")
Back to Journals » Journal of Inflammation Research » Volume 14
The Supportive Role of NSC328382, a P2X7R Antagonist, in Enhancing the Inhibitory Effect of CRID3 on NLRP3 Inflammasome Activation in Rats with Dextran Sodium Sulfate-Induced Colitis
Authors Saber S , Yahya G , Gobba NA, Sharaf H, Alshaman R, Alattar A, Amin NA, El-Shedody R, Aboutouk FH, Abd El-Galeel Y, El-Hefnawy A, Shabaka D, Khalifa A, Saleh R, Osama D, El-Zoghby G, Youssef ME
Received 17 April 2021
Accepted for publication 5 July 2021
Published 21 July 2021 Volume 2021:14 Pages 3443—3463
DOI https://doi.org/10.2147/JIR.S315938
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Sameh Saber,1 Galal Yahya,2 Naglaa A Gobba,3 Hossam Sharaf,4 Reem Alshaman,5 Abdullah Alattar,5 Noha A Amin,6 Ruwyda El-Shedody,4 Farah H Aboutouk,4 Yumna Abd El-Galeel,4 Amr El-Hefnawy,4 Dina Shabaka,4 Arwa Khalifa,4 Renad Saleh,4 Donya Osama,4 Ghada El-Zoghby,4 Mahmoud E Youssef1
1Department of Pharmacology, Faculty of Pharmacy, Delta University for Science and Technology, Gamasa, Egypt; 2Department of Microbiology and Immunology, Faculty of Pharmacy, Zagazig University, Al Sharqia, Egypt; 3Department of Pharmacology and Toxicology, College of Pharmacy, Misr University for Science and Technology, Cairo, Egypt; 4Department of Clinical Pharmacy, Faculty of Pharmacy, Delta University for Science and Technology, Gamasa, Egypt; 5Department of Pharmacology and Toxicology, Faculty of Pharmacy, University of Tabuk, Tabuk, Saudi Arabia; 6Department of Haematology, Theodor Bilharz Research Institute, Giza, Egypt
Correspondence: Mahmoud E Youssef; Sameh Saber
Department of Pharmacology, Faculty of Pharmacy, Delta University for Science and Technology, Costal International Road, P.O. Box: +11152, Mansoura, Dakahlia, Egypt
Tel +2 50 2770140; +2 01033124949
Fax +2 50 2770140
Email [email protected]; [email protected]; [email protected]
Purpose: The NLRP3 inflammasome is a substantial component of the inflammation process. The complex pathogenesis of and the implication of a vast number of components in the inflammasome-activation pathway prompted us to search for compounds that have a wide therapeutic index and act at the level of multiple cellular targets. Although CRID3 blocks NLRP3 with high specificity in the laboratory, clinical trials of the compound reported weaker potency.
Methods: We used NSC328382, a P2X7R antagonist, as an adjunctive therapy and generated a strategy to potentiate the effects of CRID3 in rats with DSS-induced colitis.
Results: NSC328382/CRID3 combined therapy exhibited a significantly increased efficacy compared with either of the monotherapies. NSC328382/CRID3 was more efficient in 1) attenuating colon shortening and disease activity; 2) improving goblet cell density and both the macroscopic and microscopic scenario of the injured colon; 3) improving the antioxidant defense mechanisms of the inflamed colon against oxidative stress; and 4) mitigating the inflammation state by downregulating the proinflammatory cytokines. Pyroptotic cell death was also conspicuously restrained. Additionally, NSC328382 interrupted the MyD88/NF-κB axis. Moreover, NSC328382/CRID3 exhibited the ability to alter Th1/Th2 dominance.
Conclusion: The clinical application of NSC328382/CRID3 may result in the generation of a novel approach for the treatment of IBDs.
Keywords: NSC328382, CRID3, P2X7R/NLRP3, MyD88/NF-κB, Colitis
Introduction
Ulcerative colitis (UC) is one of the major inflammatory bowel diseases (IBDs) that disturbs the colon and rectum and is characterized by a complex pathogenesis with a marked production of proinflammatory cytokines.1 Although the etiopathogenesis of UC remains unclear, a substantial body of evidence indicates the involvement of several factors that induce the disease development such as environmental triggers, genetic predisposition, gut microbiome, and an aberrant immune response.2,3
When activated, the purinoceptor P2X7 (P2X7R) contributes to the development of inflammation in different pathologies and has recently been acknowledged as a critical component of IBDs. In addition, P2X7R is overexpressed in the gut mucosa of patients with IBDs.4 Extracellular adenosine triphosphate (ATP), which is produced by infected or stressed cells, has been considered as an endogenous signaling molecule that is capable of mediating inflammation and immune responses through P2X7R.5 P2X7R is an ATP-gated cation channel that is expressed extracellularly on epithelial cells and on immune effector cells. It is involved in the regulation of intracellular inflammatory signaling and the formation of proinflammatory cytokines, such as IL-1β and IL-18.6 It has been reported that inflammasome activation through the NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) can be provoked in the context of ATP through P2X7R.7
NSC328382 (NSC) is a potent and selective antagonist of P2X7R with no known toxicity.8,9 NSC is effective in preventing P2X7R-induced cytotoxicity in retinal cells,10 microglia,11 and astrocytes.12 In addition, NSC attenuated the early phase of renal inflammation and interstitial fibrosis.13 Furthermore, prophylactic systemic P2X7R blockade by NSC prevented trinitrobenzene sulfonic (TNBS) acid-induced colitis in rats.5
When activated, NLRP3 oligomerizes and recruits the ASC adaptor protein, thus forming a binding site to initiate the dimerization, and activation of caspase-1, which then stimulate the cleavage and activation of the proinflammatory cytokines IL-1β and IL-18 to their active forms and mediates a type of inflammatory cell death known as pyroptosis.14 IL-1β and IL-18 probably play a crucial role in the early phase of the inflammation cascade and contribute to the pathology of the colon. NLRP3 inflammasome activation and the consequent development of the active forms IL-1β and IL-18 are a two-phase process: priming and activation.15 The stimulation of the nuclear transcription factor kappa B (NF-κB) elicits priming, leading to the production of the inactive proforms pro-IL-1β and pro-IL-18.16 During the activation phase, the assembly and activation of the NLRP3 inflammasome complex is induced by damage-associated molecular patterns or pathogen-associated molecular patterns, leading to the maturation of these proforms via caspase-1-dependent cleavage.17
There is considerable interest in the discovery of diarylsulfonylurea cytokine release inhibitory drugs (CRIDs). CRID3 is the most specific and well-characterized of the NLRP3 inhibitors reported to date and has been tested in a diverse array of NLRP3-engaging inflammatory diseases, including IBDs.18 Research efforts have been hampered by the lack of insights into the molecular targeting via which CRID3 interrupts the NLRP3 inflammasome pathway. However, it was discovered that CRID3 inhibits the NLRP3 inflammasome by directly targeting the NLRP3 NACHT domain, thus closing the active conformation of NLRP3 to an inactive state and preventing oligomerization.19 Although CRID3 blocks NLRP3 with high specificity in the laboratory, clinical trials of the compound showed weaker potency than that expected. Therefore, these efforts were discontinued. Some companies are now using CRID3 as a starting material to design safer and more effective molecules, while others are trying to identify inhibitors unrelated to the compound.
In this study, we counted on the remarkable oral availability and nontoxicity of NSC to establish a means of systemic P2X7R inhibition in the context of preventing the active conformation of NLRP3 using CRID3. We suggest that concomitant targeting of P2X7R with a downstream NLRP3 inflammasome component could be an effective strategy that might represent a possible future therapeutic approach to modulate the inflammatory response in the injured colon. The use of NSC as an adjunctive therapy may generate a novel approach to maximize the efficacy of CRID3 in patients with IBDs. In this article, we also evaluated the potential effect of NSC on other molecular components of NLRP3 inflammasome activation. We were interested in inducing UC via the administration of dextran sodium sulfate (DSS), which is toxic to colon epithelia and leads to mucosal barrier dysregulation. Consequently, a pathology resembling that of human was produced. In addition, the DSS colitis UC model is rapidly produced, and can be controlled.
Materials and Methods
Animals
Adult male Wistar rats of 250 ± 25 g weight were obtained from the Delta University for Science and Technology. Rats were acclimatized for two weeks under laboratory conditions to adopt in a new environment before initiating the experiment. They were kept in fully ventilated polycarbonate cages (6/cage) with wood-chip bedding. They were allowed pathogen-free rodent chow (Miladco Co., Giza, Egypt) and free access to water (Vittor-Société des Eaux Minerales Co., Benha, Kaliobeya, Egypt). Environmental conditions were optimized following guidelines from the Research Ethics Committee (REC) at the Delta University, REC approval number (FPDU842020).
DSS Colitis Animal Model
The induction of DSS colitis model was initiated at the third day of commencement of the experimental protocol by permitting animals ad lib access to 4% w/v DSS in water. After Seven days, DSS was halted. Animals were euthanized on day 15 after the induction of colitis. Rats received NSC or CRID3 P.O. two days prior to the induction of the model, as described by.1 Animal weight, stool consistency, and bleeding were recorded at the beginning of the experimental work and before euthanasia, on the last day of the experiment.
Experimental Design
Animals were assigned to the following 7 gps: N gp (n = 6), normal control rat group; NSC group (n = 6), animals received NSC (Sigma-Aldrich) at a dose of 50 mg/kg/day P.O. from day one to day sixteen; CRID3 gp (n = 6), animals received CRID3 (Sigma-Aldrich) at a dose of 20 mg/kg/day P.O. from day one to day sixteen; DSS gp (n = 10), animals were permitted consumption of ad lib DSS (Sigma-Aldrich) solution from day three to day nine; DSS/NSC gp (n = 8), Animals were orally administered NSC + ad lib DSS solution; DSS/CRID3 gp (n = 8), rats received CRID3 + ad lib DSS solution; DSS/NSC/CRID3 gp (n = 8), rats received NSC + CRID3 + ad lib DSS solution. Animals were then euthanized 24 h after the last drug dose, at the day seventeen (Table 1).
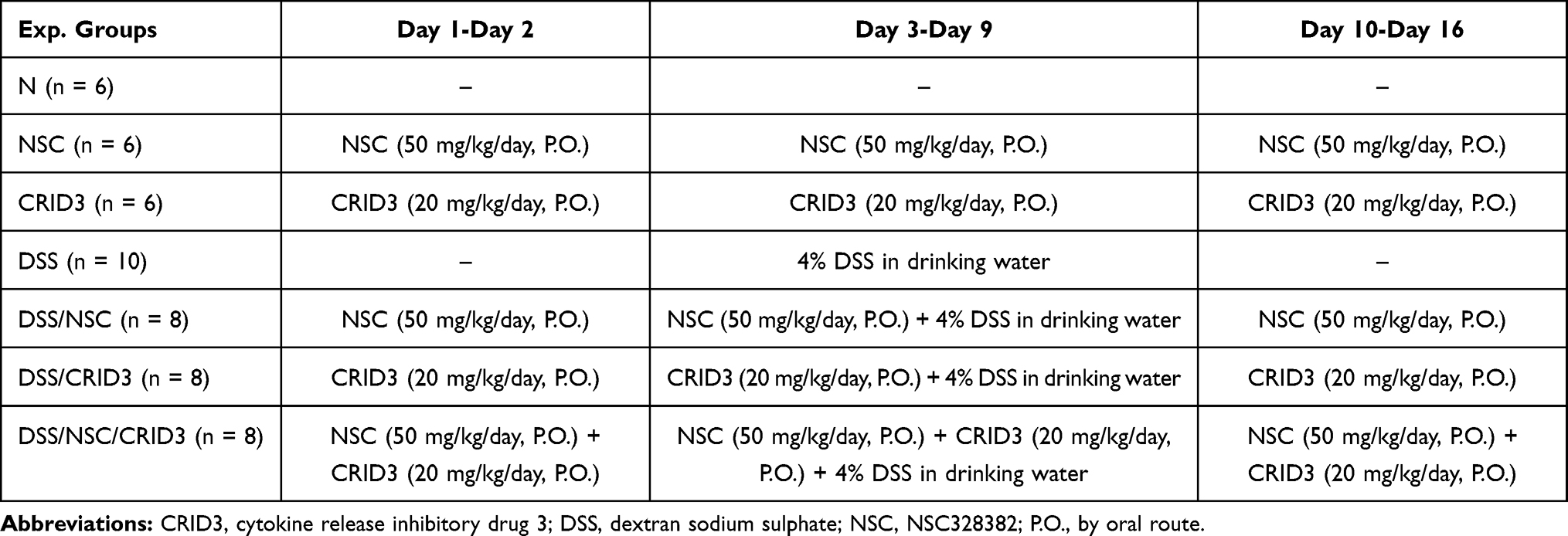 |
Table 1 Experimental Design |
Assessment of the Colon Weight/Length Ratio
Colons were excised from anus to caecum, measured, and emptied before being weighed. The weight and length of rat colons were determined and the relative colon weight/length ratio was calculated for each animal.
