Back to Journals » OncoTargets and Therapy » Volume 15
Therapeutic Targeting of FLT3 in Acute Myeloid Leukemia: Current Status and Novel Approaches
Received 10 September 2022
Accepted for publication 19 November 2022
Published 30 November 2022 Volume 2022:15 Pages 1449—1478
DOI https://doi.org/10.2147/OTT.S384293
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Melisa Tecik,1 Aysun Adan2
1Bioengineering Program, Graduate School of Engineering and Science, Abdullah Gul University, Kayseri, Turkey; 2Department of Molecular Biology and Genetics, Faculty of Life and Natural Sciences, Abdullah Gul University, Kayseri, Turkey
Correspondence: Aysun Adan, Department of Molecular Biology and Genetics, Faculty of Life and Natural Sciences, Abdullah Gul University, Kayseri, Turkey, Email [email protected]
Abstract: FMS-like tyrosine kinase 3 (FLT3) is mutated in approximately 30% of acute myeloid leukemia (AML) patients. The presence of FLT3-ITD (internal tandem duplication, 20– 25%) mutation and, to a lesser extent, FLT3-TKD (tyrosine kinase domain, 5– 10%) mutation is associated with poorer diagnosis and therapy response since the leukemic cells become hyperproliferative and resistant to apoptosis after continuous activation of FLT3 signaling. Targeting FLT3 has been the focus of many pre-clinical and clinical studies. Hence, many small-molecule FLT3 inhibitors (FLT3is) have been developed, some of which are approved such as midostaurin and gilteritinib to be used in different clinical settings, either in combination with chemotherapy or alone. However, many questions regarding the best treatment strategy remain to be answered. On the other hand, various FLT3-dependent and -independent resistance mechanisms could be evolved during FLT3i therapy which limit their clinical impact. Therefore, identifying molecular mechanisms of resistance and developing novel strategies to overcome this obstacle is a current interest in the field. In this review, recent studies of approved FLT3i and knowledge about major resistance mechanisms of clinically approved FLT3i’s will be discussed together with novel treatment approaches such as designing novel FLT3i and dual FLT3i and combination strategies including approved FLT3i plus small-molecule agents targeting altered molecules in the resistant cells to abrogate resistance. Moreover, how to choose an appropriate FLT3i for the patients will be summarized based on what is currently known from available clinical data. In addition, strategies beyond FLT3i’s including immunotherapeutics, small-molecule FLT3 degraders, and flavonoids will be summarized to highlight potential alternatives in FLT3-mutated AML therapy.
Keywords: AML, FLT3-ITD, FLT3 inhibitor, FLT3i resistance, targeted therapy, flavonoid
Introduction
Acute myeloid leukemia (AML) is an aggressive disease characterized by the accumulation of abnormal hematopoietic precursors, which are overproliferative with blocked differentiation and suppressed apoptosis in the bone marrow and peripheral blood.1 Although AML is genotypically and phenotypically heterogeneous, various chromosomal abnormalities and gene mutations have been identified, which are crucial to determine AML classification, risk groups, and treatment strategies.2
FMS-like tyrosine kinase 3 (FLT3) gene encodes for a receptor tyrosine kinase (RTK), which is mainly expressed on immature hematopoietic progenitors and hematopoietic stem cells (HSCs). Its expression is reduced when the cells complete the differentiation process.3 FLT3 signaling is initiated when FLT3 ligand (FLT3 L) binds to FLT3, inducing FLT3 dimerization and activation via autophosphorylation at tyrosine residues. PI3K/AKT, MAPK, and JAK2/STAT5 are the activated downstream signaling pathways, which lead to cell proliferation and suppression of apoptosis.4
Activating mutations in FLT3 account for 30% of all AML cases, which are FLT3 internal tandem duplication (ITD) and FLT3 tyrosine kinase domain (TKD) mutations. FLT3-ITD is observed in 20–25% of newly diagnosed AML cases while FLT3-TKD mutations represent 5–10% of all cases.5 FLT3 receptor is continuously activated as a result of these mutations irrespective of the presence of FLT3 L, leading to increased cell proliferation and decreased cell apoptosis.1,5 In addition to PI3K/AKT and MAPK signaling pathways, STAT5 pathway is found to be continuously activated in the presence of FLT3-ITD.6 Clinical impacts of FLT3-ITD mutations are associated with higher relapse rate, decreased overall survival (OS) rate, poorer treatment response and shorter disease-free survival (DFS) compared to patients with wild-type FLT3 (WT-FLT3) while adverse clinical outcomes of FLT3-TKD mutations are controversial.7
Identifying the roles of FLT3 mutations in disease pathogenesis and clinical outcomes has made it a therapeutic target, resulting in the development of FLT3i with different specificity and potency.8 Although some of these inhibitors, midostaurin and gilteritinib, have been clinically used in combination therapies or alone, respectively, for FLT3-mutated AML, responses are short-lived and patients relapse due to the emergence of resistance.9 Therefore, enlightening primary or secondary resistance mechanisms and designing novel modalities to overcome resistance are urgently needed to maximize the benefits of FLT3i. In addition to FLT3i, novel targeted therapies are currently at the stage of pre-clinical and early clinical investigation, which include novel FLT3i, dual FLT3i, FLT3 targeted CAR T cell therapy and FLT3-specific antibodies.10 Additionally, novel immunotargets have been identified with therapeutic potential. Moreover, there are studies investigating the effects of small-molecule FLT3 degraders such as HSP90 and proteasome inhibitors on FLT3 positive AML. Activities of flavonoids or their synthetic analogs on FLT3 positive AML could lead to the discovery or development of novel FLT3i or to their implementation as integrative medicine or nutraceuticals into FLT3 AML therapy.11 It seems to be the most rational strategy to combine approved FLT3i with the modulators of altered intracellular targets, resulting in the discovery of novel targets and therapeutics.
In this review, we aim to expand on the body of the literature in the management of FLT3 AML and provide an update on newly reported knowledge by specifically focusing on targeted FLT3 therapies including clinically approved FLT3is, novel FLT3i, combination approaches, immunotherapeutics, small-molecule FLT3 degraders and flavonoids. Additionally, mechanisms observed in clinical or experimental resistance to approved FLT3is will be discussed together with potential solutions to reverse the resistant phenotype.
FLT3 Structure
The FLT3 gene, encoding FMS-like tyrosine kinase 3 transmembrane receptor, is located on chromosome 13q12, containing 24 exons and 993 amino acid residues.5,12,13 FLT3, also known as fetal liver kinase 2 or human stem cell kinase-1, belongs to the type III RTK family, which also includes FMS, KIT, and PDGFR kinases that share strong sequence similarities.13 FLT3 receptor is mainly expressed on HSCs, multipotent progenitors, common myeloid and lymphoid progenitor cells, and mature dendritic cells.5,14 The expression of FLT3 is lost or reduced once the cells differentiate into mature lymphoid or myeloid cells.15–17
After translation of the receptor as a 110 KDa protein, it is sent to the endoplasmic reticulum (ER) to be transformed into 130 KDa N-glycosylated immature protein that is rich in mannose. Subsequently, it is further processed in the Golgi apparatus (GA) to become a mature 160 KDa protein which will be then directed to the cell surface.18,19 The final form of FLT3 consists of 4 different domains: an extracellular domain containing 5 immunoglobulin-like subdomains, a transmembrane domain, an intracellular juxtamembrane (JM) domain, and an intracellular C-terminal domain, comprising 2 tyrosine kinase subdomains; tyrosine kinase domain 1 and 2 (TKD1 and TKD2) connected by an activation loop (A-loop) (Figure 1).5
 |
Figure 1 FLT3 structure and common FLT3 mutations.1,42 Notes: Data from the references: ©2012 Grafone et al, Licensee PAGE Press, Italy. Grafone T, Palmisano M, Nicci C, Storti S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: biology and treatment. Oncol Rev. 2012;6(1):e8-e8 under Creative Commons CC BY-NC 3.0.1 ©2018 Staudt et al, Licensee MDPI, Basel, Switzerland. Staudt D, Murray HC, McLachlan T, et al, Targeting oncogenic signaling in mutant FLT3 acute myeloid leukemia: the path to least resistance. International Journal of Molecular Sciences. 2018;19(10):3198 under Creative Commons CC BY 4.0.42 |
FLT3 L is an extracellular ligand produced by a wide range of cells including lymphocytes, HSCs, and bone marrow stromal cells.1,14,20 FLT3 L is found as membrane-bound or in soluble form.20 The concentration of soluble FLT3 L is generally low, however, can increase exponentially due to aplasia causing only necessary activation of FLT3 via the negative-feedback mechanism.21 Binding of FLT3 L to the extracellular domain of FLT3 causes structural changes including dimerization of the monomeric receptor. The JM domain has an inhibitory function on the kinase domain. Upon binding, the JM domain changes conformation to make the kinase domain accessible for ATP binding, which eventually leads to autophosphorylation of several tyrosine residues and activation of the receptor.12,22,23 Activation of the receptor further activates the downstream signaling pathways such as PI3K and RAS cascades, resulting in hematopoietic cell maturation and proliferation.23 Thus, the function of FLT3 L is to act as a growth factor to stimulate myelopoiesis.24
FLT3 Mutations
FLT3 mutations are the most frequently identified mutations in AML patients.25 Of all cases in AML, FLT3-ITD and FLT3-TKD account for approximately 20–25% and 5–10%, respectively.5 The receptor, which stays in a monomeric form in WT-FLT3, becomes dimerized independent of the FLT3 L binding in mutated FLT3.5,26 Therefore, mutations in the receptor cause activation of the tyrosine kinase even in the absence of the ligand resulting in aberrant proliferation of the malignant cells.26
FLT3-ITD mutations are in frame gain-of-function mutations occurring in the JM domain which, in fact, is responsible for the inhibition of the receptor activation through the inhibition of the kinase domain; therefore, the mutation constitutively activates the TKD action.27–29 Patients with FLT3-ITD mutations show increased relapse and decreased OS.27 FLT3-ITD occurs as a duplication of a fragment that varies in length and position. The length of the fragment is evidently negatively correlated with the OS rate.30,31 FLT3-TKD mutations are generally single amino acid mutations such as substitution, deletion, and insertion in the A-loop of the TKD, causing loss of auto-inhibition.21,32,33 The most common point mutations in FLT3-TKD are substitution of aspartic acid of TKD2 with tyrosine or histidine at residue 835 and substitution of asparagine or phenylalanine of TKD1 at residues of 676 and 691, respectively (Figure 1).32,33
FLT3 Signaling
In WT-FLT3, several signaling pathways are activated to regulate the proliferation, differentiation, and apoptosis of the HSC.20 Upon binding of FLT3 L, trans-autophosphorylation of tyrosine residues in FLT3 is followed by binding of adaptor proteins including SHP2, GRB2, and SRC family kinases, leading to activation of mainly PI3K/Akt/mTOR and RAS/MEK/ERK pathways.34–38 FLT3-ITD and FLT3-TKD activate similar pathways with WT-FLT3. However, FLT3-ITD also induces JAK/STAT signaling through STAT5A phosphorylation.39 FLT3-TKD shows increased activation of SHP1 and SHP2, of which SHP1 is a negative reGulatory phosphatase for the JAK signaling pathway.40,41 Therefore, only low levels of JAK2 and STAT3 activation are observed in FLT3-TKD.42 Moreover, FLT3-ITD mutations can decrease the expression of c/EBPalpha and PU.1 which are associated with the differentiation of myeloid cells.39 In contrast, FLT3-TKD mutations do not suppress the c/EBPalpha and PU.1.39,43 (Figure 2).
 |
Figure 2 FLT3-ITD signal transduction.1,9 Notes: Data from the references: ©2012 Grafone et al, Licensee PAGE Press, Italy. Grafone T, Palmisano M, Nicci C, Storti S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: biology and treatment. Oncol Rev. 2012;6(1):e8-e8 under Creative Commons CC BY-NC 3.0.1 ©2017 Hospital et al, Licensee Dove Medical Press Limited. Hospital MA, Green AS, Maciel TT, et al, FLT3 inhibitors: clinical potential in acute myeloid leukemia. Onco Targets Ther. 2017;10:607-615 under Creative Commons CC BY-NC 3.0.9 |
Leukemogenesis in FLT3 Positive AML
The two-hit model of leukemogenesis suggests that class I and class II mutations must occur together. Class I mutations activate the proliferative pathways and class II mutations impair hematopoietic differentiation in AML.44,45 Along with K/NRAS, TP53, and c-KIT mutations, FLT3 mutations are classified as class I mutations. It is still controversial whether an FLT3 mutation is a passenger or driver mutation even though it seems to be an early event in disease development.46,47 Isolated FLT3-ITD mutations, when injected into transgenic mice were not sufficient to induce leukemia, suggesting that FLT3 mutation is a passenger mutation.48 However, the high incidence rate of FLT3 mutations and emergence of additional mutations along with FLT3 leading to FLT3i resistance suggests that FLT3 mutations are driver mutations.49–51 75% of the FLT3-ITD positive patients still have FLT3-ITD mutation after relapse, suggesting that FLT3 mutation is a driver mutation52 and the FLT3 mutation induced shifting from the pre-leukemic state to leukemia.53 Several important genes including MYC, GAB2, and DNMT3A have been identified to cooperate with FLT3 in promoting leukemogenesis.
MYC family genes are regulated by upstream FLT3-ITD signaling and contribute to leukemogenesis. In an FLT3-ITD mouse model, simultaneous upregulation of MYC genes and downregulation of the MYC antagonists, the MXD genes was observed, which could explain the expansion of leukemic progenitors.54 c-MYC reportedly increased the expression and stability of SIRT1 deacetylase in FLT3-ITD AML, resulting in the maintenance of leukemic stem cells (LSC).55 Telomerase reverse transcriptase (hTERT) upregulated in FLT3-ITD AML cells in a c-MYC dependent manner and inhibition of FLT3-ITD caused transcriptional repression of c-MYC.56 FLT3-ITD induced c-MYC, which increased the transcription of alternative nonhomologous end-joining (NHEJ) pathway, leading to genomic instability.57
Gab2, an amplifier protein in signaling pathways, is identified as an essential signaling molecule for leukemic transformation in FLT3-ITD AML. Bone marrow cells from Gab2-deficient and -proficient or -haploinsufficient mice were transfected with FLT3-ITD. FLT3-ITD infected cells survived when Gab2 is functional; however, FLT3-ITD infected cells could not transform in the absence of Gab2.58 In a recent study, Gab2 deficiency was shown to prevent FLT3-ITD AML development in an FLT3-ITD knock-in, DNMT3A haploinsufficient mouse model.59
FLT3-ITD knock-in and DNMT3A (a DNA methyltransferase) knock-out mice model had reduced disease onset and shortened survival as compared to either mutation alone, proving the role of DNMT3A mutations in driving leukemogenesis via enhancing self-renewal capacity of long-term-HSCs (LC-HSCs). The loss of a single allele of DNMT3A was sufficient to shorten survival and induce leukemia development.60
Prognostic and Clinical Impact of FLT3 Mutations
AML patients with an FLT3 mutation have poor prognosis compared to patients with WT-FLT3. The response rates of AML patients with or without an FLT3 mutation are similar; however, patients with an FLT3 mutation are more likely to experience relapse.61,62 Even when FLT3 mutation is not detected, the receptor can still be overexpressed, resulting in the survival and proliferation of the leukemic cell.13,24 Furthermore, chemotherapy can induce aplasia and stimulate FLT3 L production which eventually leads to recovery and expansion of the AML cells.24
The association of the FLT3-TKD mutation with the prognosis of AML is still not clear, although FLT3-TKD mutation increases the kinase activity.2,32,33 On the other hand, FLT3-ITD mutation alone is an adverse prognostic marker. The presence of FLT3-ITD is associated with high leukemic burden, poor OS, and early relapse.21,63 The prognostic impact of FLT3-ITD depends on the mutant-to-WT allelic ratio (AR), mutation insertion site, the length of the ITD duplication, karyotype, and the co-existing mutations.2,64–66 While FLT3 mutations contribute to the uncontrolled proliferation of leukemic cells, other mutations such as DNMT3A, NPM1, and IDH1/2 cause impaired differentiation and self-renewal.5,67,68
One of the most common co-mutations in FLT3-ITD AML occurs in DNMT3A. The presence of a DNMT3A co-mutation was required to consider FLT3-ITD as an adverse prognostic indicator. There was no significant difference in OS between FLT3-ITD positive and FLT3-ITD negative patients in the absence of DNMT3A co-mutation. Patients with FLT3-ITD and DNMT3A mutation had shorter OS compared to patients with either FLT3 mutation or DNMT3A mutation. After post-induction therapy, the patients with DNMT3A and FLT3-ITD co-mutation showed the highest rate of relapse.69
The nucleophosmin 1 (NPM1) gene encodes a multi-functional protein and is mostly mutated in FLT3-ITD AML. NPM1 mutations could be useful to determine the prognosis, which is dependent on the presence or absence of FLT3-ITD and FLT3-ITD AR.70 Based on the risk stratification organized by the European LeukemiaNet (ELN) and the National Comprehensive Cancer Network (NCCN), the presence of WT-NPM1 and FLT3-ITD mutation is considered as adverse risk. High FLT3-ITD AR and co-existing NPM1 mutation are classified as intermediate risk while low FLT3-ITD AR and NPM1 mutation are categorized as favorable.65,71 The effects of age and gender on NPM1 and FLT3-ITD mutations were investigated in 1570 patients younger than 75 years old. Females had more FLT3-ITD and/or NPM1 mutations compared to males. More males were double negative. FLT3-ITD caused poor survival in younger patients while NPM1 was related to good survival in older patients. FLT3-ITD/NPM1 double mutant patients’ survival was less related to age.72 In a study, patients having FLT3-ITD, DNMT3A and NPM1 triple mutations were significantly younger than patients having single or double mutations. Most of the triple-mutated patients were women having heavy disease burden and high white blood cell and bone marrow blast counts. Patients carrying triple gene mutations had the shortest OS followed by DNMT3A alone, FLT3-ITD/DNMT3A double mutation. The response to treatment was the best in patients with DNMT3A/NPM1 double mutation. Patients having triple mutation and FLT3-ITD alone showed no response to treatment. However, patients with either NPM1 mutation alone or FLT3-ITD alone had longer OS.73
Other co-mutations such as IDH1/2, CEBPA, and ASXL1 could affect the prognosis of AML patients. The prognostic analysis of isocitrate dehydrogenase (IDH1 and IDH2) mutations in newly diagnosed FLT3-ITD AML patients showed that double-mutated patients had higher white blood cell counts, increased peripheral and bone marrow blast percentages, a higher frequency of NPM1 mutations and a lower frequency of DNMT3A. There was no significant difference in OS between double-mutated and FLT3-ITD patients.74 In a recent study, CR did not differ in registered AML patients regardless of their molecular abnormalities including FLT3-ITD.75
AML Patients with additional Sex Comb-like 1 (ASXL1) and FLT3-ITD mutations had poor prognosis with a shorter OS, EFS,76 and RFS in Egyptian AML patients.77
The prognostic effect of CCAAT/enhancer binding protein A (CEBPA) involved in the proliferation and differentiation of myeloid progenitor cells depends on whether patients carry double or single mutated CEBPA. Single mutated CEBPA was seen more commonly in FLT3-ITD mutated AML patients although relatively less percentages of double mutated CEBPA were also observed.78 The effect of CEBPA mutation in patients with R/R FLT3-ITD-positive AML treated with quizartinib or SC was analyzed and CEBPA mutations were associated with high CRc rates and relatively long median OS, regardless of the treatment protocol.79
The prognostic significance of mutations in R/R FLT3-ITD AML was evaluated compared to the mutational status of newly diagnosed FLT3-ITD AML patients. The frequency of FLT3-ITD mutation increased while that of CEBPA biallelic (double) mutation decreased. NPM1, FLT3-ITD, and DNMT3A triple mutations were only found in the relapse group although their co-existence was detected in newly diagnosed, relapsed, and refractory patients. Refractory patients with NPM1 and FLT3-ITD co-mutation experienced shorter OS than the patients with FLT3-ITD mutation alone or WT-NPM1.80
Single, double and triple mutations of FLT3-ITD, NPM1, and DNMT3A were more prevalent in females. The allelic ratio of FLT3-ITD was not found to be different among the sexes. FLT3-ITD mutated female population had significantly poor outcomes. FLT3-ITD patients younger than 60 years had poor OS compared to older patients. Considering the sex and age, the female and younger population had poor OS, while there was not any significant difference in OS between the young and old male patients.81
In a recently published study, 2017 ELN risk classification has been revisited based on the current understanding of the roles of molecular targets in AML. The updated version included AML with FLT3-ITD in the intermediate risk group, irrespective of the AR or co-presence of NPM1 mutation. The absence of FLT3-ITD with WT NPM1 and the presence of mutated NPM1 with FLT3-ITD are categorized as the intermediate risk group.82
In conclusion, deciphering the mutation spectrum of FLT3-ITD AML could lead to an in-depth understanding of the pathogenesis and refine the prognostic classification of the disease. It is also possible to follow up the treatment response of the patients based on the mutational analysis, which could help reorganizing the treatment in case of relapse.
