Back to Journals » Infection and Drug Resistance » Volume 17
The Recent Transmission and Associated Risk Factor of Mycobacterium tuberculosis in Golmud City, China
Authors Song Z, He W, Cao X ![]() , Ma A, He P, Zhao B, Wang S, Liu C, Zhao Y
, Ma A, He P, Zhao B, Wang S, Liu C, Zhao Y
Received 25 August 2023
Accepted for publication 5 December 2023
Published 1 February 2024 Volume 2024:17 Pages 417—425
DOI https://doi.org/10.2147/IDR.S437026
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sandip Patil
Zexuan Song,1 Wencong He,1 Xiaolong Cao,1 Aijing Ma,1 Ping He,1 Bing Zhao,1 Shengfen Wang,1 Chunfa Liu,1,2 Yanlin Zhao1
1National Key Laboratory of Intelligent Tracking and Forecasting for Infectious Diseases, National Center for Tuberculosis Control and Prevention, Chinese Center for Disease Control and Prevention, Changping, Beijing, 102206, People’s Republic of China; 2Animal Science and Technology College, Beijing University of Agriculture, Huilongguan, Changping, Beijing, 102206, People’s Republic of China
Correspondence: Chunfa Liu; Yanlin Zhao, Email [email protected]; [email protected]
Background: Tuberculosis (TB) remains a severe public health problem globally, and it is essential to comprehend the transmission pattern to control tuberculosis. Herein, we evaluated the drug-resistant characteristics, recent transmission, and associated risk factors of TB in Golmud, Qinghai, China.
Methods: In this study, we performed a population-based study of patients diagnosed with TB in Golmud from 2013 to 2018. Drug-susceptibility testing and whole-genome sequencing were performed on 133 Mycobacterium tuberculosis strains. The genomic clustering rate was calculated to evaluate the level of recent transmission. Risk factors were identified by logistic regression analysis.
Results: Our results showed that 46.97% (62/132) of strains were phylogenetically clustered and formed into 23 transmission clusters, suggesting a high recent transmission of TB in the area. 12.78% (17/133) strains were multidrug-resistant/rifampicin tuberculosis (MDR/RR-TB), with a high drug-resistant burden. Based on drug resistance gene analysis, we found 23 strains belonging to genotype MDR/RR-TB, where some strains may have borderline mutations. Among these strains, 65.2% (15/23) were found within putative transmission clusters. Additionally, risk factor analysis showed that recent transmission of TB happened more in patients with Tibetan nationality or older age.
Conclusion: Overall our study indicates that the recent transmissions of MTB strains, especially genotypic MDR/RR strains, drive the tuberculosis epidemic in Golmud, which could contribute to developing effective TB prevention and control strategies.
Keywords: tuberculosis, recent transmission, risk factor, whole-genome sequencing
Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis (MTB), remains a major public health problem globally. The World Health Organization (WHO) estimated that 10.6 million people were infected and 1.6 million people died in 2021.1 China has the third highest TB burden worldwide, with an estimated 780,000 new TB cases in 2021.1 The emergence of drug-resistant tuberculosis and the high rate of transmission threaten the eradication of tuberculosis.
The increasing availability of whole-genome sequencing (WGS) technologies, coupled with the development of analytical methods that integrate spatial and epidemiological data, has transformed our ability to describe TB transmission in populations.2 The WGS approach has better discriminatory power compared to classical genotyping, such as spoligotyping or MIRU-VNTR typing.3 To date, WGS of MTB has been used to estimate the recent transmission of incident TB cases,4,5 and to investigate TB outbreaks and transmission chains. 6 Since phenotypic drug susceptibility testing (pDST) carried out by culture-based procedure takes a long time and stringent biosafety conditions, WGS has the potential to identify genotypic drug resistance by revealing genetic drug resistance.7 Furthermore, WGS can also provide insights into the drug resistance, diverse lineages, and genetic variants of the strains.8 In China, MTB strains are classified into four lineages (Lineage 1–Lineage 4), of which Lineage 2 is predominant, especially among multidrug-resistant (MDR) strains.9
Despite significant progress in TB control in the past few years, China remains a high-burden country in the world.10 Previous studies have shown that transmission rather than inadequate treatment was a major driving force for the actual MDR-TB epidemic.2,11,12 Thus, understanding the transmission dynamics of TB and its risk factors is particularly important for tuberculosis control and intervention. In China, genomic epidemiological approaches have been applied to characterize the recent transmission dynamics of TB in some developed provinces and cities,5,12,13 but rare reports on transmission within low population density and less developed areas.