Calculation of the Disease Activity Index (DAI)
To evaluate the severity of the developed UC, a blinded investigator calculated the DAI at the last day of the experimental procedure in compliance with a previously described method (Palla et al, 2016). The parameters of the percentage of body weight loss, diarrhea, and bloody stool were recorded, and the scoring criteria are described in Table 2. The DAI score was calculated as the sum of the scores of the aforementioned parameters.
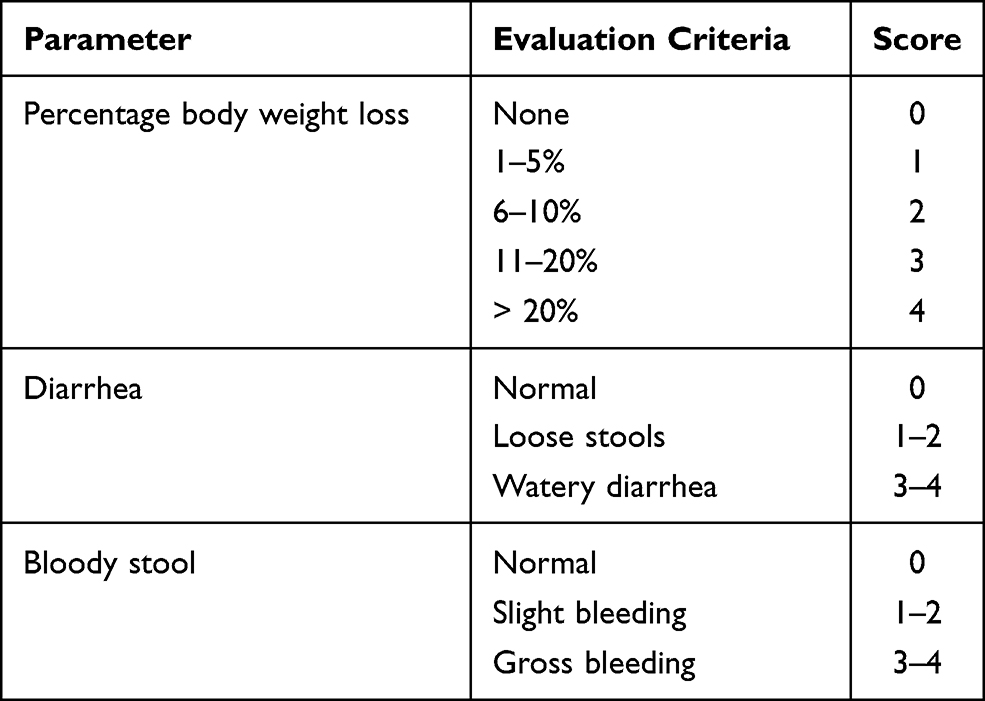 |
Table 2 Disease Activity Index (DAI) Scoring System |
Calculation of the Macroscopic Damage Index (MDI)
As shown in Table 3, the macroscopic damage criteria were applied along the colon and the MDI score was recorded individually for each animal. The scoring is built upon a single-blinded assessment technique of the colonic tissue injury.20
 |
Table 3 Macroscopic Damage Index (MDI) Scoring Criteria (Macroscopic Evaluation) |
Tissue Collection
Colons were gently excised and carefully flushed out with chilled PBS, to get rid of fecal residues, and then the excess PBS was blotted off. Portions from the distal colons were fixed in 4% neutral-buffered formalin for 24 h and embedded in paraffin for histopathological and immunohistochemical examinations. For qRT–PCR, portions were preserved in RNAlater. Other colon portions were used for the preparation of colon homogenates. In addition, fresh blood was collected for flow cytometric analysis of CD4+ve T lymphocytes and CD11b+ve monocytes. Sera were collected and stored at –80°C for further biochemical analyses.
Histological Assessment of Colitis
Standard histopathological techniques were performed sequentially by a single-blinded histologist. Tissues from the paraffin blocks were cut into 4 μm slices on a microtome, deparaffinized, rehydrated, and stained with hematoxylin and eosin (H&E).21 The microscopic features of colon lesions were assessed in reference to previously described criteria (Table 4).1
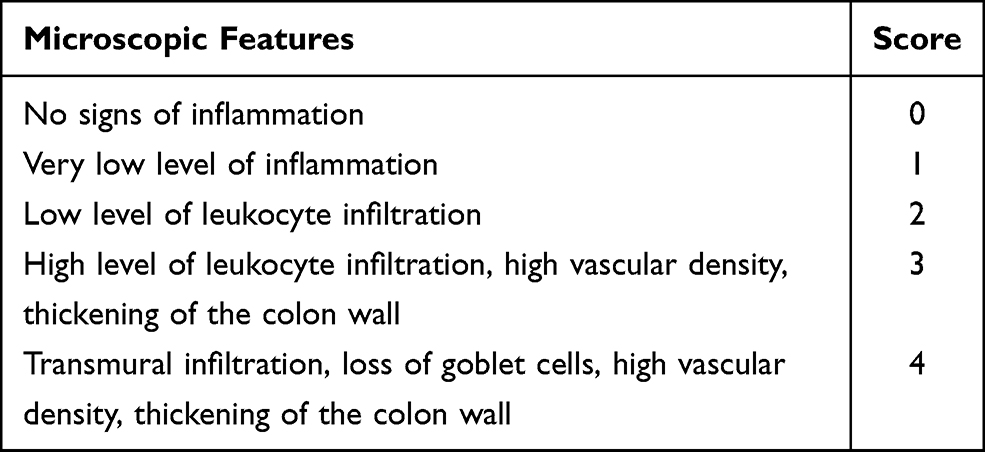 |
Table 4 Histological Scoring System (Microscopic Evaluation of Inflammation) |
Assessment of Goblet Cells in the Intestinal Mucosa
Goblet cells residing in the colonic epithelium were stained using the periodic acid-Schiff (PAS) method. Goblet cell density was assessed by the quantification of the percentage of PAS-positive cells/500 epithelial cells in the crypts and in the surface epithelium of longitudinally sectioned colonic pits.
Immunohistochemical Labeling of Caspase-1, Myeloid Differentiation Primary Response 88 (MyD88), Nuclear Transcription Factor Kappa B (NF-κB), and Caspase-3
Immunohistochemical labeling was performed in accordance with the methods described by Younis, Ghanim, Saber.22 Sections were dewaxed, hydrated, and dipped in trisodium citrate (pH = 6) as antigen-retrieval solution, Meanwhile, sections were treated with 0.3% H2O2 diluted in distilled water and subjected to protein blocking, followed by incubation with a mouse monoclonal anti-caspase-1 primary antibody (Novus Biologicals, LLC, USA; 1:100 dilution), a mouse monoclonal anti-MyD88 primary antibody (Novus Biologicals; 1:100 dilution), a mouse monoclonal anti-NF-κB p65 primary antibody (Thermo Fisher Scientific, Waltham, MA, USA; 1:100 dilution), or an anti-caspase-3 primary antibody (R&D Systems; 1:100 dilution) overnight at 4°C. All primary antibodies were diluted in specific primary antibodies diluent solution obtained from Genemed Biotechnologies (CA, USA). The slides were rinsed three times with PBS, incubated with anti-mouse IgG secondary antibodies (EnVision + System HRP; Dako) for 30 min at room temperature, visualized with di-aminobenzidine commercial kits (Liquid DAB + Substrate Chromogen System; Dako), and finally counterstained with Mayer’s hematoxylin. As a negative control procedure, the primary antibody was replaced with normal mouse serum. The labeling index was expressed as the percentage of positive cells per total 1000 counted cells in 10 high-power fields.
Determination of Reduced Glutathione (GSH), Superoxide Dismutase (SOD), and Malondialdehyde (MDA)
According to the manufacturer’s instructions, GSH and SOD were spectrophotometrically determined in colon tissue homogenates using commercial kits obtained from Bio-diagnostic (Cairo, Egypt; Cat. No. GR2523 and SD2521, respectively). MDA was spectrophotometrically determined in colon tissue homogenate using a commercial kit supplied by Bio-diagnostic (Cat. No. MD2529). In this assay, thiobarbituric acid reacts with MDA in acidic medium at a temperature of 95°C for 30 min, to form a thiobarbituric acid reactive product. The absorbance of the resultant pink product was measured at 534 nm.
Determination of the Levels of Tumor Necrosis Factor Alpha (TNF-α), IL-6, IL-1β, and IL-18 in Colon Tissues
In accordance with the manufacturer’s instructions, levels of TNFα, IL-6, and IL-1β were measured using ELISA kits supplied by R&D systems (Minneapolis, MN, USA) (Cat. No. RTA00, R6000b, and RLB00, respectively). A kit obtained from USCN Life Science Inc. (Wuhan, China; Cat. No. SEA064HU) was used to measure IL-18 levels in colon tissue.
Determination of the Levels of IL-10, IL-12 p70, Myeloperoxidase (MPO) Activity, and Caspase-1 Activity in Colon Tissues
As instructed, IL-10 was determined in colon tissue homogenates using the Quantikine Immunoassay kit purchased from R&D Systems (Cat. No. R1000). IL-12 p70 was quantified in colon tissue homogenates using a kit purchased from CUSABIO (Wuhan, China; Cat. No. CSB-E07364r). The results are expressed as IL-10/IL-12 p70 ratio. An MPO colorimetric activity assay kit purchased from Sigma-Aldrich was used to determine the activity of myeloperoxidase in the rat colon (Cat. No. MAK068). In this assay, MPO catalyzes the formation of hypochlorous acid, which reacts with taurine to form taurine chloramine. Taurine chloramine reacts with the chromophore TNB, resulting in the formation of the colorless product DTNB. One unit of MPO activity is defined as the amount of enzyme that hydrolyzes the substrate and generates taurine chloramine to consume 1.0 μmole of TNB per minute at 25°C. A colorimetric assay of caspase-1 was performed using a kit supplied by BioVision (Milpitas, CA, USA; Cat. No. K111-25). Caspase-1 activity was spectrophotometrically determined on the basis of the detection of the cleaved p-nitroanilide (p-NA) chromophore at 405 nm.