Clinically Approved FLT3 Inhibitors in Therapy
FLT3 has become an attractive target in AML given the correlations between its mutated forms and disease development, poor prognosis, high mortality rates, and insufficient therapy response.23 FLT3is have been the focus of small molecule drugs against FLT3 mutations some of which have been approved for clinical settings with favorable outcomes.30 FLT3 is are tyrosine kinase inhibitors (TKI) categorized as first- and next-generation inhibitors based on their specific ability to inhibit FLT3 and associated downstream signaling pathways.23,30 Apart from this broad classification, both first and next-generation inhibitors could be either type I or type II inhibitors in relation to their effectiveness against both FLT3-ITD and TKD mutations or only FLT3-ITD mutation, respectively.23,30,83 Type I inhibitors can bind to both active and inactive conformations of the FLT3 receptor since they bind to the FLT3 gatekeeper domain or the ATP binding pocket. Type II inhibitors bind adjacent to ATP binding domain located in the hydrophobic region when the receptor is in its inactive conformation.83 In this review, we have specifically focused on the FLT3is approved for the clinical settings (Table 1) and we will summarize the clinical trials of these FLT3is involved in novel combination studies (Table 2).
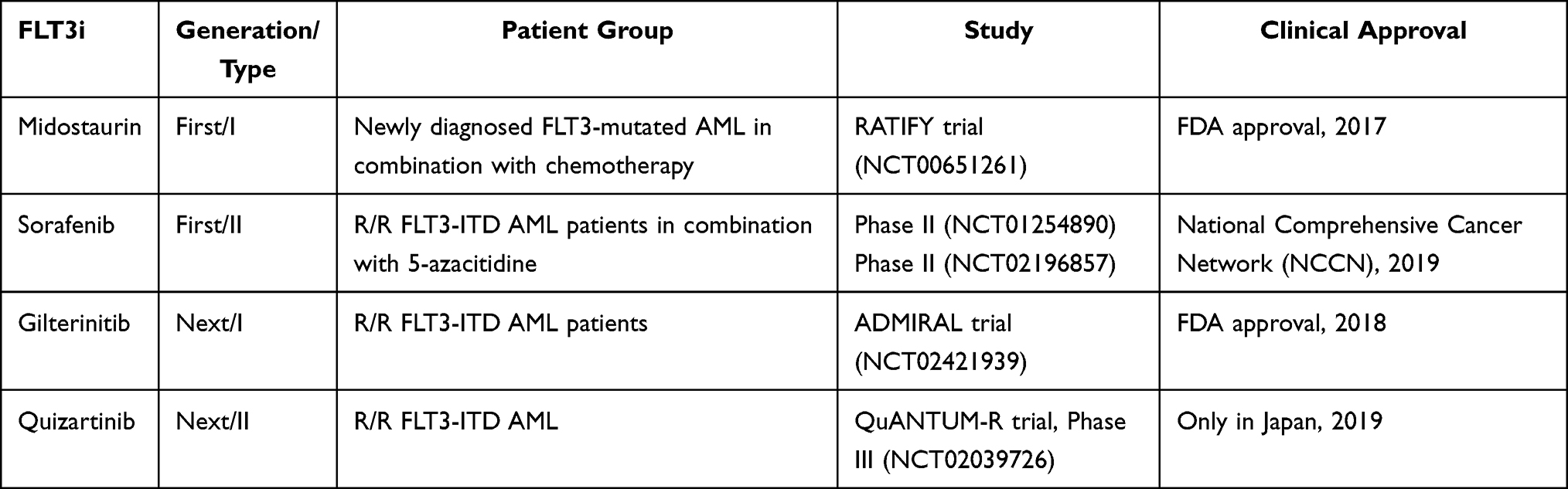 |
Table 1 Summary of the Clinical Trials Leading to Approval of FLT3 Inhibitors |
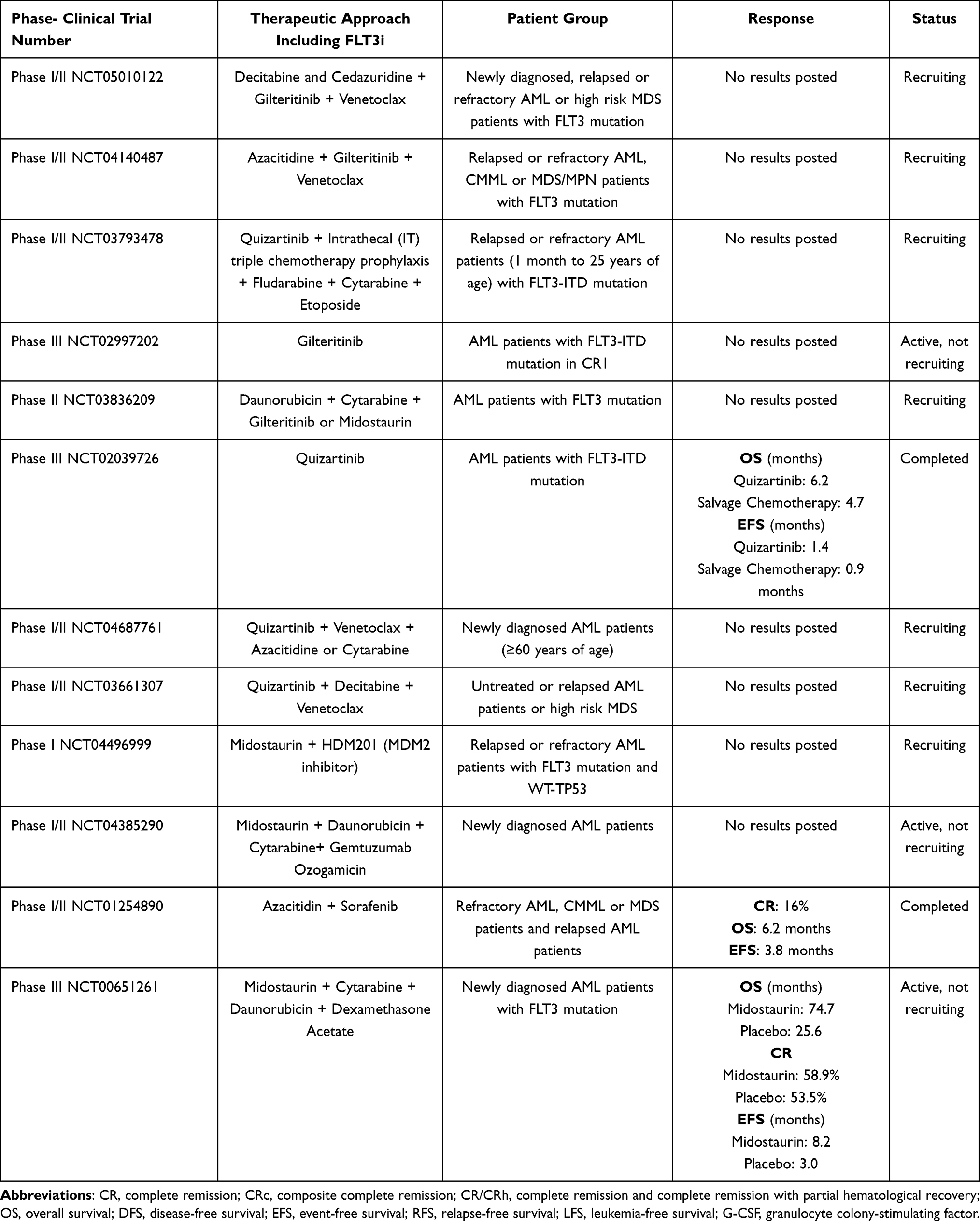 |
Table 2 Selected Clinical Trials of Approved FLT3i in Different Therapeutic Settings |
First-Generation FLT3 Inhibitors
The first-generation inhibitors show non-specific activity for FLT3 and inhibit other kinases such as KIT, PDGFR, VEGFR, RAS/RAF/MEK, and JAK kinases, hence having more off-target effects.23,30 Off-target activities could result in decreased clinical efficacy in FLT3-mutated AML with high allelic burden while resulting in increased toxicity profile and clinical benefit for WT-FLT3 AML.84 First-generation inhibitors include midostaurin and sorafenib, which are also type I and type II inhibitors, respectively.23,30,83,84
Midostaurin is active against both FLT3-ITD and TKD mutations and showed limited and transient activity in early clinical phase trials when used alone. In a clinical setting including relapsed/refractory (R/R) FLT3-mutated AML patients, midostaurin induced a 50% reduction in peripheral and bone marrow blasts.85 Another milestone study investigated the effects of midostaurin in AML patients carrying WT-FLT3 or mutated FLT3. Reduction in peripheral and bone marrow blasts was 71% in patients with mutant FLT3 and 42% in patients with WT-FLT3.86 Overall, midostaurin could not induce a complete remission (CR) and its efficacy remained limited to blast reduction due to activation of alternative survival-promoting pathways, protection of the leukemic clones in stem-cell niche and limited concentration of free midostaurin in patient’s plasma.85,86
However, its combination with several cytotoxic anti-leukemic agents including cytarabine, doxorubicin, azacitidine, or daunorubicin resulted in promising outcomes in in vitro models of FLT3 positive AML leading to its investigation in the clinic for combination therapy.87,88 Newly diagnosed younger patients with FLT3 positive AML were treated with 50 mg midostaurin/twice a day in combination with standard chemotherapy for 14 days, resulting in high CR and OS rates.47 The addition of midostaurin into standard care chemotherapy dramatically changed the course of the disease based on the results of RATIFY trial in which registered therapy-naive patients with FLT3 mutations (FLT3-ITD and FLT3-TKD) showed increased OS rates. The presence of either ITD or TKD and the ITD AR status was not distinctive factors on patients’ conditions. US Food and Drug Administration (FDA) approved midostaurin in 2017 to be used in combination with standard cytarabine and daunorubicin induction therapy and cytarabine consolidation therapy in newly diagnosed young (18–59 years) FLT3-mutated AML patients based on the findings of RATIFY trial.89 Further analysis from RATIFY trial investigated the suitability of midostaurin as a maintenance therapy while the patients were in the first CR after intensive cytarabine consolidation therapy and the analysis showed that the added effect of midostaurin during maintenance therapy was not conclusive even though overall relapses were reduced.90 The impact of midostaurin in patients having only FLT3-TKD mutations in RATIFY trial was significantly higher in terms of 5-year event-free survival (EFS) rate (45.2%) compared to the placebo arm (30.1%) while OS was similar.91
A recent study investigated the roles of midostaurin in patients’ survival who were initially treated with midostaurin plus intensive chemotherapy and then referred to allo-SCT in the first CR. Midostaurin therapy improved OS specifically in patients with high AR and only midostaurin therapy and allo-SCT in first CR were found as positive predictors for OS.92 The effect of midostaurin to prevent relapse in FLT3-ITD carrying patients (18–70 years old) subjected to allo-SCT showed that midostaurin plus standard chemotherapy could not improve relapse-free survival (RFS) (89%) significantly as compared to only chemotherapy arm (76%), concluding the addition of midostaurin as maintenance therapy following allo-SCT could be only beneficial for some patients with FLT3-ITD AML.93 Midostaurin with standard chemotherapy for older FLT3-ITD AML patients (18–70 years) was shown to be safely effective as induction therapy. Allo-SCT in the first CR after midostaurin plus chemotherapy was highly effective, irrespective of age. Maintenance with midostaurin could only be beneficial for some patients after high-dose consolidation chemotherapy or allo-SCT.94
Sorafenib is a first-generation, type II FLT3i whose safety and efficacy was established in newly diagnosed FLT3 mutant AML in combination with standard anthracycline/cytarabine induction therapy.95 The randomized SORAML trial in 2015 showed that sorafenib improved EFS and RFS and did not cause a change in OS in registered AML patients regardless of the FLT3 status compared to the placebo group.96 However, these results were updated in 2017 with increased OS.97 In a recent study, 99 patients with newly diagnosed FLT3-ITD AML were registered for sorafenib+intensive chemotherapy or placebo and EFS was not different between the two groups. However, the sorafenib group had improved OS.98 Two separate clinical trials including R/R FLT3-mutated AML patients and untreated older patients not fit for intensive chemotherapy showed the efficacy and safety of sorafenib in combination with hypomethylating agent (HMA) 5-azacitidine,99,100 which resulted in the inclusion of sorafenib+azacitidine for R/R FLT3-ITD AML patients’ treatment by the recommendation of NCCN.101 Sorafenib has been widely investigated in FLT3 mutant AML patients eligible for allo-SCT. Sorafenib increased OS as maintenance therapy after allo-SCT in FLT3 mutant AML patients.102 The SORMAIN study including 83 FLT3-ITD patients in CR after allo-SCT investigated the addition of sorafenib as maintenance therapy compared to the placebo group, resulting in a higher probability of 24-month OS in the sorafenib group.103 FLT3-ITD AML patients who underwent allo-SCT were divided randomly into sorafenib maintenance (400 mg orally twice daily) or control arms, concluding that sorafenib maintenance therapy decreased relapse.104
Next-Generation FLT3 Inhibitors
Next-generation FLT3 inhibitors are more specific and potent for FLT3 with fewer off-target effects compared to first-generation inhibitors. They could also have the capacity to induce myeloid differentiation and show greater clinical activity as monotherapeutic agents.24
Gilteritinib is a next-generation, type I FLT3i with more potency against mutated FLT3 among other TKIs even though it shows some activity for other kinases such as AXL.105 In the dose-escalation/expansion trial, 20–450 mg gilteritinib in an FLT3-mutated R/R AML patient population was studied and it was shown that its plasma concentration is higher since it is less bound to plasma protein and it inhibits the phosphorylation of FLT3 more than 85% at very low concentrations (CHRYSALIS trial).106 A landmark Phase III study called ADMIRAL investigated the effects of 120 mg gilteritinib in R/R FLT3-mutated AML patients compared to salvage chemotherapy (SC). The percentage of gilteritinib-treated patients who achieved CR with full or partial hematologic recovery (34%) was higher compared to the chemotherapy given group (15.3%) and OS was 9.3 months vs 5.6 months, respectively. The OS outcomes of ITD or TKD mutated patients were similar (9.3 vs 8 months, respectively).107 The results of this study were proof for the FDA approval of gilteritinib monotherapy in 2018 for R/R FLT3-mutated AML patients in the US. The long-term effects and safety of gilteritinib were analyzed for the patients enrolled and survived in the ADMIRAL trial, showing that 49 patients in the gilteritinib arm were alive for more than 2 years. Twenty-six patients treated with gilteritinib were alive without relapse; 18 gilteritinib given patients underwent transplantation and 16 patients were treated with gilteritinib as post-HSCT maintenance therapy. Thus, continued and post-HSCT gilteritinib maintenance therapy resulted in sustained remission with a stable safety profile.108 There are several ongoing clinical trials investigating the potential of gilteritinib versus placebo as maintenance therapy after consolidation (NCT02927262) or after allo-HCT in patients with FLT3-ITD mutations (NCT02997202). The combination of venetoclax and gilteritinib resulted in high mCRc (modified CRc) and FLT3 molecular response rates regardless of prior FLT3 inhibitor exposure. However, there is a need to determine proper doses to overcome myelosuppression.109 A randomized Phase II trial of gilteritinib versus midostaurin in combination with induction and consolidation chemotherapy is also currently recruiting (NCT03836209). The phase III LACEWING trial (NCT02752035) was designed to compare gilteritinib plus azacitidine with azacitidine alone in newly diagnosed older (65–86 years old) FLT3-mutated AML patients who were ineligible for intensive induction chemotherapy. The results showed that the gilteritinib plus azacitidine combination was safe for registered patients. CR rates were improved although there were no significant OS differences.110 However, this trial has been terminated as a result of interim analysis by an independent group of reviewers, which was due to the lack of a statistically significant increase in OS.111
Quizartinib is a next-generation, type II FLT3i active against FLT3 and, to a lesser extent (around 10-fold less), KIT, CSF1R, PDGFR, and RET kinases.112 Two sequential phase II studies including R/R FLT3-mutated AML patients treated with quizartinib monotherapy resulted in significant bone marrow remissions; however, adverse cardiac signals raised concerns about its safety even when used in lower concentrations.113,114 Recently, quizartinib monotherapy and SC were compared in a phase III study (QUANTUM-R, NCT02039726) with R/R FLT3-mutated AML patients. Quizartinib increased OS (6.2 months) as compared to SC (4.7 months).115 32% of patients in quizartinib arm could proceed to an allo-SCT (11% of patients in SC). 62% of the patients on the quizartinib arm who received allo-SCT received post-SCT quizartinib maintenance. Quizartinib is not approved by FDA in the US due to cardiac toxicities and strong myeloid suppression based on the results of QUANTUM-R; however, it is approved in Japan as monotherapy in R/R FLT3-ITD AML. A post hoc analysis of the patients on the quizartinib arm and SC therapy arm who referred to allo-SCT in QuANTUM-R showed that continuation of quizartinib after HSCT was tolerable, with no new safety signals.116 Patients with R/R FLT3 AML were treated with quizartinib plus azacitidine or low-dose cytarabine (NCT01892371). CRc rates were 64% with azacitidine and 29% with low-dose cytarabine, RFS 5.8 and 6.2 months, and OS 12.8 and 4 months, respectively. 28% of the patients from the quizartinib plus azacitidine arm underwent an allo-SCT compared to only 6% of patients from the quizartinib plus low-dose cytarabine arm.117 There is an ongoing phase I/II trial (NCT04687761) including the combination of low-dose cytarabine or azacitidine + venetoclax + quizartinib in newly diagnosed AML patients. Recently, the incorporation of quizartinib into 7+3 regime and continuation therapy of newly diagnosed FLT3-ITD AML patients aged up to 75 years old increased OS compared to those with SC.118
How to Choose an Approved FLT3i for the Patient
Clinical decisions on a specific FLT3i for a specific group of patients such as how to choose a proper concentration of FLT3i in combination approaches, and when to use FLT3i (as a frontline or maintenance therapy), before or allo-SCT should be carefully considered based on the presence of simultaneous myeloid neoplasm-related mutations, type of FLT3 mutation, FLT3-ITD insertion size and position, the use of prior FLT3i, the toxicity profile of FLT3i, patients’ age and overall health condition and eligibility for allo-SCT.83
