")
Back to Journals » Journal of Inflammation Research » Volume 14
The Potentially Therapeutic Role of EPAC in Curbing the Process of Idiopathic Pulmonary Fibrosis via Differential Cellular Pathways
Authors Cao X, Li Y, Shi J, Tang H
Received 8 December 2020
Accepted for publication 27 January 2021
Published 26 February 2021 Volume 2021:14 Pages 611—619
DOI https://doi.org/10.2147/JIR.S296382
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Xinwei Cao,1 Yajun Li,1 Jianrong Shi,2 Huifang Tang1
1Department of Pharmacology, School of Basic Medical Sciences, Zhejiang University, Hangzhou, 310058, People’s Republic of China; 2Department of Clinical Laboratory, Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center For Child Health, Hangzhou, 310003, People’s Republic of China
Correspondence: Jianrong Shi
Department of Clinical Laboratory, Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center For Child Health, Hangzhou, People’s Republic of China
Email [email protected]
Huifang Tang
Department of Pharmacology, School of Basic Medical Sciences, Zhejiang University, Hangzhou, People’s Republic of China
Email [email protected]
Abstract: Idiopathic pulmonary fibrosis (IPF) is a chronic progressive fibrosis disease caused by genetic susceptibility (causative) and other indirect risk factors such as smoking, micro-aspiration and air pollution. Repeated damage of lung epithelial cells can cause fibroblast activation and excessive collagen will lead the scar formation and severe fibrosis. It has been decades since drugs for the treatment of IPF were developed, but clinical choices were limited. Exchange Protein directly Activated by cAMP (EPAC), as a newly emerging cAMP (adenosine 3ʹ,5ʹ-cyclic monophosphate) downstream molecule, plays a vital role in the cellular pathways of IPF such as inhibiting fibroblast proliferation, stress fiber formation and epithelium cell adhesion, so it may be a novel target for drug development and treatment for curbing IPF. Here, we hypothesize that EPAC may participate in the signaling pathways related to IPF in different cell types (fibroblasts; airway smooth muscle cells; vascular endothelial cells; lung epithelial cells; macrophages; mesenchymal stem cells; T cells), thereby playing a potentially therapeutic role in resisting the process of fibrosis. We summarize the current correlation between EPAC and IPF in these different cell types, and further insights into EPAC will help to optimize the pharmacological treatment for IPF.
Keywords: EPAC, idiopathic pulmonary fibrosis, fibroblasts, cell types, cAMP, PKA
Introduction
IPF is the most common idiopathic interstitial pneumonia, the prevalence of IPF is 14–27.9 cases per 100,000 in the USA and 1.25–23 cases per 100,000 in Europe.1 Patients with IPF suffer from shortness of breath and restricted movement and continue to deteriorate, the prevalence and mortality rate will gradually increase with age.2 Notably, PF is more prevalent in men, with a male to female ratio that usually approaches 2:1.3 Occupational exposures to metal dust, plant dust or animal dust such as agriculture, hairdressing, stone cutting and polishing are closely related to IPF, especially the work of raising birds and exposure to plant or animal dust.4 These dusts contain dissociated silica which will be inhaled into the lungs along with the dust and can easily cause pulmonary silicosis (the symptoms are mainly IPF). The mechanism of pulmonary fibrosis includes repeated damage to alveolar epithelial cells (AECs) caused by various reasons, excessive accumulation of fibroblasts and activation of epithelial–mesenchymal transition (EMT).3 Due to excess deposition of extracellular matrix (ECM) in the lung, especially collagen (the ECM-related protein, including collagen Ⅰ, Ⅱ, III), leads to scar formation and loss of function such as decreased lung capacity, diffusion dysfunction and hypoxemia, which will accelerate the deterioration of the condition.5 In terms of medicine, there are few drugs with good effects. The currently recommended medicines are nintedanib and pirfenidone, but they are at a premium.6 These drugs just slow down the decline in lung capacity, but cannot reverse the fibrosis. The new treatment strategies including biomarkers guiding therapy, lung microbiome targeted therapy, lung transplantation, etc.7 are still under development. As a comorbidity of Coronavirus disease 2019 (COVID-19) pneumonia, pulmonary fibrosis (PF) disease has also received increasing attention from scientists.
EPAC can be found in various organs of the human body (lung, brain, renal) and it is an important downstream effector of cAMP, also known as cyclic adenylate binding protein or a guanine nucleotide exchange factor of Ras-like small GTPase.8 In 1998, Kawasaki et al found that EPAC has two isoforms: EPAC1 and EPAC2 (also known as cAMP-GEFⅠ and cAMP-GEFⅡ)8 (Figure 1). EPAC2 has three subtypes, namely EPAC2A, EPAC2B, EPAC2C.9,10 EPAC1 and EPAC2 are distributed in most areas of the cell, such as the cytoplasm, nuclear membrane, mitochondria and cytoskeleton.11 The distribution of EPAC in cell division cycles is dynamic, and the distribution characteristics of EPAC in cells are consistent with its biological effect diversity. In a prospective study of EPAC, by using green fluorescent protein (GFP) labeling and other technologies, researchers have found that increasing cAMP to a certain level can quickly activate EPAC and redistribute it in the cell.12 As an important bridge between cAMP and downstream signaling molecules, EPAC is of great significance for cAMP to modulate various cell biological effects in PF. For example, EPAC may cooperate with cAMP-dependent protein kinase (PKA) to achieve many effects of cAMP or achieve it independently, such as regulating macrophage inflammation, epithelium cell adhesion, fibroblast proliferation and differentiation.13 cAMP in low concentration can easily activate EPAC, which promotes fibroblast migration via repressor/activator site-binding protein 1 (Rap1), but inhibits collagen synthesis through a Rap1-independent pathway.14 In addition, EPAC1 can induce Rap1-dependent and integrin-mediated cell adhesion to fibronectin in epithelium cells.15
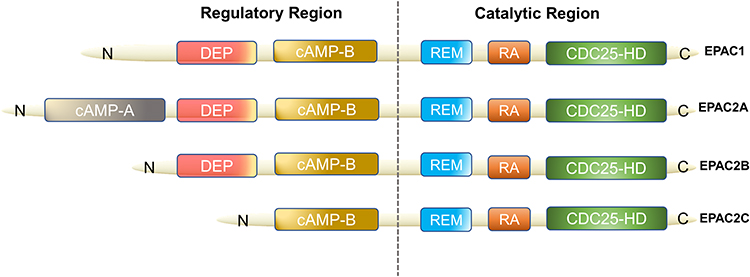 |
Figure 1 Domain architecture of EPAC isoforms. We divided them into two functional parts: regulatory region and catalytic region. The individual domains include: CDC25-HD, CDC25 homology domain (catalyzes Rap1 activation16); RA, Ras association domain; REM, Ras exchange motif; cAMP-B, cAMP-binding domains B; DEP, disheveled, EGL-10 and pleckstrin homology domain (localizes EPAC to the plasma membrane upon activation by cAMP17); cAMP-A, cAMP-binding domains A. EPAC2A has the longest sequence while EPAC2C is the shortest, EPAC2B has the resembled functional domain as EPAC1. |
In more than 20 years, EPAC has been shown to mediate a regulation in asthma and chronic obstructive pulmonary disease (COPD), and participate in various cell biological functions (airway smooth muscle cell relaxation, mesenchymal stem cell growth, etc.). One of the most classic EPAC/Rap1 functions is controlling the intercellular connections, regulating Rac1 to stabilize the function of the endothelial barrier.16 Recently, more and more evidence suggested that EPAC plays a role in IPF too. However, the mechanism of how EPAC affects the process of IPF is still unclear.
