Back to Journals » Cancer Management and Research » Volume 11
Transcriptomic analyses identify key differentially expressed genes and clinical outcomes between triple-negative and non-triple-negative breast cancer
Authors Chen B, Tang H ![]() , Chen X, Zhang G, Wang Y, Xie X
, Chen X, Zhang G, Wang Y, Xie X ![]() , Liao N
, Liao N ![]()
Received 11 September 2018
Accepted for publication 16 November 2018
Published 21 December 2018 Volume 2019:11 Pages 179—190
DOI https://doi.org/10.2147/CMAR.S187151
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Beicheng Sun
Bo Chen,1,* Hailin Tang,2,* Xi Chen,3,* Guochun Zhang,1 Yulei Wang,1 Xiaoming Xie,2 Ning Liao1
1Department of Breast Cancer, Cancer Center, Guangdong General Hospital and Guangdong Academy of Medical Sciences, Guangzhou, China; 2Department of Breast Oncology, Sun Yat-sen University Cancer Center, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Guangzhou, China; 3Department of Anatomy, Hengyang Medical College, University of South China, Hengyang, China
*These authors contributed equally to this work
Purpose: There are significant differences in the biological behavior between triple-negative breast cancer (TNBC) and non-triple-negative breast cancer (non-TNBC). In the present study, we identify key differential genes and clinical outcomes between TNBC and non-TNBC.
Materials and methods: Transcriptomic analyses used GEO datasets (GSE76275), gene ontology, KEGG pathway analysis and cBioPortal. Quantitative RT-PCR analysis (qRT-PCR) was used to validate the differentially expressed genes. We used the KM Plotter Online Tool and 240 patients with TNBC tissue microarray to assay the prognostic value of HORMAD1.
Results: The upregulated differentially expressed genes were enriched in transcription factor activity, sequence-specific DNA binding and nucleic acid binding transcription factor activity. Only 16 genes were upregulated when further screened for fold change >4-fold change. HORMAD1 and SOX8 exhibited high frequencies of change of greater than 10% (HORMAD1 was close to 20%). qRT-PCR results indicated that HORMAD1 and SOX8 mRNA levels were significantly upregulated in TNBC samples. In KM Plotter Online Tool, high HORMAD1 was associated with worse outcome. In our tissue microarray (including 240 TNBC tissues), IHC analysis revealed that 29.7% (55/240) of the tumor samples exhibited high HORMAD1 expression and 70.3% (185/240) of the tumor samples exhibited low HORMAD1 expression levels. Meanwhile, high HORMAD1 group has a bad prognosis.
Conclusion: The status of transcriptional activation is an important difference between TNBC and non-TNBC. HORMAD1 is a key differential gene associated with poor outcome in TNBC. Epigenetic therapy and agents targeting cancer/testis antigens might potentially help to customize therapies of TNBC.
Keywords: HORMAD1, triple-negative breast cancer, non-triple-negative breast cancer, prognostic factor, transcriptome
Introduction
Breast cancer, the most common malignancy in women, exhibits significant heterogeneity.1 Its molecular type is very important to guide clinical treatments and to judge the prognosis.2 Triple-negative breast cancer (TNBC) accounts for approximately 15%–20% of breast cancer cases and is defined as the absence of estrogen receptor (ER), progesterone receptor (PR) and human epidermal receptor 2 (HER2) amplification.3,4 Due to the absence of druggable molecular targets, the treatment of TNBC is very limited compared with the treatment of luminal or HER2+ subtypes.5
In the past, relevant studies emphasized the difference of clinicopathological features and prognosis of patients between TNBC and non-triple-negative breast cancer (non-TNBC). TNBC patients often exhibit younger age, increased tumor size, and more recurrence and metastasis.6 Compared with non-TNBC patients, TNBC patients exhibit a higher rate of family history of breast cancer.7,8 Blood vessels play a vital role in TNBC, leading to distant metastasis.9 The development of distant metastasis in TNBC is also associated with the presence of central fibrosis and lymphocytic infiltration.10 Moreover, the lack of effective treatment after surgery, chemotherapy and radiotherapy is also an important reason for the poor prognosis of TNBC.11
In the coming post-genomic era, attention will no longer be confined to these clinicopathological features. High-throughput technology is increasingly advanced and widespread.12 Researchers are revealing the mechanism of the disease through high-throughput technology. In the field of cancer research, transcriptional information and gene regulatory networks of malignant tumors have become hot topics. Simultaneously, numerous public databases, such as the Gene Expression Omnibus (GEO), have been developed that serve as repositories of high-throughput gene expression data and hybridization arrays, chips and microarrays.12 These public databases provide great convenience for transcriptomic research. Therefore, based on these public platforms, we explored the transcriptional differences between TNBC and non-TNBC to better understand breast cancer and provide new clues for therapy.
In this study, we compared transcriptomic differences between TNBC and non-TNBC using a GEO database that contained 265 samples. We identified differentially expressed genes, relevant pathways and upregulated genes associated with clinical outcome and used tissue microarray to confirm the prognostic value.
Materials and methods
Microarray data
We extracted gene expression profiles of TNBC and non-TNBC from a GEO database (GSE76275) that contained 265 samples, including 198 TNBC samples and 67 non-TNBC samples. The database was submitted by Burstein et al13 and based on the Agilent GPL570 platform (Affymetrix Human Genome U133 Plus 2.0 Array). Differentially expressed genes were analyzed by GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/).
Gene ontology (GO) and pathway enrichment analysis of differentially expressed genes
Gene set enrichment analyses focused on genes with minimum twofold differential expression values between both groups. GO and KEGG pathway analysis were performed using DAVID Bioinformatics Resources 6.7 (https://david.ncifcrf.gov/), which used the one-tailed Fisher’s exact probability test to determine overrepresentation of genes with altered expression within specific GO categories.14,15 A P-value <0.05 was considered to be statistically significant.
Database search
As an open-access resource, the cBio Cancer Genomics Portal (http://cbioportal.org) data from more than 5,000 tumor samples from 147 cancer studies provides interactive exploration of multidimensional cancer genomics data sets.16,17 The data set contained 9 sets of breast invasive carcinoma samples. Further details on the samples and processing of the database are provided in the cBio Cancer Genomics Portal. We selected four of these sets that contain both mutation and DNA copy number alteration (CNA) information for our study.
Cell lines and clinical samples
A normal mammary epithelial cell line (MCF-10A) and human breast cancer cell lines (MDA-MB-231, BT-549, MDA-MB-468, HCC38, MCF-7, T47D, BT-474 and Skbr3) were obtained from the American Type Culture Collection (Manassas, VA, USA). All of the cell lines were used for only up to 30 passages from authentication and were maintained according to the supplier’s instructions. All cell lines were authenticated by short tandem repeat DNA profiling before use and found to be free of mycoplasma infection. Tissue specimens (26 TNBC and the corresponding paired normal adjacent tissues [Normal 1] and from 22 non-TNBC and the corresponding paired normal adjacent tissues [Normal 2]) were diagnosed by histopathological diagnosis from November 2015 to May 2016. All clinical samples were stored in the Department of Specimens and Resources of Sun Yat-sen University Cancer Center (SYSUCC). Resected breast cancer tissues and paired matched normal mammary tissues were immediately cut and stored in RNAlater (Ambion). Sun Yat-Sen University Cancer Center Institute Research Ethics Committee approved this study (RDDB2018000454), and the collection and use of tissues followed procedures that are in accordance with the ethical standards formulated in the Declaration of Helsinki. All participants provided written informed consent prior to treatment.
