the activation of the TRPV1 increases the intracellular concentration of Ca2+ and, in turn, activates CaMKII. In addition, NMDA receptors are sensitive to calcium. Ca2+ flux triggered the activation of NMDA receptors, which contributed to the movement of NMDA receptors. NMDA receptors in the cytosol transported to the membrane (Figure 7). In our incision + remifentanil group infusion pain model, the membrane trafficking of NR1 was increased. In addition, CPZ reversed remifentanil-induced upregulation of mNR1. Findings from this study and others have shown that the activation of the TRPV1-CaMKII-NMDAR pathway is one of the major mechanisms whereby remifentanil-induced postoperative hyperalgesia occurs.46–49 Therefore, it is beneficial in preserving calcium balance by disrupting TRPV1 signaling.
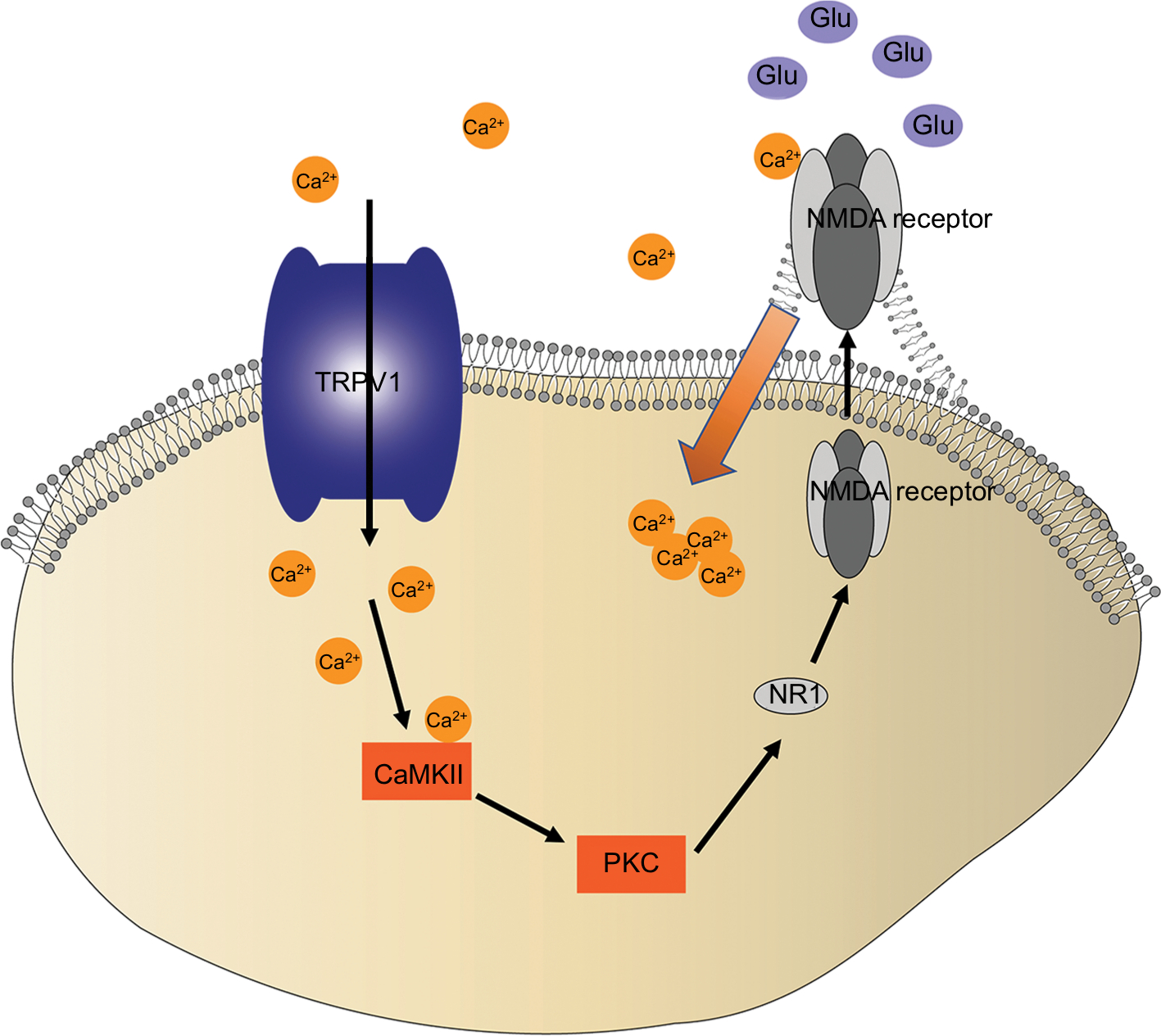 | Figure 7 TRPV1 activates a complicated pathway that converges on NR1 trafficking in remifentanil-induced postoperative hyperalgesia. Notes: Schematic illustration of proposed TRPV1-CaMKII/PKC-NR1 signaling in remifentanil-induced postoperative hyperalgesia. The activation of TRPV1 in RIH causes the cellular calcium influx, increasing CaMKII/PKC activity, which triggers NR1 membrane trafficking, and promoting a large amount of calcium influx, which triggers the release of Glu and transmits to the central neurons in the model of RIH. Abbreviations: CaMKII, calcium/calmodulin-dependent kinase II; Glu, glutamate; NMDA, N-methyl-D-aspartate; NR1, N-methyl-D-aspartate receptor subunit 1; protein kinase C; RIH, remifentanil-induced postoperative hyperalgesia; TRPV1, transient receptor potential vanilloid 1. |
The current study demonstrated that the interactions between NMDA receptors and TRPV1 in rat DRG neurons are essential for the development of thermal and mechanical hyperalgesia. Although thermal hypersensitivity is regarded as a peripheral phenomenon, the mechanical sensitization is thought to be underpinned by central mechanisms such as increased glutatmate (Glu) and NMDAR levels. The results suggest that NMDAR is involved in the effects mediated by TRPV1 activation. In addition, the activation of NMDA receptors causes an increase in the release of Glu and transmits to the central neurons.
Conclusion
Our study demonstrates that TRPV1 is involved in remifentanil-induced postoperative hyperalgesia as it contributes to the persistence of remifentanil-induced postoperative hyperalgesia through activation of the CaMKII-NMDAR signaling pathway in DRG neurons. The activation of NR1 promotes the surface trafficking of NMDA receptors and NMDA receptors insert into the membrane, which causes a large amount of calcium influx. In addition, NMDA receptors can trigger the release of Glu and transmit to the central neurons in the model of remifentanil-induced postoperative hyperalgesia. This hypothesis could be a novel target for the prevention and treatment of remifentanil-induced postoperative hyperalgesia.
Acknowledgment
This research was supported by grants from the National Natural Science Foundation of China, China (81371245, 81801107, and 81500961).