Hypothesis
We hypothesize that in idiopathic pulmonary fibrosis, EPAC may play a potential role in curbing the process of fibrosis based on the evidence that EPAC participates in the pathway of fibrosis process in different fibrosis-related cells (fibroblast; airway smooth muscle cell; endothelial cell; epithelium cell; macrophage; mesenchymal stem cell; T cell). At the same time, the immune and inflammatory response may also be involved in the linkage to affect pulmonary fibrosis.
Evaluation of the Hypothesis
EPAC Signaling in IPF Related Pulmonary Structured Cells
Fibroblasts
Pulmonary fibroblast is responsible for collagen production and ECM deposition and is directly related to the process of fibrosis. When PF is severe, the fibrosis area will become larger due to the excessive accumulation of fibroblasts and the myofibroblast production. A lot of research focuses on fibroblasts in PF, and it is vital to understand its mechanisms in this disease. Transforming growth factor-β (TGF-β) has emerged as one of the main mediators responsible for fibroblast activation,17,18 and with the stimulation of TGF-β, fibroblasts will express α-smooth muscle actin (α-SMA) and collagen, and become myofibroblasts that are more contractile, proliferative and secretory. All of this process involves the mediating role of EPAC1 and PKA. Huang et al studied in vitro the expression of EPAC at the cellular level by testing lung fibroblasts from IPF adult patients, and unveiled that EPAC1 was expressed in these cells while EPAC2 was not.19 Based on the current understanding of EPAC1 and EPAC2 in lung fibroblasts, it is possible to assign EPAC-dependent function to EPAC120.
Until now, some reports indicated that different subtypes of prostaglandins can regulate PF in different pathways, simultaneously involved in EPAC protein. Among the prostaglandin subtypes, prostaglandin E2 (PGE2) is more closely associated with fibroblasts in PF. For PGE2, it is a common substance in the study of PF. Some researchers conducted in vitro experiments on primary human fetal lung IMR-90 fibroblasts (a primary human fetal lung fibroblast line); they proved that activation of EPAC1 leads to Rap1 activation, cell migration and inhibition of proliferation, while PKA directly inhibits protein kinase C-δ (PKC-δ) and then reduces collagen expression, which are two completely different mechanisms19 (Figure 2). The common features of both are that they need to react with E-series of Prostaglandin Receptors type 2 (EP2) and sharing one signaling pathway (PGE2–EP2),21 then stimulatory G protein-α (Gαs) and adenyl cyclase (AC) are activated, together with adenosine triphosphate (ATP, produced by mitochondria) to increase the level of cAMP in fibroblasts.19 Finally, EPAC1 and PKA play a differential function, making the PGE2-related pathway relieve the progress of IPF. Besides, Haag et al testing the effectiveness of Butaprost (an EP2 receptor-special ligand) to inhibit [3H]-thymidine incorporation on the corresponding cAMP response element-binding protein (CREB) of EPAC1, EPAC2 and PKA, only found that EPAC1 can lead to a significant reduction while others cannot.22 Overall, EPAC1 is one of the key factors for EP2 receptor activation against lung fibroblast proliferation.22
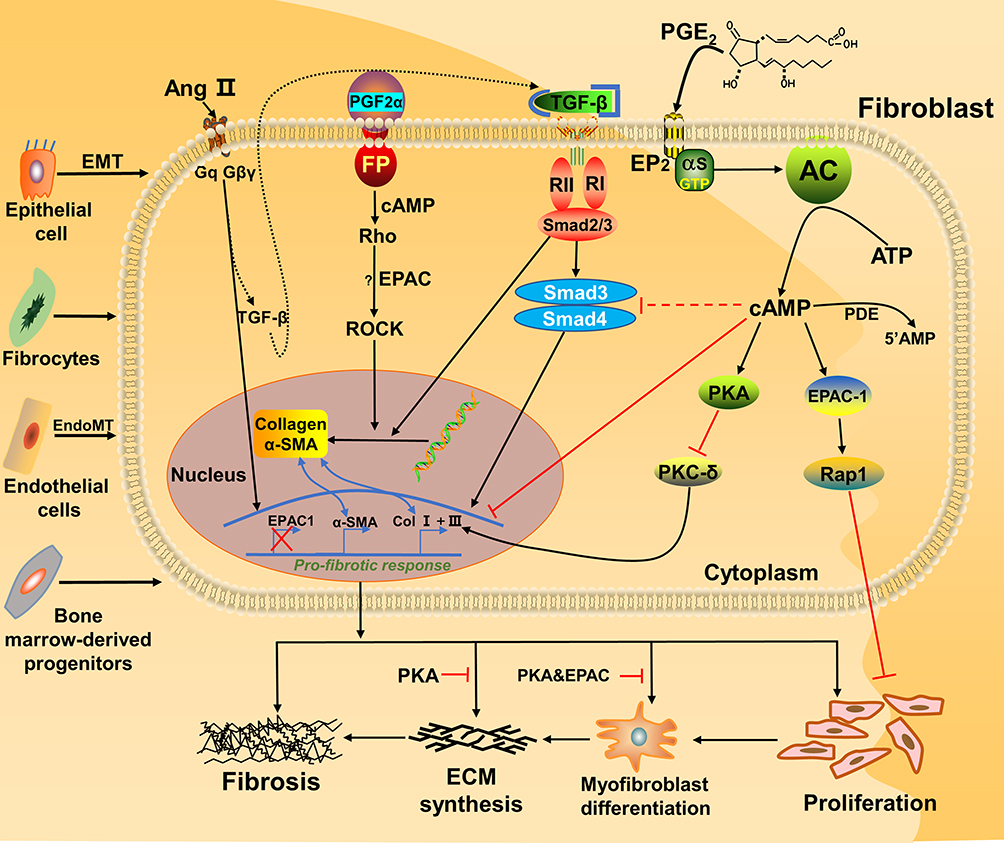 |
Figure 2 Fibroblasts involved in the EPAC pathway and connection with fibrosis. Activation of PKA and EPAC differentially inhibits specific fibroblast functions, activation of EPAC inhibits fibroblast proliferation, and both PKA and EPAC contribute to the inhibition of α-SMA expression and fibroblast transition to myofibroblasts.20 PGE2 inhibits lung fibroblast collagen expression and proliferation via independent cAMP effectors.19 Through FP receptor signals, PGF2α can enhance collagen synthesis in fibroblasts.23,56 Profibrotic effects of angiotensin Ⅱ (Ang Ⅱ) and TGF-β1 can potentially through SMAD inhibition, cAMP activation of EPAC1 and PKA lowers expression of collagens Ⅰ and III.57 Together, ECM synthesis, fibroblast proliferation and collagen production will aggravate fibrosis. |
