")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
The Role of JNk Signaling Pathway in Obesity-Driven Insulin Resistance
Authors Feng J , Lu S, Ou B , Liu Q, Dai J, Ji C, Zhou H, Huang H, Ma Y
Received 26 October 2019
Accepted for publication 18 January 2020
Published 29 April 2020 Volume 2020:13 Pages 1399—1406
DOI https://doi.org/10.2147/DMSO.S236127
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Konstantinos Tziomalos
Jia Feng,1,2 Shiyin Lu,1,2 Biqian Ou,1,2 Qian Liu,1,2 Jiaxin Dai,1,2 Chunyan Ji,1,2 Haiqing Zhou,1,2 Hongke Huang,1,2 Yi Ma1,2
1Institute of Biomedicine, Department of Cellular Biology, Jinan University, Guangzhou, People’s Republic of China; 2National Engineering Research Center of Genetic Medicine, Key Laboratory of Bioengineering Medicine of Guangdong Province, Jinan University, Guangzhou, People’s Republic of China
Correspondence: Yi Ma
Institute of Biomedicine, Department of Cellular Biology, Jinan University, 601 Huangpu Ave West, Guangzhou, Guangdong 510632, People’s Republic of China
Tel/Fax +86 20 8522 1983
Email [email protected]
Abstract: Obesity is not only closely related to insulin resistance but is one of the main factors leading to the formation of Type 2 Diabetes (T2D) too. The c-Jun N-terminal kinase (JNK) family is a member of the mitogen-activated protein kinase (MAPK) superfamily. JNK is also one of the most investigated signal transducers in obesity and insulin resistance. JNK-centric JNK signaling pathway can be activated by growth factors, cytokines, stress responses, and other factors. Many researches have identified that the activated phosphorylation JNK negatively regulates insulin signaling pathway in insulin resistance which can be simultaneously regulated by multiple signaling pathways related to the JNK signaling pathway. In this review, we provide an overview of the composition of the JNK signaling pathway, its regulation of insulin signaling pathway, and the relationship between the JNK signaling pathway and other pathways in insulin resistance.
Keywords: JNK signaling pathway, obesity, insulin resistance, type 2 diabetes
Introduction
In the past three decades, global overweight and obesity have risen rapidly because of the changes in human diets, travel and other lifestyles. Based on the current trend, the prevalence of overweight (BMI 25–29.9 kg/m2) and obesity (BMI ≥ 30 kg/m2) (World Health Organization standards) in adults worldwide is predicted to rise from 33% in 2005 to 57.8% in 2030.1 Therefore, lots of researches focus on metabolic diseases induced by obesity. In recent years, several studies have shown that chronic low-grade inflammation (e.g. IL-1α, IL-1β, IL-6, RANTES, MCP-1, TNFα) caused by obesity may be an important cause of insulin resistance and T2D.2 Generally, insulin resistance precedes the initiate of T2D, refers to a pathological state that the body, especially peripheral tissues, does not respond properly to circulating insulin. It is an important mechanism for the development of T2D and the basis of obesity and metabolic diseases including hyperlipidemia, hypertension, cardiovascular disease.3 Recently, the current evidence indicates that factors affecting the development of insulin resistance in obese subjects are extremely complex and regulated by a variety of signaling pathways. JNK affecting insulin resistance is a member of the MAPKs superfamily and plays the essential role at the core of inflammatory response and oxidative stress. In the different tissues of obese subjects, inflammatory factors can be observed to cause continuous activation of JNK. The activated JNK acts on nuclear factor-κB (NF-κB) and activator protein-1 (AP-1) to produce more inflammatory factors, further reducing the sensitivity of insulin target cells towards insulin, finally to form a vicious circle and aggravate insulin resistance. Besides, a network framed by PPARγ, NF-κB and PTP1B signaling pathways crossing with JNK signaling pathway plays an important role in regulating insulin resistance. However, to our best knowledge, a comprehensive review discussing possible effects of JNK signaling pathway crossed with PPARγ, NF-κB and PTP1B signaling pathways in obesity-driven insulin resistance is missing. Simultaneously, a better understanding of JNK signaling pathway and the relationship between PPARγ, NF-κB, PTP1B signaling pathways and the JNK signaling pathway is necessary to appreciate important drug targets for the treatment and improvement of obesity-driven insulin resistance.4
Overview of the JNK Signaling Pathway