Disclosure
The authors report no conflicts of interest in this work.
References
Chen JH, Hom J, Richman I, Asch SM, Podchiyska T, Johansen NA. Effect of opioid prescribing guidelines in primary care. Medicine. 2016;95(35):e4760. | ||
Koo CH, Yoon S, Kim BR, et al. Intraoperative naloxone reduces remifentanil-induced postoperative hyperalgesia but not pain: a randomized controlled trial. Br J Anaesth. 2017;119(6):1161–1168. | ||
Roeckel LA, Utard V, Reiss D, et al. Morphine-induced hyperalgesia involves mu opioid receptors and the metabolite morphine-3-glucuronide. Sci Rep. 2017;7(1):10406. | ||
Weber L, Yeomans DC, Tzabazis A. Opioid-induced hyperalgesia in clinical anesthesia practice: what has remained from theoretical concepts and experimental studies? Curr Opin Anaesthesiol. 2017;30(4):458–465. | ||
Liang DY, Li X, Clark JD. 5-hydroxytryptamine type 3 receptor modulates opioid-induced hyperalgesia and tolerance in mice. Anesthesiology. 2011;114(5):1180–1189. | ||
Lee M, Silverman SM, Hansen H, Patel VB, Manchikanti L. A comprehensive review of opioid-induced hyperalgesia. Pain Physician. 2011;14(2):145. | ||
Rivosecchi RM, Rice MJ, Smithburger PL, Buckley MS, Coons JC, Kane-Gill SL. An evidence based systematic review of remifentanil associated opioid-induced hyperalgesia. Expert Opin Drug Saf. 2014;13(5):587–603. | ||
Braulio G, Passos SC, Leite F, et al. Effects of transcranial direct current stimulation block remifentanil-induced hyperalgesia: a randomized, double-blind clinical trial. Front Pharmacol. 2018;9:94. | ||
Cortright DN, Szallasi A. Biochemical pharmacology of the vanilloid receptor TRPV1. An update. Eur J Biochem. 2004;271(10):1814–1819. | ||
Caterina MJ, Julius D. The vanilloid receptor: a molecular gateway to the pain pathway. Annu Rev Neurosci. 2001;24:487–517. | ||
Cavanaugh EJ, Simkin D, Kim D. Activation of transient receptor potential A1 channels by mustard oil, tetrahydrocannabinol and Ca2+ reveals different functional channel states. Neuroscience. 2008;154(4):1467–1476. | ||
Szallasi A, Cortright DN, Blum CA, Eid SR. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat Rev Drug Discov. 2007;6(5):357–372. | ||
Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389(6653):816–824. | ||
Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002; 36(1):57. | ||
Alawi K, Keeble J. The paradoxical role of the transient receptor potential vanilloid 1 receptor in inflammation. Pharmacol Ther. 2010;125(2):181–195. | ||
Cichon J, Blanck TJJ, Gan WB, Yang G. Activation of cortical somatostatin interneurons prevents the development of neuropathic pain. Nat Neurosci. 2017;20(8):1122–1132. | ||
Wang C, Li Y, Wang H, et al. Inhibition of DOR prevents remifentanil induced postoperative hyperalgesia through regulating the trafficking and function of spinal NMDA receptors in vivo and in vitro. Brain Res Bull. 2015;110:30–39. | ||
Liu H, Mantyh PW, Basbaum AI. NMDA-receptor regulation of substance P release from primary afferent nociceptors. Nature. 1997;386(6626):721–724. | ||