In contrast, it has been found that prostaglandin F2α (PGF2α) can promote the development of PF. Based on the current knowledge, we summarized the pathway diagram of PGF2α about PF in vitro (Figure 2): PGF2α binds to the prostaglandin F receptor (FP) on the cell membrane surface and dependent on the Ras homolog gene/a Rho-associated coiled coil-forming protein kinase (Rho-ROCK) pathway. By activating the small GTPase Rho signaling (PGF2α) and SMADs pathway (TGF-β), they jointly induce the increase of collagen in fibroblasts.23 In turn, since the PF-resistant function of EPAC has been proved, we infer that EPAC still plays a vital role in Rho-ROCK signaling of the PGF2α pathway.
Collectively, these recent studies in human lung fibroblasts indicate that the anti-fibrotic properties of cAMP most likely rely on the interplay of distinct cAMP-sensitive key effector proteins, and that both PKA and EPAC may (differentially) contribute to the regulation of fibroblast proliferation and collagen synthesis.20 Under limited conditions, the current understanding of EPAC in lung fibroblasts is mainly based on the study of human embryonic lung, fetal lung fibroblasts and mouse lung fibroblasts cultured in vitro, and only a few papers have studied the (functional) role of EPAC in adult human lung fibroblasts. Accordingly, many fundamental functions still need to be further verified by testing adult lung-derived fibroblasts.
Airway Smooth Muscle Cells (ASMCs)
ASMCs are generally involved in smooth muscle proliferation, relaxation and migration, their dysfunction may cause bronchiectasis and chronic purulent inflammation of surrounding lung tissue,24 and fibrogenic factors such as TGF-β, platelet derived growth factor (PDGF) and tumor necrosis factor (TNF-α) could affect the ASMCs, amplify the dysfunction effects and deteriorate the process of IPF. Recently, EPAC began to emerge and can be linked to SMCs. Chen et al proved that in vivo, EPAC inhibitor ESI-09 promoted mice ASMC proliferation and collagen deposition, suggesting a protective effect of EPAC on airway remodeling.25
It is reported that an EPAC agonist, 8-pCPT-2′-O-Me-cAMP (8-pCPT), induced relaxation of intact rat aorta precontracted with noradrenalin or phenylephrine mediated via ASMCs in vivo.26 Further experiments proved that activation of EPAC leads to relaxation of ASM Ca2+-sensitized force through activation of Rap1 accompanied by the decrease in RhoA activity and myosin phosphatase targeting subunit (MYPT1) phosphorylation.27 In addition, on guinea pig and human intact tracheal SM tissue, EPAC activation by cAMP leads to relaxation via activation of Rac, and Rac inhibitor can reverse the effect.28 In human and mouse fundus SMCs, β-adrenaline-stimulated cAMP can send a signal through EPAC1 and PKA to reduce RhoA activity and induce relaxation, and after using 8-pCPT to stimulate ASM, RhoA activity is significantly reduced as well. Further investigation found that 8-pCPT inhibition of lysophosphatidic acid (LPA)-stimulated RhoA activity is abolished in Rap1B null cells and Rap1 activation by isoproterenol was inhibited by short hairpin RNA (shRNA) down-regulation of EPAC1 expression, demonstrating that this pathway is mainly dependent on EPAC/Rap1 instead of PKA/cyclic GMP-dependent protein kinase (PKG).27 The above indicated that EPAC could regulate ASMC proliferation, migration and relaxation in IPF, then repair the dysfunction and mitigate IPF progression, which is a noticeably promising field.