Quantitative RT-PCR analysis (qRT-PCR)
Total RNA from the cells or tissues was extracted with TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Reverse transcription and qRT-PCR reactions were performed by means of a SYBR green-containing PCR kit (Qiagen NV, Venlo, the Netherlands). Each reaction mixture consisted of 2.0 ng of cDNA template, 0.3 µL each of forward and reverse primers, and 10 µL of SYBR green mix adjusted with nuclease-free water to a final volume of 20 µL. The primer sequences used are as follows: HORMAD1 forward, 5′-GCCCAGTTGCAGAGGACTC-3′ and reverse, 5′-TCTTGTTCCATAAGCGCATTCT-3′; SOX8 forward, 5′-CCACGAGTTCGACCAGTACC-3′ and reverse, 5′-CTGCAGGAACCGTAGTCGG-3′. β-Actin served as an internal reference in the mRNA quantitative measurement. All of the real-time PCR assays were performed using a Bio-Rad IQTM5 Multi-color Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA) with three biological replicates and two technical replicates.
Tissue microarray and immunohistochemistry
For the TMA, a series of 240 patients with TNBC diagnosed between 2005 and 2011 at the Sun Yat-sen University Cancer Center, China, was selected. To quantify the level of HORMAD1 (Thermo Fisher Scientific, 1:500), all sections were photographed and scored by two independent pathologists. The staining intensity was scored as follows: 0–1 (no staining), 1–2 (weak staining), 2–3 (medium staining) and 3–4 (strong staining). The semi-quantification was performed according to the staining intensity (0%–25%; 26%–50%; 51%–75% and 76%–100%) and the ratio of positively stained cells (0, low staining; 1, moderate staining; 2, high staining and 3, extremely high staining). The overall score was calculated by multiplying the intensity score and percentage score. Expression scores greater than or equal to 2 were defined as high expression, and scores less than 2 were defined as low expression.
Outcome analyses
The KM Plotter Online Tool (http://www.kmplot.com) is a public database that contains information from 3,500 patients and combined survival analysis across multiple microarray datasets, including GEO, caArray and TCGA.18 The tool was utilized to evaluate the relationship between the presence of different genes and patient clinical outcome. Definitions of breast cancer subgroups reported in the online tool are as follows: triple negative: ER-/PR-/HER2-; luminal A/B: ER+/PR±/HER2±; HER2+: ER-/PR-/HER2+ (information for ER/PR and HER2 using immunohistochemical data). The Kaplan–Meier method was used to plot overall survival (OS) curves and relapse-free survival (RFS) curves, and the log-rank test was used for comparison in Sun Yat-sen University Cancer Center cohort (SYSUCC cohort).
Results
Identification of differentially expressed genes
To analyze the differentially expressed genes between TNBC and non-TNBC, we used a GEO dataset of mRNA level data (GSE76275) that contained 198 TNBC samples and 67 non-triple-negative tumor samples (Figure 1). Based on the GEO2R analysis and using P<0.05 and fold change (FC) >2.0 criteria, a total of 643 differentially expressed genes were identified (225 upregulated and 418 downregulated).
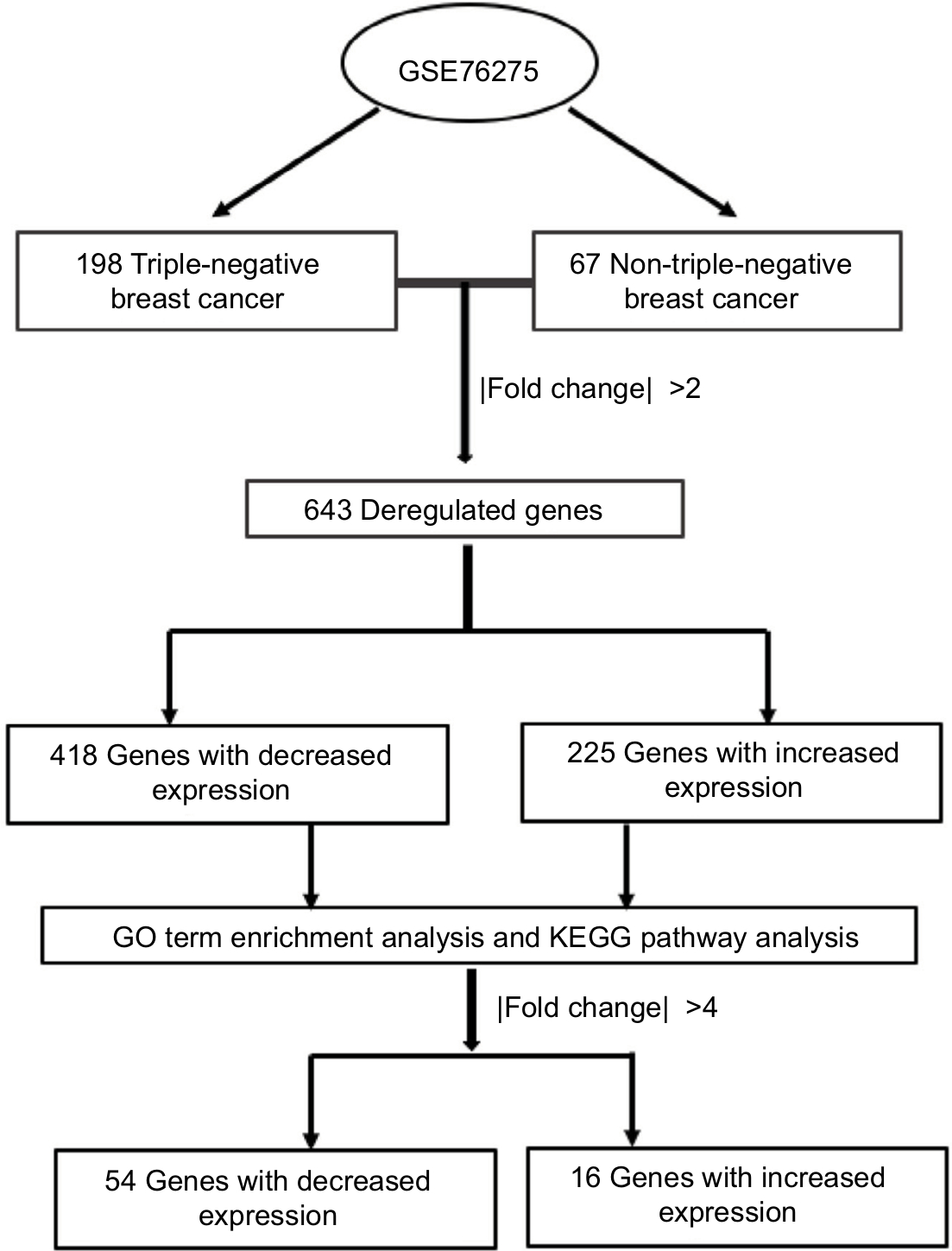 | Figure 1 Overview of the study. Abbreviation: GO, gene ontology. |
GO term enrichment analysis and KEGG pathway analysis
Next, we used DAVID Bioinformatics Resources 6.7 to identify overrepresented GO categories and KEGG pathways of the list of differentially expressed genes. As noted in Table 1, GO biological processes (BP) analysis results revealed that upregulated differentially expressed genes were significantly enriched in the regulation of gene expression, cell proliferation and cell cycle. The downregulated differentially expressed genes were significantly enriched in regulation of signaling, regulation of cell communication and cellular response to chemical stimulus. For molecular function (MF), the upregulated differentially expressed genes were enriched in transcription factor activity, sequence-specific DNA binding and nucleic acid binding transcription factor activity. The downregulated differentially expressed genes were enriched in receptor binding and identical protein binding. In addition, GO cell component (CC) analysis is also provided in Table 1. In the KEGG analysis, the most significantly enriched pathways of the upregulated differentially expressed genes were cell cycle, microRNAs in cancer and the Wnt signaling pathway, whereas the downregulated differentially expressed genes were enriched in the cGMP–PKG signaling pathway, AMPK signaling pathway and pancreatic secretion (Table 1).
