")
Back to Journals » ImmunoTargets and Therapy » Volume 13
The Role of Extracorporeal Photopheresis in the Management of Graft Versus Host Disease: Narrative Review
Authors Berhan A , Damtie S , Almaw A , Legesse B , Sharew B , Getie B , Erkihun M , Solomon Y
Received 1 February 2024
Accepted for publication 24 April 2024
Published 26 April 2024 Volume 2024:13 Pages 235—246
DOI https://doi.org/10.2147/ITT.S457366
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Michael Shurin
Ayenew Berhan, Shewaneh Damtie, Andargachew Almaw, Biruk Legesse, Bekele Sharew, Birhanu Getie, Mulat Erkihun, Yenealem Solomon
Department of Medical Laboratory Science, College of Health Science, Debre Tabor University, Debre Tabor, Ethiopia
Correspondence: Ayenew Berhan, Tel +251910613151, Email [email protected]
Abstract: Hematopoietic stem cell donation is a method used to treat both blood-related and non-blood-related malignancies. Graft-versus-host disease is a potentially life-threatening complication that can occur following a stem cell transplant from a donor. This happens after the transplanted grafts attack the recipient’s body as foreign cells, causing significant morbidity and mortality. Clinically, this condition can be classified as acute or chronic based on onset and pathophysiology. This review aims to provide an overview of recent studies on extracorporeal photopheresis as a treatment strategy option for graft-versus-host-diseased patients. It will explain how it treats graft-versus-host disease, summarize its promising effects, and provide future recommendations for its use in treating this illness. Extracorporeal photopheresis is used to treat graft-versus-host disease by collecting and separating white blood cells from the patient. This blood is fractionated into different parts, and white blood cells undergo treatment with 8-methoxy psoralen, a photoactivable drug, before exposure to ultraviolet light A. Lastly, the cells that have been treated are reinfused into the recipient’s body. It prompts the programmed cell death of lymphocytes and the engulfment of cellular debris by host antigen-presenting, leading to a subsequent rise in T regulatory cells. However, more experimental and randomized controlled studies are required to identify the best patient selection requirements, environments, and treatment regimens for graft-versus-host disease.
Keywords: graft versus host disease, hematopoietic stem cell transplantation, 8-methoxy psoralen, extracorporeal photopheresis
Introduction
Hematopoietic stem cell transplantation (HSCT) is a high-intensity therapeutic procedure for a variety of hematological and non-hematological malignancies.1–4 Currently, the sole curative option for several hematologic disorders is allogeneic HSCT.5,6 It is a blood product, bone marrow, and solid organ transplantation process that uses a half-matched donor’s human leukocyte antigen (HLA), a new approach to HLA-haploidentical blood or marrow donation.2 Over the last two decades, improved transplant techniques and supportive care have resulted in a global increase in the use of allogeneic HSCT.2,5,7 However, the scarcity of suitable donors limits its widespread use; and when allogeneic HSCT is used, the morbidity and mortality related to transplantation remain persistently high. Therefore, increased graft failure and the likelihood of grave and refractory graft-versus-host disease (GVHD) are the two drawbacks of allogeneic transplantation.1,2,7
The most common and serious complication that occurs after undergoing allogeneic HSCT is GVHD, limiting its widespread application.2,8 Immune suppressants can control the immune response and keep successfully engrafted recipients free of active GVHD. However, the recipient often has delayed immune reconstitution and remains susceptible to fatal infections. In addition, suppressing the host immune response can increase the risk of leukemic relapse.3,9
Patients with hematologic malignancies can benefit greatly from allogeneic HSCT because it replaces the recipient’s marrow with the donor’s, which contains immunocompetent cells capable of eliminating cancer cells. This process is called graft-versus leukemia (GVL) or graft-versus-tumor (GVT). Moreover, the cytotoxic properties of the conditioning regimen used in the procedure can help to prevent GVHD.4,7,8,10
Conversely, cells derived from the donor might perceive recipient organs as foreign and initiate an immune assault against the patient’s tissues.4
When the transplanted cells perceive the host’s body as foreign and mount an immune response against it, the recipient’s body may experience GVHD syndrome.2,10,11 Billingham (1966) originally characterized GVHD as a syndrome in which T cells from the donor target and attack the tissues of an immunocompromised recipient.7 GVHD affects various organ systems, encompassing the gastrointestinal tract, skin, mucosa, liver, and lungs.2,7 It is a prevalent complication following HSCT that hurts both overall survival and quality of life.12 It affects 40–60% of patients who undergo HSCT, contributing to 15–20% of deaths occurring after such transplantation.2,6,9,13
The positive effects of GVT and GVHD are both perplexing. Both are triggered by mature donor T cells in the injected graft. However, T-cell depletion (known as T-cell-depleted transplants) can diminish their occurrence.14 Improved HSCT management has increased engraftment rates and quality of life. However, GVHD remains a prominent issue,15 limiting the efficacy of allogeneic HSCT.1,10
Without any preventive measures, almost all patients will develop GVHD.10 Regardless of up-to-date preventive techniques, death and morbidity from GVHD following allogeneic HSCT are still high. Managing the development of GVHD can be challenging, with only approximately forty percent of those treated with corticosteroids responding to the therapy.11 The prevention of GVHD occurrence in allogeneic HSCT patients is crucial for improving their survival and recovery rates. Thus, this study strives to present the results of the most recent studies on extracorporeal photopheresis (ECP), a pivotal component in the treatment plans for GVHD management. ECP is a form of leukapheresis-based procedure used for the treatment of GVHD where the patient’s mononuclear cells are reintroduced into the body following treatment with 8-methoxy psoralen (8-MOP) and exposure to ultraviolet A (UVA) light. The review also seeks to elucidate the mechanism of ECP in treating GVHD, summarize its promising effects, and provide recommendations for future research.
