Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 16
Exploring the Bone Marrow Microenvironment as a Therapeutic Barrier and Targetable Source of Crosstalk in Acute Myeloid Leukemia
Authors Kolosova A, Mould IA ![]() , Pepper C
, Pepper C ![]() , Mitchell S, Pepper AG, Ladikou EE, Simoes FA
, Mitchell S, Pepper AG, Ladikou EE, Simoes FA ![]()
Received 31 January 2026
Accepted for publication 24 April 2026
Published 19 May 2026 Volume 2026:16 511758
DOI https://doi.org/10.2147/BLCTT.S511758
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Wilson Gonsalves
Aleksandra Kolosova,1,* Imogen Anna Mould,1,* Chris Pepper,1 Simon Mitchell,1 Andrea GS Pepper,1 Eleni E Ladikou,1,2 Fabio A Simoes1
1Department of Clinical and Experimental Medicine, Brighton and Sussex Medical School, University of Brighton and University of Sussex, Brighton, UK; 2King’s College Hospital NHS Foundation Trust, London, UK
*These authors contributed equally to this work
Correspondence: Fabio A Simoes, Email [email protected]
Abstract: Acute myeloid leukemia (AML) is an aggressive and genetically heterogeneous hematological malignancy characterized by the accumulation of immature myeloid blasts that disrupt healthy hematopoiesis. Despite advances in molecular profiling and targeted therapies, overcoming drug resistance and relapse remains a significant clinical challenge, resulting in poor long-term outcomes. Crucially, disease persistence is sustained not merely by intrinsic genetic lesions but by a highly adaptive bone marrow microenvironment (BMME) that functions as a therapeutic barrier. While the healthy niche tightly regulates hematopoietic stem cell maintenance, leukemic blasts co-opt stromal, vascular, and immune components to establish a sanctuary that fuels proliferation and shields the disease from cytotoxic stress. However, dissecting these reciprocal dependency mechanisms uncovers critical vulnerabilities, presenting a vital opportunity to develop novel targeted therapies. In this review, we discuss the architecture of the healthy BMME and its pathological AML-driven remodeling. We describe the role of specific signaling axes that govern AML-BMME crosstalk and evaluate targeted therapeutic strategies designed to uncouple these protective interactions. Finally, we highlight that current preclinical models lack the complexity of the BMME stromal components and its spatial organization, a limitation that continues to hinder clinical translation and delay the development of effective combination therapies.
Plain Language Summary: Acute Myeloid Leukemia (AML) is an aggressive blood cancer. It occurs when immature cells build up in the bone marrow, the soft tissue inside your bones where the body makes new blood. While many people respond well to initial treatment, the disease often returns because some AML cells survive by “hijacking” their surroundings.
This review explains how AML cells attach themselves to healthy bone marrow structures and send out chemical signals that force nearby healthy cells to protect them. This creates a shield that hides the cancer from both chemotherapy and the body’s own immune system. Importantly, the review explores how we can target this communication to prevent the disease from returning.
A major challenge remains the difficulty of accurately modeling this complex environment in the laboratory. Current models often struggle to mimic the complex human bone marrow, which is why many drugs that appear promising in tests fail to help people in clinical trials. Therefore, the review highlights the need to use advanced models which allow researchers to more reliably test new strategies to disrupt the protective bone marrow environment.
The review concludes that the bone marrow acts as a single, connected shield rather than separate pieces. Because these parts work together to protect the cancer, blocking just one pathway is often not enough. Overcoming resistance requires new strategies that simultaneously target the leukemia cells and the multiple parts of the environment that support them.
Keywords: acute myeloid leukemia, bone marrow microenvironment, therapeutic resistance, targeted therapy
Introduction
Acute myeloid leukemia (AML) is an aggressive hematological malignancy characterized by the clonal expansion of myeloid progenitor cells with a blockade in differentiation.1 This process is driven by leukemic stem cells (LSCs), a self-renewing subpopulation at the apex of the leukemic hierarchy that sustains the disease and often resists conventional therapy.2 The resulting accumulation of immature blasts in the bone marrow (BM) disrupts normal hematopoiesis, giving rise to anemia, thrombocytopenia, and increased susceptibility to infections.3
Epidemiology, Genetics and Current Treatments
AML is the most common acute leukemia in adults, with an incidence of approximately 4.4 per 100,000 individuals in the United Kingdom (UK).4 Although AML mostly affects older people, with a median diagnosis age of 72 years, it can also occur in children (pediatric AML), which generally has a much improved prognosis.4,5 However, even with induction therapy achieving complete remission in most patients (60–80% in younger adults and 45–60% in older adults), long-term outcomes remain poor, with only 16.5% surviving beyond five years in the UK, largely due to relapse and the emergence of therapy resistance.4,6
AML is a genetically heterogeneous disease, with recurrent mutations in NPM1, FLT3, DNMT3A, IDH1/2, RUNX1 and TP53.7,8 It is further characterized by translocation events that generate fusion oncogenes, most notably t(15;17) [PML-RARA], t(8;21) [RUNX1-RUNX1T1], inv(16) [CBFB-MYH11], inv(3) [GATA2-MECOM], t(6;9) [DEK-NUP214] and KMT2A rearrangements. This molecular diversity has major clinical implications for prognosis and therapy selection.9 Despite significant advances in molecular profiling and the introduction of targeted therapies, intensive induction therapy with cytarabine plus an anthracycline (“7+3”), followed by consolidation with further chemotherapy or hematopoietic stem cell transplantation (HSCT), has remained the mainstay for decades.9–11
More recently, there has been a shift towards targeted therapies that exploit specific genetic vulnerabilities. Several classes of drugs have demonstrated significant clinical efficacy, particularly when combined with hypomethylating agents or chemotherapy: FLT3 inhibitors (midostaurin,12,13 gilteritinib14); IDH1/2 inhibitors (ivosidenib,15,16 enasidenib17–19); and the B-cell lymphoma 2 (BCL-2) inhibitor venetoclax.20–26 Outcomes have also been improved in some patients through the incorporation of targeted agents into intensive chemotherapy, as seen with gemtuzumab ozogamicin (GO) in CD33-positive disease.27
However, resistance mechanisms and relapse continue to challenge durable responses.28 As a result of the significant challenges that persist, despite genetic stratification, attention is turning to the bone marrow microenvironment (BMME) as a contributor to therapeutic failure.29–32 AML blasts exploit the BMME to evade immune surveillance, gain a metabolic advantage, resist chemotherapy, and establish reservoirs of residual disease. However, despite significant promise in preclinical settings, niche-targeting strategies have yet to deliver a consistent clinical benefit (Table 1).
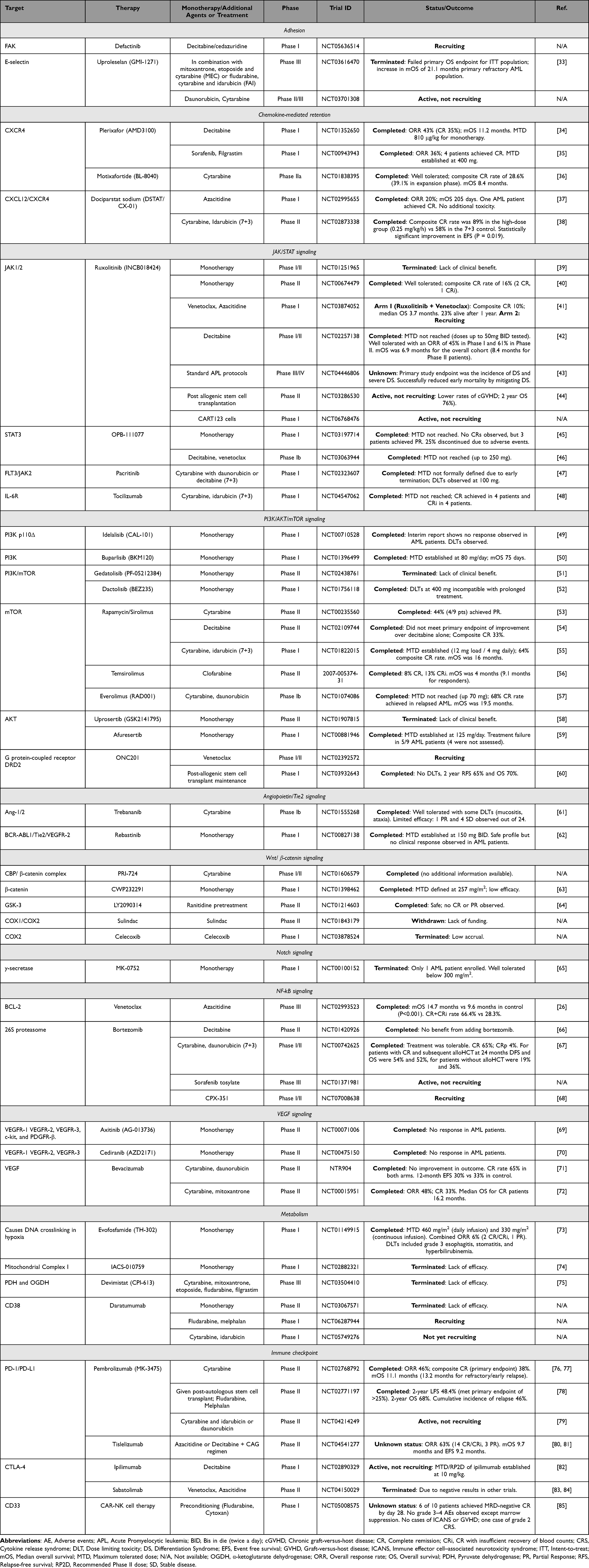 |
Table 1 Clinical Trials Targeting Crosstalk Between Acute Myeloid Leukemia (AML) Cells and the Bone Marrow Microenvironment. This Table Summarizes Key Clinical Trials Investigating Therapeutic Strategies Designed to Disrupt the Protective Interactions Between AML Blasts and the Bone Marrow Microenvironment. The Trials are Categorized by the Specific Biological Signaling Axis or Niche Component Being Targeted, Including Adhesion Molecules, Chemokine-Mediated Retention, Intracellular Signaling Pathways, Metabolism, and Immune Checkpoints. Clinical Trial Identifiers (NCT Numbers) and Status Information Were Verified via ClinicalTrials.gov or the EU clinical trials register and are Accurate as of 1 April 2026. |
Scope of This Review
In this review, we discuss how AML cells interact with and reshape the BMME, highlighting that these interactions serve as essential mechanisms for leukemic survival while simultaneously offering opportunities for new targeted therapies. We also evaluate the current barriers to clinical translation of therapies targeting interactions between AML cells and the BMME, specifically addressing the inherent complexity and redundancy of the BMME and the technical limitations of existing model systems.
Bone Marrow Microenvironment
The BM is a complex and dynamic tissue responsible for maintaining lifelong hematopoiesis through a delicate balance between hematopoietic stem cell (HSC) self-renewal, proliferation, and differentiation.86–88 This process is tightly regulated by the BMME, which provides physical scaffolding, cellular interactions, and molecular signals essential for HSC maintenance and lineage commitment. In AML, this initially healthy, HSC-supportive niche is hijacked and remodeled to create a distinctly pathogenic BMME to favor disease progression and treatment resistance, a transition that can begin with early pre-leukemic mutations that facilitate clonal expansion and leukemogenesis.89,90 This pre-leukemic remodeling creates a dysfunctional environment that facilitates the clonal expansion of mutated cells over healthy progenitors.91 By providing a selective advantage to these early clones, the remodeled niche actively contributes to the clonal evolution and eventual leukemogenic transformation of the disease.92–96 Therefore, understanding the physiological role of individual components and interactions within the BMME, and how they are manipulated in AML, can shed light on how we may re-sensitize leukemic cells to treatment.32
Healthy Niche
Two major anatomical niches are recognized within the BM: the endosteal and the vascular niches, although increasing evidence supports significant functional overlap between these compartments.97 The endosteal niche, rich in osteoblasts, supports HSC quiescence and self-renewal partly via stromal Wnt/β-catenin and Notch signaling.90,98–101 In contrast, the vascular niche, situated near sinusoids and arterioles, is formed predominantly by endothelial cells that secrete growth factors including stem cell factor (SCF), C-X-C motif chemokine 12 (CXCL12; also known as stromal cell-derived factor 1 or SDF-1), and Transforming Growth Factor Beta 2 (TGF-β2) to promote HSC activation, proliferation, and differentiation.90,98,99 Genetic lineage-tracing studies have shown that SCF derived specifically from arterial endothelial cells is essential in HSC maintenance.98,99
Within these niches, mesenchymal stromal cells (MSCs) form a central component of the stromal compartment and are multipotent progenitors capable of differentiating into osteoblasts, adipocytes, and chondrocytes, thereby contributing to the structural and functional organization of the BM niche.102,103 The majority of MSCs are situated within the perivascular space of the BMME, in close association with blood vessels, and are characterized by the expression of leptin receptor (Lepr), Nestin (Nes), or NG2 (Cspg4), which distinguish overlapping subsets with distinct HSC-supportive roles.86,99,102,104,105 CXCL12-abundant reticular (CAR) cells are an MSC subset, which largely correspond to Lepr+ perivascular MSCs. CARs form extensive structural and functional scaffolds that coordinate HSC localization, anchor them in place and mediate their quiescence.106
Fibroblasts are a mesenchymal population within the BM that is distinct from MSCs and contribute to the composition of a healthy BMME network.90,107 Although distinguishing fibroblasts from BM-MSCs remains challenging due to the absence of definitive surface markers, transcriptomic analysis has identified multiple subsets, including subsets expressing CXCL12 and ANGPT1, indicating a potential for direct regulation of the BMME.90
In addition to the key regulatory cell types mentioned above, there is an emerging picture of less well studied BMME components that may have significant functional roles in normal BMME composition and signaling. Adipocytes, once considered inert space fillers, are now recognized as active contributors to hematopoietic support through the production of factors such as SCF.108,109 Non-myelinating Schwann cells help preserve HSC dormancy by regulating sympathetic innervation and TGF-β activation.110 Megakaryocytes have also been implicated in regulating HSC proliferation; for example by releasing thrombopoietin (TPO) which can stimulate platelet production as well as supporting HSC growth.111
The immune compartment forms another integral part of the BMME, establishing a complex interplay between immune effectors and this specialized niche. Neutrophils, macrophages, T cells, and natural killer (NK) cells are distinct populations that mediate HSC fate by regulating processes such as retention, differentiation, and quiescence.112
These diverse cellular populations are surrounded by highly specialized extracellular matrix (ECM) that provides both structural support and a delivery mechanism for instructive biochemical and biomechanical cues essential for HSC regulation.113,114 The ECM is composed of glycoproteins such as laminins, fibronectin, and osteopontin, together with various collagens and proteoglycans. Collectively, these components form an organized scaffold that anchors niche cells and shapes gradients of cytokines and growth factors within the BMME. Beyond biochemical signaling, the physical properties of the ECM, such as stiffness and topography, act as potent biophysical regulators of HSC fate through mechanotransduction pathways, influencing differentiation.115
AML Niche
This balance of physical and biochemical cues that is carefully maintained in health is disrupted in malignancy. In AML, the BMME is co-opted and remodeled into a malignant niche that promotes leukemic cell survival, proliferation, and therapy resistance while impairing normal hematopoiesis.91 Recent evidence indicates that these changes can occur early in disease evolution, with a “pre-leukemic” niche emerging before overt transformation. Profiling of the pre-leukemic BMME has revealed an altered state, characterized by a loss of endothelial cells and reduced collagen deposition by fibroblasts, suggesting that microenvironmental dysfunction may precede and facilitate malignant progression.89 Once established, AML cells further manipulate cellular adhesion pathways and soluble factors within the niche to reinforce protection from chemotherapy, thereby promoting resistance and relapse.30 Examples of the extensive interactions between AML and the BMME discussed in this review are summarized in Figure 1, highlighting the complexity of this protective niche. By co-opting native BMME signaling to drive retention, proliferation, and survival, AML blasts evade therapeutic intervention, underscoring the critical need to consider this remodeled niche when assessing novel targets.
