Back to Journals » Journal of Inflammation Research » Volume 14
Transcriptome-Wide High-Throughput m6A Sequencing of Differential m6A Methylation Patterns in the Human Rheumatoid Arthritis Fibroblast-Like Synoviocytes Cell Line MH7A
Authors Jiang H, Cao K, Fan C, Cui X, Ma Y, Liu J
Received 19 December 2020
Accepted for publication 10 February 2021
Published 25 February 2021 Volume 2021:14 Pages 575—586
DOI https://doi.org/10.2147/JIR.S296006
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Hui Jiang1,2 *,* Kefeng Cao3 *,* Chang Fan,1,2 Xiaoya Cui,1,2 Yanzhen Ma,1,2 Jian Liu1
1Experimental Center of Clinical Research, First Affiliated Hospital of Anhui University of Chinese Medicine, Hefei, Anhui, People’s Republic of China; 2School of Pharmacy, Anhui University of Chinese Medicine, Hefei, Anhui, People’s Republic of China; 3Departments of Laboratory Medicine, Traditional Chinese Medical Hospital of Taihe County, Fuyang, Anhui, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jian Liu
First Affiliated Hospital of Anhui University of Chinese Medicine, 117 Meishan Road, Hefei, Anhui, People’s Republic of China
Tel +86 181 5607 8363
Email [email protected]
Introduction: N6-methyladenosine (m6A) is the most frequent internal modification in eukaryotic mRNAs and is closely related to the occurrence and development of many diseases, especially tumors. However, the relationship between m6A methylation and rheumatoid arthritis (RA) is still a mystery.
Methods: Two high-throughput sequencing methods, namely, m6A modified RNA immunoprecipitation sequence (m6A-seq) and RNA sequence (RNA-seq) were performed to identify the differentially expressed m6A methylation in human rheumatoid arthritis fibroblast-like synoviocytes cell line MH7A after stimulation with TNF-α. Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were used to obtain enriched GO terms and significant KEGG pathways. Then, four candidate genes, Wilms tumor 1-associating protein (WTAP), receptor-interacting serine/threonine protein kinase 2 (RIPK2), Janus kinase 3 (JAK3) and tumor necrosis factor receptor SF10A (TNFRSF10A) were selected to further validate the m6A methylation, mRNA and protein expression levels in MH7A cells and synovial tissues of adjuvant arthritis (AA) rats by RT-qPCR and Western blot.
Results: Using m6A-seq, we identified a total of 206 genes with differentially expressed m6A methylation, of which 118 were significantly upregulated and 88 genes were significantly downregulated. Likewise, 1207 differentially mRNA expressed mRNAs were obtained by RNA-seq, of which 793 were upregulated and 414 downregulated. Further joint analysis showed that the m6A methylation and mRNA expression levels of 88 genes changed significantly, of which 30 genes displayed increased m6A methylation and decreased mRNA expression, 57 genes displayed decreased m6A methylation and increased mRNA expression increased, and 1 gene displayed increased m6A methylation and increased mRNA expression. GO and KEGG analyses indicated that these unique genes were mainly enriched in inflammation-related pathways, cell proliferation and apoptosis. In addition, the validations of WTAP, RIPK2, JAK3 and TNFRSF10A were in accordance with the m6A and RNA sequencing results.
Conclusion: This study established the transcriptional map of m6A in MH7A cells and revealed the potential relationship between RNA methylation modification and RA related genes. The results suggested that m6A modification was associated with the occurrence and course of RA to some extent.
Keywords: m6A methylation, m6A-seq, RNA-seq, rheumatoid arthritis, MH7A cells
Introduction
N6-methyladenosine (m6A) is a common means of RNA modification, accounting for 0.1–0.4% of adenosine, and its biological significance has been gradually revealed.1,2 RNA m6A modification is widely found in the mRNAs of eukaryotes, specifically, m6A methylation means that RNA has been modified by methylation at the N6 site of the adenine base, and the modified adenosine is m6A adenine.3,4 Similar to DNA or histone methylation, RNA m6A methylation is also mainly catalyzed by methyltransferases and demethylases.5 This means that the m6A methylation modification is a dynamic reversible process.6 Various m6A methyltransferases, known as “writers,” including methyltransferase-like 3 (METTL3), METTL14 and Wilms tumor 1-associated protein (WTAP), catalyze the m6A methylation of mRNA.5,7 Various m6A demethylases are called “erasers”, including fat mass and obesity-associated protein (FTO) and ALKB family member 5 (ALKBH5), which function is to reduce the modified RNA to the original RNA.8 In addition, m6A binding protein acts as a “reader” and mainly includes the YT521-B homology (YTH) and heterogeneous nuclear ribonucleoprotein (HNRNP) families, such as HNRNPC, YTHDC1, YTHDC2, YTHDF1 and YTHDF2, which recognize m6A-modified RNA and thus regulate mRNA metabolism and function.9,10
For rheumatoid arthritis (RA), one of the most common autoimmune diseases, chronic inflammation, excessive synovial proliferation, joint swelling, and tenderness are the pathological characteristics.11,12 The differential proliferation of fibroblast-like synovial cells (FLSs) is an important factor in the progression of RA.13,14 With the deepening of research and the rapid development of technology, it has been found that there is an association between m6A methylation and RA. Luo et al15 confirmed that the expression of peripheral blood ALKBH5 and FTO was linked with autoantibody production and RA disease activity, which may be used as an indicator of activity and the inflammatory response. However, there are few studies about the effect of m6A methylation on RA FLSs.
In this study, using m6A-seq and RNA-seq, we identified differential m6A methylation and mRNA expression in MH7A cells, a type of human RA FLSs cell line that has been widely used in the study of RA. The sequencing and analysis process are shown in Figure 1.
 |
Figure 1 A schematic diagram of m6A-seq and RNA-seq analyses of MH7A cells. MH7A cells were stimulated with TNF-α, and total RNA was extracted from MH7A cells. Then, the RNA was fragmented, and m6A RNA was isolated as described by the m6A-RNA immunoprecipitation (Me-RIP) assay. The m6A-seq library and RNA-seq library of these mRNAs were constructed, and then sequenced for analysis. |
Materials and Methods
Cell Lines and Reagents
MH7A cells were provided by JENNIO Biological Technology (Guangzhou, CHN). MH7A cells were cultured in DMEM medium (Gibco), which 10% FBS (Gemini) and antibiotics (penicillin and streptomycin) were added and maintained at 37°C, 5% CO2 atmosphere. MH7A cells were stimulated with TNF-α (10 ng/mL), which was defined as the TNF-α group, and unstimulated MH7A cells were defined as the control group. After culturing for 24 h, MH7A cells were collected for the subsequent experiment.16
Animal Model
Male Sprague-Dawley rats (8 weeks old, 180–200 g) were purchased from the Anhui Experimental Animal Center. All rats were housed in the animal facility of the First Affiliated Hospital of Anhui University of Chinese Medicine and provided adequate water and food. Animal experiments in this study had been approved by the Animal Ethics Committee of Anhui University of Chinese Medicine, Ethical Approval Number: AHUCM-rats-004. The rats were adaptively fed for 1 week and then subcutaneous injection of complete Freund’s adjuvant into the left hindfoot toe to establish an adjuvant arthritis (AA) rat model. After induction of the immune response for 20 days, all rats were anesthetized by the intraperitoneal injection of 1% sodium pentobarbital (60 mg/kg) and sacrificed by exsanguination of the abdominal aorta. Then, the knee joints were removed, and the synovial tissues were separated. Experimental methods were performed according to the study of Hui Jiang et al.17 The synovial tissues of AA rats were defined as a model group. Conversely, the synovial tissues of normal rats were defined as the control group.