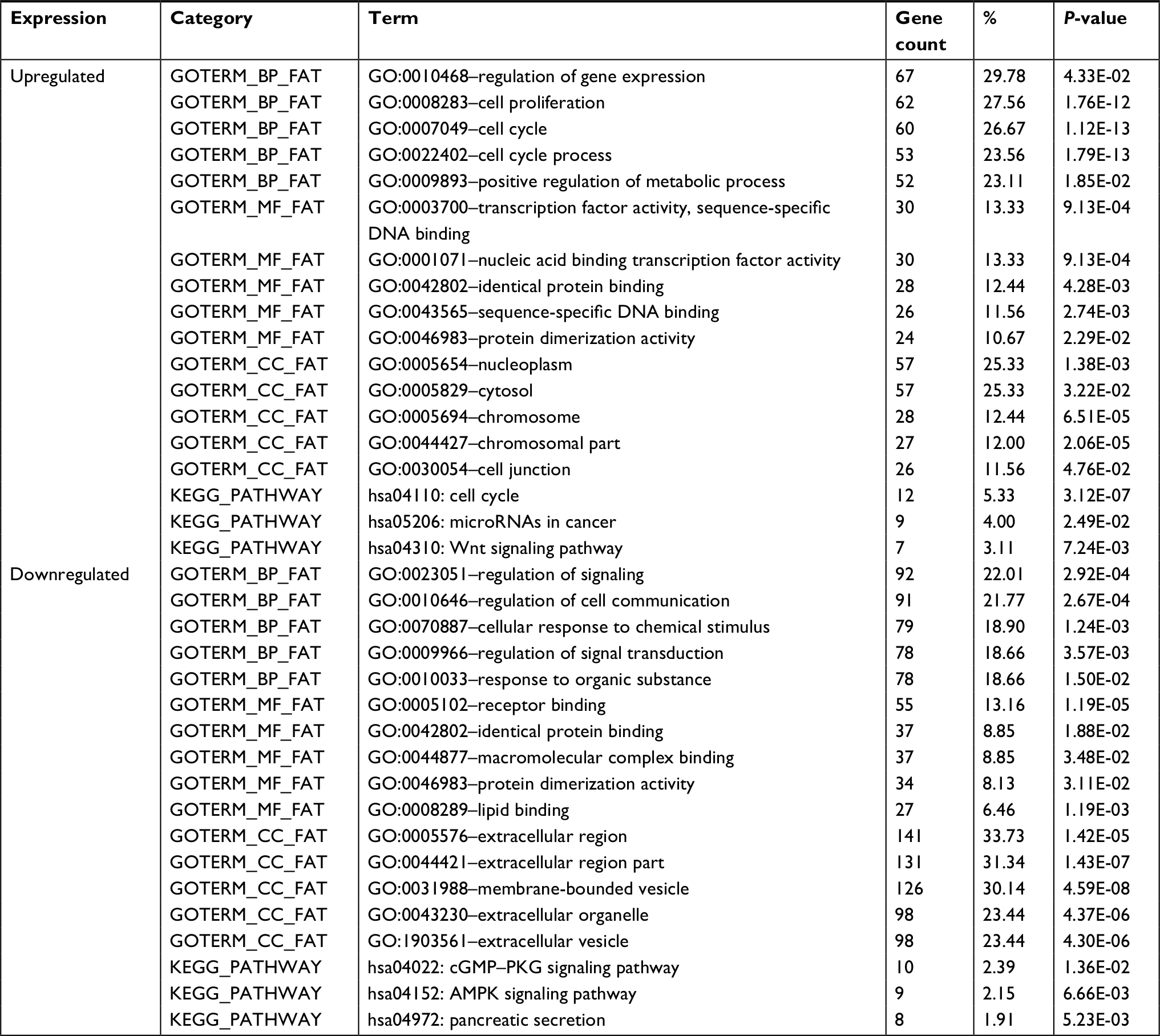 | Table 1 Gene ontology analysis and KEGG pathway analysis of differentially expressed genes |
HORMAD1 and SOX8 are key differential genes that are upregulated in TNBC
To narrow the scope to identify more important differentially expressed genes, we further screened |FC|>4.0. Among these genes, only 16 were upregulated whereas 54 were downregulated (Table S1). Then, we focused on the 16 upregulated genes (Table 2) rather than the 54 downregulated genes because we sought to identify potential therapeutic targets and not tumor suppressor genes. Four databases containing CAV and mutation information in the cBio Cancer Genomics Portal (http://Cbioportal.org) were used to determine the expression of these 16 genes in breast cancer patients (Figure 2A). As shown in Figure 2B, most of these 16 genes exhibited a frequency of alteration between 1% and 3%. Only HORMAD1 and SOX8 exhibited a frequency change of greater than 10% (HORMAD1 was close to 20%). We selected the METABRIC database to further analyze HORMAD1 and SOX8 in different molecular types of breast cancer. The results showed that the amplification ratio of HORMAD1 and SOX8 in TNBC was lower or closer to Luminal A/B, whereas the ratio of mRNA upregulation in TNBC was significantly increased compared with the Luminal A/B and HER2+ subtypes (Figure 2C). Similar results were obtained using the PAM50 classification method (Figure S1).