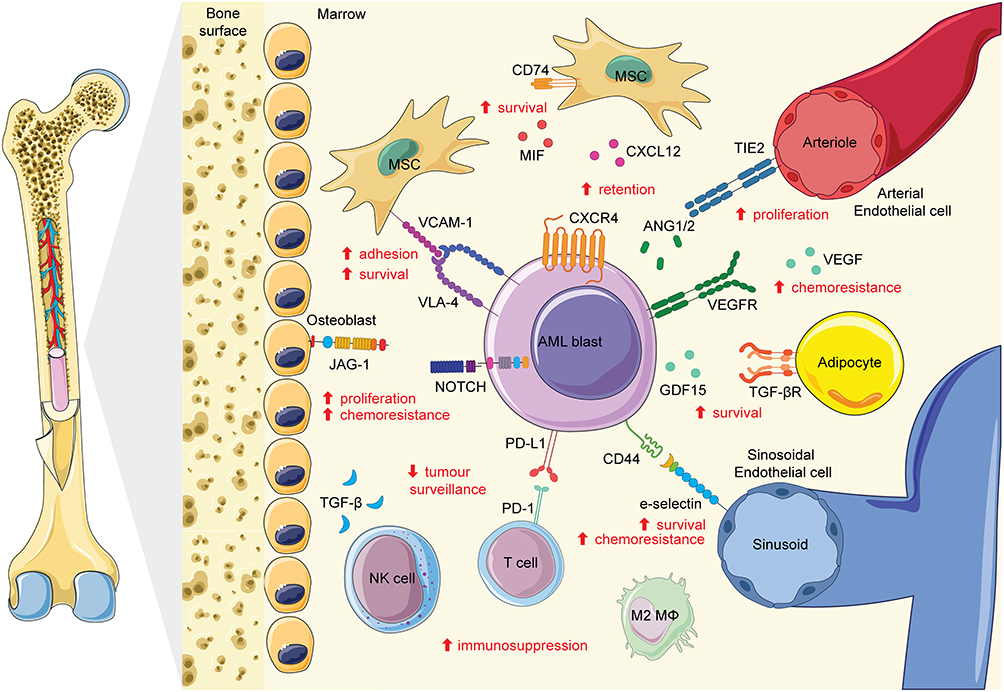 |
Figure 1 The bone marrow microenvironment as a therapeutic barrier and source of pro-survival crosstalk in acute myeloid leukemia (AML). AML blasts interact with niche-resident mesenchymal stromal cells (MSCs), osteoblasts, adipocytes and endothelial cells (arterial and sinusoidal) through adhesion and signaling axes including VCAM-1/VLA-4, CXCL12/CXCR4, E-selectin/CD44, JAG1/Notch. AML-derived factors such as MIF, ANG1/2 and GDF15 further engage receptors on BMME cells (CD74, TIE2 and TGF-βR), reinforcing reciprocal crosstalk between leukemic and stromal compartments. These interactions promote blast adhesion, retention within the niche, proliferation and resistance to chemotherapy. In parallel, the immune landscape is driven towards an immunosuppressive state; TGF-β released by AML cells impairs NK cell surveillance, while PD-L1/PD-1 checkpoint engagement and M2 macrophage polarization facilitate immune evasion. M2 macrophages abbreviated to M2 MΦ. Red arrows and text indicate the predominant functional outcomes of each interaction. Some parts of image adapted from Servier Medical Art (https://smart.servier.com/), licensed under CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). |
Adhesion-Mediated Interactions and Resistance
AML blasts interact closely with stromal and endothelial cells through adhesion molecules that activate pro-survival signaling and promote resistance to chemotherapy. Integrins represent a prominent family of these receptors, consisting of heterodimeric transmembrane proteins composed of α and β subunits.116 While leukemic cells express various integrin combinations, the β1-integrin subfamily has been shown to be essential for anchoring blasts to the extracellular matrix and stromal cells. β1-integrin signaling through the Very Late Antigen-4 (VLA-4; α4β1 integrin)–Vascular Cell Adhesion Molecule 1 (VCAM-1) axis, has been shown to enhance AML blast survival. Specifically, the overexpression of VLA-4 on AML cells and VCAM-1 on stromal cells have been shown to trigger nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-dependent signaling that protects them from cytotoxic stress.117 Engagement of CD44 enhances VLA-4 activation and avidity on AML cells, strengthening adhesion to VCAM-1-expressing stromal cells and promoting resistance to chemotherapy.118 These leukemia-supporting mechanisms can provide novel therapeutic targets, as disrupting these interactions through blocking CD44, either alone or in combination with focal adhesion kinase (FAK) blockade, re-sensitizes AML cells to treatment in vitro.119
Although direct targeting of the VLA-4–VCAM-1 axis has not yet yielded an approved therapy for AML, the anti-VLA-4 monoclonal antibody natalizumab, approved for multiple sclerosis, demonstrates the pharmacological feasibility of this approach.120 Furthermore, integrin-targeted agents have entered clinical evaluation in other malignancies,121 while dual targeting of FAK and CD44 is yet to progress further than in vitro studies,119 FAK inhibition with defactinib is currently being investigated in combination with ASTX727 (decitabine/cedazuridine) in a phase I clinical trial (NCT05636514).
To secure residence within the vascular niche, AML cells exploit endothelial adhesion molecules, such as E-selectin. In the healthy BMME, E-selectin expressed on sinusoidal endothelium regulates HSC homing and quiescence.122–124 In AML, engagement of E-selectin on BM endothelium activates pro-survival signaling in leukemic blasts and contributes to chemotherapy resistance. Conversely, uproleselan (GMI-1271), a glycomimetic which binds to E-selectin preventing cancer cell binding, reduces blast adhesion, mobilizes them into circulation, and enhances treatment efficacy in vivo.125
In a Phase III clinical trial of patients with relapsed or refractory AML (NCT03616470), uproleselan showed an acceptable safety profile and signs of clinical activity, but the study did not achieve its primary overall-survival endpoint. Pre-specified subgroup analyses suggested potential benefit in refractory disease, with few treatment-related toxicities.33 An ongoing study (NCT03701308) is evaluating daunorubicin and cytarabine with or without uproleselan as induction therapy for patients aged ≥ 60 years.
Overall, although the biological rationale for disrupting adhesion is strong, the translation into clinical benefit has proven challenging (summarized in Table 1). The inability of single-agent adhesion inhibitors to consistently improve patient outcomes indicates that AML cells rely on a complex network of retention signals rather than a single physical anchor. This suggests that the protective capacity of the BMME is not solely dependent on direct cell-to-cell contact but is likely reinforced by soluble factors. To fully understand niche-mediated retention, one must also consider the role of chemokine gradients which, much like adhesion molecules, actively tether leukemic blasts to their protective microenvironment.
Chemokine-Mediated Retention
In the normal marrow, the CXCL12/CXCR4 axis directs HSC homing;126,127 however, AML cells co-opt this signal to anchor themselves within the protective niche and maintain quiescence.128,129 Interestingly, this provides a link between AML genetic heterogeneity and the BMME as frequently occurring mutations in AML, such as TET2 and FLT3-ITD, have been correlated to aberrant CXCL12/CXCR4 signaling, effectively locking LSCs within the niche.130,131
Plerixafor (AMD3100), a CXCR4 antagonist blocking the CXCL12 signal, originally used for stem cell mobilization in lymphoma and myeloma, has been tested in clinical trials for AML.132 While it successfully mobilizes AML cells out of the protective BM niche into the peripheral blood, where they are more susceptible to cytotoxic agents, translating this mobilization into improved long-term survival has been difficult (NCT01352650 and NCT00943943).34,35 Alternative agents such as dociparstat sodium (DSTAT or CX-01), which is derived from heparin and alters the activity of the CXCL12/CXCR4 axis, has shown encouraging responses when combined with azacitidine in a Phase I trial (NCT02995655),37 as well as promising complete remission (CR) and event-free survival (EFS) rates in a recent Phase II trial when added to induction therapy (NCT02873338).38 Similarly, motixafortide (BL-8040), another high-affinity CXCR4 inhibitor, has shown efficacy in combination with cytarabine in a Phase IIa clinical trial (NCT01838395).36
Several alternative methods of drug delivery to target this axis are also being considered in preclinical research. For example, M-E5-Dox integrates a chemically synthesized CXCR4 antagonistic peptide and the cytotoxic drug, doxorubicin, using DSPE-mPEG2000 micelles, allowing CXCR4 downregulation alongside targeted doxorubicin uptake.133 These strategies aim to improve efficacy by simultaneously disrupting the protective niche and delivering cytotoxic payloads.
In summary, although pharmacological CXCR4 inhibition can transiently mobilize AML cells and enhance short-term chemosensitivity, clinical outcomes indicate that mobilization alone is insufficient to overcome durable niche protection (Table 1). The microenvironment does not merely hold AML cells in place; it engages them in reciprocal signaling that actively promotes survival and drug resistance. Therefore, to fully overcome niche protection, internal signaling cascades sustained by these stromal interactions must also be addressed.
Beyond Adhesion and Retention: Targeting Niche-Derived Signaling
While adhesion molecules physically anchor AML cells within the marrow, their long-term survival and resistance to therapy depend on a complex network of intracellular signaling pathways. AML cells do not merely passively receive these signals; they actively engage in bidirectional crosstalk with stromal components to generate a self-sustaining, protective environment.90
JAK/STAT Signaling
In AML, Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling acts as a major conduit for stromal-derived inflammatory cues, reinforcing pro-survival and anti-apoptotic responses in leukemic cells that compromise therapeutic efficacy.134–138 The JAK/STAT pathway is a central regulator of hematopoiesis, immune response, and cell survival.139 While intrinsic activating mutations (such as JAK2 V617F) are only identified in a small subset of patients, the JAK/STAT pathway is frequently activated in AML via extrinsic signals from the BMME.134 For example, leukemic blasts stimulate MSCs to upregulate the secretion of interleukin-6 (IL-6) and metalloproteinase-14 (MMP14). This leads to activation of the JAK/STAT pathway in the blasts, driven directly by IL-6 and indirectly via the MMP14-dependent release of prostaglandin E2 (PGE2) from MSCs.135–137 The resulting phosphorylation of STAT3 and STAT5 drives the expression of pro-survival BCL-2 family proteins; this axis establishes a high apoptotic threshold that confers chemoresistance.138
Strategies to disrupt JAK/STAT signaling are under active clinical investigation (Table 1). Ruxolitinib, a selective inhibitor of the AKT site on both JAK1 and JAK2, has been explored as a monotherapy, and in combination with venetoclax and/or decitabine, but showed specific potential as a second-line therapy for differentiation syndrome in acute promyelocytic leukemia (APL) (NCT03874052, NCT01251965, NCT00674479, NCT02257138 and NCT04446806).39–43 Ruxolitinib is also being investigated for limiting relapse after allogeneic stem cell transplantation (NCT03286530),44 as well as in combination with CART123 cells for relapsed or refractory disease (NCT06768476). Agents targeting this pathway less directly showed some preliminary anti-leukemic activity, including OPB-111077, which disrupts mitochondrial oxidation and STAT3 phosphorylation, as well as pacritinib, a dual FLT3/JAK2 inhibitor, which was explored for patients with FLT3 mutations (NCT03197714, NCT03063944 and NCT02323607).45–47
Tocilizumab, a humanized monoclonal antibody targeting the IL-6 receptor (IL-6R), was recently evaluated in the phase I TOCILAM study. In this trial, the addition of tocilizumab to standard “7+3” induction chemotherapy was found to be safe, with no dose-limiting toxicities observed and encouraging complete remission (CR) rates (NCT04547062).48 IL-6 blockade with tocilizumab was also explored in the post-transplant setting in a phase II trial to mitigate IL-6-driven inflammation and reduce the risk of graft-versus-host disease (GVHD) following stem cell transplantation but it did not provide significant reduction in GVDH and/or survival benefit (NCT03434730).140 Collectively, the critical role of JAK/STAT signaling in mediating stromal protection, combined with emerging signs of clinical efficacy, validates the continued pursuit of pharmacological strategies targeting this pathway.