There are recent studies investigating the impact of concurrent mutations, FLT3-ITD AR, and insertion size on the therapeutic outcome of FLT3i. An analysis of RATIFY study related to the prognostic impact of FLT3-ITD insertion site and the presence of NPM1 mutation highlighted that the presence of NPM1 mutation was correlated with the presence of insertion site in only JM domain. Insertion site in only TKD1 was evaluated as a negative prognostic factor. Midostaurin was effective for the patients carrying insertion site in only JM domain following allo-SCT in the first CR.119 Subsequent analysis of the patients with only FLT3-TKD mutations in RATIFY trial showed that the co-existence of NPM1 mutations or core binding factor (CBF)-rearrangements were considered as favorable prognostic factors in terms of midostaurin treatment.91 FLT3-ITD AR, ITD length, and the expression of hepatic leukemia factor (HLF) were checked to understand differential responses to FLT3i. High AR samples showed increased sensitivity compared to low AR samples while no association was found between ITD length and FLT3i response. RNA seq analysis displayed that there was a correlation between high AR and high HLF expression, which could determine FLT3i response.120 FLT3-ITD AR, FLT3-ITD length, or multiple FLT3-ITD mutations did not have any adverse effects on survival outcomes with gilteritinib; however, the presence of DNMT3A/NPM1 double mutations resulted in the most favorable outcomes in patients received gilteritinib.121
Prior FLT3i use or sequential FLT3i exposure could be a factor to decide which FLT3i therapy could be chosen. Sorafenib and midostaurin treated R/R FLT3-mutated AML patients involved in the CHRYSALIS and ADMIRAL trials were compared with those without prior FLT3i use after treated with gilteritinib. Similar high composite CR (CRc) rates were obtained irrespective of prior FLT3i use; however, remission duration was shorter in the prior FLT3i exposure group.122 The R/R FLT3-mutated patients previously treated with midostaurin plus intensive induction therapy showed 58% CR rate after gilteritinib treatment. However, the presence of NRAS, KRAS, and PTPN11 MAPK pathway activating mutations (known to cause gilteritinib resistance) lowered CRc and OS rates as compared to the patients without these mutations.123 In a retrospective study, 239 FLT3-mutated AML patients were exposed to sequential FLT3i and CRc rates dropped progressively and CRc rates were higher in the patients treated with sequential FLT3i exposure compared to those of FLT3i-monotherapy.124
The older or unfit adults who are not eligible for intensive chemotherapy have benefited from the combinations of FLT3i with low-intensity chemotherapy. Sorafenib or midostaurin plus azacitidine was found to be safe and feasible for those patients.99 It was shown that naive FLT3-mutated patients had the greatest benefits from the combination of midostaurin and 5-azacitidine.125 The recent study with the data from the previous clinical trial (NCT02993523) including the naive and ineligible patients (age ≥75 years and/or with comorbidities) treated with venetoclax plus azacitidine or placebo plus azacitidine groups analyzed the impact of FLT3 mutation on therapy outcomes. CRc rates were 67% and 36% and median OS was 12.5 and 8.6 months for FLT3-mutated patients in venetoclax plus azacitidine and azacitidine groups, respectively. Patients with FLT3 mutations and WT-FLT3 had similar outcomes when treated with venetoclax plus azacitidine.126 Low-dose cytarabine with or without quizartinib in older FLT3-mutated AML patients not fit for intensive chemotherapy had improved survival. Median OS was 13.7 months compared with 4.2 months with low-dose cytarabine alone.127 Based on the overall opinion from the mentioned studies, a new study analyzed the effects of triplet combination including HMA+venetoclax+FLT3i on older/unfit patients as frontline therapy, resulting in higher CR (67% vs 32% in triplet arm compared to doublet arm (HMA plus FLT3i).128 A phase II trial including 13 newly diagnosed patients (older than 60 years) or 12 R/R (older than 18 years) FLT3-mutated AML patients (without prior venetoclax exposure, some with prior FLT3i use and allo-SCT), decitabine was combined with venetoclax and an FLT3i (10 patients with gilteritinib, 10 sorafenib, and 5 midostaurin). This study revealed that even FLT3-mutated patients with prior FLT3i treatment achieved durable remissions and underwent allo-SCT consolidation after low-intensity triplet therapy.129 In conclusion, the combination of low-intensity therapy (±low dose HMA or chemotherapy±venetoclax) with FLT3i could be the best option for the patients with FLT3-mutated AML who are not eligible for intensive chemotherapy even though the combination of FLT3i with intensive chemotherapy in fit/young patients could be the preferred option.130
All mentioned key FLT3i trials in this review highlighted the impact of FLT3i in the context of allo-SCT. The presence of an FLT3i arm could make it possible to proceed to an allo-SCT either as consolidation or maintenance therapy even for older/unfit patients. Among the aforementioned FLT3is, sorafenib seems to be the most promising one as post-allo-SCT maintenance therapy in FLT3-mutated patients.103,108,116
The safety profile should be carefully monitored to decide on the proper FLT3i alone or in combination protocols. Common adverse effects of midostaurin include QTc prolongation, skin rash, and myelosuppression, which are all manageable as compared to chemotherapy except for skin rash. However, its interaction with azole derivatives has been shown to cause serious side effects such as an unpredictable increase in plasma dose level and pulmonary toxicity, which should be kept in mind while evaluating the patients’ need for azoles. Gilteritinib has a good safety profile with mild to severe alanine transaminase (ALT) and aspartate transaminase (AST) levels, QTc prolongation, and myelosuppression, which could be manageable with temporary drug suspension and dose reductions. Quizartinib also caused mild myelosuppression, gastrointestinal side effects, and QTc prolongation, which could need drug suspension and dose reductions.107,115,131
Development of Resistance Toward FLT3 Inhibitors: Possible Mechanisms and Overcoming Strategies
Primary and secondary resistance against FLT3i in mono- and combination therapies remains a significant obstacle for successful and long-lived remission. Primary resistance (innate resistance) restricts the efficacy of FLT3i at initial administration, whilst secondary resistance (acquired resistance) emerges during continuous exposure to FLT3i during treatment cycles, resulting in R/R disease.132 While primary resistance is rarely observed in FLT3-mutated AML patients, the likelihood of secondary resistance development towards FLT3i seems a major contribution. Mechanisms involved in resistance could be heterogenous and grouped as FLT3-dependent and FLT3-independent resistance mechanisms, which are not required to be equally or mutually present.132,133
FLT3-Dependent Resistance Mechanisms
The presence or emergence of FLT3-TKD or FLT3-ITD-TKD (compound) mutations before or during FLT3i treatment causes the development of resistance against FLT3i therapy. The gatekeeper mutation, F691L, in the TKD is the common one that led to resistance against all clinically used FLT3is.132,133 Gilteritinib was shown not to be effective in patients carrying F691L mutation who enrolled in the ADMIRAL study.121 While the majority of FLT3 inhibitors are effective against FLT3-ITD, especially type II inhibitors are ineffective against TKD mutations. Therefore, secondary point mutations occurring in FLT3-ITD during treatment might play a role in gaining resistance, thus rendering their original effect. Quizartinib-treated FLT3-ITD AML patients developed resistance due to the emergence of mutations at D835 and Y842 residues as well as F691 in the TKD.51 FLT3-ITD AML patients developed resistance against midostaurin via having N676K mutation in TKD.134 Association of these mutations with resistance development could be explained by their effects on drug binding, stabilization of active receptor conformation, and receptor activity or activation of downstream signaling pathways.135 For instance, F691L mutation prevents the binding of the drug to the receptor by assumably affecting several bonds, since the motions of the inhibitor are in correlation with those of the Phe 691 residue.136,137 An integrated computational approach investigated the possible mechanism of quizartinib resistance via F691 gatekeeper mutation by comparing the receptor–inhibitor interactions between FLT3 kinase domain (wild-type or F691L) and quizartinib or PLX3397 (its activity is not affected by F691L mutation). When quizartinib was bound to FLT3-FL691L, the conformational change of αC-helix and A-loop of the FLT3 protein could be induced, rendering functional receptor structure. Additionally, quizartinib dissociated more easily from FLT3-F691L than from FLT3-WT, which made quizartinib stay shorter in the mutated receptor. In contrast, there was no significant difference between the dissociation processes of WT-FLT3 and FLT3-F691L from PLX3397.138 D835F and Y842H mutations in TKD were thought to make quizartinib and sorafenib ineffective.51,139 In a particular study, multiple simulations of WT- and mutant (D835F, Y842H) FLT3 in drug-bound, drug-free, inactive or active forms were investigated.137 These mutations might shift the equilibrium towards the active state of the receptor without affecting drug–receptor interactions directly based on the results of fully atomistic molecular dynamics (MD) simulations of FLT3.137 M664I, D835N, and Y842S mutants in FLT3-TKD were highly active with enhanced autophosphorylation capacity and quizartinib-bound inactive molecules had many conformational alterations resulting in ineffective inhibition.140 FLT3-N676K mutation resulted in increased surface expression in Ba/F3 cells transfected with FLT3 N676K mutant compared to D835Y and ITD, but it was similar to WT-FLT3.141 It was found that FLT3-N676K mutation induced the phosphorylation of FLT3, MAPK, and AKT strongly compared to FLT3-ITD mutation.141,142 Leukemic cells carrying the FLT3-N676K mutant in the absence of an ITD mutation were highly sensitive to FLT3 inhibitors such as quizartinib and sorafenib.142,143 Therefore, this particular mutation seems to have a leukemogenic potential based on in vitro cell lines and in vivo mice studies. On the other hand, the FLT3-ITD-N676K compound mutation was predicted to inhibit the auto-inhibitory function of FLT3 by reducing the stability of the JM domain, resulting in midostaurin resistance.134 A novel gatekeeper mutation, N701K was detected in gilteritinib-resistant FLT3-ITD cell lines by sterically interfering with the binding of gilteritinib.144
The modulation of FLT3-ITD localization is considered as one of the on-target mechanisms behind FLT3i resistance. WT-FLT3 is mainly localized to the cell membrane while FLT3-ITD localization is mainly restricted to ER/GA due to the impaired post-translational glycosylation of FLT3-ITD caused by its constitutive kinase activity.145 The intracellular localization of FLT3-ITD activates predominantly STAT5 signaling while the one localized to the cell membrane predominantly activates AKT and MEK.43 Therefore, FLT3-ITD localized to both ER and cell membrane cooperates in cellular transformation. The impairment of FLT3-ITD maturation via inhibiting N-glycosylation could be effective and synergistic with FLT3i. Tunicamycin, an N-glycosylation inhibitor, inhibited the proliferation and induced apoptosis of FLT3-ITD expressing human and murine cell lines via partly inhibiting FLT3-ITD activated AKT and ERK signaling and its combination with quizartinib showed synergistic effects.146 A recent study also revealed the importance of different localization of FLT3-ITD mutant variants associated with the FLT3i resistance, which could be overcome by combining the inhibitors of N-glycosylation with distinct downstream signaling pathways’ inhibitors.147 FLT3-ITD was found to be S-palmitoylated by the palmitoyl acyltransferase ZDHHC6, which could be responsible for its localization to ER. The disruption of S-palmitoylation localized FLT3-ITD to the plasma membrane and activated AKT and ERK in addition to STAT5 and induced the progression of leukemia in a mice model. Inhibition of FLT3-ITD palmitoylation synergized with gilteritinib, which impaired the growth of primary FLT3-ITD+ AML cells.148
Surface expression of FLT3 due to its lower turnover rate and increased half-life was detected in MOLM-13 FLT3-ITD AML cells with acquired midostaurin resistance.149 Majority of FLT3-mutated AML has both WT-FLT3 and mutant-FLT3 expression and this heterogenicity could be responsible for limited response to FLT3i. In a study, the effects of quizartinib and sorafenib were decreased in 32D cells co-expressing WT- and FLT3-ITD as compared to 32D cells with only FLT3-ITD expression,150 which is partially explained by the activation of WT-FLT3 by FLT3 ligand secreted by stromal cells.