Vascular Endothelial Cells (VECs)
Hashimoto et al discovered for the first time that a considerable number of fibroblasts were transformed by TGF-β induced endothelial cells, named EndoMT (endothelial–mesenchymal transition), and excessive fibroblasts would accelerate the process of fibrosis.29 In addition, endothelial cells are rich in pulmonary vasculature and related to the endothelial barrier. Once barrier integrity is destroyed, it is easy to lead the flooding of underlying tissues,30 thereby causing collagen deposition and thickening alveolar capillary spaces, aggravating the course of inflammation and fibrosis. Sands et al found in vitro, a novel PKA-independent anti-inflammatory effect of cAMP elevation in vascular endothelial cells, which involves EPAC-mediated accumulation of GTP-bound Rap1a, leading to the ERK-dependent up-regulation of Suppressor of Cytokine Signaling 3 (SOCS-3) and the inhibition of Signal Transducer and Activator of Transcription 3 (STAT3) phosphorylation.31 Another study demonstrated that the EPAC agonist enhanced extracellular signal-regulated kinase (ERK) phosphorylation in endothelial cells, and in contrast, inhibition of mitogen-activated protein kinase/ERK kinase (MEK)/ERK signaling can increase the endothelial barrier stabilizing effect of the EPAC agonist.32 Additionally, in cell endothelial barrier function, after conducting EPAC stimulation experiments on human umbilical vein endothelial cells (HUVECs), the researchers found that EPAC-selective cAMP analog 8CPT-2Me-cAMP improves endothelial barrier function in repopulated lung scaffolds.33 Similarly, after treatment with EPAC agonists, the barrier function of induced pluripotent stem cell (iPSC) derived endothelial colony forming cells is enhanced, and remains for at least 60 hours.33 Moreover, in the dynamic regulation of the endothelial barrier, cAMP in the sub-plasma membrane cavity acts through EPAC to tighten cell adhesion, and reduce thrombin-induced hyperpermeability.30 Therefore, EPAC could indirectly resist fibrosis by regulating inflammation and endothelial barrier dysfunction in VECs.
Lung Epithelial Cells (LECs)
In the current study of the PF disease mechanism, repeated injury/stress/apoptosis of LECs, subsequent abnormal repairs can lead the EMT and matrix accumulation, eventually causing IPF and loss of pulmonary function.34,35 Within mink lung epithelial cells (Mv1Lu), EPAC1 signal transduction is involved in inhibiting TGF-β-dependent cell migration and adhesion, and endogenous TGFRI and EPAC1 can form a complex.36 EPAC inhibitor ESI-09 can cause increased stress fiber formation and junctional degradation on MLE-12, mouse alveolar epithelial cells,37 so EPAC may have a beneficial effect to help resist the IPF in LECs. Besides, EPAC is also involved in the anti-inflammatory effect of Quercetin such as inhibiting the release of keratinocyte-derived chemokine (KC) from MLE-12.38 However, in epithelial tumor cells, EPAC expression is increased: scientists found that EPAC plays a novel role in promoting PGE2-induced EMT in vitro, subsequent activation of β-catenin in non-small cell lung carcinoma (NSCLC) cells via Ezrin.39 Lim and Juhnn also reported that EPAC (by activating Rap1A) signaling mediates the ISO-induced inhibition of c-Raf and increases in histone deacetylase 6 (HDAC6) expression, eventually causing A549 cell migration.40 Although there is no direct evidence to support the key role in lung fibrosis, all this evidence suggested that EPAC may play an important role in regulating the function of epithelial cells.
Mesenchymal Stem Cells (MSCs)
MSCs are pluripotent stem cells and can be isolated from various tissues (bone marrow, blood, placenta, etc.), their migration, adhesion and itself contributing to myofibroblast formation, eventually promoting the process of PF, for example, lung resident perivascular GLI1+MSC-like cells significantly lead the myofibroblast production during bleomycin-induced PF in mice.41 In the study of human MSCs (hMSCs), Yu et al revealed EPAC activation can enhance hMSC migration and adhesion via SDF-1 (stromal cell derived factor 1).42 Willis et al found that exosomes derived from MSCs can improve lung morphology and growth, reduce lung fibrosis and remodel pulmonary vascular.43 These exosomes associated with apoptosis, cell growth, inflammation and match with miRNA in fibrosis-related signal transduction pathways such as Ras, Hippo and EPAC pathways, eventually play the role of anti-inflammatory, anti-fibrosis and promote the regeneration of bronchial stem cells in mice.44 In addition, Fujita et al found that in vivo, extracellular vesicles (EV) derived from MSCs improved lung tissue remodeling and fibrosis by inhibiting the inflammatory response and reducing oxidative stress.45 Shentu et al have proved it can interact with integrin β to reduce the level of fibronectin and collagen III, suppress the differentiation from lung fibroblasts into myofibroblasts and finally relieve PF.46 Due to the correlation of EPAC and cytokines mentioned above, the EPAC-related exosomes and EVs derived from MSCs are the hot topics in the PF field and further research is on the way.
EPAC Signaling in IPF Related Immune Cells
In the IPF process, different types of macrophages play multi-faceted roles in different phases. M2 macrophages or M2-like macrophages may recruit and regulate the transition from inflammation to fibrosis.47 Meantime, macrophages secrete various cytokines (such as TGF-β; IGF-1; PDGF) that can promote collagen production of fibroblast and lead the PF when excessive secretion happens.48 Calcitonin gene-related peptide (CGRP) reduces inflammation and limits fibrosis by inducing cAMP/EPAC1 and cAMP/PKA pathways in vivo,47 and EPAC may help Quercetin suppress inflammation and delay the transition of inflammation to fibrosis via the CGRP system in macrophages. Meanwhile, the signaling of intracellular cAMP in macrophages, up-regulation of EPAC1, not PKA, can particularly suppress phagocytosis.49 Together, EPAC showed a protective function to help curb the fibrosis. Because the M2 macrophages did not express CGRP receptors, we speculated that the cAMP pathway may reverse the macrophage M2 polarization process. In monocytes, activation of EPAC1 enhances monocyte adhesion to VECs via fibronectins,50 and the impact of monocytes can regulate inflammation and its transition to IPF. Therefore, macrophages also could be used as an entry point to cut off the correlation between inflammation and fibrosis.
