Back to Journals » International Journal of Nanomedicine » Volume 16
tLyP-1 Peptide Functionalized Human H Chain Ferritin for Targeted Delivery of Paclitaxel
Authors Ma Y, Li R, Dong Y ![]() , You C, Huang S, Li X, Wang F, Zhang Y
, You C, Huang S, Li X, Wang F, Zhang Y ![]()
Received 7 November 2020
Accepted for publication 18 January 2021
Published 4 February 2021 Volume 2021:16 Pages 789—802
DOI https://doi.org/10.2147/IJN.S289005
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yan Shen
Yuanmeng Ma,1 Ruike Li,1 Yixin Dong,1 Chaoqun You,1,2 Shenlin Huang,1 Xun Li,1 Fei Wang,1 Yu Zhang1
1College of Chemical Engineering, Jiangsu Key Laboratory for the Chemistry and Utilization of Agro-Forest Biomass, Jiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, Jiangsu Key Laboratory of Biomass-Based Green Fuels and Chemicals, Nanjing Forestry University, Nanjing, 210037, People’s Republic of China; 2School of Chemistry and Chemical Engineering, Southeast University, Nanjing, 210089, People’s Republic of China
Correspondence: Yu Zhang Tel +86-25-85427635
Fax +86-25-85418873
Email [email protected]
Purpose: The aims of this study were to test the feasibility, targeting specificity and anticancer therapeutic efficacy of CendR motif tLyP-1 functionalized at the N-terminal of ferritin for paclitaxel (PTX) delivery.
Methods: A tumor homing and penetrating peptide tLyP-1 was fused to the N-terminal of human H chain ferritin (HFtn) to generate a dual-targeting nanoparticle delivery system. PTX molecules were encapsulated into the HFtn nanocage using the disassembly/assembly method by adjusting pHs. Cellular uptake was examined by confocal laser scanning microscopy (CLSM) and flow cytometry. The MTT assay was used to test the cytotoxicity of various PTX-loaded NPs against MDA-MB-231 and SMMC-7721 tumor cells. The wound healing and cell migration assays were conducted to assess the inhibitory effect on cell motility and metastasis. The inhibition effect on the SMMC-7721 tumor spheroids was studied and penetration ability was evaluated by CLSM. The antitumor efficacy of PTX-loaded NPs was assessed in MDA-MB-231 breast cancer xenografted in female BALB/c nude mice.
Results: Compared with HFtn-PTX, in vitro studies demonstrated that the tLyP-1-HFtn-PTX displayed enhanced intracellular delivery and better cytotoxicity and anti-invasion ability against both SMMC-7721 and MDA-MB-231 cells. The better penetrability and growth inhibitory effect on SMMC-7721 tumor spheroids were also testified. In vivo distribution and imaging demonstrated that the tLyP-1-HFtn-PTX NPs were selectively accumulated and penetrated at the tumor regions. Verified by the breast cancer cells model in BABL/c nude mice, tLyP-1-HFtn-PTX displayed higher in vivo therapeutic efficacy with lower systemic toxicity.
Conclusion: Ferritin decorated with tumor-homing penetration peptide tLyP-1 at the N terminal could deliver PTX specifically inside the cell via receptor-mediated endocytosis with better efficacy. The peptide tLyP-1 which is supposed to work only at the C terminus showed enhanced tumor tissue penetration and antitumor efficacy, demonstrating that it also worked at the N-terminal of HFtn.
Keywords: ferritin, tLyP-1 peptide, paclitaxel, neuropilin-1, transferrin receptor 1
Introduction
Poor site-specificity and incapable deep-penetration into tumors are the most challenging problems of the chemotherapy.1 Paclitaxel (PTX) is widely used to treat patients with various cancers, such as ovarian, breast cancer.2,3 However, its extremely poor water solubility and serious side effects restrict its clinical application.4
Active targeting nanoparticulate drug-delivery systems (DDS) have been widely developed in recent years and some have been applied in clinical trials.5–10 With excellent biocompatibility and well-defined architectures, protein cages that can encapsulate drugs into the inner cavity and self-assembled in an appropriate size range have been widely used.11–15 Atomic-resolution structural information of the cages allows the accurate design and modification of the desired positions on the protein cage either chemically or genetically to confer functionality. Since the protein shell can act as a barrier for the encapsulated drug, the nanoparticles can be accumulated to the tumor region with targeted ligands or peptides displayed on the outer surface of the cage. Furthermore, protein cage nanoparticles can target tumors through a passive or active process. Passive targeting refers to the fact that NPs can enter the endothelial tissue through enhanced permeation and retention (EPR) effect.15,16 Active targeting can directly interact with complementary receptors on the surface of the target sites. Compared with passive targeting, the advantage of active targeting is that the drugs can be accumulated within the tumor regions for a longer time due to the binding with the receptors, preventing drugs from re-entering the systemic circulation.17–20
Dysfunctional tumor vessels and high interstitial pressure often prevent drugs and nanoparticles from entering the tumor tissue, limiting the effectiveness of treatment.20,21 Peptides, ubiquitous in nature, are composed of a short chain of amino acids and can perform specific biological functions. Due to their unique properties such as the inherent biological origins, bioactivity, biocompatibility and biodegradability, peptides have promising applications in the fields of drug delivery.22–26 The modification of nanocarriers with cell-penetrating peptides (CPPs) provides an alternative strategy to solve the problem of impermeable tumor tissue. The modified CPPs can dramatically improve the drug penetration efficiency, promote endosomal escape, enhance the stability of the drug in the blood, improve the specific targeting of the tumor, and provide the controlled drug release by using the stimulation response mechanism in tumor microenvironments.27–29
Tumor penetrating peptides, which usually contain the sequence motif R/KXXR/K (also known as the C-end Rule motif, or CendR, X represents any amino acid), have previously been used to improve tumor penetration and thereby antitumor efficacy of chemotherapeutics.30–32 The receptor for the CendR motif is neuropilin-1 (NRP-1), which is a transmembrane protein overexpressed in tumor vascular endothelial cells. The tLyP-1 peptide (CGNKRTR) has been reported to improve tumor penetration through an NRP-1-dependent internalization pathway.33 This makes the tLyP-1 peptide an effective targeting moiety, which mediates tumor site-specific delivery and penetration of nanocarriers into solid tumors.