FLT3-Independent Resistance Mechanisms
Additional mutations in the FLT3 receptor account for a small proportion of resistant cases. Studies elucidating FLT3-independent resistance mechanisms such as the impact of the tumor microenvironment, metabolism of FLT3i, and modulation of alternative intracellular signaling pathways have been paid attention.151
Bone marrow stromal cells with elevated CYP3A4 expression, a cytochrome P450 enzyme, impaired the activity of sorafenib, quizartinib, and gilteritinib in FLT3-ITD-positive AML by reducing the plasma concentrations of the inhibitors.152 Another resistance-causing mechanism in the bone marrow microenvironment is the release of fibroblast growth factor 2 (FGF2) from the stromal cells, which caused quizartinib resistance in patients.153 Pim kinase-2 overexpression was detected in sorafenib-resistant FLT3-ITD AML patients compared to untreated samples at the time of diagnosis, which resulted in sequestration of anti-apoptotic BAD in the cytoplasm and suppression of apoptosis.154 AXL kinase expression and activation increased in midostaurin and quizartinib-resistant primary blasts from FLT3-ITD AML patients by constantly activating the RAS/MAPK and PI3K/AKT/mTOR pathways.155 In in vitro models of midostaurin and sorafenib resistance, cytokine CCL5 was found to be elevated, which also increased phosphorylated AKT and STAT5 levels.156 Also, midostaurin-resistant blast cells from the patient showed increased CCL5 transcript levels. Activating JAK mutations were identified in both in vitro models (cell lines) and patient samples of FLT3-ITD AML with midostaurin or sorafenib resistance. JAK1 V658F mutation specifically was found to activate CSF2RB-JAK-STAT5 pathway to eliminate the effects of FLT3i.157
In a recent study, it was shown that actin filaments, one of the cytoskeletal components, are remodeled in a RAC-1-dependent manner, causing the development of midostaurin resistance in FLT3-ITD AML cell lines and primary patient samples. Anti-apoptotic BCL-2 expression and activity increased as a result of RAC-1 activation.158 Early resistance and late resistance mechanisms were well defined in gilteritinib-resistant cell lines and patient samples by using whole-exome sequencing, CRISPR-Cas9, metabolomics, proteomics, and pharmacologic approaches.159 Early resistant cells were protected by the bone marrow microenvironment to evolve into late resistant cells with newly gained intrinsic alterations. Late resistant cells were derived from the subclones already carrying NRAS mutations. Aurora kinase B was activated in early resistant cells while both early and late resistance cells underwent metabolic reprogramming. Activating mutations in RAS/MAPK pathway were also shown in gilteritinib-resistant FLT3-ITD AML patients using targeted-next generation sequencing (NGS).160 NGS data from bone marrow samples of FLT3-ITD AML patients collected after type I or type II FLT3i treatment (secondary resistance group) showed mutational differences at relapse.161 Detected mutations against FLT3i were found in epigenetic modifiers such as DNMT3A and RAS/MAPK pathway in addition to FLT3-D835 mutation. The most common secondary mutation was in RAS/MAPK pathway against type I FLT3i while FLT3-D835 was the most emerged mutation against type II FLT3i.161 FLT3-ITD cell lines with acquired sorafenib resistance and primary patient samples from sorafenib-resistant FLT3-ITD AML showed activation of PI3K/mTOR pathway.162 Clonal evaluation of midostaurin resistance in patients in RATIFY trial revealed different molecular profiles at the time of diagnosis and relapse. Midostaurin resistance emerged due to acquired mutations in signaling pathways such as MAPK while they became FLT3-ITD negative based on whole genome sequencing.163 Transcriptome analysis of the samples from patients with FLT3-ITD/D835 mutations in comparison to those with FLT3-ITD only revealed the overexpression of anti-apoptotic BCL2A1 in FLT3-ITD/D835 patient samples, which was associated with decreased quizartinib sensitivity. The combination of quizartinib with venetoclax specifically in FLT3-ITD/D835 cell lines showed that the presence of this compound mutation could be responsible for quizartinib and venetoclax resistance.164 MCL-1, an anti-apoptotic BCL-2 family member was shown to be upregulated in FLT3-ITD AML through STAT5 activation,165 and responsible for leukemia relapse, poor therapeutic outcomes, and venetoclax resistance.166–168 Hence, several MCL-1 inhibitors such as VU661013, S63845, and MIK665 have been developed. MCL-1 inhibition together with BCL-2 inhibition could show synergistic effects and overcome venetoclax resistance.168,169 There are recruiting clinical trials using the combination of S64315 with venetoclax (NCT03672695) and the combination of a novel BCL-2 inhibitor S65487/VOB560 with an MCL-1 inhibitor MIK665 (NCT04702425) in R/R FLT3 AML. In FLT3-ITD AML models, multikinase inhibitor olverembatinib (HQP1351) induced apoptosis via MCL-1 downregulation which was enhanced in the presence of BCL-2 inhibitor lisaftoclax (APG-2575). The elevated expression of the TEK-family kinase, BMX, in gilteritinib-unresponsive patients was detected using single-cell RNA sequencing, which mediated gilteritinib resistance via upregulation of cell-cycle, DNA/RNA metabolic processes, and protein translation.170
Overall, these studies underlined the importance of differences in resistance profiles of type I and type II inhibitors to design an appropriate treatment strategy after identifying the genetic makeup of each patient.
Approaches to Overcome FLT3i Resistance
Common strategies to overcome FLT3i resistance could be discussed under two general groups, including the development of novel FLT3is and dual-inhibitors and combinational approaches with the inhibitors of altered signaling molecules, anti-apoptotic molecules, epigenetic targets, or other molecular targets (Figure 3). Ongoing clinical trials of novel treatment strategies in FLT3 positive AML including novel FLT3is, dual FLT3 inhibitors, FLT3 antibody, CAR-T cell therapy or novel targeted therapies beyond FLT3is are summarized in Table 3.
 |
Table 3 Ongoing Clinical Trials of Novel Treatment Approaches in FLT3 Positive AML |
 |
Figure 3 Selected FLT3i resistance mechanisms and strategies to overcome resistance. |
Designing Novel FLT3i
Secondary mutations against type II FLT3is tend to occur on FLT3 itself while FLT3-independent resistance commonly occurs against type I FLT3is based on the results from previously discussed pre-clinical and clinical studies. However, the F691L mutation is still responsible for resistance to all clinically approved FLT3is. Hence, the development of novel FLT3is could be a possible way to overcome such resistance.
FF-10101 is a newly designed type I FLT3i, which binds covalently to C695 residue on FLT3 in an irreversible manner. It binds to both active and inactive FLT3 and is active against various FLT3 mutations, including FLT3-ITD, D835, Y842, and even F691L mutations.171 FF-10101 is recently shown to reduce bone marrow stromal cell-mediated protection of FLT3-ITD AML observed in resistance against other FLT3is such as quizartinib.172 FF-10101 is currently evaluated in phase I/IIa studies to identify its pharmacokinetics, toxicity profile, and safety in R/R AML (NCT03194685). HM43239, a novel reversible small-molecule type I FLT3 inhibitor with activity against WT-FLT3, FLT3-ITD, FLT3-TKD, and also FLT3 ITD/TKD double mutations showed in vitro (WT-FLT3 and mutant FLT3 cell lines) and in vivo (FLT3 ITD/TKD double mutated xenograft mouse model) efficacy via inhibiting STAT5, ERK, SYN, JAK1/2, and TAK1 kinases, which resulted in a currently recruiting phase I/II clinical trial (NCT03850574) to evaluate its appropriate doses and safety in R/R FLT3 AML.173 MZH29, a novel FLT3i, showed inhibitory activity against WT, FLT3-ITD, FLT3-D835H/Y/V, and FLT3-K663Q mutants and FLT3-ITD/F691L double mutation.174 LT-171-861, a novel FLT3i, bound to FLT3 strongly and inhibited the growth of FLT3 mutant cell lines such as FLT3-D835Y, FLT3-ITD-N676D, FLT3-ITD-D835Y, FLT3-ITD-F691L, FLT3-ITD-Y842C and AML blasts from patients with FLT3-ITD. In vivo effects also led to tumor regression and prolonged survival.175
Rationalized design of dual inhibitors targeting both FLT3 and another target such as cyclin-dependent kinase 4/6 (CDK4/6), JAK, Aurora A, tubulin, and PIM is thought to reverse resistance by simultaneously inhibiting two or more signaling pathways.176 CCT24571, a dual FLT3/Aurora A inhibitor showed cytotoxic and apoptotic effects on FLT3-ITD carrying MOLM-13 and MV4-11 AML cells and reversed D835Y resistance in vitro.177 Another FLT3/Aurora A inhibitor, CCT241736, inhibited tumor growth of FLT3-ITD and FLT3-ITD-TKD human tumor xenograft models and also showed efficacy in primary patient samples with quizartinib resistance.178 AMG925, a novel dual inhibitor of FLT3/CDK4-6 induced apoptosis in both WT and mutant FLT3 AML cell lines and primary blasts.179 KX2-391 is a recently identified dual FLT3/Tubulin inhibitor with promising resistance-reversion activity against FLT3-ITD and FLT3-ITD-D835/F691 mutation in in vitro cell lines, a murine model, and patient blasts.180 Inhibition of JAK2 and FLT3 at the same time could be a possible strategy to eradicate resistant cells with FLT3 mutations. Momelotinib, a potential dual FLT3/JAK2 inhibitor, gave better responses against D835, D839, and Y842 mutations and growth factor-mediated resistance in mouse and human primary cells expressing FLT3-ITD.181 The dual JAK/FLT3 inhibitor pacritinib in combination with chemotherapy showed clinical activity in FLT3-ITD and FLT3-TKD AML patients.182 Dual targeting of FLT3 and PIM kinase by SEL24 showed anti-proliferative and apoptotic activities against MOLM-13, MV4-11 FLT3-ITD positive cells, and primary FLT3-ITD cells compared to FLT3 inhibitor or PIM kinase inhibitor alone. Its activity was not lowered by FTL3-ITD, certain FLT3-TKD, or FLT3-ITD-TKD mutations.183 A dose escalation trial for SEL24 (NCT03008187) is recruiting in R/R FLT3 AML with no available treatment strategy.
Innate immune pathway activation via the interleukin-1 receptor-associated kinase 1 and 4 (IRAK1/4) complex contributed to adaptive quizartinib resistance in FLT3-mutant AML cells via restoring RAS/MAPK signaling along with NF-κB, which could be overcome by a small molecule-dual inhibitor, NCGC1481, targeting both FLT3 and IRAK1/4 kinases.184 MRX-2843, a dual MERTK/FLT3 inhibitor, showed activity against quizartinib-resistant FLT3-ITD-D835 or F691 mutants and prolonged survival in xenograft models of quizartinib-resistant AML.185 A674563, a dual inhibitor targeting both FLT3-ITD and AKT was active against FLT3-D835Y mutant-expressing cells and could overcome FLT3 ligand-induced drug resistance.186 CG-806, dual BTK (Bruton’s tyrosine kinase)/FLT3 inhibitor is being evaluated in a trial (NCT04477291) including R/R patients or ineligible patients for other treatments. FGFR/FLT3 dual inhibitor, MAX-040279, is under evaluation in a Phase I (NCT04187495) study for R/R FLT3 AML due to the role of FGF2/FGFR signaling in FLT3i resistance.
To sum up, there are many newly designed FLT3is or dual inhibitors to reduce drug resistance and increase responses. However, most novel FLT3is and dual-target inhibitors are only tested in pre-clinical studies at present or some of them are undergoing early-stage of phase studies.
Novel FLT3i Combinatorial Treatment Approaches
Apart from combinations of FLT3i with conventional chemotherapy as mentioned in critical trials, inhibiting altered signaling pathway molecules or other players could be synergistically effective in FLT3i-resistant FLT3-ITD AML.
MEK inhibitor trametinib combined with midostaurin had a synergistic effect to overcome midostaurin resistance in FLT3 mutated AML.187 The combination of venetoclax, a BCL-2 inhibitor, with midostaurin or gilteritinib showed synergism in MOLM-13 and MV4-11 FLT3-ITD AML cell lines.188 MOLM-13 xenograft treated with gilteritinib plus venetoclax had improved survival compared to gilteritinib alone. Simultaneous downregulation of MCL-1 by midostaurin or gilteritinib and inhibition of BCL-2 by venetoclax made BIM free, resulting in synergistic induction of apoptosis in FLT3-ITD AML cell lines and patient samples.189 Cotreatment with venetoclax and quizartinib had greater anti-leukemic activity in pre-clinical models of FLT3-ITD AML and a patient-derived FLT3-ITD AML xenograft model.190 There is a recently completed Phase IB/Phase II trial investigating the side effects and appropriate dose of venetoclax in combination with quizartinib in R/R FLT3-ITD AML patients (NCT03735875). Triple combination including decitabine (DNA methyltransferase inhibitor) + venetoclax + quizartinib was shown to be highly active in R/R FLT3-ITD mutated AML patients, with CR rates of 69% and the median OS of 7.1 months, which is under clinical phase I/II study (NCT03661307).191 The combination of gilteritinib with venetoclax specifically in FLT3-ITD/D835 cell lines had synergistic effects in contrast to quizartinib plus venetoclax via downregulating MCL-1.164 Homoharringtonine, as a STAT inhibitor, was evaluated in vitro and in vivo in combination with sorafenib, quizartinib, and gilteritinib in FLT3-ITD AML. Sorafenib in combination with low-dose homoharringtonine induced synergism in an R/R FLT3-AML patient, which is evaluated in phase II trial (NCT03170895) in R/R FLT3-ITD AML.192 Homoharringtonine plus quizartinib triggered apoptosis via upregulating BIM and BAX and downregulating MCL-1 in FLT3-ITD AML cell lines and increased OS in mice model.193 The combination of homoharringtonine with gilteritinib also decreased MCL-1 by UBE2L6-mediated proteasomal degradation.194 There is a phase II trial (NCT03135054) evaluating if quizartinib plus omacetaxine mepesuccinate (homoharringtonine) results in durable CRc in patients with newly diagnosed or R/R AML carrying FLT3-ITD. Activating JAK mutation, JAK1 V658F, was related to midostaurin resistance and combination of JAK1/2 inhibitor, ruxolitinib, with midostaurin was able to sensitize FLT3-ITD AML resistant cells to midostaurin.157 Inhibition of RAC-1 or BCL-2 using pharmacological inhibitors together with midostaurin overcame midostaurin resistance.158 BTK is identified as a novel target in FLT3-ITD AML patient blasts and cell lines. Inhibition of BTK by ibrutinib blocked the survival and proliferation of FLT3-ITD primary AML blasts and AML cell lines by inhibiting MAPK, AKT, and STAT5.195 Ibrutinib might specifically target FLT3-ITD in addition to BTK via decreasing FLT3-ITD autophosphorylation. c-MYC expression and STAT5 phosphorylation were also decreased in response to ibrutinib in FLT3-ITD AML cell lines and it showed synergistic or additive effects in combination with FLT3i.196,197 There is a phase II/III trial (NCT03642236) accepting registration to investigate the efficacy and safety of the combination of BTK inhibitor, ibrutinib with chemotherapy with/without FLT3 inhibitor in refractory/relapsed FLT3 mutant AML. The combination of MDM2, a negative regulator of the tumor suppressor p53, inhibitor milademetan with quizartinib showed synergistic apoptotic effects in FLT3-ITD positive and p53 WT AML cell lines and murine cell lines with FLT3-ITD+F691L and D835Y mutations via suppression of MCL-1 and upregulation of p53, PUMA and p21. In vivo mice model treated with this combination had prolonged survival.198 Milademetan (MDM2 inhibitor) plus quizartinib combination study in FLT3-ITD mutant AML patients is recently completed (NCT03552029, results not shared yet). MDM2 inhibitor NVP-HDM201 and midostaurin combination showed synergistic effects in AML cells with high FLT3-ITD AR and WT TP53 and NPM1,199 which is under a phase I trial (NCT04496999). The combination of gilteritinib with a pharmaceutical inhibitor of the NFKB family inhibited the secretion of tumor-promoting cytokines from gilteritinib-treated leukemic blasts.200 Apoptosis induced in FLT3-ITD AML cell lines and primary patient samples treated with gilteritinib in combination with CUDC-907, a dual inhibitor of PI3K and histone deacetylases via FLT3 downregulation, inhibition of MAPK/ERK and JAK/STAT pathways, reduction of MCL-1 and c-MYC and induction of BIM.201 Protein arginine methyltransferase 1 (PRMT1) was upregulated and defined as an important target involved in the maintenance of FLT3-ITD+ AML. PRMT1 methylates FLT3-ITD at R972/973 residues which enhanced Y969 phosphorylation to recruit downstream SH2 domain-containing adaptor molecules. Thus, R972/973 methylation promoted STAT5 and AKT phosphorylation. Inhibition of PRMT1 using MS023 in combination with quizartinib enhanced the elimination of FLT3-ITD cells.202 Translation initiation factor eIF4a was inhibited with rohinitib (RHT) via downregulation of transcription factor heat shock factor 1 (HSF1) in FLT3-ITD AML cells, resulting in an anti-leukemic effect. Knockdown of HSF1 sensitized FLT3-mutant AML cells with both ITD and TKD mutations to clinical FLT3i.203
Targeted FLT3 AML Therapeutics Beyond FLT3 Inhibitors
Even though there are small FLT3is with FDA approval such as midostaurin and gilteritinib, they have their own limitations for effective treatment such as drug resistance, toxicities, limited and short-lived responses. Therefore, there is still a need to search for novel treatment modalities for FLT3-ITD AML including immunotherapy, small-molecule FLT3 degraders, and flavonoids.
FLT3 Antibodies
Monoclonal antibodies (mAb) with high specificity and affinity for FLT3 have been developed and their effects have been evaluated in pre-clinical and early clinical phase studies. IMC-EB10, a human anti-FLT3 antibody, targeted both WT-FLT3 and FLT3-ITD mutant in a ligand-dependent (via blocking the binding of FLT3 L to FLT3) and -independent manner and inhibited downstream signaling pathways such as MAPK and AKT in both cell lines and mice model.204 IMC-NC7 is another human anti-FLT3 antibody sharing similar working mechanism with IMC-EB10.205 In this case, IMC-EB10 initiated antibody-dependent cell-mediated cell toxicity in FLT3 expressing cells and decreased engraftment of FLT3 expressing cells into non-obese diabetic/severe combined immunodeficient mice more effectively.205 Although a phase I study (NCT00887926) was initiated based on this result, it is recently terminated due to lack of efficacy in 26 registered R/R AML patients. 4G8SDIEM is the first reported Fc-optimized antibody targeting FLT3 and induced cellular toxicity on both FLT3 expressing cell lines and AML blasts,206 which led to the current phase I/II clinical trial (NCT02789254) for AML patients with minimal residual disease (MRD). To target a wider population of AML patients, an immunoglobulin G-based bispecific antibody (7370) with an affinity for both FLT3 and CD3 has been developed recently.207 It was shown to have a long half-life and target FLT3 irrespective of mutation profile in AML blasts and it gave promising results in cynomolgus monkeys via inducing depletion of peripheral FLT3+ dendritic cells and bone marrow FLT3+ stem cells and progenitors. CLN-049, a CD4+ and CD8+ T cell activating bispecific antibody targeting FLT3 and CD3 is just developed.208 In mouse xenograft models established using human leukemic cell lines and patient-derived AML blasts, CLN-049 was highly active.