Determination of the Levels of P2X7R, MyD88, NLRP3, and p-p65 (Ser 536)/p65 in Colon Tissues
Following instructions, the levels of P2X7R, MyD88, and NLRP3 in colon tissues were measured using ELISA kits purchased from MyBioSource (CA, USA; Cat. No. MBS9902691, MBS2703631, and MBS2033695, respectively). The ratio of NF-κB p-p65/p65 was calculated with a kit obtained from Abcam (Cambridge, MA, USA; Cat. No. AB176663).
qRT–PCR Analysis of the Expression of the P2X7R, MyD88, NLRP3, and p65 mRNAs
Colon tissues stored in RNAlater were used to extract total RNA using a kit supplied by Qiagen (Venlo, the Netherlands), according to the manufacturer’s instructions. The quality and purity of RNA were confirmed spectrophotometrically at 260 nm using a Nano Drop 2000 spectrophotometer (Thermo Fisher Scientific, USA). RNA was reverse transcribed using the RevertAid First Strand cDNA synthesis kit. qRT–PCR was performed on a StepOne™ Real-Time PCR System (Thermo Fisher Scientific). The thermal cycling conditions were as follows: an initial activation of DNA polymerase at 95°C for 15 min, followed by a three-step cycling including 40 cycles of denaturation at 95°C for 15 s, annealing at 60°C for 20 s, and elongation at 72°C for 20 s. The relative expression was calculated using the Ct (2−ΔΔCT) method. All values were normalized to the GAPDH gene as an invariant endogenous control in the same sample. The sequences of the PCR primers used in this experiment are described in Table 5.
 |
Table 5 Primer Sequences for qPCR |
Flow Cytometric Determination of CD4+ve T Lymphocytes and CD11b+ve Monocytes in Peripheral Blood
Ten microliter of the appropriate conjugated anti-rat antibodies (CD4/FITC, CD11b/PE; Becton Dickinson, CA, USA) were added to 100 µL of peripheral blood from each animal and incubated at room temperature for 15 min in the dark. Erythrocytes were eliminated by adding 500 µL of RBC lysing buffer (ebioscience, USA) for 10 min. After incubation, leukocytes were washed twice in PBS. The washed cells were re-suspended in 0.3 mL of PBS. Cells were analyzed using a Beckman Coulter instrument equipped for four-color flow cytometry (Coulter, Coultronic, Margency, France), and up to 10000 events were obtained. Data were analyzed using the Kaluza analysis software (Beckman Coulter Inc., USA).
Statistical Analysis
GraphPad Prism/two way ANOVA followed by Tukey’s post-hoc test were applied for multiple comparison between different groups. Kruskal–Wallis test followed by Dunn’s as a post-hoc test was used to analyse differences between groups for the histology score, DAI and MDI. Values are expressed as the mean ± standard deviation (SD). A value of P < 0.05 was considered statistically significant.
Results
Effect on the Histological Features of the Rat Colon
As displayed in Figure 1, sections from the N, NSC, or CRID3 rats showed normal colonic mucosa, crypts, and glands. Additionally, sections from the DSS-treated rats showed severe colitis associated with complete necrosis of the crypts and edema mixed with inflammatory cell infiltration. Conversely, specimens from the DSS/NSC and DSS/CRID3 groups showed an improved histological picture with decreased superficial ulceration of the intestinal mucosa associated with marked decrease in interstitial inflammatory cell infiltration. Additionally, specimens from the DSS/NSC/CRID3 group of rats show markedly decreased erosions and superficial de-epithelialization associated with distinct decreased interstitial inflammatory cell infiltration. Furthermore, upon histological evaluation of intestinal damage, we revealed that DSS induced a significant increase in the histological score, which was significantly repressed in the DSS/NSC (P < 0.01), DSS/CRID3 (P < 0.01), and DSS/NSC/CRID3 (P < 0.0001) groups of rats.
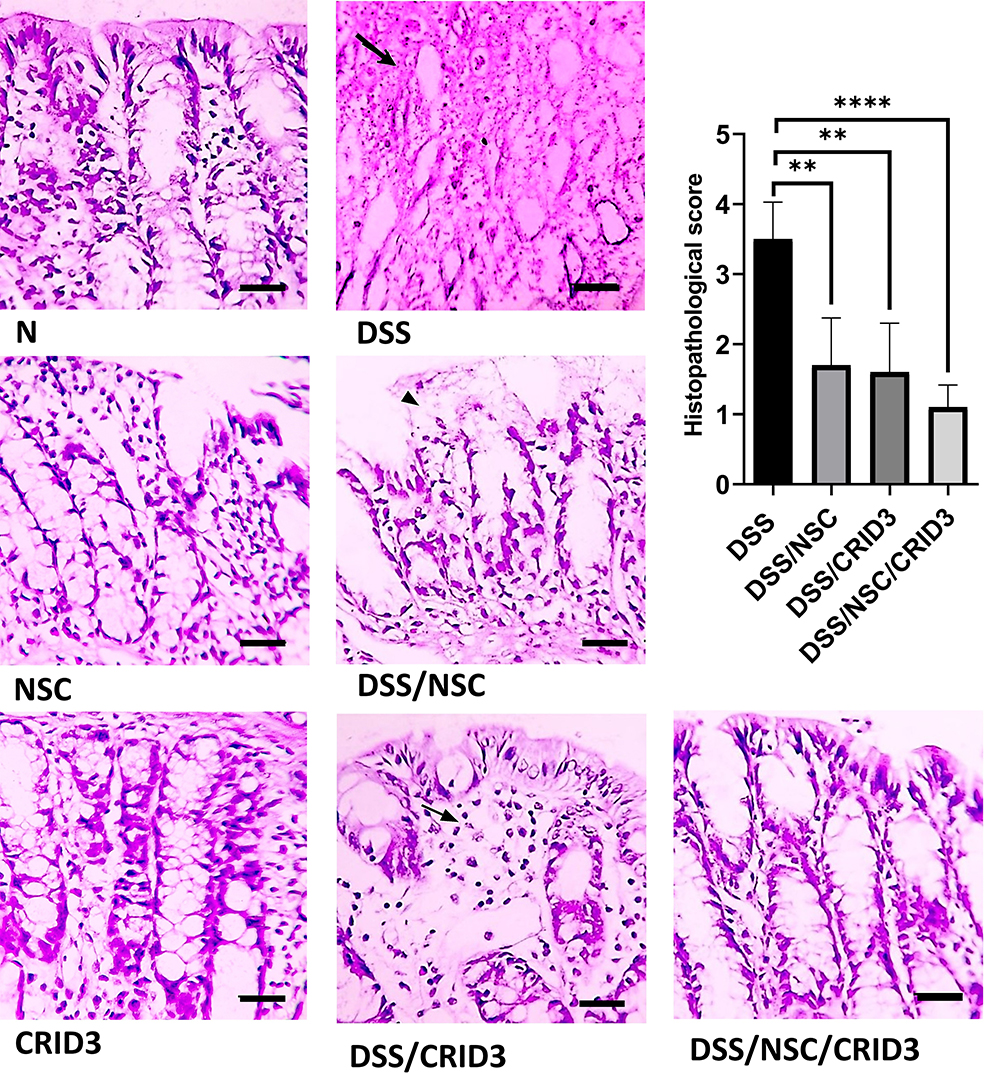 |
Figure 1 Histopathological examination. Photomicrographs of colon sections from: (N), (NSC), and (CRID3) display normal mucosa and crypts; (DSS), shows diffuse mucosal necrosis and damaged crypts (open arrow); (DSS/NSC), shows superficial necrosis of surface epithelium (filled arrowhead); (DSS/CRID3), shows mucosal edema with few inflammatory cell infiltration (filled arrow); (DSS/NSC/CRID3), displays improved histology and retained normal appearance. As shown in histological score, the DSS gp has the highest score. Additionally, the DSS/NSC/CRID3 gp show marked decrease in the score compared with that of the DSS gp. H&E stain, X 400, bar = 50 µm. **P < 0.01 vs DSS, ***P < 0.0001 vs DSS. |
Effect on Goblet Cell Density in the Rat Colon
As presented in Figure 2, sections from N, NSC, or CRID3 rats showed normal goblet cell density within at least 500 epithelial cells in the crypts and in the surface epithelium of longitudinally sectioned colonic pits. Specimens from the DSS group of rats showed a scattered and excessive loss of goblet cell density in the crypts and in the surface epithelium. Conversely, specimens from the DSS/NSC and DSS/CRID3 groups showed a focal decrease in the goblet cell density in the crypts and in the surface epithelium. In addition, sections from the DSS/NSC/CRID3 group of rats showed an appearance that was comparatively similar to the normal goblet cell density. Furthermore, upon evaluation of goblet cell density in the rat colon, we found that DSS induced a significant decrease in the goblet cell density score, which was significantly reversed in the DSS/NSC and DSS/CRID3, and more pronouncedly in the DSS/NSC/CRID3 groups of rats.
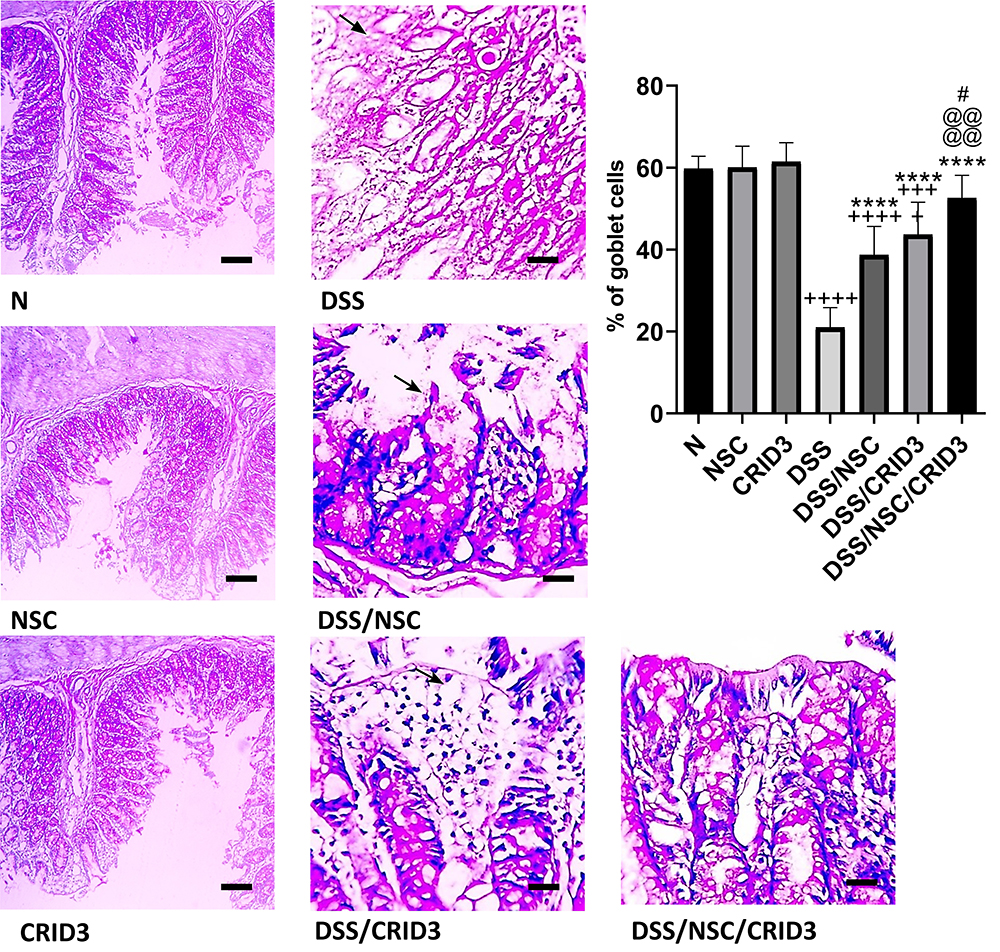 |
Figure 2 Effect on goblet cells percentage. Photomicrographs of PAS-stained colon sections from: (N), (NSC), and (CRID3) show normal goblet cell density, X 100; (DSS), shows diffuse loss of goblet cells (filled arrow), X 400; (DSS/NSC), shows focal decrease in goblet cell density (filled arrow), X 400; (DSS/CRID3), shows focal decrease in goblet cell density (filled arrow), X 400; (DSS/NSC/CRID3), shows marked increase in goblet cell density, X 400. As shown in % goblet cell density, the DSS rat colons have the lowest percentage of goblet cell density. In addition, the DSS/NSC/CRID3 gp show marked increase in the percentage of goblet cell density compared to that of the DSS gp. +++P < 0.001 vs N,++++P < 0.0001 vs N, ****P < 0.0001 vs DSS, @@@@P < 0.0001 vs DSS/NSC, #P < 0.05 vs DSS/CRID3. |
Effect on the Immunohistochemical Labeling of Caspase-1
As displayed in Figure 3, sections from the N, NSC, or CRID3 rats showed scarce caspase-1 cytosolic expression within the epithelium and the interstitial tissue. The tissue sections from untreated DSS rats displayed a distinct caspase-1 immunolabeling within the desquamated covering epithelia and in the interstitial tissues. Specimens from the DSS/NSC and DSS/CRID3 groups displayed a moderate reduction in caspase-1 expression either in the epithelia of glands or in the interstitial tissues. Specimens from the DSS/NSC/CRID3 group displayed a more pronounced decrease in caspase-1 expression. Furthermore, a significant decrease in the expression of caspase-1 was observed in rats treated with NSC and CRID3, and more significantly in those treated with DSS/NSC/CRID3 compared with the untreated DSS rats.