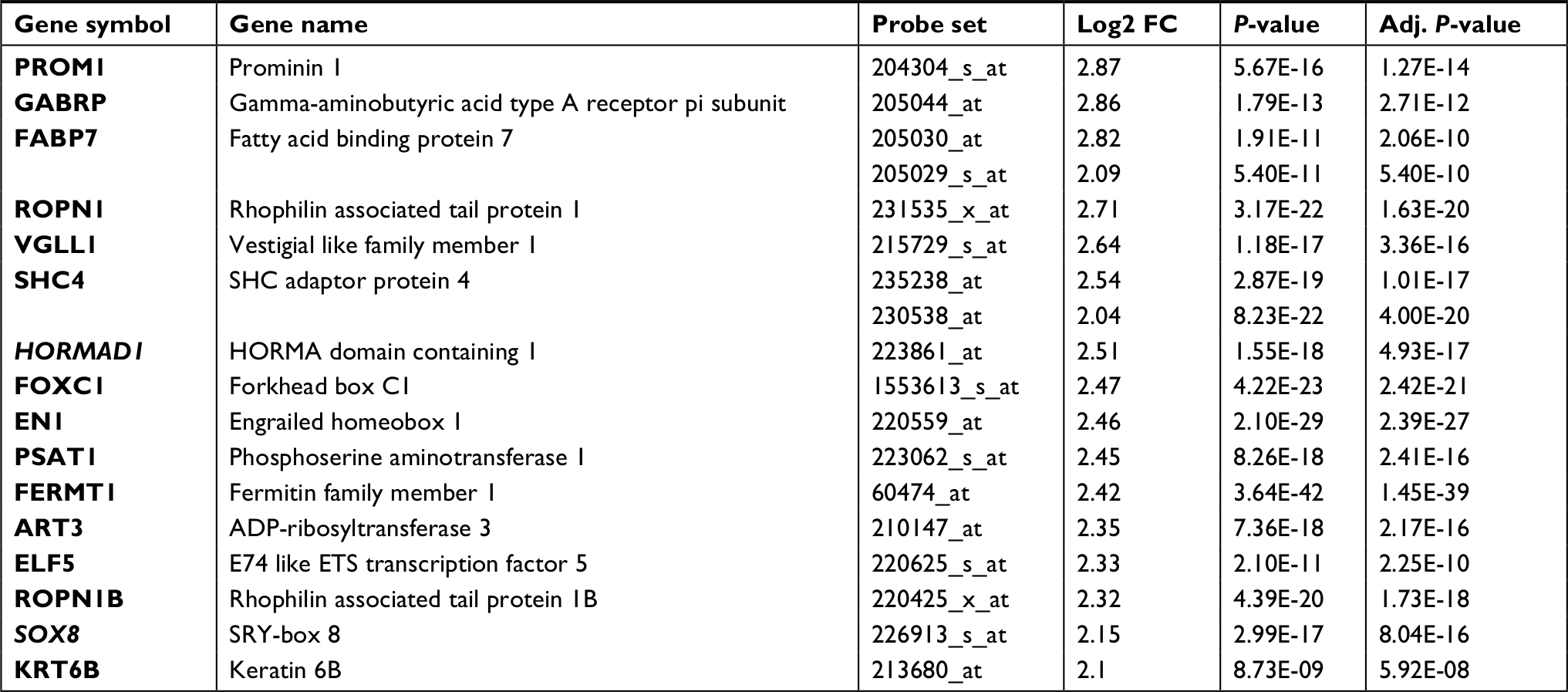 | Table 2 The 16 upregulation identified genes Abbreviation: FC, fold change. |
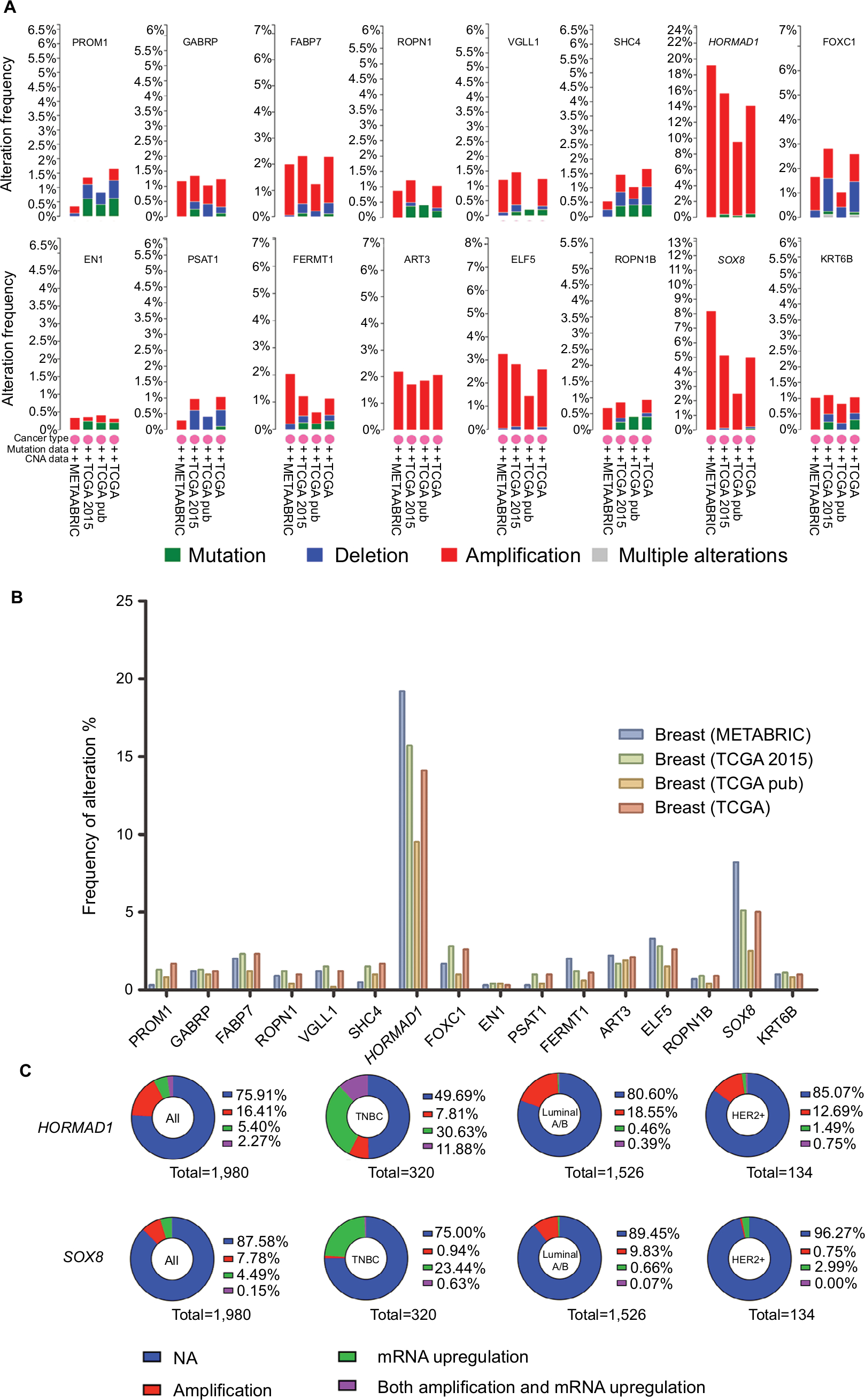 | Figure 2 The 16 upregulation identified genes. Notes: (A) Details of the frequency alteration of the 16 identified upregulated genes in the cBio Cancer Genomics Portal. (B) The frequency of alteration. (C) HORMAD1 and SOX8 status in different subtypes of breast cancer. Abbreviations: HER2, human epidermal receptor 2; TNBC, triple-negative breast cancer. |
qRT-PCR validation of HORMAD1 and SOX8 in breast cancer cell lines and clinical samples
To verify the expression of HORMAD1 and SOX8 in breast cancer, we measured HORMAD1 and SOX8 mRNA levels in 1 normal mammary epithelial cell line and 8 human breast cancer cell lines using a qRT-PCR method. As shown in Figure 3A, D, compared with the normal mammary epithelial cell line, both HORMAD1 and SOX8 mRNA levels are upregulated in 7 human breast cancer cell lines, especially in TNBC cell lines (including MDA-MB-231, BT549, MDA-MB-468 and HCC38). To confirm these results, we tested HORMAD1 and SOX8 mRNA levels in tissue samples from 26 TNBC patients and 22 non-TNBC patients. qRT-PCR results revealed that HORMAD1 (Figure 3B, C) and SOX8 (Figure 3E, F) mRNA levels were significantly upregulated in TNBC samples.