Pathophysiology and Classification of GVHD
Achieving better clinical outcomes from allogeneic HSCT and the results of more effective GVHD treatments require a complete understanding of the disease’s biology. This will lead to leveraging the role of allogeneic HSCT in preserving the GVL effect after transplantation.8 In 1956, Barnes and Loutit discovered GVHD in a mouse model by exposing them to radiation and then injecting them with allogenic bone marrow and spleen cells. The mice initially showed recovery from both radiation injuries and aplasia. However, over time, they manifested symptoms such as diarrhea, weight loss, skin changes, and liver abnormalities, ultimately leading to secondary diseases and death- a condition known as GVHD.15,16
Billingham (1966) identified three critical pre-requirements for the event of GVHD.9 First, the graft must have immunologically competent cells, typically including T cells. Second, the patient’s tissue must contain antigens that are absent in the graft. Therefore, the host appears foreign to the graft, thereby eliciting antigenic stimulation. Finally, the recipient’s immune system must be unable to eliminate the transplanted cells. GVHD can arise when tissues containing T-cells, such as blood, bone marrow, and solid organs, are transferred from one individual to another who is unable to eliminate these cells. Individuals with impaired immune systems are especially vulnerable to GVHD if they receive a graft containing white blood cells from another person.15,17
GVHD Classically Develops Through Five Steps
Immune System Preparation
During the initial stage of acute GVHD produced by conditioning chemotherapy, damaged host tissues release tumor necrosis factor (TNF) and interleukin (IL)-1, contributing to a “cytokine storm”. This triggers an enhanced release of adhesion molecules, costimulatory molecules, major histocompatibility complex (MHC) antigens, and chemokine gradients. This generates the “red alert” in host tissue cells, particularly in the antigen-presenting cells (APC). In this stage, conditioning chemotherapy also harms the gastrointestinal tract, allowing for the systemic translocation of lipopolysaccharide and direct activation of APC. As a result, some human randomized trials have linked intensive conditioning regimens to a higher risk of GVHD.4
T Cell Activation and Co-Stimulation
This phase constitutes a crucial component of the immune response in graft-versus-host interactions. When T cells from the donor come in contact with the APCs, they start to proliferate and mature into different types of cells. A group of self-renewing clusters of differentiation (CD) 44lo/CD62Lhi/CD8+ T cells, which are post-mitotic, are in charge of producing and maintaining all allogeneic T-cell subsets during GVHD reactions. This activation is further intensified by the “red alert” induced in the initial phase. The level of mismatch between HLAs is closely linked with the incidence of acute GVHD. Although HLA-identical grafts can be used for transplantation, the recipient might continue to experience acute GVHD because of genetic variations outside the MHC loci responsible for encoding minor histocompatibility antigens. The interaction between CD28/cytotoxic T-lymphocyte antigen 4 (CD152) and B7 molecules can initiate GVHD through a stimulatory and an inhibitory pathway. Inducible costimulatory (ICOS; CD278), another member of the B7 supergene family, binds to the ligand B7h (CD275) articulated on patient APCs. This leads to the enhancement of T effector responses. Reducing ICOS on the T cell of the donor can help reduce GVHD in the gut and liver. Likewise, TNF receptor family members act as costimulatory molecules that control GVHD. Once T cells become activated and cause tissue damage, the body responds by activating inhibitory pathways to prevent further harm. The immune response is suppressed by two proteins, Cytotoxic T-lymphocyte-associated protein-4 and programmed death-1 (CD279), which are typically present in the CD4+CD25+ regulatory T cells and cytoplasm of activated T cells. Along with such markers, interferon (IFN) activates the tryptophan catabolic pathway known as indole amine 2, 3-dioxygenase in targeted organs. This pathway reduces damage to T-effector cells through local mechanisms, leading to increased T-cell death and reduced expansion.4
Allo Reactive T Cell Expansion and Differentiation
GVHD is a medical illness that arises when various subsets of T lymphocytes attack the recipient’s organs. Studies on animals have revealed that T cells proliferate rapidly in lymph nodes and Peyer patches. Research has shown that regulatory T cells identified by the antigens CD4, CD25, and Foxp3 possess suppressor activities in both in vitro and in vivo investigations. In animal models, GVHD can occur after HSCT when CD25+ T cells are removed from the donor or the recipient. However, it still maintains the GVL result. Besides, immunosuppressive drugs can affect the growth and activity of regulatory T cells. Calcineurin inhibitors like cyclosporine reduce the secretion of IL-2, which causes a decline in regulatory T cells. However, in ex vivo culture systems, rapamycin has been shown to either preserve or increase the population of murine and human regulatory T cells in terms of total cell yield. The natural killer T (NKT) cell is an immune cell type that shares surface characteristics of both NK and T cells. Studies have shown that NKT cells can inhibit GVHD. Recent research in humans undergoing HSCTs has indicated that a higher proportion of donor NKT cells in a graft with a higher amount of mobilized T cells can minimize the frequency of GVHD. 17 helper T (Th17) cells, recently discovered, are associated with GVHD. This subset of the T-cell has been observed in animal models of GVHD on inflammatory bowel disease, lung disease, and skin.4
Activated T-Cell Trafficking
The process of T-cell migration towards secondary lymphoid organs during GVHD and other inflammatory responses is widely recognized. Fortunately, the passage of leukocytes toward parenchymal organs is not completely comprehended. In addition, changes in vascular permeability have been described, and the specific interactions required for this process among selectin-ligand, chemokine receptor, and integrin-ligand are not yet fully understood. In the course of the GVHD reaction, the donor T cells first travel to the peripheral lymphoid tissue and spleen. These tissues are crowded with naive donor T cells. Meanwhile, APCs send activation signals to allo-reactive T cells, which move towards a particular target organ where GVHD occurs. Although transplant antigens are expressed in all tissues of the recipient, acute GVHD is limited to specific organs, such as secondary lymphoid organs, the skin, the gastrointestinal system, the thymus, and the liver. The onset of acute colitis and the entry of CD4+ donor T cells into mesenteric nodes, resulting in GVHD of the gut, requires the expression of both CD62L and beta7 integrins. Donor T cells that cause GVHD infiltrate specific organs by binding to corresponding receptors on host tissues and opposing receptors articulated on donor T cells, with the assistance provided by members of the chemokine family, such as macrophage inflammatory protein 1a and other chemokines like CCL2, CCL5, CXCL2, and so on.4
Destruction of the Target Tissues by Effector T Cells
Effector T cells initiate tissue destruction by recruiting leukocytes to GVHD target tissues and exerting cytotoxic activity. GVHD is a Th1/T cytotoxic-type disease due to the numerousness of cytotoxic T cell-mediated disease and amplified expression of Th1 cytokines, such as IL-12, IL-2, and IFN-γ. For the occurrence of GVHD, timing and cytokine amounts released into target organs are critical. IL-10 induces a Th2 and Type 1 regulatory T cell response, promoting allograft resilience. T cells exhibit the effects of GVHD via multiple pathways. Fas and Fas ligand expression boosts on both CD8+ and CD4+ donor T cells. The perforin-granzyme route has essential significance in GVHD pathogenesis. TNF-α gene polymorphisms in transplant recipients contribute to an elevated risk of GVHD due to increased levels of TNF-α production.4
Clinically, GVHD comprises two syndromes: acute and chronic.1 These syndromes affect various organs and tissues, resulting in different signs and symptoms. Post-allogenic HSCT patients may experience either one or both types of GVHD, or they may not experience either of these conditions. Formerly, the classification of GVHD was primarily determined by the timing of its occurrence. GVHD developed within 100 days after HSCT was known as acute, while GVHD occurred after 100 days known as chronic. However, new research implies that these two entities of GVHD are two separate pathophysiological disorders.10,18
Acute GVHD