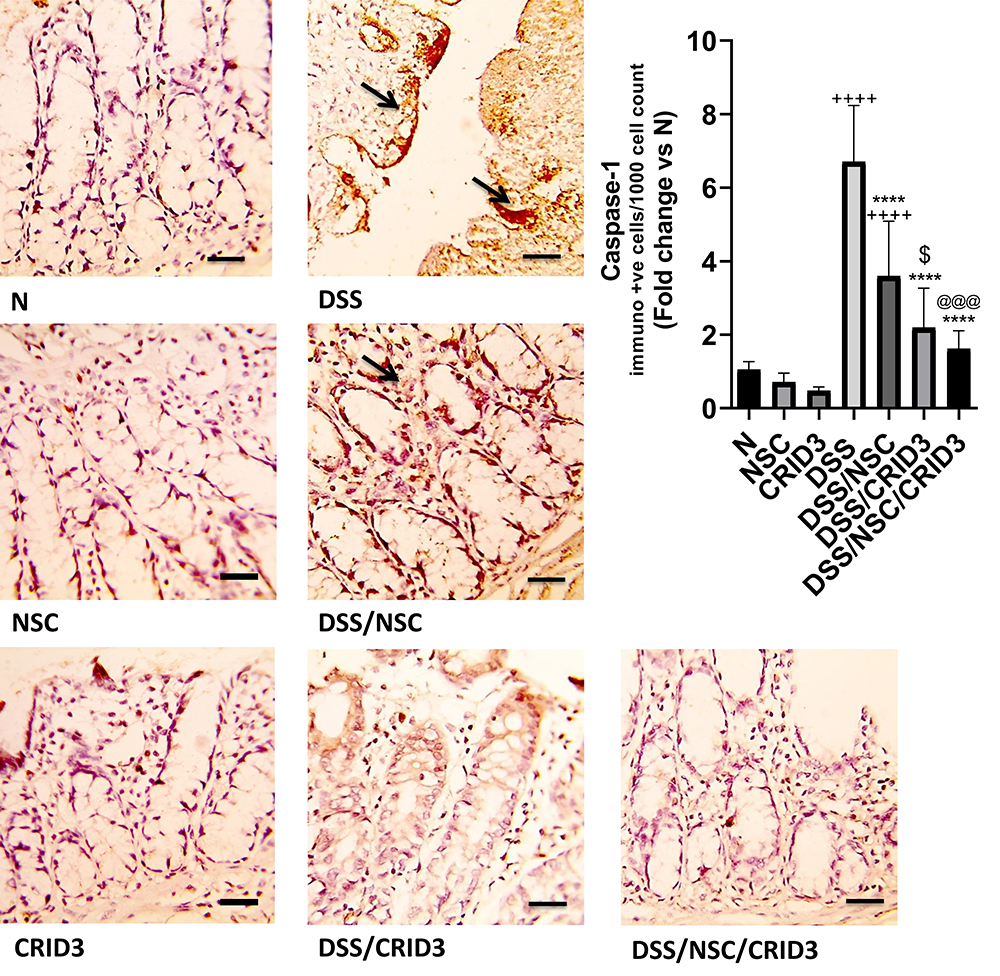 |
Figure 3 Immunostaining of caspase-1. Photomicrographs of colon specimens from: (N), (NSC), and (CRID3) show scarce caspase-1 cytosolic expression within the glandular epithelia and the interstitial tissue; (DSS), shows marked expression of caspase-1 within the desquamated covering epithelium and within the interstitial tissues (open arrows); (DSS/NSC), shows moderate decrease in the caspase-1 expression either within epithelial lining of the intestinal glands and within the interstitial tissues (open arrow); (DSS/CRID3), shows moderate reduction in the caspase-1 expression either in epithelia of the glands and in the interstitial tissues (open arrow); (DSS/NSC/CRID3), shows a more pronounced decrease in the caspase-1 expression. As shown in caspase-1 immunoreactivity evaluation figure, a significant reduction in the percentage of caspase-1 immunopositive cells/1000 counted cells was detected in rats with ulcerative colitis that were treated with NSC, CRID3, and NSC/CRID3 compared with the untreated DSS-insulted rats. Caspase-1 IHC, bar = 50 µm. ++++P < 0.0001 vs N, ****P < 0.0001 vs DSS, @@@P < 0.001 vs DSS/NSC, $P < 0.05 DSS/CRID3 vs DSS/NSC. |
Effect on the Immunohistochemical Labeling of MyD88
As displayed in Figure 4, sections of the N, NSC, or CRID3 rat groups showed scanty MyD88 expression. The tissue sections of untreated DSS rats displayed a marked expression of MyD88 in the degenerated crypts and inflammatory cells, such as polymorphonuclear leukocytes and monocytes. Specimens from the DSS/NSC group displayed a moderate reduction in MyD88 expression. In addition, specimens from the DSS/NSC/CRID3 rats displayed a marked reduction in MyD88 expression in the epithelia of glands and crypts. However, specimens from the DSS/CRID3 rat group displayed increased expression of MyD88. Furthermore, a significant decrease in the percentage of MyD88-immunopositive cells/1000 counted cells was observed in rats treated with DSS/NSC and DSS/NSC/CRID3 compared with the untreated DSS rats. An insignificant change in the percentage of MyD88-immunopositive cells/1000 counted cells was observed in rats treated with CRID3.
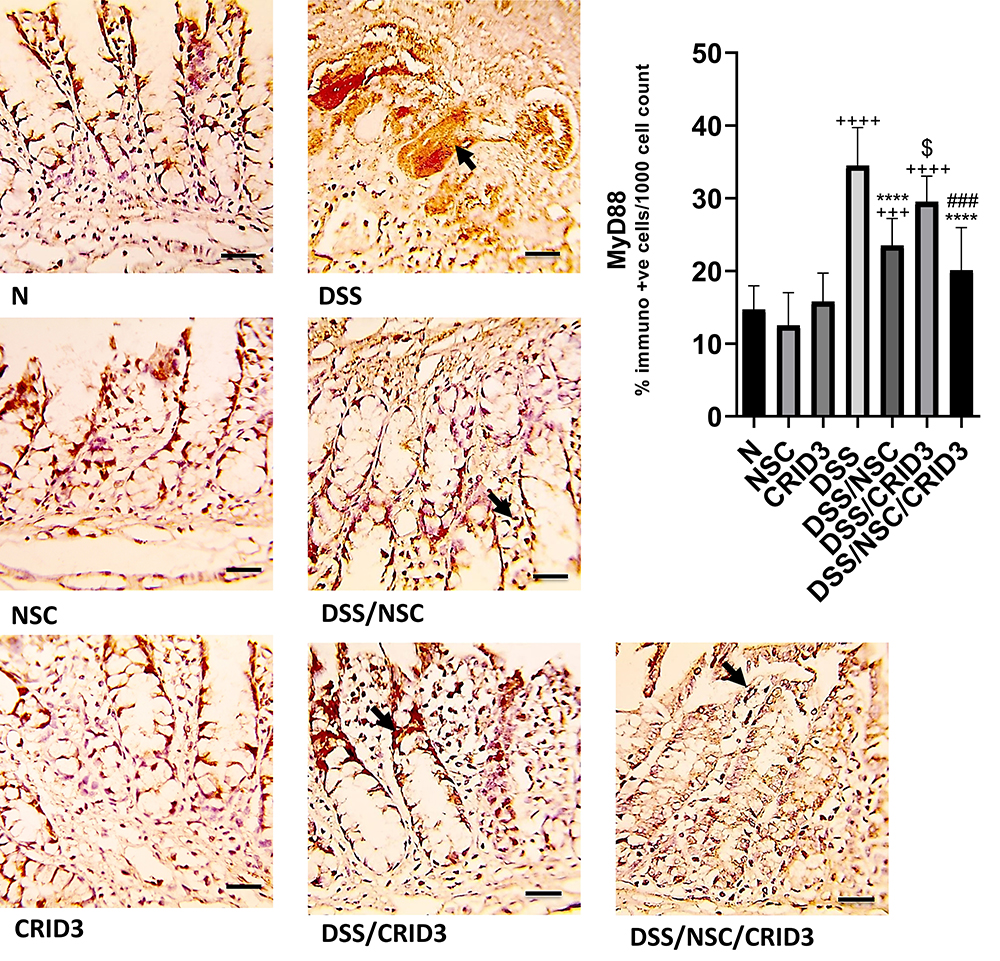 |
Figure 4 Immunostaining of MyD88. Photomicrographs of specimens from: (N), (NSC), and (CRID3) show scanty MyD88 expression; (DSS), shows marked expression of MyD88 in the damaged crypts and inflammatory cells (filled arrow), such as polymorphonuclear leukocytes and monocytes; (DSS/NSC), shows moderate decrease in the MyD88 expression (filled arrow); (DSS/CRID3), shows increased expression of MyD88 (filled arrow); (DSS/NSC/CRID3), displays significant decrease in the MyD88 expression in the epithelia of glands and crypts (filled arrow). As shown in MyD88 immunoreactivity evaluation figure, a significant decrease in the percentage of MyD88 immunopositive cells/1000 counted cells was detected in rats treated with DSS/NSC and DSS/NSC/CRID3 compared with the untreated DSS rats. The insignificant change in the percentage of MyD88 immunopositive cells/1000 counted cells was observed in rats treated with CRID3. MyD88 IHC, bar = 50 µm. ++++P < 0.0001 vs N, +++P < 0.001 vs N, ****P < 0.0001 vs DSS, ###P < 0.001 vs DSS/CRID3, $P < 0.05 DSS/CRID3 vs DSS/NSC. |
Effect on the Immunohistochemical Labeling of NF-κB p65
As displayed in Figure 5, sections from the N, NSC, or CRID3 groups showed scanty NF-κB p65 expression. Tissue sections from the untreated DSS rats displayed marked expression of both cytoplasmic and nuclear NF-κB P65 in the degenerated crypts and inflammatory cells, such as polymorphonuclear leukocytes and monocytes. Specimens from the DSS/NSC rat group displayed a moderate decrease in the NF-κB P65 expression. Moreover, specimens from the DSS/NSC/CRID3 rats display a distinct reduction in NF-κB P65 expression in the epithelia of glands and crypts. However, specimens from the DSS/CRID3 rat group display increased expression of both cytoplasmic and nuclear NF-κB P65. Furthermore, a significant decrease in the percentage of NF-κB P65-immunopositive cells/1000 counted cells was detected in rats treated with DSS/NSC and DSS/NSC/CRID3 compared with the untreated DSS rats. An insignificant change in the percentage of NF-κB P65-immunopositive cells/1000 counted cells was observed in rats treated with CRID3.