Coggeshall RE, Carlton SM. Ultrastructural analysis of NMDA, AMPA, and kainate receptors on unmyelinated and myelinated axons in the periphery. J Comp Neurol. 2015;391(1):78–86. | ||
Sato K, Kiyama H, Park HT, Tohyama M. AMPA, KA and NMDA receptors are expressed in the rat DRG neurones. Neuroreport. 1993;4(11):1263–1265. | ||
Tan PH, Chia YY, Chow LH, et al. Gene knockdown of the N-methyl-D-aspartate receptor NR1 subunit with subcutaneous small interfering RNA reduces inflammation-induced nociception in rats. Anesthesiology. 2010;112(6):1482–1493. | ||
Traynelis SF, Wollmuth LP, McBain CJ, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62(3):405–496. | ||
Liu Y, Liang Y, Hou B, et al. The inhibitor of calcium/calmodulin-dependent protein kinase II KN93 attenuates bone cancer pain via inhibition of KIF17/NR2B trafficking in mice. Pharmacol Biochem Behav. 2014;124:19–26. | ||
Veldhuis NA, Poole DP, Grace M, McIntyre P, Bunnett NW. The G protein-coupled receptor-transient receptor potential channel axis: molecular insights for targeting disorders of sensation and inflammation. Pharmacol Rev. 2015;67(1):36–73. | ||
Backes TM, Rössler OG, Hui X, Grötzinger C, Lipp P, Thiel G. Stimulation of TRPV1 channels activates the AP-1 transcription factor. Biochem Pharmacol. 2018;150:160–169. | ||
Mellström B, Savignac M, Gomez-Villafuertes R, Naranjo JR. Ca2+-operated transcriptional networks: molecular mechanisms and in vivo models. Physiol Rev. 2008;88(2):421–449. | ||
Petho˝ G, Reeh PW. Sensory and signaling mechanisms of bradykinin, eicosanoids, platelet-activating factor, and nitric oxide in peripheral nociceptors. Physiol Rev. 2012;92(4):1699–1775. | ||
Yuan Y, Wang JY, Yuan F, Xie KL, Yu YH, Wang GL. Glycogen synthase kinase-3β contributes to remifentanil-induced postoperative hyperalgesia via regulating N-methyl-D-aspartate receptor trafficking. Anesth Analg. 2013;116(2):473–481. | ||
Milligan ED, Hinde JL, Mehmert KK, Maier SF, Watkins LR. A method for increasing the viability of the external portion of lumbar catheters placed in the spinal subarachnoid space of rats. J Neurosci Methods. 1999;90(1):81–86. | ||
Li Y, Wang H, Xie K, et al. Inhibition of glycogen synthase kinase-3β prevents remifentanil-induced hyperalgesia via regulating the expression and function of spinal N-methyl-D-aspartate receptors in vivo and vitro. PLoS One. 2013;8(10):e77790. | ||
Xiao X, Zhao XT, Xu LC, et al. Shp-1 dephosphorylates TRPV1 in dorsal root ganglion neurons and alleviates CFA-induced inflammatory pain in rats. Pain. 2015;156(4):597–608. | ||
Bevan S, Hothi S, Hughes G, et al. Capsazepine: a competitive antagonist of the sensory neurone excitant capsaicin. Br J Pharmacol. 1992;107(2):544–552. | ||
Shi C, Liu Y, Zhang W, et al. Intraoperative electroacupuncture relieves remifentanil-induced postoperative hyperalgesia via inhibiting spinal glial activation in rats. Mol Pain. 2017;13:174480691772563. | ||