T cells play different roles in different stages of PF. In the early stage of PF, differentiated regulatory T cell (Tregs) promote fibrosis by secreting the pro-fibrotic factors (PDGF-B, TGF-β); while in the advanced stage, blocking the activity of Tregs will result in the aggregation of effector T cells and the continuous expression of pro-fibrotic T effector cytokines (IL-4, IFN-γ, IL-13, IL-17, etc.), ultimately aggravating PF.51,52 Previous research showed that in mice expressing active Rap1 in their T cell compartment both the thymocytes and mature T cells exhibited increased integrin-mediated cell adhesion, in addition, these cells showed enhanced T cell receptor–mediated responses.53 Based on this result in vivo, we speculated that the increase in cAMP level can regulate EPAC and Rap1 to affect integrin-mediated cell adhesion to fibronectin. Notably, EPAC is particularly highly expressed in the ovary, thyroid, kidney, adrenal gland and brain,8,54 and it is expected that the cAMP–EPAC pathway leading to integrin activation may operate particularly in these tissues. Meanwhile, it has been reported that in vivo, the deletion of EPAC1 in T cells could increase the level of Smad7 protein, reduce the expression of Smad4 and inhibit the phosphorylation of Smad2. Consequently, CD4+ T cells were desensitized to TGF-β1,55 which means resistant to the fibrosis and inflammation. Besides, inhibition of EPAC1 leads to resistance of Teff (activation and proliferation) to Treg suppression and concurrently diminishes the suppressive potency of the latter,55 so EPAC may have a harmful effect on resisting fibrosis through the immune system in the early stage while on the contrary for the advanced stage. In summary, restoring the activation of Treg cells and regulating EPAC in T cells may become a new strategy for the prevention and treatment of IPF.
A summary of the above seven cell types’ correlation with EPAC and IPF is shown in Figure 3.
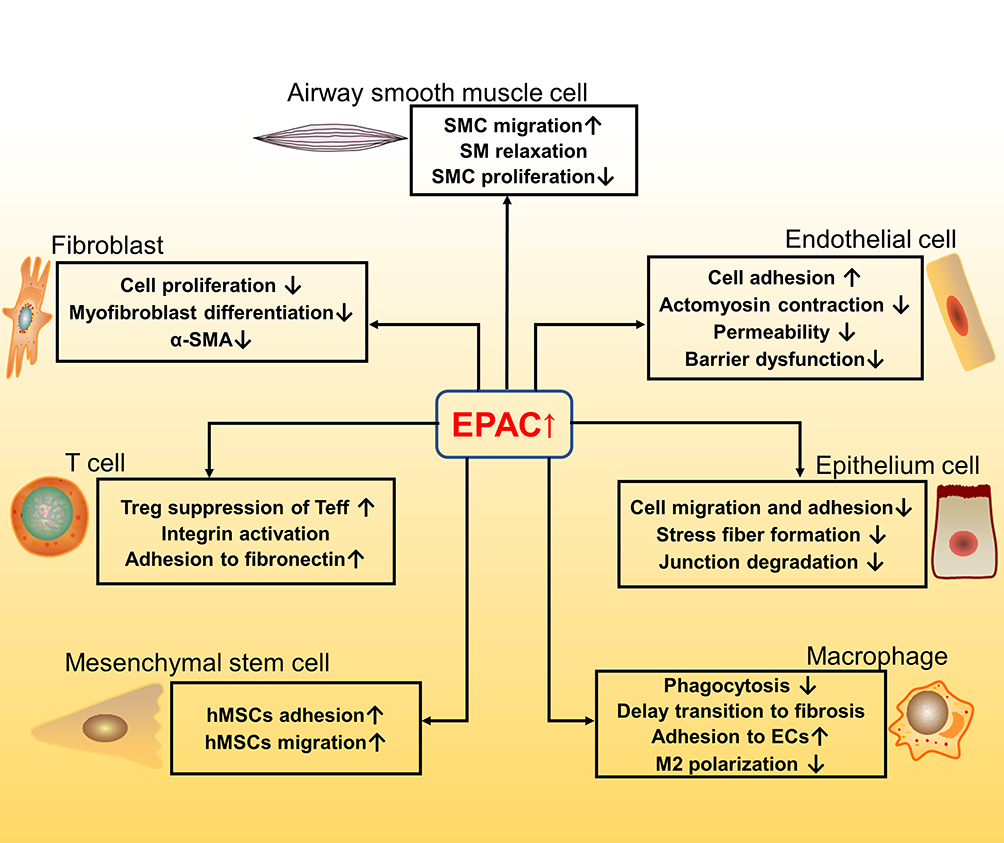 |
Figure 3 Pathways and function of EPAC related to fibrosis in different cell types (fibroblast; airway smooth muscle cell; endothelial cell; epithelium cell; macrophage; mesenchymal stem cell; T cell). ↑ means increase or up-regulated; ↓ means decrease or down-regulated. |
Consequences of Hypothesis and Discussion
The discovery of EPAC enriches the role of cAMP signaling and many physiological processes (anti-inflammatory, anti-fibrosis, regulating cell differentiation, airway remodeling, endothelial dysfunction, etc.). EPAC can be used to inhibit fibroblast proliferation through Rap1/EP2 receptors and work with SMAD pathways to regulate cell migration as a way of treatment. In addition, different subtypes of prostaglandins can play opposite roles in fibroblasts through EPAC: PGE2 can delay the development of PF, while PGF2α promotes it. EPAC-related pathways have been found in various cell types such as epithelial cells (decrease stress fiber formation), macrophages (delay transition to fibrosis) and endothelial cells (repair barrier dysfunction), all related to IPF. Therefore, based on the current references and data, EPAC can achieve anti-fibrosis function through the IPF-related signaling pathways of different cell types mentioned above. Meantime, the immune and inflammatory systems also play a role in it; we believe that this is a complex, comprehensive regulation process that is linked by multiple cells, proteins, cytokines and various biochemical pathways together.
