Back to Journals » OncoTargets and Therapy » Volume 14
TP53 Co-Mutational Features and NGS-Calibrated Immunohistochemistry Threshold in Gastric Cancer
Authors Yu R ![]() , Sun T, Zhang X, Li Z, Xu Y, Liu K, Shi Y, Wu X, Shao Y, Kong L
, Sun T, Zhang X, Li Z, Xu Y, Liu K, Shi Y, Wu X, Shao Y, Kong L
Received 10 June 2021
Accepted for publication 17 September 2021
Published 1 October 2021 Volume 2021:14 Pages 4967—4978
DOI https://doi.org/10.2147/OTT.S321949
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
Ruili Yu,1 Tingyi Sun,1 Xianwei Zhang,1 Zhen Li,1 Yang Xu,2 Kaihua Liu,2 Yuqian Shi,2 Xue Wu,2 Yang Shao,2,3 Lingfei Kong1
1Department of Pathology, Henan Provincial People’s Hospital, People’s Hospital of Zhengzhou University, Zhengzhou, Henan, People’s Republic of China; 2Geneseeq Research Institute, Nanjing Geneseeq Technology Inc., Nanjing, Jiangsu, People’s Republic of China; 3School of Public Health, Nanjing Medical University, Nanjing, Jiangsu, People’s Republic of China
Correspondence: Lingfei Kong
Department of Pathology, Henan Provincial People’s Hospital, People’s Hospital of Zhengzhou University, 7 Weiwu Road, Zhengzhou, 450003, Henan Province, People’s Republic of China
Tel +86 138 0386 8286
Email [email protected]
Purpose: TP53 is the most frequently mutated gene in gastric cancer and it can be potentially used for gastric cancer diagnosis and screening. However, standardized clinical approaches that could accurately and cost-effectively detect TP53 mutations in gastric cancer are largely lagged behind.
Patients and Methods: We conducted next-generation sequencing (NGS) analysis of 425 cancer-related genes in 42 gastric cancer patients in our cohort. A 1313-patient cohort derived from the cBioPortal database was used for validation. We performed immunohistochemistry (IHC) staining with four commonly used p53 antibodies, and the NGS results were used as the gold standard to optimize the IHC threshold for each antibody.
Results: By NGS analysis, we found that around 80% of gastric cancer patients in our cohort harbored TP53 alterations. Genetic alterations of BRCA1/ 2 or KMT2B were mostly exclusive with TP53 mutations, so were the MSI status or low grade of tumors. These results were further validated using the data from cBioPortal. We then used the NGS-derived TP53 status to optimize four commonly used IHC antibodies for detecting TP53 mutations. We showed that all antibodies could achieve more than 93% accuracy when proper IHC positivity thresholds were used, especially for the SP5 antibody that could reach 100% sensitivity and specificity with the 20% threshold.
Conclusion: Our results indicated that exclusivity between TP53 and BRCA mutations could be potentially used as a cost-effective way to predict BRCA status. Also, setting proper IHC thresholds for each specific antibody is critical to accurately detect TP53 mutations and facilitate disease diagnosis.
Keywords: TP53, gastric cancer, next-generation sequencing, co-mutation, immunohistochemistry threshold
Introduction
Tumor protein p53 (TP53) is a well-known tumor suppressor gene and its encoded protein p53 is involved in DNA repair, cell cycle arrest, apoptosis, metabolism, differentiation, and various developmental processes.1 TP53 is the most frequently altered gene in cancer, and one of the main outcomes of TP53 mutations is disrupting the normal function of p53 through loss-of-function or nonsense mutations (~10%).2 In addition, a substantial amount of TP53 missense mutations (~75%), primarily located at the DNA-binding domain, lose their ability to activate the canonical downstream target genes; instead, they could promote the oncogenic process by repressing the wild-type (WT) p53 or other p53 family members, such as p63 and p73.3 Multiple studies have shown that tumors could acquire selective advantages by retaining the mutated TP53, thus enabling their adaption to stress conditions, metabolic imbalance, and tumor microenvironments.3 Therefore, the TP53 mutation can serve as a diagnostic/prognostic biomarker and could be potentially used as a treatment target for various cancers.
Gastric cancer is the sixth most common cancer and the second cancer-related mortality globally,4 with TP53 being one of the most frequently mutated genes. Busuttil et al found that the intestinal metaplasia, a premalignant lesion of gastric cancer, was associated with elevated WT p53 expression while TP53 mutations nearly exclusively occurred in the gastric tumors,5 implying the involvement of TP53 mutations during the premalignancy to cancer transition and their clinical values for cancer diagnosis. On the other hand, conflicting results were observed between TP53 mutation status and gastric patient prognosis,6–9 and several possible explanations have been proposed to address this discrepancy. First, due to the tumor heterogeneity and the complexity of p53-related pathways, TP53 mutation itself is not enough to accurately predict patient outcomes, and coupling TP53 mutations with other mutations might better stratify patients with gastric cancer.10,11 Second, many studies assessed the TP53 status using immunohistochemistry (IHC), which may detect the accumulation of WT p53 that was induced during stress conditions.7 In addition, un-optimized IHC could improperly assign the TP53 status, resulting in inconclusive or contradictive results among studies. It has been estimated that the false positive rate for using p53 IHC ranged from 2% to 45%, and Roshandel et al showed that the traditional threshold of 10% IHC positivity led to only 42% specificity in glioblastoma patients.12 Although several researchers investigated the correlation between p53 IHC positivity and TP53 missense mutations in various cancers including glioma and astrocytomas, optimization of the p53 IHC threshold in gastric cancer is largely lagged behind.
In this study, we aimed to further explore the potential usage of the TP53 mutation as a biomarker for gastric cancer. We performed targeted next-generation sequencing (NGS) to investigate TP53 mutations and their co-occurred genetic alterations in 42 gastric cancer patients, and the results were further confirmed using the cBioPortal database. In order to achieve reliable p53 IHC results in gastric cancer, we used the TP53 status derived from the NGS data to optimize the p53 IHC threshold, which could be potentially used for cost-effective and accurate detection of TP53 mutations and facilitate gastric cancer diagnosis.
Patients and Methods
Patients and Samples
Forty-three patients who were diagnosed with gastric cancer in Henan Provincial People’s Hospital were included in this study. Each patient had 1 gastric tumor tissue sample, and the adjacent tumor tissues were used as normal controls to filter out germline mutations. A total of 1313 gastric cancer samples from cBio Cancer Genomics Portal (http://cbioportal.org) were used as the validation cohort.13–16 This study was approved by the ethical board of Henan Provincial People’s Hospital (ethical number: 2020–89), and this study was conducted in accordance with the Declaration of Helsinki. All patients have signed written informed consent forms prior to sample collection and consented to the publication of related clinical information and data.