Ferritin is a highly symmetrical protein cage composed of 24 subunits and self-assembled into a hollow structure with an outer diameter of 12 nm and an inner diameter of 8 nm. With the exceptional characteristics, such as water solubility, biodegradability, functional versatility and size uniformity, human H chain ferritin (HFtn) has attracted more and more attention in the field of drug delivery.11,34–36 Furthermore, it has been reported that the HFtn binds to tumor cells by interacting with the transferrin receptor 1 (TfR1).37–39 Although it is well known that the TfR1 is overexpressed in human cancer cells and used as a targeting moiety, HFtn-based nanoparticles still rely on recognition ligand functionalization to achieve targeting.15,37
In this study, we functionalized the N-terminal of HFtn with the C-end rule peptide tLyP-1 as the targeting moiety for PTX drug delivery. To prove its effectiveness was attributed to the N-terminal peptide functionalization, the corresponding mutation M-tLyP-1-HFtn was generated as a control that the key amino acid residues of the CendR peptide, arginine and lysine, were mutated to alanine, respectively (Scheme 1). The aims of this study were to test the feasibility of tLyP-1 peptide fusion at the N-terminus and evaluate its targeting specificity and anticancer therapeutic efficacy.
 |
Scheme 1 (A) Schematic diagrams of HFtn functionalized with tLyP-1 or M-tLyP-1 peptide. (B) The loading of PTX nanoparticles was prepared using the disassembly/reassembly method. (C) Schematic illustration of the tLyP-1 peptide functionalized HFtn for targeted delivery. For the tLyP-1-HFtn-PTX delivery system, it binds tumor cells by interacting with TfR1 or NRP-1, and then transfers into endosome and lysosome to release PTX into the cytoplasm. Finally, the free PTX acts on tubulin to inhibit cancer cell mitosis and trigger cell apoptosis. |
Materials and Methods
Materials
T4 polynucleotide kinase and T4 DNA ligase were purchased from Takara Biotech, Co., Ltd. (Dalian, China). DMT enzyme was obtained from TransGen Biotech Co., Ltd. (Beijing, China). Paclitaxel (>97% purity) was purchased from Energy Chemical (Shanghai, China). Anti-transferrin receptor rabbit pAb and anti-neuropilin-1 rabbit mAb were purchased from Beyotime Biotechnology (Shanghai, China). Anti-GAPDH rabbit pAb and HRP-labeled goat anti-rabbit secondary antibody were purchased from Servicebio (Wuhan, China). MTT was purchased from Guangzhou Saiguo Biotech Co., Ltd (Guangzhou, China). Penicillin-streptomycin solution (100×) and 0.25% trypsin-EDTA (1×) solution were purchased from Biosharp Life Sciences. Fetal bovine serum (FBS) was purchased from Zhejiang Tianhang Biotechnology Co., Ltd. Dulbecco’s modified Eagle’s medium (DMEM) was purchased from HyClone. Fluorescein isothiocyanate (FITC) was purchased from Shanghai Yuanye Biotechnology Co., Ltd (Shanghai, China). DAPI was purchased from Nanjing KeyGen Biotech. Co., Ltd. (Nanjing, China). All cell lines were purchased from the cell bank of the Chinese Academy of Sciences (Shanghai, China).
Cloning of tLyP-1-HFtn and the Mutant M-tLyP-1-HFtn
The recombinant plasmid for the expression of tLyP-1-HFtn was constructed with pET20b (+). The tLyP-1-HFtn gene was amplified by PCR using HFtn gene as the template with the following primers: 5ʹ-GTACCCGTGGTGGTGGTGGTAGCATGACTACCGCATCGACAAGTC-3ʹ and reverse primer: 5ʹ-GTTTATTACCACACATATGTATATCTCCTTCTTAAAGTTAAACAA-3ʹ. The template was removed by DMT enzyme, followed by the phosphorylation reaction using T4 polynucleotide kinase. Finally, the ligation reaction was performed using T4 DNA ligase. The M-tLyP-1-HFtn gene was constructed by PCR site-directed mutagenesis using tLyP-1-HFtn gene as the template with the following primers: 5ʹ-GCAGGTGGTGGTGGTAGCATG-3ʹ and reverse primer: 5ʹ-GGTACGTGCATTACCACACATATGTATATCTC-3ʹ. The mutated sequence of tLyP-1 peptide was changed to CGARTA. The resulting plasmid was verified by DNA sequencing and was transformed into the expression strain E. coli BL21 (DE3).
Preparation and Characterization of PTX-Loaded NPs
Proteins HFtn, tLyP-1-HFtn and the mutant M-tLyP-1-HFtn were produced in E. coli and purified as previously described.39 The protein purity was determined by SDS-PAGE electrophoresis (Figure S1). The loading of paclitaxel (PTX) nanoparticles was prepared as described previously.39 Briefly, PTX was dissolved in ethyl alcohol with a final concentration of 15 mM. PTX solution was added to HFtn, tLyP-1-HFtn and M-tLyP-1-HFtn solution with a protein/PTX ratio of 1:200, 1:500 and 1:500, respectively. The mixture was adjusted to pH at 2.5 by 0.1 M HCl to disassemble ferritin cages into subunits. After 30 min, the solution pH was adjusted to 7.4 by 0.1 M NaOH. The solution was dialyzed against 0.1 M PBS (pH 7.4) overnight using the 6–8 kDa MWCO dialysis bag to remove free PTX. After centrifugation, a clear PTX-loaded NPs solution was obtained and the PTX concentration was determined by HPLC.
Characterization of the PTX-Loaded NPs
The PTX-loaded NPs were purified and analyzed using a size-exclusion chromatography (SEC) system (GE Healthcare) on a Superdex 200 10/300 GL column equilibrated with running buffer (50 mM NaH2PO4, 150 mM NaCl, pH 7.0). SEC analysis of protein before and after PTX encapsulation was monitored at 280 nm. The column was calibrated as previously described.40
Particle size and the morphology of HFtn-PTX, tLyP-1-HFtn-PTX and M-tLyP-1-HFtn-PTX NPs were examined by transmission electron microscopy (TEM) (JEM-1400, JOEL, Japan) using 2% uranyl acetate as the negative stain.
To examine whether the secondary structure would be changed due to the PTX encapsulation within the cage cavity, circular dichroism was performed to measure the secondary structure of samples by using MOS-500 circular dichroism spectrometer (Bio-Logic Science Instruments, France). The samples were dialyzed against phosphate buffer (50 mM NaH2PO4, pH 7.2) and the protein concentrations were determined by BCA (Novagen) and the samples were measured at the concentration of 0.14 μM.
Encapsulation Efficiency and Drug Loading Capacity
The PTX content of the drug was determined by HPLC as described previously.39 PTX molecules were released by adjusting the pH value of HFtn-PTX or tLyP-1-HFtn-PTX sample (500 μL) to 2.5 with 1 M HCl. Encapsulation efficiency (%) and drug loading capacity (%) were calculated using the following equations: encapsulation efficiency (%) = weight of encapsulated PTX/weight of PTX added ×100%; loading capacity (%) = weight of encapsulated PTX/weight of PTX-loaded NP × 100%.