Acute GVHD is a severe medical condition that arises when transplanted donor T cells rapidly target and harm the patient’s skin, liver, and gastrointestinal tract relatively soon after transplantation. The patients develop a skin rash, diarrhea, jaundice, and weight loss. Unfortunately, many patients with severe acute GVHD do not survive.1,3 The frequency of acute GVHD varies from 26% to 32% in individuals receiving grafts from sibling donors and from 42% to 52% in those receiving grafts from unrelated donors. Depending on the degree (stage) of involvement of the liver, skin, gastrointestinal tract, or liver, acute GVHD can be graded as I (mild), II (moderate), III (severe), and IV (very severe), representing its overall severity.8 Overall, nearly 30% to 50% of individuals undergoing transplantation encounter acute GVHD.19,20 Acute GVHD frequency following allogeneic HSCT varies based on factors such as the level of HLA difference, the cutting-edge age of the recipient or donor, the advanced malignant condition at transplantation, the donor hematopoietic cell source, and other factors.8
Staging systems have been verified and proven effective in predicting mortality. The pathophysiology of acute GVHD involves an intricate series of immune events that can be categorized into distinct stages. Firstly, as the remaining host cells, capable of presenting antigens, become activated, they engage with donor T cells, producing inflammatory cytokines. This further attracts and stimulates donor cells, leading to direct cytotoxicity against the recipient tissue. This process is enhanced by IL-1, lipopolysaccharide, and TNF-a mediators. Eventually, this leads to the destruction of tissue and the clinical manifestation of acute GVHD.8,21 Although it commonly manifests within the initial 100 days following transplantation, patients undergoing non-myeloablative conditioning or exhibiting mixed-donor chimerism may experience its onset at a later stage.10,21
Chronic GVHD
The biggest challenges to improving outcomes after allogeneic HSCT are the return of the underlying malignancy and the occurrence of GVHD.10 Chronic GVHD is a severe and common complication that occurs after HSCT. It is a major cause of long-term illness and death in these patients.22,23 Chronic GVHD may develop in up to 50% of patients, and in moderate to severe cases, systemic immunosuppressive therapy is necessary for individuals who are undergoing allogeneic HSCT.1,22,24
Individuals who have survived allogeneic HSCT for more than two years face the risk of non-relapse-related illness and death, with chronic being the predominant cause. Even as procedures for transplantation improve, there is a potential increase in the occurrence of chronic GVHD. Several risk variables have been identified through research for chronic GVHD, including the use of peripheral blood grafts, transplants from donors other than matched siblings, and older recipients.23,25
Chronic GVHD pathogenesis is quite intricate, reflecting its variability in clinical appearance. Its pathogenesis is primarily based on allo-reactive T-cells, dysregulated interactions with B-cells, and the involvement of innate immune effectors like macrophages, dendritic cells (DC), and neutrophils.25 Chronic GVHD can induce several symptoms affecting several body systems, including the mucocutaneous, myofascial, pulmonary, and other systems. Chronic inflammation can lead to various issues affecting the eyes, mouth, esophagus, skin, joints, fascia, and genital tissues. Fibrosis, an accumulation of extra fibrous tissue, can affect several organs in seriously affected patients, eventually leading to clinically significant cases. While uncommon, grave clinical manifestations of chronic GVHD may comprise polyserositis, characterized by pleural and pericardial effusions, and polymyositis, characterized by serious weaknesses in muscles and raised levels of muscle enzymes.24,25
The mechanisms of chronic GVHD partially overlap with those of acute GVHD, especially in cases where chronic GVHD develops from pre-existing acute GVHD. Allo-reactive T-cells and deregulated B-cells primarily drive chronic GVHD pathogenesis. Additionally, macrophages, DC, and neutrophils, which are innate immunity effectors, also contributed to the development of chronic GVHD.9,25
Managements of GVHD
The occurrence of moderate or severe GVHD can lead to critical illness and reduced survival rates. Unfortunately, it is challenging to treat once it has occurred. Therefore, it is essential to take preventative measures before and after transplantation to decrease the likelihood and severity of GVHD. Every precaution should be taken to avoid GVHD altogether. To prevent GVHD, donor T-cell function must be suppressed with immunosuppressive drugs. These medications are given before and after the HSCT. Because there is no standard GVHD prophylaxis regimen, diverse drug combinations are employed at various institutions. Some commonly used medications for preventing GVHD include Methotrexate (Trexall®), Cyclosporine, Tacrolimus (Prograf®), Mycophenolate Mofetil (CellCept®), Sirolimus (Rapamune®), Corticosteroids (methylprednisolone or prednisone), Antithymocyte Globulin (ATG), Alemtuzumab (Campath®), and Cyclophosphamide (Cytoxan®). The main approach to managing both acute and chronic GVHD involves suppressing the immune system with corticosteroids. Managing GVHD using corticosteroids is a challenging task because this approach yields favorable outcomes in less than half of acute GVHD patients and approximately 40–50% of chronic GVHD patients. The treatment’s success is greatly affected based on the severity of the situation at the time of onset.26
When immunosuppressants are used to suppress the immune response, patients who have successfully received a transplant and are free of active GVHD may experience delayed immune reconstitution and be in danger of fatal infections. Additionally, suppressing the recipient’s immune response may increase the chances of leukemia relapse.3
Despite numerous advances in HSCT, the prevention and management of GVHD still face challenges. Many patients may not respond to standard first-line drugs, resulting in a high mortality rate, particularly when first-line steroid treatment fails. Although numerous next-line therapies are available, they lack more standardization. They are associated with various complications, such as severe suppression of immunity, harm to end-organs, an increased risk of secondary disease, and metabolic disorders. An approach that regulates the immune system without directly reducing its function is promising for preventing GVHD. Regardless of GVHD preventive measures, new strategies targeting other pathways involved in GVHD growth are strongly advised. An example of such an approach is ECP, which has demonstrated effects on regulatory T cells and dendritic cells (DCs). It is widely accessible and utilized for various medical conditions. It is the sole form of photopheresis therapy approved by the Food and Drug Administration (FDA) for cancer treatment. Its application has broadened to encompass other T-cell-mediated diseases, including GVHD and rejection following solid-organ transplantation. Even with its extensive utilization, the optimal function of ECP in the context of GVHD requires a clearer definition.21
Role of Photopheresis in the Management of GVHD
Overview About Photopheresis
ECP is thought to initiate immunomodulatory effects, which help mitigate the immune response responsible for GVHD, all without the need for traditional medications. The process involves separating plasma containing white blood cells, then treating them with 8-MOP and exposing them to UVA light irradiation. The treated blood is then returned to the patient.21,27–29, the treatment is believed to work by triggering apoptosis in the patient’s lymphocytes and modulating the immune response. This can help improve the patient’s condition through the elevation of regulatory T lymphocytes (regulatory T cells, CD4+, CD25+, and FoxP3+), along with the secretion of IL-10 from B lymphocytes and of anti-inflammatory cytokines (eg, IL-10 and TGF-β). These components are primarily responsible for controlling immune responses. Due to these considerations, ECP is regarded as an immunomodulatory treatment rather than an immunosuppressive therapy.30–32 The therapy was developed as an alternative for those individuals who showed unresponsiveness to their primary treatment options. It is a leukapheresis-based therapy. Originally employed to treat therapy-refractory cutaneous T cell lymphoma/leukemia (CTCL), specifically Sezary syndrome (a form of CTCL). Dr. Richard Edelson developed the procedure in 1987, and it was approved for palliative care of skin manifestations in CTCL patients in the late 1980s. Edelson et al found a response rate exceeding 70%.31,33
This procedure has been used for over three decades to treat erythrodermic CTCL and for over two decades to treat chronic and acute GVHD, as well as solid organ transplant rejection and some other kinds of autoimmune disorders, like scleroderma, lupus erythematosus, Crohn disease, and type 1 diabetes. The ECP procedure comprises three stages:
- leukapheresis,
- Photo activation with 8-MOP/UVA and
- Re-infusion of buffy coat.