Qinghai province is located in western China, with a high burden of tuberculosis.14 Previous studies have investigated that the spatial epidemiological characteristics and transmission of tuberculosis in Qinghai province, but rarely report on the genomic transmission characteristics and drug resistance of MTB strains. In this study, we conducted a population-based study to characterize transmission dynamics and associated risk factors of TB based on MTB strains in Golmud city, which is a large city of Qinghai province. These findings could understand the characterization of MTB strains and develop effective strategies to control and further eliminate TB.
Methods
Sample Collection
This was a retrospective study based on routine national drug resistance surveillance work in Qinghai province.15 The samples included all MTB strains isolated from suspected pulmonary tuberculosis patients with sputum smears positive, who attended local designated hospitals or dispensaries in the surveillance site (Golmud) of Qinghai province between January 2013 and December 2018. Cultures with growing colonies were sent to the National Tuberculosis Reference Laboratory (NTRL) in China CDC for identification and WGS analysis. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MADLI-TOF-MS) was used to differentiate Mycobacterium tuberculosis and nontuberculous mycobacteria (NTM).
Epidemiological Investigation
The epidemiological information of each patient including demographic, sexual, educational, job-seeking characteristics and national was collected from the national drug resistance surveillance database, which was collected from patients after written informed consent. The study received ethical approval, which complies with the Declaration of Helsinki, from the Ethics Committee of the Chinese Center for Disease Control and Prevention (202223).
Drug Susceptibility Testing
The drug susceptibility test (DST) of MTB strains against isoniazid (INH), rifampicin (RIF), ethambutol (EMB), kanamycin (KAN), amikacin (AMI), levofloxacin (LFX) and moxifloxacin (MXF) were determined according to the standard operating protocol defined by CRyPTIC.16 Briefly, 0.5 McFarland suspensions of MTB isolates prepared from fresh colonies (no longer than 14 days old) grown on Lowenstein-Jensen tubes were diluted 100-fold in 10 mL of 7H9 broth prior to plate inoculation. The semi-automated Sensititre TM Auto-inoculator (Thermo Fisher, Scientific Inc., USA) was used to aliquots100μL into each well of the UKMYC6 microdilution plate. Then, all the plates were sealed and incubated for 14 days at 37°C. The DST results for each drug were performed separately by two trained laboratory operators using the Thermo Fisher Sensititre™ Vizion™ digital MIC viewing system. The minimum inhibitory concentration (MIC) is the lowest antibiotic concentration that inhibits observable microorganism growth. Quality control runs with reference M. tuberculosis H37Rv ATCC 27294 was performed on the plate on a regular basis. The critical concentration value of anti-TB drugs included in this study refers to our previous report.17
DNA Extraction and Sequencing
All MTB strains were recovered successfully and were scraped from L-J slant, a genomic DNA sequence extract by the Cetyltrimethylammonium bromide (CTAB) method according to the previous report.18 Whole-genome sequencing of strains was performed on purified DNA using the Illumina HiSeq PE150 technique by Annoroad Gene Technology company (Beijing, China).
Phylogenetic Analysis
The overall quality of sequence reads was checked using FastQC (v0.11.8). Verified paired-end reads were filtered with Trimmomatic with default values and a minimum Phred Quality score of 20. The variants calling was performed as previously reported.17 Briefly, the paired reads were mapped to the reference genome H37Rv (NC_000962.3) using BWA-MEM (v0.7.17). SAMtools (v1.3.1) and GATK (v3.8.0) were used to call variants, and each variant could satisfy the requirements of a minimum coverage depth of 10X, a minimum quality score of Q20, and an allele frequency of more than 75%. SNPs located in known drug resistance-related genes, mobile genetic elements, PE or PPE regions were excluded from phylogenetic analysis. A recombination core SNP alignment was constructed, and a maximum likelihood phylogenetic tree was constructed by IQ-TREE with 1000 bootstraps. The phylogenetic tree was visualized and modified by ChiPlot.19 Pairwise SNP distance of the strains performed by SNP-dist (v0.7.0). According to the previous report, clusters were defined as isolates within pairwise genetic distance of less than 12 SNPs.17 The lineage of the strains was performed by fast-lineage-caller (v0.3).
Core Genome MLST (cgMLST) Analysis
The cg-MLST analysis was performed by Ridom SeqSphere+ software (v7.2.3) with the default settings.20 Each strain sequence was aligned to the M. tuberculosis cg-MLST scheme consisting of 2891 core genes of the seed genome H37Rv (NC_000962.3), the default settings including the removal of the shorter of two genes overlapping by more than four bases and the genes with an internal stop codon. The full cgMLST analysis was performed on strain sequences that imparted >90% good target, and the minimum spanning tree was calculated for the allelic profiles of the targets. According to previous studies, the isolates sharing less than 12 allelic differences are classed into a cluster, suggesting the occurrence of recent transmission.21,22
Antimicrobial Resistance Prediction
Drug resistance profiles of the strains were performed using the TB Profiler (https://github.com/jodyphelan/TBProfiler), which could detect drug resistance variants and predict drug resistance genotypes.23
Statistical Analysis
The chi-square test or Fisher's exact test was used for categorical data. Logistic regression analysis was used to calculate the odds ratios (OR) and 95% confidence intervals (CI) for risk factors associated with MDR/RR-TB and genomic clustering. P value less than 0.05 were considered statistically significant. All statistical analysis was performed in the SPSS (v18.0).