Since the 21st century, protein kinases have been an important research objective, and their roles are phosphorylation by other kinases, transferring amino acid residues to the ATP-terminal phosphate group for phosphorylation, or autophosphorylation. Based on the type of amino acid residue type of phosphorylated substrate protein, the protein kinases were classified into five categories: serine/threonine (Ser/Thr) protein kinase; histidine (His) protein kinase; tyrosine (Tyr) protein kinase; tryptophan (Trp) protein kinase; aspartyl/glutamyl protein kinase. MAPK, a class of Ser/Thr protein kinases, are widely found in mammalian cells. Currently, there are four members of the MAPK family, such as JNK/stress-activated protein kinase (SAPK), extracellular signal-regulated kinase (ERK1/2), p38, and ERK5. There are three JNK isoforms encoded by three different genes, JNK1 (Mapk8) and JNK2 (Mapk9) express ubiquitously, and JNK3 (Mapk10) expression is restricted to the brain, testis, heart, and pancreatic β-cells.5 Obesity, alcohol and radiation may activate the JNK signaling pathway to regulate the expression of inflammation, oxidative stress and proliferation-related genes and proteins.4 The JNK protein can be activated by two MAP2K (MKK4 and MKK7) which are located upstream of JNK. Both MKK4 and MKK7 can simultaneously phosphorylate JNK at threonine-183 and tyrosine-185 sites, but the difference is that MKK4 prefers to phosphorylate tyrosine, while MKK7 prefers to phosphorylate threonine. Besides, MKK4 and MKK7 are activated in turn by a series of upstream MAP3K which are regulated by plenty of different upstream factors, including TNFα and FFA.6–8 JNK was named c-Jun N-terminal kinase because it was originally discovered to be a kinase that specifically phosphorylates the nuclear transcription factor c-Jun, but the later studies indicated that many substrates are located downstream of the JNK signaling pathway, including the jun family, AP-1, P53, Bcl-2, c-myc, Mcl-1, Sab, ATF-2, insulin receptor substrate-1 (IRS-1), JNK interacting protein 1 (JIP1). Upon activation of the JNK signaling pathway, JNK is translocated from the cytoplasm into the nucleus and activates the c-Jun transcription factor by transphosphorylation of Ser63 and Ser73 in the amino-terminal active domain of the c-Jun transcription factor. The c-Jun transcription factor is activated and binds to the binding site of transcription factor activator protein-1 (AP-1) locating at the gene promoter, promoting the expression of proinflammatory genes and protein synthesis (e.g. TNFα, IL-1β, IL-6 and IL-8),9,10 which plays an important role in impairing glucose tolerance by obesity-induced and insulin resistance. In JNK signaling pathway, JNK activity mainly regulates metabolism by phosphorylating its substrate and plays a crucial role in the occurrence and development of human diseases. Altogether, these pieces of evidence indicate that JNK has a significant effect on metabolism, especially in inflammation and obesity complications.
Obesity-Driven Insulin Resistance
The current view is that obesity is closely related to insulin resistance which usually occurs in the liver, muscle, and adipose tissue. Importantly, it shows that during obesity, activated JNK is elevated in mouse muscle, fat, and liver. JNK is one of the most investigated molecules in obesity models of insulin resistance, and studies have led to a new paradigm of how excess adiposity may cause insulin resistance.11 Obesity induces chronic low-level inflammation, produces inflammatory factors and FFA to activate JNK signaling pathway in insulin target cells.2 The inflammatory factor TNFα binds to its receptor to activate the JNK signaling pathway, which in turn regulates the expression of the TNF gene to form a vicious circle.6,12,13 Biopsy of subcutaneous adipose tissue in obese individuals showed an increased activation of phosphorylated JNK that further amplified the inflammatory response and impaired the insulin signaling pathway.14,15 Yang et al found that Liver-specific knockdown of JNK1 resulted in significant reductions in circulating insulin and glucose levels, by 57% and 16%, respectively. At the molecular level, JNK1 knockdown mice had a sustained and significant increase of hepatic Akt phosphorylation. Furthermore, knockdown of JNK1 enhanced insulin signaling in vitro.16 Prada et al found that TNFα and IL-6 levels were consistent with JNK activity, in skeletal muscle.17 The cause of insulin resistance in target organs is that inflammatory factors interfere with the IRS-1/PI3K/Akt/GLUT1-4 insulin signaling pathway, then attenuate glucose uptake and subsequent disposal mediated by insulin, and increase production and output of glycogen. Finally, insulin resistance occurs in insulin receptor cells.18 The energy supply for inflammatory cells comes from glucose. In an inflammatory state, with the increase of macrophages in adipose tissue, the demand of immune system for glucose increases. The body needs to provide a substrate for gluconeogenesis and increase the supply of glucose by increasing the breakdown of fat, which promotes lipolysis, and glucose and lipid metabolic disorder. On the other hand, inflammation in obese patients can damage the endothelial function and cause endothelial insulin resistance. At the same time, increased expression of inflammatory factors can increase the production of ROS (Reactive Oxygen Species, ROS), decrease the transcription of uncoupled protein (UCP) S, resulting in insufficient ability to scavenge oxygen-free radicals, which is the main cause of oxidative stress. Increased ROS content in obese people causes oxidative stress, in turn activates JNK and NF-κB signaling pathways. With the JNK signaling pathway activated, JNK proteins regulate a variety of nuclear substrates and extranuclear substrates, for example, JNK regulates AP-1 and NF-KB, controls the expression of many genes including pro-inflammatory cytokines to interfere with Insulin signaling pathway and GLU-T4 translocation, which results in insulin resistance.19