This process leads to the cross-linking of pyrimidine bases in Deoxy Ribonucleic Acid (DNA), resulting in significant mortality of the treated (patient) cells.1,14,24
In ECP therapy, the patient’s entire blood is subjected to leukapheresis. Then a leukocyte-enriched buffy coat is generated through centrifugation. Typically, 3–6 cycles of apheresis are required to remove enough whole blood to yield around 240 mL of buffy coat. Subsequently, the buffy coat layer is infused with a concentration of 20 µg/mL of soluble 8-MOP (Uvadex®), photosensitizer agent. It is then exposed to UVA light for 25–45 minutes (1.5 J/cm2) and re-infused into the patient.34 It is supposed that the ECP causes lymphocyte apoptosis and inhibits the production of pro-inflammatory cytokines. This leads to a decrease in the motivation of effector T cells, variations in DC roles, and stimulation of regulatory T cells, ultimately leading to the energy gain of T41 cells. It is frequently employed as a second-line treatment for mucocutaneous chronic GVHD. This is due to its efficacy exceeding 80% and substantial enhancement in chronic GVHD with sclerosis.13,24,32
Each procedure targets 5% to 10% of circulating blood cells. Several patient variables must be considered when considering ECP treatment. These include the patient’s body weight, allowable extracorporeal volume, the device employed for cell collection, clinical condition during the procedure, various laboratory values such as platelet counts, bilirubin levels, total white blood cells, hematocrit, and potential requirements for central venous access. It is critical to consider these variables to determine the most appropriate treatment plan for each patient.21
Closed (inline) and open (offline) techniques are currently available as conventional techniques for ECP treatment.35,36 Closed ECP system (“one-step” process): This technique involves the process of separating cells, activating drugs through photoactivation, and re-infusing the treated cells within the fully automated and integrated device using a single instrument. The components used in this process have been tested and validated together for use with 8-MOP. When used as directed, there is no danger of an incorrect return of blood. Additionally, the likelihood of infection and contamination linked to the medical device is minimal.33,37 The procedure can be conducted using the Therakos CELLEX Photopheresis System (Mallinckrodt Pharmaceuticals in New Jersey, USA). Using centrifugal force, the device differentiates and collects a buffy coat rich in monocytes and lymphocytes before returning the remaining plasma and red cells to the patient. The buffy coat remains within the system, undergoing treatment with 8-MOP, followed by exposure to UVA light. The instrument determines the photoactivation time, taking into account factors such as volume, hematocrit of the lymphomonocytic fraction, and the remaining power of the UVA lamps.38
The open ECP system (two-step method), which uses distinct equipment for separating cells and drug light activation, is also referred to as the “two-step” method. Distinct devices are employed for each step in this system, and they have not undergone validation as a combined unit. Therefore, closed systems are the preferred treatment technique.33,37 The offline method demands several devices and comprises three distinct phases. The initial stage involves the collection of mononuclear cells using various separation systems. In the second stage, these cells undergo treatment with 8-MOP, and the resulting buffy coat is transferred to a specialized bag permeable to UVA. Finally, in the third stage, the irradiated mononuclear cells are reintroduced into the patient.39 The closed system, the only FDA-approved system, adheres to the initial design developed by Edelson et al. In contrast, the open system involves the usage of various separation instruments.31 Due to the multifaceted process of administering therapy using the offline method, there exists a risk of infection, cross-contamination, and patient re-infusion error. Therefore, certified centers are limited to using open systems, which require separate handling of blood components. Conversely, closed systems do not face this constraint and are the recommended method for ECP treatment.33
Mechanism Action of ECP
The mechanism underlying ECP’s therapeutic activity is still being studied.19,31,33 However, several theories attempt to explain the impact of ECP on the collected lymphocytes. ECP primarily affects the T-cell section of immunological action.35 This results in an increase in the activity of the immune system. However, in situations following transplantation with GVHD and rejection of solid organ allografts, it suppresses the immune system. Therefore, ECP affects the immune system for several reasons. Firstly, the harvested cells undergo environmental changes that induce several changes in the mononuclear cells. Secondly, combining psoralen treatment and UVA ray exposure alters cellular structure. Lastly, alterations in the function of immune cells and the recipient’s cytokine environment occur after the reinfusion of the treated cells.19,33 APCs subsequently phagocytose circulating apoptotic cells. This results in an increase in regulatory T cells and a decrease in proinflammatory cytokines.29,35
Early studies have shown that ECP can have a therapeutic effect on initiating apoptosis in lymphoid cells. To do this, UVA light exposure (320–400 nm) paired with 8-MOP is used. Despite an essential difference, this concept initially originated from oral psoralen plus UVA therapy. In the ECP procedure, an 8-MOP-incubated buffy coat is exposed to UVA light rather than 8-MOP-photosensitized skin (as in traditional oral plus UVA therapy). After 8-MOP incubation, UVA light irradiation of cells results in DNA crosslinking. Subsequently, primarily NK and T cells undergo apoptosis following reinfusion of the treated cells.31