Although the current research on EPAC has made progress in the field of fibrosis, but the specific cell pathways and pharmacological basis of Epac in IPF are still unknown. Meanwhile, almost presented data come from cell, and there is a lack of animal data and clinical data. Therefore, it is necessary to continue the EPAC research in IPF, and the conclusion also needs to be confirmed by animal model and clinical trials. A more systematic understanding of EPAC's role in IPF can help to develop diagnostic and therapeutic methods specifically for this type of protein as soon as possible, and provide a pharmacological basis for the development of targeted drugs.
Abbreviations
AC, adenyl cyclase; AECs, alveolar epithelial cells; Ang II, angiotensin II; ASMCs, airway smooth muscle cells; ATP, adenosine triphosphate; cAMP, adenosine 3ʹ,5ʹ-cyclic monophosphate; CGRP, calcitonin gene-related peptide; COPD, chronic obstructive pulmonary disease; COVID-19, Coronavirus disease 2019; CREB, cAMP response element-binding protein; ECM, extracellular matrix; ECs, endothelial cells; EMT, epithelial–mesenchymal transition; EndoMT, endothelial–mesenchymal transition; EP2, E prostanoid 2; EPAC, Exchange Protein directly Activated by cAMP; ERK, extracellular signal-regulated kinase; EV, extracellular vesicles; FP, prostaglandin F receptor; GEF, guanine nucleotide exchange factor; GFP, green fluorescent protein; GTPase, guanosine triphosphatase; Gαs, stimulatory G protein-α; HDAC6, histone deacetylase 6; HUVECs, human umbilical vein endothelial cells; IFN-γ, interferon-γ; IGF-1, insulin-like growth factor-1; IL, interleukin; IP, prostacyclin receptor; IPF, idiopathic pulmonary fibrosis; KC, keratinocyte-derived chemokine; LPA, lysophosphatidic acid; MEK, mitogen-activated protein kinase/ERK kinase; MLE12, murine lung epithelial 12; MSCs, mesenchymal stem cells; MYPT1, myosin phosphatase targeting subunit; NSCLC, non-small cell lung carcinoma; PDGF, platelet derived growth factor; PF, pulmonary fibrosis; PGE2, prostaglandin E2; PGF2α, prostaglandin F2α; PKA, cAMP-dependent protein kinase; PKC-δ, protein kinase C-δ; PKG, cyclic GMP-dependent protein kinase; Rac, ras-related C3 botulinum toxin substrate; Rap, repressor/activator site-binding protein; Rho-ROCK, Ras homolog gene/a Rho-associated coiled coil-forming protein kinase; SDF-1, stromal cell derived factor 1; shRNA, short hairpin RNA; SMAD, Drosophila mothers against decapentaplegic; SOCS-3, Suppressor of Cytokine Signaling 3; STAT3, Signal Transducer and Activator of Transcription 3; TGF-β, transforming growth factor-β; TNF, tumor necrosis factor; Treg, regulatory T cell; USA, United States of America; α-SMA, α-smooth muscle actin.
Acknowledgments
The study was supported by the National Natural Science Foundation of China (81402989 and 81570056), and National Natural Science Foundation of Zhejiang provincial (LY18H310002 and LGD20H010002).
Disclosure
The authors declare no conflict of interest.
References
1. Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev. 2012;21(126):355–361. doi:10.1183/09059180.00002512
2. Ahluwalia N, Shea BS, Tager AM. New therapeutic targets in idiopathic pulmonary fibrosis. Aiming to rein in runaway wound-healing responses. Am J Respir Crit Care Med. 2014;190(8):867–878. doi:10.1164/rccm.201403-0509PP
3. Meyer KC. Pulmonary fibrosis, part I: epidemiology, pathogenesis, and diagnosis. Expert Rev Respir Med. 2017;11(5):343–359. doi:10.1080/17476348.2017.1312346
4. Trethewey SP, Walters GI. The role of occupational and environmental exposures in the pathogenesis of idiopathic pulmonary fibrosis: a narrative literature review. Medicina (Kaunas). 2018;54(6).