PI3K/AKT/mTOR Signaling
Activated by direct contact with stromal cells or soluble factors such as CXCL12, Phosphoinositide 3-kinase (PI3K), protein kinase B (AKT) and mechanistic target of rapamycin (mTOR) (PI3K/AKT/mTOR) signaling contributes to the therapeutic barrier maintained by the BMME, promoting AML survival and resistance.129,141 Evidence highlights a reciprocal interaction where AML blasts induce the downregulation of methyltransferase-like 3 (METTL3) in MSCs; this triggers AKT activation in the stroma and directs MSCs toward adipogenic differentiation, creating a lipid-rich niche that fuels cancer cell survival.142,143
Effective targeting of the pathway in a clinical setting has proven challenging (Table 1). First-generation PI3K inhibitors, such as idelalisib (CAL-101), which inhibits the PI3Kδ isoform, showed no meaningful response in AML, while the pan-PI3K inhibitor buparlisib (BKM120) demonstrated only modest efficacy with significant toxicity (NCT00710528 and NCT01396499).49,50 Similarly, dual targeting with inhibitors of both PI3K and mTOR (eg gedatolisib, dactolisib) have largely failed as single agents due to lack of therapeutic response and poor tolerance (NCT02438761 and NCT01756118).51,52
Complexes of mTOR, such as mTORC1, are activated within osteoblastic niches, acting via IL-6 to promote AML cell growth, providing rational for mTOR-directed therapies to target this interaction.144 The mTOR inhibitors rapamycin/sirolimus, temsirolimus and everolimus have shown limited single-agent activity, though some benefit has been observed in combination with chemotherapy (NCT00235560, NCT02109744, NCT01822015, 2007–005374-31 and NCT01074086).53–57 Similarly, direct AKT inhibitors (eg uprosertib, afuresertib) have also been largely ineffective (NCT01907815 and NCT00881946).58,59 However, novel strategies are emerging such as ONC201, an imipridone that antagonizes DRD2 and activates mitochondrial ClpP to downstream inactivate AKT and ERK, is currently being evaluated in combination with venetoclax (NCT02392572). It was found to be well tolerated and had encouraging early responses as maintenance therapy to prevent post-transplant relapse (NCT03932643).60 Together, these findings indicate that PI3K/AKT/mTOR signaling in AML reflects a microenvironmentally reinforced survival state, driven by convergent niche-derived signals rather than a discrete oncogenic dependency, helping to explain the limited clinical efficacy of direct pathway inhibition and underscoring the need for combinatorial or context-dependent therapeutic strategies.
Angiopoietin/Tie2 Signaling
Angiopoietin/Tie2 signaling serves as a vascular therapeutic barrier in AML within the BMME, stabilizing endothelial niches and reinforcing pro-survival cues, which drives disease progression and protects leukemic cells from cytotoxic and targeted therapies.145–147 Leukemic blasts have been shown to overexpress angiopoietin-1 (Ang-1), which engages Tie2 receptors on endothelial cells to subvert the vascular niche and promote cancer cell proliferation.145
Consequently, disruption of this signaling network has been explored as a therapeutic strategy (Table 1).145,148 In preclinical studies, the dual Tie2/p38 MAPK inhibitor pexmetinib (ARRY-614) demonstrated the ability to abolish AML cell proliferation.149 However, whilst this agent has been well tolerated and has shown efficacy in myelodysplastic syndromes (MDS), it has not yet progressed to the clinical setting for AML (NCT00916227).150 Conversely, other inhibitors of this pathway have entered clinical evaluation but yielded modest results. For instance, trebananib, a peptibody that neutralizes Ang-1 and Ang-2, and the switch-control tyrosine kinase inhibitor, that specifically targets Tie2, as well as VEGFR-2 and BCR-ABL, have both been tested in clinical trials for AML, but demonstrated limited efficacy as monotherapies or in combination with chemotherapy (NCT01555268 and NCT00827138).61,62 Collectively, these findings suggest that Angiopoietin/Tie2 signaling functions primarily to stabilize a permissive vascular niche rather than acting as a leukemia-specific driver, helping to explain the limited clinical efficacy of Tie2-targeted monotherapies in AML.
Wnt/β-Catenin Signaling
Wnt/β-catenin signaling represents a niche-remodeling component of the BMME as a therapeutic barrier in AML, promoting osteogenic skewing and stromal states that indirectly reinforce leukemic cell survival and chemoresistance.151 Osteoblasts derived from patients with AML exhibit elevated levels of β-catenin activation, a feature clinically correlated with poor prognosis and reduced treatment response.152 Mechanistically, the interaction between AML blasts and MSCs leads to the release of the pro-inflammatory mediator PGE2 from MSCs, which subsequently drives β-catenin expression.153
Therapeutic targeting of this pathway has proved challenging (Table 1). Direct inhibition strategies have failed to demonstrate efficacy in clinical trials (eg NCT01606579 and NCT01398462)63 and alternative approaches aimed at stabilizing β-catenin through inhibition of Glycogen Synthase Kinase-3 (GSK-3) with inhibitors such as LY2090314, have likewise been explored with limited efficacy in patients with AML (NCT01214603).64,154,155 Targeting upstream regulators has also been explored, but the inhibition of the cyclooxygenase-1/2 (COX1/2) axis with the non-steroidal anti-inflammatory drugs sulindac and celecoxib did not translate clinically, with trials ultimately abandoned due to lack of funding and low accrual (NCT01843179 and NCT03878524).156,157 Despite promising preclinical data, this consistent lack of translational success highlights that Wnt signaling within the leukemic niche operates predominantly through microenvironmental remodeling rather than direct leukemic cell dependency, underscoring why tumor-intrinsic pathway inhibition has failed to overcome this component of the therapeutic barrier.158–160
Notch Signaling
Notch signaling acts as another niche-remodeling element creating a therapeutic barrier, where AML cells activate this pathway within MSCs to drive aberrant osteogenic differentiation, which results in the accumulation of osteoprogenitors and pre-osteoblasts.161 This remodeled niche not only enhances leukemic cell growth and therapy resistance, but also compromises the support of normal HSCs.161 Consistent with this, BM-MSCs derived from patients with AML exhibit upregulated Notch1 and its ligand Jagged1 (JAG1), as well as increased expression of the downstream target gene HES1. Together, these findings confirm active Notch signaling within components of the AML BMME. Concurrently, activation of Notch1 within the AML blasts themselves further supports leukemic proliferation and chemoresistance.162,163
Therapeutic strategies targeting this axis have shown promise in preclinical models. In particular, synergistic apoptosis induction has been observed when FLT3 tyrosine kinase inhibitors (TKIs) are combined with γ-secretase inhibitors (GSIs), such as DAPT and RO4929097, which block Notch activation.164 However, clinical translation has been limited (Table 1); only GSI MK-0752 has been tested in AML patients, where it yielded poor response rates (NCT00100152).65 These limitations have promoted interest in alternative approaches to targeting notch ligand-specific interactions. For instance, JAG1 inhibition has been shown in vitro to restore healthy hematopoiesis and ameliorate anemia, thrombocytopenia, and immune dysregulation driven by the β-catenin/JAG1/Notch1 axis in AML osteoblasts.165 Similarly, inhibitors of JAG1 and JAG2, such as CTX014, show preclinical promise; they have been shown to sensitize tumors to T-cell-mediated killing.166 In summary, the Notch pathway represents an attractive therapeutic target for disrupting the leukemic niche, yet further clinical evaluation is required to translate this into therapeutic benefit.
NF-κB Signaling
The NF-κB pathway is a critical mediator of inflammation and cell survival, that is frequently hijacked through stromal interactions within the leukemic niche.117,167,168 Constitutive activation of NF-κB in AML cells drives the expression of anti-apoptotic proteins, including BCL-2 and B-cell lymphoma-extra large (BCL-XL), thereby promoting cell survival and resistance to cytotoxic stress.169 Due to the on-target toxicities associated with NF-κB-directed treatment, focusing on downstream mediators is a more attractive option.170 The NF-κB-driven increase in anti-apoptotic proteins has been successfully exploited clinically, through the BCL-2 inhibitor, venetoclax, in combination with the hypomethylating agent azacitidine; a regimen that has established a new standard of care for patients who are ineligible for intensive chemotherapy (NCT02993523).26
Beyond direct BCL-2 inhibition, strategies targeting upstream regulators of NF-κB have also been explored (Table 1). For example, stabilization of the NF-κB inhibitor, IκBα, using the proteasome inhibitor MG-132, either alone or in combination with idarubicin, effectively promoted leukemic cell death in preclinical models; however, this specific approach has not yet translated to clinical practice due to low bioavailability, specificity and stability of the drug.171–173 An alternative proteosome inhibitor bortezomib, which inhibits the degradation of IκBα, reducing NF-kB activity has been evaluated in several AML trials with mixed results in older populations.173 While its combination with decitabine did not enhance outcomes (NCT01420926),66 adding it to daunorubicin and cytarabine (NCT00742625) successfully improved remission rates.67 Currently, bortezomib is being assessed in newly diagnosed patients: a Phase III trial is investigating its combination with sorafenib tosylate (NCT01371981), and a Phase I/II study (NCT07008638) is evaluating it with CPX-351 for patients harboring TP53 mutations.68 In summary, the notable success of venetoclax, alongside encouraging findings of manipulating the upstream members of the NF-kB, as well as its critical role in AML pathogenesis, highlights this pathway as a compelling target for further clinical investigation.
TGF-β
Transforming growth factor-beta (TGF-β) plays a multifaceted role in the leukemic niche, contributing to chemoresistance while actively remodeling microenvironmental components, including endothelial and Natural Killer (NK) cells.174–176 Notably, the activity of TGF-β is significantly elevated in the BM of patients with relapsed AML, impairing NK cell function and compromising innate tumor surveillance.174 Niche remodeling extends further to BM adipocytes. AML cells secrete growth differentiation factor 15 (GDF15), which signals through the GDF15/TGFBRII/FOXC1/TRPV4 axis to alter adipocyte metabolism by reducing Ca2+ influx, and ultimately enhance leukemic cell survival.177,178
Pharmacological targeting of the TGF-β pathway has shown promise in related myeloid malignancies. For example, the TGF-β1 receptor inhibitor, galunisertib, has demonstrated clinical efficacy for MDS (NCT02008318);179 however, application in AML remains to be established. TGF-β receptor targeting molecules have also been explored as a way of disrupting adipocyte-mediated survival support within the AML BMME, although results have been variable. For example, the TGF-β receptor I inhibitor LY-2109761 has shown mixed effects in preclinical AML models.178,180,181 Conversely, restoring downstream signaling through pharmacological activation of TRPV4 in BM adipocytes using the agonist 4aPDD has yielded promising preclinical results,178 suggesting that targeting downstream effectors of the TGF-β axis may represent a more tractable therapeutic strategy.
IL-8
Interleukin-8 (IL-8) expression within the leukemic niche is driven by complex bidirectional crosstalk between AML blasts and stromal cells.182,183 AML-derived migration inhibitory factor (MIF) activates CD74 and downstream PKCβ signaling in MSCs, triggering IL-8 overexpression. This promotes MSC migration towards AML cells, releasing CXCL12 and facilitating leukemic cell survival.184 This inflammatory state is further reinforced by osteoblast-derived signals and the hypoxic conditions characteristic of the BMME.182,185 Pre-clinical pharmacological disruption of this axis, either through PKCβ inhibition (using agents such as Ro-31-8220, Go6976, or enzastaurin) or direct blockade of CD74 and IL-8, has been shown in vitro to abolish this stromal-mediated protection.184
Clinically, activation of the IL-8/CXCR2 pathway correlates with adverse outcomes in AML. Preclinical investigation of SB332235, which binds to and blocks CXCR2 activation, shows that it can suppress AML cell proliferation and induces cell cycle arrest.186 Although several CXCR1/CXCR2 inhibitors, including reparixin and SCH-527123, as well as IL-8-neutralising antibodies, have been evaluated in other disease settings,187–189 their therapeutic efficacy in AML has not yet been tested clinically.
VEGF
Vascular Endothelial Growth Factor (VEGF), a signaling protein involved in angiogenesis, is markedly upregulated in endothelial cells following exposure to the cytotoxic drug, cytarabine.190 In co-culture AML blasts develop resistance to cytarabine, which can be partly resensitized through targeting the VEGF receptor.190 Despite the rationale that this endothelial support limits treatment efficacy for AML blasts, clinical trials evaluating VEGF receptor inhibitors (such as axitinib and cediranib), or direct VEGF sequestration with bevacizumab, have provided limited benefit (Table 1), regardless of whether they were administered concurrently with or following chemotherapy (NCT00071006, NCT00475150, NTR904 and NCT00015951).69–72,191
Although multi-targeted tyrosine kinase inhibitors, including sunitinib and sorafenib, possess anti-VEGF activity, their clinical and pre-clinical utility in AML has been largely restricted to FLT3-mutant disease rather than inhibition of angiogenic signaling (NCT00783653, NCT01254890 and NCT02728050).192–194 Similarly, aflibercept, a decoy VEGF receptor that prevents ligand binding, demonstrated potent anti-leukemic synergy with chemotherapy in preclinical models but failed to successfully translate into the clinical setting.195 This may suggest that VEGF is a bystander rather than offering a causative change in therapy resistance.
Metabolic Crosstalk Within the Leukemic Niche
As indicated above, the bidirectional relationship between AML cells and the BMME extends beyond structural and signaling support to include a complex metabolic interplay. AML cells actively remodel the niche to promote their own metabolic reprogramming, thereby supporting survival, proliferation, and resistance to therapy.196 These metabolic interactions are currently being exploited through various niche-targeted therapeutic interventions undergoing evaluation in both preclinical studies and clinical trials (Table 1).
