")
Back to Journals » Pathology and Laboratory Medicine International » Volume 14
The Magnitude of Hereditary Spherocytosis Among Human Immunodeficiency Virus-Infected Adults Attending University of Gondar Comprehensive Specialized Hospital Northwest Ethiopia 2021 GC, Cross-Sectional Study Design
Authors Sahile Kebede S , Yalew A, Yesuf T, Mesfin Bambo G , Duguma T , Woldu B
Received 21 March 2022
Accepted for publication 2 September 2022
Published 12 September 2022 Volume 2022:14 Pages 15—23
DOI https://doi.org/10.2147/PLMI.S366451
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Paul Zhang
Samuel Sahile Kebede,1 Aregawi Yalew,2 Tesfaye Yesuf,3 Getachew Mesfin Bambo,1 Tadesse Duguma,1 Berhanu Woldu2
1Department of Medical Laboratory Sciences, College of Medicine and Health Sciences, Mizan Tepi University, Mizan, Ethiopia; 2Department of Hematology and Immunohematology, School of Biomedical and Laboratory Sciences, University of Gondar, Gondar, Ethiopia; 3Department of Internal Medicine School of Medicine, University of Gondar, Gondar, Ethiopia
Correspondence: Samuel Sahile Kebede, Tel +251917768545, Email [email protected]
Background: Hereditary spherocytosis is a type of hemolytic anemia characterized by a clinically heterogeneous, genetically defined red blood cell membrane abnormality that results in hemolytic crisis. The inheritance of HS is autosomal dominant in 80% of affected individuals and recessive genes or sporadic type in the remaining 20%.
Objective: The main aim of this study was to determine the magnitude of immune hereditary spherocytosis among human immunodeficiency virus-infected adults at the University of Gondar comprehensive specialized hospital in northwest Ethiopia from March to April 2021.
Methods: An institution-based cross-sectional study was conducted on 358 human immunodeficiency virus-infected adults selected by systematic random sampling at the University of Gondar comprehensive specialized hospital from March to April 2021. Data for socio-demographic data were collected by structured pretested questionnaire. Five ml of venous blood was drawn from each participant and analyzed by Unicel DHX 800 hematology analyzer, and blood film examination and antihuman globulin test were performed to exclude immune hemolytic anemia. Data was entered into Epidata version 4.6 and analyzed by STATA version 14. Descriptive statistics were computed and drawn in charts and graphs.
Results: The prevalence of hereditary spherocytosis was 2 of 358 participants. Both individuals who developed hereditary spherocytosis were females and in the age group of 22 to 35. The overall prevalence of anemia was 91 (25.42%). Of those anemic study population 3 (3.29%), 28 (30.77%), and 60 (65.93%), respectively, had severe, moderate, and mild anemia.
Conclusion and Recommendation: Hereditary spherocytosis is a less frequent condition in human immunodeficiency virus-infected adults. In these patients, early detection and treatment are necessary at the familial level by using a test algorithm.
Keywords: anemia, hereditary spherocytosis, spherocytosis, HIV/AIDS
Introduction
Hereditary spherocytosis is hemolytic anemia characterized by a clinically heterogeneous, genetically defined red blood cell membrane abnormality. Hemolytic crisis due to hereditary spherocytosis is a condition that can be inherited through families and result from destructed red blood cells.1 It is widely common in European Caucasians with the estimated prevalence in the Caucasian population ranging from 1:2000 to 1:5000.2,3 Although a vast number of asymptomatic individuals are unaware of their condition and most patients have well-compensated hemolytic anemia, symptoms range from asymptomatic forms to very severe conditions requiring splenectomy and regular transfusions.4
Hemolytic crisis due to hereditary spherocytosis can occur due to defects in Spectrin or spectrin-ankyrin which are more frequent in HS, suggesting that they could be the root cause of spherocyte development. Lack of or instability of ankyrin, which acts as Spectrin’s major membrane attachment point, is another possible cause of Spectrin shortage. There is a deficiency in the binding of Spectrin to protein 4.1, the primary component of the skeletal structure, in the red-cell membrane skeleton. This results in red cells being trapped in the spleen cords becoming more fragile and deformable.5,6