8-MOP can enter cells passively and intercalate into DNA within just two minutes. 8-MOP causes DNA cross-linking when cells are exposed to UVA light, resulting in disruption in the cell duplication machinery. Conversely, ECP can inhibit cell proliferation, simultaneously triggering the apoptosis of T-cells and the release of immunomodulatory cytokines. It causes the death of T-cells while releasing substances that regulate the immune system. DC, activated by irradiation, transforms into APCs. These APCs can recognize and engulf apoptotic T-cells treated with ECP. This process results in the activation of regulatory T cells. In GVHD, regulatory T-cells suppress immune responses by releasing tolerogenic cytokines. However, in CTCL, ECP-induced regulatory T-cells secrete inflammatory cytokines that stimulate the immune response in neoplastic cells.40 ECP is thought to have two opposing immune effects: in CTCL, immune stimulation targets neoplastic cells, whereas immunosuppression addresses T-cell-mediated disorders like GVHD. It is improbable that the immune modulatory effects of ECP are directly attributed to the apoptosis of lymphocytes. This is because only a slim percentage of circulating leukocytes undergo ECP treatment. In addition, the observed responses have lasted longer than what could be attributed to apoptosis alone. Instead, it is thought that the way these apoptotic cells are processed and the subsequent immunological responses they produce are the clinical benefits of ECP that it provides.34
When the treated cells are reintroduced into the patient, APCs identify the cell surface markers associated with apoptosis, leading to a modification in their response. Apoptotic T-cells engulfed by APCs engage with cells from the identical antigen-specific lineage in the spleen. Regulatory T-cell production contributes to the inhibitory impact on cellular immunity. These regulatory T cells exhibit antigen-specificity, corresponding to the identical clones of cytotoxic T cells found in the product of ECP’s apoptotic cell. Because the majority of the T-cells harvested in the whole blood cell product for ECP come from expanded clones that exist in circulating blood during an active immune response, ECP specifically exerts suppressive effects on cellular immune responses that re-active at the time of its administration.38
Treatment of GVHD Using ECP
ECP has been increasingly used for the control of GVHD.33 According to case reports and small uncontrolled research, patients afflicted with acute GVHD, and showing resistance to traditional immunosuppressive medication were almost entirely treated with ECP.29 Although there is more excellent experience with ECP in chronic GVHD, it is primarily in the form of case reports and small, uncontrolled trials. The preferred timeline for ECP involves two sessions on consecutive days, repeated every two weeks, for a minimum of three months. Patient evaluation is performed in the 3rd month and thereafter, repeated every three months to ascertain the appropriate course for continuing the treatment. The ECP can be decreased to two treatments every three to four weeks after the therapy begins to show results. However, after the treatment has been proven to be effective, the standard procedure is to begin by reducing immunosuppressive agents, particularly steroids. If there is no response in 3 or 4 months, the procedure should be discontinued. Skin abnormalities due to Sclera dermatosis improve extremely slowly; it may take six to twelve months of treatment before tapering is needed.14,21,34 A study done by Malagola et al showed that among 94 GVHD patients undergoing ECP treatment, the overall response rate was greater than 80%.32 In another study, it was revealed that 75% of patients achieved an overall response, with 53% experiencing complete responses following the treatment of 99 GVHD patients.29 In summary, ECP has been approved for treating GVHD due to its lower viral reactivation and risk of infection compared to systemic immunosuppressants.41
ECP has demonstrated efficacy as a treatment for challenging cases of acute GVHD in children, especially those who have not responded to multiple immunosuppressive approaches. This procedure is both safe and well-tolerated, even among very young patients, resulting in significant reductions in the need for daily steroid intake.42 Of the two commonly used ECP instruments, CELLEX® is preferred over UVAR-XTS® due to its near-continuous flow system, which reduces fluid shifts and the occurrence of hypotension, making it suitable for use in small pediatric patients.43,44
Several short studies were conducted to investigate the significance of ECP in GVHD, demonstrating diverse responses in disease outcomes. Table 1 summarizes the studies on the outcomes of ECP in the second-line treatment of GVHD, it is notable that the assessment of responses exhibited variability among the studies.
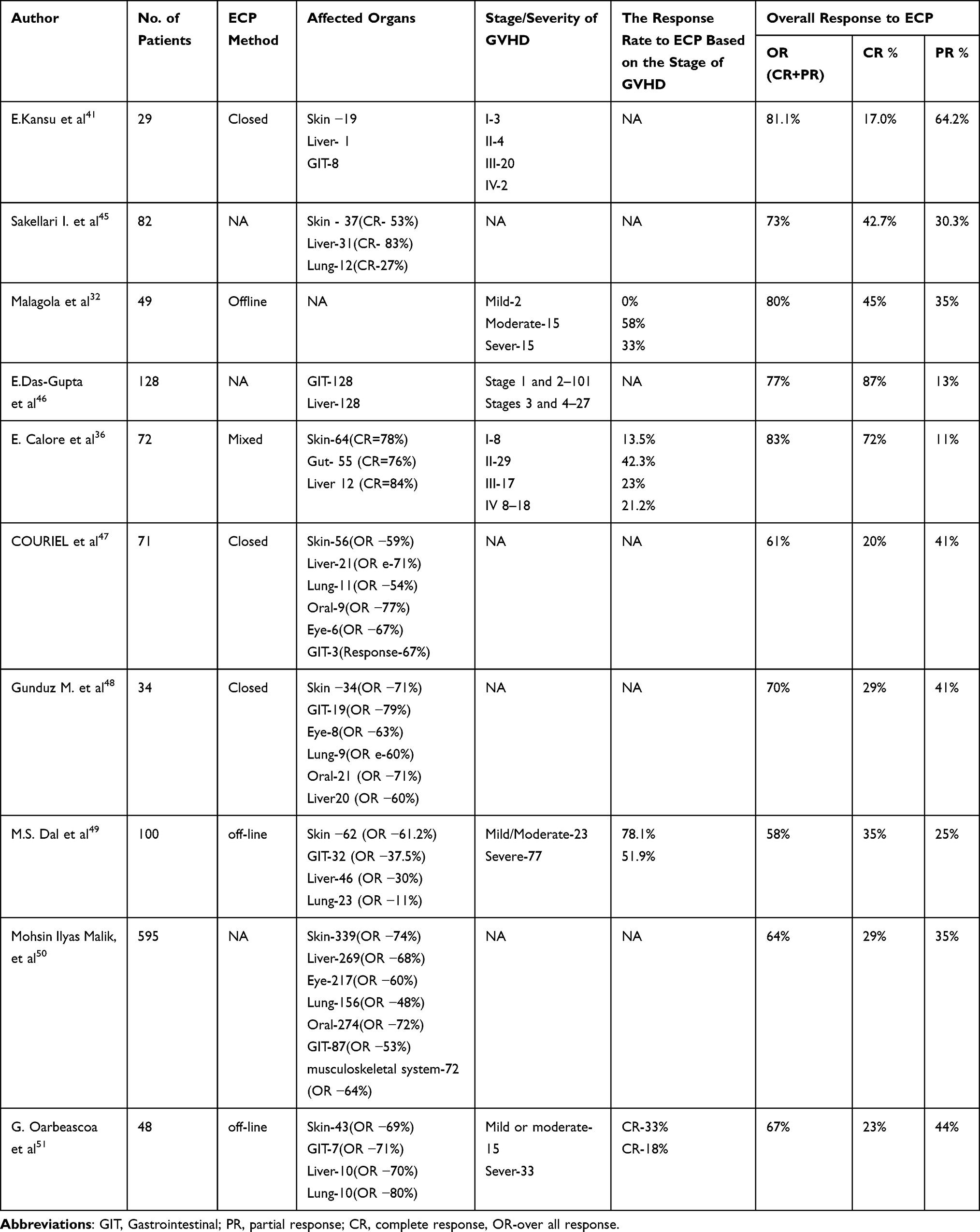 |
Table 1 Result of ECP Treatment for GVHD by Different Authors |
Contraindications of ECP