Construction of Library RNA-Seq and m6A-Seq
MH7A cells were digested with trypsin and centrifuged to collect cell precipitates, and the total RNA was extracted by the Total RNA Rapid Extraction Kit (TR205-200, Tianmo, China). The RNA after processing was determined by an Agilent 2100 Bioanalyzer (Agilent Technologies, United States), and the RNA concentration was detected by a NanoDrop one (Thermo Fisher Scientific, United States). VAHTS mRNA Capture Beads was used for capturing poly (A) RNA, which was fragmented by VAHTS 2X Frag/Prime Buffer. Some of the fragments were used for constructing the RNA-seq library with the VAHTS mRNA-seq V2 Library Prep Kit for Illumina, and others were used for the MeRIP-seq library following the instructions of the EpiMark N6-Methyladenosine Enrichment Kit. The obtained libraries were quantified by Qubit 3.0 Fluorometer (Life Technologies) and then sequenced on an Illumina NovaSeq 6000 (Illumina), and the sequencing process was entrusted to Origin-Biotech Inc (Ao-Ji Biotech, Shanghai, China).
Differential Gene Expression Analysis
The quality visualization of RNA-seq reads was accomplished by FastQC (version. 0.11.3).18 Trimming was performed by fastp for known Illumina TruSeq adapter sequences, poor reads, and ribosomal RNA reads.19 The trimmed reads were then mapped to the human reference genome (hg38) using Hisat2.20,21 And p-value < 0.05 and an absolute value of a fold change >2 were used as criteria for screening differentially expressed genes.
Differential m6A Peaks Analysis
Similar to the RNA-seq analysis process, the raw reads were subjected to QC, and trimmed, and m6A-enriched peaks were obtained by mapped to hg38. StringTie (version: 1.3.0) was performed for each gene count from trimmed reads.21 Gene counts were normalized by TMM,22 and FPKM was performed using Perl script.23 EdgeR was used to determine differential gene expression,24 with significance determined by a p-value <0.05 and an absolute value of a fold change >2.25
Functional Enrichment Analysis
Based on the crucial differentially expressed genes obtained by m6A-seq and RNA-seq, Gene Ontology (GO)26 and Kyoto Encyclopedia of Genes and Genomes (KEGG)26 analyses were performed by the clusterProfifiler of R package. Then, the top 30 GO terms and pathways were selected according to the P-value and the degree of enrichment.
PPI Network
The differentially expressed genes (DEGs) were imported into the STRING database, which contains comprehensive information about interactions between proteins;27 thus, the interaction relationship between DEGs was obtained. The protein–protein interaction (PPI) network was constructed based on importing the data into Cytoscape 3.5.1 software, and then, the network was analyzed by Network Analyzer. The DEGs with interactions with combined scores greater than 0.4 were selected to construct a protein–protein interaction network diagram.28
RT-qPCR Validation
RT-qPCR was used to detect candidate gene m6A methylation and mRNA expression levels. Total RNA from MH7A cells and synovial tissues of AA rats was extracted by Mireasy Mini Kit (cat 217004, Qiagen). A Nanodrop nd-2000 spectrophotometer (Thermo Fisher Science, Walsham, Massachusetts, USA) was used to determine the concentration and purity of RNA. Then, cDNA reverse transcription and RT-qPCR reactions were performed using StepOne SYBR Green Real-time PCR Master Mix (ABclonal, RK20404). Reactions proceeded using the following conditions: 95°C for 1 min, followed by 40 cycles of 95°C for 5 s, 60°C for 30 s. Melting curves ranging from 60°C to 95°C were included in each run.
Western Blot
Total protein of MH7A cells and synovial tissues of AA rats were extracted. SDS loading buffer was added to the extracted protein and the mixture was heated in boiling water for 10 min to denature the protein. A 10% polyacrylamide gel was prepared by an SDS-PAGE gel preparation kit (cat P0012A, Beyotime), and then, the protein samples were added to each well for electrophoretic separation. The protein was transferred to a polyvinylidene difluoride membrane and placed in 5% skim milk to seal at room temperature for 2 h. The membranes were incubated with primary antibody against WTAP (cat ab195380, Abcam, 1:2000), RIPK2 (cat bs-3546R, Bioss, 1:1000), JAK3 (cat ab45141, Abcam, 1:1000) and TNFRSF10A (cat bs-0591R, Bioss, 1:1000) overnight at 4°C with slow shaking. Then, the membranes were transferred to horseradish peroxidase-labeled secondary antibodies (1:20000) and incubated continuously at room temperature for 1 h. The protein was developed by a multifunction chemiluminescence imaging system and quantitatively analyzed by ImageJ software.
Statistical Analysis
The experimental data are presented as the mean ± standard deviation (SD). Statistical analysis was performed by using SPSS 23.0 software. Paired Student’s t-tests were used to detect the differences between the two groups. When the p-value < 0.05, the results were considered to be statistically significant.
Results
Transcriptome-Wide m6A-Seq Revealed Global m6A Modification Patterns in MH7A Cells
We compared m6A methylation peaks at each site in MH7A cells after stimulation with TNF-α. The differences and overlaps in m6A methylation between the individuals are shown by the Venn diagram in Figure 2A. We found 13,763 m6A peaks in the control group and 13,621 m6A peaks in the TNF-α group, of which 12,896 m6A peaks were common between the two groups. Compared with the control group, 725 peaks appeared and 867 peaks disappeared in the TNF-α group, indicating that there was a significant difference in the m6A modification pattern of MH7A cells after stimulation with TNF-α.