As familial hemolytic anemia, the hallmarks of this family hemolytic illness include anemia, recurrent jaundice, and splenomegaly, all of which are caused by increased red blood cell destruction, which has a wide range of clinical manifestations.5,7 HS is the most prevalent hemolytic anemia due to a red cell membrane abnormality and caused by mutations in the genes ankyrin 1 gene (ANK1), erythrocyte protein band 3 (EPB3), erythroblasts and protein 4.2 (ELB4.2), Spectrin alpha erythrocyte 1 (SPTA1), and SPTB. The illness due to HS varies clinically, biochemically, and genetically. However, it generally occurs at which osmotically fragile cells are selectively trapped and killed in the spleen.8,9
The inheritance of HS is autosomal dominant in 80% of affected individuals and recessive genes or sporadic type in the remaining 20%.2,10 There was no evidence of a link between HS and HIV co-infection that could be located in the literature. HS is caused by a particular genetic abnormality, and there is no link between HIV and HS.3 Furthermore, HIV is linked to autoimmune hemolytic anemia (AIHA), which, like HS, is characterized by hemolytic and spherocyte on a peripheral smear, but unlike HS, AIHA has a positive Coombs test.11
Although there is no direct relationship between HIV and HS, HIV aggravating HS has significant clinical implications and therapeutic choices. This adds to the difficulty, and the practitioner should be aware of it, especially in places with high HIV seroprevalence, where this uncommon relationship may occur.12 This study aims to determine the prevalence of HS among HIV-infected adults attending UOGCSH, North West Ethiopia.
Materials and Methods
Study Design and Period
An institutional-based cross-sectional study was conducted to determine the magnitude of hereditary spherocytosis among HIV-infected adults attending UOGCSH North West Ethiopia from March to April 2021.
Population
Source population
All HIV-infected adult individuals attending ART clinic in UOGCSH, North West Ethiopia.
Study Population
All HIV-infected adult individuals attending ART clinic at UOGCSH during a time of data collection can be used as the study population.
Inclusion Criteria and Exclusion Criteria
Inclusion Criteria
All HIV-infected individuals who were greater than or equal to 15 years or older, who had a confirmed HIV infection upon follow-up at the UOGCSH, and who had clinical data and laboratory data such as viral load and CD4 counts in the record within the last six months of data collection time were included in the study.
Exclusion Criteria
Individuals who had been seriously ill and unable to respond and give blood specimens were excluded from the study.
Sample Size Calculation and Sampling Technique
The required sample size for this study was calculated using the single population proportion formula and by considering the following assumptions. Since there is no study conducted to show the prevalence of HS, the sample size can be calculated by using 50% proportion with 95% confidence interval and 5% marginal error, sample size (n) was determined using the following statistical formula. This gives 384 since the total population was less than ten thousand, the population correction factor was used then gives 358. A convenient sampling technique was used to select study participants.
Data Collection Tools and Methods
Sociodemographic and Clinical Data Collection
The data was collected by structured questionnaire, and collection was performed by trained expert nurses. The questionnaire had three parts including sociodemographic and clinical for HS. The questionnaire was translated into the Amharic language. Sociodemographic data such as age, sex, residence, marital status, education, and religion were collected via face-to-face interview with study subjects. The clinical data such as CD4 result, and viral load were collected from records.
Sample Collection Procedures and Hematological Analysis
Blood Collection Procedures
About 5mL of blood was collected with sterile syringe and needle by expert medical laboratory technologist into study participant code number labeled EDTA anticoagulant test tube. The collected blood sample was delivered to the hematology laboratory for analysis of hematological parameters, DAT, and blood film preparation. The blood was transported to the hematology laboratory within 1 to 2 hours and the analysis was performed. From the collected blood sample, hematological analysis was performed, and then, blood film was prepared from the remnant sample. Finally, DAT was performed on the rest blood sample to exclude immune hemolytic anemia.