Library Preparation and Sequencing
The sample and library preparation procedures follow the methods described by Yang et al.17 Briefly, the formalin-fixed paraffin-embedded (FFPE) samples were used for DNA extraction using the QIAamp DNA FFPE Kit (QIAGEN, Valencia, CA, USA) following the manufacturer’s instructions. DNA quality was assessed by spectrophotometry and quantified by Qubit 2.0. Libraries were prepared as previously described.17 Briefly, 1 μg of fragmented genomic DNA underwent end-repairing, A-tailing and ligation with indexed adapters sequentially, followed by size selection using Agencourt AMPure XP beads (Beckman Coulter). Hybridization-based target enrichment was carried out with GeneseeqOne™ pancancer gene panel (425-cancer-relevant genes, Geneseeq Technology Inc.), and xGen Lockdown Hybridization and Wash Reagents Kit (Integrated DNA Technologies). Captured libraries by Dynabeads M-270 (Life Technologies) were amplified in KAPA HiFi HotStart ReadyMix (KAPA Biosystems) and quantified by qPCR using the KAPA Library Quantification kit (KAPA Biosystems). Target enriched libraries were sequenced on the HiSeq4000 platform (Illumina) with 2×150 bp pair-end reads.
Mutation Calling and Copy Number Alteration Analysis
Sequencing data were demultiplexed by bcl2fastq (v2.19), analyzed by Trimmomatic18 to remove low-quality (quality<15) or N bases, and mapped to the reference hg19 genome (Human Genome version 19) using the Burrows-Wheeler Aligner.19 PCR duplicates were removed by Picard (available at https://broadinstitute.github.io/picard/). The Genome Analysis Toolkit (GATK)20 was used to perform local realignments around indels and base quality reassurance. Single-nucleotide polymorphisms (SNPs) and indels were called by VarScan221 and HaplotypeCaller/UnifiedGenotyper in GATK, with 0.5% mutant allele frequency as the cutoff for tissue samples and a minimum of three unique mutant reads. Common variants were removed using dbSNP and the 1000 Genome project. Germline mutations were filtered out by comparing to patient’s normal controls. The resulting somatic variants were further filtered through an in-house list of recurrent sequencing errors that were generated from over 10,000 normal control samples on the same sequencing platform. Gene fusions were identified by FACTERA22 and copy number variations (CNVs) were analyzed with ADTEx.23 The log2 ratio cut-off for copy number gain was defined as 2.0 for tissue samples. A log2 ratio cut-off of 0.67 was used for copy number loss detection in all sample types. The thresholds were determined from previous assay validation using the absolute CNVs detected by droplet digital PCR (ddPCR).
Immunohistochemistry (IHC)
Unstained FFPE sections from tumor specimens collected at diagnosis were subjected to IHC staining with p53 antibodies of SP5 (rabbit monoclonal antibody; Thermo Fisher Scientific), MX008 (mouse monoclonal antibody; MXB® Biotechnologies), BP-53-12 (mouse monoclonal antibody; Thermo Fisher Scientific), or DO-7 (mouse monoclonal antibody; Thermo Fisher Scientific), according to the current protocols of the Department of Pathology of Henan Provincial People’s Hospital. FFPE sections were also stained with Ki-67 antibody, the clone of which was MIB-1 (Dako), according to the protocol.
The Concordance Analysis Between p53 IHC and NGS Data
We analyzed the relationship between the TP53 missense mutations derived from the NGS data and the p53 IHC data using four different antibodies (SP5, MX008, BP-53-12, and DO-7) for a total of 42 samples from the discovery cohort. For different IHC positivity thresholds (0%, 10%, 20%, 30%, 50%, 60%, 90%, and 100%), we calculated their sensitivity and specificity using the NGS results as the reference, and the one with the highest accuracy was selected as the optimal IHC threshold for the specific antibody.12
Statistical Analysis
The TP53 co-occurring and mutually exclusive mutations were analyzed using the somatic Interactions function available in maftools; specifically, odds ratios (Fisher’s exact test) were used to distinguish the co-occurring and mutually exclusive mutations, with odds ratios larger than 1 indicating co-mutation while odds ratios smaller than 1 indicating mutual exclusivity.24 Comparisons of mutation frequency between different groups were done using Fisher’s exact test. For survival data, Kaplan–Meier curves were analyzed using the Log rank test. Two-sided p values of less than 0.05 was considered as statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). All statistical analyses were done in R (R 3.6.2).
Results
Patient and Sample Characteristics
A total of 43 gastric cancer patients were included in our study and their tumor tissue samples underwent targeted NGS of 425 cancer-related genes. One sample did not pass the NGS quality control, leaving 42 patient samples for further analysis (discovery cohort). We also analyzed NGS data from 1313 gastric cancer patients, which were obtained from the cBioPortal database and used as the validation cohort. Of the 42-patient discovery cohort, the median age was 60.5 years old, which is comparable with the validation cohort (67 years) (Table 1 and Supplementary Table 1). There are more male patients than female patients (78.6% vs 21.4%), and similar gender bias was also seen in both previous studies25 and the validation cohort (Supplementary Table 1). Around half of the 42 gastric cancer patients had a smoking history and one-third of them had a drinking history (Table 1). Also, the majority of their tumors were at stage III, with high Ki-67 expression,26 stable microsatellite, and lymph node metastasis (Table 1). According to the Cancer Genome Atlas (TCGA) molecular subtyping approach, 2.4%, 7.1%, 19%, and 71.4% of the patients in the discovery cohort were classified as Epstein-Barr virus-positive (EBV-positive), microsatellite instable (MSI), chromosomal instable (CIN), and genomically stable (GS), respectively (Table 1), whereas the majority of gastric cancer subtype (71.7%) in the validation cohort was not available (Supplementary Table 1).