Although the BMME maintains high vascular density, it remains inherently hypoxic.197 This condition is further exacerbated in AML through increased vascular leakiness.198 This low oxygen state is not merely a byproduct of the disease but a functional advantage, as it actively promotes AML cell survival and proliferation. Attempts to exploit this environment have led to the development of TH-302 (evofosfamide), a hypoxia-activated prodrug.199 TH-302 induces DNA damage upon metabolism in low-oxygen conditions and showed promise in preclinical models, but this success failed to translate into clinical efficacy (NCT01149915).73,199
The environmental constraints of the BMME, coupled with the differentiation status of the cells, dictate the specific metabolic dependencies of AML cells. While rapidly dividing blasts tend to rely on glycolysis, primitive LSCs are more dependent on mitochondrial oxidative phosphorylation (OXPHOS) for survival.200–203 Indeed, co-culture of AML cells with BM-MSCs induced metabolic shift toward glycolysis in AML cells, via CXCL12/CXCR4/mTOR signaling, inducing protection against cytarabine.204 Targeting this pathway with plerixafor or directly inhibiting glycolysis using 2-Deoxy-D-glucose (2-DG) and diclofenac increased chemosensitivity.
Similar challenges have emerged when targeting AML cells dependent on mitochondrial OXPHOS. Utilising inhibitors such as IACS-010759 (a BH3 mimetic) and CPI-613 (devimistat, a non-redox active analog of the OXPHOS cofactor lipoic acid) has yielded disappointing results in humans.75,205 A phase I clinical trial for IACS-010759 was terminated due to ineffectiveness and adverse effects (NCT02882321).74 While CPI-613 failed to demonstrate additional benefit (NCT03504410).75 Crucially, there is evidence that interactions between AML cells and MSCs within the BM can actively enhance OXPHOS, further driving chemoresistance.206
This metabolic flexibility is fuelled by a bidirectional exchange of metabolites between the BM stroma and AML cells, which may be an additional source of therapeutic targets. For instance, AML cells induce MSCs to secrete acetate which they take up via gap junctions to fuel the tricarboxylic acid cycle (TCA) cycle and lipid biosynthesis.207 Similarly, AML blasts trigger metabolic reprogramming in adipocytes to activate lipolysis.208,209 This generates fatty acids that are utilised by leukemic cells via fatty acid-oxidation (FAO) to produce the energy required for proliferation. While a detailed review of BMME adipocyte-driven FAO is beyond the current scope, its role as a therapeutic target has been documented extensively elsewhere.210
In addition to the exchange of metabolites, there is also active exchange of mitochondria from MSCs through tunnelling nanotubes to AML cells, increasing proliferation and resistance to therapy.211,212 AML cells are also able to increase mitochondrial biogenesis within MSCs.213 The anti-CD38 monoclonal antibody daratumumab has been shown to inhibit this mitochondrial transfer; however, it has demonstrated limited efficacy as a monotherapy in a clinical trial (NCT03067571).214 Further trials combining daratumumab with chemotherapy are currently recruiting (NCT06287944 and NCT05749276). Evidence further suggests that mitochondrial transfer is not limited to MSCs, as it also occurs between AML blasts and other BM resident cells, including endothelial cells and macrophages.215,216
Immune Evasion in the BMME
Within the BM, AML blasts actively drive immune niche remodeling to shield themselves from immune surveillance, thereby contributing a critical immunological component to the therapeutic barrier. This protection is orchestrated through the coordinated activity of stromal cells and immune-modulatory pathways that suppress anti-leukemic immune responses and promote disease persistence, representing potential targets for personalized therapy.217
Immune Checkpoint Inhibition
AML cells and LSCs exploit immune checkpoint pathways, such as PD-1/PD-L1, CTLA-4 and TIM-3 to evade immune detection, primarily through suppression of B and T cell activity.217 Beyond immune suppression, recent evidence suggests that PD-L1 also exerts intrinsic effects within AML cells, regulating cell cycle progression, proliferation, and apoptosis via downstream signaling pathways including PI3K/AKT.218
Therapeutic targeting of the PD-1/PD-L1 axis, using monoclonal antibodies such as pembrolizumab, nivolumab, and atezolizumab, is well-established in solid tumors and is now being actively evaluated in AML (Table 1). Early clinical studies have demonstrated potential benefits, including reductions in graft-versus-host disease (GVHD) following allogenic transplantation and improved responses in relapsed/refractory disease settings (NCT02768792, NCT02771197 and NCT04541277).76–78,80,81 A phase II trial is currently investigating the addition of pembrolizumab to standard induction chemotherapy (cytarabine plus idarubicin or daunorubicin) (NCT04214249).79
Alternative immune checkpoint targets are also being explored (Table 1). Ipilimumab, a CTLA-4 blocking antibody, was investigated in a successful phase I trial in combination with nivolumab219 and is currently being assessed in combination with decitabine for relapsed/refractory AML (NCT02890329).82 However, not all checkpoint-directed strategies have shown the same promise. For instance, while sabatolimab (a TIM-3 receptor inhibitor) demonstrated acceptable safety and tolerability in combination with venetoclax and azacitidine, its phase II trial (NCT04150029) was terminated following the failure of other TIM-3 inhibitor trials to meet their primary endpoints.83,84,217
Blockade of growth differentiation factor 15 (GDF-15) has been shown to synergize with PD-1 inhibition in solid tumors (NCT04725474).220 Similarly, antibodies targeting T cell immunoreceptor with Ig and ITIM domains (TIGIT), an inhibitory immune checkpoint receptor expressed on T cells and NK cells that suppresses cytotoxic function through engagement of ligands such as CD155, have demonstrated clinical benefit in combination with atezolizumab in non-small cell lung cancer (NCT03563716)221 However, evaluation in the context of AML remains at an early stage. Looking forward, emerging targets such as these, from solid tumor oncology, may hold relevance for AML.
BMME Shaping Response to Cellular Immunotherapies
The efficacy of cellular immunotherapies in AML, such as Chimeric Antigen Receptor (CAR) T-cells, is frequently constrained by the immunosuppressive BMME, which limits effector cell persistence, function and target engagement. To overcome this barrier, novel “dual-targeting” CAR-T cells strategies have been developed that simultaneously target AML blasts and immunosuppressive niche components. Examples include CAR constructs directed against CD123 and NKG2D ligands, designed to not only eliminate leukemic cells but also monocyte-like myeloid-derived suppressor cells (M-MDSCs) and alternatively activated (M2) macrophages that contribute to immune evasion.222
Despite these advances, CAR-T cell therapy in relapsed/refractory AML has historically been associated with limited efficacy and significant off-target toxicity, reflecting the lack of tumor-specific antigens and persistent microenvironmental immunosuppression.223 CAR-engineered natural killer (CAR-NK) cells offer a potential alternative, by retaining anti-leukemic activity while exhibiting a more favorable toxicity profile. Notably, CD33-targeted CAR-NK cells, adapted from CAR-T constructs, have recently demonstrated preliminary efficacy and safety in a phase I clinical trial (NCT05008575).85
Challenges in Modeling the BMME and AML Interactions
The intricate and dynamic crosstalk between AML blasts and the protective BMME represents a significant barrier to the successful translation of targeted therapies. While the biological rationale for disrupting microenvironmental support is compelling, faithfully reproducing these complex interactions in preclinical models remains a formidable challenge, which contributes to the gap between experimental promise and clinical efficacy. The various experimental platforms used to study these interactions, ranging from simplified cell cultures to advanced humanized systems, are summarized in Figure 2.
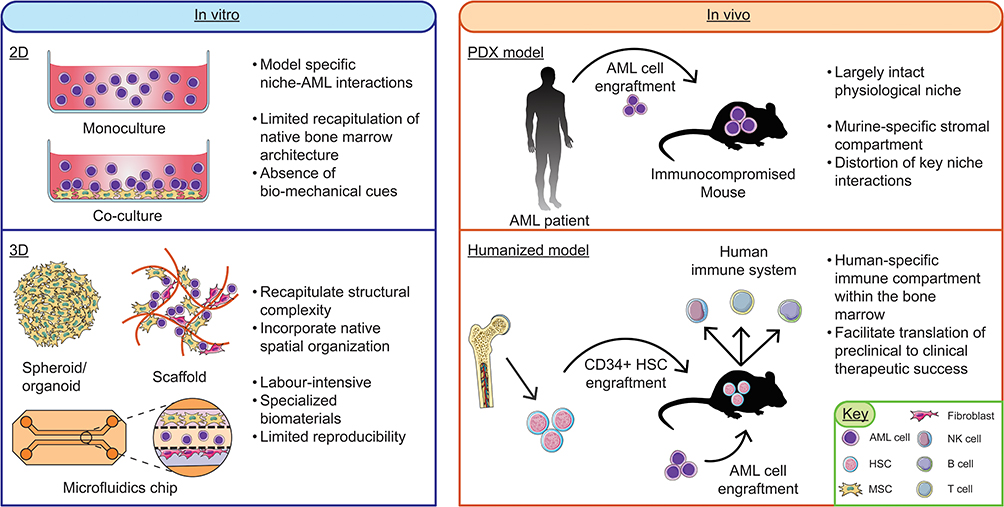 |
Figure 2 Methodological landscape of preclinical AML models. Left: In vitro platforms ranging from 2D monocultures and co-cultures (with mesenchymal stromal cells (MSCs) or fibroblasts) to 3D systems. While 2D models offer high reproducibility, 3D platforms (spheroids, organoids, scaffolds, and microfluidic chips) are utilized to better recapitulate the structural complexity, extracellular matrix (ECM) organization, and biomechanical cues of the bone marrow microenvironment (BMME). Right: In vivo murine models for translational validation. Patient-derived xenograft (PDX) models utilize immunocompromised mice to facilitate engraftment, though murine-specific stroma can distort human niche-mediated resistance. Humanized models involve the engraftment of human CD34+ hematopoietic stem cells (HSCs) to generate a human-specific immune compartment (T, B, and NK cells), facilitating more accurate clinical translation of novel therapeutics. Some parts of image adapted from Servier Medical Art (https://smart.servier.com/), licensed under CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). |
Standard preclinical studies historically relied on immortalized AML cell lines cultured in suspension. Although these models facilitate the high-throughput interrogation of defined AML subtypes based on morphology, genetic mutations, and immunophenotypes, they fail to capture the cellular heterogeneity, spatial organization, and context dependence observed in patient disease.224,225 Two-dimensional (2D) co-culture systems incorporating stromal components (such as MSCs or endothelial cells) have therefore been developed to model niche-mediated protection more accurately.226 However, even these adaptations remain limited by the absence of native three-dimensional (3D) architecture and bio-mechanical cues that are integral to the BMME.
To better recapitulate this structural complexity, a range of 3D culture models have been introduced. While these systems offer important conceptual advances, their widespread adoption has been limited by significant technical barriers; they often require labor-intensive preparation, specialized biomaterials, and lengthy optimization periods. As a result, reproducibility across laboratories remains limited, reflected in the relative paucity of follow-up studies utilizing these specific models once initially described.227–232
In vivo murine models remain the gold standard for investigating leukemogenesis within a largely intact physiological niche.226 Nevertheless, fundamental species-specific differences impose important limitations. The reliance on immunocompromised hosts to permit human AML engraftment, together with the presence of a murine-specific stromal compartment, results in the absence or distortion of key immunological, cytokine and cell-cell interactions. These discrepancies likely contribute to the high attrition rate of therapies that show promise in murine models but fail to translate into meaningful clinical benefit.34,233,234
To address these challenges, increasing efforts are focused on the development of more representative, “humanized” preclinical models. Approaches such as mice engrafted with functional human immune systems, aim to restore aspects of the immune context lacking in conventional xenografts.235 Ultimately, progress in this area will depend on the generation of experimental platforms that more accurately reflect both the intrinsic heterogeneity of AML, and the multifaceted, adaptive nature of the human BMME thereby enabling more reliable evaluation of niche-targeted strategies.
Conclusion
AML is sustained not only by intrinsic genetic lesions but also by a highly dynamic and adaptable BMME that actively promotes leukemic cell survival, immune evasion, and therapeutic resistance. As outlined in this review, AML blasts engage in extensive bidirectional crosstalk with stromal, vascular, and immune compartments through a complex network of adhesion-dependent interactions, soluble factors, and reciprocal signaling pathways, collectively establishing a permissive niche that undermines the efficacy of otherwise rational targeted therapies.
Despite substantial advances in delineating these mechanisms, clinical translation of strategies aimed at disrupting niche-mediated support has remained limited (Table 1). This reflects both inherent redundancy and plasticity of microenvironmental support networks and the ongoing difficulty of modeling their complexity in preclinical systems. Together, these observations indicate that the BMME functions as an integrated therapeutic barrier rather than a collection of independent, targetable pathways. Consequently, effective disruption of niche-mediated protection is unlikely to be achieved through single-pathway inhibition alone. Future therapeutic strategies will instead need to employ rational, context-aware combination strategies that simultaneously target leukemic cells and key components of their supportive microenvironment, informed by disease stage, molecular subtype and treatment timing. Such strategies represent a critical step toward eliminating the reservoirs of residual disease that drive clinical relapse, thereby improving long-term survival and cure rates for patients with AML.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Döhner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015;373(12):1136–25. doi:10.1056/NEJMra1406184
2. Stelmach P, Trumpp A. Leukemic stem cells and therapy resistance in acute myeloid leukemia. Haematologica. 2023;108(2):353–366. doi:10.3324/haematol.2022.280800
3. DiNardo CD, Erba HP, Freeman SD, Wei AH. Acute myeloid leukaemia. Lancet. 2023;401(10393):2073–2086. doi:10.1016/S0140-6736(23)00108-3
4. Haematological Malignancy Research Network (HMRN). Survival statistics: acute myeloid leukaemias. 2025, Available from: https://hmrn.org/statistics/survival.
5. Lonetti A, Pession A, Masetti R. Targeted Therapies for Pediatric AML: gaps and Perspective. Front Pediatr. 2019;7:463. doi:10.3389/fped.2019.00463
6. Murphy T, KWL Y. Cytarabine and daunorubicin for the treatment of acute myeloid leukemia. Expert Opin Pharmacother. 2017;18(16):1765–1780. doi:10.1080/14656566.2017.1391216
7. Network TCGAR. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N Engl J Med. 2013;368(22). doi:10.1056/NEJMoa1301689
8. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016;374(23):2209–2221. doi:10.1056/NEJMoa1516192
9. Döhner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140(12):1345–1377. doi:10.1182/blood.2022016867
10. Yates JW, Wallace Jr HJ, Ellison RR, Holland JF. Cytosine arabinoside (NSC-63878) and daunorubicin (NSC-83142) therapy in acute nonlymphocytic leukemia. Cancer Chemother Rep. 1973;57(4):485–488.