Results
Study Population and Samples
Between January 2013 and December 2018, a total of 343 suspected tuberculosis patients attended the local designated hospitals in Golmud, Qinghai. 134 MTB strains isolated from pulmonary tuberculosis patients with sputum smear-positive. In addition, one strain was excluded due to belonging to NTM. Finally, 133 MTB strains from Golmud city were included in the follow-up study (Figure 1).
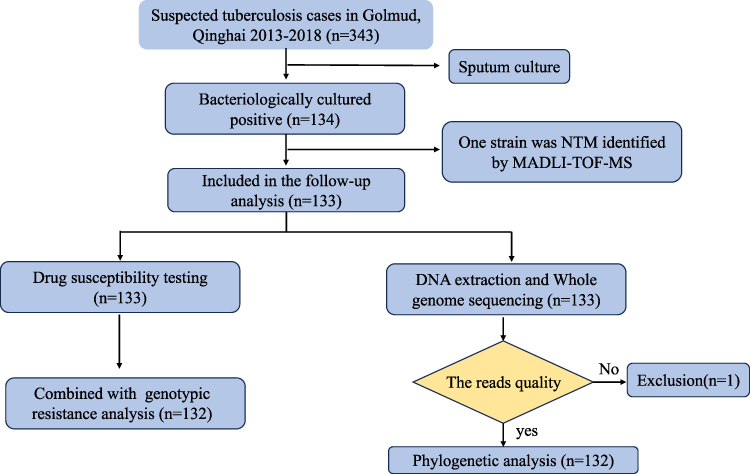 |
Figure 1 Diagram of M. tuberculosis strains included in the study. |
Phenotypic Drug-Resistant Profile
Among all the 133 MTB strains, 12.78% (17/133) strains were resistant to rifampicin, 12.03% (16/133) and 3.01% (4/133) strains were resistant to isoniazid and ethambutol, respectively. Lower fluoroquinolone resistance rates were observed, 0.75% (1/133) and 1.50% (2/133) were moxifloxacin and levofloxacin resistance, respectively. No strains were resistant to aminoglycoside drugs (Figure S1). Based on previous drug-resistant patterns,17 12.78% (17/133) of MTB belong to MDR/RR strains in this study.
Genotypic Drug-Resistant Characteristics
Out of the remaining 132 isolates with WGS, 17.42% (23/132) of strains belong to MDR/RR-TB (Figure 2A). In this study, we have only described and examined the molecular resistance of rifampicin and isoniazid due to the small number of other resistant strains (Tables S1 and S2). The most prevalent rifampicin-resistant mutation was rpoB_S450L (52.17%, 12/23), followed by rpoB_H445D (17.39%, 4/23). We also found that 18 strains (13.63%, 18/132) have detectable mutations related to isoniazid resistance and the majority mutation was katG_S315T (88.89%, 16/18). Notably, our results show that some strains with associated resistance genetic mutations do not exhibit phenotypic resistance, which may be due to some genetic mutations belonging to borderline mutations, such as rpoB_L452P, rpoB_L430P and fabG1_ C-15T.7,24
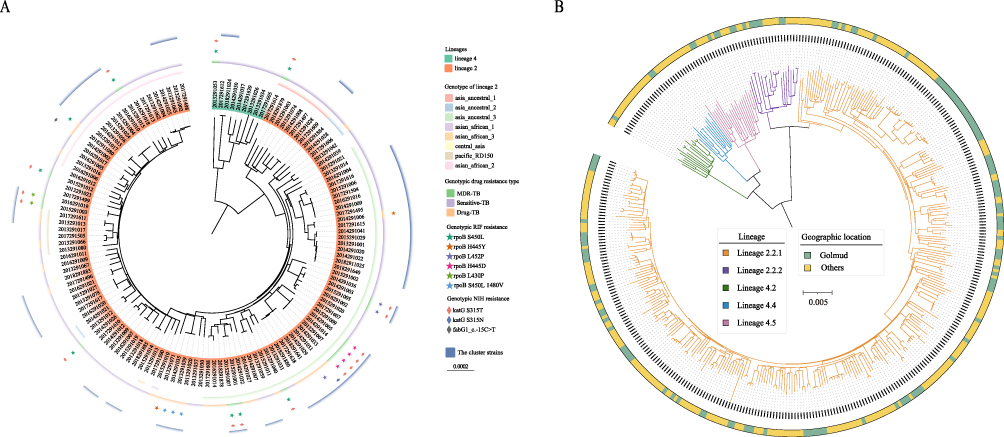 |
Figure 2 The phylogenetic tree of MTB strains in this study. (A) The midpoint-rooted maximum likelihood phylogenetic tree of 132 strains in this study. The genotype of lineage 2, genotype drug resistant, rifampicin and isoniazid resistant genotypic resistance are shown on the tree from inner to outer circles. The purple outer circle indicates clusters of strains. (B) The Phylogenetic tree of 132 strains from Golmud and 251 strains detected in other areas of China. The colors of the branches represent different lineages, and the colors of the outer rings showed the geographic location of the strains. |