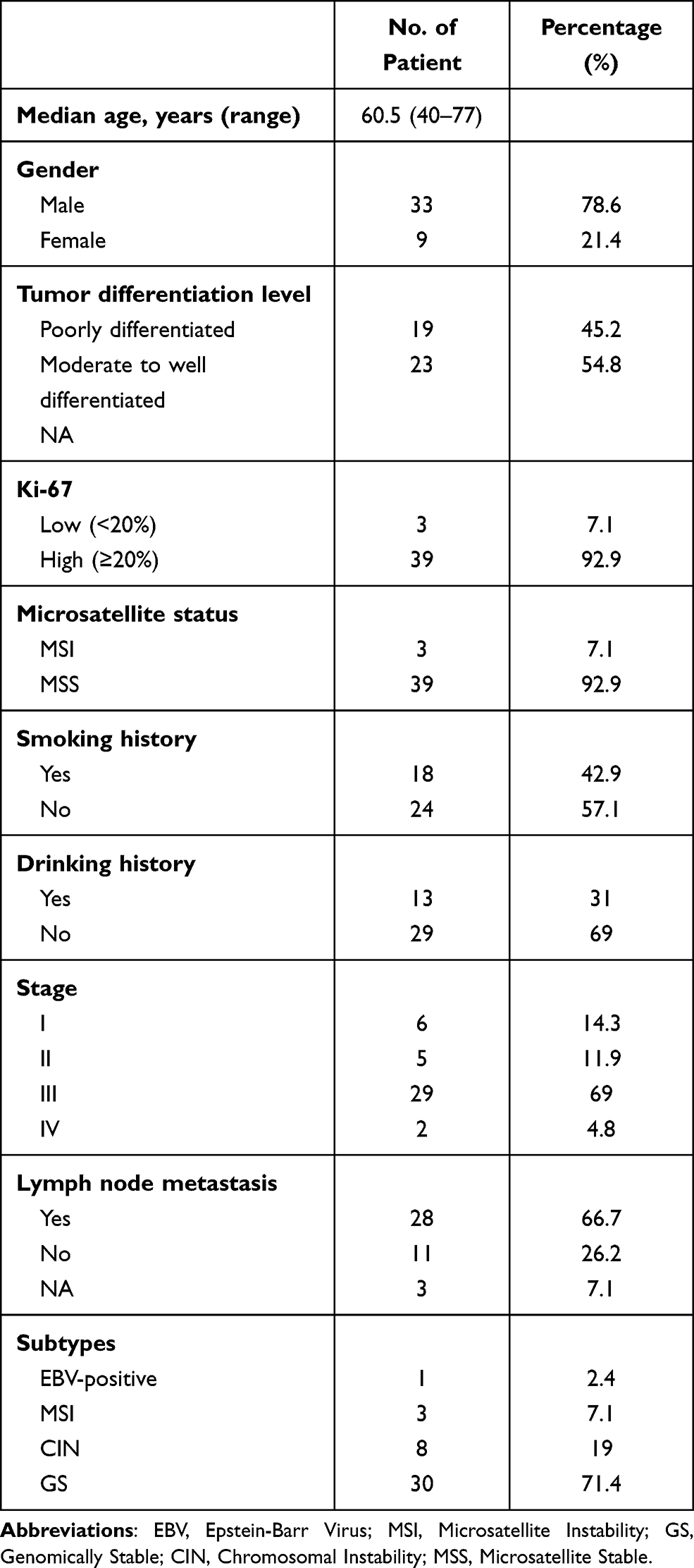 |
Table 1 The Demographic Characteristics of 42 Gastric Cancer Patients in the Discovery Cohort |
Mutation Profile and TP53 Mutation Features
As shown in Figure 1A, TP53 was the most frequently altered gene within the discovery cohort (78.6%) while the TP53 mutation frequency in the validation cohort was 47%, and this difference might be due to the variation in the tumor stage, tumor differentiation level, ethnicity, gastric cancer subtype, and treatment history between the two patient cohorts.27,28 The majority of TP53 mutations were missense mutations, followed by some nonsense mutations including frameshift and stop gain mutations (Figure 1A). By examining details of these TP53 mutations, we found that most of them were located within the p53 DNA-binding domains in both discovery and validation cohorts, and a large proportion of them were TP53 hotspot mutations9 that were associated with tumor progression and/or recurrences, such as R175, G245, R248, R273, and R282 (Figure 1B and Supplementary Table 2). In addition to TP53 mutations, multiple other genes were also frequently mutated in our cohort, including mutations in LRP1B, ARID1A, and GNAS, as well as amplification of ERBB2, CCNE1, MYC, and KRAS (Figure 1A), which is in line with previous reports.29
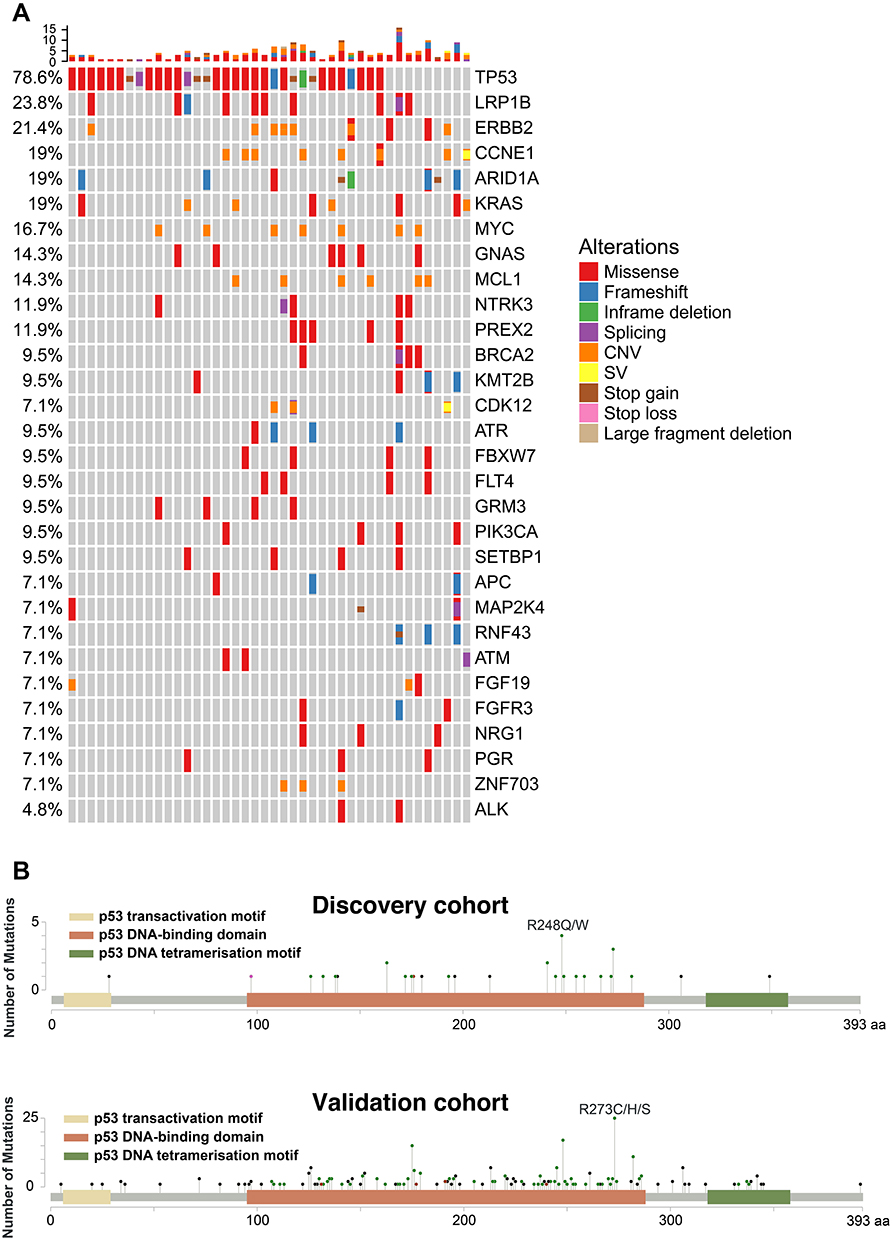 |
Figure 1 The genetic alterations in gastric cancer patients. (A) The co-mutation plot of the top 30 mutated genes from the discovery gastric cancer patient cohort was illustrated and the genes were ranked based on their mutational frequency. CNV: copy-number variation; SV: structural variation. (B) Lollipop plot for TP53 mutations identified in the discovery gastric cancer patient cohort (upper panel) or the validation gastric cancer patient cohort (lower panel). Abbreviations: CNV, copy number variation; SV, structural variant; aa, amino acid. |
Characterizing TP53 Co-Occurring and Exclusive Mutations in Gastric Cancer
We then investigated the co-occurrence of TP53 mutations with other genetic alterations. As shown in Supplementary Table 3, three mutated genes were found to be exclusive with TP53 mutations in the discovery cohort, including RNF43, BRCA2, and KMT2B. To eliminate any sampling bias of the discovery cohort, we validated these results using the 1313-patient results derived from the cBioPortal database. We also included BRCA1 in the analysis because it is closely related to BRCA2 in the DNA double-strand break repair pathway and its mutations were also relatively common in gastric cancer. TP53 mutations demonstrated statistically significant exclusivity with mutations in both BRCA1 and BRCA2 in the validation cohort (p value=0.012 and 0.048, respectively) (Figure 2A), implying that there might be some pathway-overlaps between TP53 and BRCA in terms of gastric cancer tumorigenesis and progression. Similarly, the exclusivity between TP53 mutations and KMT2B mutations was confirmed in the validation cohort (p value=0.001) (Figure 2B). On the other hand, the result for TP53 and RNF43 mutations were not