Hematological Analysis
The hematological analysis was performed on a blood sample in an EDTA anti-coagulated test tube to confirm the presence of HS by following standard operating procedures. Hb measurement, mean reticulocyte volume, RBC count, and RBC indices such as MCV, MCH, and MCHC were performed by using an automated hematology analyzer (Unicel DxH800, Danaher Corporation, Beckman Coulter, United States of America (USA)). Unicel DxH800 determines RBC count, reticulocyte count, immature reticulocyte fraction, and nucleated RBC on whole blood by impedance principle and leukocyte 5-part differential (Diff) and platelet by flow cytometry or light scattering principle the blood sample is suspended in diluent and passes through the apparatus causing direct current resistance.13 Change in blood cell size is detected as the electrical pulse and blood cell count is measured by counting pulse.14
Blood Film Examination
After the hematological parameter was performed, the remaining EDTA blood was used for blood film examination. A thin blood smear was prepared by wedge method by putting a drop of blood on the slide about 1–2cm from the end of the slide and making smear by another smooth-edged slide as spreader at an approximate angle of 30° on three-fourth (¾) of the length of the slide. The prepared smear was air-dried by placing the smear film side up on a staining rack. The dried smear was covered with filtered undiluted wright stain, left for 1minute, washed then dried, and examined by using oil immersion (100x objective) on the microscope. The morphology was examined by a trained technologist for the presence of features of hemolysis. In blood film, the presence of spherocyte, and nucleated RBC were evidence of hemolysis, and in the meantime for cross-checking for morphology with analyzer result was performed.
Coombs Test (DAT)
Three percent of washed blood suspension was used for direct anti-globin test (DAT) for detection of coated antibody on the surface of red cells that results in immune hemolysis. Coombs test (DAT) was performed based on the principle of a hemagglutination test. Two drops of the anti-globulin reagent were added to two drops of the three percent of red cell suspension into the test tube. Polyspecific anti-human globulin antiIgG-C3d acts as a link between the antibodies and the complement coating of neighboring RBC and induces agglutination. The test tube would be immediately centrifuged after thorough mixing, and finally, reading for the presence of agglutination was examined microscopically and then reported as positive or negative for DAT.
Hereditary Spherocytosis
Finally, hereditary spherocytosis was diagnosed from the result of hematological parameters, blood smear, and Coombs test results. It was defined as low Hb, normocytic or macrocytic red cell, the feature of hemolysis on blood film such as spherocytosis, increased MCH, RDW, and negative for direct anti-human globulin test. The test was finally confirmed for the presence of HS by a laboratory technologist.
Quality Management of Laboratory Tests and Data
Quality Assurance for Sociodemographic Data
Before data collection, training was given to the data collectors to ensure the reliability and validity of the data to reduce technical and observation bias. The questionnaires were tested (pretest) on randomly selected patients from the study site for reliability and validity before they were used for actual data collection. To check language translation information quality, the translated questionnaire was reviewed by three individuals and retranslated back to the English language from Amharic. The validity of the information was checked again.
Quality Control for Hematology Analyzer
Quality control for working equipment and reagents was ensured using standard controls as well as standard operating procedures. For Unicel DHX800 hematology analyzer normal background reading was checked daily, and the performance was checked by low, normal, and high controls. The result of each test was properly recorded.
Quality Control for Coombs Test
The quality control was done for DAT, by using an Rh-positive blood sample coated with an anti-D for positive control, and negative control by Rh-negative blood was used. The results of both controls were then properly recorded (annex v).
Quality Control for Microscopy and Wright Stain Reagent
The preventive maintenance was performed for the microscope to prevent the entrance of abnormal artifacts in the morphological examination. The microscope was cleaned daily for quality examination of blood smear morphology and reticulocyte count. A microscopic smear review was performed to check the functionality of the microscope, quality of slide, and staining by using previously examined and confirmed slides. To make quality staining, the solution was filtered before staining the smear. The quality control for wright staining solution was performed by using a patient sample with normal MCV, MCH, MCHC, and total white blood cell count (annex v).
Data Management and Analysis
Data entry was entered into Epi data version 4.6 (Epidata, Inc. Redwood City, CA, United States), and analysis was done by using STATA (Software for statistics and data science) statistical software version 14 developed by StataCorp for data. Every day the collected data was checked for completeness and accuracy by the principal investigator. During the entry of data, it was cross-checked to ensure the right data was entered and cleaned for accuracy. Descriptive statistics such as frequency, charts, tables, and percentages were used to summarize the data. The results were presented in words and tables. Based on the result, conclusions and recommendations were done.
Dissemination of Results
The study result would be submitted to the Department of Clinical Hematology and Immunohematology, the School of BMLS and CMHS, UOG, and also the results would be submitted to the study site. The abstract would be submitted to local concerning bodies such as EMLA and libraries. The result would be communicated with the research community through a presentation on conferences and publication on peer-reviewed reputable journals to communicate with the international community.