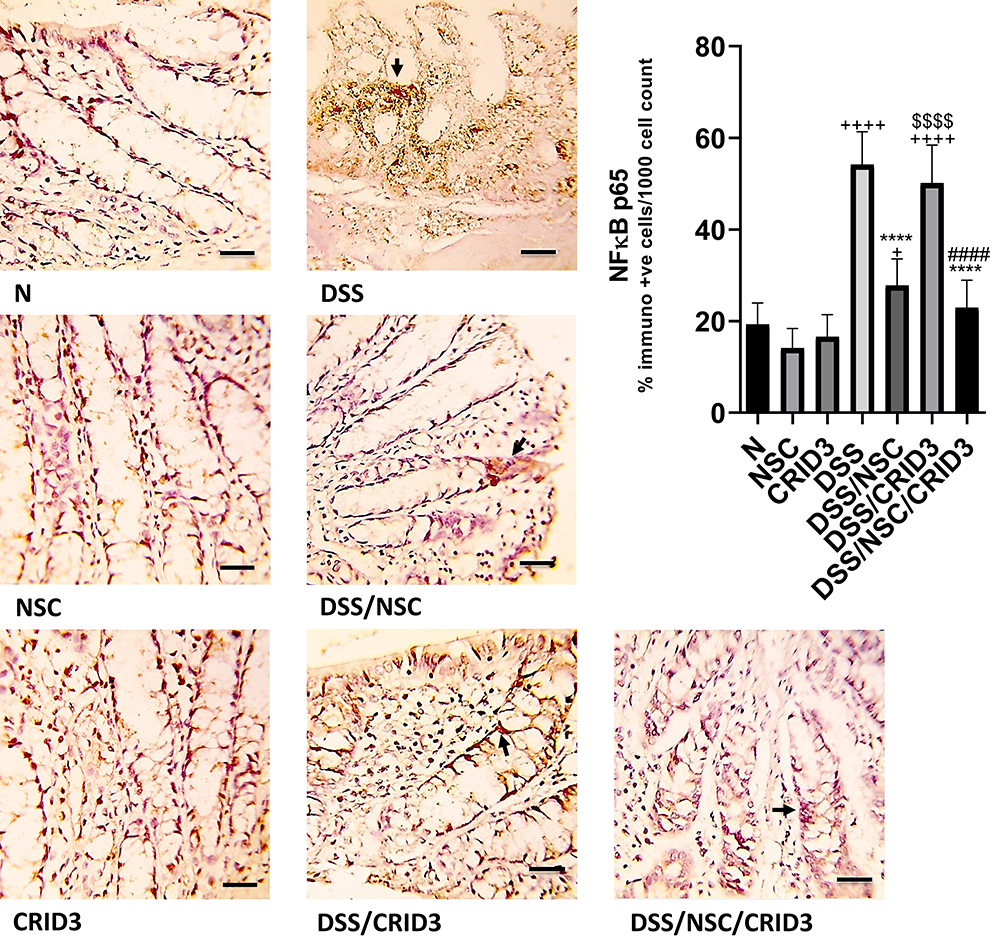 |
Figure 5 Immunostaining of NF-κB. Photomicrographs of specimens from: (N), (NSC), and (CRID3) show scanty NF-κB p65 expression; (DSS), shows marked expression of NF-κB P65 in the damaged crypts and inflammatory cells, such as polymorphonuclear leukocytes and monocytes (filled arrow); (DSS/NSC), shows moderate decrease in the NF-κB P65 expression (filled arrow); (DSS/CRID3), shows increased expression of NF-κB P65 (filled arrow); (DSS/NSC/CRID3), shows marked reduction in the NF-κB P65 expression in the epithelia of glands and crypts (filled arrow). As shown in NF-κB p65 immunoreactivity evaluation figure, a significant decrease in the percentage of NF-κB P65 immunopositive cells/1000 counted cells was detected in rats treated with DSS/NSC and DSS/NSC/CRID3 compared with the untreated DSS rats. The insignificant change in the percentage of NF-κB P65 immunopositive cells/1000 counted cells was observed in rats treated with CRID3. NF-κB p65 IHC, bar = 50 µm. +P < 0.05 vs N, ++++ P < 0.0001 vs N, ****P < 0.0001 vs DSS, ####P < 0.0001 vs DSS/CRID3, $$$$P < 0.0001 DSS/CRID3 vs DSS/NSC. |
Effect on the Immunohistochemical Labeling of Caspase-3
As displayed in Figure 6, sections from the normal treated or untreated rats showed mild caspase-3 cytosolic expression within the lining epithelium of the intestinal crypts. Tissue sections from the untreated DSS rats displayed marked expression of caspase-3 mostly in the degenerated crypts. Specimens from the DSS/NSC, and particularly from the DSS/CRID3 and DSS/NSC/CRID3 groups displayed a marked decrease in caspase-3 expression. Furthermore, the most significant decrease in caspase-3 expression was observed in rats treated with CRID3 and NSC/CRID3 compared with the untreated DSS rats. The least significant change in the percentage of caspase-3-immunopositive cells/1000 counted cells was detected in rats treated with NSC.
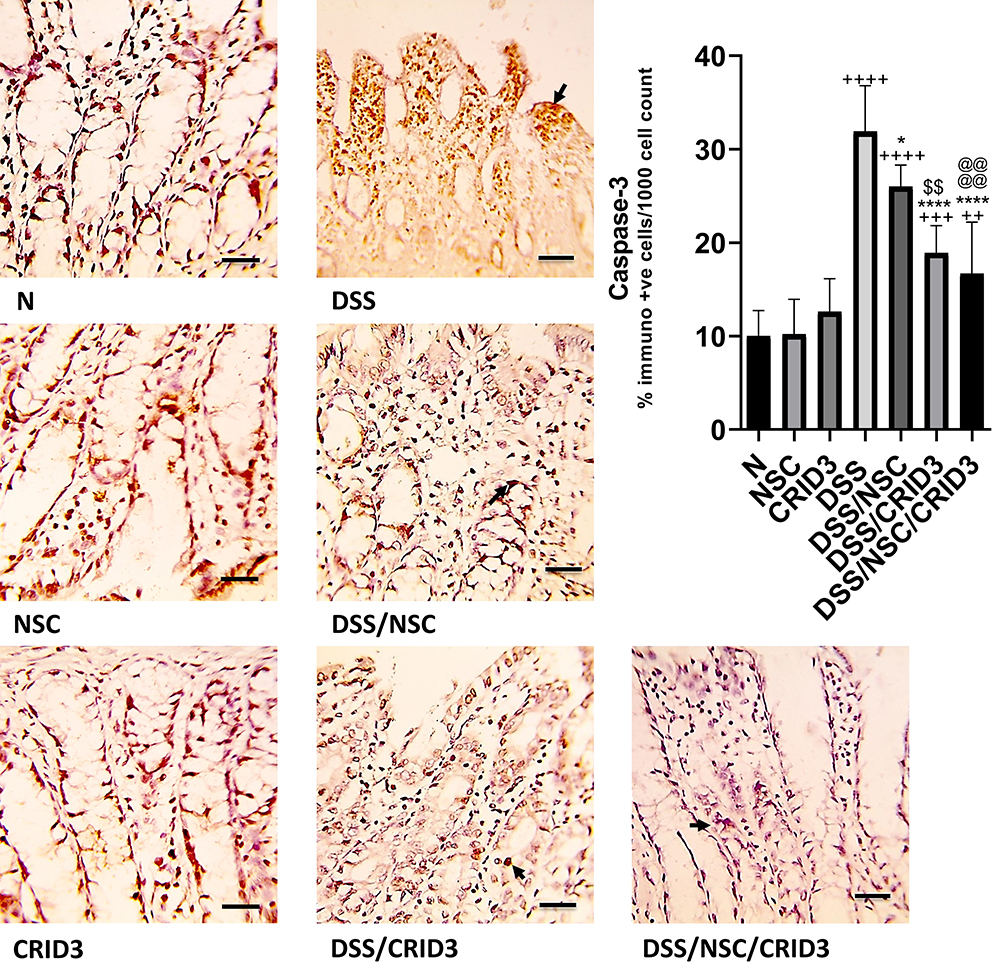 |
Figure 6 Immunostaining of caspase-3. Photomicrographs of specimens from: (N), (NSC), and (CRID3) show mild caspase-3 cytosolic expression in the epithelium of the crypts; (DSS), displays marked expression of caspase-3 mostly in the degenerated crypts (filled arrow); (DSS/NSC) and (DSS/CRID3), show a reduction in the caspase-3 expression (filled arrow); (DSS/NSC/CRID3), shows marked decrease in the caspase-3 expression (filled arrow). As shown in caspase-3 immunoreactivity evaluation figure, a significant reduction in the percentage of caspase-3 immunopositive cells/1000 counted cells was detected in rats treated with NSC, and more significantly with CRID3 and DSS/NSC/CRID3 compared with the untreated DSS rats. Caspase-3 IHC, bar = 50 µm. ++P < 0.01 vs N, +++P < 0.001 vs N, ++++P < 0.0001 vs N, * P < 0.05 vs DSS, ****P < 0.0001 vs DSS, @@@@P < 0.0001 vs DSS/NSC, $$P < 0.0001 DSS/CRID3 vs DSS/NSC. |
Effect on the Colon Weight/Length Ratio
The typical histopathological lesion of UC is the crypt abscess, in which the epithelia of crypts are broken down. The lamina propria becomes infiltrated with polymorphonuclear leukocytes. As the crypts are damaged, the colon mucosal architecture is lost and the substantial scarring that ensues shortens the colon. This in turn increases the ratio of colon weight to colon length. In this regard, we observed that NSC (50 mg/kg/day P.O.) and CRID3 (20 mg/kg/day P.O.) significantly reduced the DSS-induced increase in the colon weight/length ratio. This finding was more remarkable among rats from the DSS/NSC/CRID3 group (Figure 7A).
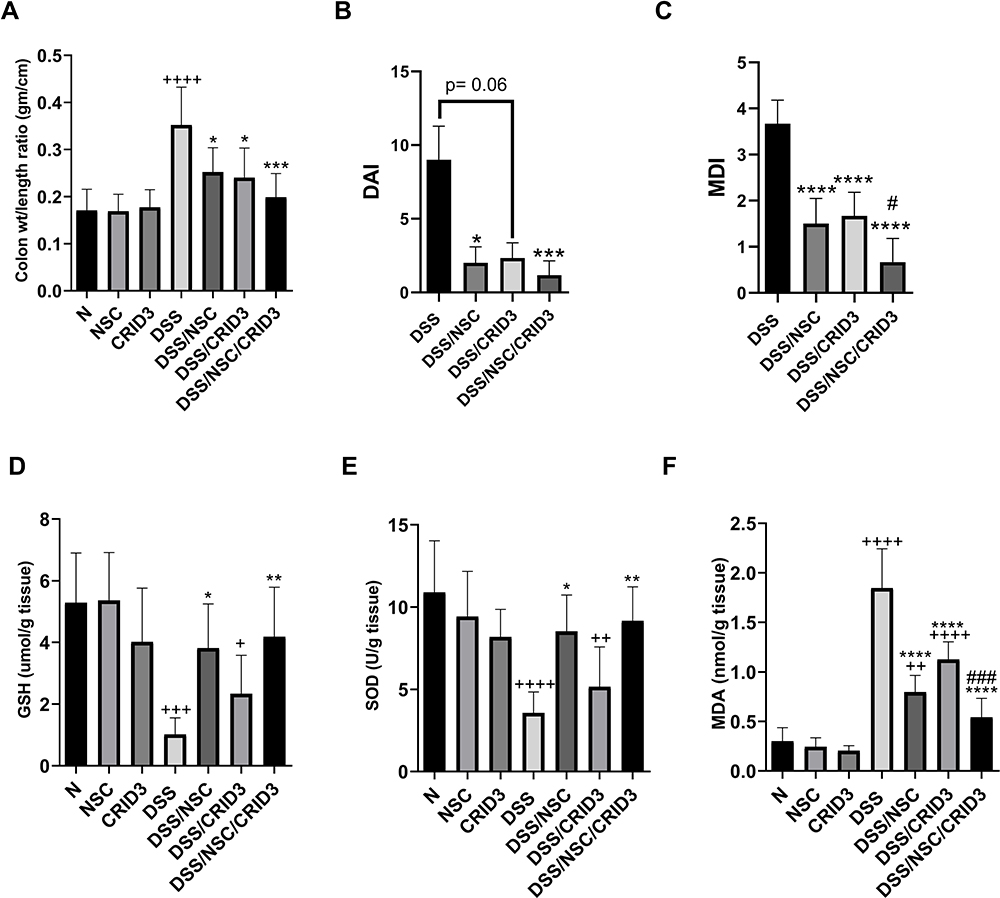 |
Figure 7 Effect on colon/weight ratio, DAI, MDI, GSH, SOD, and MDA. Effect of NSC, CRID3 and NSC/CRID3 on (A), colon weight/length ratio; (B), disease activity index (DAI); (C), macroscopic damage index (MDI); (D), reduced glutathione (GSH); (E), superoxide dismutase (SOD) and (F), malondialdehyde (MDA). +P < 0.05 vs N, ++P < 0.01 vs N, +++P < 0.001 vs N, ++++P < 0.0001 vs N, *P < 0.05 vs DSS, **P < 0.01 vs DSS, ***P < 0.001 vs DSS, ****P < 0.0001 vs DSS, #P < 0.05 vs DSS/CRID3, ###P < 0.001 vs DSS/CRID3. |
Effect on the DAI
We adapted a DAI to quantify the severity of UC in rats. This research tool informed us regarding whether or not the pathology progressed on a daily basis. On the last day of the experiment, we found that DSS induced a significant elevation in the DAI compared with that of the Normal group, which showed a DAI of zero. However, UC rats treated with NSC and NSC/CRID3 exhibited a significant decline in the DAI compared with that of the DSS group. In addition, UC rats treated with CRID3 showed a strong tendency toward statistical significance (P = 0.06) compared with the untreated DSS rats (Figure 7B).