statistically significant (p value=0.082), thus being excluded for further analysis. Besides gene mutations, we also checked the co-existence of TP53 mutations with other molecular and clinical features. Microsatellite instability (MSI) was usually associated with impaired DNA mismatch repair pathways, and we found MSI was more likely to be found in patients with WT TP53, which is statistically significant in both the discovery cohort and the validation cohort (p value=0.0073 and 0.0139, respectively) (Figure 2C). Intriguingly, we also found that well-differentiated tumors tended to be associated with more TP53 mutations in both patient cohorts (Supplementary Figure 1), although the result was not statistically significant as most samples in the validation cohort had unknown tumor differentiation status (Supplementary Table 1). Intriguingly, because both MSI and well-differentiation were associated with better prognosis in gastric cancer,30,31 their opposite relationship to TP53 mutation frequency might be one of the reasons for the conflicting results of using TP53 mutation as the prognostic biomarker.
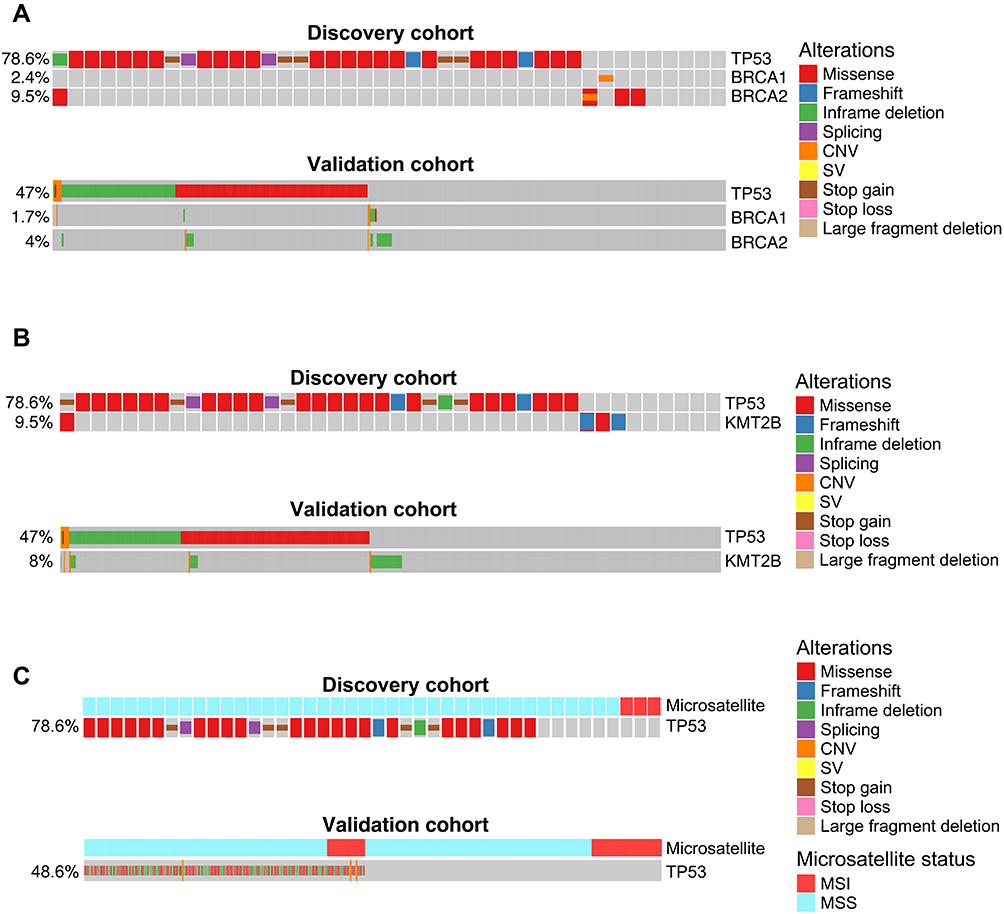 |
Figure 2 Cross-validated TP53 co-occurring or mutually exclusive genetic alterations in gastric cancer. (A–C) Co-mutation plot of TP53 mutations with BRCA1/BRCA2 mutations (A), KMT2B mutations (B), or microsatellite stability status (C) in the discovery patient cohort (upper panel) or the validation patient cohort (lower panel). For the validation patient cohort, only the samples with known BRCA1/BRCA2/KMT2B mutation status or microsatellite stability status were included in the analysis. Abbreviations: MSI, microsatellite instability; MSS, microsatellite stable; CNV, copy number variation; SV, structural variant. |
As patient overall survival data is available for the validation cohort, we examined whether different TP53 co-mutated genes would have distinct clinical outcomes. As illustrated in Supplementary Figure 2A, gastric cancer patients with mutated TP53 had similar overall survival to those with WT TP53. Patients with only BRCA1 mutations had worse survival rates, while patients with both TP53 and BRCA1 mutations, although very rare, had the worst overall survival among all the gastric cancer patients in the validation cohort (Supplementary Figure 2A). On the other hand, patients with only BRCA2 mutations or with co-occurred TP53 and BRCA2 mutations had indistinguishable overall survival compared with the rest of gastric cancer patients (Log-rank p value=0.19; Supplementary Figure 2B). Besides, although KMT2B-mutant patients had similar clinical outcomes when compared with patients with only TP53 mutations or with TP53-WT/ KMT2B-WT, patients with co-existed TP53 and KMT2B mutations had better overall survival (Supplementary Figure 2C). Therefore, these results indicated that even though some mutations tended to be excluded in TP53 mutated gastric tumors, a few patients could still harbor both mutations and their clinical outcomes seem to be various and dependent on the specific genetic context.