In vitro PTX Release
Dialysis method was used to study the in vitro PTX release from HFtn-PTX or tLyP-1-HFtn-PTX. Briefly, about 1.0 mL sample in a dialysis bag (6–8 kDa, MWCO) was placed into 10 mL PBS buffer (0.1 M, pH 7.4 or pH 6.0) and acetate buffer (0.1 M, pH 5.0) at the shaking speed of 100 rpm, 37°C, respectively. At the time intervals of 0.5, 1, 2, 3, 4, 6, 8, 12, 24, 48 and 60 h, 1.0 mL sample of the release solution was extracted and replaced with fresh buffer. The released amount of PTX into the solution was determined by HPLC analysis.
Cell Experiments
Cell Culture
Human umbilical vein endothelial cell (HUVEC) line, human breast cancer line MDA-MB-231 and hepatic cancer line SMMC-7721 were cultured in DMEM containing 10% FBS and 1% (v/v) penicillin-streptomycin. Cells were maintained in an incubator with 5% CO2 at 37°C.
TfR1 and NRP-1 Expression Level
The expression levels of TfR1 and NRP-1 on MDA-MB-231 and SMMC-7721 cells were measured via Western blot analyses. Approximately 3 ×105 cells were lysed by RIPA cell lysis buffer containing 1% PMSF. Protein levels of cell lysis solution were measured by BCA kit and protein samples were resolved on 10% SDS-PAGE gels. The proteins on gels were transferred to a PVDF membrane blocked with 5% BSA for 1 h, and then incubated with anti-transferrin receptor rabbit pAb, anti-neuropilin-1 rabbit mAb and anti-GAPDH rabbit pAb overnight at 4°C, respectively. After incubation with HRP-labeled goat anti-rabbit secondary antibody, the membranes were visualized on chemical films using ECL reagents.
Cellular Uptake
The uptake of PTX-loaded NPs in cells was quantitatively analyzed and measured by confocal laser scanning microscopy (CLSM) (Zeiss LSM710) and flow cytometry. The fluorescent dye FITC was used to label the NPs. Typically, MDA-MB-231 cells were seeded at 1 × 105 cells/well in 24-well plates and cultured for 24 h in 5% CO2 atmosphere at 37°C. FITC-labeled HFtn, tLyP-1-HFtn, M-tLyP-1-HFtn NPs were added into the plates at the final FITC concentration of 10 μg mL−1. After 4 h incubation, the cells were washed three times with PBS, trypsinized, and finally resuspended in 0.5 mL PBS. In the competitive receptor study, cells were pre-incubated with excess HFtn protein for 1 h before adding FITC-labeled HFtn or tLyP-1-HFtn. Finally, the fluorescent intensity of FITC was analyzed by flow cytometry. The results were the average of three repeats.
The cellular uptake efficiency of NPs was evaluated by CLSM. Briefly, cells were seeded on glass-bottom dishes at 3 × 104 cells/well and incubated for 24 h at 37°C. Then, the cells were exposed to FITC-labeled HFtn, tLyP-1-HFtn, M-tLyP-1-HFtn NPs for 4 h. The final FITC concentration was 10 μg mL−1. After 4 h incubation, the cells were washed with PBS for three times and fixed with 4% paraformaldehyde for 15 min, followed by the addition of DAPI (10 μg mL−1) for nuclei staining. The fluorescence of cells was visualized by CLSM.
Cytotoxicity of PTX-Loaded NPs
In vitro cytotoxicity of tLyP-1-HFtn and HFtn were tested to verify their safety by the MTT-based assay using HUVEC cells. HUVEC cells were seeded at 5 × 103 cells/well into a 96-well plate and incubated for 24 h. The culture medium was discarded and the cells were treated with a series of concentrations of tLyP-1-HFtn and HFtn for 48 h at 37°C. After treatment, the cells were washed three times with PBS and 20 μL of 5 mg/mL MTT solution was added in each well. After 4 h incubation, the culture medium was replaced with 200 μL DMSO to dissolve formazan. The absorbance was measured at 570 nm using a 96-well plate reader (Cytation5, BioTek). Each experiment was repeated for three times.
The cytotoxicity of tLyP-1-HFtn-PTX or HFtn-PTX against cells was tested using MDA-MB231 and SMMC-7721 cells. Typically, cells were seeded at 5 × 103 cells/well in 96-well plates and incubated for 24 h. The culture medium was discarded and the cells were treated with PTX, HFtn-PTX and tLyP-1-HFtn-PTX at a series of PTX concentrations (1–5000 ng/mL) for 48 h at 37°C, using the medium as a control. The cytotoxicity of tLyP-1-HFtn-PTX or HFtn-PTX was assessed by the MTT assay. Each experiment was carried out in triplicate.
Would Healing and Cell Migration Inhibition Assay
Cells were seeded at 1 × 105 cells/well in 12-well plates and cultured for 24 h. The confluent monolayer cells were wounded by a 10-μL pipette tip, followed by washing with PBS twice. After incubation for 24 h, the cells were treated with free PTX, HFtn-PTX, tLyP-1-HFtn-PTX NPs and blank medium (as control) at a concentration of 0.5 μg PTX/mL, as well as the corresponding tLyP-1-HFtn NPs, respectively. Reparation of the wounding area was observed under a microscope (Olympus, Japan).
For cell migration assay, cells were seeded at a density of 3 × 105 cells/mL with 100 μL of the serum-free medium in the membrane inserts of transwell chambers (Corning, USA). Then, 500 μL culture medium containing 10% FBS was added to the bottom of the transwell chamber. Subsequently, the cells were incubated for 24 h with free PTX, HFtn-PTX NPs, tLyP-1-HFtn-PTX NPs and blank medium (as control) at a concentration of 0.5 μg PTX/mL, as well as the corresponding tLyP-1-HFtn NPs, respectively. Cells on the upper surface of the membrane were then wiped. The cells on the bottom of the chamber were fixed, stained, and viewed under a microscope (Olympus, Japan).
Cell Apoptosis Assay in vitro
Cell apoptosis was determined using Annexin V-FITC/PI apoptosis detection kit. Briefly, cells were seeded at a density of 4 × 105 cells/well in a 6-well plate and allowed to attach for 24 h. Cells were then treated with HFtn-PTX, tLyP-1-HFtn-PTX and M-tLyP-1-HFtn-PTX NPs for 24 h at 37°C with the PTX final concentration at 5 μg/mL. After that, the cells were harvested and suspended in the provided binding buffer. Subsequently, 5 μL of Annexin V-FITC was added to the cell suspensions and incubated for 15 min in the dark followed by adding 10 μL of PI. The stained cells were analyzed by flow cytometry.