11. Yates J, Glidewell O, Wiernik P, et al. Cytosine Arabinoside With Daunorubicin or Adriamycin for Therapy of Acute Myelocytic Leukemia: a CALGB Study. Blood. 1982;60(2):454. doi:10.1182/blood.V60.2.454.454
12. Stone RM, Fischer T, Paquette R, et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia. 2012;26(9):115. doi:10.1038/leu.2012.115
13. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med. 2017;377(5):454–464. doi:10.1056/NEJMoa1614359
14. Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3 -Mutated AML. N Engl J Med. 2019;381(18):1728–1740. doi:10.1056/NEJMoa1902688
15. DiNardo CD, Stein EM, Botton S, et al. Durable Remissions with Ivosidenib in IDH1 -Mutated Relapsed or Refractory AML. N Engl J Med. 2018;378(25):2386–2398. doi:10.1056/NEJMoa1716984
16. Montesinos P, Recher C, Vives S, et al. Ivosidenib and Azacitidine in IDH1 -Mutated Acute Myeloid Leukemia. N Engl J Med. 2022;386(16):1519–1531. doi:10.1056/NEJMoa2117344
17. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–731. doi:10.1182/blood-2017-04-779405
18. Pollyea DA, Tallman MS, de Botton S, et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia. 2019;33(11):2575–2584. doi:10.1038/s41375-019-0472-2
19. Stein EM, DiNardo CD, Fathi AT, et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood. 2019;133(7):676–687. doi:10.1182/blood-2018-08-869008
20. DiNardo CD, Pratz KW, Letai A, et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, Phase 1b study. Lancet Oncol. 2018;19(2):216–228. doi:10.1016/S1470-2045(18)30010-X
21. Pollyea DA, Pratz KW, Jonas BA, et al. Venetoclax in Combination with Hypomethylating Agents Induces Rapid, Deep, and Durable Responses in Patients with AML Ineligible for Intensive Therapy. Blood. 2018;132(Supplement 1):285. doi:10.1182/blood-2018-99-117179
22. DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7–17. doi:10.1182/blood-2018-08-868752
23. Wei AH, Montesinos P, Ivanov V, et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: a Phase 3 randomized placebo-controlled trial. Blood. 2020;135(24):2137–2145. doi:10.1182/blood.2020004856
24. Wei AH, Panayiotidis P, Montesinos P, et al. Long-term follow-up of VIALE-C in patients with untreated AML ineligible for intensive chemotherapy. Blood. 2022;140(25):2754–2756. doi:10.1182/blood.2022016963
25. Pratz KW, Jonas BA, Pullarkat V, et al. Long-term follow-up of VIALE-A: venetoclax and azacitidine in chemotherapy-ineligible untreated acute myeloid leukemia. Am J Hematol. 2024;99(4):615–624. doi:10.1002/ajh.27246
26. DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N Engl J Med. 2020;383(7):617–629. doi:10.1056/NEJMoa2012971
27. Norsworthy KJ, Ko CW, Lee JE, et al. FDA approval summary: mylotarg for treatment of patients with relapsed or refractory CD33-positive acute myeloid leukemia. oncologist. 2018;23(9):1103–1108. doi:10.1634/theoncologist.2017-0604
28. Song F, Lin S, Xu T, et al. Targeted therapy in acute myeloid leukemia: resistance and overcoming strategy. Drug Resist Updates. 2025;83:101286. doi:10.1016/j.drup.2025.101286
29. Skelding KA, Barry DL, Theron DZ, et al. Bone Marrow Microenvironment as a Source of New Drug Targets for the Treatment of Acute Myeloid Leukaemia. Int J Mol Sci. 2022;24(1):563. doi:10.3390/ijms24010563
30. Allert C, Müller-Tidow C, Blank MF. The relevance of the hematopoietic niche for therapy resistance in acute myeloid leukemia. International Journal of Cancer. 2024;154(2):197–209. doi:10.1002/ijc.34684
31. Tettamanti S, Pievani A, Biondi A, et al. Catch me if you can: how AML and its niche escape immunotherapy. Leukemia. 2021;36(1):13–22. doi:10.1038/s41375-021-01350-x
32. Ladikou EE, Sivaloganathan H, Pepper A, et al. Acute Myeloid Leukaemia in Its Niche: the Bone Marrow Microenvironment in Acute Myeloid Leukaemia. Current Oncology Reports. 2020;22(3). doi:10.1007/s11912-020-0885-0
33. DeAngelo DJ, Schuh AC, Jonas BA, et al. Efficacy and Safety of Uproleselan Combined with Chemotherapy Vs. Chemotherapy Alone in Relapsed/Refractory Acute Myeloid Leukemia: findings from an International Phase 3 Trial. Blood. 2024;144(Supplement 1):733. doi:10.1182/blood-2024-209097
34. Roboz GJ, Ritchie EK, Dault Y, et al. Phase I trial of plerixafor combined with decitabine in newly diagnosed older patients with acute myeloid leukemia. Haematologica. 2018;103(8):1308. doi:10.3324/haematol.2017.183418
35. Borthakur G, Zeng Z, Cortes JE, et al. Phase 1 study of combinatorial sorafenib, G-CSF, and plerixafor treatment in relapsed/refractory, FLT3-ITD-mutated acute myelogenous leukemia patients. Am J Hematol. 2020;95(11):1296–1303. doi:10.1002/ajh.25943
36. Borthakur G, Ofran Y, Tallman MS, et al. BL-8040 CXCR4 antagonist is safe and demonstrates antileukemic activity in combination with cytarabine for the treatment of relapsed/refractory acute myelogenous leukemia: an open-label safety and efficacy Phase 2a study. Cancer. 2021;127(8):1246–1259. doi:10.1002/cncr.33338
37. Huselton E, Rettig MP, Campbell K, et al. Combination of dociparstat sodium (DSTAT), a CXCL12/CXCR4 inhibitor, with azacitidine for the treatment of hypomethylating agent refractory AML and MDS. Leukemia Res. 2021;110:106713. doi:10.1016/j.leukres.2021.106713
38. Kovacsovics T, Levy MY, Cook RJ, et al. A randomized phase II trial of CX-01 with standard therapy in elderly patients with acute myeloid leukemia (AML). Am Soc Clin Oncol. 2019;37(15_suppl):7001. doi:10.1200/JCO.2019.37.15_suppl.7001
39. Pemmaraju N, Kantarjian H, Kadia T, et al. A Phase I/II Study of the Janus Kinase (JAK)1 and 2 Inhibitor Ruxolitinib in Patients With Relapsed or Refractory Acute Myeloid Leukemia. Clin Lymphoma Myeloma Leukemia. 2015;15(3):171–176. doi:10.1016/j.clml.2014.08.003
40. Eghtedar A, Verstovsek S, Estrov Z, et al. Phase 2 study of the JAK kinase inhibitor ruxolitinib in patients with refractory leukemias, including postmyeloproliferative neoplasm acute myeloid leukemia. Blood. 2012;119(20):4614–4618. doi:10.1182/blood-2011-12-400051
41. Borate UM, Madanat YF, Tognon C, et al. Results of a Phase 1 Trial Testing the Novel Combination Therapy of Venetoclax and Ruxolitinib in Relapsed/Refractory Acute Myeloid Leukemia Patients. Blood. 2023;142(Supplement 1):1515. doi:10.1182/blood-2023-191166
42. Bose P, Verstovsek S, Cortes JE, et al. A phase 1/2 study of ruxolitinib and decitabine in patients with post-myeloproliferative neoplasm acute myeloid leukemia. Leukemia. 2020;34(9):2489–2492. doi:10.1038/s41375-020-0778-0
43. Wu Q, Yang X, Zhang J, et al. Improved prevention and treatment strategies for differentiation syndrome contribute to reducing early mortality in patients with acute promyelocytic leukemia. Blood Cancer J. 2024;14(1):113. doi:10.1038/s41408-024-01074-y
44. DeFilipp Z, Kim HT, Knight LW, et al. Low rates of chronic graft-versus-host disease with ruxolitinib maintenance following allogeneic HCT. Blood. 2025;145(20):2312–2316. doi:10.1182/blood.2024028005
45. Martínez-López J, Montesinos P, López-Muñoz N, et al. Biomarker‑driven phase Ib clinical trial of OPB‑111077 in acute myeloid leukemia. Med Int. 2022;2(2):7. doi:10.3892/mi.2022.32
46. Wilde L, Martinez-Outschoorn U, Palmisiano N, Keiffer G, Kasner M. Results of the Phase 1b Dose Escalation Study of OPB-111077, Decitabine, and Venetoclax for the Treatment of Newly Diagnosed or Relapsed/Refractory AML. Blood. 2020;136(Supplement 1):10. doi:10.1182/blood-2020-140729
47. Jeon JY, Zhao Q, Buelow DR, et al. Preclinical activity and a pilot phase I study of pacritinib, an oral JAK2/FLT3 inhibitor, and chemotherapy in FLT3-ITD-positive AML. Invest New Drugs. 2020;38(2):340–349. doi:10.1007/s10637-019-00786-4
48. Peterlin P, Garnier A, Le Bourgeois A, et al. Tocilizumab in combination with a standard induction chemotherapy in acute myeloid leukaemia patients (TOCILAM study): a single-centre, single-arm, phase 1 trial. EClinicalMedicine. 2023;64:1 doi:10.1016/j.eclinm.2023.102254.
49. Flinn IW, Byrd JC, Furman RR, et al. Evidence of Clinical Activity in a Phase 1 Study of CAL-101, An Oral P110Δ Isoform-Selective Inhibitor of Phosphatidylinositol 3-Kinase, in Patients with Relapsed or Refractory B-Cell Malignancies. Blood. 2009;114(22):922. doi:10.1182/blood.V114.22.922.922
50. Ragon BK, Kantarjian H, Jabbour E, et al. Buparlisib, a PI3K inhibitor, demonstrates acceptable tolerability and preliminary activity in a phase I trial of patients with advanced leukemias. Am J Hematol. 2017;92(1):7–11. doi:10.1002/ajh.24568
51. Vargaftig J, Farhat H, Ades L, et al. Phase 2 Trial of Single Agent Gedatolisib (PF-05212384), a Dual PI3K/mTOR Inhibitor, for Adverse Prognosis and Relapse/Refractory AML: clinical and Transcriptomic Results. Blood. 2018;132(Supplement 1):5233. doi:10.1182/blood-2018-99-117485
52. Lang F, Wunderle L, Badura S, et al. A phase I study of a dual PI3-kinase/mTOR inhibitor BEZ235 in adult patients with relapsed or refractory acute leukemia. BMC Pharmacol Toxicol. 2020;21(1):70. doi:10.1186/s40360-020-00446-x
53. Récher C, Beyne-Rauzy O, Demur C, et al. Antileukemic activity of rapamycin in acute myeloid leukemia. Blood. 2005;105(6):2527–2534. doi:10.1182/blood-2004-06-2494
54. Liesveld JL, Baran A, Azadniv M, et al. A phase II study of sequential decitabine and rapamycin in acute myelogenous leukemia. Leukemia Res. 2022;112:106749. doi:10.1016/j.leukres.2021.106749
55. Palmisiano N, Jeschke G, Wilde L, et al. A Phase I Trial of Sirolimus with “7&3” Induction Chemotherapy in Patients with Newly Diagnosed Acute Myeloid Leukemia. Cancers. 2023;15(21):5129. doi:10.3390/cancers15215129
56. Amadori S, Venditti A, Ammatuna E, et al. Temsirolimus, An mTOR Inhibitor, In Combination with Low-Dose Clofarabine in Older Patients with Advanced Acute Myeloid Leukemia: results of a Phase 2 GIMEMA Study (AML-1107). Blood. 2010;116(21):510. doi:10.1182/blood.V116.21.510.510
57. Park S, Chapuis N, Saint Marcoux F, et al. A phase Ib GOELAMS study of the mTOR inhibitor RAD001 in association with chemotherapy for AML patients in first relapse. Leukemia. 2013;27(7):1479–1486. doi:10.1038/leu.2013.17
58. Ragon BK, Odenike O, Baer MR, et al. Oral MEK 1/2 Inhibitor Trametinib in Combination With AKT Inhibitor GSK2141795 in Patients With Acute Myeloid Leukemia With RAS Mutations: a Phase II Study. Clinical Lymphoma Myeloma Leukemia. 2019;19(7):431–440.e13. doi:10.1016/j.clml.2019.03.015
59. Spencer A, Yoon -S-S, Harrison SJ, et al. The novel AKT inhibitor afuresertib shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. Blood. 2014;124(14):2190–2195. doi:10.1182/blood-2014-03-559963
60. Bhatt V, Wichman C, Bouska A, et al. Phase I trial of a novel first-in-class drug ONC201 as a post-transplant maintenance for acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). Blood. 2025;146(Supplement 1):
61. ES Wang, GJ Fetterly, L Pitzonka, et al. Phase 1 study of the angiopoietin 1/2 neutralizing peptibody, trebananib, in acute myeloid leukemia. J Clin Oncol. 2014;32:5s. doi:10.1200/jco.2014.32.15_suppl.7082
62. Cortes J, Talpaz M, Smith HP, et al. Phase 1 dose-finding study of rebastinib (DCC-2036) in patients with relapsed chronic myeloid leukemia and acute myeloid leukemia. Haematologica. 2017;102(3):519–528. doi:10.3324/haematol.2016.152710