Optimization of the IHC Antibody for Detecting TP53 Missense Mutations
Given that IHC is still widely used in clinical and experimental practices when determining the TP53 mutation status, we tried to optimize the TP53 IHC results using the NGS data as the reference. For 42 patients within the discovery cohort, each patient sample underwent IHC staining with four commonly used p53 antibodies, including SP5, MX008, BP-53-12, and DO-7. Multiple TP53 missense mutations have been reported to promote tumor survival, and cancer cells tended to selectively retain or amplify the mutant TP53. On the other side, cells with WT TP53 might have various p53 protein expression levels depending on the cell stress conditions, and cells with nonsense TP53 mutations (eg, frameshifts and stop gains) usually had various TP53 DNA copy number and very low to none p53 protein expression levels.3 As a result, it is potentially possible for IHC to distinguish different types of TP53 mutations. As we investigated the relationship between p53 IHC positivity and mutant allele frequency for different TP53 mutations, we found that all four p53 antibodies could generally separate most TP53 missense mutations from other TP53 genotypes (Figure 3A). In addition, the SP5 antibody could clearly divide TP53 mutations into three groups, that is, WT, missense mutations, and other mutations, which is superior to the other three p53 antibodies (Figure 3A and Supplementary Figure 3). This indicates that the SP5 antibody might have higher specificity to missense mutated p53 compared with the WT p53 or other truncated p53 that resulted from frameshift and stop gain mutations. Also, as the TP53 missense mutations frequently occurred in exon 5 to exon 8, we verified that the four p53 antibodies could detect mutations located in all of these exons (Figure 3B). Lastly, we used the TP53 NGS data to select the IHC positivity threshold in order to get the highest accuracy of detecting TP53 missense mutations. All four antibodies could achieve more than 93% accuracy with the optimized IHC threshold, especially for the SP5 antibody that had 100% sensitivity and specificity when setting the positivity threshold to 20% (Table 2). Overall, these results suggest that a proper IHC positivity threshold is crucial to accurately predict the TP53 mutation status and SP5 was the best p53 IHC antibody among all the tested antibodies in our patient cohort.
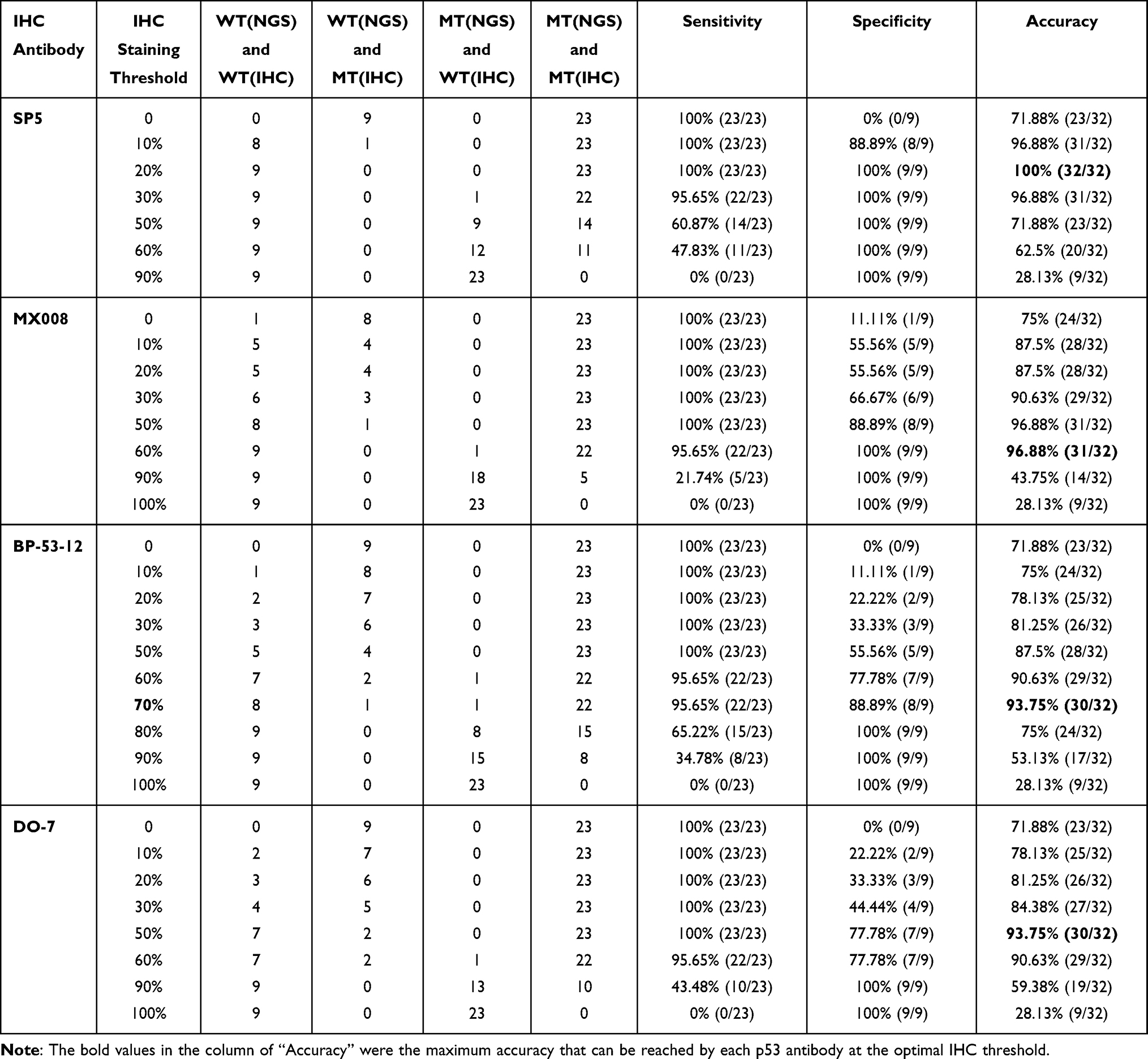 |
Table 2 The Sensitivity, Specificity, and Accuracy of Various p53 IHC Antibodies |
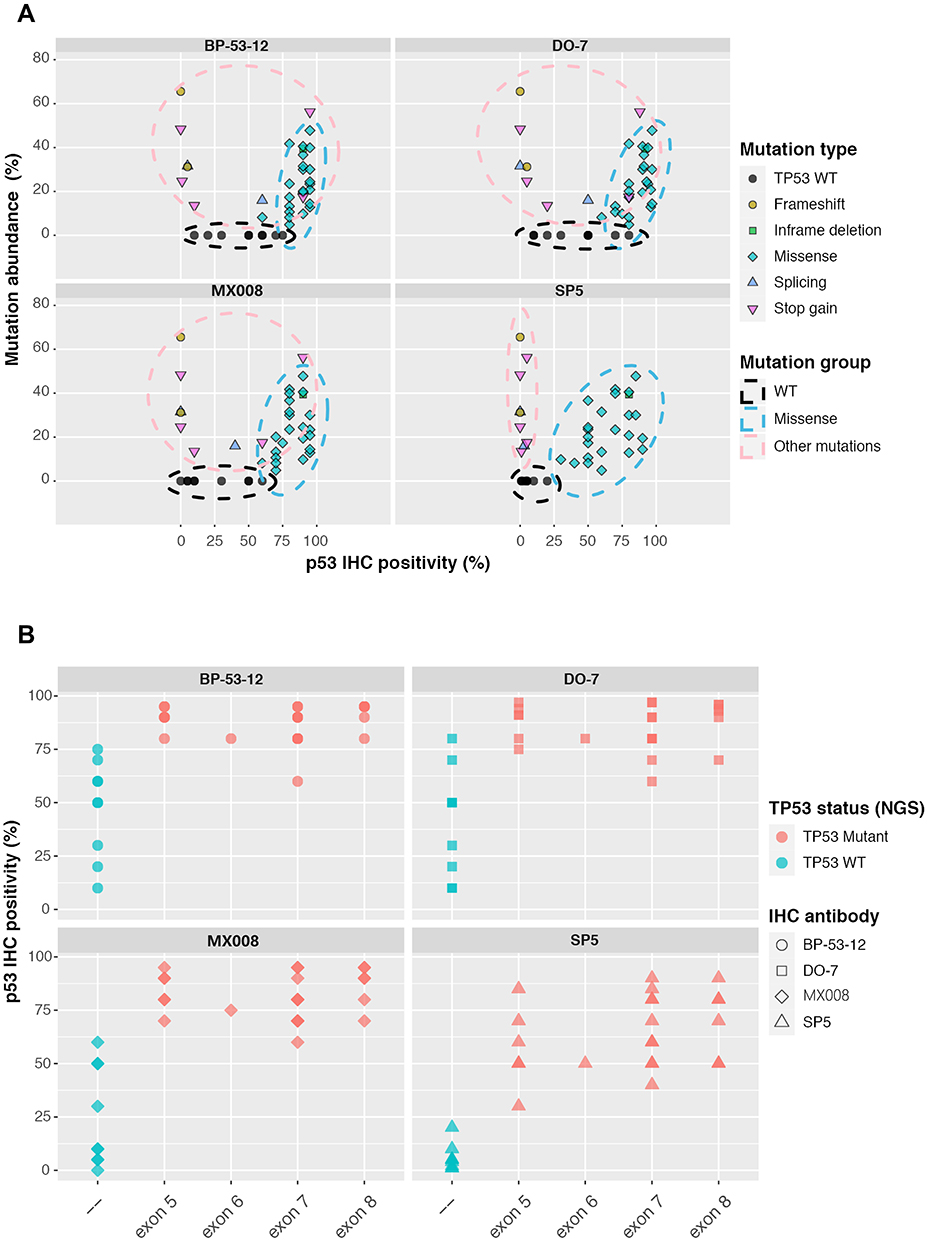 |
Figure 3 Characterizing different p53 IHC antibodies. (A) The relationship between the TP53 mutated allele frequency and p53 IHC positivity for 4 commonly used p53 antibodies, including SP5, MX008, BP-53-12, and DO-7. Each dot represents a TP53 genetic change from the 42-patient cohort. (B) p53 IHC positivity in different TP53 exon regions for the 4 tested p53 IHC antibodies. Based on the NGS results, TP53 WT samples were shown in green while samples harboring TP53 missense mutations were shown in red. WT, wild type. |
Discussion