Growth Inhibition on Three-Dimensional Tumor Spheroids
Spheroids of SMMC-7721 cells were established to evaluate the tumor-inhibiting ability of tLyP-1-HFtn-PTX. Cells were seeded at 2 × 103 cells/well in 48-well plates pre-coated with 100 μL of agarose-based PBS (2% w/v). Afterwards, the SMMC-7721 tumor spheroids were incubated for 1 week with tLyP-1-HFtn-PTX, HFtn-PTX, Taxol® and DMEM at a concentration of 0.5 μg mL−1. The spheroid size was measured using an inverted phase microscope. The major (dmax) and minor (dmin) diameters of each tumor spheroid were measured, and the volume was calculated with the equation: V = 0.5 × dmax × dmin2. The change ratio of the tumor spheroid volume was calculated as follows: R = (Vi/V0) × 100%, where Vi is the tumor spheroid volume after treatment and V0 is the tumor spheroid volume prior to treatment.
A spheroid model of SMMC-7721 cells was established to evaluate the tumor penetrating ability of tLyP-1-HFtn-PTX. Tumor spheroids were incubated for 4 h with free FITC, FITC-labeled HFtn and tLyP-1-HFtn. The final FITC concentration was 10 μg mL−1. After washing with cold PBS for three times, the tumor spheroids were imaged by CLSM.
In vivo Experiments
Mice Model Establishment
Female BABL/c nude mice (4–5 weeks old) were purchased from Shanghai SLAC Laboratory Animal Co. Ltd. (Shanghai, China) and housed in dedicated pathogen-free animal facilities. Mice were fed water and food ad libitum and kept under standard conditions (temperature 25 ± 2°C, humidity 55 ± 15%, 12 h light/12 h night cycle).
In order to evaluate the performance of PTX-loaded NPs in vivo, the female BABL/c nude mice (20 g) were used. Mice bearing MDA-MB-231 breast cancer xenografts were prepared by injecting 0.1 mL of cell suspension in saline (1×107 cells mL−1) subcutaneously into the right flank of nude mice. Mice were cultured under the standard condition for 18 days and used for subsequent evaluation when the tumor size had grown to ~100 mm3.
In vivo Imaging
To investigate the in vivo biodistribution and tumor-targeting ability of tLyp-1 peptide decorated ferritin, samples were labeled using the near-infrared fluorescent probe DiR. The female BABL/c nude mice were administrated with 200 μL of various DiR formulations (saline, free DiR, DiR-labeled HFtn and tLyP-1-HFtn) through the tail vein with the final DiR concentration of 10 μg/mL. After 1, 4, 12 and 24 h injection, the biodistribution of protein nanocarrier was observed by an in vivo imaging system (IVIS® Lumina LT Series III, PerkinElmer). After 24 h administration, the mice were sacrificed with major organs harvested and imaged.
In vivo Antitumor Efficacy
The MDA-MB-231 breast cancer xenografted in female BALB/c nude mice were used for in vivo therapeutic efficacy study. The MDA-MB-231 tumor-bearing mice (6/group) were intravenously administrated at a dose of 4 mg, 1 mg and 1 mg PTX/kg with Taxol®, HFtn-PTX and tLyP-1-HFtn-PTX, respectively, every other day for 20 days using saline as a control. The tumor volume and body weight were measured at a two-day interval. Tumor volume was calculated using the following equation: V = length × width2/2. Finally, mice were sacrificed and all tumors were dissected, photographed and weighed.
Statistical Analysis
All data were expressed as means ± SD. Student’s t-test was performed for comparisons. P values were acquired using SPSS 18.0 (SPSS, IL, USA). A P-value less than 0.05 was statistically significant, and a P-value less than 0.01 was considered as highly significant.
Results and Discussion
Preparation and Characterization of PTX-Loaded NPs
The tLyP-1 peptide was introduced at the N-terminal of the HFtn (tLyP-1-HFtn) as the targeting moiety. Since tLyP-1 peptide was reported to obey the C-end rule and in order to test its effectiveness, the peptide was mutated by replacing K and R residues to A to generate M-tLyP-1-HFtn (with the N-terminal sequence of CGNARTA). Proteins were expressed and purified as described previously.39 The SDS-PAGE gels demonstrated that the proteins were expressed successfully and the purified protein bands were the corrected sizes (Figure S1). The loading of PTX into the protein cage was carried out by adjusting the pH of the solution as previously described. Approximately 18 PTX molecules per protein molecule could be encapsulated into the protein cage. The encapsulation efficiency of PTX for all the three proteins, HFtn, tLyP-1-HFtn and M-tLyP-1-HFtn were similar (Table S1, around 19%).
Purified HFtn-PTX, tLyP-1-HFtn-PTX and M-tLyP-1-HFtn-PTX NPs were analyzed by transmission electron microscopy (TEM) to examine their ability of self-assembling into a cage before and after the loading of drug. The micrographs showed that they were monodispersed with well-defined spherical morphology (Figure 1A). The sizes of HFtn-PTX, tLyP-1-HFtn-PTX and M-tLyP-1-HFtn-PTX NPs were 12.0 ± 0.50 nm, 12.6 ± 0.25 nm, and 12.8 ± 0.32 nm, respectively (Table S1).
 |
Figure 1 Characterizations of PTX-loaded NPs. (A) TEM micrographs of HFtn, tLyP-1-HFtn, and PTX-loaded NPs. (B) and (C) are size-exclusion chromatography and circular dichroism of PTX-loaded NPs, respectively. |
Size-exclusion chromatography (SEC) analysis displayed that the elution volume of the PTX-loaded NPs was almost identical after the drug encapsulation except for the peak around the void volume of the column. It has been previously reported that they were clusters of PTX-loaded NPs, which is examined by the TEM analysis.39 The results also indicated that the sizes of protein cages were almost the same that were consistent with the results from TEM analyses (Figure 1B).
The CD spectra of PTX-loaded NPs showed that the secondary structure had no much change to that of their parent protein, suggesting that PTX encapsulation did not affect the structural conformation of the protein cage (Figure 1C).