Researchers found that fatty acids were converted into acyl-CoA when free fatty acids increased in the blood, then increased DAG, activated PKCθ, changed the phosphorylation site of IRS-1, interfered the activation of PI3K, and affected GLUT4 translocation to cell surface; therefore, glucose utilization of insulin-mediated was reduced. Another research indicates that free fatty acids inhibit uptake glucose in various tissues after insulin stimulation, which reduces glucose oxidation and glycolysis, decreases insulin sensitivity of tissues.20
Regulation of Insulin Signaling Pathway by the JNK Signaling Pathway
Insulin is an important hormone, regulating the metabolism of carbohydrate and fat and the physiological functions of cells through PI3K-AKT signaling pathway. After insulin activates Akt via PI3K, Akt was proposed to participate in the metabolic regulation of insulin by four mechanisms: promoting glycogen synthesis through glycogen synthase kinase 3 (GSK3);3 regulating vesicles translocation in intracellular glucose carriers (mainly GLUT4); inhibiting fatty acid oxidation and hepatocyte gluconeogenesis through Akt/PKB phosphorylates transcription factor PGC-1α; insulin decreases cAMP levels and inhibits PKA to activate hormone-sensitive lipase via PI3K and Akt-dependent signaling pathways, finally, lipolysis is inhibited.
Chronic low-grade inflammation and FFA induced by obesity can activate JNK to phosphorylate insulin receptor substrates (IRS) 1 and 2 at Ser/Thr residues18 and dephosphorylate IRS-1 Tyr residues,21 which not only prevents IRS from activating downstream PI3K also the interaction of IRS with its upstream insulin receptor (IR). After the conduction of the PI3K-AKT insulin signaling pathway is blocked, the sensitivity of target cells to insulin decreases, which stimulates the body to continuously produce insulin to eliminate excess glucose in the blood. However, in islet β cells, compensatory insulin synthesis format hyperinsulinemia and add the burden of islet β-cell secretion,22 eventually, leading to hyperproliferation and the trend of apoptosis to form insulin resistance even T2D.23 This is one of the mechanisms that JNK activity links to insulin resistance during obesity. This view is supported by a series of studies. Yang et al used gene knockout technology to selectively silence the JNK1 gene in mouse adipose tissue.16 They found that the lack of JNK1 in adipose tissue inhibited insulin resistance induced by a high-fat diet in the liver. More importantly, inactivation of the JNK2 gene also enhanced the sensitivity of the cell to insulin.4 To test the role of JNK, Han et al established a macrophage-selective JNK deficiency mouse model feeding with a high-fat diet and found that only JNK-deficient mouse macrophages were still sensitive to insulin.24 JNK gene silencing decreases IRS-1 Ser 307 phosphorylation, increases Tyr phosphorylation, enhances insulin signaling, and increase cell sensitivity to insulin. Meanwhile, JNK gene silencing inhibits the expression of inflammatory genes, alleviates insulin resistance. For instance, δ-tocotrienol can inhibit the activity of JNK to reduce the transcriptional activities of inflammation-related factors AP-1 and NF-κB, which alleviates insulin resistance. In conclusion, these studies indicate that JNK activity may cause insulin resistance; therefore, JNK is a potential target for insulin resistance therapy.25
The Relationship Between JNK and Other Signaling Pathways in Insulin Resistance
In cell, multiple signal pathways crossing and interacting with each other form an extremely complex network, which is called Signaling Crosstalk. Signaling Crosstalk occurs when the transducer is stimulated by one or more extracellular factors, possibly in the cytosol or nucleus. In addition, it reflects at all levels of the signal path, ultimately, to enhance or suppress signaling pathways. With advanced technology and thorough researches of signal pathways, reports on Signaling Crosstalk of the JNK signal path have sharply increased in recent years. The JNK signaling pathway in obesity-driven insulin resistance may be activated by inflammatory factors and FFA, which results in more inflammatory factors to form a vicious cycle and aggravate insulin resistance. In this progression, PPARγ, NF-κB, and PTP1B signaling pathways crossing with the JNK signaling pathway contribute to regulate the conduction of the insulin signaling pathway in target cells and the level of inflammatory factors in various tissues. Importantly, clarifying the relationship between the JNK signaling pathway and other signaling pathways is of great significance for finding targets for drug therapy and treating diabetes.