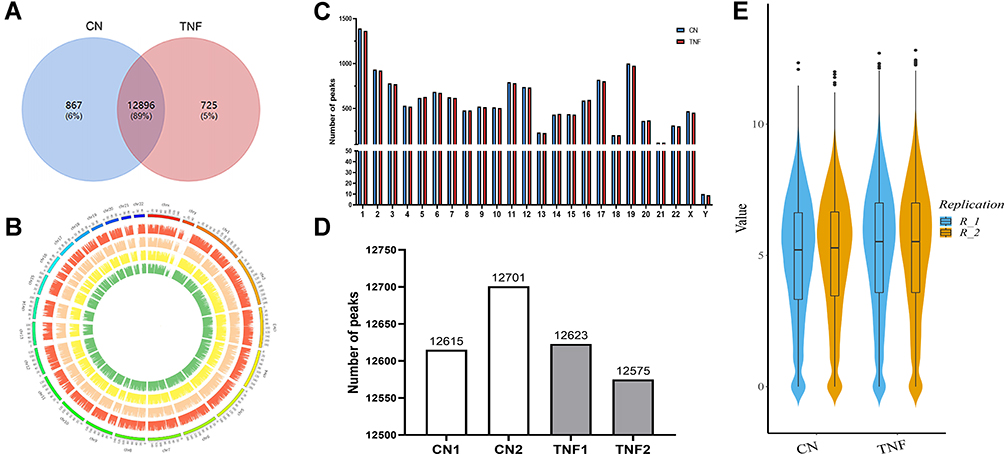 |
Figure 2 Transcriptome-wide m6A-Seq revealed global m6A modification patterns in MH7A cells after stimulation with TNF-α. (A) Venn diagram of m6A modified genes in the control group and the TNF-α group. (B) The distribution patterns of m6A peaks in different chromosomes. (C) The number of m6A peaks in per chromosome. (D) The number of m6A peaks in each group. (E), Violin plot of the relative abundance of m6A peaks in each group. |
The analysis of the m6A methylation distribution at different chromosome loci found that the chromosomes with the most m6A methylation were chromosome 1 with 1365 m6A methylation peaks, chromosome 2 with 922 m6A methylation peaks and chromosome 19 with 976 m6A methylation peaks, respectively (Figure 2B and C). Then, by comparing the m6A methylation levels, we found an average of 12,658 peaks in the control group and 12,599 peaks in the TNF‐α group, respectively (Figure 2D). The analysis of the degree of enrichment of m6A methylation showed that the average logarithmic fold-enrichment of the control group was 4.91, while that of the TNF-α group was 5.19 (Figure 2E).
Differential m6A Methylation of Genes in MH7A Cells After Stimulation with TNF-α
Using the filtering criteria of fold change > 2 and p-value < 0.05, 206 genes with differential m6A methylation genes in MH7A cells were identified after stimulation with TNF-α, of which 118 m6A hypermethylated genes and 88 m6A hypomethylated genes were identified (Figure 3A and B). The top 10 hypermethylated genes and the top 10 hypomethylated genes are shown in Table 1.
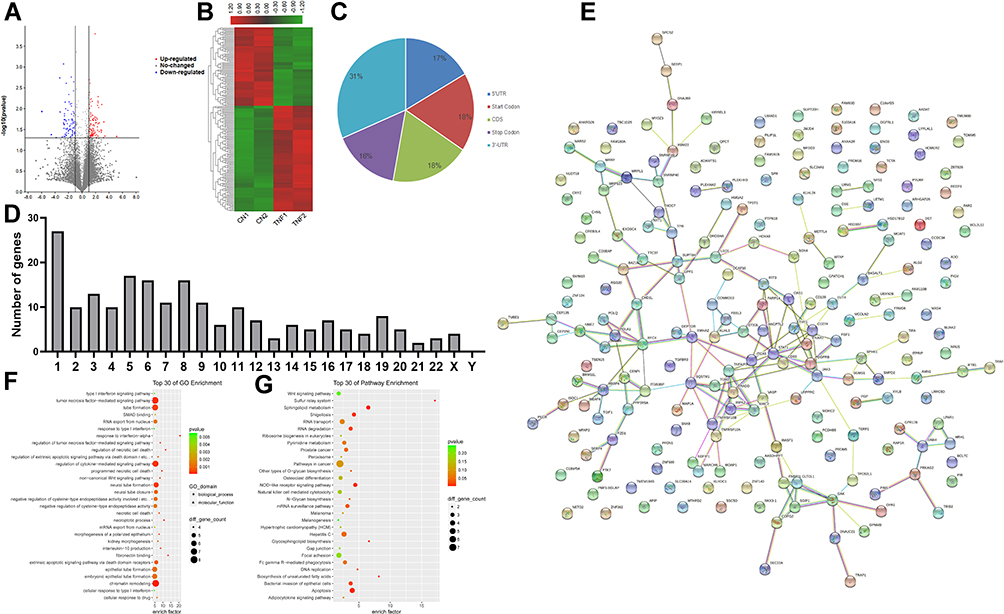 |
Figure 3 Genes with differential m6A methylation modification genes in MH7A cells after stimulation with TNF-α. (A) Volcano plot representation of microarray data on the differentially expressed m6A methylation genes between the control group and TNF-α group. The blue and red dots to the left and to the right of the two vertical lines indicate more than a 1.5 fold change and represent the differentially expressed m6A methylation genes with statistical significance. Statistical significance was defined as a fold change of 1.5 and a p-value of 0.05 between the control and TNF-α groups. (B) Hierarchical cluster analysis of differentially expressed m6A methylation genes between the control group and TNF-α group. Hierarchical clustering showed that the differentially expressed m6A methylation genes ultimately clustered into two major branches, including hypermethylated genes, which are labeled in red, and hypomethylated genes, which are labeled in green. The darker the color, the more significant the difference. (C) The sector graph shows the proportion of m6A methylation distribution in the indicated regions. (D) The number of genes with differential m6A methylation in per chromosome. (E) PPI of differentially expressed m6A methylation genes. (F) The top 30 enriched GO terms of differentially expressed m6A methylated genes in MH7A cells. (G) The top 30 pathway enrichments of differentially expressed m6A methylated genes in MH7A cells. |