In vitro PTX Release
PTX-loaded NPs were incubated at pH 5.0, 6.0 and 7.4 at 37°C for 60 h and the release of PTX was determined by HPLC. As the targeting peptide locates at the N terminus and points out of the cage, the release profile of M-tLyP-1-HFtn-PTX would not be different from its parent protein tLyP-1-HFtn-PTX. HFtn-PTX and tLyP-1-HFtn-PTX presented almost the same release profile. At pH 7.4, no significant release from tLyP-1-HFtn and HFtn nanocages even after 60 h incubation, indicating that the NPs were relatively stable. At pH 6.0, the cumulative release rate of PTX at 60 h were 34% and 33% from tLyP-1-HFtn-PTX and HFtn-PTX, respectively. It indicated that PTX was released to a certain extent at pH 6.0 before reaching endosome/lysosome. In contrast, at pH 5.0, the obvious release profiles were observed for both HFtn-PTX and tLyP-1-HFtn-PTX, around 73% and 76%, respectively, after 60 h (Figure 2). According to these results, it is evidenced that HFtn delivery vehicle is based on the endo/lysosome drug release mechanism.34 The biphasic release pattern of PTX may attribute to the aggregated protein cages eluted at the void volume of the column.39
 |
Figure 2 PTX release from PTX-loaded NPs at pH 7.4, pH 6.0 and 5.0 at 37°C. |
Cellular Uptake
The cellular uptake properties of the proteins labeled with FITC were analyzed by flow cytometry. The uptake of HFtn by tumor cells was mediated by TfR1 has been proved previously. As shown in Figure 3, cellular uptake of HFtn was much higher than that of free FITC, which is probably attributed to the TfR1 receptor-mediated endocytosis. Significantly, the mean fluorescence intensity of FITC-tLyP-1-HFtn was approximately three-fold higher than FITC-HFtn, suggesting that the enhanced intracellular uptake of tLyP-1-HFtn was probably attributed to TfR1 and NRP-1 dual receptor-mediated endocytosis. In contrast, FITC-M-tLyP-1-HFtn showed similar mean fluorescence intensity as the FITC-HFtn, indicating that the mutation at the tLyP-1 peptide sequence resulted in the inactivation of the NRP-1 receptor-mediated endocytosis.
 |
Figure 3 (A) Confocal microscope images of cellular distribution and uptake of free FITC, FITC-HFtn, FITC-tLyP-1-HFtn. FITC-HFtn+HFtn and tLyP-1-HFtn+HFtn were the results of pretreatment with HFtn for 1 h prior to the analyses. The concentration of FITC was 10 μg/mL. (B) Flow cytometric measurement of the cellular uptake in MDA-MB-231 cells. Cells were treated with FITC-labeled proteins at 37°C for 4 h (n=3). (C) Graph of mean fluorescent intensity based on flow cytometric analysis. Note: *P < 0.05; ***P < 0.001. |
To further demonstrate the role of the functional tLyP-1 peptide, we first measured receptor expression levels of cells using Western blot analysis. As shown in Figure S2, both receptors (TfR1 and NRP-1) were highly expressed in MDA-MB-231 cells. Competitive experiments were then carried out. Cells were pretreated with excess HFtn protein to cover the tumor cell surface receptor (TfR1) for 1 h before the association with their FITC-labeled proteins. As shown in Figure 3, the cellular uptake of FITC-HFtn decreased significantly after pre-incubation with HFtn. In contrast, after the treatment of HFtn, the mean fluorescence intensity of FITC-tLyP-1-HFtn was almost three-fold higher than FITC-HFtn, indicating the function of tLyP-1 and the corresponding NRP-1 mediated endocytosis. Although FITC-HFtn exhibited less cellular uptake than that of FITC-tLyP-1-HFtn, it also demonstrated that both TfR1 and NRP-1 receptors were highly expressed in MDA-MB-231 cells, which was consistent with the Western blot analysis (Figure S2). Moreover, the similar fluorescent intensity of FITC-M-tLyP-1-HFtn and FITC-HFtn further proved the function of tLyP-1. More importantly, it suggested that tumor-specific penetrating peptide, the CendR motif tLyP-1, functionalized at the N terminal of HFtn protein carrier also worked and could be internalized by tumor cells.
Cytotoxicity of the PTX-Loaded NPs
The MTT assay was employed to assess the biocompatibility of tLyP-1-HFtn and HFtn using HUVEC cells. As shown in Figure 4A, the viabilities of cells incubated with HUVECs for 48 h were all above 95%, indicating that these NPs had quite good biocompatibility. In addition, compared with the control group, the cell morphology also exhibited no significant changes after 48 h incubation with tLyP-1-HFtn at a high dose of 200 µg/mL protein (Figure 4B).
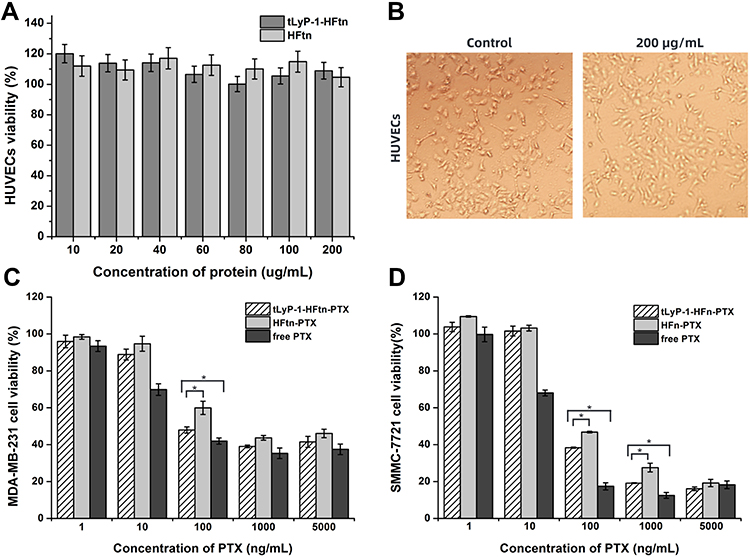 |
Figure 4 (A) In vitro biocompatibility of HUVECs with different concentrations of tLyP-1-HFtn and HFtn. (B) The HUVEC morphology after 48 h treatment with 200 µg/mL of tLyP-1-HFtn. (C, D) Cell viability of MDA-MB-231 and SMMC-7721 after 48 h incubation with free PTX, HFtn-PTX and tLyP-1-HFtn-PTX. Note: *P < 0.05. |
We further evaluated the cytotoxicity of various PTX-loaded NPs against two human cancer cell lines of MDA-MB-231 and SMMC-7721 overexpressing both NRP-1 and TfR1 receptors, as compared with free PTX at equivalent doses.37,41–43 As the mutant M-tLyP-1-HFtn resulted in the inactivation of the NRP-1 receptor-mediated endocytosis and displayed similar cellular uptake properties as the parent HFtn, its following in vitro and in vivo evaluations were not carried out further. Figure 4C and D showed the viability of the two cancer cell lines after incubation with various PTX concentrations. The IC50 value of tLyP-1-HFtn-PTX was 1.4± 0.02 μg/mL in MDA-MB-231, lower than that of HFtn-PTX. It showed similar results in the SMMC-7721 cells (Table S2). Except at 5000 ng mL−1 for SMMC-7721 cancer cells, free PTX demonstrated the strongest cytotoxicity against both cancer cells. tLyP-1-HFtn-PTX NPs showed higher cytotoxicity than the HFtn-PTX NPs at all concentrations from 1 to 5000 ng mL−1, suggesting that the tLyP-1-modified HFtn enhanced the cytotoxicity of tLyP-1-HFtn-PTX NPs. These results again verified that tLyP-1 peptide functionalized at N terminal of HFtn protein also worked and tLyP-1-HFtn-PTX had enhanced cytotoxic effect on cancer cell than HFtn-PTX.