Effect on the MDI
The MDI is a visual tool that provides evidence of the presence of mucosal erythema, edema, erosions, bleeding, and tissue necrosis. We revealed that the MDI was significantly higher in the DSS rats, and found it was significantly reduced in rat-groups treated with NSC, CRID3, and NSC/CRID3 compared with that of the DSS rats (Figure 7C).
Effect on Reduced Glutathione (GSH), Superoxide Dismutase (SOD), and Malondialdehyde (MDA)
DSS administration resulted in a sharp and significant decline in the expression of GSH and SOD in the colon compared with that of the normal group of rats. Treatment of UC rats with NSC and more distinctly with NSC/CRID3 resulted in increased GSH (Figure 7D) and SOD (Figure 7E) levels in colon tissues compared with those detected in the DSS rats. However, CRID3 showed only a tendency toward a significant elevation in GSH and SOD levels with respect to that of the DSS rats (Figure 7D and E, respectively). Therefore, we suggest that NSC (50 mg/kg) increases the antioxidant defense machinery of colon tissues, which is augmented upon dual administration with CRID3. MDA has been widely used as a marker of oxidative stress and of the antioxidant status of colon tissues. An increase in free radicals causes overproduction of MDA. Our results revealed that treatment of UC rats with NSC, CRID3, or their combination resulted in decreased oxidative stress in colonic tissue, as indicated by the significant decline in colonic MDA levels compared with that of the untreated DSS rats (Figure 7F).
Effect on the Colon Tissue Levels of Tumor Necrosis Factor Alpha (TNF-α), IL-6, IL-1β, and IL-18
As shown in Figure 8, DSS induced a significant increase in the colon tissue levels of TNF-α, IL-6, IL-1β, and IL-18 with respect to those of the normal group of rats. Treatment with NSC and, more pronouncedly, NSC/CRID3 significantly decreased the DSS-induced increase in the colon tissue levels of TNF-α (Figure 8A) and IL-6 (Figure 8B). These effects might be attributable to the ability of NSC to inactivate the NF-κB signaling pathway, in addition to the effect of disrupting the P2X7R/NLRP3 interactions. A less pronounced significant change in the colon tissue levels of TNF-α and IL-6 was observed in DSS/CRID3 rats compared with the DSS group of rats. Treatment with CRID3 and, more pronouncedly, NSC/CRID3 significantly reduced the DSS-induced increase in the colon tissue levels of IL-1β (Figure 8C), and IL-18 (Figure 8D). These effects might be attributable to the ability of CRID3 to interrupt the NLRP3/caspase-1 interaction. These effects were confirmed by the decreased expression and activity of caspase-1 in the rat colons. A less pronounced significant change in the colon tissue levels of IL-1β and IL-18 was observed in DSS/NSC rats compared with the DSS group of rats.
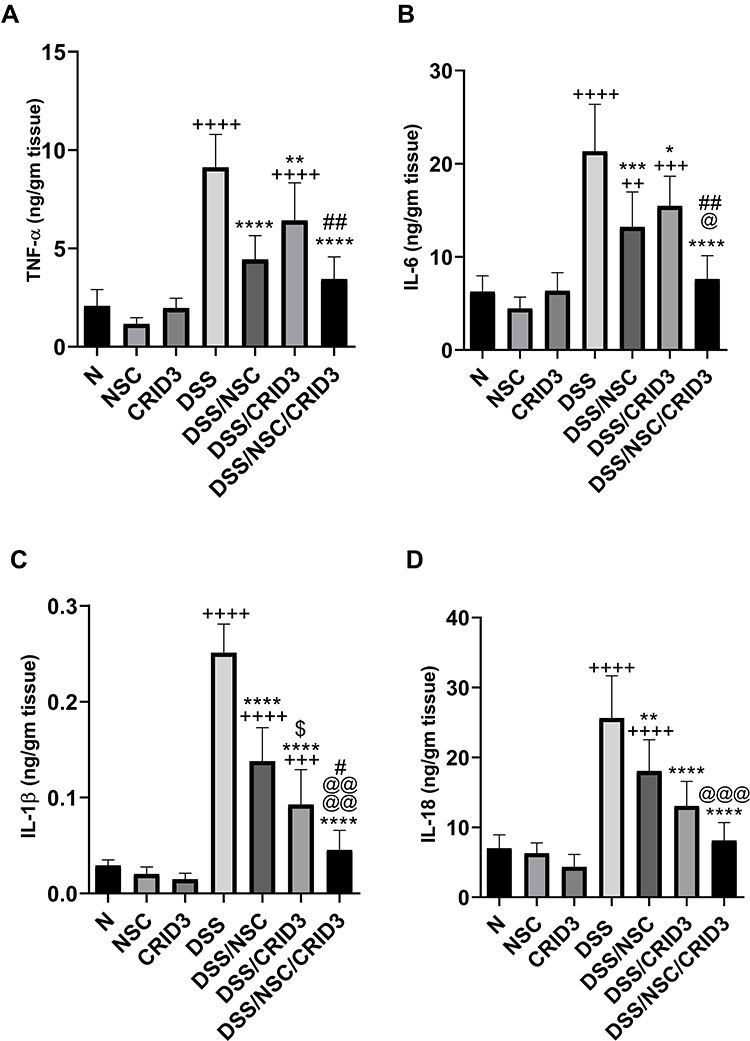 |
Figure 8 Effect on TNF-α, IL-6, IL-1β, and IL-18. Effect of NSC, CRID3 and NSC/CRID3 on (A), tumor necrosis factor alpha (TNF-α); (B), IL-6; (C), IL-1β and (D), IL-18. +P < 0.05 vs N, ++P < 0.01 vs N, +++P < 0.001 vs N, ++++P < 0.0001 vs N, *P < 0.05 vs DSS, **P < 0.01 vs DSS, ***P < 0.001 vs DSS, ****P < 0.0001 vs DSS, #P < 0.05 vs DSS/CRID3, ##P < 0.01 vs DSS/CRID3, @P < 0.05 vs DSS/NSC, @@@P < 0.001 vs DSS/NSC, @@@@P < 0.0001 vs DSS/NSC. |
Effect on the Colon Tissue Levels of IL-10, IL-12 p70, Myeloperoxidase Activity (MPO), and Caspase-1 Activity
For the assessment of the immune status of the injured colon, we calculated the ratio of IL-10/IL-12 p70 as a quantitative mean of the balance between an anti-inflammatory and a proinflammatory state (or Th1 vs Th2 dominance). As displayed in Figure 9A, DSS induced a sharp and significant decline in the ratio of IL-10/IL-12 p70 compared with that of the N group of rats. Conversely, treatment with NSC, CRID3, and, more distinctly, NSC/CRID3 repressed the DSS-induced decline in that ratio. Regarding this finding, we suggest that the IL-10/IL-12 p70 ratio is an important predictive marker of disease severity. Elevated activity of myeloperoxidase is a characteristic feature of UC. MPO is a heme-containing enzyme that catalyzes the oxidation of halide ions to hypochlorous acid by regulating hydrogen peroxidase enzymatic activity. It is a believed that MPO is highly expressed in neutrophils and plays an essential role in the neutrophilic antimicrobial activity. Our results revealed that UC rats treated with NSC, CRID3, and their combination showed a significant decrease in MPO activity compared with the untreated UC rats. These data revealed that NSC and CRID3 induced decreased neutrophil infiltration in the injured colonic tissue (Figure 9B). In addition, NSC, CRID3, and their combination induced a significant reduction in caspase-1 activity compared with that of the untreated DSS rats (Figure 9C), an effect that can be attributed to the disruption of P2X7R/NLRP3 (NSC) and NLRP3/caspase-1 (CRID3) interactions. Herein, treatment with NSC, CRID3, and, more effectively, their combination repressed caspase-1-induced pyroptotic cell death in the injured colon.
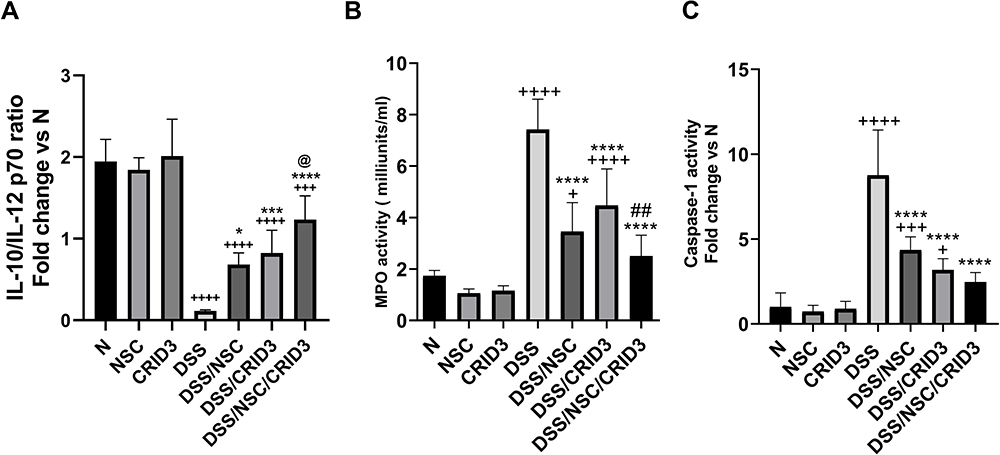 |
Figure 9 Effect on IL-10/IL-12 ratio, MPO, and caspase-1 activity. Effect of NSC, CRID3 and NSC/CRID3 on (A), IL-10/1L-12 p70; (B), myeloperoxidase (MPO) activity and (C), caspase-1 activity. +P < 0.05 vs N, +++P < 0.001 vs N, ++++P < 0.0001 vs N, *P < 0.05 vs DSS, ***P < 0.001 vs DSS, ****P < 0.0001 vs DSS, @P < 0.05 vs DSS/NSC, ##P < 0.01 vs DSS/CRID3. |
Effect on the mRNA Expression of P2X7R, MyD88, NLRP3, and p65; and the Ratio of p-p65 (Ser 536)/p65
As shown in Figure 10, DSS treatment resulted in a significant increase in the mRNA expression and colon tissue levels of P2X7R, MyD88, and NLRP3; the mRNA expression of p65; and p-p65 (Ser 536)/p65 with respect to normal rats. Treatment with NSC and, more effectively, with NSC/CRID3 resulted in a significant decline in the mRNA expression and colon tissue levels of P2X7R and MyD88; in the mRNA expression of p65; and in the p-p65 (Ser 536)/p65 ratio. In this regard, monotherapy with CRID3 did not provoke a significant change in the values of the aforementioned parameters compared with those of the DSS group of rats. The mRNA expression and colon tissue levels of NLRP3 were significantly downregulated in the DSS/NSC, DSS/CRID3, and DSS/NSC/CRID3 groups of rats compared to the DSS rats. In this regard, a greater level of significance was observed among the CRID3-administered colitic rats.