ECP treatment is strictly prohibited for people who have a documented sensitivity to psoralen compounds and aphakia (the absence of a lens in the eye) due to a significantly increased risk of retinal damage, uncontrolled infections, and pregnancy. In addition, the following conditions are the minimum restriction for administrating ECP: unsteady respiratory or circulatory condition, low white blood cell count (1×109/L); patients who have low platelet count, low hematocrit, risk of bleeding or active bleeding, unsatisfactory cardiovascular function, and low body mass index active infection should be treated with caution.33,52,53 Stable patients may not need to provide additional samples in addition to their current course of medication, even though a recent complete blood count is recommended. Further pretreatment blood tests may be considered individually or as part of a standardized ECP routine. Total blood count; in patients receiving acid citrate dextrose-A, calcium, and potassium; in patients receiving warfarin, international normalization ratio; additional blood tests may be conducted concurrently to assess the overall health status of the patient or to evaluate their GVHD status.52 If there is a hemodynamic disturbance arising from conditions like heart failure, sepsis, or positive blood cultures, the ECP procedure would be canceled.54
Safety of ECP
Given careful patient selection, ECP therapy demonstrates a favorable safety profile, with nearly no notable adverse events reported to date. The primary side effects are generally mild, including heightened photosensitivity due to 8-MOP and patient-related challenges associated with volume shifts and repeated venous punctures during the procedure. No reports have indicated a greater likelihood of disease relapse or infection. However, a cautionary statement has been issued specifically for patients who have had a splenectomy. As a result of heightened photosensitivity, patients are advised to use eye and skin protection for a duration of 24 hours following treatment of ECP.55
In contrast to other existing immunosuppressive therapies used for GVHD treatment, ECP does not show associations with the development of opportunistic infections, treatment-related adverse events, organ toxicities, underlying disease relapse, or treatment-related adverse events.56 Adverse responses may be linked to leukapheresis, including temporary hypotension resulting from blood volume shifts in the extracorporeal circuit, citrate toxicity due to the anticoagulant employed, mild anemia, and thrombocytopenia following multiple treatments, or bleeding from the cannula sites utilized for venous access. Exposure to psoralen may cause various reactions, including an increment in urination, a metallic taste, itchiness that appears 6–8 hours after treatment, possibly increased sensitivity to light, and increased skin redness. After reinfusion of ECP products, certain patients may report experiencing mild fever within 2–12 hours post-treatment, along with feelings of fatigue and hematuria caused by the reintroduction of red blood cells following exposure to 8-MOP.57 In addition, studies have explored the toxic characteristics of long-wave ultraviolet light therapies, finding that treatments like PUVA or narrowband UV-B exhibit mutagenic and carcinogenic effects on skin cells.58
Conclusion
HSCT is the primary therapeutic choice for a wide range of hematological conditions as well as nonhematological malignant disorders. Despite numerous advancements in transplant methodologies, post-transplant care, and enhanced strategies for preventing and treating GVHD, it continues to be the most formidable challenge throughout the allogeneic HSCT procedure. While steroids are used as the first line of treatment, people with steroid-resistant GVHD have a poor prognosis. Based on current evidence, ECP emerges as a reasonable first option for managing patients with GVHD who do not respond to corticosteroid treatment. In comparison with other medications, it has a remarkably safe profile and no higher risk of infection. ECP is a therapy that is well-tolerated and safe, that acts on the cellular immune system to reduce T-cell action.
To better understand and use ECP as a primary treatment choice for preventing and treating GVHD, additional multicenter clinical studies with a sufficient number of patients, appropriate therapeutic control groups, and clinically relevant endpoints to evaluate ECP’s efficacy are highly recommended. In addition, clinical trials should determine the ideal ECP dosage and duration as well as investigate new biomarkers for predicting and tracking ECP treatment.
Abbreviations
APCs, Antigen Presenting Cells; CD, Cluster of Differentiation; CTCL, Cutaneous T cell Lymphoma/Leukemia; DNA, Deoxy Ribonucleic Acid; ECP, Extra Corporeal Photopheresis; FDA, Food and Drug Administration; GVHD, Graft versus Host Disease; GVT, Graft versus Tumor; HLA, Human Leukocyte Antigen; HSCT, Hematopoietic Stem Cell Transplantation; IL, Interleukin; INF, Interferon; MHC, Major Histocompatibility Complex; MOP, Methoxy Psoralen; NKT, Natural Killer T, Cell; TNF, Tumor Necrosis Factor; UV, Ultra Violet; WBC, White Blood Cell.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Mankarious M, Matthews NC, Snowden JA, Alfred A. Extracorporeal photopheresis (ECP) and the potential of novel biomarkers in optimizing management of acute and chronic graft vs. host disease (GvHD). Front Immunol. 2020;11:81. doi:10.3389/fimmu.2020.00081
2. Zhao L, Chen S, Yang P, Cao H, Li L. The role of mesenchymal stem cells in hematopoietic stem cell transplantation: prevention and treatment of graft-versus-host disease. Stem Cell Res Ther. 2019;10(1):1–13. doi:10.1186/s13287-019-1287-9
3. Iwasaki T. Recent advances in the treatment of graft-versus-host disease. Clin Med Res. 2004;2(4):243–252. doi:10.3121/cmr.2.4.243
4. Sung AD, Chao NJ. Concise review: acute graft-versus-host disease: immunobiology, prevention, and treatment. Stem Cells Translat Med. 2013;2(1):25–32. doi:10.5966/sctm.2012-0115
5. Calleja CH, Hidalgo DM, Curto CR, et al. Graft versus host disease-related eosinophilic fasciitis: cohort description and literature review. Adv Rheumatol. 2022;2022:62.