Wound Healing and Cell Migration Assay
The wound healing and cell migration assays were operated to assess the inhibitory effect of PTX-loaded NPs on cell motility and metastasis. As shown in Figure 5, both SMMC-772 cells and MDA-MB-231 cells exhibited good motility and high metastasis for control groups. All the PTX formulations significantly inhibited wound healing and migratory activities of cells, especially for SMMC-7721 cells. Interestingly, for both the two cancer cell lines, the blank tLyP-1-HFtn protein carrier showed no anti-motile activities, suggesting that the tLyP-1-HFtn protein itself had no effects on the cell motility.
 |
Figure 5 Graphs of wound edge (A) and cell migration (B) of SMMC-772 cells and MDA-MB-231 cells after incubation with free PTX, PTX-loaded NPs, and blank tLyP-1-HFtn formulations for 24 h, respectively. |
Cell Apoptosis Assay
To further evaluate the cell apoptosis property, Annexin V-FITC/PI staining assay was performed. As shown in Figure S3, the apoptotic rate of the control, HFtn-PTX, tLyP-1-HFtn-PTX and M-tLyP-1-HFtn-PTX were 12.92%, 32.24%, 44.82% and 13.71% in MDA-MB-231 cells, respectively. It demonstrated that tLyP-1-HFtn-PTX NPs significantly improved the apoptotic feature on MDA-MB-231 cells, which was 1.4-fold and 3.3-fold higher than that of HFtn-PTX and M-tLyP-1-HFtn-PTX, respectively. It showed similar results in SMMC-7721 cells. The cell apoptosis rate for tLyP-1-HFtn-PTX NPs was almost 1.5-fold higher than that of HFtn-PTX. The higher apoptotic feature further demonstrated that the functionalization of the tLyP-1 peptide also worked at the N-terminal and enhanced its antitumoral activity.
Growth Inhibition Effect on Tumor Spheroids
The inhibition effects on the SMMC-7721 tumor spheroids were evaluated by incubation with different PTX formulations. The tumor spheroid volume ratio for the control group was 179.49 ± 21.34% after a week, indicating the dynamic tumor spheroids growth. The tumor spheroids volume ratios of SMMC-7721 cells were 98.99 ± 9.96%, 95.90 ± 7.62% and 81.15 ± 1.76%, respectively, after the treatment with Taxol, HFtn-PTX and tLyP-1-HFtn-PTX at a concentration of 0.5 μg PTX/mL (Figure 6). All the PTX formulations suppressed the tumor spheroids growth efficiently, while tLyP-1-HFtn-PTX NPs exhibited the highest efficiency as compared with Taxol and HFtn-PTX NPs. The cell-penetrating peptide modified nanocarrier and the resulted tLyP-1-HFtn-PTX NPs played a dominant role in the inhibition effects on the tumor spheroids. Therefore, tLyP-1-HFtn possessed better penetration ability than HFtn and tLyP-1-HFtn-PTX may be possible to solve the problem of insufficient drug administration to the solid tumor which probably causes the chemotherapeutics less efficient and side effects due to the lack of targeting.
 |
Figure 6 Inhibitory effects of PTX formulations on the growth of SMMC-7721 tumor spheroids (n = 3). Note: ***P < 0.001. |
The 3D spheroid model of SMMC-7721 cells was established to evaluate the penetrating ability of FITC-labeled proteins by confocal microscopy. As shown in Figure 7, FITC-tLyP-1-HFtn displayed stronger fluorescence and deeper penetration ability than free FITC and FITC-HFtn, indicating that FITC-tLyP-1-HFtn not only increased uptake by cells but also effectively enhanced the tumor penetration ability.
 |
Figure 7 Penetration ability of free FITC, FITC-HFtn and FITC-tLyP-1-HFtn into SMMC-7721 tumor spheroids imaged using confocal microscopy, with a 5 μm interval between consecutive slides. Original magnification, 200×. |
In vivo Imaging Study
In order to evaluate the biodistribution and tumor-targeting ability of the NPs in vivo, DiR-labeled HFtn and tLyP-1-HFtn NPs were given intravenously into MDA-MB-231 tumor-bearing mice through the tail veil. The time-dependent biodistribution of NPs was observed by NIRF imaging. As shown in Figure 8A, there was no fluorescence in tumor tissue for the free DiR group but a fraction in the liver. The fluorescence signal in tumor tissue appeared 4 h after injection of HFtn and tLyP-1-HFtn NPs and reached a maximum at 24 h. As shown in Figure 8B, the results obtained in vivo were further confirmed by the ex vivo fluorescence images of the tumors.