Phylogenetic Analysis
To better understand the genetic structure of MTB strains in Golmud, a phylogenetic tree was constructed based on non-convergent SNPs (Figure 2A). Most (94.18%, 123/132) strains belong to Lineage 2 (L2), while others (5.82%, 9/132) were Lineage 4 (L4), which is in line with previous reports about the main lineages in China.25 In this study, we further classified the strains of lineage 2 according to Shitikov’s schemes,26 and the results showed that 73.17% (90/123) of the L2 strains were divided into five genotypes. The majority of the L2 strains belong to Asia ancestral 3 (47/123), followed by Asian African 2 (21/123), Asia ancestral 1(8/123), Pacific RD150 (6/123). Three isolates belong to Asian African 1, Asian African 3 and Central Asia, respectively. Our results indicate that MTB strains have high genetic diversity.
Furthermore, we constructed phylogenetic trees based on WGS data of these strains together with the strains collected from other areas of China9 (Figure 2B). The results showed that mostly strains in Golmud and other areas were distributed across the phylogenetic tree and do not cluster into distinct clades, suggesting that the MTB strains have a shared evolutionary history.
Transmission Analysis
Genomic transmission clusters were defined using 12 SNPs as a cutoff value, and there are 23 clusters identified by SNP-barcoding analysis (Figure 2A). To better investigate the transmission dynamics of TB, we confirmed the relationship between MTB strains using cgMLST method, which has previously been used to look at TB outbreaks.21 The minimum spanning tree of 132 strains was built by cg-MLST analysis, which showed that 46.97% (62/132) of strains consist of 23 genomic clusters in our study, with the size ranging from 2 to 5 strains (Figure 3). Among all the clusters, 82.60% (19/23) of clusters in this study have an interval time of fewer than 3 years, which suggests that most of the recent transmission events have happened within 3 or fewer years.
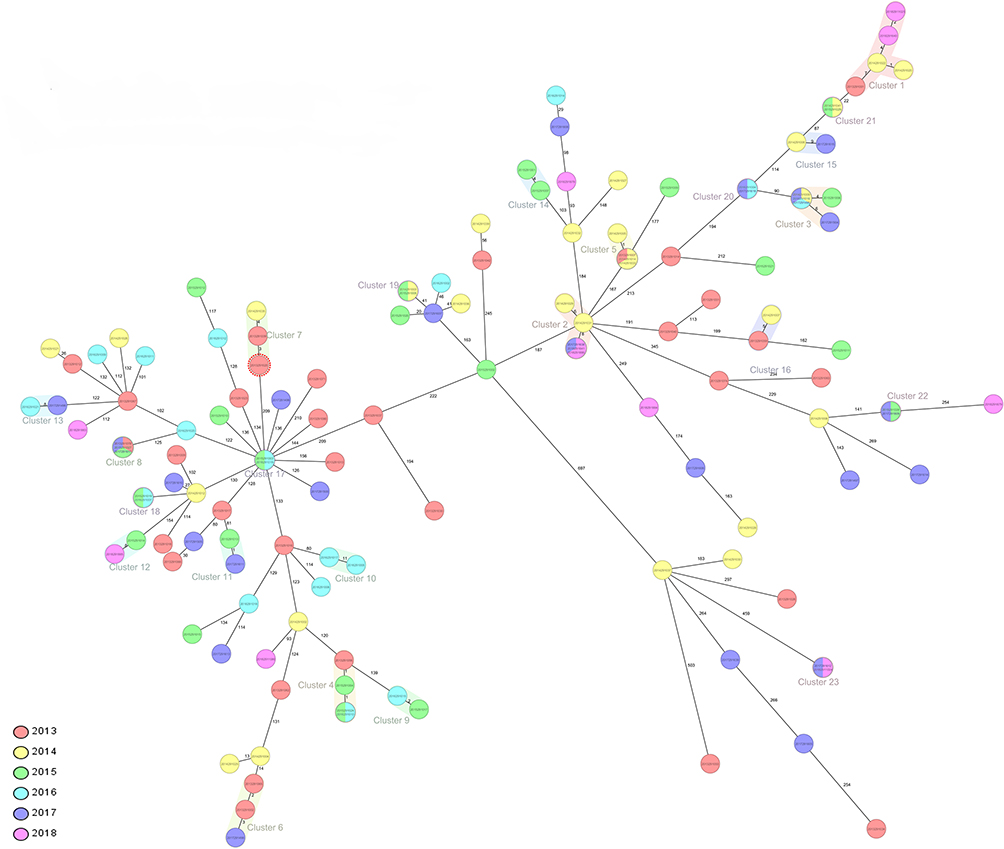 |
Figure 3 The minimum distance spanning tree of strains in this study. The tree was constructed by Ridom SeqSphere+ with cg-MLST analysis. Pairwise SNP distance of the strains and the cluster were shown in the Tree, the clusters were defined as strains with 12 or fewer SNPs. According to the legend, the color represents the isolation times of the strains. |
We note that the clustering of strains obtained using the SNP and cgMLST method was consistent in this study, further indicating the presence of recent transmission of these clusters in Golmud city. Furthermore, the high clustering rate of MTB strains indicates that transmission is likely to be a major driver of the local TB epidemic.