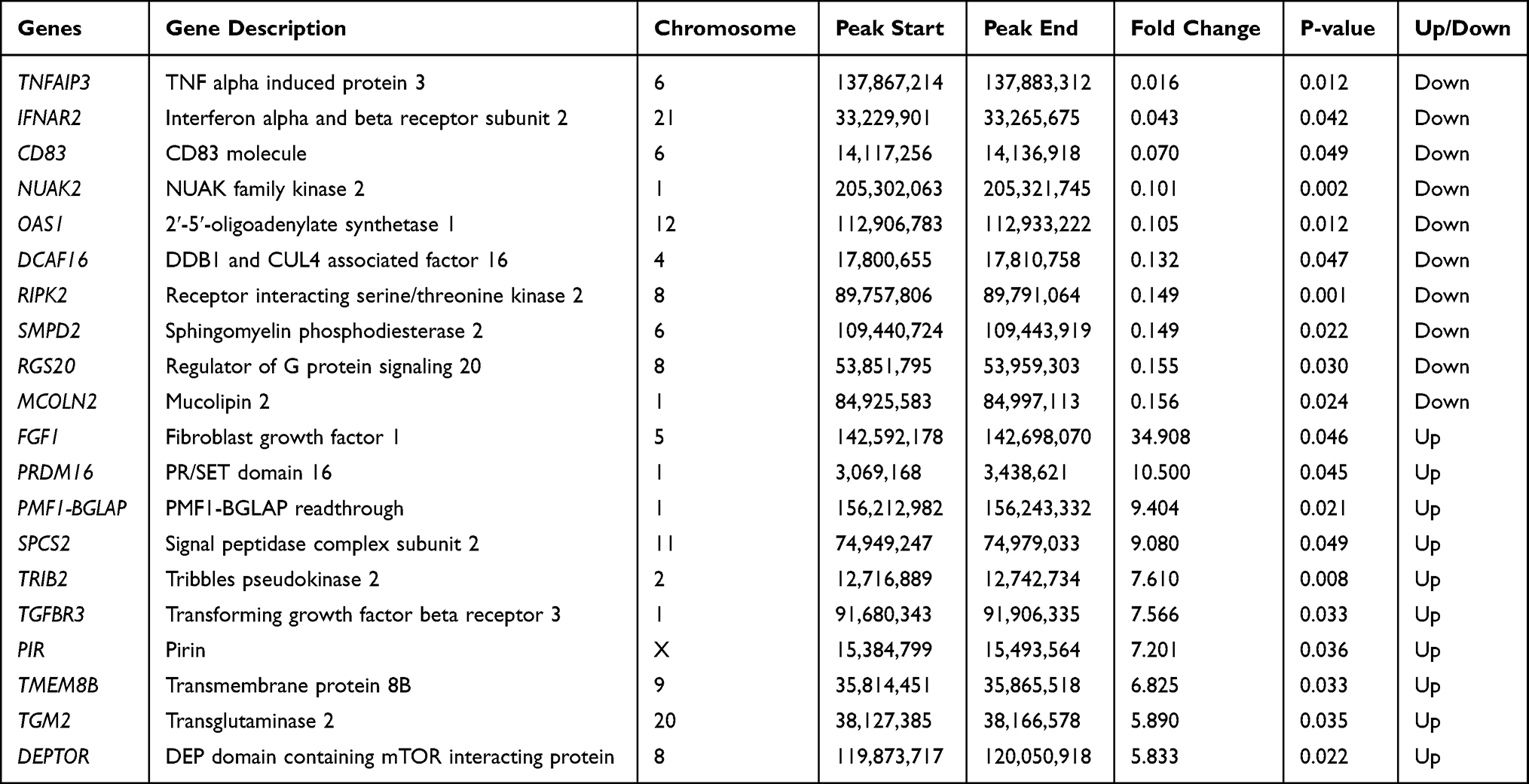 |
Table 1 List of the Top 10 Hypermethylated Genes and the Top 10 Hypomethylated Genes |
Then, we analyzed the number of m6A methylation modifications in different gene structures. As shown in Figure 3C the differentially expressed genes were mainly distributed in 3ʹ-UTR (31%), with similar percentages in CDS (18%), promoter (18%), 5ʹ-UTR (17%) and terminator (16%). In addition, we also analyzed the distribution of genes with differential m6A methylation genes in chromosomes (Figure 3D). We found that most chromosomes had more than one differentially methylated gene, including 27 on chromosome 1, 17 genes were on chromosome 5 and 16 genes were on chromosomes 6 and 8. Based on interactions with combined scores ≥ 0.4,27 the PPI network analysis constructed interaction networks for genes with differential m6A modification, as shown in Figure 3E.
Simultaneously, the results of GO analysis showed that 854 enriched GO terms in the biological process, cellular component and molecular function categories, particularly in the tumor necrosis factor-mediated signaling pathway, tube formation, regulation of cytokine-mediated signaling pathway, and more. Similarly, KEGG analysis found that 93 pathways were enriched, especially RNA degradation, the NOD-like receptor signaling pathway and apoptosis. The top 30 enriched GO terms and pathways are shown in Figure 3F and G.
Differences in mRNA Expression of MH7A Cells After Stimulation with TNF-α
As shown in Figure 4A and B, a fold change >2 and p-value <0.05 were the screening conditions, and 1207 differentially expressed mRNAs in MH7A cells after stimulation with TNF-α were found, of which 793 were upregulated genes and 414 were downregulated. The top 10 upregulated mRNAs and the top 10 down-regulated mRNAs are shown in Table 2. Based on interactions with combined scores ≥ 0.4,27 the PPI network analysis constructed interaction networks with differentially expressed mRNA genes, as shown in Figure 4C.
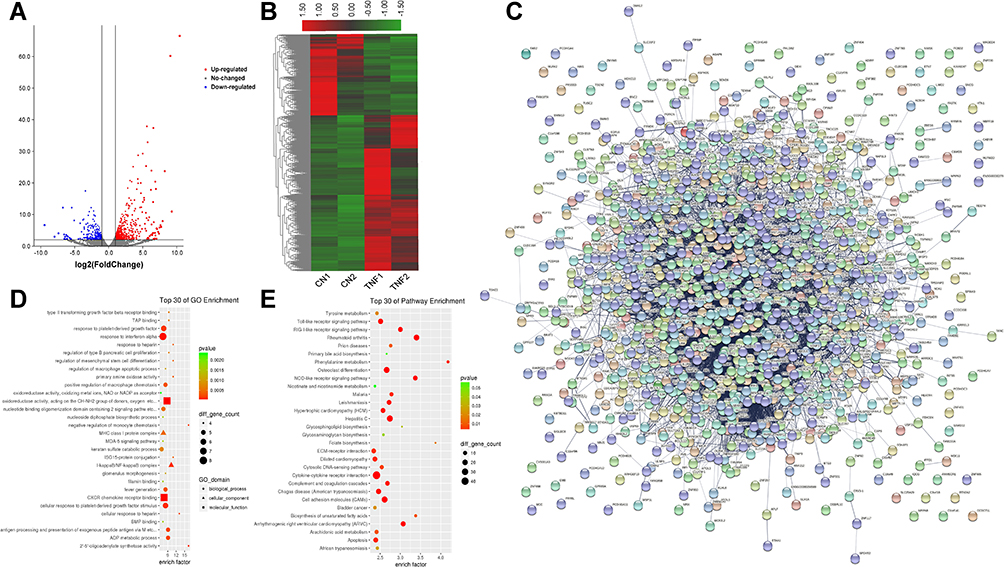 |
Figure 4 Differences in the mRNA expression genes in MH7A cells after stimulation with TNF-α. (A) Volcano plot representation of microarray data on the differentially expressed mRNA genes between the control group and TNF-α group. The blue and red dots to the left and to the right of the two vertical lines indicate more than a 1.5-fold change and represent the differentially expressed genes with statistical significance. Statistical significance was defined as a fold change of 1.5 and a p-value of 0.05 between the control and TNF-α groups. (B) Hierarchical cluster analysis of differentially expressed mRNA genes between the control group and TNF-α group. Hierarchical clustering showed that the differentially expressed genes ultimately clustered into two major branches, including upregulated genes, which are labeled in red, and downregulated genes, which are labeled in green. The darker the color, the more significant the difference. (C) PPI of differentially expressed mRNA genes. (D) The top 30 enriched GO terms of differentially expressed mRNA genes in MH7A cells. (E) The top 30 pathway enrichments of differentially expressed mRNA genes in MH7A cells. |