 |
Figure 8 (A) Fluorescence imaging of MDA-MB-231 tumor-bearing nude mice in vivo after intravenous injection of free DiR, DiR-loaded HFtn and tLyP-1-HFtn NPs. (B) Ex vivo fluorescence images of excised tissues at 24 h post injection. |
In vivo Antitumor Efficacy
To estimate the in vivo anti-tumor efficacy of tLyP-1-HFtn NPs, female BALB/c mice were applied and formulations of Taxol®, PTX-loaded HFtn and tLyP-1-HFtn NPs were administered at 4 mg/kg, 1 mg/kg and 1 mg/kg does of PTX, respectively, the other day for 20 days. The changes in tumor volumes and the body weights were monitored at a two-day interval for 20 days. As shown in Figure 9A, compared with the tumor volume (2.26 cm3) in PBS group, tumors in Taxol® and PTX-loaded HFtn exhibited a much lower increase in the volume to around 0.8 cm3. The tumor volume treated with tLyP-1-HFtn-PTX NPs showed the lowest, around 0.6 cm3. Compared with the body weight changes during the 20-day experimental period in the saline group, the body weight of mice in Taxol® group decreased significantly (P < 0.01) (Figure 9B). While the tLyP-1 peptide functionalized nanocarrier, the tLyP-1-HFtn NPs group exhibited almost no difference compared to the PBS control group, suggesting that we prepared tLyP-1-HFtn-PTX NPs could inhibit tumor growth without significant side effects on mice. The tumors in each group were dissected and it showed that the sizes of PTX formulations were significantly smaller than the control group, indicating a significant therapeutic effect in vivo (Figure 9C). Although the PTX doses of the HFtn-PTX and tLyP-1-HFtn-PTX formulations were 4 times less than that of the Taxol® group, the enhanced in vivo therapeutic efficacy of the two NPs can be ascribed to tumor-targeting efficiency. Importantly, tLyP-1-HFtn-PTX NPs displayed a better antitumor effect and lower systemic toxicity in the MDA-MB-231 xenografts mice. The functionalized penetrating peptide tLyP-1 may help the tLyP-1-HFtn-PTX NPs to achieve efficient tumor targeting, internalization and drug delivery into tumor tissues. Surprisingly, the specific binding peptide tLyP-1, the C end Rule motif R/KXXR/K, which was reported to work only at the C terminus showed enhanced tumor cell recognition and tumor tissue penetration by binding to MDA-MB-231 cells that overexpressed NRP-1 receptors.
 |
Figure 9 (A) Tumor growth trend chart of mice in different treatment groups (normalized to the initial sizes). (B) Changes of mouse weight in different groups. (C) Tumor weight of each group after 20 days treatment. Data are presented as the means ± SD (n = 6, **P < 0.01 vs PBS group). |
Conclusion
We have developed a dual-targeting nanoparticle delivery system decorated with a tumor homing and penetrating peptide tLyP-1, which was fused to the N-terminal of human H chain ferritin that has been reported to bind to tumor cells via TfR1. The tLyP-1-HFtn-PTX NPs were successfully prepared and characterized. Compared with HFtn-PTX NPs and Taxol, tLyP-1-HFtn-PTX NPs displayed stronger intracellular uptake efficiency and anti-invasion ability, which was due to the dual receptor-mediated endocytosis process. The NIRF imaging in vivo demonstrated higher tumor-targeting capacity than HFtn-PTX NPs. The BALB/c mice model also showed that tLyP-1-HFtn-PTX had lower systemic toxicity and higher therapeutic efficacy in vivo. The decreased cellular uptake of M-tLyP-1-HFtn demonstrated that the NRP-1-dependent internalization was invalid due to the mutation of the key residues in the tLyP-1 peptide sequence, which in turn verified the feasibility of the N-terminal fusion of tLyP-1 peptide to the HFtn nanocarrier. Therefore, we claimed that tLyP-1 peptide also worked when functionalized at the N terminal of protein nanocage like HFtn and showed better anticancer therapeutic efficacy.
Ethics Approval
All animal experiments in this research work complied with the Regulations on the Administration of Laboratory Animals and the relevant national laws and regulations, and were performed under the guidance approved by the Laboratory Animal Ethics Committee at School of Medicine, Southeast University (Nanjing, China).
Acknowledgments
The work was supported by the National Key Research and Development Program (2016YFD0600801), the Natural Science Foundation of Jiangsu Province (BK20181401) and the National Natural Science Foundation of China (31200564).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Liang D-S, Su H-T, Liu Y-J, Wang A-T, Qi X-R. Tumor-specific penetrating peptides-functionalized hyaluronic acid- d -α-tocopheryl succinate based nanoparticles for multi-task delivery to invasive cancers. Biomaterials. 2015;71:11–23. doi:10.1016/j.biomaterials.2015.08.035
2. Kim J-H, Kim Y-S, Kim S, et al. Hydrophobically modified glycol chitosan nanoparticles as carriers for paclitaxel. J Controlled Release. 2006;111(1–2):228–234. doi:10.1016/j.jconrel.2005.12.013
3. Kingston DGI. The chemistry of taxol. Pharmacol Ther. 1991;52(1):1–34. doi:10.1016/0163-7258(91)90085-Z
4. Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37(13):1590–1598.
5. Meyers JD, Doane TL, Burda C, Basilion JP. Nanoparticles for imaging and treating brain cancer. Nanomedicine. 2013;8(1):123–143.
6. Li Y, Song K, Cao Y, Peng C, Yang G. Keratin-Templated Synthesis of Metallic Oxide Nanoparticles as MRI Contrast Agents and Drug Carriers. ACS Appl Mater Interfaces. 2018;10(31):26039–26045.
7. Miraftab R, Xiao H. Feasibility and Potential of Graphene and Its Hybrids with Cellulose as Drug Carriers: a Commentary. J Bioresour Bioprod. 2019;4(4):200–201.
8. Wu Y, Zeng Q, Qi Z, Deng T, Liu F. Recent Progresses in Cancer Nanotherapeutics Design Using Artemisinins as Free Radical Precursors. Front Chem. 2020;8:472.
9. Lin C, Zhang X, Chen H, et al. Dual-ligand modified liposomes provide effective local targeted delivery of lung-cancer drug by antibody and tumor lineage-homing cell-penetrating peptide. Drug Deliv. 2018;25(1):256–266.