Globally, more than one million people were diagnosed with gastric cancer annually, and gastric cancer accounts for around 782,685 patient deaths in the year 2018.4 The poor overall survival in gastric cancer is partially attributed to the late diagnosis of the disease. TP53 is the most frequently mutated gene in gastric cancer, and TP53 mutations are important for the premalignancy to cancer transition, suggesting the great potential of using TP53 mutations as a diagnostic marker for gastric cancer. The frequently used techniques to detect TP53 mutations include NGS and IHC. The NGS approach has a high detection accuracy, but it is also relatively expensive and time-consuming. On the other hand, the traditional IHC approach could rapidly detect TP53 aberrations at a very low cost; however, IHC cannot easily distinguish between the TP53 missense mutation and the accumulation of WT p53 under stress conditions, thus limiting its detection accuracy and clinical utility. In the current study, we used NGS-determined TP53 missense mutations as the gold standard to optimize the IHC threshold for four commonly used p53 antibodies. Of note, all antibodies achieved more than 93% accuracy after threshold optimization, especially for the SP5 antibody. Our results suggest that setting proper IHC thresholds and choosing appropriate p53 antibodies are important to accurately detect TP53 missense mutations, thereby, facilitating early gastric cancer diagnosis.
Recently, Schoop et al32 found that p53 immunohistochemical evaluation cannot be used to predict TP53 mutations in gastric cancer, which seems to be different from our conclusion; however, there were several crucial differences between the two studies. Firstly, Schoop et al tested only one p53 antibody (ie DO-7), while we tested four commonly used p53 antibodies. Based on our results, by choosing the optimal IHC threshold, the accuracy of DO-7 could reach as high as 93.75%, which is similar to that of BP-53-12 antibody (accuracy=93.75%) but lower than that of MX008 (accuracy=96.88%) or SP5 (accuracy=100%) antibodies. Therefore, the p53 antibody used in Schoop et al’s study was not the best one to predict the mutational status of TP53. Secondly, Schoop et al pre-selected IHC threshold and/or optimized the thresholds of IHC staining by their correlation with clinical characteristics (eg, EBV positivity, microsatellite instability, and HER2 and MET status); they then used sequencing data to validate their IHC algorithms and found the prediction results were not ideal. On the other hand, we directly used NGS data to optimize the IHC threshold. According to our results of the DO-7 antibody, the accuracy of mutation prediction ranged from 59.38% to 93.75%, so choosing an improper IHC threshold could lead to a significant decrease in accuracy. Thirdly, Schoop et al’s algorithm tempted to distinguish WT TP53 from all of the mutated TP53, including missense, frameshift, inframe deletion, stop gain, etc. As shown in Figure 3A, TP53 missense mutations had the highest level of IHC positivity and they separated well from other genotypes, so it is possible to choose a proper IHC threshold for each specific p53 to identify TP53 missense mutations. However, it seems to be unrealistic to separate WT TP53 from other non-missense TP53 mutations as their levels of IHC positivity were largely overlapped (Figure 3A). Overall, all of these differences in methods and research scopes are likely to contribute to the different conclusions between Schoop et al’s study and our study.