T-Cell Based Approaches
Autologous T cells could be modified and given back to the patient to target FLT3-ITD with increased specificity for cancer cell killing. The most common strategy is chimeric antigen receptor (CAR) T cell therapy for FLT3 positive AML.
CARs have an extracellular domain to recognize cancer-specific antigens and an intracellular signaling domain for T cell proliferation and activation to kill cancer cells.209 T cells from healthy donors were engineered to recognize FLT3 with CD28 co-stimulatory signaling domain and CD3ζ activation domain.210 Its cytotoxicity was evaluated in FLT3 expressing cell lines including FLT3-ITD positive MOLM-13 and MV4-11 cells and WT-FLT3 expressing cells EOL1. FLT3 CAR T cells killed the FLT3 expressing cells and secreted IFN-γ and IL-2. Similar results were also obtained using FLT3 expressing AML blasts. In vivo xenograft mice models established via engraftment of MOLM-13 cells and FLT3 positive AML patient blasts, FLT3 CAR T cells showed anti-leukemic activity with 100% survival rate compared to the controls.210 There is a phase I study evaluating the safety, tolerability, and efficacy of FLT3 CAR T cell therapy (AMG 553) in FLT3 R/R AML (NCT03904069).211
FLT3 L CAR T cells were constructed using FLT3 L as an extracellular recognizing domain with 4–1BB and CD3ζ intracellular signaling domains.212 FLT3 L CAR T cells were co-cultured with FLT3 positive cell lines and patient blasts and showed cytotoxicity via secreting IFN-γ and TNF alpha. In vivo xenograft model treated with FLT3 L CAR T cells had prolonged survival. Newly designed FLT3 L CAR T cells with a combined ICOS and 4–1BB co-stimulatory domain and a CD3ζ activating domain were effective against WT-FLT3 carrying THP-1 cells.213 The combination of FLT3 CAR T cell therapy with FLT3i, midostaurin or quizartinib, showed promising results.214 These FLT3is increased surface expression of FLT3, then FLT3 CAR T cells recognized MOLM-13 and MV4-11 FLT3-ITD cells effectively and improved response rate in mice model. There is a pre-clinical study investigating the effect of allogeneic FLT3 CAR T cell therapy with a rituximab on-off switch mechanism to eliminate some challenges observed in autologous therapy such as limited or nonfunctional peripheral T cells in patients after treatment, which renders effective production of patient-based T cells for CAR T therapy.215 This approach resulted in promising results in vitro and in vivo, however, there was also the elimination of human HSC and progenitor cells leading to myelotoxicity. In this case, rituximab switch was useful to remove FLT3 CAR T cells after depleting AML blasts and allowing bone marrow to recover. The synergistic effect of dual-target FLT3 single-chain fragment variable (scFv)/NKG2D (Natural killer group 2 member D ligands) CAR T cells with gilteritinib associated with the lysis of R/R FLT3-ITD AML cell lines and mouse model. Gilteritinib increased the efficacy of bispecific CAR T cells via increasing the expression of NKG2DL.216
Novel Immunotargets in FLT3 AML
Higher expression of the IL3 receptor α-chain (CD123) and MIC-2 (CD99) in combination with the IL2 receptor α-chain (CD25) within the CD34+ cell population was detected in minor FLT3-ITD subclones at diagnosis and also in 83% of cases with FLT3-ITD relapse. The presence of the CD34/CD25/CD123/CD99+ population was significantly associated with ITD mutation in the FLT3 gene, which could be the LSCs.217,218 A higher FLT3-ITD load was observed within CD34/CD123/CD25/CD99+LSCs. Treatment with an anti-CD99 mAb was cytotoxic on LSCs in two patients, whereas there was no effect on CD34+cells from healthy donors.219 Anti-CD99 mAb showed more apoptotic effects on FLT3-ITD AML patient samples and cell lines compared to those expressing WT-FLT3 via upregulating both intrinsic and extrinsic pathways of apoptosis with a specific emphasis on the p53-mediated pathway. CD99 targeting also reduced glycolysis and mitochondrial respiration.220 Targeting CD33 with gemtuzumab ozogamicin (GO), a calicheamicin-conjugated mAb, in combination with induction chemotherapy in pediatric patients with high FLT3-ITD AR reduced relapse.221 An active clinical trial (NCT04385290) is investigating the safety and efficacy of midostaurin plus GO as frontline therapy in newly diagnosed FLT3 mutated patients. The role of programmed cell death 1 (PD-1) and PD-1 ligand (PD-L1) is not clear for AML; however, the high expression of PD-L1 could be responsible for worse prognosis in NPM1/FLT3 double mutant AML patients.222 A clinical study of atezolizumab, PD-L1 targeting mAb in combination with gilteritinib in R/R FLT3 mutated AML is recently completed (NCT03730012). CSF2RB was phosphorylated directly by FLT3-ITD via direct interaction and its knockdown increased sensitivity against midostaurin via decreasing STAT5 phosphorylation, hence, targeting CSF2RB-FLT3-ITD interaction using small peptides could be a strategy to improve FLT3i’ efficacy.223
Small-Molecule FLT3 Degraders
The first report about the degradation of FLT3 was the inhibition of HSP90, a molecular chaperone, in FLT3-ITD expressing leukemia cells, which proved FLT3-ITD as a client kinase for HSP90.224 HSP90 inhibitor 17-AAG was cytotoxic to primary AML cells carrying FLT3 mutants, but not for WT-FLT3 via inhibiting JAK/STAT, MAPK, and PI3K/AKT pathways and 17-AGG dissociated FLT3-ITD from HSP90, hence inducing FLT3-ITD degradation.225 The patients with FLT3-ITD expression also had high HSP90 levels to stabilize FLT3-ITD. HSP90 inhibition had a stronger pro-apoptotic effect on FLT3-ITD AML cells compared to those with WT-FLT3.226 Inhibition of HSP90 in FLT3i resistant FLT3-D835Y and several FLT3-ITD/TKD mutants by HSP90 inhibitors geldanamycin 17-AAG or 17-DMAG resulted in the degradation of mutant FLT3 in addition to FLT3-ITD and inhibition of STAT5 and ERK1/2.227 c-Cbl and Cbl-b are reported as E3 ubiquitin ligases for FLT3-ITD and are involved in the degradation process via the ubiquitin-proteasomal pathway induced by 17-AAG.228 A loss-of-function mutation in the E3 ligase domain of c-Cbl was found to eliminate its degradative function in a mice model established using HCS with FLT3-ITD mutation and c-Cbl mutation since mice developed AML.229 The combination of 17-AAG and midostaurin showed high activity against AML cells with FLT3 mutations through downregulating FLT-3, p-FLT-3, p-AKT, p-ERK1/2, and p-STAT5 and inducing apoptosis.230 Ba/F3 cells expressing FLT3-ITD+D835V cells were very sensitive to HSP90 inhibitors which resulted in degradation via autophagy. Quizartinib-resistant MV4-11 cells with FLT3-ITD+D835H and FLT3-ITD+D835V were also sensitive to HSP90 inhibitors.231 USP10 was identified as an FLT3-specific deubiquitinase to stabilize FLT3, hence its inhibition by small USP10 inhibitors gave promising results in cell lines, patient samples, and a mouse model of FLT3-ITD AML.232 A novel USP10, Wu-5, induced both WT-FLT3 and FLT3-ITD degradation and induced apoptosis.233 HSP70 could be a new therapeutic target that was shown to interact with FLT3-ITD, leading to its stabilization. The inhibition of HSP70 by QL47 induced degradation in both FLT3-ITD and drug-resistant mutants including F691L, N676D, and D835Y.234
Proteasome inhibitor, bortezomib triggered apoptosis specifically in FLT3-ITD AML cell lines and patient samples compared to WT-FLT3 AML cells by down-regulating PI3K/AKT, STAT5, and MAPK/ERK and the degradation of FLT3-ITD was related to autophagy. Bortezomib treatment overcame acquired quizartinib resistance in MOLM-14 FLT3-ITD-D835Y double mutant.235 Recently, proteaphagy, a degradation system activated after proteasome inhibition was identified in FLT3-ITD AML cells after bortezomib treatment via activation of autophagy, which suggested the inhibition of proteasome together with autophagy (using bafilomycin A) could be synergistic.236 In a mouse model of FLT3-ITD AML, arsenic trioxide (ATO) induced autophagic degradation of FLT3-ITD, which resulted in decreases in leukemic burden.237 Treatment with ATO resulted in the degradation of FLT3-ITD via partly decreasing its interaction with USP10, causing poly-ubiquitination and proteasomal degradation. ATO showed synergistic effects with sorafenib and quizartinib via inhibiting FLT3 autophosphorylation and downstream STAT5, AKT, and ERK signaling pathways.238
Polyphenols from green tea, (–)-epigallocatechin-3-gallate, (–)-epigallocatechin, and (–)-epicatechin-3-gallate, inhibited the proliferation and suppressed the FLT3 expression in FLT3 mutated cells which eventually led to inhibition of the downstream pathways such as PI3K, MAPK, and STAT5.239 The reason behind the FLT3 suppression was the disruption of the interaction between of HSP90 and FLT3-ITD, resulting in degradation of FLT3-ITD. These flavonoids also induced apoptosis in FLT3 mutated cells synergistically in combination with midostaurin.239 PROTAC, proteolysis targeting chimera, are bifunctional small molecules designed to bind target protein and E3 ubiquitin ligase simultaneously, which induce target protein ubiquitylation and then degradation by the proteasome.240 Several PROTAC molecules were synthesized based on the binding model of dovitinib and FLT3 among which molecules 101 and 102 showed anti-proliferative effects against MOLM-13 and MV4-11 cells and induced the degradation of FLT3-ITD.241 A promising PROTAC PF15 was synthesized recently and degraded FLT3 and inhibited downstream STAT5 signaling.242 All degraders showed efficacy in vivo models. Quizartinib was converted into a PROTAC with more selectivity and enhanced apoptotic activity via FLT3-ITD degradation in both in vitro and in vivo models although its kinase inhibitory function was partly abolished.243
Flavonoids in FLT3 AML
Flavonoids are natural products commonly found in plants.11 These compounds work as anti-oxidant, anti-bacterial, anti-viral, anti-inflammatory, and anti-cancer agents.244 Only flavones and flavonols are suggested to be anti-cancer agents among flavonoids due to their ability to induce mitochondria-mediated apoptosis and suppress multiple signaling pathways including MAPK, PI3K, and NF-κB.11 Hispidulin, luteolin, acacetin, and eupatin, subgroups of flavones and flavonols, are shown to have inhibitory properties towards FLT3.245,246
Natural chalcones were more potent toward FLT3-ITD cell lines and inhibited cell growth. All chalcones inhibited the FLT3 and reduced the phosphorylation of downstream pathways including STAT5 and ERK.247
O-methylated flavonol, a precursor of fisetin, inhibited the activity of FLT3-ITD and FLT3-D835Y kinases.244 Moreover, FLT3-ITD MOLM-13 and MV4-11 cells, when treated with this flavonol, arrested cell cycle at the G0/G1 phase and activated apoptotic proteins such as PARP, caspase 3, and BAX.244
Isoliquiritigenin extracted from licorice root also demonstrated anti-cancer activity by targeting FLT3. The proliferation of FLT3-ITD AML MOLM-13 and MV4-11 cells was selectively inhibited by isoliquiritigenin. In vivo studies on isoliquiritigenin revealed that it inhibited tumor growth in the MV4-11 xenograft mouse model and extended the survival time. The inhibitory activity of isoliquiritigenin towards resistant FLT3-ITD/F691L cell line makes it a promising natural compound in FLT3-ITD mutant AML.248
Resveratrol, an anti-carcinogenic plant-derived polyphenol was shown to inhibit the proliferation of FLT3-ITD mutated MV4-11 and MOLM-13 cells via modulation of sphingolipid metabolism enzymes including anti-apoptotic sphingosine kinase-1 and glucosylceramide synthase and apoptotic serine palmitoyltransferase.249,250 Although the studies investigating the roles of flavonoids in FLT3 positive AML are limited, natural products or their derivatives seem to possess the potential to develop new therapeutic agents targeting FLT3 or to be used as an integrative medicine in combination with FLT3i.
Conclusion
The discovery of FLT3 and its mutations has dramatically changed the AML course and treatment. The patients with FLT3 mutations have better options to be treated due to the approval of FLT3i in the clinical settings, which should be carefully selected based on patients’ current health condition, previous treatment history, genetic characterization, and eligibility for induction, consolidation or maintenance therapy as discussed extensively. On the other hand, the emergence of primary and mainly secondary resistance is a major limiting factor for effective FLT3i treatment, therefore understanding the detailed molecular mechanisms behind resistance will obviously increase the design of novel FLT3i or dual FLT3i and combinational treatment strategies. Additionally, emerging treatments involve immunotherapeutics such as FLT3 targeted antibodies, FLT3-specific T cell therapy, and novel potential immunotargets such as CD99 and PD-L1 and small molecule FLT3 degraders including HSP90 and proteasome inhibitors and PROTACs which have resulted in some initial success in pre-clinical and early phase clinical studies. However, more mechanistic studies or larger randomized trials are still needed to prove the advantages of these novel treatment modalities over approved treatments. Their specificity, safety profile, and appropriate dose should be carefully discussed based on the data which will be accumulating in the future. It is also inevitable that the development of novel therapeutics after identifying specific molecular targets in FLT3-ITD AML will be shaping the treatment strategies. To be noted, flavonoids are being investigated for their potential as adjuvants in FLT3-mutated AML with increasing anti-leukemic potentials. Based on all the studies investigating the targeting of FLT3, it would be possible to find a more specific and durable FLT3-ITD therapy approach even for a specific group of patients in the near future most probably in combination with already approved therapies.
Abbreviations
FLT3, FMS-like tyrosine kinase 3; AML, acute myeloid leukemia; ITD, internal tandem duplication; TKD, tyrosine kinase domain; FLT3i, FLT3 inhibitor; WT-FLT3, wild-type FLT3; RTK, receptor tyrosine kinase; HSC, hematopoietic stem cell; LSC, leukemic stem cell; LC-HSC, long-term hematopoietic stem cell; GA, Golgi apparatus; ER, endoplasmic reticulum; A-loop, activation loop; FLT3 L, FLT3 ligand; OS, overall survival; DFS, disease free survival; JM, juxtamembrane domain; TKI, tyrosine kinase inhibitor; R/R, relapsed/refractory; CR, complete remission; CRc, composite complete remission; mCRc, modified composite complete remission; EFS, event-free survival; RFS, relapse-free survival; CDK4/6, cyclin-dependent kinase 4/6; MRD, minimal residual disease; CAR, chimeric antigen receptor; hTERT, telomerase reverse transcriptase; NHEJ, nonhomologous end-joining; NPM1, nucleophosmin 1; IDH, isocitrate dehydrogenase; ASXL1, additional Sex Comb-like 1;CEBPA, CCAAT/enhancer binding protein A; HMA, hypomethylating agent; SC, salvage chemotherapy; CBF, core binding factor; HLF, hepatic leukemia factor; ALT, alanine transaminase; AST, aspartate transaminase; FGF2, fibroblast growth factor 2; NGS, next generation sequencing; IRAK1/4, interleukin-1 receptor-associated kinase 1 and 4; HSF1, heat shock factor 1; mAb, monoclonal antibodies; scFv, single-chain fragment variable; NKG2D, Natural killer group 2 member D ligands; GO, gemtuzumab ozogamicin; PD-1, programmed cell death 1; ATO, arsenic trioxide.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. Aysun Adan supervised the study.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Grafone T, Palmisano M, Nicci C, Storti S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: biology and treatment. Oncol Rev. 2012;6(1):e8–e8. doi:10.4081/oncol.2012.e8
2. Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33(2):299–312.
3. Small D, Levenstein M, Kim E, et al. STK-1, the human homolog of Flk-2/Flt-3, is selectively expressed in CD34+ human bone marrow cells and is involved in the proliferation of early progenitor/stem cells. Proc Natl Acad Sci U S A. 1994;91(2):459–463.
4. Leick MB, Levis MJ. The future of targeting FLT3 activation in AML. Curr Hematol Malig Rep. 2017;12(3):153–167.
5. Kiyoi H, Kawashima N, Ishikawa Y. FLT3 mutations in acute myeloid leukemia: therapeutic paradigm beyond inhibitor development. Cancer Sci. 2020;111(2):312–322.
6. Nitika WJ, Hui AM. Role of biomarkers in FLT3 AML. Cancers. 2022;14(5):85.
7. Daver N, Venugopal S, Ravandi F. FLT3 mutated acute myeloid leukemia: 2021 treatment algorithm. Blood Cancer J. 2021;11(5):104.
8. Wu M, Li C, Zhu X. FLT3 inhibitors in acute myeloid leukemia. J Hematol Oncol. 2018;11(1):133.
9. Hospital MA, Green AS, Maciel TT, et al. FLT3 inhibitors: clinical potential in acute myeloid leukemia. Onco Targets Ther. 2017;10:607–615.
10. Mahalleh M, Shabani M, Rayzan E, Rezaei N. Reinforcing the primary immunotherapy modulators against acute leukemia; monoclonal antibodies in AML. Immunotherapy. 2019;11(18):1583–1600. doi:10.2217/imt-2019-0043
11. Chin YW, Kong JY, Han SY. Flavonoids as receptor tyrosine kinase FLT3 inhibitors. Bioorg Med Chem Lett. 2013;23(6):1768–1770.