10. Tian X, Zhang L, Yang M, et al. Functional magnetic hybrid nanomaterials for biomedical diagnosis and treatment.. Wiley interdisciplinary Reviews. Nanomedicine and Nanobiotechnology. 2018;10(1):1. doi:10.1002/wnan.1476
11. Khoshnejad M, Parhiz H, Shuvaev VV, Dmochowski IJ, Muzykantov VR. Ferritin-based drug delivery systems: hybrid nanocarriers for vascular immunotargeting. J Controlled Release. 2018;282:13–24. doi:10.1016/j.jconrel.2018.02.042
12. Zhang Y, Ardejani MS, Orner BP. Design and applications of protein-cage-based nanomaterials. Chem Asian J. 2016;11(20):2814–2828. doi:10.1002/asia.201600769
13. Yang R, Zhou Z, Sun G, et al. Synthesis of homogeneous protein-stabilized rutin nanodispersions by reversible assembly of soybean (Glycine max) seed ferritin. RSC Adv. 2015;5(40):31533–31540. doi:10.1039/C5RA03542B
14. Falvo E, Tremante E, Arcovito A, et al. Improved doxorubicin encapsulation and pharmacokinetics of ferritin–fusion protein nanocarriers bearing proline, serine, and alanine elements. Biomacromolecules. 2016;17(2):514–522. doi:10.1021/acs.biomac.5b01446
15. He J, Fan K, Yan X. Ferritin drug carrier (FDC) for tumor targeting therapy. J Controlled Release. 2019;311–312:288–300. doi:10.1016/j.jconrel.2019.09.002
16. Nakamura Y, Mochida A, Choyke PL, Kobayashi H. Nanodrug Delivery: is the enhanced permeability and retention effect sufficient for curing cancer? Bioconjugate Chem. 2016;27(10):2225–2238. doi:10.1021/acs.bioconjchem.6b00437
17. Cui Y, Xu Q, Chow PK-H, Wang D, Wang C-H. Transferrin-conjugated magnetic silica PLGA nanoparticles loaded with doxorubicin and paclitaxel for brain glioma treatment. Biomaterials. 2013;34(33):8511–8520. doi:10.1016/j.biomaterials.2013.07.075
18. Du B, Jia S, Wang Q, et al. A self-targeting, dual ROS/ph-responsive apoferritin nanocage for spatiotemporally controlled drug delivery to breast cancer. Biomacromolecules. 2018;19(3):1026–1036. doi:10.1021/acs.biomac.8b00012
19. Wang X, Li S, Shi Y, et al. The development of site-specific drug delivery nanocarriers based on receptor mediation. J Controlled Release. 2014;193:139–153. doi:10.1016/j.jconrel.2014.05.028
20. Khawar IA, Kim JH, Kuh H-J. Improving drug delivery to solid tumors: priming the tumor microenvironment. J Controlled Release. 2015;201:78–89. doi:10.1016/j.jconrel.2014.12.018
21. Heldin C-H, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure – an obstacle in cancer therapy. Nat Rev Cancer. 2004;4(10):806–813. doi:10.1038/nrc1456
22. Li J, Xing R, Bai S, Yan X. Recent advances of self-assembling peptide-based hydrogels for biomedical applications. Soft Matter. 2019;15(8):1704–1715. doi:10.1039/C8SM02573H
23. Tesauro D, Accardo A, Diaferia C, et al. Peptide-based drug-delivery systems in biotechnological applications: recent advances and perspectives. Molecules. 2019;24(2):2. doi:10.3390/molecules24020351
24. Jelonek K, Zajdel A, Wilczok A, et al. Dual-targeted biodegradable micelles for anticancer drug delivery. Mater Lett. 2019;241:187–189. doi:10.1016/j.matlet.2019.01.081
25. Daglioglu C. Environmentally responsive dual-targeting nanoparticles: improving drug accumulation in cancer cells as a way of preventing anticancer drug efflux. J Pharm Sci. 2018;107(3):934–941. doi:10.1016/j.xphs.2017.10.029
26. Qiao Y, Wan J, Zhou L, et al. Stimuli-responsive nanotherapeutics for precision drug delivery and cancer therapy.. Wiley interdisciplinary Reviews. Nanomedicine and Nanobiotechnology. 2019;11(1):1. doi:10.1002/wnan.1527
27. Cheng Y, Sun C, Liu R, et al. A multifunctional peptide-conjugated AIEgen for efficient and sequential targeted gene delivery into the nucleus. Angew Chem Int Ed. 2019;58(15):5049–5053. doi:10.1002/anie.201901527
28. Silva S, Almeida AJ, Vale N. Combination of cell-penetrating peptides with nanoparticles for therapeutic application: a review. Biomolecules. 2019;9(1):1. doi:10.3390/biom9010022
29. Zhao P, Xu Q, Tao J, et al. Near infrared quantum dots in biomedical applications: current status and future perspective. Nanomed Nanobiotechnology. 2018;10(3):e1483. doi:10.1002/wnan.1483
30. Teesalu T, Sugahara KN, Kotamraju VR, Ruoslahti E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc Natl Acad Sci U S A. 2009;106(38):16157–16162.
31. Wang J, Lei Y, Xie C, et al. Retro-inverso CendR peptide-mediated polyethyleneimine for intracranial glioblastoma-targeting gene therapy. Bioconjugate Chem. 2014;25(2):414–423.
32. Teijeiro-Valino C, Novoa-Carballal R, Borrajo E, et al. A multifunctional drug nanocarrier for efficient anticancer therapy. J Controlled Release. 2019;294:154–164.
33. Ruoslahti E. Tumor penetrating peptides for improved drug delivery. Adv Drug Delivery Rev. 2017;110:3–12.
34. Liang M, Fan K, Zhou M, Duan D, Yan X. H-ferritin-nanocaged doxorubicin nanoparticles specifically target and kill tumors with a single-dose injection. Proc Natl Acad Sci U S A. 2014;111(41):14900–14905.
35. Fan K, Jia X, Zhou M, et al. Ferritin nanocarrier traverses the blood brain barrier and kills glioma. ACS Nano. 2018;12(5):4105–4115.
36. Pandolfi L, Bellini M, Vanna R, et al. H-ferritin enriches the curcumin uptake and improves the therapeutic efficacy in triple negative breast cancer cells. Biomacromolecules. 2017;18(10):3318–3330.
37. Fan K, Cao C, Pan Y, et al. Magnetoferritin nanoparticles for targeting and visualizing tumour tissues. Nat Nanotechnol. 2012;7(7):459–464.
38. Li L, Fang CJ, Ryan JC, Niemi EC, Seaman WE. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc Natl Acad Sci U S A. 2010;107(8):3505–3510.
39. Li R, Ma Y, Dong Y, et al. Novel paclitaxel-loaded nanoparticles based on human h chain ferritin for tumor-targeted delivery. ACS Biomater Sci Eng. 2019;5(12):6645–6654.
40. Zhang Y, Zhou J, Ardejani M, Li X, Wang F, Orner B. Designability of aromatic interaction networks at e. coli bacterioferritin B-Type Channels. Molecules. 2017;22(12):2184.
41. Crich SG, Cadenazzi M, Lanzardo S, et al. Targeting ferritin receptors for the selective delivery of imaging and therapeutic agents to breast cancer cells. Nanoscale. 2015;7(15):6527–6533.
42. Song H, Zhang J, Wang W, et al. Acid-responsive PEGylated doxorubicin prodrug nanoparticles for neuropilin-1 receptor-mediated targeted drug delivery. Colloids Surf B. 2015;136:365–374.
43. Zhang Y, Liu P, Jiang Y, et al. High expression of neuropilin-1 associates with unfavorable clinicopathological features in hepatocellular carcinoma. Pathol Oncol Res. 2016;22(2):367–375.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.