According to the IHC results (Figure 3 and Supplementary Figure 3), although all four antibodies could achieve high accuracy of detecting TP53 mutations when appropriate IHC thresholds were chosen, there were still some subtle differences in their IHC staining patterns. Unlike some detection assays where proteins are usually denatured and the structure of the proteins are disrupted (eg, Western blot), p53 protein in the IHC assay has been fixed. As a result, p53 protein in IHC staining will reserve most of its original protein structures, either WT or mutated p53. Based on previous studies,33 mutated p53 had some structural changes that were different from the WT counterparts, which could confer the differential binding affinity of various p53 antibodies. Although none of the four antibodies we used were originally designed to specifically recognize mutated p53 structures, our IHC results provided indirect evidence of the differential binding capacity of these p53 antibodies to various forms of p53. For example, compared with the other three antibodies, SP5 antibodies had the lowest level of WT p53 staining, whereas the four antibodies stained the p53 missense mutation sections similarly (Supplementary Figure 3). Nevertheless, our results need to be further investigated by future structural studies.
Multiple studies have reported the insignificant impact of TP53 mutation status on treatment response or overall survival.6,7 On the other hand, some researchers found TP53-mutated patients had improved response to chemotherapy,8 while others reported that patients harboring TP53 hotspot mutations had worse overall survival and recurrence-free survival.9 Given that these contradictive results might be partially due to unreliable detection of TP53 mutations, properly determining TP53 mutations status is important for predicting patient prognosis. In addition, most TP53 missense mutations gained capacities to repress some tumor suppressors, such as WT p53, p63, and p73, and could promote tumorigenesis and progression. As a result, targeting mutated TP53 becomes a hot area for anti-cancer therapies. Multiple small molecule drugs have been found to promote proper folding of mutant p53 and restore normal p53 function, and some of these drugs have achieved promising pre-clinical and clinical results.34 Therefore, accurately detecting the TP53 mutation has potential therapeutic values to direct targeted therapies in gastric cancer.
We characterized the co-existence of TP53 mutations with other clinical and molecular features. We found that TP53 mutations tended to be associated with microsatellite stable (MSS) and more differentiated tumors, which might partially explain the conflicting clinical results of using TP53 mutations as a prognostic biomarker because patients with MSS and well-differentiated gastric tumors were found to have opposing expectations in prognosis.30,31 Besides the tumor differentiation level and MSI status, we found that TP53 mutations occurred mutually exclusive with BRCA1 mutations in gastric tumors. Previous studies demonstrated that BRCA1 and p53 could exist in the same protein complex and p53 mediated homologous recombination through inhibiting BRCA1 over-function in response to DNA repair.35,36 Also, BRCA1 mutations were found to co-occur with TP53 mutations in breast cancer,37,38 and restoring the WT p53 function could potentially target these tumors.39 The difference in BRCA1 and TP53 co-mutational behavior in gastric and breast cancer might be due to the fact that the co-mutation is crucial for the progression and/survival of breast cancer cells while BRCA1 and TP53 mutation could independently promote gastric cancer progression, thus resulting in less co-occurring frequency. Given that PARP inhibitors showed promising clinical responses in patients with BRCA mutations and detection of BRCA mutation status is relatively expensive,40 our results imply that TP53-positive gastric cancer patients were less likely to harbor BRCA mutations, thus providing a cost-effective approach to direct the treatment of anti-PARP drugs. In addition, in some rare cases where BRCA1 and TP53 mutations co-occurred in the same gastric tumor, these patients tended to have worse overall survival. On the other hand, patients harboring both KMT2B and TP53 mutations were likely to have better overall survival. All these results suggest that combining TP53 mutations with other clinical and molecular features might better predict the prognosis of gastric cancer patients. Similar results were also observed by Park et al who found that co-mutation of NRXN1 and TP53 were associated with distinct drug responses in gastric cancer patients.11
Our study had several limitations. First of all, we optimized the p53 IHC threshold using the discovery cohort; however, as the IHC results for the validation cohort were not available, we cannot further confirm these results. Secondly, the cohort size of the discovery cohort was limited. Thereby, future large cohort studies are still needed to further validate the IHC thresholds identified in our study. Thirdly, although TP53/BRCA1 co-mutations and TP53/KMT2B co-mutations were found to be correlated with patient survival, they were mutually exclusive mutation pairs in our cohort. As a result, there were a limited number of gastric cancer patients harboring these co-mutations, which limited the clinical utility of these results.
Conclusion
Overall, our results illustrated that TP53 mutations tended to co-occur with certain molecular and clinical features in gastric tumors, such as WT BRCA and WT KMT2B as well as stable microsatellite and high level of tumor differentiation. We also optimized the IHC threshold for commonly used p53 antibodies to enable feasible and reliable detection of TP53 missense mutations, which could further promote the diagnostic, prognostic, and therapeutic values of using the TP53 mutation as a gastric cancer biomarker.
Acknowledgments
We owe thanks to the patients in our study and their family members. We acknowledge the staff for their assistance in our study.
Disclosure
Yang Xu, Kaihua Liu, Yuqian Shi, Xue Wu, and Yang Shao are employees of Nanjing Geneseeq Technology Inc. Xue Wu reports personal fees from Nanjing Geneseeq Technology Inc., outside the submitted work. The authors report no other potential conflicts of interest for this work.
References
1. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137(3):413–431. doi:10.1016/j.cell.2009.04.037
2. Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. doi:10.1038/nature12634
3. Mantovani F, Collavin L, Del Sal G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019;26(2):199–212. doi:10.1038/s41418-018-0246-9
4. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
5. Busuttil RA, Zapparoli GV, Haupt S, et al. Role of p53 in the progression of gastric cancer. Oncotarget. 2014;5(23):12016–12026. doi:10.18632/oncotarget.2434
6. Fenoglio-Preiser CM, Wang J, Stemmermann GN, Noffsinger A. TP53 and gastric carcinoma: a review. Hum Mutat. 2003;21(3):258–270. doi:10.1002/humu.10180
7. Ott K, Vogelsang H, Mueller J, et al. Chromosomal instability rather than p53 mutation is associated with response to neoadjuvant cisplatin-based chemotherapy in gastric carcinoma. Clin Cancer Res. 2003;9(6):2307–2315.