Lenz H, Raeder J, Draegni T, Heyerdahl F, Schmelz M, Stubhaug A. Effects of COX inhibition on experimental pain and hyperalgesia during and after remifentanil infusion in humans. Pain. 2011;152(6):1289–1297. | ||
Lahtinen P, Kokki H, Hynynen M. Remifentanil infusion does not induce opioid tolerance after cardiac surgery. J Cardiothorac Vasc Anesth. 2008;22(2):225–229. | ||
Ishii H, Petrenko AB, Kohno T, Baba H. No evidence for the development of acute analgesic tolerance during and hyperalgesia after prolonged remifentanil administration in mice. Mol Pain. 2013;9:11. | ||
Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat Rev Immunol. 2014;14(4):217–231. | ||
Ghilardi JR, Röhrich H, Lindsay TH, et al. Selective blockade of the capsaicin receptor TRPV1 attenuates bone cancer pain. J Neurosci. 2005;25(12):3126–3131. | ||
McGaraughty S, Chu KL, Brown BS, et al. Contributions of central and peripheral TRPV1 receptors to mechanically evoked and spontaneous firing of spinal neurons in inflamed rats. J Neurophysiol. 2008;100(6):3158–3166. | ||
Hillery CA, Kerstein PC, Vilceanu D, et al. Transient receptor potential vanilloid 1 mediates pain in mice with severe sickle cell disease. Blood. 2011;118(12):3376–3383. | ||
Brenneis C, Kistner K, Puopolo M, et al. Phenotyping the function of TRPV1-expressing sensory neurons by targeted axonal silencing. J Neurosci. 2013;33(1):315–326. | ||
Chung MK, Lee J, Joseph J, Saloman J, Ro JY. Peripheral group I metabotropic glutamate receptor activation leads to muscle mechanical hyperalgesia through TRPV1 phosphorylation in the rat. J Pain. 2015;16(1):67–76. | ||
Mickle AD, Shepherd AJ, Loo L, Mohapatra DP. Induction of thermal and mechanical hypersensitivity by parathyroid hormone-related peptide through upregulation of TRPV1 function and trafficking. Pain. 2015;156(9):1620–1636. | ||
Shepherd AJ, Mickle AD, Kadunganattil S, Hu H, Mohapatra DP. Parathyroid Hormone-Related Peptide Elicits Peripheral TRPV1-dependent Mechanical Hypersensitivity. Front Cell Neurosci. 2018;12:38. | ||
Du J, Zhou S, Coggeshall RE, Carlton SM. N-methyl-D-aspartate-induced excitation and sensitization of normal and inflamed nociceptors. Neuroscience. 2003;118(2):547–562. | ||
Kawasaki Y, Kohno T, Zhuang ZY, et al. Ionotropic and metabotropic receptors, protein kinase A, protein kinase C, and Src contribute to C-fiber-induced ERK activation and cAMP response element-binding protein phosphorylation in dorsal horn neurons, leading to central sensitization. J Neurosci. 2004;24(38):8310–8321. | ||
Yao CY, Weng ZL, Zhang JC, Feng T, Lin Y, Yao S. Interleukin-17A acts to maintain neuropathic pain throughactivation of CaMKII/CREB signaling in spinal neurons. Mol Neurobiol. 2016;53(6):3914–3926. | ||
Crown ED, Gwak YS, Ye Z, et al. Calcium/calmodulin dependent kinase II contributes to persistent central neuropathic pain following spinal cord injury. Pain. 2012;153(3):710–721. | ||
He Y, Chen Y, Tian X, et al. CaMKIIα underlies spontaneous and evoked pain behaviors in Berkeley sickle cell transgenic mice. Pain. 2016;157(12):2798–2806. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.