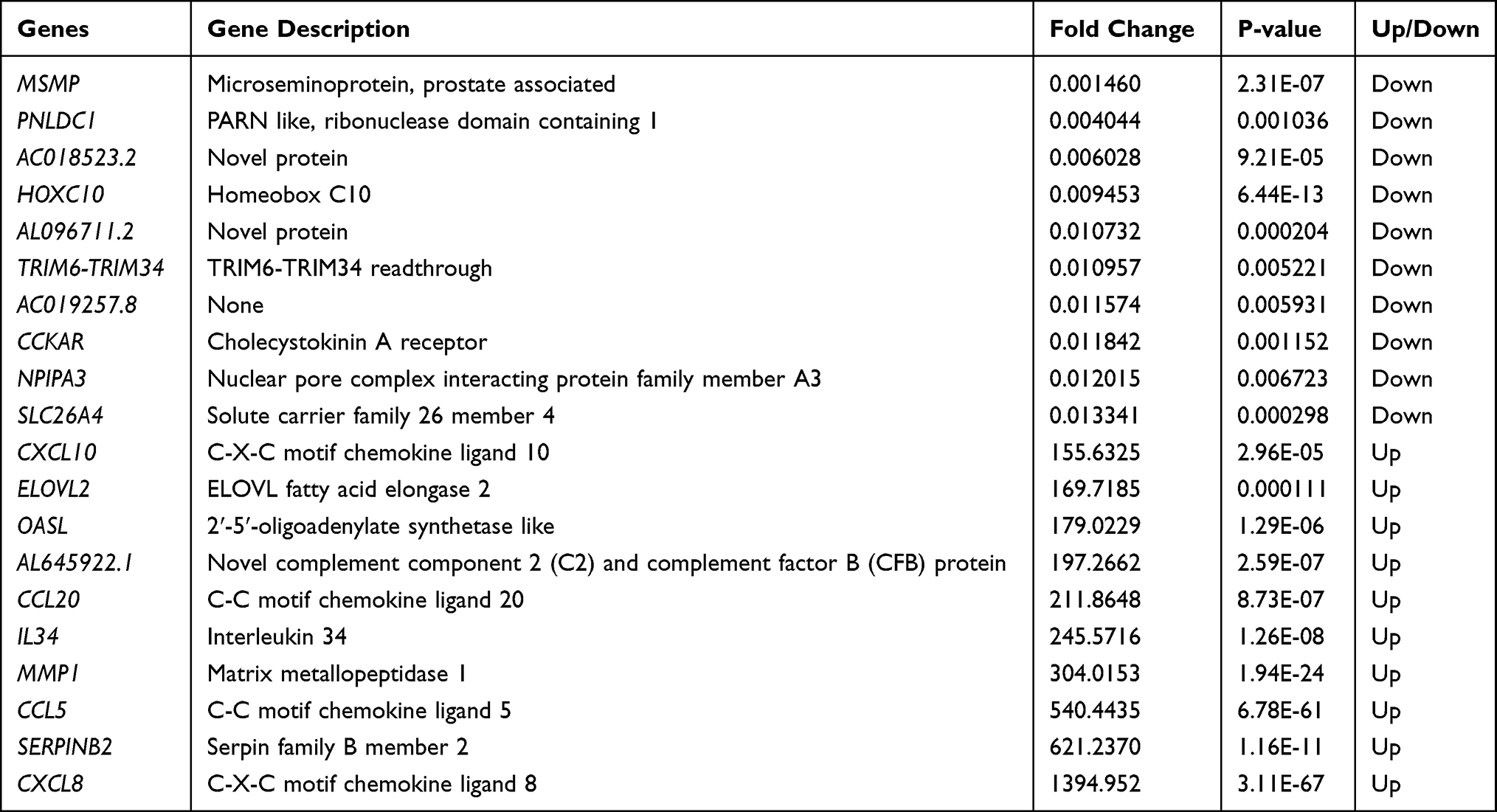 |
Table 2 List of the Top 10 Up-Regulated mRNAs and the Top 10 Down-Regulated mRNAs |
Meanwhile, the results of GO analysis showed that 2980 enriched GO terms in the biological process, cellular component and molecular function categories, particularly in response to interferon-alpha, I-kappa B/NF-kappa B complex and CXCR chemokine receptor binding, and more. Similarly, KEGG analysis found that 191 pathways were enriched, particularly the Toll-like receptor signaling pathway, rheumatoid arthritis and cell apoptosis, and more. The top 30 enriched GO terms and pathways are shown in Figure 4D and E.
Conjoint Analysis of m6A-Seq and RNA-Seq Data of MH7A Cells After Stimulation with TNF-α
A conjoint analysis was conducted for m6A-seq and RNA-seq data. We found 88 genes that commonly had differential m6A methylation levels and differentially expressed mRNA levels. Among them, there were 57 genes with m6A hypomethylation and downregulated mRNA expression, 30 genes with m6A hypermethylation and downregulated mRNA expression and 1 gene with m6A hypermethylation and upregulated mRNA expression (Figure 5A and B). Based on interactions with combined scores ≥ 0.4,27 the PPI network analysis constructed interaction networks for these common genes, as shown in Figure 5C.
 |
Figure 5 Joint analysis of m6A methylation and mRNA expression in MH7A cells after stimulation with TNF-α. (A) Four quadrant graph of genes with differential m6A methylation and differentially expressed mRNA levels. (B) Venn diagram of genes with differential m6A methylation and differentially expressed mRNA levels. (C) PPI of genes with differentially expressed m6A methylation and differential expressed mRNA. (D) The top 30 GO enrichments of genes with differentially expressed m6A methylation and differentially expressed mRNA in MH7A cells. (E) The top 30 pathway enrichments of genes with differentially expressed m6A methylation and differentially expressed mRNA in MH7A cells. |
Then, we analyzed these common genes by GO and KEGG assays. The GO analysis showed that there were 532 enriched GO terms and these genes were particularly enriched in the tumor necrosis factor-mediated signaling pathway, I-kappaB kinase/NF-kappaB signaling, and cellular response to tumor necrosis factor (Figure 5C and E). Similarly, KEGG analysis found that 59 pathways were enriched, and these genes were closely related to inflammation-related pathways such as the Toll-like receptor signaling pathway, NOD-like receptor signaling pathway, Jak-STAT signaling pathway and cell apoptosis. The top 30 enriched GO terms and pathways are shown in Figure 5D and E.