6. Saidu NEB, Bonini C, Dickinson A, et al. New approaches for the treatment of chronic graft-versus-host disease: current status and future directions. Front Immunol. 2020;11:578314. doi:10.3389/fimmu.2020.578314
7. Jamil MO, Mineishi S. State-of-The-art acute and chronic GVHD treatment. Int J Hematol. 2015;101:452–466. doi:10.1007/s12185-015-1785-1
8. Choi SW, Levine JE, Ferrara JL. Pathogenesis and management of graft-versus-host disease. Immunol Allergy Clin. 2010;30(1):75–101.
9. Kuba A, Raida L. Graft versus host disease: from basic pathogenic principles to DNA damage response and cellular senescence. Mediators Inflammation. 2018;2018:1–13. doi:10.1155/2018/9451950
10. Rezvani AR, Storb RF. Prevention of graft-vs.-host disease. Expert Opin Pharmac. 2012;13(12):1737–1750. doi:10.1517/14656566.2012.703652
11. Hamilton BK. Current approaches to prevent and treat GVHD after allogeneic stem cell transplantation. Hematology. 2018;2018(1):228–235.
12. Fatobene G, Cordeiro A, Mariano L, et al. GVHD treatment with extracorporeal photopheresis in Brazil: a national survey. Hematol Transf Cell Therap. 2022;44:450–452.
13. Funke VA, Moreira MCR, Vigorito AC. Acute and chronic Graft-versus-host disease after hematopoietic stem cell transplantation. Rev Assoc Med Bras. 2016;62:44–50. doi:10.1590/1806-9282.62.suppl1.44
14. Klassen J. The role of photopheresis in the treatment of graft-versus-host disease. Current Oncol. 2010;17(2):55–58. doi:10.3747/co.v17i2.565
15. Ghimire S, Weber D, Mavin E, Wang XN, Dickinson AM, Holler E. Pathophysiology of GvHD and other HSCT-related major complications. Front Immunol. 2017;8:79. doi:10.3389/fimmu.2017.00079
16. Naserian S, Leclerc M, Shamdani S, Uzan G. Current preventions and treatments of aGVHD: from pharmacological prophylaxis to innovative therapies. Front Immunol. 2020;11:607030. doi:10.3389/fimmu.2020.607030
17. Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. 2009;373(9674):1550–1561. doi:10.1016/S0140-6736(09)60237-3
18. Zhang L, Chu J, Yu J, Wei W. Cellular and molecular mechanisms in graft-versus-host disease. J Leucoc Bio. 2016;99(2):279–287. doi:10.1189/jlb.4RU0615-254RR
19. Ali MM, Gedde-Dahl T, Osnes LT, et al. Extracorporeal photopheresis as graft-versus-host disease prophylaxis: a randomized controlled trial. Transplantat Cell Ther. 2023;29(6):
20. Zhang L, Yu J, Wei W. Advance in targeted immunotherapy for graft-versus-host disease. Front Immunol. 2018;9:1087. doi:10.3389/fimmu.2018.01087
21. Schneiderman J. Extracorporeal photopheresis: cellular therapy for the treatment of acute and chronic graft-versus-host disease. Hematology. 2017;2017(1):639–644.
22. Anand S, Sarantopoulos S. Chronic graft-versus-host disease: a long road ahead. Biol Blood Marrow Transplant. 2018;24(3):423. doi:10.1016/j.bbmt.2018.01.010
23. Arai S, Arora M, Wang T, et al. Increasing incidence of chronic graft-versus-host disease in allogeneic transplantation: a report from the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant. 2015;21(2):266–274. doi:10.1016/j.bbmt.2014.10.021
24. Vargas DF, Pereira MP, Silva TS, David C, Paz AA, Astigarraga CC. Extracorporeal photopheresis in chronic graft-versus-host disease: clinical description and economic study. Hematol Transf Cell Therap. 2023;45:182–187. doi:10.1016/j.htct.2021.08.014
25. Cooke KR, Luznik L, Sarantopoulos S, et al. The biology of chronic graft-versus-host disease: a task force report from the national institutes of health consensus development project on criteria for clinical trials in chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2017;23(2):211–234. doi:10.1016/j.bbmt.2016.09.023
26. Garnett C, Apperley JF, Pavlů J. Treatment and management of graft-versus-host disease: improving response and survival. Therap Advanc Hematol. 2013;4(6):366–378. doi:10.1177/2040620713489842
27. Ling Y-L, Huang X, Mitri G, et al. Real-world use of extracorporeal photopheresis for patients with cutaneous T-cell lymphoma in the United States: 2010–2015. J Dermatological Treat. 2020;31(1):91–98. doi:10.1080/09546634.2019.1587144
28. Niittyvuopio R, Juvonen E, Heiskanen J, et al. Extracorporeal photopheresis in the treatment of acute graft‐versus‐host disease: a single‐center experience. Transfusion. 2018;58(8):1973–1979. doi:10.1111/trf.14649
29. Worel N, Lehner E, Führer H, et al. Extracorporeal photopheresis as second‐line therapy for patients with acute graft‐versus‐host disease: does the number of cells treated matter? Transfusion. 2018;58(4):1045–1053. doi:10.1111/trf.14506
30. Batgi H, Dal MS, Erkurt MA, et al. Extracorporeal photopheresis in the treatment of acute graft-versus-host disease: a multicenter experience. Transfus Apheresis Sci. 2021;60(5):103242. doi:10.1016/j.transci.2021.103242
31. Cho A, Jantschitsch C, Knobler R. Extracorporeal photopheresis—an overview. Front Med. 2018;5:236. doi:10.3389/fmed.2018.00236
32. Malagola M, Cancelli V, Skert C, et al. Extracorporeal photopheresis for treatment of acute and chronic graft versus host disease: an Italian multicentric retrospective analysis on 94 patients on behalf of the gruppo italiano trapianto di midollo osseo. Transplantation. 2016;100(12):e147–e55. doi:10.1097/TP.0000000000001466
33. Knobler R, Berlin G, Calzavara‐Pinton P, et al. Guidelines on the use of extracorporeal photopheresis. J Eur Acad Dermatol Venereol. 2014;28:1–37. doi:10.1111/jdv.12311
34. Hart JW, Shiue LH, Shpall EJ, Alousi AM. Extracorporeal photopheresis in the treatment of graft-versus-host disease: evidence and opinion. Therap Advanc Hematol. 2013;4(5):320–334. doi:10.1177/2040620713490316