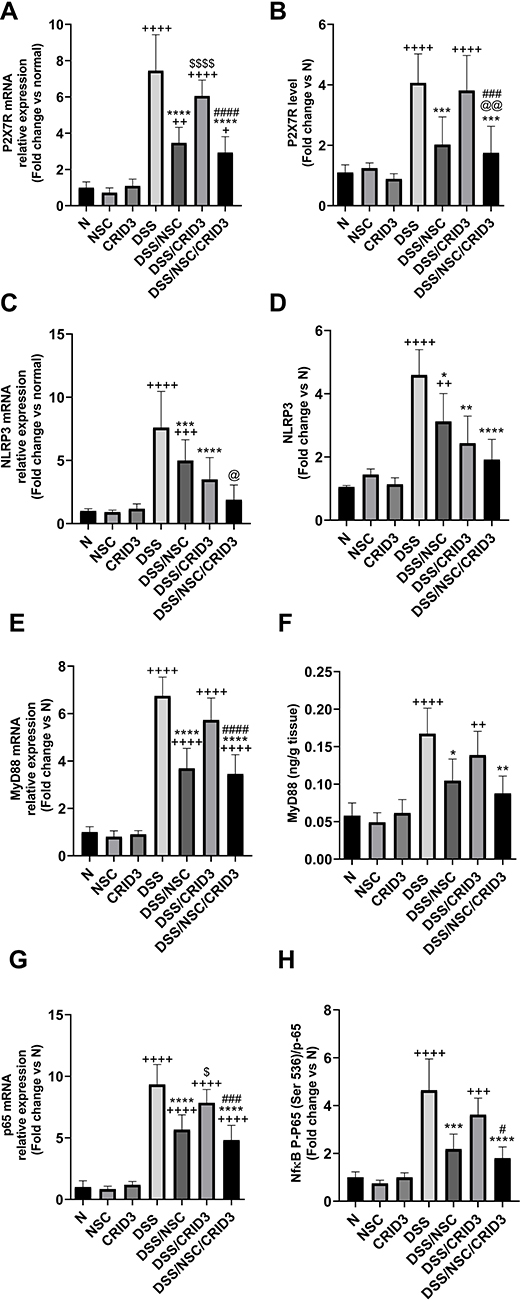 |
Figure 10 Effect on NLRP3, MyD88, and p65 expression. Effect of NSC, CRID3 and NSC/CRID3 on (A), P2X7R mRNA; (B), P2X7R; (C), NLRP3 mRNA; (D), NLRP3; (E), MyD88 mRNA; (F), MyD88; (G), p65 mRNA and (H), p-p65 (Ser 536)/p65. +P < 0.05 vs N, ++P < 0.001 vs N, +++P < 0.001 vs N, ++++P < 0.0001 vs N, *P < 0.05 vs DSS, **P < 0.01 vs DSS, ***P < 0.001 vs DSS, ****P < 0.0001 vs DSS, @P < 0.05 vs DSS/NSC, @@P < 0.01 vs DSS/NSC, #P < 0.05 vs DSS/CRID3, ###P < 0.001 vs DSS/CRID3, ####P < 0.0001 vs DSS/CRID3, $P < 0.05 DSS/CRID3 vs DSS/NSC, P < 0.0001 DSS/CRID3 vs DSS/NSC. |
Effect on the Flow Cytometric Determination of CD4+ve T Lymphocytes and CD11b+ve Monocytes in the Peripheral Blood
As shown in Figure 11, the percentage of CD4+ve T lymphocytes and CD11b+ve monocytes in the peripheral blood were significantly higher among the DSS-treated group compared with the N group of rats. Treatment of UC rats with NSC, CRID3, and their combination significantly reduced the elevation in the percentage of CD4+ve T lymphocytes and CD11b+ve monocytes in the peripheral blood compared to that of the DSS-treated rats. Interestingly, the DSS/NSC/CRID3 group of rats exhibited significantly decreased percentages of CD4+ve T lymphocytes and CD11b+ve monocytes compared with the N group. These effects expose a compromised innate immunity; therefore, future investigations are warranted to evaluate the immune modulatory effects of such drug combination.
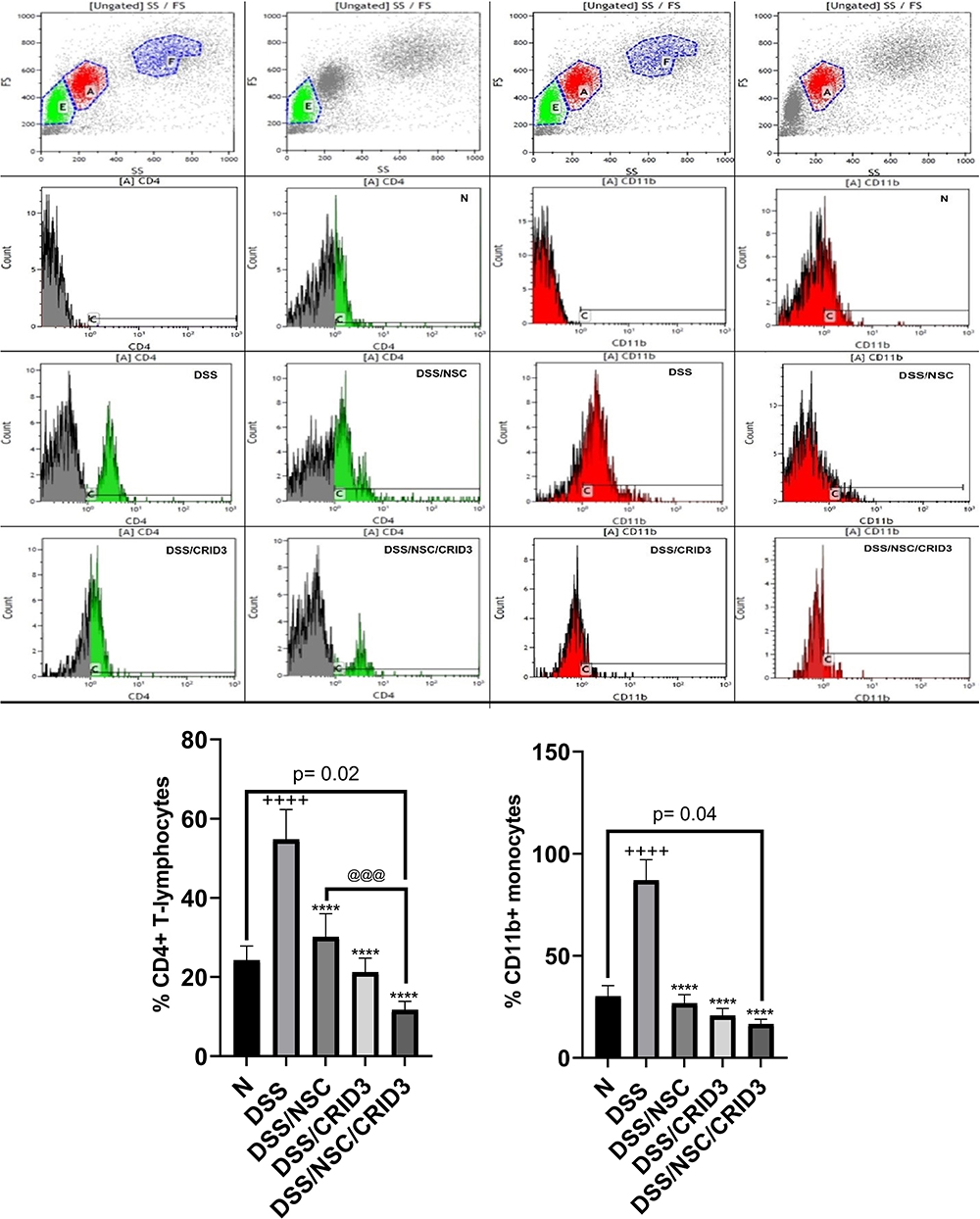 |
Figure 11 Flowcytometric determination of CD4+ve and CD11b cells. Effect of NSC, CRID3 and NSC/CRID3 on the % of CD4+ve T-lymphocytes (left half panel) and % of CD11b+ve monocytes (right half panel) in peripheral blood. The flow cytometric histograms depict flow cytometric gating according to cell size and granularity. A: monocytes population and E: lymphocytes population. @@@P < 0.001 vs DSS/NSC, ****P < 0.0001 vs DSS, ++++ P < 0.0001 vs N |
Discussion
Inflammasome activation plays an essential role in inducing the body’s innate immune response to microbial and injury-related signals through the stimulation and production of IL-1β and IL-18.23 The oligomerization of ASC is initiated upon the activation of NLRP3, leading to the eventual assembly of the NLRP3 inflammasome complex. This is followed by a catalytic cleavage of pro-caspase-1 into the active caspase-1 and the subsequent secretion of IL-1β and IL-18. The interactions among these protein components tightly regulate inflammasome function to ensure immune activity only when appropriate.
Targeting NLRP3 might present certain advantages over the biological inhibitors of IL-1β. It should be noted that IL-1β maturation can be mediated by several enzymes, including serine proteases and caspase-8.24 Moreover, CRID3 does not block the major antimicrobial inflammasomes NLRC4 and NLRP1. Thus, specific targeting of NLRP3 will not result in the complete blockade of IL-1β. As a consequence, the antimicrobial responses could remain intact. Therefore, CRID3 might have less immunosuppressive effects compared with biologics such as canakinumab.25
The specific inhibitor CRID3 blocks NLRP3-induced ASC oligomerization, ultimately relieving the NLRP3-induced colon inflammation. The molecular target of CRID3 has not been completely identified. However, CRID3 does not repress NLRP3 through the inhibition of inflammasome priming.24 Conversely, in the current study, NSC, a P2X7R antagonist, effectively interrupted the MyD88/NF-κB axis, which is believed to be a priming signal in the cascade of NLRP3 inflammasome assembly and activation. A growing body of evidence highlights a proinflammatory contribution of P2X7R to colitis pathology. Additionally, it has been discovered that P2X7R might positively regulate NF-κB activation by stimulating the MyD88-dependent pathway.26 Thus, NSC has multiple cellular targets, including both the priming and activation (second) signals of the NLRP3 inflammasome. In contrast, our results revealed that CRID3 did not affect the MyD88/NF-κB axis. Therefore, we are in agreement with a report in which authors stated that CRID3 does not inhibit NLRP3 priming or toll-like receptor signaling.25
In addition to inducing caspase-1 activity, prolonged NLRP3 activation induces proapoptotic molecules, such as cleaved caspase-3. In this regard, we revealed that both NSC and CRID3 significantly downregulated the expression of caspase-3 in the injured colon. However, the most significant effect was observed for CRID3, rather than NSC, probably because of the ability of NSC to inactivate the anti-apoptotic NF-κB. Herein, NF-κB inactivation was confirmed by the ratio of p-p65 (Ser 536)/p65, immunolabeling of MyD88 and NF-κB p65, the mRNA expression of p65, and the mRNA and protein expression of MyD88.
Using NSC in the setting of CRID3 effectively potentiated the CRID3 inhibitory effect on IL-1β by interfering NF-κB signaling, leading to further repression of the production of pro-IL-1β. This avenue might contribute to the enhancement of local immunosuppressive effects on colon tissue. Pyroptosis, a caspase-1-dependent programmed cell death, is characterized by plasma membrane rupture, cell lysis, and release of IL-1β and IL-18.27 IL-1β, which is a pyroptosis product, has been considered a key component of inflammation and colon tissue damage.28 Moreover, emerging evidence has pinpointed pyroptosis as an important trigger and endogenous regulator of colon inflammation. Similarly, IL-18 has been shown to contribute to the breakdown of the mucosal barrier, triggering inflammation and amplifying the damage elicited in the intestinal epithelium during disease.29 Furthermore, a clinical study reported a strong correlation between increased epithelial secretion of IL-18 and increased severity of IBD.30 These findings showed that pyroptosis plays a prominent role in IBD. Therefore, therapeutic strategies targeting the activation of the NLRP3 inflammasome and pyroptosis hold promise for the treatment of IBDs. In the present study, the NSC/CRID3 combined therapy efficiently inhibited the caspase-1-dependent release of IL-1β and IL-18, thus keeping a minimal level of pyroptotic cell lysis.