Risk Factors for Genomic Cluster
Finally, we identify the risk factors associated with the genomic cluster by the logistic regression. Our data showed that the years, occupation and nationality were associated with clustering (Table 1). Multivariate analysis indicated that recent transmission of TB was more happened in patients with Tibetan nationality (P = 0.02) or elder age (35–44 or ≥65 years, P = 0.01 and P = 0.04). Also, genotypic RR/MDR-TB had a higher risk of clustering (P = 0.01) (Table 1).
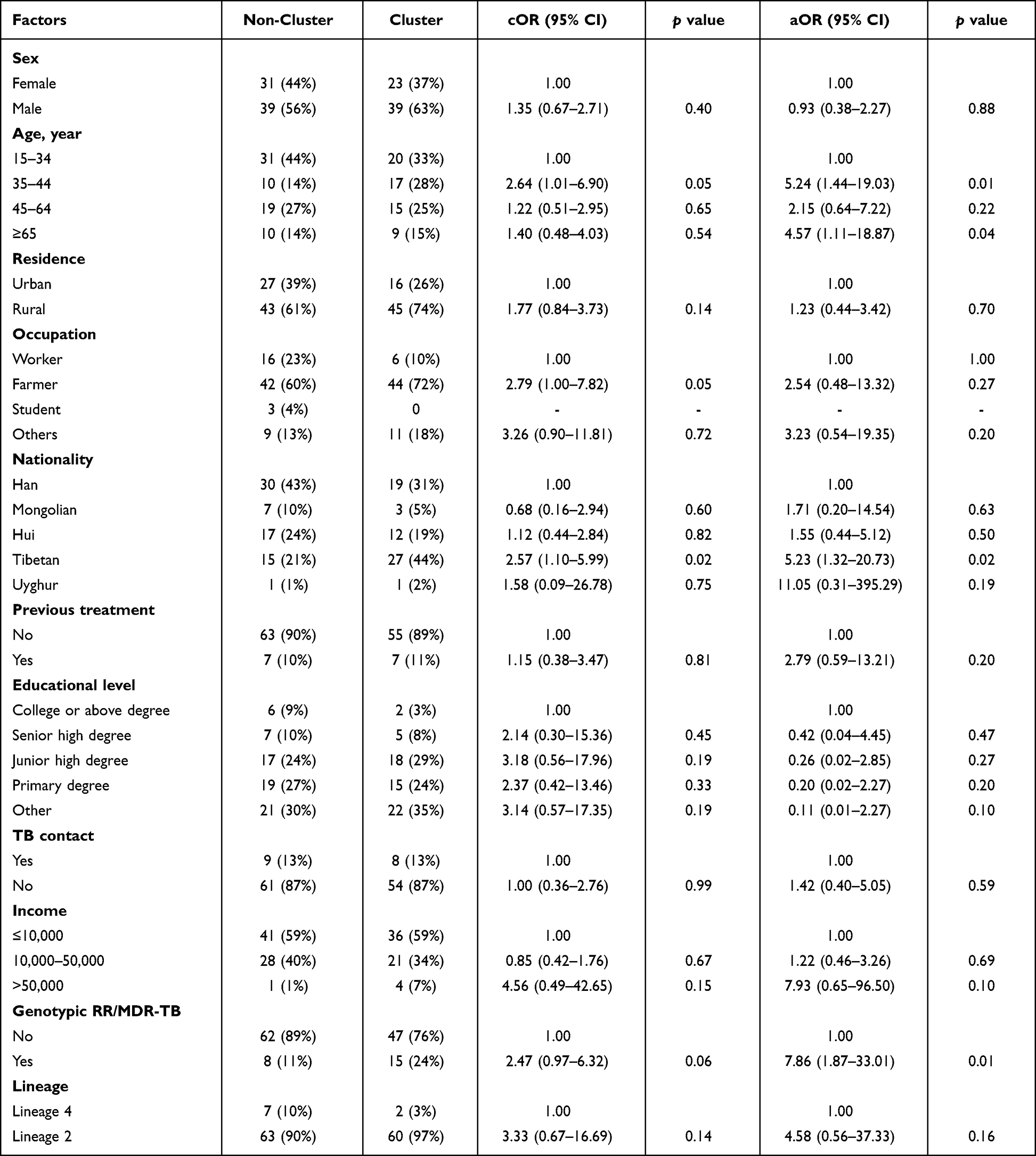 |
Table 1 Univariate and Multivariate Logistic Regression on the Risk Factors of MTB Clustering (n=132) |
Discussion
In our study, we analyzed the genetic diversity and drug resistance of MTB strains, and described the TB transmission pattern with risk factors in Golmud city of Qinghai province, China. The results showed that more than 10% of the strains belong to MDR/RR-TB, which is higher than the national level in China (7.42%),7 but similar to the level of Yichang city (11.3%).27 In addition, low fluoroquinolone resistance rate was detected in this study, which is inconsistent with reports from other provinces in China.17,22,27 The conflicting result might be due to the frequency and dosage of fluoroquinolone antibiotics used in different regions or the strains selected for the research. However, this finding favors future applications of fluoroquinolones in TB treatment regimens of shorter duration.
WGS methods can currently be combined with epidemiological data to effectively study the transmission dynamics of MTB strains,2,28–30 either using cg-MLST3,21,29,31 or SNP barcode methods.5,32 In this study, we provided a snapshot of tuberculosis transmission based on cg-MLST analysis and SNP calling methods, both of them get the same results. Our data show that nearly half (46.97%, 62/132) of MTB strains in possible transmission clusters, higher than in rural counties in China (31.4%)11 and Yichang city (30.1%),27 indicating recent transmission is a noticeable cause of TB patients in Golmud. The role of recent transmission was even more pronounced for genotype MDR/RR-TB, among which 65.2% (15/23) were found within putative transmission clusters. This phenomenon also occurs in rural areas in China, which showed that 81.4% of the MDR-TB cases were likely from transmission of MDR strains.11 This may be due to the fact that most cases of MDR/RR-TB are undetected or inappropriately treated, and the transmission of these strains can lead to a more serious MDR/RR-TB epidemic. Therefore, increasing the management of TB patients is a powerful measure to reduce the local TB burden.