35. Hautmann A, Wolff D, Hahn J, et al. Extracorporeal photopheresis in 62 patients with acute and chronic GVHD: results of treatment with the COBE Spectra System. Bone Marrow Transplant. 2013;48(3):439–445. doi:10.1038/bmt.2012.156
36. Calore E, Marson P, Pillon M, et al. Treatment of acute graft-versus-host disease in childhood with extracorporeal photochemotherapy/photopheresis: the Padova experience. Biol Blood Marrow Transplant. 2015;21(11):1963–1972. doi:10.1016/j.bbmt.2015.07.007
37. Alfred A, Taylor PC, Dignan F, et al. The role of extracorporeal photopheresis in the management of cutaneous T‐cell lymphoma, graft‐versus‐host disease, and organ transplant rejection: a consensus statement updates from the UK Photopheresis Society. Br J Hematol. 2017;177(2):287–310. doi:10.1111/bjh.14537
38. Ward DM. Extracorporeal photopheresis: how, when, and why. J clin apher. 2011;26(5):276–285. doi:10.1002/jca.20300
39. De Waure C, Capri S, Veneziano MA, et al. Extracorporeal photopheresis for second-line treatment of chronic graft-versus-host diseases: results from a health technology assessment in Italy. Value Health. 2015;18(4):457–466. doi:10.1016/j.jval.2015.01.009
40. Laulhe M, Lefebvre S, Le Broc-Ryckewaert D, Pierre M, Ferry A, Delorme B. A standardized methodical approach to characterize the influence of key parameters on the in vitro efficacy of extracorporeal photopheresis. PLoS One. 2019;14(3):e0212835. doi:10.1371/journal.pone.0212835
41. Kitko CL, Abdel-Azim H, Carpenter PA, et al. A prospective, multicenter study of closed-system extracorporeal photopheresis for children with steroid-refractory acute graft-versus-host disease. Transplantat Cell Ther. 2022;28(5):
42. Nelson AS, Pinkard SL, Carey P, Khandelwal P, Marsh RA, Davies SM. Extracorporeal Photopheresis (ECP) for treatment of refractory acute graft-versus-host disease in children (GVHD) after allogeneic hematopoietic stem cell transplantation (HSCT). Biol Blood Marrow Transplant. 2016;22(3):S255–6. doi:10.1016/j.bbmt.2015.11.684
43. Kapadia E, Wong E, Perez‐Albuerne E, Jacobsohn D. Extracorporeal photopheresis performed on the CELLEX® compared with the UVAR‐XTS® instrument is more efficient and better tolerated in children with steroid‐refractory graft‐versus‐host disease. Pediatr Blood Cancer. 2015;62(8):1485–1488. doi:10.1002/pbc.25487
44. Rangarajan HG, Punzalan RC, Camitta BM, Talano JA. The use of novel Therakos™ Cellex® for extracorporeal photopheresis in the treatment of graft‐versus‐host disease in paediatric patients. Br J Haematol. 2013;163(3):357–364. doi:10.1111/bjh.12535
45. Sakellari I, Gavriilaki E, Batsis I, et al. Favorable impact of extracorporeal photopheresis in acute and chronic graft versus host disease: prospective single‐center study. J clin apher. 2018;33(6):654–660. doi:10.1002/jca.21660
46. Das-Gupta E, Greinix H, Jacobs R, et al. Extracorporeal photopheresis as second-line treatment for acute graft-versus-host disease: impact on six-month freedom from treatment failure. Haematologica. 2014;99(11):1746. doi:10.3324/haematol.2014.108217
47. Couriel DR, Hosing C, Saliba R, et al. Extracorporeal photochemotherapy for the treatment of steroid-resistant chronic GVHD. Blood. 2006;107(8):3074–3080. doi:10.1182/blood-2005-09-3907
48. Gunduz M, Atilla E, Atilla P, Topcuoglu P, Ilhan O. Early initiation of extracorporeal photo chemotherapy increases response for chronic graft versus host disease following steroid failure. Transfus Clin Biol. 2019;26(1):32–37. doi:10.1016/j.tracli.2018.03.005
49. Dal MS, Batgi H, Erkurt MA, et al. Extracorporeal photopheresis in steroid-refractory chronic graft-versus-host disease: a retrospective multicenter study. Transfus Apheresis Sci. 2021;60(5):103243. doi:10.1016/j.transci.2021.103243
50. Malik MI, Litzow M, Hogan W, et al. Extracorporeal photopheresis for chronic graft-versus-host disease: a systematic review and meta-analysis. Blood Res. 2014;49(2):100. doi:10.5045/br.2014.49.2.100
51. Oarbeascoa G, Lozano ML, Guerra LM, et al. Retrospective multicenter study of extracorporeal photopheresis in steroid-refractory acute and chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2020;26(4):651–658. doi:10.1016/j.bbmt.2019.12.769
52. Nygaard M, Wichert S, Berlin G, Toss F. Extracorporeal photopheresis for graft‐vs‐host disease: a literature review and treatment guidelines proposed by the Nordic ECP Quality Group. Eur J Haematol. 2020;104(5):361–375. doi:10.1111/ejh.13381
53. Dunbar NM, Raval JS, Johnson A, et al. Extracorporeal photopheresis practice patterns: an international survey by the ASFA ECP subcommittee. J clin apher. 2017;32(4):215–223. doi:10.1002/jca.21486
54. Sanford KW, Balogun RA. Therapeutic apheresis in critically ill patients. J clin apher. 2011;26(5):249–251. doi:10.1002/jca.20304
55. Drexler B, Buser A, Infanti L, Stehle G, Halter J, Holbro A. Extracorporeal photopheresis in graft-versus-host disease. Transfusion Med Hemotherapy. 2020;47(3):214–225. doi:10.1159/000508169
56. Knobler R, Arenberger P, Arun A, et al. European Dermatology forum–updated guidelines on the use of extracorporeal photopheresis 2020–part 1. J Eur Acad Dermatol Venereol. 2020;34(12):2693–2716. doi:10.1111/jdv.16890
57. Vieyra-Garcia PA, Wolf P. Extracorporeal photopheresis: a case of immunotherapy ahead of its time. Transfusion Med Hemotherapy. 2020;47(3):226–235. doi:10.1159/000508479
58. Samur BM, Karakukcu C, Ozcan A, Unal E, Yilmaz E, Karakukcu M. Assessment of extracorporeal photopheresis related cell damage. Transfus Apher Sci. 2022;61(6):103472. doi:10.1016/j.transci.2022.103472
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.