12. Rosnet O, Schiff C, Pebusque M, et al. Human FLT3/FLK2 gene: cDNA cloning and expression in hematopoietic cells. Blood. 1993;82(4):1110–1119.
13. Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100(5):1532–1542.
14. Tsapogas P, Mooney CJ, Brown G, Rolink A. The cytokine Flt3-Ligand in normal and malignant hematopoiesis. Int J Mol Sci. 2017;18(6):659.
15. Gabbianelli M, Pelosi E, Montesoro E, et al. Multi-level effects of flt3 ligand on human hematopoiesis: expansion of putative stem cells and proliferation of granulomonocytic progenitors/monocytic precursors. Blood. 1995;86(5):1661–1670.
16. Ratajczak MZ, Ratajczak J, Ford J, Kregenow R, Marlicz W, Gewirtz AM. FLT3/FLK-2 (STK-1) ligand does not stimulate human megakaryopoiesis in vitro. Stem Cells. 1996;14(1):146–150.
17. Turner AM, Lin NL, Issarachai S, Lyman SD, Broudy VC. FLT3 receptor expression on the surface of normal and malignant human hematopoietic cells. Blood. 1996;88(9):3383–3390.
18. Schmidt-Arras DE, Böhmer A, Markova B, Choudhary C, Serve H, Böhmer FD. Tyrosine phosphorylation regulates maturation of receptor tyrosine kinases. Mol Cell Biol. 2005;25(9):3690–3703.
19. Schmidt-Arras D, Böhmer SA, Koch S, et al. Anchoring of FLT3 in the endoplasmic reticulum alters signaling quality. Blood. 2009;113(15):3568–3576.
20. Hannum C, Culpepper J, Campbell D, et al. Ligand for FLT3/FLK2 receptor tyrosine kinase regulates growth of haematopoietic stem cells and is encoded by variant RNAs. Nature. 1994;368(6472):643–648.
21. Zhao JC, Agarwal S, Ahmad H, Amin K, Bewersdorf JP, Zeidan AM. A review of FLT3 inhibitors in acute myeloid leukemia. Blood Rev. 2022;52:100905.
22. Dosil M, Wang S, Lemischka IR. Mitogenic signalling and substrate specificity of the Flk2/Flt3 receptor tyrosine kinase in fibroblasts and interleukin 3-dependent hematopoietic cells. Mol Cell Biol. 1993;13(10):6572–6585.
23. Antar AI, Otrock ZK, Jabbour E, Mohty M, Bazarbachi A. FLT3 inhibitors in acute myeloid leukemia: ten frequently asked questions. Leukemia. 2020;34(3):682–696.
24. Ambinder AJ, Levis M. Potential targeting of FLT3 acute myeloid leukemia. Haematologica. 2021;106(3):671–681.
25. Sandhöfer N, Bauer J, Reiter K, et al. The new and recurrent FLT3 juxtamembrane deletion mutation shows a dominant negative effect on the wild-Type FLT3 receptor. Sci Rep. 2016;6:548.
26. Nakao M, Yokota S, Iwai T, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10(12):1911–1918.
27. Janke H, Pastore F, Schumacher D, et al. Activating FLT3 mutants show distinct gain-of-function phenotypes in vitro and a characteristic signaling pathway profile associated with prognosis in acute myeloid leukemia. PLoS One. 2014;9(3):e89560.
28. Schnittger S, Bacher U, Haferlach C, Alpermann T, Kern W, Haferlach T. Diversity of the juxtamembrane and TKD1 mutations (Exons 13–15) in the FLT3 gene with regards to mutant load, sequence, length, localization, and correlation with biological data. Genes Chromosomes Cancer. 2012;51(10):910–924.
29. De Freitas T, Marktel S, Piemontese S, et al. High rate of hematological responses to sorafenib in FLT3-ITD acute myeloid leukemia relapsed after allogeneic hematopoietic stem cell transplantation. Eur J Haematol. 2016;96(6):629–636.
30. Wang Z, Cai J, Cheng J, et al. FLT3 inhibitors in acute myeloid leukemia: challenges and recent developments in overcoming resistance. J Med Chem. 2021;64(6):2878–2900.
31. Patnaik MM. The importance of FLT3 mutational analysis in acute myeloid leukemia. Leuk Lymphoma. 2018;59(10):2273–2286.
32. Yamamoto Y, Kiyoi H, Nakano Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97(8):2434–2439.
33. Abu-Duhier FM, Goodeve AC, Wilson GA, Care RS, Peake IR, Reilly JT. Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br J Haematol. 2001;113(4):983–988.
34. Rottapel R, Turck CW, Casteran N, et al. Substrate specificities and identification of a putative binding site for PI3K in the carboxy tail of the murine Flt3 receptor tyrosine kinase. Oncogene. 1994;9(6):1755–1765.
35. Heiss E, Masson K, Sundberg C, et al. Identification of Y589 and Y599 in the juxtamembrane domain of Flt3 as ligand-induced autophosphorylation sites involved in binding of Src family kinases and the protein tyrosine phosphatase SHP2. Blood. 2006;108(5):1542–1550.
36. Zhang S, Mantel C, Broxmeyer HE. Flt3 signaling involves tyrosyl-phosphorylation of SHP-2 and SHIP and their association with Grb2 and Shc in Baf3/Flt3 cells. J Leukoc Biol. 1999;65(3):372–380.
37. Zhang S, Broxmeyer HE. Flt3 ligand induces tyrosine phosphorylation of Gab1 and Gab2 and their association with Shp-2, Grb2, and PI3 kinase. Biochem Biophys Res Commun. 2000;277(1):195–199.
38. Zhang S, Fukuda S, Lee Y, et al. Essential role of signal transducer and activator of transcription (Stat)5a but not Stat5b for Flt3-dependent signaling. J Exp Med. 2000;192(5):719–728.
39. Choudhary C, Schwäble J, Brandts C, et al. AML-associated Flt3 kinase domain mutations show signal transduction differences compared with Flt3 ITD mutations. Blood. 2005;106(1):265–273.
40. Zhang Y, Askenazi M, Jiang J, et al. Model for iTRAQ quantification reveals divergent signaling between oncogenic FLT3 mutants in acute myeloid leukemia. Mol Cell Proteomics. 2010;9(5):780–790.
41. Klingmüller U, Lorenz U, Cantley LC, Neel BG, Lodish HF. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell. 1995;80(5):729–738.
42. Staudt D, Murray HC, McLachlan T, et al. Targeting oncogenic signaling in mutant FLT3 acute myeloid leukemia: the path to least resistance. Int J Mol Sci. 2018;19(10):3198.
43. Choudhary C, Olsen JV, Brandts C, et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol Cell. 2009;36(2):326–339.
44. Takahashi S. Current findings for recurring mutations in acute myeloid leukemia. J Hematol Oncol. 2011;4:36.
45. Kihara R, Nagata Y, Kiyoi H, et al. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia. 2014;28(8):1586–1595.
46. Knapper S, Russell N, Gilkes A, et al. A randomized assessment of adding the kinase inhibitor lestaurtinib to first-line chemotherapy for FLT3-mutated AML. Blood. 2017;129(9):1143–1154.
47. Stone RM, Fischer T, Paquette R, et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia. 2012;26(9):2061–2068.
48. Lee BH, Williams IR, Anastasiadou E, et al. FLT3 internal tandem duplication mutations induce myeloproliferative or lymphoid disease in a transgenic mouse model. Oncogene. 2005;24(53):7882–7892.
49. Ley TJ, Miller C, et al; Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074.
50. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Eng J Med. 2016;374(23):2209–2221.
51. Smith CC, Wang Q, Chin C-S, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485(7397):260–263.
52. Krönke J, Bullinger L, Teleanu V, et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood. 2013;122(1):100–108.
53. Daver N, Strati P, Jabbour E, et al. FLT3 mutations in myelodysplastic syndrome and chronic myelomonocytic leukemia. Am J Hematol. 2013;88(1):56–59.
54. Basit F, Andersson M, Hultquist A. The Myc/Max/Mxd network is a target of mutated Flt3 signaling in hematopoietic stem cells in Flt3-ITD-induced myeloproliferative disease. Stem Cells Int. 2018;2018:3286949.
55. Li L, Osdal T, Ho Y, et al. SIRT1 activation by a c-MYC oncogenic network promotes the maintenance and drug resistance of human FLT3-ITD acute myeloid leukemia stem cells. Cell Stem Cell. 2014;15(4):431–446.
56. Zhang X, Li B, Yu J, et al. MYC-dependent downregulation of telomerase by FLT3 inhibitors is required for their therapeutic efficacy on acute myeloid leukemia. Ann Hematol. 2018;97(1):63–72.
57. Muvarak N, Kelley S, Robert C, et al. c-MYC generates repair errors via increased transcription of alternative-NHEJ factors, LIG3 and PARP1, in tyrosine kinase–activated leukemias. Mol Cancer Res. 2015;13(4):699–712.
58. Sies K, Spohr C, Gründer A, et al. Gab2 is essential for transformation by FLT3-ITD in acute myeloid leukemia. HemaSphere. 2019;3(2):e184.
59. Spohr C, Poggio T, Andrieux G, et al. Gab2 deficiency prevents Flt3-ITD driven acute myeloid leukemia in vivo. Leukemia. 2022;36(4):970–982.
60. Poitras JL, Heiser D, Li L, et al. Dnmt3a deletion cooperates with the Flt3-ITD mutation to drive leukemogenesis in a murine model. Blood. 2014;124(21):3568.
61. Ali B, Gesine B, Frederic B, et al. Clinical practice recommendation on hematopoietic stem cell transplantation for acute myeloid leukemia patients with FLT3-internal tandem duplication: a position statement from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Haematologica. 2020;105(6):1507–1516.
62. Schlenk RF, Döhner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Eng J Med. 2008;358(18):1909–1918.
63. Patel U, Luthra R, Medeiros LJ, Patel KP. Diagnostic, prognostic, and predictive utility of recurrent somatic mutations in myeloid neoplasms. Clin Lymphoma Myeloma Leuk. 2017;17:S62–S74.
64. Whitman SP, Archer KJ, Feng L, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytoGenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res. 2001;61(19):7233–7239.
65. Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447.
66. Kottaridis PD, Gale RE, Langabeer SE, Frew ME, Bowen DT, Linch DC. Studies of FLT3mutations in paired presentation and relapse samples from patients with acute myeloid leukemia: implications for the role of FLT3mutations in leukemogenesis, minimal residual disease detection, and possible therapy with FLT3 inhibitors. Blood. 2002;100(7):2393–2398.
67. Kelly LM, Kutok JL, Williams IR, et al. PML/RARα and FLT3-ITD induce an APL-like disease in a mouse model. Proc Natl Acad Sci U S A. 2002;99(12):8283–8288.
68. Meyer SE, Qin T, Muench DE, et al. DNMT3A haploinsufficiency transforms FLT3ITD myeloproliferative disease into a rapid, spontaneous, and fully penetrant acute myeloid leukemia. Cancer Discov. 2016;6(5):501–515.
69. Ma J, Dunlap J, Paliga A, et al. DNMT3A co-mutation is required for FLT3-ITD as an adverse prognostic indicator in intermediate-risk cytogenetic group AML. Leuk Lymphoma. 2018;59(8):1938–1948.
70. Hindley A, Catherwood MA, McMullin MF, Mills KI. Significance of NPM1 gene mutations in AML. Int J Mol Sci. 2021;22(18):10040.
71. Gale RE, Green C, Allen C, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. 2008;111(5):2776–2784.
72. Juliusson G, Jädersten M, Deneberg S, et al. The prognostic impact of FLT3-ITD and NPM1 mutation in adult AML is age-dependent in the population-based setting. Blood Advances. 2020;4(6):1094–1101.
73. Elrhman HAEA, El-Meligui YM, Elalawi SM. Prognostic impact of concurrent DNMT3A, FLT3 and NPM1 gene mutations in acute myeloid leukemia patients. Clin Lymphoma Myeloma Leuk. 2021;21(12):e960–e969.
74. Boddu P, Takahashi K, Pemmaraju N, et al. Influence of IDH on FLT3-ITD status in newly diagnosed AML. Leukemia. 2017;31(11):2526–2529.
75. Lu J, Chen M, Hua H, et al. Additional mutations in IDH1/2-mutated patients with acute myeloid leukemia. Int J Lab Hematol. 2021;43(6):1483–1490.
76. Fan Y, Liao L, Liu Y, et al. Risk factors affect accurate prognosis in ASXL1-mutated acute myeloid leukemia. Cancer Cell Int. 2021;21(1):526.
77. Ebian HF, Elshorbagy S, Mohamed H, et al. Clinical implication and prognostic significance of FLT3-ITD and ASXL1 mutations in Egyptian AML patients: a single-center study. Cancer Biomarkers. 2021;32:379–389.
78. He J, Liu J, Shen H, et al. Companion gene mutations and their clinical significance in AML with double or single mutant CEBPA. Int J Hematol. 2022;116(1):71–80.
79. Perl AE, Cortes JE, Ganguly S, et al. Effect of co-mutations and FLT3-ITD variant allele frequency (VAF) on response to quizartinib or salvage chemotherapy (SC) in relapsed/refractory (R/R) acute myeloid leukemia (AML). Blood. 2019;134(Supplement_1):737.
80. Wang M, Wang R, Wang H, et al. Difference in gene mutation profile in patients with refractory/relapsed versus newly diagnosed acute myeloid leukemia based on targeted next-generation sequencing. Leuk Lymphoma. 2021;62(10):2416–2427.
81. Hellesøy M, Engen C, Grob T, Löwenberg B, Valk PJM, Gjertsen BT. Sex disparity in acute myeloid leukaemia with FLT3 internal tandem duplication mutations: implications for prognosis. Mol Oncol. 2021;15(9):2285–2299.
82. Döhner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140(12):1345–1377.
83. Senapati J, Kadia TM. Which FLT3 inhibitor for treatment of AML? Curr Treat Options Oncol. 2022;23(3):359–380.
84. Weisberg E, Roesel J, Furet P, et al. Antileukemic effects of novel first- and second-generation FLT3 inhibitors: structure-affinity comparison. Genes Cancer. 2010;1(10):1021–1032.
85. Stone RM, DeAngelo DJ, Klimek V, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105(1):54–60.
86. Fischer T, Stone RM, Deangelo DJ, et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28(28):4339–4345.
87. Furukawa Y, Vu HA, Akutsu M, et al. Divergent cytotoxic effects of PKC412 in combination with conventional antileukemic agents in FLT3 mutation-positive versus -negative leukemia cell lines. Leukemia. 2007;21(5):1005–1014.
88. Möllgård L, Deneberg S, Nahi H, et al. The FLT3 inhibitor PKC412 in combination with cytostatic drugs in vitro in acute myeloid leukemia. Cancer Chemother Pharmacol. 2008;62(3):439–448.
89. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Eng J Med. 2017;377(5):454–464.
90. Larson RA, Mandrekar SJ, Huebner LJ, et al. Midostaurin reduces relapse in FLT3-mutant acute myeloid leukemia: the Alliance CALGB 10603/RATIFY trial. Leukemia. 2021;35(9):2539–2551.
91. Voso MT, Larson RA, Jones D, et al. Midostaurin in patients with acute myeloid leukemia and FLT3-TKD mutations: a subanalysis from the RATIFY trial. Blood Advances. 2020;4(19):4945–4954.
92. Ofran Y, Leiba R, Frisch A, et al. Midostaurin in combination with chemotherapy is most effective in patients with acute myeloid leukemia presenting with high FLT3-ITD allelic ratio who proceed to allogeneic stem cell transplantation while in first complete remission. Eur J Haematol. 2021;106(1):64–71.
93. Maziarz RT, Levis M, Patnaik MM, et al. Midostaurin after allogeneic stem cell transplant in patients with FLT3-internal tandem duplication-positive acute myeloid leukemia. Bone Marrow Transplant. 2021;56(5):1180–1189.
94. Schlenk RF, Weber D, Fiedler W, et al. Midostaurin added to chemotherapy and continued single-agent maintenance therapy in acute myeloid leukemia with FLT3-ITD. Blood. 2019;133(8):840–851.
95. Ravandi F, Cortes JE, Jones D, et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol. 2010;28(11):1856–1862.
96. Röllig C, Serve H, Hüttmann A, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, Phase 2, randomised controlled trial. Lancet Oncol. 2015;16(16):1691–1699.
97. Rollig C, Serve H, Hüttmann A, et al. The addition of sorafenib to standard AML treatment results in a substantial reduction in relapse risk and improved survival. Trial Blood. 2017;130:721.
98. Wei AH, Kennedy GA, Morris KL, et al. Results of a phase 2, randomized, double-blind study of sorafenib versus placebo in combination with intensive chemotherapy in previously untreated patients with FLT3-ITD acute myeloid leukemia (ALLG AMLM16). Blood. 2020;136(Supplement 1):36–38.
99. Ohanian M, Garcia-Manero G, Levis M, et al. Sorafenib combined with 5-azacitidine in older patients with untreated FLT3-ITD mutated acute myeloid leukemia. Am J Hematol. 2018;93(9):1136–1141.
100. Ravandi F, Alattar ML, Grunwald MR, et al. Phase 2 study of azacitidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. 2013;121(23):4655–4662. doi:10.1182/blood-2013-01-480228
101. Tallman MS, Wang ES, Altman JK, et al. Acute Myeloid Leukemia, Version 3.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2019;17(6):721–749.