Correlation Analysis, RT-qPCR and Western Blot Verification of Genes with Differential m6A Modification and Differential mRNA Expression
As shown in Figure 6A and B, joint m6A modification and mRNA expression joint analysis showed changes in m6A methylation abundance and mRNA abundance in WTAP, RIPK2, JAK3 and TNFRSF10A. Compared with the control group, the m6A methylation abundance of WTAP, RIPK2 and JAK3 in MH7A cells after TNF-α stimulation was significantly decreased, while the mRNA abundance were increased. In contrast, the m6A methylation abundance of TNFRSF10A was significantly increased, while the mRNA abundance was significantly decreased. To validate the sequencing results, RT-qPCR and Western blot were applied to detect the mRNA and protein expression levels of WTAP, RIPK2, JAK3 and TNFRSF10A in MH7A cells and synovial tissues of AA rats. The results showed that the mRNA and protein expression trends of these genes were in accordance with the sequencing results (Figure 6C–G).
 |
Figure 6 Correlation analysis, RT-qPCR and Western blot verification of genes with differential m6A modification and differential mRNA expression. (A) Integrative genomics viewer (IGV) plots of m6A methylation abundances and expression abundances in the control group and the TNF-α group. The x-axis represents the genomic position and the y-axis represents the sequence read number. a, the m6A methylation abundances and expression abundances of WTAP. b, the m6A methylation abundances and expression abundances of RIPK2. c, the m6A methylation abundances and expression abundances of JAK3. d, the m6A methylation abundances and expression abundances of TNFRSF10A. (B) The m6A methylation levels were measured by RT-qPCR in the control group and the TNF-α group. a, the m6A methylation level of WTAP. b, the m6A methylation level of RIPK2. c, the m6A methylation level of JAK3. d, the m6A methylation level of TNFRSF10A. (C) The mRNA expression levels in MH7A cells were measured by RT-qPCR. a, the mRNA expression level of WTAP. b, the mRNA expression level of RIPK2. c, the mRNA expression level of JAK3. d, the mRNA expression level of TNFRSF10A. #p < 0.05, ##p < 0.01 compared with the control group. (D) The mRNA expression levels were measured by RT-qPCR in synovial tissues of AA rats. a, the mRNA expression level of WTAP. b, the mRNA expression level of RIPK2. c, the mRNA expression level of JAK3. d, the mRNA expression level of TNFRSF10A. #p < 0.05, ##p < 0.01 compared with the control group. (E) The protein expression levels were measured by Western blot in MH7A cells and synovial tissues of AA rats. a, the protein expression level of WTAP in MH7A cells. b, the protein expression level of WTAP in synovial tissues of AA rats. c, the protein expression level of RIPK2 in MH7A cells. d, the protein expression level of RIPK2 in synovial tissues of AA rats. e, the protein expression level of JAK3 in MH7A cells. f, the protein expression level of JAK3 in synovial tissues of AA rats. g, the protein expression level of TNFRSF10A in MH7A cells. h, the protein expression level of TNFRSF10A in synovial tissues of AA rats. (F), Semiquantitative analysis of protein expression in MH7A cells. a, semiquantitative analysis of WTAP protein. b, semiquantitative analysis of RIPK2 protein. c, semiquantitative analysis of JAK3 protein. d, semiquantitative analysis of TNFRSF10A protein. #p < 0.05, ##p < 0.01 compared with the control group. (G) Semiquantitative analysis of protein expression in synovial tissues of AA rats. a, semiquantitative analysis of WTAP protein. b, semiquantitative analysis of RIPK2 protein. c, semiquantitative analysis of JAK3 protein. d, semiquantitative analysis of TNFRSF10A protein. #p < 0.05, ##p < 0.01 compared with the control group. |
Discussion
As an important method of RNA modification, m6A methylation reversibly changes the local structure of RNA and affects the biological function such as the transcription and translation of RNA, thus affecting the expression of downstream target genes.29,30 Therefore, it has an intimate connection to the occurrence and development of many diseases. In recent years, an increasing number of studies on m6A and diseases have been carried out.31,32 However, the transcriptome-wide distributions of m6A modifications in MH7A cells and the specific function of m6A methylation differences in the pathogenesis of RA are not clear.
The m6A and RNA sequence approaches were utilized to construct the m6A methylation library and RNA library to investigate differentially m6A methylation and gene expression in MH7A cells. Our results showed that there were 206 differentially expressed m6A methylation genes and 1207 differentially mRNAs in MH7A cells after stimulation with TNF-α. By analyzing the transcriptional region with the m6A peaks, the differentially expressed m6A methylated genes were mainly concentrated in the 3ʹ-UTR, which was quite consistent with the previous findings of Dominissini et al.33 On the other hand, according to the joint analysis of genes with differentially expressed m6A methylation and mRNA, there were significant differences between the m6A methylation and mRNA expression levels of 88 genes. We suspected that these potential genes, through m6A methylation, are involved in the occurrence and development of RA.
We conducted GO and KEGG analyses of differentially expressed m6A methylated genes. Biological processes and pathways related to inflammation, cell characteristics and cell fate, especially the NOD-like receptor single pathway, NF-κB pathway and cell apoptosis were enriched. NOD‐like receptor family, pyrin domain containing 3 (NLRP3) gene polymorphisms have a direct relationship with inflammation in the course of RA. Guo et al34 found that NLRP3 inflammatory bodies were highly activated in the synovium of RA patients and CIA rats, and the inhibition of NLRP3 could significantly improve the pathological symptoms. The NF-κB pathway plays an important role in the regulation of immune and inflammatory responses. Roman JA et al35 found that NF-κB is differentially activated in RA synovium, and the NF-κB pathway regulates the expression of many inflammatory factors, such as cytokines and cell adhesion molecules, in the synovium and synovial fluid of RA patients. Apoptosis is an active programmed death of cells that maintains the stability of the body.36 Both apoptosis and cell proliferation are precisely regulated by genes to ensure the dynamic balance of the number of cells in the body.37 It has been found that the damage to articular cartilage and bone tissue in the process of RA is closely related to the excessive proliferation of synovial cells and insufficient apoptosis.38,39
Then, according to biological function, we selected WTAP, RIPK2, JAK3 and TNFRSF10A for further analysis. The sequencing, RT-qPCR and Western blot results in MH7A cells after stimulation with TNF-α all clearly indicated that the m6A methylation levels of WTAP, RIPK2 and JAK3 were significantly reduced, while the mRNA and protein expression levels were significantly increased. Interestingly, the m6A methylation as well as mRNA and protein expression levels of TNFRSF10A presented the completely opposite trends compared with the above three genes. To further validate the above experimental results, we established and detected the mRNA and protein expression levels of the above genes in synovial tissues of AA model rats. The expression levels of WTAP, RIPK2, JAK3 and TNFRSF10A showed the same trends between the cellular level and animal level.