63. Lee JH, Faderl S, Pagel JM, et al. Phase 1 study of CWP232291 in patients with relapsed or refractory acute myeloid leukemia and myelodysplastic syndrome. Blood Adv. 2020;4(9):2032–2043. doi:10.1182/bloodadvances.2019000757
64. Rizzieri DA, Cooley S, Odenike O, et al. An open-label phase 2 study of glycogen synthase kinase-3 inhibitor LY2090314 in patients with acute leukemia. Leukemia Lymphoma. 2016;57(8):1800–1806. doi:10.3109/10428194.2015.1122781
65. Deangelo DJ, Stone RM, Silverman LB, et al. A phase I clinical trial of the notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. J clin oncol. 2006;24(18_suppl):6585. doi:10.1200/jco.2006.24.18_suppl.6585
66. Roboz GJ, Mandrekar SJ, Desai P, et al. Randomized trial of 10 days of decitabine ± bortezomib in untreated older patients with AML: CALGB 11002 (Alliance). Blood Adv. 2018;2(24):3608–3617. doi:10.1182/bloodadvances.2018023689
67. Attar EC, Johnson JL, Amrein PC, et al. Bortezomib added to daunorubicin and cytarabine during induction therapy and to intermediate-dose cytarabine for consolidation in patients with previously untreated acute myeloid leukemia age 60 to 75 years: CALGB (Alliance) study 10502. J Clin Oncol. 2013;31(7):923–929. doi:10.1200/jco.2012.45.2177
68. Norton J, Sumransub N, Cordner K, et al. Phase I/II clinical trial of bortezomib in combination with CPX-351 for the treatment of newly-diagnosed TP53-mutated Acute Myeloid Leukemia (NCT07008638). Blood. 2025;146(Supplement 1):3433. doi:10.1182/blood-2025-3433
69. Giles FJ, Bellamy WT, Estrov Z, et al. The anti-angiogenesis agent, AG-013736, has minimal activity in elderly patients with poor prognosis acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS). Leukemia Res. 2006;30(7):801–811. doi:10.1016/j.leukres.2005.10.024
70. Mattison R, Jumonville A, Flynn PJ, et al. A phase II study of AZD2171 (cediranib) in the treatment of patients with acute myeloid leukemia or high-risk myelodysplastic syndrome. Leuk Lymphoma. 2015;56(7):2061–2066. doi:10.3109/10428194.2014.977886
71. Ossenkoppele GJ, Stussi G, Maertens J, et al. Addition of bevacizumab to chemotherapy in acute myeloid leukemia at older age: a randomized phase 2 trial of the Dutch-Belgian Cooperative Trial Group for Hemato-Oncology (HOVON) and the Swiss Group for Clinical Cancer Research (SAKK). Blood. 2012;120(24):4706–4711. doi:10.1182/blood-2012-04-420596
72. Karp JE, Gojo I, Pili R, et al. Targeting Vascular Endothelial Growth Factor for Relapsed and Refractory Adult Acute Myelogenous Leukemias: therapy with Sequential 1-β-d-Arabinofuranosylcytosine, Mitoxantrone, and Bevacizumab. Clin Cancer Res. 2004;10(11):3577–3585. doi:10.1158/1078-0432.Ccr-03-0627
73. Badar T, Handisides DR, Benito JM, et al. Phase I study of evofosfamide, an investigational hypoxia-activated prodrug, in patients with advanced leukemia. Am J Hematol. 2016;91(8):800–805. doi:10.1002/ajh.24415
74. Yap TA, Daver N, Mahendra M, et al. Complex I inhibitor of oxidative phosphorylation in advanced solid tumors and acute myeloid leukemia: phase I trials. Nat Med. 2023;29(1):115–126. doi:10.1038/s41591-022-02103-8
75. Pardee TS, Powell BL, Larson RA, et al. Devimistat plus chemotherapy vs chemotherapy alone for older relapsed or refractory patients with AML: results of the ARMADA trial. Blood Neoplasia. 2024;1(2):100009. doi:10.1016/j.bneo.2024.100009
76. Tschernia NP, Kumar V, Moore DT, et al. Safety and efficacy of pembrolizumab prior to allogeneic stem cell transplantation for acute myelogenous leukemia. Transplantat Cell Ther. 2021;27(12):1021.e1–1021.e5. doi:10.1016/j.jtct.2021.08.022
77. Zeidner JF, Vincent BG, Ivanova A, et al. Phase II trial of pembrolizumab after high-dose cytarabine in relapsed/refractory acute myeloid leukemia. Blood Cancer Discovery. 2021;2(6):616–629. doi:10.1158/2643-3230.BCD-21-0070
78. Solomon SR, Solh M, Morris JLE, et al. Phase 2 study of PD-1 blockade following autologous transplantation for patients with AML ineligible for allogeneic transplant. Blood Adv. 2023;7(18):5215–5224. doi:10.1182/bloodadvances.2023010477
79. Zeidan AM, Boddu PC, Wood BL, et al. Blast MRD AML-1 Trial: blockade of PD-1 Added to Standard Therapy to Target Measurable Residual Disease in Acute Myeloid Leukemia (AML) 1- an Investigator-Initiated, CTEP-Sponsored, Randomized Phase 2 Study of the Anti-PD-1 Antibody Pembrolizumab in Combination with Conventional Intensive Chemotherapy (IC) As Frontline Therapy in Patients with Acute Myeloid Leukemia (AML). Blood. 2020;136(Supplement 1):15. doi:10.1182/blood-2020-139668
80. Zhou H-S, Su Y-F, Wang J, et al. Updates from a single-center phase 2 study of PD-1 inhibitor combined with hypomethylating agent plus CAG regimen in patients with relapsed/refractory acute myeloid leukemia. Front Immunol. 2025;16:1533467. doi:10.3389/fimmu.2025.1533467
81. Gao X-N, Su Y-F, M-y L, et al. Single-center phase 2 study of PD-1 inhibitor combined with DNA hypomethylation agent+ CAG regimen in patients with relapsed/refractory acute myeloid leukemia. Cancer Immunol Immunother. 2023;72(8):2769–2782. doi:10.1007/s00262-023-03454-y
82. Garcia JS, Flamand Y, Penter L, et al. Ipilimumab plus decitabine for patients with MDS or AML in posttransplant or transplant-naïve settings. Blood. 2023;141(15):1884–1888. doi:10.1182/blood.2022017686
83. Zeidan AM, Westermann J, Kovacsovics T, et al. AML-484 first results of a phase II study (STIMULUS-AML1) investigating sabatolimab+ azacitidine+ venetoclax in patients with newly diagnosed acute myeloid leukemia (ND AML). Clin Lymphoma Myeloma Leukemia. 2022;22:S255. doi:10.1016/S2152-2650(22)01303-9
84. Zeidan A, Kovacsovics T, Adriano V. PS1483: primary results STIMULUS-AML1: a large, international, phase II study of sabatolimab combined with azacitidine and venetoclax as frontline therapy for unfit acute myeloid leukemia (AML) patients (pts). HemaSphere. 2025;9(suppl 1):2433–2435.
85. Huang R, Wang X, Yan H, et al. Safety and efficacy of CD33-targeted CAR-NK cell therapy for relapsed/refractory AML: preclinical evaluation and phase I trial. Exp Hematol Oncol. 2025;14(1):1. doi:10.1186/s40164-024-00592-6
86. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327–334. doi:10.1038/nature12984
87. Kwon M, Kim BS, Yoon S, Oh S-O, Lee D. Hematopoietic stem cells and their niche in bone marrow. Int J Mol Sci. 2024;25(13):6837. doi:10.3390/ijms25136837
88. Fröbel J, Landspersky T, Percin G, et al. The hematopoietic bone marrow niche ecosystem. Front Cell Develop Biol. 2021;9:705410. doi:10.3389/fcell.2021.705410
89. Goda C, Kulkarni R, Bustos Y, et al. Cellular taxonomy of the preleukemic bone marrow niche of acute myeloid leukemia. Leukemia. 2025;39(1):51–63. doi:10.1038/s41375-024-02415-3
90. Baryawno N, Przybylski D, Kowalczyk MS, et al. A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell. 2019;177(7):1915–1932.e16. doi:10.1016/j.cell.2019.04.040
91. Duarte D, Hawkins ED, Akinduro O, et al. Inhibition of endosteal vascular niche remodeling rescues hematopoietic stem cell loss in AML. Cell Stem Cell. 2018;22(1):64–77.e6. doi:10.1016/j.stem.2017.11.006
92. Raaijmakers MH, Mukherjee S, Guo S, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464(7290):852–857. doi:10.1038/nature08851
93. Walkley CR, Olsen GH, Dworkin S, et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell. 2007;129(6):1097–1110. doi:10.1016/j.cell.2007.05.014
94. Kode A, Manavalan JS, Mosialou I, et al. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature. 2014;506(7487):240–244. doi:10.1038/nature12883
95. Dong L, Yu WM, Zheng H, et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature. 2016;539(7628):304–308. doi:10.1038/nature20131
96. Mian SA, Ngo S, Bonnet D. Is the bone marrow microenvironment the hidden catalyst in malignant haematopoiesis? Leukemia. 2025;39(7):1589–1592. doi:10.1038/s41375-025-02630-6
97. Levesque J, Helwani F, Winkler I. The endosteal ‘osteoblastic’niche and its role in hematopoietic stem cell homing and mobilization. Leukemia. 2010;24(12):1979–1992. doi:10.1038/leu.2010.214
98. Xu C, Gao X, Wei Q, et al. Stem cell factor is selectively secreted by arterial endothelial cells in bone marrow. Nat Commun. 2018;9(1):2449. doi:10.1038/s41467-018-04726-3
99. Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481(7382):457–462. doi:10.1038/nature10783
100. Calvi L, Adams G, Weibrecht K, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425(6960):841–846. doi:10.1038/nature02040
101. Kim J-A, Kang Y-J, Park G, et al. Identification of a Stroma-Mediated Wnt/β-Catenin Signal Promoting Self-Renewal of Hematopoietic Stem Cells in the Stem Cell Niche. Stem Cells. 2009;27(6):1318–1329. doi:10.1002/stem.52
102. Méndez-Ferrer S, Michurina TV, Ferraro F, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. nature. 2010;466(7308):829–834. doi:10.1038/nature09262
103. Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. science. 1999;284(5411):143–147. doi:10.1126/science.284.5411.143
104. Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495(7440):231–235. doi:10.1038/nature11885
105. Kunisaki Y, Bruns I, Scheiermann C, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502(7473):637–643. doi:10.1038/nature12612
106. Omatsu Y, Sugiyama T, Kohara H, et al. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 2010;33(3):387–399. doi:10.1016/j.immuni.2010.08.017
107. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–598. doi:10.1038/nrc.2016.73
108. Mattiucci D, Maurizi G, Izzi V, et al. Bone marrow adipocytes support hematopoietic stem cell survival. J Cell Physiol. 2018;233(2):1500–1511. doi:10.1002/jcp.26037
109. Zhou BO, Yu H, Yue R, et al. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat Cell Biol. 2017;19(8):891–903. doi:10.1038/ncb3570
110. Yamazaki S, Ema H, Karlsson G, et al. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. 2011;147(5):1146–1158. doi:10.1016/j.cell.2011.09.053
111. Pinho S, Marchand T, Yang E, Wei Q, Nerlov C, Frenette PS. Lineage-biased hematopoietic stem cells are regulated by distinct niches. Dev Cell. 2018;44(5):634–641.e4. doi:10.1016/j.devcel.2018.01.016
112. Xu H, Li Y, Gao Y. The role of immune cells settled in the bone marrow on adult hematopoietic stem cells. Cell Mol Life Sci. 2024;81(1):420. doi:10.1007/s00018-024-05445-3
113. Lee-Thedieck C, Schertl P, Klein G. The extracellular matrix of hematopoietic stem cell niches. Adv Drug Delivery Rev. 2022;181:114069. doi:10.1016/j.addr.2021.114069
114. Gattazzo F, Urciuolo A, Bonaldo P. Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochim Biophys Acta. 2014;1840(8):2506–2519. doi:10.1016/j.bbagen.2014.01.010
115. Shi G, Chang Z, Zhang P, et al. Heterogeneous stiffness of the bone marrow microenvironment regulates the fate decision of haematopoietic stem and progenitor cells. Cell Prolif. 2024;57(12):e13715. doi:10.1111/cpr.13715
116. Grenier JMP, Testut C, Fauriat C, Mancini SJC, Aurrand-Lions M. Adhesion Molecules Involved in Stem Cell Niche Retention During Normal Haematopoiesis and in Acute Myeloid Leukaemia. Front Immunol. 2021;12:756231. doi:10.3389/fimmu.2021.756231
117. Jacamo R, Chen Y, Wang Z, et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood J Am Soc Hematol. 2014;123(17):2691–2702 doi:10.1182/blood-2013-06-511527.