5. Lederer DJ, Martinez FJ, Longo DL. Idiopathic pulmonary fibrosis. N Engl J Med. 2018;378(19):1811–1823. doi:10.1056/NEJMra1705751
6. Raghu G, Rochwerg B, Zhang Y, et al., European Respiratory, S. Japanese Respiratory, S. Latin American Thoracic, A. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. an update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192(2):e3–19. doi:10.1164/rccm.201506-1063ST
7. Somogyi V, Chaudhuri N, Torrisi SE, Kahn N, Muller V, Kreuter M. The therapy of idiopathic pulmonary fibrosis: what is next? Eur Respir Rev. 2019;28(153):153. doi:10.1183/16000617.0021-2019
8. Kawasaki H, Springett GM, Mochizuki N, et al. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282(5397):2275–2279. doi:10.1126/science.282.5397.2275
9. Niimura M, Miki T, Shibasaki T, Fujimoto W, Iwanaga T, Seino S. Critical role of the N-terminal cyclic AMP-binding domain of Epac2 in its subcellular localization and function. J Cell Physiol. 2009;219(3):652–658. doi:10.1002/jcp.21709
10. Hoivik EA, Witsoe SL, Bergheim IR, et al. DNA methylation of alternative promoters directs tissue specific expression of Epac2 isoforms. PLoS One. 2013;8(7):e67925. doi:10.1371/journal.pone.0067925
11. Qiao J, Mei FC, Popov VL, Vergara LA, Cheng X. Cell cycle-dependent subcellular localization of exchange factor directly activated by cAMP. J Biol Chem. 2002;277(29):26581–26586. doi:10.1074/jbc.M203571200
12. Ponsioen B, Gloerich M, Ritsma L, Rehmann H, Bos JL, Jalink K. Direct spatial control of Epac1 by cyclic AMP. Mol Cell Biol. 2009;29(10):2521–2531. doi:10.1128/MCB.01630-08
13. Roscioni SS, Elzinga CR, Schmidt M. Epac: effectors and biological functions. Naunyn Schmiedebergs Arch Pharmacol. 2008;377(4–6):345–357. doi:10.1007/s00210-007-0246-7
14. Yokoyama U, Patel HH, Lai NC, Aroonsakool N, Roth DM, Insel PA. The cyclic AMP effector Epac integrates pro- and anti-fibrotic signals. PNAS. 2008;105(17):6386–6391. doi:10.1073/pnas.0801490105
15. Rangarajan S, Enserink JM, Kuiperij HB, et al. Cyclic AMP induces integrin-mediated cell adhesion through Epac and Rap1 upon stimulation of the beta 2-adrenergic receptor. J Cell Biol. 2003;160(4):487–493. doi:10.1083/jcb.200209105
16. Aslam M, Tanislav C, Troidl C, Schulz R, Hamm C, Gunduz D. cAMP controls the restoration of endothelial barrier function after thrombin-induced hyperpermeability via Rac1 activation. Physiol Rep. 2014;2(10):10. doi:10.14814/phy2.12175
17. Bartram U, Speer CP. The role of transforming growth factor β in lung development and disease. Chest. 2004;125(2):754–765. doi:10.1378/chest.125.2.754
18. Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18(7):816–827. doi:10.1096/fj.03-1273rev
19. Huang SK, Wettlaufer SH, Chung J, Peters-Golden M. Prostaglandin E2 inhibits specific lung fibroblast functions via selective actions of PKA and Epac-1. Am J Respir Cell Mol Biol. 2008;39(4):482–489. doi:10.1165/rcmb.2008-0080OC
20. Dekkers BG, Racke K, Schmidt M. Distinct PKA and Epac compartmentalization in airway function and plasticity. Pharmacol Ther. 2013;137(2):248–265.
21. White ES, Atrasz RG, Dickie EG, et al. Prostaglandin E2 inhibits fibroblast migration by E-prostanoid 2 receptor–mediated increase in PTEN activity. Am J Respir Cell Mol Biol. 2005;32(2):135–141. doi:10.1165/rcmb.2004-0126OC
22. Haag S, Warnken M, Juergens UR, Racke K. Role of Epac1 in mediating anti-proliferative effects of prostanoid EP(2) receptors and cAMP in human lung fibroblasts. Naunyn Schmiedebergs Arch Pharmacol. 2008;378(6):617–630. doi:10.1007/s00210-008-0334-3
23. Oga T, Matsuoka T, Yao C, et al. Prostaglandin F(2alpha) receptor signaling facilitates bleomycin-induced pulmonary fibrosis independently of transforming growth factor-beta. Nat Med. 2009;15(12):1426–1430. doi:10.1038/nm.2066
24. Amrani Y, Panettieri RA, Ramos-Ramirez P, Schaafsma D, Kaczmarek K, Tliba O. Important lessons learned from studies on the pharmacology of glucocorticoids in human airway smooth muscle cells: too much of a good thing may be a problem. Pharmacol Ther. 2020;213:107589. doi:10.1016/j.pharmthera.2020.107589
25. Chen YF, Huang G, Wang YM, et al. Exchange protein directly activated by cAMP (Epac) protects against airway inflammation and airway remodeling in asthmatic mice. Respir Res. 2019;20(1):285. doi:10.1186/s12931-019-1260-2
26. Sukhanova IF, Kozhevnikova LM, Popov EG, Podmareva ON, Avdonin PV. Activators of Epac proteins induce relaxation of isolated rat aorta. Dokl Biol Sci. 2006;411(1):441–444. doi:10.1134/S0012496606060044
27. Zieba BJ, Artamonov MV, Jin L, et al. The cAMP-responsive Rap1 guanine nucleotide exchange factor, Epac, induces smooth muscle relaxation by down-regulation of RhoA activity. J Biol Chem. 2011;286(19):16681–16692. doi:10.1074/jbc.M110.205062
28. Roscioni SS, Maarsingh H, Elzinga CR, et al. Epac as a novel effector of airway smooth muscle relaxation. J Cell Mol Med. 2011;15(7):1551–1563. doi:10.1111/j.1582-4934.2010.01150.x
29. Hashimoto N, Phan SH, Imaizumi K, et al. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2010;43(2):161–172. doi:10.1165/rcmb.2009-0031OC
30. Sayner SL. Emerging themes of cAMP regulation of the pulmonary endothelial barrier. Am J Physiol Lung Cell Mol Physiol. 2011;300(5):L667–78. doi:10.1152/ajplung.00433.2010
31. Sands WA, Woolson HD, Milne GR, Rutherford C, Palmer TM. Exchange protein activated by cyclic AMP (Epac)-mediated induction of suppressor of cytokine signaling 3 (SOCS-3) in vascular endothelial cells. Mol Cell Biol. 2006;26(17):6333–6346. doi:10.1128/MCB.00207-06