102. Ali B, Myriam L, Giorgia B, et al. Allogeneic stem cell transplantation for FLT3-mutated acute myeloid leukemia: in vivo T-cell depletion and posttransplant sorafenib maintenance improve survival. A Retrospective Acute Leukemia Working Party-European Society for Blood and Marrow Transplant Study. Clin Hematol Int. 2019;1(1):58–74.
103. Burchert A, Bug G, Fritz LV, et al. Sorafenib maintenance after allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia with FLT3–internal tandem duplication mutation (SORMAIN). J Clin Oncol. 2020;38(26):2993–3002.
104. Xuan L, Wang Y, Huang F, et al. Sorafenib maintenance in patients with FLT3-ITD acute myeloid leukaemia undergoing allogeneic haematopoietic stem-cell transplantation: an open-label, multicentre, randomised Phase 3 trial. Lancet Oncol. 2020;21(9):1201–1212.
105. Nuhoğlu kantarcı E, Eşkazan AE. Gilteritinib in the management of acute myeloid leukemia: current evidence and future directions. Leuk Res. 2022;114:106808.
106. Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, Phase 1-2 study. Lancet Oncol. 2017;18(8):1061–1075.
107. Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Eng J Med. 2019;381(18):1728–1740.
108. Perl AE, Larson RA, Podoltsev NA, et al. Follow-up of patients with R/R FLT3-mutation–positive AML treated with gilteritinib in the phase 3 ADMIRAL trial. Blood. 2022;139(23):3366–3375.
109. Daver N, Perl AE, Maly J, et al. Venetoclax plus gilteritinib for FLT3-mutated relapsed/refractory acute myeloid leukemia. J Clin Oncol. 2022.
110. Wang ES, Montesinos P, Minden MD, et al. Phase 3, open-label, randomized study of gilteritinib and azacitidine vs azacitidine for newly diagnosed FLT3-mutated acute myeloid leukemia in patients ineligible for intensive induction chemotherapy. Blood. 2021;138:700.
111. T-Cells CA. (Astellas Reports XOSPATA® (gilteritinib) in combination with azacitidine did not meet endpoint of overall survival in newly diagnosed FLT3 mutation-positive acute myeloid leukemia patients ineligible for intensive induction chemotherapy [news release]. 2020. Tokyo: https://www.astellas.com/us/news/5306.
112. Zarrinkar PP, Gunawardane RN, Cramer MD, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood. 2009;114(14):2984–2992.
113. Cortes J, Perl AE, Döhner H, et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: an open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2018;19(7):889–903.
114. Cortes JE, Tallman MS, Schiller GJ, et al. Phase 2b study of 2 dosing regimens of quizartinib monotherapy in FLT3-ITD-mutated, relapsed or refractory AML. Blood. 2018;132(6):598–607.
115. Cortes JE, Khaled S, Martinelli G, et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019;20(7):984–997.
116. Ganguly S, Cortes JE, Krämer A, et al. Clinical outcomes in patients with FLT3-ITD-mutated relapsed/refractory acute myelogenous leukemia undergoing hematopoietic stem cell transplantation after quizartinib or salvage chemotherapy in the QuANTUM-R trial. Transplantation Cell Therapy. 2021;27(2):153–162.
117. Swaminathan M, Kantarjian HM, Levis M, et al. A phase I/II study of the combination of quizartinib with azacitidine or low-dose cytarabine for the treatment of patients with acute myeloid leukemia and myelodysplastic syndrome. Haematologica. 2021;106(8):2121–2130.
118. Erba H, Montesinos P, Vrhovac R, et al. AML-029 quizartinib prolonged overall survival (OS) vs placebo plus intensive induction and consolidation therapy followed by single-agent continuation in patients aged 18-75 years with newly diagnosed FLT3–internal tandem duplication positive (FLT3-ITD+) acute myeloid leukemia (AML). Clin Lymphoma Myeloma Leuk. 2022;22:S208–S209.
119. Rücker FG, Du L, Luck TJ, et al. Molecular landscape and prognostic impact of FLT3-ITD insertion site in acute myeloid leukemia: RATIFY study results. Leukemia. 2022;36(1):90–99.
120. Kivioja J, Malani D, Kumar A, et al. FLT3-ITD allelic ratio and HLF expression predict FLT3 inhibitor efficacy in adult AML. Sci Rep. 2021;11(1):23565.
121. Smith CC, Levis MJ, Perl AE, Hill JE, Rosales M, Bahceci E. Molecular profile of FLT3-mutated relapsed/refractory patients with AML in the phase 3 ADMIRAL study of gilteritinib. Blood Adv. 2022;6(7):2144–2155.
122. Perl AE, Hosono N, Montesinos P, et al. Clinical outcomes in patients with relapsed/refractory FLT3-mutated acute myeloid leukemia treated with gilteritinib who received prior midostaurin or sorafenib. Blood Cancer J. 2022;12(5):84.
123. Numan Y, Abdel Rahman Z, Grenet J, et al. Gilteritinib clinical activity in relapsed/refractory FLT3 mutated acute myeloid leukemia previously treated with FLT3 inhibitors. Am J Hematol. 2022;97(3):322–328.
124. Yilmaz M, Alfayez M, DiNardo CD, et al. Correction to: outcomes with sequential FLT3-inhibitor-based therapies in patients with AML. J Hematol Oncol. 2021;14(1):34.
125. Strati P, Kantarjian H, Ravandi F, et al. Phase I/II trial of the combination of midostaurin (PKC412) and 5-azacitidine for patients with acute myeloid leukemia and myelodysplastic syndrome. Am J Hematol. 2015;90(4):276–281.
126. Konopleva M, Thirman MJ, Pratz KW, et al. Impact of FLT3 mutation on outcomes after venetoclax and azacitidine for patients with treatment-naïve acute myeloid leukemia. Clin Cancer Res. 2022;28(13):2744–2752.
127. Dennis M, Thomas IF, Ariti C, et al. Randomized evaluation of quizartinib and low-dose ara-C vs low-dose ara-C in older acute myeloid leukemia patients. Blood Advances. 2021;5(24):5621–5625.
128. Yilmaz M, Kantarjian H, Short NJ, et al. Hypomethylating agent and venetoclax with FLT3 inhibitor “triplet” therapy in older/unfit patients with FLT3 mutated AML. Blood Cancer J. 2022;12(5):77.
129. Maiti A, DiNardo CD, Daver NG, et al. Triplet therapy with venetoclax, FLT3 inhibitor and decitabine for FLT3-mutated acute myeloid leukemia. Blood Cancer J. 2021;11(2):25.
130. Kucukyurt S, Eskazan AE. New drugs approved for acute myeloid leukaemia in 2018. Br J Clin Pharmacol. 2019;85(12):2689–2693.
131. Marconi G, Giannini MB, Bagnato G, et al. The safety profile of FLT3 inhibitors in the treatment of newly diagnosed or relapsed/refractory acute myeloid leukemia. Expert Opin Drug Saf. 2021;20(7):791–799.
132. Eguchi M, Minami Y, Kuzume A, Chi S. Mechanisms underlying resistance to FLT3 inhibitors in acute myeloid leukemia. Biomedicines. 2020;8(8):245.
133. Lam SSY, Leung AYH. Overcoming resistance to FLT3 inhibitors in the treatment of FLT3-mutated AML. Int J Mol Sci. 2020;21(4):1537.
134. Heidel F, Solem FK, Breitenbuecher F, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107(1):293–300.
135. Friedman R. The molecular mechanisms behind activation of FLT3 in acute myeloid leukemia and resistance to therapy by selective inhibitors. Biochimica et Biophysica Acta. 2022;1877(1):188666.
136. Dávila-Rodríguez MJ, Freire TS, Lindahl E, Caracelli I, Zukerman-Schpector J, Friedman R. Is breaking of a hydrogen bond enough to lead to drug resistance? Chem Commun. 2020;56(49):6727–6730.
137. Friedman R. The molecular mechanism behind resistance of the kinase FLT3 to the inhibitor quizartinib. Structure Function Bioinformatics. 2017;85(11):2143–2152.
138. Sun Y, Xia Z, Zhao Q, Zheng B, Zhang M, Ying Y. Insights into the resistance mechanisms of inhibitors to FLT3 F691L mutation via an integrated computational approach. Front Pharmacol. 2019;10:85.
139. Man CH, Fung TK, Ho C, et al. Sorafenib treatment of FLT3-ITD+ acute myeloid leukemia: favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood. 2012;119(22):5133–5143.
140. Georgoulia PS, Bjelic S, Friedman R. Deciphering the molecular mechanism of FLT3 resistance mutations. FEBS J. 2020;287(15):3200–3220.
141. Opatz S, Polzer H, Herold T, et al. Exome sequencing identifies recurring FLT3 N676K mutations in core-binding factor leukemia. Blood. 2013;122(10):1761–1769.
142. Huang K, Yang M, Pan Z, et al. Leukemogenic potency of the novel FLT3-N676K mutant. Ann Hematol. 2016;95(5):783–791.
143. Daver N, Price A, Benton CB, et al. First report of sorafenib in patients with acute myeloid leukemia harboring non-canonical FLT3 mutations. Front Oncol. 2020;10:89.
144. Joshi SK, Sharzehi S, Pittsenbarger J, et al. A noncanonical FLT3 gatekeeper mutation disrupts gilteritinib binding and confers resistance. Am J Hematol. 2021;96(7):E226–E229.
145. Schmidt-Arras D-E, Böhmer A, Markova B, Choudhary C, Serve H, Böhmer F-D. Tyrosine phosphorylation regulates maturation of receptor tyrosine kinases. Mol Cell Biol. 2005;25(9):3690–3703.
146. Tsitsipatis D, Jayavelu AK, Müller JP, et al. Synergistic killing of FLT3ITD-positive AML cells by combined inhibition of tyrosine-kinase activity and N-glycosylation. Oncotarget. 2017;8:16.
147. Fleischmann M, Fischer M, Schnetzke U, et al. Modulation of FLT3-ITD localization and targeting of distinct downstream signaling pathways as potential strategies to overcome FLT3-inhibitor resistance. Cells. 2021;10(11):2992.
148. Lv K, Ren J-G, Han X, Gui J, Gong C, Tong W. Depalmitoylation rewires FLT3-ITD signaling and exacerbates leukemia progression. Blood. 2021;138(22):2244–2255.
149. Weisberg E, Ray A, Nelson E, et al. Reversible resistance induced by FLT3 inhibition: a novel resistance mechanism in mutant FLT3-expressing cells. PLoS One. 2011;6(9):e25351–e25351.
150. Chen F, Ishikawa Y, Akashi A, Naoe T, Kiyoi H. Co-expression of wild-type FLT3 attenuates the inhibitory effect of FLT3 inhibitor on FLT3 mutated leukemia cells. Oncotarget. 2016;7(30):47018–47032.
151. Ghiaur G, Levis M. Mechanisms of resistance to FLT3 inhibitors and the role of the bone marrow microenvironment. Hematol Oncol Clin North Am. 2017;31(4):681–692.
152. Chang Y-T, Hernandez D, Alonso S, et al. Role of CYP3A4 in bone marrow microenvironment-mediated protection of FLT3/ITD AML from tyrosine kinase inhibitors. Blood Adv. 2019;3(6):908–916.
153. Javidi-Sharifi N, Martinez J, English I, et al. FGF2-FGFR1 signaling regulates release of Leukemia-Protective exosomes from bone marrow stromal cells. eLife. 2019;8:e40033.
154. Green AS, Maciel TT, Hospital M-A, et al. Pim kinases modulate resistance to FLT3 tyrosine kinase inhibitors in FLT3-ITD acute myeloid leukemia. Sci Adv. 2015;1(8):e1500221.
155. Park IK, Mundy-Bosse B, Whitman SP, et al. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia. 2015;29(12):2382–2389.
156. Waldeck S, Rassner M, Keye P, et al. CCL5 mediates target-kinase independent resistance to FLT3 inhibitors in FLT3-ITD-positive AML. Mol Oncol. 2020;14(4):779–794.
157. Rummelt C, Gorantla SP, Meggendorfer M, et al. Activating JAK-mutations confer resistance to FLT3 kinase inhibitors in FLT3-ITD positive AML in vitro and in vivo. Leukemia. 2021;35(7):2017–2029.
158. Garitano-Trojaola A, Sancho A, Götz R, et al. Actin cytoskeleton deregulation confers midostaurin resistance in FLT3-mutant acute myeloid leukemia. Communications Biol. 2021;4(1):799.
159. Joshi SK, Nechiporuk T, Bottomly D, et al. The AML microenvironment catalyzes a stepwise evolution to gilteritinib resistance. Cancer Cell. 2021;39(7):999–1014.e1018.
160. McMahon CM, Ferng T, Canaani J, et al. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 2019;9(8):1050–1063.
161. Alotaibi AS, Yilmaz M, Kanagal-Shamanna R, et al. Patterns of resistance differ in patients with acute myeloid leukemia treated with type I versus type II FLT3 inhibitors. Blood Cancer Discov. 2021;2(2):125–134.
162. Lindblad O, Cordero E, Puissant A, et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene. 2016;35(39):5119–5131.
163. Schmalbrock LK, Dolnik A, Cocciardi S, et al. Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood. 2021;137(22):3093–3104.
164. Yamatani K, Ai T, Saito K, et al. Inhibition of BCL2A1 by STAT5 inactivation overcomes resistance to targeted therapies of FLT3-ITD/D835 mutant AML. Transl Oncol. 2022;18:101354.
165. Yoshimoto G, Miyamoto T, Jabbarzadeh-Tabrizi S, et al. FLT3-ITD up-regulates MCL-1 to promote survival of stem cells in acute myeloid leukemia via FLT3-ITD–specific STAT5 activation. Blood. 2009;114(24):5034–5043.
166. Kaufmann SH, Karp JE, Svingen PA, et al. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood. 1998;91(3):991–1000.
167. Li XX, Zhou JD, Wen XM, et al. Increased MCL-1 expression predicts poor prognosis and disease recurrence in acute myeloid leukemia. Onco Targets Ther. 2019;12:3295–3304.
168. Ramsey HE, Fischer MA, Lee T, et al. A novel MCL1 inhibitor combined with venetoclax rescues venetoclax-resistant acute myelogenous leukemia. Cancer Discov. 2018;8(12):1566–1581.
169. Hormi M, Birsen R, Belhadj M, et al. Pairing MCL-1 inhibition with venetoclax improves therapeutic efficiency of BH3-mimetics in AML. Eur J Haematol. 2020;105(5):588–596.
170. Buelow DR, Bhatnagar B, Orwick SJ, et al. BMX kinase mediates gilteritinib resistance in FLT3-mutated AML through microenvironmental factors. Blood Advances. 2022;6(17):5049–5060.
171. Yamaura T, Nakatani T, Uda K, et al. A novel irreversible FLT3 inhibitor, FF-10101, shows excellent efficacy against AML cells with FLT3 mutations. Blood. 2018;131(4):426–438.
172. Ferng TT, Terada D, Ando M, et al. The Irreversible FLT3 Inhibitor FF-10101 Is Active Against a Diversity of FLT3 Inhibitor Resistance Mechanisms. Mol Cancer Ther. 2022;21(5):844–854.
173. Daver NG, Lee KH, Yoon -S-S, et al. HM43239, a novel potent small molecule FLT3 inhibitor, in acute myeloid leukemia (AML) with FMS-like tyrosine kinase 3 (FLT3) mutations: phase 1 /2 study. Blood. 2019;134(Supplement_1):1331.
174. Xu B, Zhao Y, Wang X, Gong P, Ge W. MZH29 is a novel potent inhibitor that overcomes drug resistance FLT3 mutations in acute myeloid leukemia. Leukemia. 2017;31(4):913–921.
175. Yu Z, Du J, Hui H, et al. LT-171-861, a novel FLT3 inhibitor, shows excellent preclinical efficacy for the treatment of FLT3 mutant acute myeloid leukemia. Theranostics. 2021;11(1):93–106.
176. Yuan T, Qi B, Jiang Z, et al. Dual FLT3 inhibitors: against the drug resistance of acute myeloid leukemia in recent decade. Eur J Med Chem. 2019;178:468–483.
177. Tariq MU, Furqan M, Parveen H, et al. CCT245718, a dual FLT3/Aurora A inhibitor overcomes D835Y-mediated resistance to FLT3 inhibitors in acute myeloid leukaemia cells. Br J Cancer. 2021;125(7):966–974.
178. Moore AS, Faisal A, Mak GWY, et al. Quizartinib-resistant FLT3-ITD acute myeloid leukemia cells are sensitive to the FLT3-Aurora kinase inhibitor CCT241736. Blood Advances. 2020;4(7):1478–1491.
179. Zeng Z, Ly C, Daver N, et al. High-throughput proteomic profiling reveals mechanisms of action of AMG925, a dual FLT3-CDK4/6 kinase inhibitor targeting AML and AML stem/progenitor cells. Ann Hematol. 2021;100(6):1485–1496.
180. Wang P, Xiao X, Zhang Y, et al. A dual inhibitor overcomes drug-resistant FLT3-ITD acute myeloid leukemia. J Hematol Oncol. 2021;14(1):105.
181. Azhar M, Kincaid Z, Kesarwani M, et al. Momelotinib is a highly potent inhibitor of FLT3-mutant AML. Blood Adv. 2022;6(4):1186–1192.
182. Jeon JY, Zhao Q, Buelow DR, et al. Preclinical activity and a pilot phase I study of pacritinib, an oral JAK2/FLT3 inhibitor, and chemotherapy in FLT3-ITD-positive AML. Invest New Drugs. 2020;38(2):340–349.
183. Czardybon W, Windak R, Gołas A, et al. A novel, dual pan-PIM/FLT3 inhibitor SEL24 exhibits broad therapeutic potential in acute myeloid leukemia. Oncotarget. 2018;9(24):16917–16931.