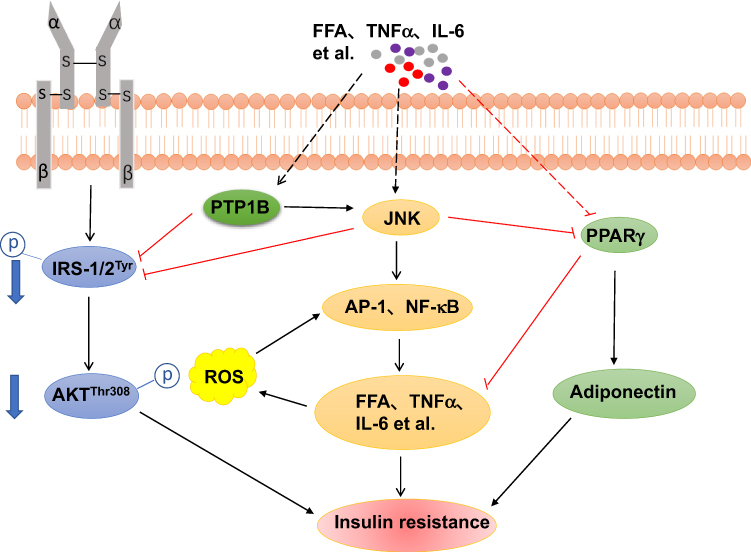 |
Figure 1 A general model for the relationship between JNK signaling pathway and the others, in the obesity-driven insulin resistance. JNK plays a critical role in obesity-induced pro-inflammatory state, leading to enhanced production of pro-inflammatory factors such as IL-1β, TNFα and IL-6. These factors are likely to induce impaired glucose metabolism and insulin resistance in a number of ways (see text for details). For example, JNK inhibits insulin signaling pathway and increases the production of inflammatory cytokines. Pro-inflammatory factors regulate PPARγ to control the expression of adiponectin and pro-inflammatory state. NF-κB signaling pathway can be affected by JNK to regulate the expression of inflammation cytokines. PTP1B activated by inflammation cytokines may block the insulin signaling pathway and activate JNK signaling pathway to aggravate insulin resistance. |
JNK Signaling Pathway and PPARγ Signaling Pathway
Peroxisome proliferator-activated receptors (PPARs) are a kind of ligand-activated nuclear transcription factors composed of α, β (also known as δ) and γ subtypes, belonging to II type nuclear receptor superfamily. Currently, PPAR members that have been recognized are PPARα, PPARβ/δ, PPARγ. There are so many researches indicating that PPARγ plays a central role in adipocyte differentiation, sugar and lipid metabolism, inflammatory response, atherosclerosis, and the improvement of insulin resistance.26,27 PPARγ ligands inhibit the expression of various inflammation-related genes, including inducible nitric oxide synthase, matrix metalloproteinases, and interleukins. These behaviors may also be associated with obesity-related insulin resistance, because macrophage accumulation and gene expression in adipose tissue have been proven to play an important role in the pathogenesis of obesity-driven insulin resistance.28,29 Therefore, it is meaningful to illuminate that the PPARγ signaling pathway regulates obesity-driven insulin resistance in improving insulin resistance and treating T2D.