118. Gutjahr JC, Bayer E, Yu X, et al. CD44 engagement enhances acute myeloid leukemia cell adhesion to the bone marrow microenvironment by increasing VLA-4 avidity. haematologica. 2020;106(8):2102. doi:10.3324/haematol.2019.231944
119. Ladikou EE, Sharp K, Simoes FA, et al. A Novel In Vitro Model of the Bone Marrow Microenvironment in Acute Myeloid Leukemia Identifies CD44 and Focal Adhesion Kinase as Therapeutic Targets to Reverse Cell Adhesion-Mediated Drug Resistance. Cancers. 2025;17(1):135. doi:10.3390/cancers17010135
120. Chaves B, Santose Silva JC, Nakaya H, et al. In vitro morphological profiling of T cells predicts clinical response to natalizumab therapy in patients with multiple sclerosis. Nat Commun. 2025;16(1):5533. doi:10.1038/s41467-025-60224-3
121. Ogana HA, Hurwitz S, Wei N, et al. Targeting integrins in drug-resistant acute myeloid leukaemia. Br J Pharmacol. 2024;181(2):295–316. doi:10.1111/bph.16149
122. Winkler IG, Barbier V, Nowlan B, et al. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nature Med. 2012;18(11):1651–1657. doi:10.1038/nm.2969
123. Hidalgo A, Weiss LA, Frenette PS. Functional selectin ligands mediating human CD34(+) cell interactions with bone marrow endothelium are enhanced postnatally. J Clin Invest. 2002;110(4):559–569. doi:10.1172/JCI14047
124. Katayama Y, Hidalgo A, Furie BC, Vestweber D, Furie B, Frenette PS. PSGL-1 participates in E-selectin-mediated progenitor homing to bone marrow: evidence for cooperation between E-selectin ligands and alpha4 integrin. Blood. 2003;102(6):2060–2067. doi:10.1182/blood-2003-04-1212
125. Barbier V, Erbani J, Fiveash C, et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat Commun. 2020;11(1):2042. doi:10.1038/s41467-020-15817-5
126. Peled A, Petit I, Kollet O, et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science. 1999;283(5403):845–848. doi:10.1126/science.283.5403.845
127. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977–988. doi:10.1016/j.immuni.2006.10.016
128. Tavor S, Petit I, Porozov S, et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004;64(8):2817–2824. doi:10.1158/0008-5472.can-03-3693
129. Zeng Z, Shi YX, Samudio IJ, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113(24):6215–6224. doi:10.1182/blood-2008-05-158311
130. Li Y, Xue M, Deng X, et al. TET2-mediated mRNA demethylation regulates leukemia stem cell homing and self-renewal. Cell Stem Cell. 2023;30(8):1072–1090.e10. doi:10.1016/j.stem.2023.07.001
131. Anderson NR, Sheth V, Li H, et al. Microenvironmental CXCL12 deletion enhances Flt3-ITD acute myeloid leukemia stem cell response to therapy by reducing p38 MAPK signaling. Leukemia. 2023;37(3):560–570. doi:10.1038/s41375-022-01798-5
132. Slater S. Plerixafor. J Adv Pract Oncol. 2012;3(1):49.
133. Zhang M, Ge Y, Xu S, et al. Nanomicelles co-loading CXCR4 antagonist and doxorubicin combat the refractory acute myeloid leukemia. Pharmacol Res. 2022;185:106503. doi:10.1016/j.phrs.2022.106503
134. Lee HJ, Daver N, Kantarjian HM, Verstovsek S, Ravandi F. The role of JAK pathway dysregulation in the pathogenesis and treatment of acute myeloid leukemia. Clin Cancer Res. 2013;19(2):327–335. doi:10.1158/1078-0432.CCR-12-2087
135. Wu J, Li X, Liu Y, et al. MMP14 from BM-MSCs facilitates progression and Ara-C resistance in acute myeloid leukemia via the JAK/STAT pathway. Exp Hematol Oncol. 2025;14(1):43. doi:10.1186/s40164-025-00635-6
136. Lu J, Dong Q, Zhang S, Feng Y, Yang J, Zhao L. Acute myeloid leukemia (AML)-derived mesenchymal stem cells induce chemoresistance and epithelial–mesenchymal transition-like program in AML through IL-6/JAK2/STAT3 signaling. Cancer Science. 2023;114(8):3287–3300. doi:10.1111/cas.15855
137. Hou D, Wang B, You R, et al. Stromal cells promote chemoresistance of acute myeloid leukemia cells via activation of the IL-6/STAT3/OXPHOS axis. Ann Transl Med. 2020;8(21):1346. doi:10.21037/atm-20-3191
138. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. 2021;6(1):402. doi:10.1038/s41392-021-00791-1
139. Owen KL, Brockwell NK, Parker BS. JAK-STAT Signaling: a Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers. 2019;11(12):2002. doi:10.3390/cancers11122002
140. Politikos I, Brown S, Fein JA, et al. Phase 2 trial of cyclosporine-A, mycophenolate mofetil, and tocilizumab GVHD prophylaxis in cord blood transplantation. Blood Adv. 2025;9(10):2570–2584. doi:10.1182/bloodadvances.2024014177
141. Chen P, Jin Q, Fu Q, et al. Induction of Multidrug Resistance of Acute Myeloid Leukemia Cells by Cocultured Stromal Cells via Upregulation of the PI3K/Akt Signaling Pathway. Oncol Res. 2016;24(4):215–223. doi:10.3727/096504016x14634208143021
142. Pan Z-P, Wang B, Hou D-Y, et al. METTL3 mediates bone marrow mesenchymal stem cell adipogenesis to promote chemoresistance in acute myeloid leukaemia. FEBS Open Bio. 2021;11(6):1659–1672. doi:10.1002/2211-5463.13165
143. Zhang L, Zhao Q, Cang H, et al. Acute myeloid leukemia cells educate mesenchymal stromal cells toward an adipogenic differentiation propensity with leukemia promotion capabilities. Adv Sci. 2022;9(16):2105811. doi:10.1002/advs.202105811
144. Fukasawa K, Tokumura K, Yoshimoto M, et al. Regulatory role of mTORC1 signaling in osteoblasts in acute myeloid leukemia progression and steady-state hematopoiesis. iScience. 2025;29(1). doi:10.1016/j.isci.2025.114533.
145. Hatfield K, Øyan AM, Ersvaer E, et al. Primary human acute myeloid leukaemia cells increase the proliferation of microvascular endothelial cells through the release of soluble mediators. Br J Haematol. 2009;144(1):53–68. doi:10.1111/j.1365-2141.2008.07411.x
146. Brindle NP, Saharinen P, Alitalo K. Signaling and functions of angiopoietin-1 in vascular protection. Circ Res. 2006;98(8):1014–1023. doi:10.1161/01.RES.0000218275.54089.12
147. Fiedler U, Augustin HG. Angiopoietins: a link between angiogenesis and inflammation. Trends Immunol. 2006;27(12):552–558. doi:10.1016/j.it.2006.10.004
148. Reikvam H, Hatfield KJ, Lassalle P, Olsnes Kittang A, Ersvær E, Bruserud Ø. Targeting the angiopoietin (Ang)/Tie-2 pathway in the crosstalk between acute myeloid leukaemia and endothelial cells: studies of Tie-2 blocking antibodies, exogenous Ang-2 and inhibition of constitutive agonistic Ang-1 release. Expert Opin Invest Drugs. 2010;19(2):169–183. doi:10.1517/13543780903485659
149. Bachegowda L, Morrone K, Winski SL, et al. Pexmetinib: a Novel Dual Inhibitor of Tie2 and p38 MAPK with Efficacy in Preclinical Models of Myelodysplastic Syndromes and Acute Myeloid Leukemia. Cancer Res. 2016;76(16):4841–4849. doi:10.1158/0008-5472.Can-15-3062
150. Garcia-Manero G, Khoury HJ, Jabbour E, et al. A phase I study of oral ARRY-614, a p38 MAPK/Tie2 dual inhibitor, in patients with low or intermediate-1 risk myelodysplastic syndromes. Clin Cancer Res. 2015;21(5):985–994. doi:10.1158/1078-0432.Ccr-14-1765
151. Palani HK, Ganesan S, Balasundaram N, et al. Ablation of Wnt signaling in bone marrow stromal cells overcomes microenvironment-mediated drug resistance in acute myeloid leukemia. Sci Rep. 2024;14(1):8404. doi:10.1038/s41598-024-58860-8
152. Mosialou I, Ali AM, Labella R, et al. A niche driven mechanism determines response and a mutation-independent therapeutic approach for myeloid malignancies. Cancer Cell. 2025;43(6):1007–1024.e13. doi:10.1016/j.ccell.2025.03.007
153. Minciacchi VR, Karantanou C, Bravo J, et al. Differential inflammatory conditioning of the bone marrow by acute myeloid leukemia and its impact on progression. Blood Adv. 2024;8(19):4983–4996. doi:10.1182/bloodadvances.2024012867
154. Gupta K, Stefan T, Ignatz-Hoover J, et al. GSK-3 Inhibition Sensitizes Acute Myeloid Leukemia Cells to 1,25D-Mediated Differentiation. Cancer Res. 2016;76(9):2743–2753. doi:10.1158/0008-5472.Can-15-2290
155. Gupta K, Gulen F, Sun L, et al. GSK3 is a regulator of RAR-mediated differentiation. Leukemia. 2012;26(6):1277–1285. doi:10.1038/leu.2012.2
156. Steinert G, Oancea C, Roos J, et al. Sulindac Sulfide Reverses Aberrant Self-Renewal of Progenitor Cells Induced by the AML-Associated Fusion Proteins PML/RARα and PLZF/RARα. PLoS One. 2011;6(7):e22540. doi:10.1371/journal.pone.0022540
157. Lu Y, Liu X-F, Liu T-R, et al. Celecoxib exerts antitumor effects in HL-60 acute leukemia cells and inhibits autophagy by affecting lysosome function. Biomed Pharmacother. 2016;84:1551–1557. doi:10.1016/j.biopha.2016.11.026
158. Pan M, Jiao C, Sun M, et al. Venetoclax synergizes with Wnt/β-catenin inhibitor C-82 in acute myeloid leukemia by increasing the degradation of Mcl-1 protein. Can Cell Inter. 2025;25(1):197. doi:10.1186/s12935-025-03825-8
159. Jiang X, Mak PY, Mu H, et al. Disruption of Wnt/β-Catenin Exerts Antileukemia Activity and Synergizes with FLT3 Inhibition in FLT3-Mutant Acute Myeloid Leukemia. Clin Cancer Res. 2018;24(10):2417–2429. doi:10.1158/1078-0432.Ccr-17-1556
160. Hurwitz S, Ogana H, Wan Z, Navid F, Bhojwani D, Kim Y-M. TBL1 Inhibitor, Tegavivint, Supresses Cell Survival and Promotes Sensitivity to Chemotherapy in ALL and AML. Blood. 2022;140(Supplement 1):5995. doi:10.1182/blood-2022-167445
161. Tomasoni C, Arsuffi C, Donsante S, et al. AML alters bone marrow stromal cell osteogenic commitment via Notch signaling. Front Immunol. 2023;14:1320497. doi:10.3389/fimmu.2023.1320497
162. Tian Y, Xu Y, Xue T, et al. Notch activation enhances mesenchymal stem cell sheet osteogenic potential by inhibition of cellular senescence. Cell Death Dis. 2017;8(2):e2595. doi:10.1038/cddis.2017.2
163. Takam Kamga P, Bassi G, Cassaro A, et al. Notch signalling drives bone marrow stromal cell-mediated chemoresistance in acute myeloid leukemia. Oncotarget. 2016;7(16):21713–21727. doi:10.18632/oncotarget.7964
164. Li D, Li T, Shang Z, et al. Combined inhibition of Notch and FLT3 produces synergistic cytotoxic effects in FLT3/ITD+ acute myeloid leukemia. Signal Transduc Target Ther. 2020;5(1):21. doi:10.1038/s41392-020-0108-z
165. Mosialou I, Ali AM, Adams R, et al. Therapeutic Anti-Jagged1 Antibody Targeting Osteoblast-Related Myeloid Dysplasia to Overcome Standard of Care Resistance. Blood. 2022;140(Supplement 1):2902. doi:10.1182/blood-2022-167042
166. Sierra RA, Trillo-Tinoco J, Mohamed E, et al. Anti-jagged immunotherapy inhibits MDSCs and overcomes tumor-induced tolerance. Cancer Res. 2017;77(20):5628–5638. doi:10.1158/0008-5472.CAN-17-0357
167. Zinatizadeh MR, Schock B, Chalbatani GM, Zarandi PK, Jalali SA, Miri SR. The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Genes Dis. 2021;8(3):287–297. doi:10.1016/j.gendis.2020.06.005
168. Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transduc Target Ther. 2017;2(1):17023. doi:10.1038/sigtrans.2017.23
169. Chen C, Edelstein LC, Gélinas C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol Cell Biol. 2000;20(8):2687–2695. doi:10.1128/mcb.20.8.2687-2695.2000
170. Di Francesco B, Verzella D, Capece D, et al. NF-κB: a druggable target in acute myeloid leukemia. Cancers. 2022;14(14):3557. doi:10.3390/cancers14143557
171. Guzman ML, Neering SJ, Upchurch D, et al. Nuclear factor-κB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001;98(8):2301–2307. doi:10.1182/blood.V98.8.2301
172. Guzman ML, Swiderski CF, Howard DS, et al. Preferential induction of apoptosis for primary human leukemic stem cells. Proc Natl Acad Sci. 2002;99(25):16220–16225. doi:10.1073/pnas.252462599
173. Csizmar CM, Kim DH, Sachs Z. The role of the proteasome in AML. Blood Cancer J. 2016;6(12):e503. doi:10.1038/bcj.2016.112
174. Wang D, Sun Z, Zhu X, et al. GARP-mediated active TGF-β1 induces bone marrow NK cell dysfunction in AML patients with early relapse post–allo-HSCT. Blood J Am Soc Hematol. 2022;140(26):2788–2804 doi:10.1182/blood.2022015474.
175. Wang Z-K, Zhang Z-W, Lyu Z-S, et al. Inhibition of TGF-β signaling in bone marrow endothelial cells promotes hematopoietic recovery in acute myeloid leukemia patients. Cancer Lett. 2024;605:217290. doi:10.1016/j.canlet.2024.217290
176. Liang X, Zhou J, Li C, et al. The roles and mechanisms of TGFB1 in acute myeloid leukemia chemoresistance. Cell Signal. 2024;116:111027. doi:10.1016/j.cellsig.2023.111027
177. Lu W, Wan Y, Li Z, et al. Growth differentiation factor 15 contributes to marrow adipocyte remodeling in response to the growth of leukemic cells. J Exp Clin Cancer Res. 2018;37(1):66. doi:10.1186/s13046-018-0738-y
178. Shaoxin Y, Wei L, Chong Z, et al. Leukemia cells remodel marrow adipocytes via TRPV4-dependent lipolysis. Haematologica. 2020;105(11):2572–2583. doi:10.3324/haematol.2019.225763
179. Santini V, Valcárcel D, Platzbecker U, et al. Phase II study of the ALK5 inhibitor galunisertib in very low-, low-, and intermediate-risk myelodysplastic syndromes. Clin Cancer Res. 2019;25(23):6976–6985. doi:10.1158/1078-0432.CCR-19-1338
180. Xu Y, Tabe Y, Jin L, et al. TGF-beta receptor kinase inhibitor LY2109761 reverses the anti-apoptotic effects of TGF-beta1 in myelo-monocytic leukaemic cells co-cultured with stromal cells. Br J Haematol. 2008;142(2):192–201. doi:10.1111/j.1365-2141.2008.07130.x
181. Chen J, Mu Q, Li X, et al. Homoharringtonine targets Smad3 and TGF-β pathway to inhibit the proliferation of acute myeloid leukemia cells. Oncotarget. 2017;8(25):3 doi:10.18632/oncotarget.16956.