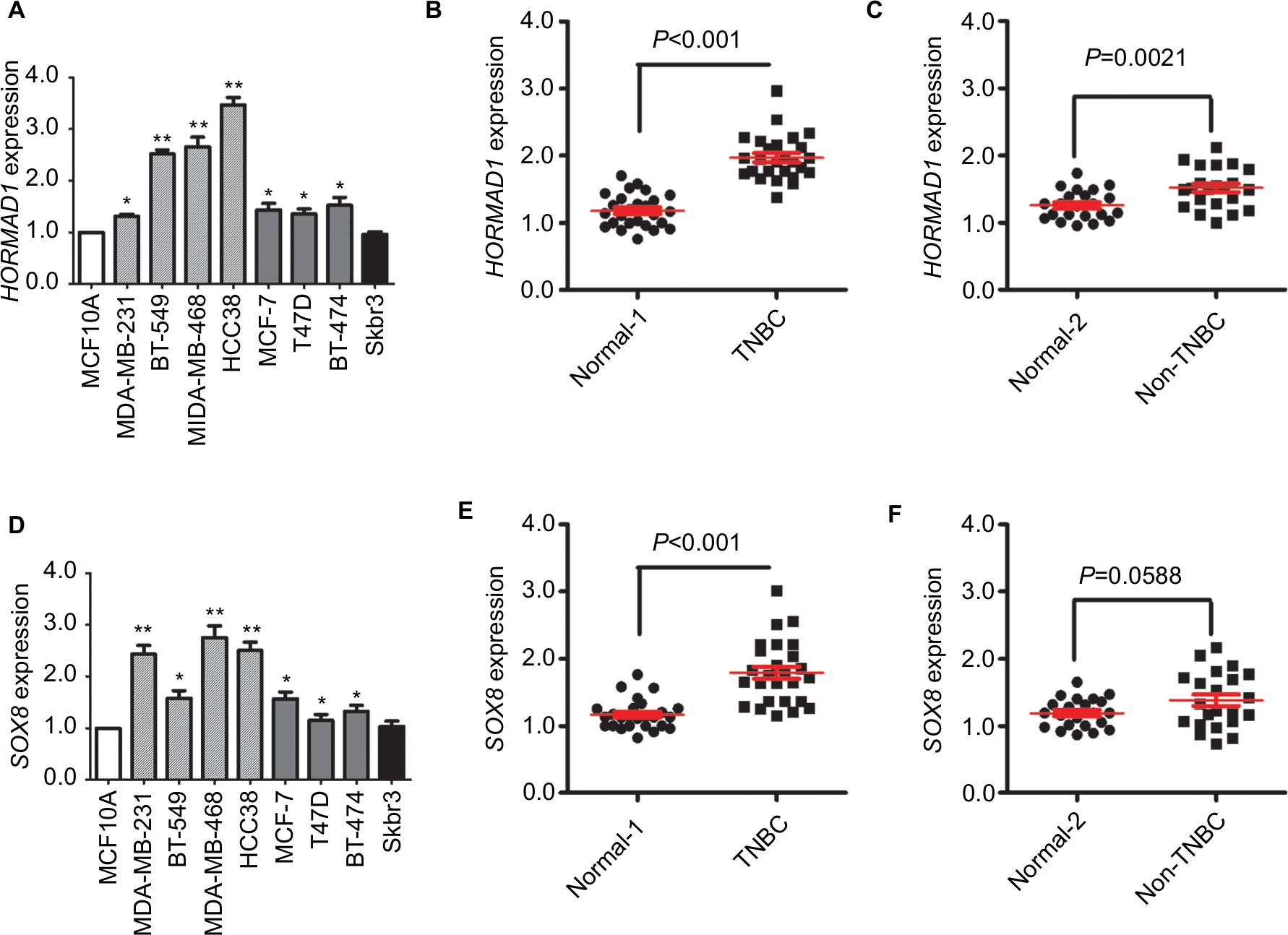 | Figure 3 qRT-PCR validation of HORMAD1 and SOX8 expression in breast cancer. Notes: HORMAD1 (A) and SOX8 (D) mRNA levels determined by qRT-PCR in a normal mammary cell line and in breast cancer cell lines. β-actin serves as the internal reference. The error bars represent the standard deviations from triplicates of one representative experiment. *P<0.05 and **P<0.01. (B) HORMAD1 mRNA levels in TNBC and the corresponding paired normal adjacent tissues (Normal 1). (C) HORMAD1 mRNA levels in non-TNBC and the corresponding paired normal adjacent tissues (Normal 2). (E) SOX8 mRNA levels in TNBC and the corresponding paired normal adjacent tissues (Normal 1). (F) SOX8 mRNA levels in non-TNBC and the corresponding paired normal adjacent tissues (Normal 2). Abbreviations: qRT-PCR, quantitative RT-PCR analysis; TNBC, triple-negative breast cancer. |
HORMAD1 predicts prognosis in TNBC
Next, we explored whether HORMAD1 and SOX8 were associated with worse RFS and OS in different subtypes of breast cancer. As described in the “Materials and methods” section, we used the KM Plotter Tool to perform survival analysis. Only HORMAD1 was associated with worse outcome (P=0.0023 for RFS, Figure 4A), and SOX8 was not useful to assess breast cancer patient prognosis (Figure S2). Based on the molecular subtype of breast cancer, only HORMAD1 was associated with worse RFS in TNBC (P=0.019, Figure 4B), but not in luminal A/B (P=0.19, Figure 4C) or HER2+ (P=0.12, Figure 4D). As shown in Figure 4E, HORMAD1 was also associated with worse OS in breast cancer (Figure 4E left). Given the limited data for OS, we only compared ER-positive and ER-negative breast cancer patients in the hierarchical analysis of OS. As expected, HORMAD1 was associated with worse OS in ER-negative breast cancer (Figure 4E middle) but not ER-positive breast cancer (Figure 4E right).
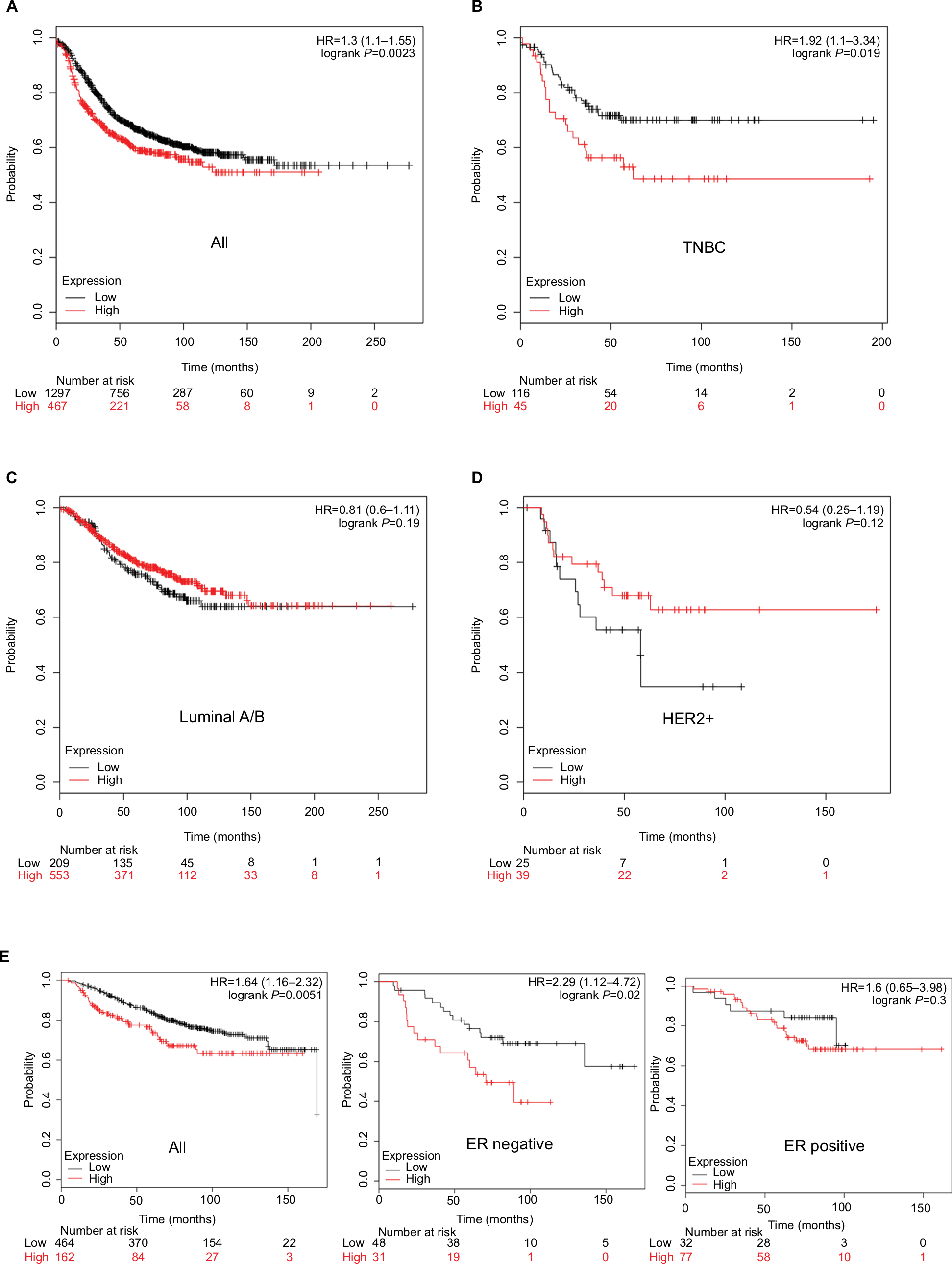 | Figure 4 Association of HORMAD1 with RFS and OS in breast cancer (KM Plotter Tool cohort). Notes: Kaplan–Meier survival curves of (A) RFS for breast cancer, (B) RFS for TNBC, (C) RFS for Luminal A/B, (D) RFS for HER2+ and (E) OS for breast cancer (left), ER-negative breast cancer (middle) and ER-positive breast cancer (right). Red line = high HORMAD1 group, black line = low HORMAD1 group. The cutoff value between high and low is computed by KM Plotter Tool, and the best performing threshold is used as a cutoff. Number at risk table below graph shows the number of cases that reached the endpoint (number of events), the number of cases that did not reach the endpoint (number censored) and the total number of cases. Abbreviations: HER2, human epidermal receptor 2; OS, overall survival; RFS, relapse-free survival; TNBC, triple-negative breast cancer. |