TNF-α inhibits the transcriptional activity of PPARγ by activating ERK and JNK in chronic low-grade inflammation caused by obesity.30,31 ERK and JNK inhibit PPARγ function through directly phosphorylating PPARγ at Ser112 residue, and this phosphorylation leads to inhibition of PPARγ transactivation.32 Similarly, histone deacetylase 3 is a co-repressor of PPARγ in the presence of inflammatory factors, such as TNF-α.33 Histone deacetylase 3 binds to heterodimers of PPARγ and retinoid X receptors, which reduces PPARγ transcriptional activity.34 Inflammation-induced downregulation of PPARγ has an important consequence that adiponectin (a hormone produced exclusively by adipocytes) expression is decreased. Whether in vitro and in vivo, PPARγ agonists can increase the expression and secretion of adiponectin.35 This adipocyte-specific cytokine adiponectin is involved in the regulation of insulin sensitivity in target cells. The lack of PPARγ in fat cells, adiponectin expression, the ability of storing fat, and insulin sensitivity will decrease.36 Gavrilova et al found that the selective loss of PPARγ in mouse skeletal muscle and liver resulted in severe systemic insulin resistance, which also confirmed the above results (Figure 1).37
Activated PPAR forms a heterodimer with the retinoic acid x receptor, binding to specific regions of the DNA sequence to regulate transcriptional activation of downstream target genes. PPARγ improve insulin resistance by two ways: The promotion of glucose and fatty acid metabolism will accelerate fatty acid consumption, lower the TG content in the liver and muscle, improve the high-fat diet-induced obesity and insulin resistance;38 Adipose cells reduce the secretion of TNF-α and other signaling molecules to alleviate the body’s inflammatory state and activation of the JNK signaling pathway. The expression level of PPARγ in the diabetic group was significantly lower than the normal group, but the expression of AP-1 and NF-κB increase. Activated AP-1 and NF-κB have functions to regulate the gene expression of various inflammatory factors, which plays an important role in pathophysiological processes, including cell proliferation, differentiation, apoptosis, and inflammatory response (Figure 1). Moreover, AP-1 and NF-κB can activate JNK transcriptional activity and induce insulin resistance in obese patients. However, elevated PPARγ activity will be accompanied by a decrease in AP-1 and NF-κB expression, reduced in inflammation of the body, and blocking JNK signaling pathway.39,40 PPARγ is an essential molecule in obesity-induced inflammation, intersects with the JNK signaling pathway and regulates the expression of adiponectin, which may indirectly reduce the expression of inflammatory factors to improve insulin resistance.
JNK Signaling Pathway and NF-κB Signaling Pathway
Nearly all cells have IKK/NF-κB signaling system, including NF-κB family, inhibitor IκB family, and IKK multi-component protein kinase complex. The IκB protein family is an inhibitor of NF-κB, and mainly includes IκBα, IκBβ, and IκB∑. Under normal conditions, IκBα can covalently bind to NF-κB dimers P65 and P50 in the cytoplasm to mask the nuclear localization signal (NLS) of NF-κB, preventing its nuclear translocation. Due to the presence of IκBα, NF-κB is quiescent in the cytoplasm, but a variety of extracellular stimuli can activate IκKβ that can phosphorylate IκBα to dissociate IκBα from NF-κB. Therefore, NF-κB is transferred to the nucleus to regulate the expression of inflammation-related factors. However, its overexpression is an important factor leading to insulin resistance.
Inflammation induced by obesity activates JNK and NF-κB signaling pathways in insulin target cells, which plays a key role in insulin resistance.41,42 At the cellular level, Inflammation induced by obesity may be involved in phosphorylation of the transcription factor NF-κB leading to an increase of the expression of TNFα and IL-6 genes associated with insulin resistance.43 Inflammatory cytokines may inhibit IRS-1 Tyr phosphorylation in the insulin signaling pathway, also lead to increase in JNK phosphorylation, and decrease Akt phosphorylation. In addition, IRS-1, Akt and JNK are three upstream targets of NF-κB. Activated JNK and inhibited Akt activity can increase NF-κB activation, resulting in inflammatory factor synthesis and aggravating insulin resistance (Figure 1).44–46 Therefore, Inflammation induced by obesity can activate various inflammatory mechanisms through JNK and NF-κB signaling pathways, which promote the expression of inflammatory factors, inhibiting insulin signal transduction to form insulin resistance.47
JNK Signaling Pathway and PTP1B Signaling Pathway
Protein tyrosine phosphatase 1B (PTP1B) is the first isolated and purified PTP and its structure has been elucidated by X-ray crystallography. Studies have shown that the expression and activation of PTP1B are increased in obese subjects and PTP1B negatively regulates insulin signaling pathways.48 PTP1B can dephosphorylate Tyr residue of IR or IRS to attenuate insulin signaling pathway (Figure 1).49,50 In muscle, transgene overexpression of PTP1B results in impairing insulin signaling pathway, lowering glucose uptake in tissues,thus forming insulin resistance. In contrast, PTP1B deficient mice were resistant to gain weight, showed increased insulin sensitivity, and still maintained insulin sensitivity during a high-fat diet, suggesting that insulin signaling can be enhanced by PTP1B inhibition.51
Obese subjects have double muscle ceramide content of lean subjects.52 At the molecular level, ceramide is capable of inhibiting several different intermediates in the insulin signaling pathway. Treatment of cells with short-chain ceramide analogs or bacterial sphingomyelinase block insulin-stimulated Tyr phosphorylation of IRS-1 as well as subsequent recruitment and activation of PI3K.53 Also, knockdown of PTP1B lentivirus in myocytes prevents ceramide from reducing IRS-1 and Akt phosphorylation, suggesting that PTP1B can be considered as one of the mediators of ceramide-induced insulin resistance in myocytes. Moreover, Taheripak et al found that PTP1B inhibition activates Akt by increased signaling through insulin receptor substrates/PI3K or decreased signaling through ROS/JNK pathways.50 In skeletal muscle cells, hyperglycemia, FFA, and inflammation contribute to the overexpression of PTP1B and the activation of PTP1B is mediated by the ceramide-JNK-NF-κB signaling pathway. Activated NF-kB enters the nucleus and binds to DNA leading to the expression of the inflammatory factor-related gene. Excessive inflammatory factors induce PTP1B overexpression impairing the insulin signaling pathway, which causes disorder of glucose and lipid metabolism, finally, to form insulin resistance.49 These findings offer a novel insight into the beneficial effects of the regulation of insulin-sensitivity by PTP1B, and promote the potential application of molecular targets toward the management of insulin resistance in obesity and diabetes.