Risk factors of TB transmission analysis indicated that patients with older age had higher transmission incidence, a result in line with previous research in Shanghai, China, which showed that being older than 45 years is at higher risk of transmission.12 Thus, the management of the elderly population should be emphasized in the process of TB prevention and control. In addition, a meta-analysis about risk factors of MDR-TB in China showed that history of tuberculosis treatment was considered to be at risk of MDR-TB.33,34 Effective TB control relies on early diagnosis and subsequent rapid DST to provide appropriate treatment in order to interrupt further transmission.35 We noted that 40% MDR/RR-TB cases are previously treated in Golmud city, indicating that some patients are not receiving appropriate and adequate treatments. Therefore, rapid diagnosis and rational drug regimen are essential to control MDR/RR-TB. Previous studies have demonstrated that whole-genome sequence could predict the susceptibility of MTB to first-line drugs with high specificity and sensitivity,7,36 suggesting WGS or rapidly targeted sequencing for drug resistance diagnosis of TB in the future.
Our study has several limitations. First, it is possible that we did not collate all of the culture positive TB cases and strains in the population. The sample size of included strains is relatively small, resulting in an under-representation of the region. Second, strains could be misclassified as unique if they were in fact clustered with strains outside the study period and geographical setting. In the future, a larger sample size of its neighboring provinces and cities should be included for a systematic examination of TB transmission in the area. In addition, the field of epidemiological investigation was insufficient that have missed some transmission settings and epidemiological links.
In conclusion, our study helped to investigate the transmission dynamics of TB based on genomic epidemiological approaches in Golmud city. Transmission appears to occur principally among Tibetan nationality, or those 35–44 years old patients, and therefore more attention should be paid to this targeted population. Further improvement in drug-resistant identification, especially in patients who were previously treated, is important for reducing transmission and improving TB control in Golmud, China.