Moreover, we analyzed the significance of HORMAD1 by IHC and in terms of clinical prognosis, and a Kaplan–Meier survival analysis was conducted using patient disease-free survival (DFS) and OS in 240 TNBC patients’ tumor tissue samples. IHC analysis revealed that 29.7% (55/240) of the tumor samples exhibited high HORMAD1 expression and 70.3% (185/240) of the tumor samples exhibited low HORMAD1 expression levels. As shown in Table 3, HORMAD1 expression was positively correlated with the tumor size (P<0.001), infiltrated lymph nodes (P=0.002) and TNM stage (I–II vs III–IV; P<0.001) of TNBC. In addition, the survival analysis showed that patients with high HORMAD1 expression presented shorter mean months of DFS and OS than patients with low HORMAD1 expression (P<0.001 for both DFS and OS; Figure 5A, B).
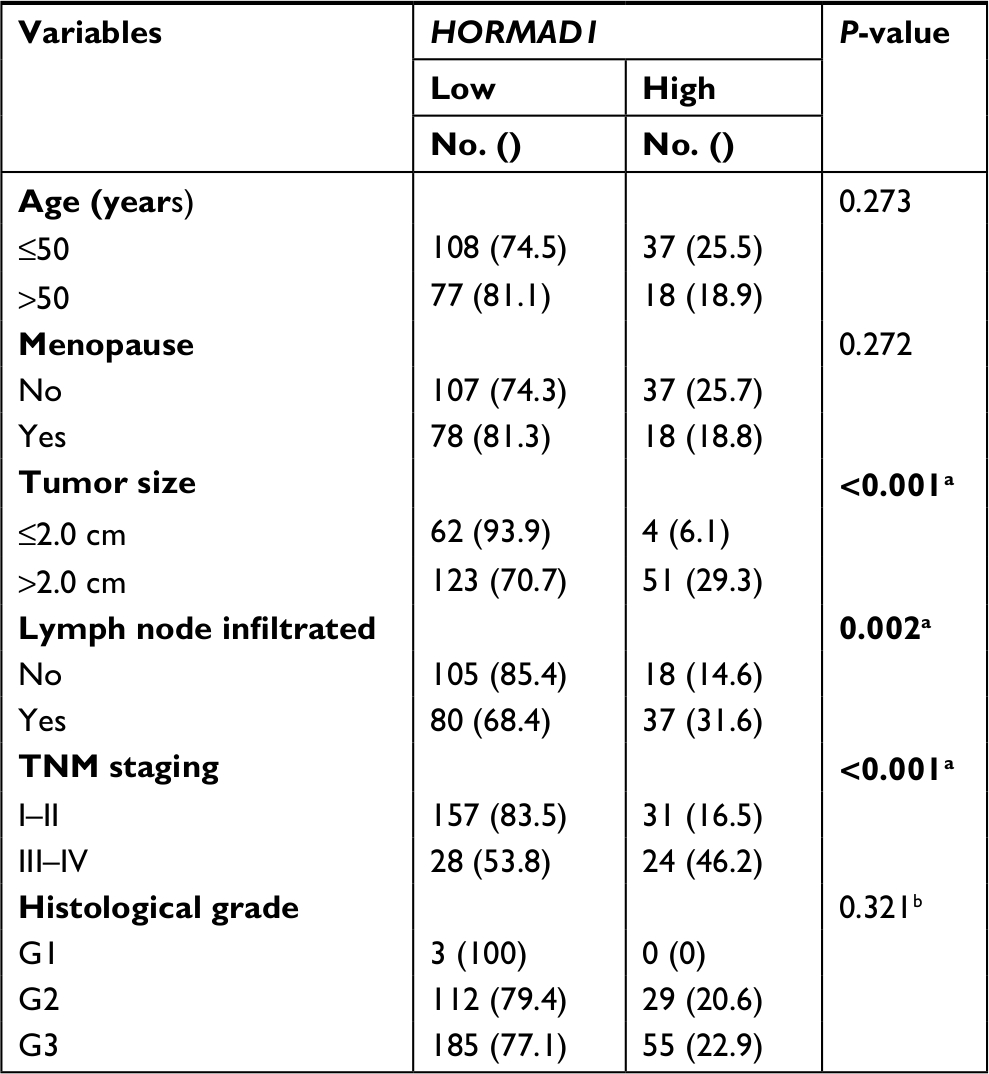 | Table 3 Association between HORMAD1 and clinicopathological characteristics in triple-negative breast cancer Notes: Bold figures indicate statistically significant. aP<0.05. bUsing Fisher’s exact test. |
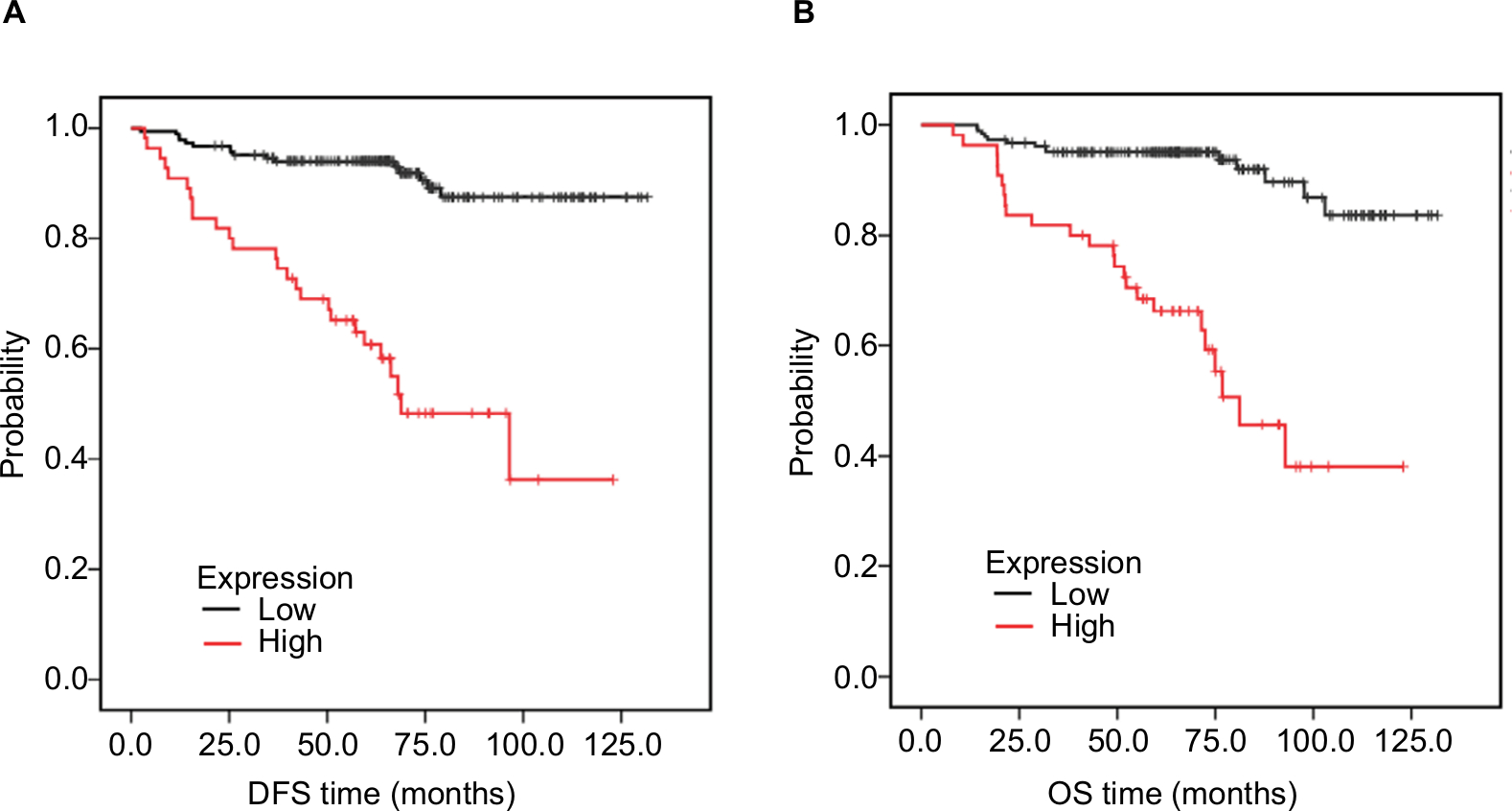 | Figure 5 Association of HORMAD1 with prognosis in 240 TNBC patients from SYSUCC (SYSUCC cohort). Notes: (A) DFS curves for 240 TNBC patients with high or low HORMAD1 levels. (B) OS curves for 240 TNBC patients with high or low HORMAD1 levels. Red line = high HORMAD1 group, black line = low HORMAD1 group. Abbreviations: DFS, disease-free survival; OS, overall survival; SYSUCC, Sun Yat-sen University Cancer Center; TNBC, triple-negative breast cancer. |
Discussion
The subtypes of breast cancer have not only distinct clinical presentations but also distinct prognostic implications.19 Due to high-risk biological characteristics and the lack of the benefit of a specific therapy, TNBC has attracted more attention both clinically and experimentally.6 It is likely that the improvements in survival have currently plateaued for TNBC patients. By contrast, other subtypes of breast cancer patients seem to exhibit better survival. In the present study, we used a GEO database to compare the differences in transcriptomic expression between TNBC and non-TNBC, and these results were validated in METABRIC and TCGA.20 We found that HORMAD1 was a key differentially expressed gene in which mRNA levels might specifically reflect the prognosis of TNBC.
Using GO analysis, we found that the difference between TNBC and non-TNBC is at least partially due to the status of transcriptional activation. The upregulated differentially expressed genes were significantly enriched in the regulation of gene expression and for MF. The upregulated differentially expressed genes were enriched in transcription factor activity, sequence-specific DNA binding and nucleic acid binding transcription factor activity. Analyzing two significant differentially expressed genes, HORMAD1 and SOX8, we observed no significant difference in gene copy number, but the proportion of the mRNA upregulation in TNBC was significantly increased compared with non-TNBC. This phenomenon might suggest that the transcription of TNBC is more active than non-TNBC due to epigenetic regulation. We hypothesize that TNBC and non-TNBC are like identical twins; the main source of the difference is epigenetic rather than genetic sequence differences. If this assumption is correct, it may not be difficult to explain why the tumor molecular