8. Xu HY, Xu WL, Wang LQ, Chen MB, Shen HL. Relationship between p53 status and response to chemotherapy in patients with gastric cancer: a meta-analysis. PLoS One. 2014;9(4):e95371. doi:10.1371/journal.pone.0095371
9. Tahara T, Shibata T, Okamoto Y, et al. Mutation spectrum of TP53 gene predicts clinicopathological features and survival of gastric cancer. Oncotarget. 2016;7(27):42252–42260. doi:10.18632/oncotarget.9770
10. Robles AI, Harris CC. Clinical outcomes and correlates of TP53 mutations and cancer. Cold Spring Harb Perspect Biol. 2010;2(3):a001016. doi:10.1101/cshperspect.a001016
11. Park S, Lee J, Kim YH, Park J, Shin JW, Nam S. Clinical relevance and molecular phenotypes in gastric cancer, of TP53 mutations and gene expressions, in combination with other gene mutations. Sci Rep. 2016;6(1):34822. doi:10.1038/srep34822
12. Roshandel AK, Busch CM, Mullekom JV, et al. The predictive capability of immunohistochemistry and DNA sequencing for determining TP53 functional mutation status: a Comparative Study of 41 glioblastoma patients. Oncotarget. 2019;10(58):6204–6218. doi:10.18632/oncotarget.27252
13. Wang K, Yuen ST, Xu J, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014;46(6):573–582. doi:10.1038/ng.2983
14. Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513(7517):202–209. doi:10.1038/nature13480
15. Hoadley KA, Yau C, Hinoue T, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018;173(2):291–304e296.
16. Ellrott K, Bailey MH, Saksena G, et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 2018;6(3):271–281e277.
17. Yang Z, Yang N, Ou Q, et al. Investigating novel resistance mechanisms to third-generation EGFR tyrosine kinase inhibitor osimertinib in non-small cell lung cancer patients. Clin Cancer Res. 2018;24(13):3097–3107. doi:10.1158/1078-0432.CCR-17-2310
18. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi:10.1093/bioinformatics/btu170
19. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi:10.1093/bioinformatics/btp324
20. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi:10.1038/ng.806
21. Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–576. doi:10.1101/gr.129684.111
22. Newman AM, Bratman SV, Stehr H, et al. FACTERA: a practical method for the discovery of genomic rearrangements at breakpoint resolution. Bioinformatics. 2014;30(23):3390–3393. doi:10.1093/bioinformatics/btu549
23. Amarasinghe KC, Li J, Halgamuge SK. CoNVEX: copy number variation estimation in exome sequencing data using HMM. BMC Bioinform. 2013;14(Suppl 2):S2. doi:10.1186/1471-2105-14-S2-S2
24. Gerstung M, Pellagatti A, Malcovati L, et al. Combining gene mutation with gene expression data improves outcome prediction in myelodysplastic syndromes. Nat Commun. 2015;6(1):5901. doi:10.1038/ncomms6901
25. Yang L, Zheng R, Wang N, et al. Incidence and mortality of stomach cancer in China, 2014. Chin J Cancer Res. 2018;30(3):291–298. doi:10.21147/j.issn.1000-9604.2018.03.01
26. Ko GH, Go SI, Lee WS, et al. Prognostic impact of Ki-67 in patients with gastric cancer-the importance of depth of invasion and histologic differentiation. Medicine (Baltimore). 2017;96(25):e7181. doi:10.1097/MD.0000000000007181
27. Wu MS, Shun CT, Wang HP, et al. Genetic alterations in gastric cancer: relation to histological subtypes, tumor stage, and Helicobacter pylori infection. Gastroenterology. 1997;112(5):1457–1465. doi:10.1016/S0016-5085(97)70071-4
28. Wu MS, Shun CT, Sheu JC, et al. Overexpression of mutant p53 and c-erbB-2 proteins and mutations of the p15 and p16 genes in human gastric carcinoma: with respect to histological subtypes and stages. J Gastroenterol Hepatol. 1998;13(3):305–310. doi:10.1111/j.1440-1746.1998.01560.x
29. Verma R, Sharma PC. Next generation sequencing-based emerging trends in molecular biology of gastric cancer. Am J Cancer Res. 2018;8(2):207–225.
30. Cristescu R, Lee J, Nebozhyn M, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med. 2015;21(5):449–456. doi:10.1038/nm.3850
31. Feng F, Liu J, Wang F, et al. Prognostic value of differentiation status in gastric cancer. BMC Cancer. 2018;18(1):865. doi:10.1186/s12885-018-4780-0
32. Schoop I, Maleki SS, Behrens HM, Kruger S, Haag J, Rocken C. p53 immunostaining cannot be used to predict TP53 mutations in gastric cancer: results from a large Central European cohort. Hum Pathol. 2020;105:53–66. doi:10.1016/j.humpath.2020.09.006
33. Joerger AC, Fersht AR. Structure-function-rescue: the diverse nature of common p53 cancer mutants. Oncogene. 2007;26(15):2226–2242. doi:10.1038/sj.onc.1210291
34. Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. 2018;18(2):89–102. doi:10.1038/nrc.2017.109
35. Dong C, Zhang F, Luo Y, et al. p53 suppresses hyper-recombination by modulating BRCA1 function. DNA Repair (Amst). 2015;33:60–69. doi:10.1016/j.dnarep.2015.06.005
36. Arizti P, Fang L, Park I, et al. Tumor suppressor p53 is required to modulate BRCA1 expression. Mol Cell Biol. 2000;20(20):7450–7459. doi:10.1128/MCB.20.20.7450-7459.2000
37. Greenblatt MS, Chappuis PO, Bond JP, Hamel N, Foulkes WD. TP53 mutations in breast cancer associated with BRCA1 or BRCA2 germ-line mutations: distinctive spectrum and structural distribution. Cancer Res. 2001;61(10):4092–4097.
38. Peng L, Xu T, Long T, Zuo H. Association between BRCA status and p53 status in breast cancer: a meta-analysis. Med Sci Monit. 2016;22:1939–1945.
39. Na B, Yu X, Withers T, et al. Therapeutic targeting of BRCA1 and TP53 mutant breast cancer through mutant p53 reactivation. NPJ Breast Cancer. 2019;5(1):14. doi:10.1038/s41523-019-0110-1
40. Yi M, Dong B, Qin S, Chu Q, Wu K, Luo S. Advances and perspectives of PARP inhibitors. Exp Hematol Oncol. 2019;8(1):29. doi:10.1186/s40164-019-0154-9
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.