Conclusion
It is intriguing that chronic inflammation caused by obesity activates JNK and its related pathways, leading to impair glucose and lipid metabolism, block insulin signaling, and lower insulin sensitivity of insulin target cells. It is one of the important causes of T2D. In recent years, people have made great progress in the research of the JNK signaling pathway. A large number of JNK substrates and regulatory molecules have been discovered, which provides new ideas for the further study, such as the role of JNK signaling pathways and others crossed with it in obesity-driven insulin resistance. At the same time, we should recognize that JNK can be regulated by many factors and it is a conserved and extremely complex signaling pathway. Explaining these problems as soon as possible will help people understand JNK signaling pathway, provide important drug targets for the treatment and improvement of obesity-driven insulin resistance, and accelerate the basic research results of the JNK signaling pathway to clinical application.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No 81373314, 81741130), the Guangdong Basic and Applied Basic Research Foundation (No 2019A1515011866), Guangzhou Municipal Science and Technology Program (No 201707010245, 201704020117), the Cooperation project of Guangzhou Jinan Meisu Biotechnology Co., Ltd and Jinan University (Grant Nos/GZJDA2019-02, GZJDA2020-01), and Key Scientific Research Projects of Regular Universities of Guangdong Province in 2019 (Grant No. 2019KZDXM011).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chen L, Magliano DJ, Zimmet PZ. The worldwide epidemiology of type 2 diabetes mellitus–present and future perspectives. Nat Rev Endocrinol. 2011;8(4):228–236. doi:10.1038/nrendo.2011.183
2. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115(5):1111–1119. doi:10.1172/JCI25102
3. Lee BC, Lee J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim Biophys Acta. 2014;1842(3):446–462. doi:10.1016/j.bbadis.2013.05.017
4. Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. doi:10.1038/nature01137
5. Seki E, Brenner DA, Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143(2):307–320. doi:10.1053/j.gastro.2012.06.004
6. Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Semin Immunol. 2014;26(3):237–245. doi:10.1016/j.smim.2014.02.009
7. Evans JL, Goldfine ID, Maddux BA, et al. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. 2002;23(5):599–622. doi:10.1210/er.2001-0039
8. Xu H, Xiong C, He L, et al. Trans-resveratrol attenuates high fatty acid-induced P2X7 receptor expression and IL-6 release in PC12 cells: possible role of P38 MAPK pathway. Inflammation. 2015;38(1):327–337. doi:10.1007/s10753-014-0036-6
9. Trop-Steinberg S, Azar Y. AP-1 expression and its clinical relevance in immune disorders and cancer. Am J Med Sci. 2017;353(5):474–483. doi:10.1016/j.amjms.2017.01.019
10. Bumrungpert A, Kalpravidh RW, Chitchumroonchokchai C, et al. Xanthones from mangosteen prevent lipopolysaccharide-mediated inflammation and insulin resistance in primary cultures of human adipocytes. J Nutr. 2009;139(6):1185–1191. doi:10.3945/jn.109.106617
11. Hotamisligil GS, Davis RJ. Cell signaling and stress responses. Cold Spring Harb Perspect Biol. 2016;8(10):a006072. doi:10.1101/cshperspect.a006072
12. Bennett BL, Satoh Y, Lewis AJ. JNK: a new therapeutic target for diabetes. Curr Opin Pharmacol. 2003;3(4):420–425. doi:10.1016/S1471-4892(03)00068-7