subtype of some patients changes after treatment.21 As a result of external intervention, such as chemotherapy and endocrine therapy, the tumor microenvironment and a number of epigenetic genetic changes cause the tumor to change to another phenotype.22–24 Although this point of view should be confirmed by more experimental evidence, epigenetic therapy is a promising therapy in TNBC.25 In KEGG analysis, we found that cell cycle, microRNAs in cancer and the Wnt signaling pathway were upregulated in TNBC, whereas the cGMP–PKG and AMPK signaling pathways were downregulated. Therefore, focusing on these signaling pathways will help us to discover new therapeutic approaches for TNBC.
After we identified key upregulated genes, we validated HORMAD1 and SOX8 mRNA levels in both breast cancer cell lines and clinical samples using qRT-PCR. The results revealed that these two genes were increased in TNBC compared with non-TNBC and normal tissues. HORMA domains are involved in chromatin binding and participate in cell-cycle regulation.26,27 In previous studies, HORMAD1 was identified in developing mouse and mammalian gonads, and expression was restricted to germ cells.28 In mitosis and meiosis, HORMAD proteins act as adaptor proteins to recruit other proteins for DNA repair and checkpoint regulation.29,30 Through the analysis of gene expression, some studies revealed that HORMAD1 might function as an oncogene in several cancers, such as gastric cancer.31,32 However, the clinical significance of HORMAD1 in TNBC has not been fully explored. Our findings suggested that HORMAD1 is one of the most important differently expressed genes between TNBC and non-TNBC. HORMAD1 expression was positively correlated with the tumor size (P<0.001), infiltrated lymph nodes (P=0.002) and TNM stage (I–II vs III–IV; P<0.001) of TNBC. Furthermore, HORMAD1 was a prognostic marker of TNBC in subsequent survival analyses (KM Plotter Tool cohort and SYSUCC cohort). Recently, a study demonstrated that HORMAD1 contributed to homologous recombination deficiency in TNBC, which supports our findings.33 HORMAD1 is also known as cancer/testis antigen 46 (CT46) and belongs to a group of germ cell genes that are expressed in cancer.34 Cancer/testis antigens are exclusively expressed in germ cells and exhibit restricted expression in normal tissues.35,36 This characteristic may allow cancer/testis antigens, including HORMAD1, to be used for tumor-specific therapies with minimal side effects. Therefore, further research on the HORMAD1 gene and agents involved in cancer/testis antigens might offer hope for TNBC patients. Our work has some limitations. The GSE76275 dataset is mainly derived from Caucasian and Asian individuals; thus, we do not know whether the results are applicable to other races. Meanwhile, the sample size of KM Plotter Tool cohort is small and the log-rank test for comparing subclasses of tumors according to gene expression might not have enough power to detect differences. The results should be confirmed in large sample size of homogeneous prospective and retrospective studies.