Funding
This work was supported by the National Key R&D Program of China (NO. 2022YFC2305200).
Disclosure
The authors report no conflicts of interest in this work.
References
1. World Health Organization. Global Tuberculosis Report 2022. Geneva: World Health Organization; 2022.
2. Yang C, Sobkowiak B, Naidu V, et al. Phylogeography and transmission of M. tuberculosis in Moldova: a prospective genomic analysis. PLoS Med. 2022;19(2):e1003933. doi:10.1371/journal.pmed.1003933
3. Kohl TA, Harmsen D, Rothgänger J, Walker T, Diel R, Niemann S. Harmonized genome wide typing of tubercle bacilli using a web-based gene-by-gene nomenclature system. EBioMedicine. 2018;34:131–138. doi:10.1016/j.ebiom.2018.07.030
4. Guerra-Assunção JA, Crampin AC, Houben RM, et al. Large-scale whole genome sequencing of M. tuberculosis provides insights into transmission in a high prevalence area. Elife. 2015;4. doi:10.7554/eLife.05166
5. Jiang Q, Liu Q, Ji L, et al. Citywide transmission of multidrug-resistant tuberculosis under China’s Rapid Urbanization: a retrospective population-based genomic spatial epidemiological study. Clin Infect Dis. 2020;71(1):142–151. doi:10.1093/cid/ciz790
6. Lalor MK, Casali N, Walker TM, et al. The use of whole-genome sequencing in cluster investigation of a multidrug-resistant tuberculosis outbreak. Europ resp J. 2018;51(6):1702313.
7. Liu D, Huang F, Zhang G, et al. Whole-genome sequencing for surveillance of tuberculosis drug resistance and determination of resistance level in China. Clin Microbiol Infect. 2021. doi:10.1016/j.cmi.2021.09.014
8. Cirillo DM, Cabibbe AM, De Filippo MR, et al. Use of WGS in Mycobacterium tuberculosis routine diagnosis. Int J Mycobacteriol. 2016;5(Suppl 1):S252–S253. doi:10.1016/j.ijmyco.2016.09.053
9. Song Z, Liu C, He W, et al. Insight into the drug-resistant characteristics and genetic diversity of multidrug-resistant Mycobacterium tuberculosis in China. Microbiol Spectr. 2023;11(5):e0132423. doi:10.1128/spectrum.01324-23
10. World Health Organization. Global Tuberculosis Report 2021. Geneva: World Health Organization; 2021.
11. Li M, Guo M, Peng Y, et al. High proportion of tuberculosis transmission among social contacts in rural China: a 12-year prospective population-based genomic epidemiological study. Emerg Microbes Infect. 2022;11(1):2102–2111. doi:10.1080/22221751.2022.2112912
12. Yang C, Luo T, Shen X, et al. Transmission of multidrug-resistant Mycobacterium tuberculosis in Shanghai, China: a retrospective observational study using whole-genome sequencing and epidemiological investigation. Lancet Infect Dis. 2017;17(3):275–284. doi:10.1016/s1473-3099(16)30418-2
13. Yang C, Lu L, Warren JL, et al. Internal migration and transmission dynamics of tuberculosis in Shanghai, China: an epidemiological, spatial, genomic analysis. Lancet Infect Dis. 2018;18(7):788–795. doi:10.1016/s1473-3099(18)30218-4
14. Rao HX, Zhang X, Zhao L, et al. Spatial transmission and meteorological determinants of tuberculosis incidence in Qinghai Province, China: a spatial clustering panel analysis. Infect Dis Poverty. 2016;5(1):45. doi:10.1186/s40249-016-0139-4
15. Zhao Y, Xu S, Wang L, et al. National survey of drug-resistant tuberculosis in China. N Engl J Med. 2012;366(23):2161–2170. doi:10.1056/NEJMoa1108789
16. The CC. A data compendium associating the genomes of 12,289 Mycobacterium tuberculosis isolates with quantitative resistance phenotypes to 13 antibiotics. PLoS Biol. 2022;20(8):e3001721. doi:10.1371/journal.pbio.3001721
17. He W, Tan Y, Liu C, et al. Drug-resistant characteristics, genetic diversity, and transmission dynamics of rifampicin-resistant Mycobacterium tuberculosis in Hunan, china, revealed by whole-genome sequencing. Microbiol Spectr. 2022;10(1):e0154321. doi:10.1128/spectrum.01543-21
18. Somerville W, Thibert L, Schwartzman K, Behr MA. Extraction of Mycobacterium tuberculosis DNA: a question of containment. J Clin Microbiol. 2005;43(6):2996–2997. doi:10.1128/jcm.43.6.2996-2997.2005
19. Xie J, Chen Y, Cai G, Cai R, Hu Z, Wang H. Tree Visualization By One Table (tvBOT): a web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023. doi:10.1093/nar/gkad359
20. Guo Q, Pan Y, Yang Z, et al. Epidemiology and Clinical Characteristics of Pediatric Drug-Resistant Tuberculosis in Chongqing, China. PLoS One. 2016;11(3):e0151303. doi:10.1371/journal.pone.0151303
21. Jones RC, Harris LG, Morgan S, et al. Phylogenetic analysis of Mycobacterium tuberculosis strains in Wales by use of core genome multilocus sequence typing to analyze whole-genome sequencing data. J Clin Microbiol. 2019;57(6):10–128.