184. Melgar K, Walker MM, Jones LM, et al. Overcoming adaptive therapy resistance in AML by targeting immune response pathways. Sci Transl Med. 2019;11(508):eaaw8828.
185. Minson KA, Smith CC, DeRyckere D, et al. The MERTK/FLT3 inhibitor MRX-2843 overcomes resistance-conferring FLT3 mutations in acute myeloid leukemia. JCI Insight. 2016;1:3.
186. Wang A, Wu H, Chen C, et al. Dual inhibition of AKT/FLT3-ITD by A674563 overcomes FLT3 ligand-induced drug resistance in FLT3-ITD positive AML. Oncotarget. 2016;7:20.
187. Morales ML, Arenas A, Ortiz-Ruiz A, et al. MEK inhibition enhances the response to tyrosine kinase inhibitors in acute myeloid leukemia. Sci Rep. 2019;9(1):18630.
188. Brinton LT, Zhang P, Williams K, et al. Synergistic effect of BCL2 and FLT3 co-inhibition in acute myeloid leukemia. J Hematol Oncol. 2020;13(1):139.
189. Ma J, Zhao S, Qiao X, et al. Inhibition of Bcl-2 synergistically enhances the antileukemic activity of midostaurin and gilteritinib in preclinical models of FLT3-mutated acute myeloid leukemia. Clin Cancer Res. 2019;25(22):6815–6826.
190. Singh Mali R, Zhang Q, DeFilippis RA, et al. Venetoclax combines synergistically with FLT3 inhibition to effectively target leukemic cells in FLT3-ITD+ acute myeloid leukemia models. Haematologica. 2021;106(4):1034–1046.
191. Yilmaz M, Kantarjian HM, Muftuoglu M, et al. Quizartinib with decitabine and venetoclax (triplet) is highly active in patients with FLT3-ITD mutated acute myeloid leukemia (AML). J Clin Oncol. 2021;39(15_suppl):e19019.
192. Xu G, Mao L, Liu H, Yang M, Jin J, Qian W. Sorafenib in combination with low-dose-homoharringtonine as a salvage therapy in primary refractory FLT3-ITD-positive AML: a case report and review of literature. Int J Clin Exp Med. 2015;8(11):19891–19894.
193. Wang F, Huang J, Guo T, et al. Homoharringtonine synergizes with quizartinib in FLT3-ITD acute myeloid leukemia by targeting FLT3-AKT-c-Myc pathway. Biochem Pharmacol. 2021;188:114538.
194. Cai J, Huang H, Hu X, et al. Homoharringtonine synergized with gilteritinib results in the downregulation of myeloid cell leukemia-1 by upregulating UBE2L6 in FLT3-ITD-mutant acute myeloid (leukemia) cell lines. J Oncol. 2021;2021:3766428.
195. Pillinger G, Abdul-Aziz A, Zaitseva L, et al. Targeting BTK for the treatment of FLT3-ITD mutated acute myeloid leukemia. Sci Rep. 2015;5(1):12949.
196. Wu H, Hu C, Wang A, et al. Ibrutinib selectively targets FLT3-ITD in mutant FLT3-positive AML. Leukemia. 2016;30(3):754–757.
197. Oellerich T, Mohr S, Corso J, et al. FLT3-ITD and TLR9 use Bruton tyrosine kinase to activate distinct transcriptional programs mediating AML cell survival and proliferation. Blood. 2015;125(12):1936–1947.
198. Andreeff M, Zhang W, Kumar P, et al. Synergistic anti-leukemic activity with combination of FLT3 inhibitor quizartinib and MDM2 inhibitor milademetan in FLT3-ITD Mutant/p53 wild-type acute myeloid leukemia models. Blood. 2018;132(Supplement 1):2720.
199. Katja S, Miguel ATM, Corinne S, Beatrice UM, Thomas P. MDM2- and FLT3-inhibitors in the treatment of FLT3-ITD acute myeloid leukemia, specificity and efficacy of NVP-HDM201 and midostaurin. Haematologica. 2018;103(11):1862–1872.
200. Cao H, Tadros V, Hiramoto B, et al. Targeting TKI-activated NFKB2-MIF/CXCLs-CXCR2 signaling pathways in FLT3 mutated acute myeloid leukemia reduced blast viability. Biomedicines. 2022;10(5):1038.
201. Qiao X, Ma J, Knight T, et al. The combination of CUDC-907 and gilteritinib shows promising in vitro and in vivo antileukemic activity against FLT3-ITD AML. Blood Cancer J. 2021;11(6):111.
202. He X, Zhu Y, Lin Y-C, et al. PRMT1-mediated FLT3 arginine methylation promotes maintenance of FLT3-ITD+ acute myeloid leukemia. Blood. 2019;134(6):548–560.
203. Nishida Y, Zhao R, Heese LE, et al. Inhibition of translation initiation factor eIF4a inactivates heat shock factor 1 (HSF1) and exerts anti-leukemia activity in AML. Leukemia. 2021;35(9):2469–2481.
204. Li Y, Li H, Wang M-N, et al. Suppression of leukemia expressing wild-type or ITD-mutant FLT3 receptor by a fully human anti-FLT3 neutralizing antibody. Blood. 2004;104(4):1137–1144.
205. Piloto O, Levis M, Huso D, et al. Inhibitory anti-FLT3 antibodies are capable of mediating antibody-dependent cell-mediated cytotoxicity and reducing engraftment of acute myelogenous leukemia blasts in nonobese diabetic/severe combined immunodeficient mice. Cancer Res. 2005;65:1514–1522.
206. Hofmann M, Große-Hovest L, Nübling T, et al. Generation, selection and preclinical characterization of an Fc-optimized FLT3 antibody for the treatment of myeloid leukemia. Leukemia. 2012;26(6):1228–1237.
207. Yeung YA, Krishnamoorthy V, Dettling D, et al. An optimized full-length FLT3/CD3 bispecific antibody demonstrates potent anti-leukemia activity and reversible hematological toxicity. Mol Ther. 2020;28(3):889–900.
208. Mehta NK, Pfluegler M, Meetze K, et al. A novel IgG-based FLT3xCD3 bispecific antibody for the treatment of AML and B-ALL. J Immunother Cancer. 2022;10(3):e003882.
209. Jackson HJ, Rafiq S, Brentjens RJ. Driving CAR T-cells forward. Nat Rev Clin Oncol. 2016;13(6):370–383.
210. Chen L, Mao H, Zhang J, et al. Targeting FLT3 by chimeric antigen receptor T cells for the treatment of acute myeloid leukemia. Leukemia. 2017;31(8):1830–1834.
211. Karbowski C, Goldstein R, Frank B, et al. Nonclinical safety assessment of AMG 553, an investigational chimeric antigen receptor T-cell therapy for the treatment of acute myeloid leukemia. Toxicol Sci. 2020;177(1):94–107.
212. Wang Y, Xu Y, Li S, et al. Targeting FLT3 in acute myeloid leukemia using ligand-based chimeric antigen receptor-engineered T cells. J Hematol Oncol. 2018;11(1):60.
213. Maiorova V, Mollaev MD, Vikhreva P, et al. Natural Flt3Lg-based chimeric antigen receptor (Flt3-CAR) T cells successfully target Flt3 on AML cell lines. Vaccines. 2021;9(11):1238.
214. Jetani H, Garcia-Cadenas I, Nerreter T, et al. CAR T-cells targeting FLT3 have potent activity against FLT3−ITD+ AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia. 2018;32(5):1168–1179.
215. Sommer C, Cheng H-Y, Nguyen D, et al. Allogeneic FLT3 CAR T cells with an off-switch exhibit potent activity against AML and can be depleted to expedite bone marrow recovery. Mol Ther. 2020;28(10):2237–2251.
216. Li KX, Wu HY, Pan WY, et al. A novel approach for relapsed/refractory FLT3mut+ acute myeloid leukaemia: synergistic effect of the combination of bispecific FLT3scFv/NKG2D-CAR T cells and gilteritinib. Mol Cancer. 2022;21(1):66.
217. Ottone T, Zaza S, Divona M, et al. Identification of emerging FLT3 ITD-positive clones during clinical remission and kinetics of disease relapse in acute myeloid leukaemia with mutated nucleophosmin. Br J Haematol. 2013;161(4):533–540.
218. Angelini DF, Ottone T, Guerrera G, et al. A leukemia-associated CD34/CD123/CD25/CD99+ immunophenotype identifies FLT3-mutated clones in acute myeloid leukemia. Clin Cancer Res. 2015;21(17):3977–3985.
219. Travaglini S, Angelini DF, Alfonso V, et al. Characterization of FLT3-ITDmut acute myeloid leukemia: molecular profiling of leukemic precursor cells. Blood Cancer J. 2020;10(8):85.
220. Travaglini S, Ottone T, Angelini DF, et al. CD99 as a novel therapeutic target on leukemic progenitor cells in FLT3-ITDmut AML. Leukemia. 2022;36(6):1685–1688.
221. Tarlock K, Alonzo TA, Gerbing RB, et al. Gemtuzumab ozogamicin reduces relapse risk in FLT3/ITD acute myeloid leukemia: a report from the children’s oncology group. Clin Cancer Res. 2016;22(8):1951–1957.
222. Brodská B, Otevřelová P, Šálek C, Fuchs O, Gašová Z, Kuželová K. High PD-L1 expression predicts for worse outcome of leukemia patients with concomitant NPM1 and FLT3 mutations. Int J Mol Sci. 2019;20(11):2823.
223. Charlet A, Kappenstein M, Keye P, et al. The IL-3, IL-5, and GM-CSF common receptor beta chain mediates oncogenic activity of FLT3-ITD-positive AML. Leukemia. 2022;36(3):701–711.
224. Yao Q, Nishiuchi R, Li Q, Kumar AR, Hudson WA, Kersey JH. FLT3 expressing leukemias are selectively sensitive to inhibitors of the molecular chaperone heat shock protein 90 through destabilization of signal transduction-associated kinases. Clin Cancer Res. 2003;9(12):4483–4493.
225. Al Shaer L, Walsby E, Gilkes A, et al. Heat shock protein 90 inhibition is cytotoxic to primary AML cells expressing mutant FLT3 and results in altered downstream signalling. Br J Haematol. 2008;141(4):483–493.
226. Reikvam H, Hatfield KJ, Ersvær E, et al. Expression profile of heat shock proteins in acute myeloid leukaemia patients reveals a distinct signature strongly associated with FLT3 mutation status – consequences and potentials for pharmacological intervention. Br J Haematol. 2012;156(4):468–480.
227. Yu C, Kancha RK, Duyster J. Targeting oncoprotein stability overcomes drug resistance caused by FLT3 kinase domain mutations. PLoS One. 2014;9(5):e97116.
228. Oshikawa G, Nagao T, Wu N, Kurosu T, Miura O. c-Cbl and Cbl-b ligases mediate 17-allylaminodemethoxygeldanamycin-induced degradation of autophosphorylated Flt3 kinase with internal tandem duplication through the ubiquitin proteasome pathway *. J Biol Chem. 2011;286(35):30263–30273.
229. Taylor SJ, Thien CBF, Dagger SA, et al. Loss of c-Cbl E3 ubiquitin ligase activity enhances the development of myeloid leukemia in FLT3-ITD mutant mice. Exp Hematol. 2015;43(3):191–206.e191.
230. George P, Bali P, Cohen P, et al. Cotreatment with 17-Allylamino-Demethoxygeldanamycin and FLT-3 Kinase Inhibitor PKC412 is highly effective against human acute myelogenous leukemia cells with mutant FLT-3. Cancer Res. 2004;64(10):3645–3652.
231. Katayama K, Noguchi K, Sugimoto Y. Heat shock protein 90 inhibitors overcome the resistance to Fms-like tyrosine kinase 3 inhibitors in acute myeloid leukemia. Oncotarget. 2018;9:76.
232. Weisberg EL, Schauer NJ, Yang J, et al. Inhibition of USP10 induces degradation of oncogenic FLT3. Nat Chem Biol. 2017;13(12):1207–1215.
233. Yu M, Fang Z, Wang W, et al. Wu-5, a novel USP10 inhibitor, enhances crenolanib-induced FLT3-ITD-positive AML cell death via inhibiting FLT3 and AMPK pathways. Acta Pharmacol Sin. 2021;42(4):604–612.
234. Hu C, Zou F, Wang A, et al. Targeting chaperone protein HSP70 as a novel therapeutic strategy for FLT3-ITD-positive acute myeloid leukemia. Signal Transduction Targeted Therapy. 2021;6(1):334.
235. Larrue C, Saland E, Boutzen H, et al. Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood. 2016;127(7):882–892.
236. Lopez-Reyes RG, Quinet G, Gonzalez-Santamarta M, Larrue C, Sarry J-E, Rodriguez MS. Inhibition of the proteasome and proteaphagy enhances apoptosis in FLT3-ITD-driven acute myeloid leukemia. FEBS Open Bio. 2021;11(1):48–60.
237. Liu X-J, Wang L-N, Zhang Z-H, et al. Arsenic trioxide induces autophagic degradation of the FLT3-ITD mutated protein in FLT3-ITD acute myeloid leukemia cells. J Cancer. 2020;11(12):3476–3482.
238. Nagai K, Hou L, Li L, et al. Combination of ATO with FLT3 TKIs eliminates FLT3/ITD+ leukemia cells through reduced expression of FLT3. Oncotarget. 2018;9:68.
239. Ly BTK, Chi HT, Yamagishi M, et al. Inhibition of FLT3 expression by green tea catechins in FLT3 mutated-AML cells. PLoS One. 2013;8(6):e66378–e66378.
240. He M, Cao C, Ni Z, et al. PROTACs: great opportunities for academia and industry (an update from 2020 to 2021). Signal Transduction Targeted Therapy. 2022;7(1):181.
241. Cao S, Ma L, Liu Y, et al. Proteolysis-targeting chimera (PROTAC) modification of dovitinib enhances the antiproliferative effect against FLT3-ITD-positive acute myeloid leukemia cells. J Med Chem. 2021;64(22):16497–16511.
242. Chen Y, Yuan X, Tang M, et al. Degrading FLT3-ITD protein by proteolysis targeting chimera (PROTAC). Bioorg Chem. 2022;119:105508.
243. Burslem GM, Song J, Chen X, Hines J, Crews CM. Enhancing antiproliferative activity and selectivity of a FLT-3 inhibitor by proteolysis targeting chimera conversion. J Am Chem Soc. 2018;140(48):16428–16432.
244. Yen S-C, Wu Y-W, Huang -C-C, et al. O-methylated flavonol as a multi-kinase inhibitor of leukemogenic kinases exhibits a potential treatment for acute myeloid leukemia. Phytomedicine. 2022;100:154061.
245. Chen L-C, Huang H-L, HuangFu W-C, et al. Biological evaluation of selected flavonoids as inhibitors of MNKs targeting acute myeloid leukemia. J Nat Prod. 2020;83(10):2967–2975.
246. Yen S-C, Chen L-C, Huang H-L, et al. Investigation of selected flavonoid derivatives as potent FLT3 inhibitors for the potential treatment of acute myeloid leukemia. J Nat Prod. 2021;84(1):1–10.
247. Malik HS, Bilal A, Ullah R, et al. Natural and semisynthetic chalcones as dual FLT3 and microtubule polymerization inhibitors. J Nat Prod. 2020;83(10):3111–3121.
248. Cao Z-X, Wen Y, He J-L, et al. Isoliquiritigenin, an orally available natural FLT3 inhibitor from licorice, exhibits selective anti-acute myeloid leukemia efficacy in vitro and in vivo. Mol Pharmacol. 2019;96(5):589–599.
249. Ersöz NŞ, Adan A. Differential in vitro anti-leukemic activity of resveratrol combined with serine palmitoyltransferase inhibitor myriocin in FMS-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD) carrying AML cells. Cytotechnology. 2022;74(2):271–281.
250. Ersöz NŞ, Adan A. Resveratrol triggers anti-proliferative and apoptotic effects in FLT3-ITD-positive acute myeloid leukemia cells via inhibiting ceramide catabolism enzymes. Medical Oncol. 2022;39(3):35.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Impact of HOXB4 and PRDM16 Gene Expressions on Prognosis and Treatment Response in Acute Myeloid Leukemia Patients
El-Meligui YM, Hassan NM, Kassem AB, Gouda NA, Mohanad M, Hamouda MA, Salahuddin A
Pharmacogenomics and Personalized Medicine 2022, 15:663-674
Published Date: 25 June 2022
Health Disparities in Acute Myeloid Leukemia Patients Undergoing Treatment with Tyrosine Kinase Inhibitor (TKI) Therapy Targeting FLT3, IDH1, or IDH2
Espinoza-Gutarra MR, Jarrett BA, Wang X, Afghahi A, Bae S
Blood and Lymphatic Cancer: Targets and Therapy 2026, 16:559759
Published Date: 12 March 2026
Exploring the Bone Marrow Microenvironment as a Therapeutic Barrier and Targetable Source of Crosstalk in Acute Myeloid Leukemia
Kolosova A, Mould IA, Pepper C, Mitchell S, Pepper AG, Ladikou EE, Simoes FA
Blood and Lymphatic Cancer: Targets and Therapy 2026, 16:511758
Published Date: 19 May 2026
CD71-Targeted and ROS-Responsive Micelles for Homoharringtonine Delivery to Enhance Therapeutic Efficiency Against FLT3-ITD Acute Myeloid Leukemia
Tang Y, Li J, Ye W, Du Y, Zhang Y, Yang Y, Ye Y, Gong Y
International Journal of Nanomedicine 2026, 21:588865
Published Date: 27 May 2026