Conclusion
We showed that the status of transcriptional activation is an important difference between TNBC and non-TNBC. In addition, HORMAD1 is a key differentially expressed gene that is associated with poor outcome in TNBC. Epigenetic therapy and agents involved in cancer/testis antigens might have potential utility when customizing therapies for TNBC.
Acknowledgment
This work was supported by funds from the National Natural Science Foundation of China (81472575, 81672598, Xiaoming Xie; 81472469, Hailin Tang) and the Science and Technology Planning Project of Guangzhou and Guangdong (2015B090901050, 2015B020211002, Xiaoming Xie; 2014A020212079, 2016A020214009, Hailin Tang).
Disclosure
The authors report no conflicts of interest in this work.
References
Zardavas D, Irrthum A, Swanton C, Piccart M. Clinical management of breast cancer heterogeneity. Nat Rev Clin Oncol. 2015;12(7):381–394. | ||
Vaz-Luis I, Ottesen RA, Hughes ME, et al. Outcomes by tumor subtype and treatment pattern in women with small, node-negative breast cancer: a multi-institutional study. J Clin Oncol. 2014;32(20):2142–2150. | ||
Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol. 2010;7(12):683–692. | ||
Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer. 2007;109(9):1721–1728. | ||
De Laurentiis M, Cianniello D, Caputo R, et al. Treatment of triple negative breast cancer (TNBC): current options and future perspectives. Cancer Treat Rev. 2010;36(Suppl 3):S80–S86. | ||
Qiu J, Xue X, Hu C, et al. Comparison of Clinicopathological Features and Prognosis in Triple-Negative and Non-Triple Negative Breast Cancer. J Cancer. 2016;7(2):167–173. | ||
Li CY, Zhang S, Zhang XB, Wang P, Hou GF, Zhang J. Clinicopathological and prognostic characteristics of triple- negative breast cancer (TNBC) in Chinese patients: a retrospective study. Asian Pac J Cancer Prev. 2013;14(6):3779–3784. | ||
Zhou T, Yang L, Ma GM, et al. Clinicopathologic features and prognosis of triple negative breast cancer. Zhonghua Yi Xue Za Zhi. 2009;89(32):2261–2264. | ||
Perentes JY, Kirkpatrick ND, Nagano S, et al. Cancer cell-associated MT1-MMP promotes blood vessel invasion and distant metastasis in triple-negative mammary tumors. Cancer Res. 2011;71(13):4527–4538. | ||
Linn SC, Van ‘t Veer LJ. Clinical relevance of the triple-negative breast cancer concept: genetic basis and clinical utility of the concept. Eur J Cancer. 2009;45(Suppl 1):11–26. | ||
Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13(11):674–690. | ||
Leek JT, Scharpf RB, Bravo HC, et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet. 2010;11(10):733–739. | ||
Burstein MD, Tsimelzon A, Poage GM, et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2015;21(7):1688–1698. | ||
Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. | ||
Huang DW, Sherman BT, Tan Q, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35(Web Server issue):W169–W175. | ||
Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. | ||
Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. | ||
Györffy B, Lanczky A, Eklund AC, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123(3):725–731. | ||
Weigelt B, Reis-Filho JS. Histological and molecular types of breast cancer: is there a unifying taxonomy? Nat Rev Clin Oncol. 2009;6(12):718–730. | ||
Pereira B, Chin SF, Rueda OM, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479. | ||
Beitsch P, Whitworth P, Baron P, et al. Genomic impact of neoadjuvant therapy on breast cancer: incomplete response is associated with altered diagnostic gene signatures. Ann Surg Oncol. 2016;23(10):3317–3323. | ||
Marks DL, Olson RL, Fernandez-Zapico ME. Epigenetic control of the tumor microenvironment. Epigenomics. 2016;8(12):1671–1687. | ||
Dey P. Epigenetic changes in tumor microenvironment. Indian J Cancer. 2011;48(4):507–512. | ||
Seftor EA, Meltzer PS, Kirschmann DA, Margaryan NV, Seftor RE, Hendrix MJ. The epigenetic reprogramming of poorly aggressive melanoma cells by a metastatic microenvironment. J Cell Mol Med. 2006;10(1):174–196. | ||
Treppendahl MB, Kristensen LS, Grønbæk K. Predicting response to epigenetic therapy. J Clin Invest. 2014;124(1):47–55. | ||
Wojtasz L, Daniel K, Roig I, et al. Mouse HORMAD1 and HORMAD2, two conserved meiotic chromosomal proteins, are depleted from synapsed chromosome axes with the help of TRIP13 AAA-ATPase. PLoS Genet. 2009;5(10):e1000702. | ||
Fukuda T, Daniel K, Wojtasz L, Toth A, Höög C. A novel mammalian HORMA domain-containing protein, HORMAD1, preferentially associates with unsynapsed meiotic chromosomes. Exp Cell Res. 2010;316(2):158–171. | ||
Pangas SA, Yan W, Matzuk MM, Rajkovic A. Restricted germ cell expression of a gene encoding a novel mammalian HORMA domain-containing protein. Gene Expr Patterns. 2004;5(2):257–263. | ||
Kogo H, Tsutsumi M, Ohye T, Inagaki H, Abe T, Kurahashi H. HORMAD1-dependent checkpoint/surveillance mechanism eliminates asynaptic oocytes. Genes Cells. 2012;17(6):439–454. | ||
Shin YH, McGuire MM, Rajkovic A. Mouse HORMAD1 is a meiosis I checkpoint protein that modulates DNA double- strand break repair during female meiosis. Biol Reprod. 2013;89(2):29. | ||
Aung PP, Oue N, Mitani Y, et al. Systematic search for gastric cancer-specific genes based on SAGE data: melanoma inhibitory activity and matrix metalloproteinase-10 are novel prognostic factors in patients with gastric cancer. Oncogene. 2006;25(17):2546–2557. | ||
Shahzad MM, Shin YH, Matsuo K, et al. Biological significance of HORMA domain containing protein 1 (HORMAD1) in epithelial ovarian carcinoma. Cancer Lett. 2013;330(2):123–129. | ||
Watkins J, Weekes D, Shah V, et al. Genomic complexity profiling reveals that HORMAD1 overexpression contributes to homologous recombination deficiency in triple-negative breast cancers. Cancer Discov. 2015;5(5):488–505. | ||
Chen YT, Venditti CA, Theiler G, et al. Identification of CT46/HORMAD1, an immunogenic cancer/testis antigen encoding a putative meiosis-related protein. Cancer Immun. 2005;5:9. | ||
Costa FF, Le Blanc K, Brodin B. Concise review: cancer/testis antigens, stem cells, and cancer. Stem Cells. 2007;25(3):707–711. | ||
Chen YT, Ross DS, Chiu R, et al. Multiple cancer/testis antigens are preferentially expressed in hormone-receptor negative and high-grade breast cancers. PLoS One. 2011;6(3):e17876. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.