13. Nie J, Chang Y, Li Y, et al. Caffeic acid phenethyl ester (Propolis extract) ameliorates insulin resistance by inhibiting JNK and NF-kappaB inflammatory pathways in diabetic mice and HepG2 cell models. J Agric Food Chem. 2017;65(41):9041–9053. doi:10.1021/acs.jafc.7b02880
14. Boden G, Duan X, Homko C, et al. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes. 2008;57(9):2438–2444. doi:10.2337/db08-0604
15. Carvalho BM, Oliveira AG, Ueno M, et al. Modulation of double-stranded RNA-activated protein kinase in insulin sensitive tissues of obese humans. Obesity (Silver Spring, Md). 2013;21(12):2452–2457. doi:10.1002/oby.20410
16. Yang R, Wilcox DM, Haasch DL, et al. Liver-specific knockdown of JNK1 up-regulates proliferator-activated receptor gamma coactivator 1 beta and increases plasma triglyceride despite reduced glucose and insulin levels in diet-induced obese mice. J Biol Chem. 2007;282(31):22765–22774. doi:10.1074/jbc.M700790200
17. Prada PO, Hirabara SM, Souza CT, et al. L-glutamine supplementation induces insulin resistance in adipose tissue and improves insulin signalling in liver and muscle of rats with diet-induced obesity. Diabetologia. 2007;50(9):1949–1959. doi:10.1007/s00125-007-0723-z
18. Lee YH, Giraud J, Davis RJ, et al. c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J Biol Chem. 2003;278(5):2896–2902. doi:10.1074/jbc.M208359200
19. Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440(7086):944–948. doi:10.1038/nature04634
20. Randle PJ, Garland PB, Hales CN, et al. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet (London, England). 1963;1(7285):785–789. doi:10.1016/S0140-6736(63)91500-9
21. Aguirre V, Uchida T, Yenush L, et al. The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem. 2000;275(12):9047–9054. doi:10.1074/jbc.275.12.9047
22. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840–846. doi:10.1038/nature05482
23. Solinas G, Becattini B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol Metab. 2017;6(2):174–184. doi:10.1016/j.molmet.2016.12.001
24. Han MS, Jung DY, Morel C, et al. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science. 2013;339(6116):218–222. doi:10.1126/science.1227568
25. Shen J, Yang T, Xu Y, et al. Delta-tocotrienol, isolated from rice bran, exerts an anti-inflammatory effect via MAPKs and PPARs signaling pathways in lipopolysaccharide-stimulated macrophages. Int J Mol Sci. 2018;19(10):3022. doi:10.3390/ijms19103022
26. Boughanem H, Cabrera-Mulero A, Millan-Gomez M, et al. Transcriptional analysis of FOXO1, C/EBP-alpha and PPAR-gamma 2 genes and their association with obesity-related insulin resistance. Genes. 2019;10(9):706. doi:10.3390/genes10090706
27. Zhang B, Shu M, Xu C, et al. Virtual screening, docking, synthesis and bioactivity evaluation of thiazolidinediones as potential PPAR gamma partial agonists for preparation of antidiabetic agents. Lett Drug Des Discov. 2019;16(6):608–617. doi:10.2174/1570180815666180827123512
28. Blaschke F, Takata Y, Caglayan E, et al. Obesity, peroxisome proliferator-activated receptor, and atherosclerosis in type 2 diabetes. Arterioscler Thromb Vasc Biol. 2006;26(1):28–40. doi:10.1161/01.ATV.0000191663.12164.77
29. Kumar S, Sinha K, Sharma R, et al. Phloretin and phloridzin improve insulin sensitivity and enhance glucose uptake by subverting PPAR gamma/Cdk5 interaction in differentiated adipocytes. Exp Cell Res. 2019;383(1):111480. doi:10.1016/j.yexcr.2019.06.025
30. Camp HS, Tafuri SR. Regulation of peroxisome proliferator-activated receptor gamma activity by mitogen-activated protein kinase. J Biol Chem. 1997;272(16):10811–10816. doi:10.1074/jbc.272.16.10811
31. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–445.