WTAP, as an important component of the m6A methyltransferase complex, does not have N6-methyladenine methyltransferase activity but is required for efficient RNA methylation in vivo and for the localization of METTL3 and METTL14 to nuclear speckles.40,41 WTAP has been found to be strongly related to the development of many diseases, including autoimmune diseases.42,43 Tong et al44 found that mice with the specific deletion of m6A methyltransferases in regulatory T cells would develop severe autoimmune diseases such as enlarged lymph nodes and an enlarged spleen, indicating that WTAP and other methyltransferases are closely related to T cell function and involved in the occurrence of immune diseases. TNFRSF10A, also known as TRAIL-R1 or DR4, is a member of the tumor necrosis factor receptor family, which can regulate cell function and so on.45,46 TNFRSF10A can not only induce apoptosis but also inhibit inflammation, and its ligands can activate the NF-κB pathway and promote cell proliferation and migration.47,48 Li et al49 found that TNFRSF10A was involved in apoptosis induced by endoplasmic reticulum stress, and TNFRSF10A knockout reduced the expression level of TNFRSF10B, thereby protecting cells from apoptosis. RIPK2 is the downstream target of the pattern recognition receptors NOD1 and NOD2 and is closely related to inflammatory diseases.50,51 Usluoglu N et al52 found that RIPK2 knockout could reduce the phosphorylation of p38MAPK, ERK and Ikappa B, leading to a reduction in the inflammatory factor IL-12. In addition, Krieg A et al53 also found that the inhibition of endogenous RIPK2 significantly reduced cell apoptosis. JAK3 belongs to the intracellular nonreceptor tyrosine kinase family, which mediates the signal produced by cytokines and is transmitted through the JAK-STAT signal pathway.54 Some studies have reported that JAK3 plays an extensive role in the pathogenesis of many autoimmune diseases.55,56 Byung-Hak Kim et al57 found that the upregulation of JAK3/STATs is closely associated with arthritic inflammation, and JAK3 antagonists inhibit JAK3 activity to reduce inflammation in the body. From our verification results, there were divergences in m6A methylation and mRNA expression levels in these genes. This suggests that the pathogenesis of RA is related to changes in the RNA methylation levels of these genes.
Conclusion
In summary, our study revealed the differential expression of m6A methylation and mRNA genes in MH7A cells, provided the m6A transcriptome map of MH7A cells, and revealed the potential relationship between RNA methylation modification and RA related genes. Nevertheless, the specific and mechanism of m6A methylation in RA remains to be further studied in the future.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (grant no. 81873139), the Key Research and Development Project of Anhui Province (201904a07020004) and the 12th Batch of “115” Innovation Team of Anhui Province (Anhui Talent Office [2019] No. 1). We are grateful to Mr. Qiang Fan (Ao Ji Bio-tech Co. Ltd. Shanghai, China) for providing sequencing service.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet. 2014;15(5):293–306. doi:10.1038/nrg3724
2. Burgess A, David R, Searle IR. Deciphering the epitranscriptome: a green perspective. J Integr Plant Biol. 2016;58(10):822–835. doi:10.1111/jipb.12483
3. Zhang X-Z, Liu H, Chen S-R. Mechanisms of long non-coding RNAs in cancers and their dynamic regulations. Cancers. 2020;12(5):5. doi:10.3390/cancers12051245
4. Li D, Cai L, Meng R, Feng Z, Xu Q. METTL3 Modulates Osteoclast Differentiation and Function by Controlling RNA Stability and Nuclear Export. Int J Mol Sci. 2020;21:5.
5. Li E, Wei B, Wang X, Kang R. METTL3 enhances cell adhesion through stabilizing integrin β1 mRNA via an m6A-HuR-dependent mechanism in prostatic carcinoma.. Am J Cancer Res. 2020;10(3):1012–1025.
6. Qin Y, Li L, Luo E, et al. Role of m6A RNA methylation in cardiovascular disease (Review). Int J Mol Med. 2020;46(6):1958–1972. doi:10.3892/ijmm.2020.4746
7. Reichel M, Köster T, Staiger D. Marking RNA: m6A writers, readers, and functions in Arabidopsis. Journal of Molecular Cell Biology. 2019;11(10):899–910. doi:10.1093/jmcb/mjz085
8. Xu S, Li Y, Chen J-P, et al. Oxygen glucose deprivation/re-oxygenation-induced neuronal cell death is associated with Lnc-D63785 m6A methylation and miR-422a accumulation. Cell Death Dis. 2020;2020(9):816. doi:10.1038/s41419-020-03021-8
9. Fei Q, Zou Z, Roundtree IA, Sun HL, He C. YTHDF2 promotes mitotic entry and is regulated by cell cycle mediators. PLoS Biol. 2020;18(4):e3000664.
10. Hazra D, Chapat C, Graille M. m6A mRNA Destiny: chained to the rhYTHm by the YTH-Containing Proteins. Genes. 2019;10(1):1. doi:10.3390/genes10010049
11. Sparks JA. Rheumatoid Arthritis. Ann Intern Med. 2019;170(1):Itc1–itc16. doi:10.7326/AITC201901010
12. Adawi M, Firas S, Blum A. Rheumatoid Arthritis and Atherosclerosis.. Isr Med Assoc J. 2019;21(7):460–463.
13. Liu T, Wang X, He YL, et al. In Vivo and In Vitro Anti-Arthritic Effects of Cardenolide-Rich and Caffeoylquinic Acid-Rich Fractions of Periploca forrestii. Molecules. 2018;23:8.
14. Fan S, Li C, Ai R, Wang M, Firestein GS, Wang W. Computationally expanding infinium HumanMethylation450 BeadChip array data to reveal distinct DNA methylation patterns of rheumatoid arthritis. Bioinformatics. 2016;32(12):1773–1778. doi:10.1093/bioinformatics/btw089
15. Luo Q, Gao Y, Zhang L, et al. Decreased ALKBH5, FTO, and YTHDF2 in Peripheral Blood Are as Risk Factors for Rheumatoid Arthritis. Biomed Res Int. 2020;2020:5735279. doi:10.1155/2020/5735279
16. Zheng J, Zeng P, Zhang H, et al. Long noncoding RNA ZFAS1 silencing alleviates rheumatoid arthritis via blocking miR-296-5p-mediated down-regulation of MMP-15. Int Immunopharmacol. 2020;107061.
17. Jiang H, Liu J, Wang T, et al. Urinary metabolite profiling provides potential differentiation to explore the mechanisms of adjuvant-induced arthritis in rats. Biomed Chromatography. 2016;30(9):1397–1405. doi:10.1002/bmc.3697
18. Brown J, Pirrung M, McCue LA, Dashboard: FQC, Wren J. FQC Dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics. 2017;33(19):3137–3139. doi:10.1093/bioinformatics/btx373
19. Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–i890. doi:10.1093/bioinformatics/bty560
20. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–360. doi:10.1038/nmeth.3317
21. Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11(9):1650–1667. doi:10.1038/nprot.2016.095
22. Oshlack A, Robinson MD, Young MD. From RNA-seq reads to differential expression results. Genome Biol. 2010;11(12):220. doi:10.1186/gb-2010-11-12-220
23. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi:10.1038/nmeth.1226
24. Nikolayeva O, Robinson MD. edgeR for differential RNA-seq and ChIP-seq analysis: an application to stem cell biology. Methods Mol Biol. 2014;1150:45–79.
25. Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125(1–2):279–284. doi:10.1016/S0166-4328(01)00297-2
26. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi:10.1038/75556
27. Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–d368. doi:10.1093/nar/gkw937
28. Wang X, Liu X, Liu N, Chen H. Prediction of crucial epigenetically‑associated, differentially expressed genes by integrated bioinformatics analysis and the identification of S100A9 as a novel biomarker in psoriasis.. Int J Mol Med. 2020;45(1):93–102. doi:10.3892/ijmm.2019.4392
29. Zhou KI, Shi H, Lyu R, et al. Regulation of Co-transcriptional Pre-mRNA Splicing by m6A through the Low-Complexity Protein hnRNPG. Mol Cell. 2019;76(1):70–81.e79. doi:10.1016/j.molcel.2019.07.005
30. Zheng H, Li S, Zhang X, Sui N. Functional Implications of Active N6-Methyladenosine in Plants. Front Cell Dev Biol. 2020;8:291. doi:10.3389/fcell.2020.00291
31. Zhang B, Wu Q, Li B, Wang D, Wang L, Zhou YL. m6A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol Cancer. 2020;19(1):53. doi:10.1186/s12943-020-01170-0
32. Qu N, Qin S, Zhang X, et al. Multiple m6A RNA methylation modulators promote the malignant progression of hepatocellular carcinoma and affect its clinical prognosis. BMC Cancer. 2020;20(1):165. doi:10.1186/s12885-020-6638-5
33. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–206. doi:10.1038/nature11112
34. Guo C, Fu R, Wang S, et al. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin Exp Immunol. 2018;194(2):231–243. doi:10.1111/cei.13167
35. Roman-Blas JA, Jimenez SA. NF-κB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthritis Cartilage. 2006;14(9):839–848. doi:10.1016/j.joca.2006.04.008
36. Zhao S-Y, Liao L-X, Tu P-F, Li -W-W, Zeng K-W. Icariin Inhibits AGE-Induced Injury in PC12 cells by directly targeting apoptosis regulator bax. Oxid Med Cell Longev. 2019;2019:7940808. doi:10.1155/2019/7940808
37. Guan X, Lu J, Sun F, Li Q, Pang Y. The molecular evolution and functional divergence of lamprey programmed cell death genes. Front Immunol. 2019;10:1382. doi:10.3389/fimmu.2019.01382
38. Garg NK, Tyagi RK, Singh B, et al. Nanostructured lipid carrier mediates effective delivery of methotrexate to induce apoptosis of rheumatoid arthritis via NF-κB and FOXO1. Int J Pharm. 2016;499(1–2):301–320. doi:10.1016/j.ijpharm.2015.12.061
39. Hellvard A, Zeitlmann L, Heiser U, et al. Inhibition of CDK9 as a therapeutic strategy for inflammatory arthritis. Sci Rep. 2016;6(1):31441. doi:10.1038/srep31441
40. Sorci M, Ianniello Z, Cruciani S, et al. METTL3 regulates WTAP protein homeostasis. Cell Death Dis. 2018;9(8):796. doi:10.1038/s41419-018-0843-z
41. Ping X-L, Sun B-F, Wang L, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–189. doi:10.1038/cr.2014.3
42. Li H, Su Q, Li B, et al. High expression of WTAP leads to poor prognosis of gastric cancer by influencing tumour-associated T lymphocyte infiltration. J Cell Mol Med. 2020;24(8):4452–4465. doi:10.1111/jcmm.15104
43. Wang L-J, Xue Y, Li H, et al. Wilms‘ tumour 1-associating protein inhibits endothelial cell angiogenesis by m6A-dependent epigenetic silencing of desmoplakin in brain arteriovenous malformation. J Cell Mol Med. 2020;24(9):4981–4991. doi:10.1111/jcmm.15101
44. Tong J, Cao G, Zhang T, et al. m6A mRNA methylation sustains Treg suppressive functions. Cell Res. 2018;28(2):253–256. doi:10.1038/cr.2018.7
45. Chen L, Qiu Y, Hao Z, et al. A novel humanized anti-tumor necrosis factor-related apoptosis-inducing ligand-R2 monoclonal antibody induces apoptotic and autophagic cell death. IUBMB Life. 2017;69(9):735–744. doi:10.1002/iub.1659
46. Duiker EW, Dijkers EC, Lambers Heerspink H, et al. Development of a radioiodinated apoptosis-inducing ligand, rhTRAIL, and a radiolabelled agonist TRAIL receptor antibody for clinical imaging studies. Br J Pharmacol. 2012;165(7):2203–2212. doi:10.1111/j.1476-5381.2011.01718.x
47. Bi R, Deng Y, Tang C, et al. Andrographolide sensitizes human renal carcinoma cells to TRAIL‑induced apoptosis through upregulation of death receptor 4. Oncol Rep. 2020;44(5):1939–1948. doi:10.3892/or.2020.7737
48. Dai X, Zhang J, Arfuso F, et al. Targeting TNF-related apoptosis-inducing ligand (TRAIL) receptor by natural products as a potential therapeutic approach for cancer therapy. Exp Biol Med (Maywood). 2015;240(6):760–773. doi:10.1177/1535370215579167
49. Li T, Su L, Lei Y, Liu X, Zhang Y, Liu X. DDIT3 and KAT2A Proteins Regulate TNFRSF10A and TNFRSF10B expression in endoplasmic reticulum stress-mediated apoptosis in human lung cancer cells. J Biol Chem. 2015;290(17):11108–11118. doi:10.1074/jbc.M115.645333
50. Kapoor A, Fan Y-H, Arav-Boger R. Bacterial Muramyl Dipeptide (MDP) restricts human cytomegalovirus replication via an IFN-β-Dependent Pathway. Sci Rep. 2016;6(1):20295. doi:10.1038/srep20295
51. Duggan BM, Foley KP, Henriksbo BD, Cavallari JF, Tamrakar AK, Schertzer JD. Tyrosine kinase inhibitors of Ripk2 attenuate bacterial cell wall-mediated lipolysis, inflammation and dysglycemia. Sci Rep. 2017;7(1):1578. doi:10.1038/s41598-017-01822-0
52. Usluoglu N, Pavlovic J, Moelling K, Radziwill G. RIP2 mediates LPS-induced p38 and IκBα signaling including IL-12 p40 expression in human monocyte-derived dendritic cells. Eur J Immunol. 2007;37(8):2317–2325. doi:10.1002/eji.200636388
53. Krieg A, Le Negrate G, Reed JC. RIP2-β: a novel alternative mRNA splice variant of the receptor interacting protein kinase RIP2. Mol Immunol. 2009;46(6):1163–1170. doi:10.1016/j.molimm.2008.11.002
54. Santos J, Hubert T, Milthorpe BK. Valproic acid promotes early neural differentiation in adult mesenchymal stem cells through protein signalling pathways. Cells. 2020;9(3):3. doi:10.3390/cells9030619
55. Rokosz LL, Beasley JR, Carroll CD, et al. Kinase inhibitors as drugs for chronic inflammatory and immunological diseases: progress and challenges. Expert Opin Ther Targets. 2008;12(7):883–903. doi:10.1517/14728222.12.7.883
56. Pei H, He L, Shao M, et al. Discovery of a highly selective JAK3 inhibitor for the treatment of rheumatoid arthritis. Sci Rep. 2018;8(1):5273. doi:10.1038/s41598-018-23569-y
57. Kim B-H, Kim M, Yin C-H, et al. Inhibition of the signalling kinase JAK3 alleviates inflammation in monoarthritic rats. Br J Pharmacol. 2011;164(1):106–118. doi:10.1111/j.1476-5381.2011.01353.x
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.