For the assessment of the immune status at the colon tissue level, we calculated the ratio of IL-10/IL-12. This ratio represents a measure of the balance between the anti-inflammatory and proinflammatory states31 or Th1 vs Th2 dominance.32 It is well acknowledged that IL-12 plays a significant role in the activation of the Th1 immune response and that, concomitantly, IL-10 plays a negative regulatory function in this process. Herein, NSC/CRID3 combined therapy significantly reversed the sharp decline in that ratio, indicating inhibition of the cellular-mediated immunity.
In addition, flow cytometric analysis of the percentages of peripheral blood CD11b+ve monocytes and CD4+ve cells revealed significant inhibition of the cellular-mediated immunity with respect to normal rats. These data were further confirmed by the efficient inhibition of IL-1β, decreased activity of MPO, and the sharp increase in the ratio of IL-10/IL-12 after NSC/CRID3 combined therapy. Therefore, because of the observed impairment in the innate immune response, we suggest that NSC/CRID3 combined therapy is more suitable for evaluation in a subject in the acute phase of colitis vs a subject in the chronic phase. Furthermore, NSC/CRID3 combined therapy could be used to alter the outcome of some diseases in which a hyperactive Th1 arm is implicated because, in some cases, reduction of the Th1 response or a Th2 type bias may alleviate such conditions.33
This research represents the first report describing the potential therapeutic effects of using NSC and CRID3 combination for the treatment of inflamed colon. The utmost noteworthy findings were that, on the one hand, NSC interrupted the MyD88/NF-κB axis (priming signal) and, on the other hand, it interrupted ATP/P2X7R-induced NLRP3 activation (second signal). These two effects lead to the maximization of the efficacy of CRID3. Accordingly, the severity of the DSS-induced colitis was distinctly decreased after the simultaneous administration of NSC and CRID3. However, innate immune responses might be impaired should a complete inhibition of IL-1β occur. Therefore, further investigations are needed to evaluate the immunosuppressive effects of the combination therapy of CRID3 with NSC using different dose levels. It should be recalled that the complex pathogenesis and the implication of a vast number of components in the inflammasome activation pathway prompted us to search for compounds that act at the level of multiple cellular targets and have a wide therapeutic index. Therefore, the future clinical application of NSC/CRID3 combined therapy may result in the generation of novel approach in the treatment of IBDs in addition to other more complex diseases, such as type 2 diabetes, multiple sclerosis, atherosclerosis, and Alzheimer’s disease in which NLRP3 inflammasome activation has been identified as a substantial component of the inflammatory process. Moreover, it is worth noting that NSC and CRID3 may also be more cost effective than biological-agents.34 Furthermore, using NSC and CRID3 at lower doses might act to decrease the likelihood of off-target effects and prevent the prospective impairment of the innate immunity. In the latter instance, this combination therapy could be applied in future investigations to patients with chronic inflammatory conditions.
Conclusion
In conclusion, compared with the effects of NSC and CRID3 as monotherapeutic agents, our data revealed that NSC/CRID3 combined therapy effectively attenuated colon shortening and disease activity. NSC/CRID3 also effectively improved both the macroscopic and microscopic scenarios and increased goblet cell density in the injured colon. Moreover, NSC/CRID3 efficiently improved the antioxidant defense machinery of the injured colon against oxidative stress. Furthermore, NSC/CRID3 mitigated the inflammation state by downregulating the proinflammatory cytokines. Pyroptotic cell death is also significantly restrained at the pathological state of the colon, as indicated by lower levels of caspase-1 tissue expression and activity and the consequent downregulation in the colon tissue IL-1β and IL-18. Therefore, the broadly available, inexpensive, well-tolerated, and systemically potent P2X7R inhibitor, NSC, has multiple cellular targets and may be added as an adjunctive therapy to CRID3. Such combination therapy may enhance the possibility of optimal recovery of patients with UC by increasing the efficacy of CRID3.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Saber S, Basuony M, Eldin AS. Telmisartan ameliorates dextran sodium sulfate-induced colitis in rats by modulating NF-κB signalling in the context of PPARγ agonistic activity. Arch Biochem Biophys. 2019;671:185–195. doi:10.1016/j.abb.2019.07.014
2. Saber S, El-Kader EMA. Novel complementary coloprotective effects of metformin and MCC950 by modulating HSP90/NLRP3 interaction and inducing autophagy in rats. Inflammopharmacology. 2021;29:237–251. doi:10.1007/s10787-020-00730-6
3. Saber S, Abd El-Kader EM, Sharaf H, et al. Celastrol augments sensitivity of NLRP3 to CP-456773 by modulating HSP-90 and inducing autophagy in dextran sodium sulphate-induced colitis in rats. Toxicol Appl Pharmacol. 2020;400:115075. doi:10.1016/j.taap.2020.115075
4. Figliuolo VR, Savio LEB, Safya H, et al. P2X7 receptor promotes intestinal inflammation in chemically induced colitis and triggers death of mucosal regulatory T cells. Biochim Biophys Acta Mol Basis Dis. 2017;1863(6):1183–1194. doi:10.1016/j.bbadis.2017.03.004
5. Marques CC, Castelo-Branco MT, Pacheco RG, et al. Prophylactic systemic P2X7 receptor blockade prevents experimental colitis. Biochim Biophys Acta Mol Basis Dis. 2014;1842(1):65–78. doi:10.1016/j.bbadis.2013.10.012
6. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 receptor in infection and inflammation. Immunity. 2017;47(1):15–31. doi:10.1016/j.immuni.2017.06.020
7. Martínez-García JJ, Martínez-Banaclocha H, Angosto-Bazarra D, et al. P2X7 receptor induces mitochondrial failure in monocytes and compromises NLRP3 inflammasome activation during sepsis. Nat Commun. 2019;10(1):2711. doi:10.1038/s41467-019-10626-x
8. Remy M, Thaler S, Schumann RG, et al. An in vivo evaluation of Brilliant Blue G in animals and humans. Br J Ophthalmol. 2008;92(8):1142. doi:10.1136/bjo.2008.138164
9. Peng W, Cotrina ML, Han X, et al. Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proc Natl Acad Sci. 2009;106(30):12489. doi:10.1073/pnas.0902531106
10. Zhang X, Zhang M, Laties AM, Mitchell CH. Stimulation of P2X7 receptors elevates Ca2+ and kills retinal ganglion cells. Invest Ophthalmol Vis Sci. 2005;46(6):2183–2191. doi:10.1167/iovs.05-0052
11. Suzuki T, Hide I, Ido K, Kohsaka S, Inoue K, Nakata Y. Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J Neurosci. 2004;24(1):1. doi:10.1523/JNEUROSCI.3792-03.2004
12. Jacques-Silva MC, Rodnight R, Lenz G, et al. P2X7 receptors stimulate AKT phosphorylation in astrocytes. Br J Pharmacol. 2004;141(7):1106–1117. doi:10.1038/sj.bjp.0705685
13. Pereira JMS, Barreira AL, Gomes CR, et al. Brilliant blue G, a P2X7 receptor antagonist, attenuates early phase of renal inflammation, interstitial fibrosis and is associated with renal cell proliferation in ureteral obstruction in rats. BMC Nephrol. 2020;21(1):206. doi:10.1186/s12882-020-01861-2
14. Boucher D, Monteleone M, Coll RC, et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med. 2018;215(3):827–840. doi:10.1084/jem.20172222
15. Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP 3 inflammasome activation and assembly. Immunol Rev. 2015;265(1):35–52. doi:10.1111/imr.12286
16. Saber S, Khodir AE, Soliman WE, et al. Telmisartan attenuates N-nitrosodiethylamine-induced hepatocellular carcinoma in mice by modulating the NF-κB-TAK1-ERK1/2 axis in the context of PPARγ agonistic activity. Naunyn Schmiedebergs Arch Pharmacol. 2019;392(12):1591–1604. doi:10.1007/s00210-019-01706-2
17. Saber S, Ghanim AMH, El-Ahwany E, El-Kader EMA. Novel complementary antitumour effects of celastrol and metformin by targeting IκBκB, apoptosis and NLRP3 inflammasome activation in diethylnitrosamine-induced murine hepatocarcinogenesis. Cancer Chemother Pharmacol. 2020;85(2):331–343. doi:10.1007/s00280-020-04033-z
18. Perera AP, Fernando R, Shinde T, et al. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci Rep. 2018;8(1):8618. doi:10.1038/s41598-018-26775-w
19. Tapia-Abellán A, Angosto-Bazarra D, Martínez-Banaclocha H, et al. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat Chem Biol. 2019;15(6):560–564. doi:10.1038/s41589-019-0278-6
20. Jagtap A, Shirke S, Phadke A. Effect of polyherbal formulation on experimental models of inflammatory bowel diseases. J Ethnopharmacol. 2004;90(2–3):195–204. doi:10.1016/j.jep.2003.09.042
21. Khalil R, Shata A, Abd El-Kader EM, et al. Vildagliptin, a DPP-4 inhibitor, attenuates carbon tetrachloride-induced liver fibrosis by targeting ERK1/2, p38α, and NF-κB signaling. Toxicol Appl Pharmacol. 2020;407:115246. doi:10.1016/j.taap.2020.115246
22. Younis NS, Ghanim AMH, Saber S. Mebendazole augments sensitivity to sorafenib by targeting MAPK and BCL-2 signalling in n-nitrosodiethylamine-induced murine hepatocellular carcinoma. Sci Rep. 2019;9(1):19095. doi:10.1038/s41598-019-55666-x
23. Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10(3):241–247. doi:10.1038/ni.1703
24. Coll RC, Robertson AAB, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248–255. doi:10.1038/nm.3806
25. Dinarello CA, van der Meer JWM. Treating inflammation by blocking interleukin-1 in humans. Semin Immunol. 2013;25(6):469–484.
26. Liu Y, Xiao Y, Li Z. P2X7 receptor positively regulates MyD88-dependent NF-κB activation. Cytokine. 2011;55(2):229–236. doi:10.1016/j.cyto.2011.05.003
27. Ma Y, Jiang J, Gao Y, et al. Research progress of the relationship between pyroptosis and disease. Am J Transl Res. 2018;10(7):2213–2219.
28. Saber S, Abd El-Fattah EE, Yahya G, et al. A novel combination therapy using rosuvastatin and lactobacillus combats dextran sodium sulfate-induced colitis in high-fat diet-fed rats by targeting the TXNIP/NLRP3 interaction and influencing gut microbiome composition. Pharmaceuticals. 2021;14(4):341. doi:10.3390/ph14040341
29. Mao L, Kitani A, Strober W, Fuss IJ. The role of NLRP3 and IL-1β in the pathogenesis of inflammatory bowel disease. Front Immunol. 2018;9:2566.
30. Pizarro TT, Michie MH, Bentz M, et al. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: expression and localization in intestinal mucosal cells. J Immunol. 1999;162(11):6829.
31. van Hemert S, Meijerink M, Molenaar D, et al. Identification of Lactobacillus plantarum genes modulating the cytokine response of human peripheral blood mononuclear cells. BMC Microbiol. 2010;10:293. doi:10.1186/1471-2180-10-293
32. Bien E, Balcerska A, Adamkiewicz-Drozynska E, Rapala M, Krawczyk M, Stepinski J. Pre-treatment serum levels of interleukin-10, interleukin-12 and their ratio predict response to therapy and probability of event-free and overall survival in childhood soft tissue sarcomas, Hodgkin’s lymphomas and acute lymphoblastic leukemias. Clin Biochem. 2009;42(10–11):1144–1157. doi:10.1016/j.clinbiochem.2009.04.004
33. Abdel-Ghany R, Rabia I, El-Ahwany E, et al. Blockade of PGE2, PGD2 receptors confers protection against prepatent schistosomiasis mansoni in mice. J Egypt Soc Parasitol. 2015;45(3):511–520. doi:10.12816/0017911
34. López-Castejón G, Pelegrín P. Current status of inflammasome blockers as anti-inflammatory drugs. Expert Opin Investig Drugs. 2012;21(7):995–1007. doi:10.1517/13543784.2012.690032
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.