32. Hosooka T, Noguchi T, Kotani K, et al. Dok1 mediates high-fat diet-induced adipocyte hypertrophy and obesity through modulation of PPAR-gamma phosphorylation. Nat Med. 2008;14(2):188–193. doi:10.1038/nm1706
33. Ye JP. Regulation of PPAR gamma function by TNF-alpha. Biochem Biophys Res Commun. 2008;374(3):405–408. doi:10.1016/j.bbrc.2008.07.068
34. Sun Z, Miller RA, Patel RT, et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat Med. 2012;18(6):
35. Ruan H, Pownall HJ, Lodish HF. Troglitazone antagonizes tumor necrosis factor-alpha-induced reprogramming of adipocyte gene expression by inhibiting the transcriptional regulatory functions of NF-kappa B. J Biol Chem. 2003;278(30):28181–28192. doi:10.1074/jbc.M303141200
36. Trujillo ME, Scherer PE. Adiponectin - journey from an adipocyte secretory protein to biomarker of the metabolic syndrome. J Intern Med. 2005;257(2):167–175. doi:10.1111/j.1365-2796.2004.01426.x
37. Gavrilova O, Haluzik M, Matsusue K, et al. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem. 2003;278(36):34268–34276. doi:10.1074/jbc.M300043200
38. Olefsky JM. Treatment of insulin resistance with peroxisome proliferator-activated receptor gamma agonists. J Clin Invest. 2000;106(4):467–472. doi:10.1172/JCI10843
39. Poulsen L, Siersbaek M, Mandrup S. PPARs: fatty acid sensors controlling metabolism. Semin Cell Dev Biol. 2012;23(6):631–639. doi:10.1016/j.semcdb.2012.01.003
40. Li P-Y, Hsu -C-C, Yin M-C, et al. Protective effects of red guava on inflammation and oxidative stress in streptozotocin-induced diabetic mice. Molecules. 2015;20(12):22341–22350. doi:10.3390/molecules201219831
41. Mangali S, Bhat A, Udumula MP, et al. Inhibition of protein kinase R protects against palmitic acid-induced inflammation, oxidative stress, and apoptosis through the JNK/NF-kB/NLRP3 pathway in cultured H9C2 cardiomyocytes. J Cell Biochem. 2019;120(3):3651–3663. doi:10.1002/jcb.27643
42. Ruelas Cinco EDC, Ruiz Madrigal B, Dominguez Rosales JA, et al. Expression of the receptor of advanced glycation end-products (RAGE) and membranal location in peripheral blood mononuclear cells (PBMC) in obesity and insulin resistance. Iran J Basic Med Sci. 2019;22(6):623–630. doi:10.22038/ijbms.2019.34571.8206
43. Ormazabal P, Scazzocchio B, Vari R, et al. Effect of protocatechuic acid on insulin responsiveness and inflammation in visceral adipose tissue from obese individuals: possible role for PTP1B. Int J Obes. 2018;42(12):2012–2021. doi:10.1038/s41366-018-0075-4
44. Yang R, Trevillyan JM. c-Jun N-terminal kinase pathways in diabetes. Int J Biochem Cell Biol. 2008;40(12):2702–2706. doi:10.1016/j.biocel.2008.06.012
45. Romeo G, Liu WL, Asnaghi V, et al. Activation of nuclear factor-kappa B induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes. 2002;51(7):2241–2248. doi:10.2337/diabetes.51.7.2241
46. Gadang V, Gilbert W, Hettiararchchy N, et al. Dietary bitter melon seed increases peroxisome proliferator-activated receptor-gamma gene expression in adipose tissue, down-regulates the nuclear factor-kappa B expression, and alleviates the symptoms associated with metabolic syndrome. J Med Food. 2011;14(1–2):86–93. doi:10.1089/jmf.2010.0010
47. Kiechl S, Wittmann J, Giaccari A, et al. Blockade of receptor activator of nuclear factor-kappaB (RANKL) signaling improves hepatic insulin resistance and prevents development of diabetes mellitus. Nat Med. 2013;19(3):358–363. doi:10.1038/nm.3084
48. Dodd GT, Xirouchaki CE, Eramo M, et al. Intranasal targeting of hypothalamic PTP1B and TCPTP reinstates leptin and insulin sensitivity and promotes weight loss in obesity. Cell Rep. 2019;28(11):2905–22 e5. doi:10.1016/j.celrep.2019.08.019
49. MohammadTaghvaei N, Taheripak G, Taghikhani M, et al. Palmitate-induced PTP1B expression is mediated by ceramide-JNK and nuclear factor kappaB (NF-kappaB) activation. Cell Signal. 2012;24(10):1964–1970. doi:10.1016/j.cellsig.2012.04.019
50. Taheripak G, Bakhtiyari S, Rajabibazl M, et al. Protein tyrosine phosphatase 1B inhibition ameliorates palmitate-induced mitochondrial dysfunction and apoptosis in skeletal muscle cells. Free Radic Biol Med. 2013;65:1435–1446.
51. Elchebly M, Payette P, Michaliszyn E, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science (New York, NY). 1999;283(5407):1544–1548. doi:10.1126/science.283.5407.1544
52. Straczkowski M, Kowalska I, Nikolajuk A, et al. Relationship between insulin sensitivity and sphingomyelin signaling pathway in human skeletal muscle. Diabetes. 2004;53(5):1215–1221. doi:10.2337/diabetes.53.5.1215
53. Peraldi P, Hotamisligil GS, Buurman WA, et al. Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. J Biol Chem. 1996;271(22):13018–13022. doi:10.1074/jbc.271.22.13018
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.