182. Kuett A, Rieger C, Perathoner D, et al. IL-8 as mediator in the microenvironment-leukaemia network in acute myeloid leukaemia. Sci Rep. 2015;5(1):18411. doi:10.1038/srep18411
183. Picinich SC, Glod JW, Banerjee D. Protein kinase C zeta regulates interleukin-8-mediated stromal-derived factor-1 expression and migration of human mesenchymal stromal cells. Exp Cell Res. 2010;316(4):593–602. doi:10.1016/j.yexcr.2009.11.011
184. Abdul-Aziz AM, Shafat MS, Mehta TK, et al. MIF-Induced Stromal PKCβ/IL8 Is Essential in Human Acute Myeloid Leukemia. Cancer Res. 2017;77(2):303–311. doi:10.1158/0008-5472.Can-16-1095
185. Bruserud O, Ryningen A, Wergeland L, Glenjen NI, Gjertsen BT. Osteoblasts increase proliferation and release of pro-angiogenic interleukin 8 by native human acute myelogenous leukemia blasts. Haematologica. 2004;89(4):391–402.
186. Schinke C, Giricz O, Li W, et al. IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood. 2015;125(20):3144–3152. doi:10.1182/blood-2015-01-621631
187. Ginestier C, Liu S, Diebel ME, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest. 2010;120(2):485–497. doi:10.1172/JCI39397
188. Ning Y, Labonte MJ, Zhang W, et al. The CXCR2 antagonist, SCH-527123, shows antitumor activity and sensitizes cells to oxaliplatin in preclinical colon cancer models. Mol Cancer Ther. 2012;11(6):1353–1364. doi:10.1158/1535-7163.MCT-11-0915
189. Mahler DA, Huang S, Tabrizi M, Bell GM. Efficacy and Safety of a Monoclonal Antibody Recognizing Interleukin-8 in COPD: a Pilot Study. CHEST. 2004;126(3):926–934. doi:10.1378/chest.126.3.926
190. Drusbosky L, Gars E, Trujillo A, et al. Endothelial cell derived angiocrine support of acute myeloid leukemia targeted by receptor tyrosine kinase inhibition. Leuk Res. 2015;39(9):984–989. doi:10.1016/j.leukres.2015.05.015
191. Hegde M, Ahmad MH, Mulet Lazaro R, et al. The co-receptor Neuropilin-1 enhances proliferation in inv(16) acute myeloid leukemia via VEGF signaling. Leukemia. 2025;39(2):360–370. doi:10.1038/s41375-024-02471-9
192. Roskoski R. Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun. 2007;356(2):323–328. doi:10.1016/j.bbrc.2007.02.156
193. Fiedler W, Kayser S, Kebenko M, et al. A phase I/II study of sunitinib and intensive chemotherapy in patients over 60 years of age with acute myeloid leukaemia and activating FLT3 mutations. Br J Haematol. 2015;169(5):694–700. doi:10.1111/bjh.13353
194. Auclair D, Miller D, Yatsula V, et al. Antitumor activity of sorafenib in FLT3-driven leukemic cells. Leukemia. 2007;21(3):439–445. doi:10.1038/sj.leu.2404508
195. Lal D, Park JA, Demock K, et al. Aflibercept Exerts Antivascular Effects and Enhances Levels of Anthracycline Chemotherapy In vivo in Human Acute Myeloid Leukemia Models. Mol Cancer Ther. 2010;9(10):2737–2751. doi:10.1158/1535-7163.Mct-10-0334
196. Tirado HA, Balasundaram N, Laaouimir L, Erdem A, van Gastel N. Metabolic crosstalk between stromal and malignant cells in the bone marrow niche. Bone Report. 2023;18:101669. doi:10.1016/j.bonr.2023.101669
197. Spencer JA, Ferraro F, Roussakis E, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. 2014;508(7495):269–273. doi:10.1038/nature13034
198. Passaro D, Di Tullio A, Abarrategi A, et al. Increased Vascular Permeability in the Bone Marrow Microenvironment Contributes to Disease Progression and Drug Response in Acute Myeloid Leukemia. Cancer Cell. 2017;32(3):324–341e6. doi:10.1016/j.ccell.2017.08.001
199. Portwood S, Lal D, Hsu YC, et al. Activity of the hypoxia-activated prodrug, TH-302, in preclinical human acute myeloid leukemia models. Clin Cancer Res. 2013;19(23):6506–6519. doi:10.1158/1078-0432.CCR-13-0674
200. Chen WL, Wang JH, Zhao AH, et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood. 2014;124(10):1645–1654. doi:10.1182/blood-2014-02-554204
201. Jones CL, Stevens BM, D’Alessandro A, et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell. 2018;34(5):724–740e4. doi:10.1016/j.ccell.2018.10.005
202. Panuzzo C, Jovanovski A, Pergolizzi B, et al. Mitochondria: a Galaxy in the Hematopoietic and Leukemic Stem Cell Universe. Int J Mol Sci. 2020;21(11):3928. doi:10.3390/ijms21113928
203. Lagadinou ED, Sach A, Callahan K, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12(3):329–341. doi:10.1016/j.stem.2012.12.013
204. Braun M, Qorraj M, Buttner M, et al. CXCL12 promotes glycolytic reprogramming in acute myeloid leukemia cells via the CXCR4/mTOR axis. Leukemia. 2016;30(8):1788–1792. doi:10.1038/leu.2016.58
205. Liu F, Kalpage HA, Wang D, et al. Cotargeting of Mitochondrial Complex I and Bcl-2 Shows Antileukemic Activity against Acute Myeloid Leukemia Cells Reliant on Oxidative Phosphorylation. Cancers. 2020;12(9):2400. doi:10.3390/cancers12092400
206. You R, Hou D, Wang B, et al. Bone marrow microenvironment drives AML cell OXPHOS addiction and AMPK inhibition to resist chemotherapy. J Leukoc Biol. 2022;112(2):299–311. doi:10.1002/JLB.6A0821-409RR
207. Vilaplana-Lopera N, Cuminetti V, Almaghrabi R, et al. Crosstalk between AML and stromal cells triggers acetate secretion through the metabolic rewiring of stromal cells. Elife. 2022;11:e75908 doi:10.7554/eLife.75908.
208. Shafat MS, Oellerich T, Mohr S, et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood. 2017;129(10):1320–1332. doi:10.1182/blood-2016-08-734798
209. Tabe Y, Yamamoto S, Saitoh K, et al. Bone Marrow Adipocytes Facilitate Fatty Acid Oxidation Activating AMPK and a Transcriptional Network Supporting Survival of Acute Monocytic Leukemia Cells. Cancer Res. 2017;77(6):1453–1464. doi:10.1158/0008-5472.Can-16-1645
210. Tabe Y, Konopleva M, Andreeff M. Fatty Acid Metabolism, Bone Marrow Adipocytes, and AML. Front Oncol. 2020;10:155. doi:10.3389/fonc.2020.00155
211. Moschoi R, Imbert V, Nebout M, et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood. 2016;128(2):253–264. doi:10.1182/blood-2015-07-655860
212. Saito K, Zhang Q, Yang H, et al. Exogenous mitochondrial transfer and endogenous mitochondrial fission facilitate AML resistance to OxPhos inhibition. Blood Adv. 2021;5(20):4233–4255. doi:10.1182/bloodadvances.2020003661
213. Marlein CR, Zaitseva L, Piddock RE, et al. PGC-1alpha driven mitochondrial biogenesis in stromal cells underpins mitochondrial trafficking to leukemic blasts. Leukemia. 2018;32(9):2073–2077. doi:10.1038/s41375-018-0221-y
214. Mistry JJ, Moore JA, Kumar P, et al. Daratumumab inhibits acute myeloid leukaemia metabolic capacity by blocking mitochondrial transfer from mesenchymal stromal cells. Haematologica. 2021;106(2):589–592. doi:10.3324/haematol.2019.242974
215. Dhakal N, Perez FS, Hernandez RG, et al. Endothelial cell-derived mitochondrial transfer to Acute Myeloid Leukemia alters global gene expression to confer chemoresistance. Blood. 2025;146(Supplement 1):5015. doi:10.1182/blood-2025-5015
216. Nwarunma E, Miari K, Papadopoulou A, et al. M2-like macrophages transfer mitochondria to acute myeloid leukaemia cells via tunnelling nanotubes promoting therapy resistance. Blood. 2024;144(Supplement 1):2761. doi:10.1182/blood-2024-200028
217. Reinhardt C, Ochsenbein AF. Immune checkpoints regulate acute myeloid leukemia stem cells. Leukemia. 2025;39(6):1277–1293. doi:10.1038/s41375-025-02566-x
218. Wang F, Yang L, Xiao M, et al. PD-L1 regulates cell proliferation and apoptosis in acute myeloid leukemia by activating PI3K-AKT signaling pathway. Sci Rep. 2022;12(1):11444. doi:10.1038/s41598-022-15020-0
219. Alatrash G, Knape C, Whited L, et al. A Phase I Study of Nivolumab in Combination with Ipilimumab for the Treatment of Patients with High Risk or Refractory/Relapsed Acute Myeloid Leukemia Following Allogeneic Stem Cell Transplantation. Blood. 2024;144(Supplement 1):1047. doi:10.1182/blood-2024-211343
220. Melero I, de Miguel Luken M, de Velasco G, et al. Neutralizing GDF-15 can overcome anti-PD-1 and anti-PD-L1 resistance in solid tumours. Nature. 2025;637(8048):1218–1227. doi:10.1038/s41586-024-08305-z
221. Guan X, Hu R, Choi Y, et al. Anti-TIGIT antibody improves PD-L1 blockade through myeloid and Treg cells. Nature. 2024;627(8004):646–655. doi:10.1038/s41586-024-07121-9
222. Jin X, Xie D, Sun R, et al. CAR-T cells dual-target CD123 and NKG2DLs to eradicate AML cells and selectively target immunosuppressive cells. Oncoimmunology. 2023;12(1):2248826. doi:10.1080/2162402X.2023.2248826
223. Vanhooren J, Dobbelaere R, Derpoorter C, et al. CAR-T in the Treatment of Acute Myeloid Leukemia: barriers and How to Overcome Them. Hemasphere. 2023;7(9):e937. doi:10.1097/HS9.0000000000000937
224. Skopek R, Palusińska M, Kaczor-Keller K, et al. Choosing the Right Cell Line for Acute Myeloid Leukemia (AML) Research. Int J Mol Sci. 2023;24(6):5377. doi:10.3390/ijms24065377
225. Pettirossi V, Venanzi A, Spanhol-Rosseto A, et al. The gene mutation landscape of acute myeloid leukemia cell lines and its exemplar use to study the BCOR tumor suppressor. Leukemia. 2023;37(2):473–477. doi:10.1038/s41375-022-01788-7
226. Skelding KA, Barry DL, Lincz LF. Modeling the Bone Marrow Microenvironment to Better Understand the Pathogenesis, Progression, and Treatment of Hematological Cancers. Cancers. 2025;17(15):2571. doi:10.3390/cancers17152571
227. Borella G, Da Ros A, Porcù E, et al. Acute Myeloid Leukemia (AML) in a 3D Bone Marrow Niche Showed High Performance for in Vitro and In Vivo Drug Screenings. Blood. 2019;134(Supplement_1):544. doi:10.1182/blood-2019-128382
228. Cheung HL, Wong YH, Li YY, et al. Microenvironment matters: in vitro 3D bone marrow niches differentially modulate survival, phenotype and drug responses of acute myeloid leukemia (AML) cells. Biomaterials. 2025;312:122719. doi:10.1016/j.biomaterials.2024.122719
229. Blanco TM, Mantalaris A, Bismarck A, Panoskaltsis N. The development of a three-dimensional scaffold for ex vivo biomimicry of human acute myeloid leukaemia. Biomaterials. 2010;31(8):2243–2251. doi:10.1016/j.biomaterials.2009.11.094
230. Aljitawi OS, Li D, Xiao Y, et al. A novel three-dimensional stromal-based model for in vitro chemotherapy sensitivity testing of leukemia cells. Leuk Lymphoma. 2014;55(2):378–391. doi:10.3109/10428194.2013.793323
231. Bray LJ, Binner M, Körner Y, von Bonin M, Bornhäuser M, Werner C. A three-dimensional ex vivo tri-culture model mimics cell-cell interactions between acute myeloid leukemia and the vascular niche. Haematologica. 2017;102(7):1215–1226. doi:10.3324/haematol.2016.157883
232. Duval K, Grover H, Han LH, et al. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology. 2017;32(4):266–277. doi:10.1152/physiol.00036.2016
233. Nervi B, Ramirez P, Rettig MP, et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113(24):6206–6214. doi:10.1182/blood-2008-06-162123
234. Cooper TM, Sison EAR, Baker SD, et al. A phase 1 study of the CXCR4 antagonist plerixafor in combination with high-dose cytarabine and etoposide in children with relapsed or refractory acute leukemias or myelodysplastic syndrome: a Pediatric Oncology Experimental Therapeutics Investigators’ Consortium study (POE 10-03). Pediatr Blood Cancer. 2017;64(8). doi:10.1002/pbc.26414
235. Chen D-W, Huang T, Huang Y, et al. Developing a Humanized Murine Model for Co-Transplantation of Acute Myeloid Leukemia. Blood. 2022;140(Supplement 1):5776–5777. doi:10.1182/blood-2022-158615
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Therapeutic Targeting of FLT3 in Acute Myeloid Leukemia: Current Status and Novel Approaches
Tecik M, Adan A
OncoTargets and Therapy 2022, 15:1449-1478
Published Date: 30 November 2022
Health Disparities in Acute Myeloid Leukemia Patients Undergoing Treatment with Tyrosine Kinase Inhibitor (TKI) Therapy Targeting FLT3, IDH1, or IDH2
Espinoza-Gutarra MR, Jarrett BA, Wang X, Afghahi A, Bae S
Blood and Lymphatic Cancer: Targets and Therapy 2026, 16:559759
Published Date: 12 March 2026