32. Gunduz D, Troidl C, Tanislav C, Rohrbach S, Hamm C, Aslam M. Role of PI3K/Akt and MEK/ERK signalling in cAMP/Epac-mediated endothelial barrier stabilisation. Front Physiol. 2019;10:1387. doi:10.3389/fphys.2019.01387
33. Yuan Y, Engler AJ, Raredon MS, et al. Epac agonist improves barrier function in iPSC-derived endothelial colony forming cells for whole organ tissue engineering. Biomaterials. 2019;200:25–34. doi:10.1016/j.biomaterials.2019.02.005
34. Selman M, Rojas M, Mora AL, Pardo A. Aging and interstitial lung diseases: unraveling an old forgotten player in the pathogenesis of lung fibrosis. Semin Respir Crit Care Med. 2010;31(5):607–617. doi:10.1055/s-0030-1265901
35. Selman M, Pardo A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am J Respir Crit Care Med. 2014;189(10):1161–1172. doi:10.1164/rccm.201312-2221PP
36. Conrotto P, Yakymovych I, Yakymovych M, Souchelnytskyi S. Interactome of transforming growth factor-beta type I receptor (TbetaRI): inhibition of TGFbeta signaling by Epac1. J Proteome Res. 2007;6(1):287–297. doi:10.1021/pr060427q
37. Young BM, Shankar K, Tho CK, Pellegrino AR, Heise RL. Laminin-driven Epac/Rap1 regulation of epithelial barriers on decellularized matrix. Acta Biomater. 2019;100:223–234. doi:10.1016/j.actbio.2019.10.009
38. Wang XF, Song SD, Li YJ, et al. Protective effect of quercetin in LPS-induced murine acute lung injury mediated by cAMP-Epac pathway. Inflammation. 2018;41(3):1093–1103. doi:10.1007/s10753-018-0761-3
39. Jansen SR, Poppinga WJ, de Jager W, et al. Epac1 links prostaglandin E2 to beta-catenin-dependent transcription during epithelial-to-mesenchymal transition. Oncotarget. 2016;7(29):46354–46370. doi:10.18632/oncotarget.10128
40. Lim JA, Juhnn YS. Isoproterenol increases histone deacetylase 6 expression and cell migration by inhibiting ERK signaling via PKA and Epac pathways in human lung cancer cells. Exp Mol Med. 2016;48(1):e204. doi:10.1038/emm.2015.98
41. El Agha E, Kramann R, Schneider RK, et al. Mesenchymal stem cells in fibrotic disease. Cell Stem Cell. 2017;21(2):166–177. doi:10.1016/j.stem.2017.07.011
42. Yu JL, Deng R, Chung SK, Chan GC. Epac activation regulates human mesenchymal stem cells migration and adhesion. Stem Cells. 2016;34(4):948–959. doi:10.1002/stem.2264
43. Willis GR, Fernandez-Gonzalez A, Anastas J, et al. Mesenchymal stromal cell exosomes ameliorate experimental bronchopulmonary dysplasia and restore lung function through macrophage immunomodulation. Am J Respir Crit Care Med. 2018;197(1):104–116. doi:10.1164/rccm.201705-0925OC
44. Tan JL, Lau SN, Leaw B, et al. Amnion epithelial cell-derived exosomes restrict lung injury and enhance endogenous lung repair. Stem Cells Transl Med. 2018;7(2):180–196. doi:10.1002/sctm.17-0185
45. Fujita Y, Kadota T, Araya J, Ochiya T, Kuwano K. Clinical application of mesenchymal stem cell-derived extracellular vesicle-based therapeutics for inflammatory lung diseases. J Clin Med. 2018;7(10):10. doi:10.3390/jcm7100355
46. Shentu TP, Huang TS, Cernelc-Kohan M, et al. Thy-1 dependent uptake of mesenchymal stem cell-derived extracellular vesicles blocks myofibroblastic differentiation. Sci Rep. 2017;7(1):18052. doi:10.1038/s41598-017-18288-9
47. Hartopo AB, Emoto N, Vignon-Zellweger N, et al. Endothelin-converting enzyme-1 gene ablation attenuates pulmonary fibrosis via CGRP-cAMP/EPAC1 pathway. Am J Respir Cell Mol Biol. 2013;48(4):465–476. doi:10.1165/rcmb.2012-0354OC
48. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–737. doi:10.1038/nri3073
49. Aronoff DM, Canetti C, Serezani CH, Luo M, Peters-Golden M. Cutting edge: macrophage inhibition by cyclic AMP (cAMP): differential roles of protein kinase A and exchange protein directly activated by cAMP-1. J Immunol. 2005;174(2):595–599. doi:10.4049/jimmunol.174.2.595
50. Lorenowicz MJ, van Gils J, de Boer M, Hordijk PL, Fernandez-Borja M. Epac1-Rap1 signaling regulates monocyte adhesion and chemotaxis. J Leukoc Biol. 2006;80(6):1542–1552. doi:10.1189/jlb.0506357
51. Cao Q, Wang Y, Zheng D, et al. IL-10/TGF-beta-modified macrophages induce regulatory T cells and protect against adriamycin nephrosis. J Am Soc Nephrol. 2010;21(6):933–942. doi:10.1681/ASN.2009060592
52. Lo R, Lecocq S, Uwambayinema M, et al. Platelet-derived growth factor-producing CD4+ Foxp3+ regulatory T lymphocytes promote lung fibrosis. Am J Respir Crit Care Med. 2011;184(11):1270–1281. doi:10.1164/rccm.201103-0516OC
53. Sebzda E, Bracke M, Tugal T, Hogg N, Cantrell DA. Rap1A positively regulates T cells via integrin activation rather than inhibiting lymphocyte signaling. Nat Immunol. 2002;3(3):251–258. doi:10.1038/ni765
54. de-Rooij J, Zwartkruis F, Verheijen M, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP (see comments). Nature. 1998;396(6710):474–477. doi:10.1038/24884
55. Almahariq M, Mei FC, Wang H, et al. Exchange protein directly activated by cAMP modulates regulatory T-cell-mediated immunosuppression. Biochem J. 2015;465(2):295–303. doi:10.1042/BJ20140952
56. Olman MA. Beyond TGF-beta: a prostaglandin promotes fibrosis. Nat Med. 2009;15(12):1360–1361. doi:10.1038/nm1209-1360
57. Insel PA, Murray F, Yokoyama U, et al. cAMP and Epac in the regulation of tissue fibrosis. Br J Pharmacol. 2012;166(2):447–456. doi:10.1111/j.1476-5381.2012.01847.x
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.