22. Zhao B, Liu C, Fan J, et al. Transmission and drug resistance genotype of multidrug-resistant or rifampicin-resistant Mycobacterium tuberculosis in Chongqing, China. Microbiol Spectr. 2022;10(5):e0240521. doi:10.1128/spectrum.02405-21
23. Coll F, McNerney R, Preston MD, et al. Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med. 2015;7(1):51. doi:10.1186/s13073-015-0164-0
24. Xia H, Song Y, Zheng Y, et al. Detection of Mycobacterium tuberculosis rifampicin resistance conferred by borderline rpoB mutations: xpert MTB/RIF is superior to phenotypic drug susceptibility testing. Infect Drug Resist. 2022;15:1345–1352. doi:10.2147/idr.S358301
25. Liu Q, Ma A, Wei L, et al. China’s tuberculosis epidemic stems from historical expansion of four strains of Mycobacterium tuberculosis. Nat Ecol Evol. 2018;2(12):1982–1992. doi:10.1038/s41559-018-0680-6
26. Shitikov E, Kolchenko S, Mokrousov I, et al. Evolutionary pathway analysis and unified classification of East Asian lineage of Mycobacterium tuberculosis. Sci Rep. 2017;7(1):9227. doi:10.1038/s41598-017-10018-5
27. Ji L, Tao FX, Yu YF, et al. Whole-genome sequencing to characterize the genetic structure and transmission risk of Mycobacterium tuberculosis in Yichang city of China. Front Public Health. 2022;10:1047965. doi:10.3389/fpubh.2022.1047965
28. Roetzer A, Diel R, Kohl TA, et al. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study. PLoS Med. 2013;10(2):e1001387. doi:10.1371/journal.pmed.1001387
29. Meehan CJ, Moris P, Kohl TA, et al. The relationship between transmission time and clustering methods in Mycobacterium tuberculosis epidemiology. EBioMedicine. 2018;37:410–416. doi:10.1016/j.ebiom.2018.10.013
30. Nikolayevskyy V, Niemann S, Anthony R, et al. Role and value of whole genome sequencing in studying tuberculosis transmission. Clin Microbiol Infect. 2019;25(11):1377–1382. doi:10.1016/j.cmi.2019.03.022
31. Jajou R, Kohl TA, Walker T, et al. Towards standardisation: comparison of five whole genome sequencing (WGS) analysis pipelines for detection of epidemiologically linked tuberculosis cases. Euro Surveill. 2019;24(50). doi:10.2807/1560-7917.Es.2019.24.50.1900130
32. Nikolayevskyy V, Kranzer K, Niemann S, Drobniewski F. Whole genome sequencing of Mycobacterium tuberculosis for detection of recent transmission and tracing outbreaks: a systematic review. Tuberculosis. 2016;98:77–85. doi:10.1016/j.tube.2016.02.009
33. Feng M, Xu Y, Zhang X, et al. Risk factors of multidrug-resistant tuberculosis in China: a meta-analysis. Public Health Nurs. 2019;36(3):257–269. doi:10.1111/phn.12582
34. Cai X, Zhang D, Yan Y, Tan D, Xu Y. [Meta-analysis on risk factors of multidrug resistant tuberculosis in China]. Zhonghua liu xing bing xue za zhi. 2015;36(12):1424–1429. Chinese.
35. Shi W, Davies Forsman L, Hu Y, et al. Improved treatment outcome of multidrug-resistant tuberculosis with the use of a rapid molecular test to detect drug resistance in China. Int J Infect Dis. 2020;96:390–397. doi:10.1016/j.ijid.2020.04.049
36. Allix-Béguec C, Arandjelovic I, Bi L, et al. Prediction of susceptibility to first-line tuberculosis drugs by DNA sequencing. N Engl J Med. 2018;379(15):1403–1415. doi:10.1056/NEJMoa1800474
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.