Result
The study was done on a total of 384 participants. Among study participants 142 (39.66%) of them were male and 216 (60.34%) of them were females. All-female participants were non-pregnant during the study time. The mean age of the study participants was 39.17 ± with an SD of 9 years, 116 (30.40%) being in the age group 36–42 years; age and sex distribution of the study population (Table 1)
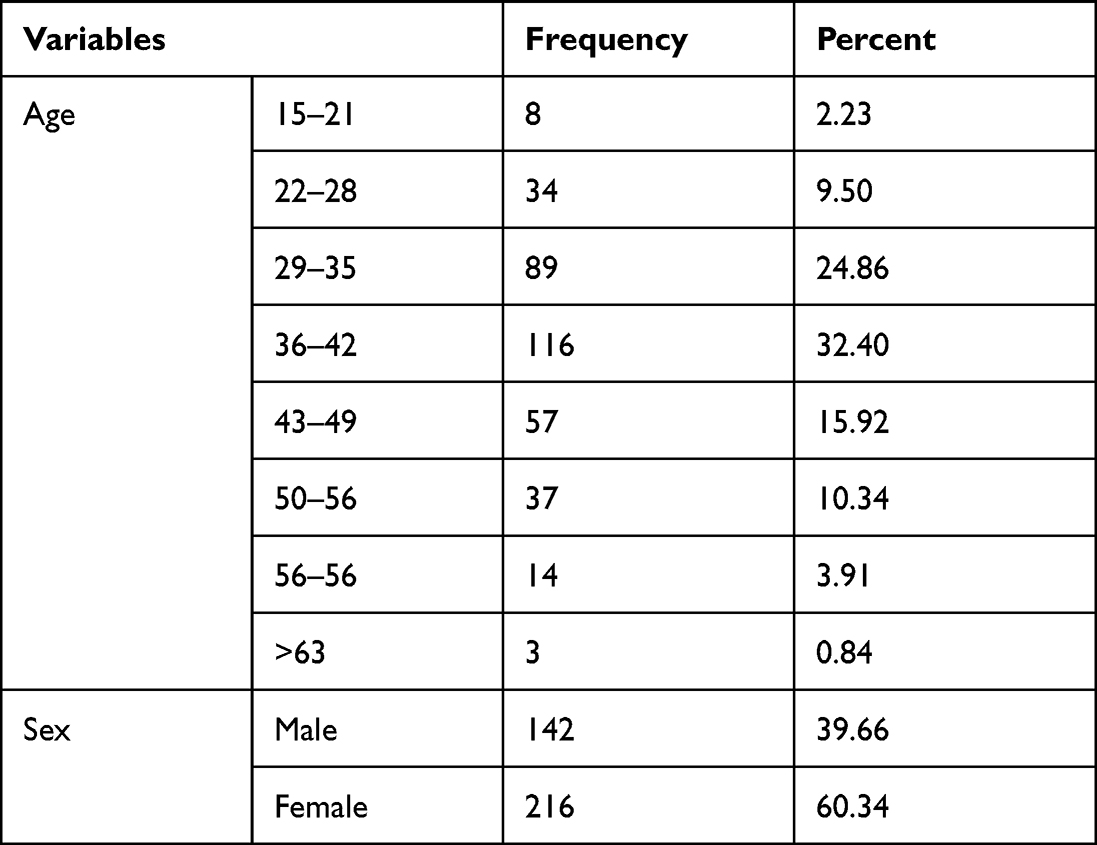 |
Table 1 Age and Sex Distribution of Study Participants |
Anemia and Its Classification
Among study participants, 28 (7.82%) of them had high MCHC and 26 (7.26%) of them had high red cell distribution width. Based on morphological characters and red cell indices, of all anemic participants 57 (62.63%), 19 (20.87%), 15 (16.48%) were normocytic normochromic, microcytic, hypochromic, and macrocytic, respectively. Of those anemic study population, 3 (3.29%), 28 (30.77%), and 60 (65.93%), respectively, had severe, moderate, and mild anemia. In a morphological examination, 18 (19.78%) had spherocyte with normocytic and macrocytic features and 24 (26.37%) had schistocytes and bite cells. Of all study participants of the study, 2 of them were found to be positive for hereditary spherocytosis, 76 (21.23) of them had CD4 count less than 200 and the overall prevalence of anemia was 91 (25.42%) (Table 2).
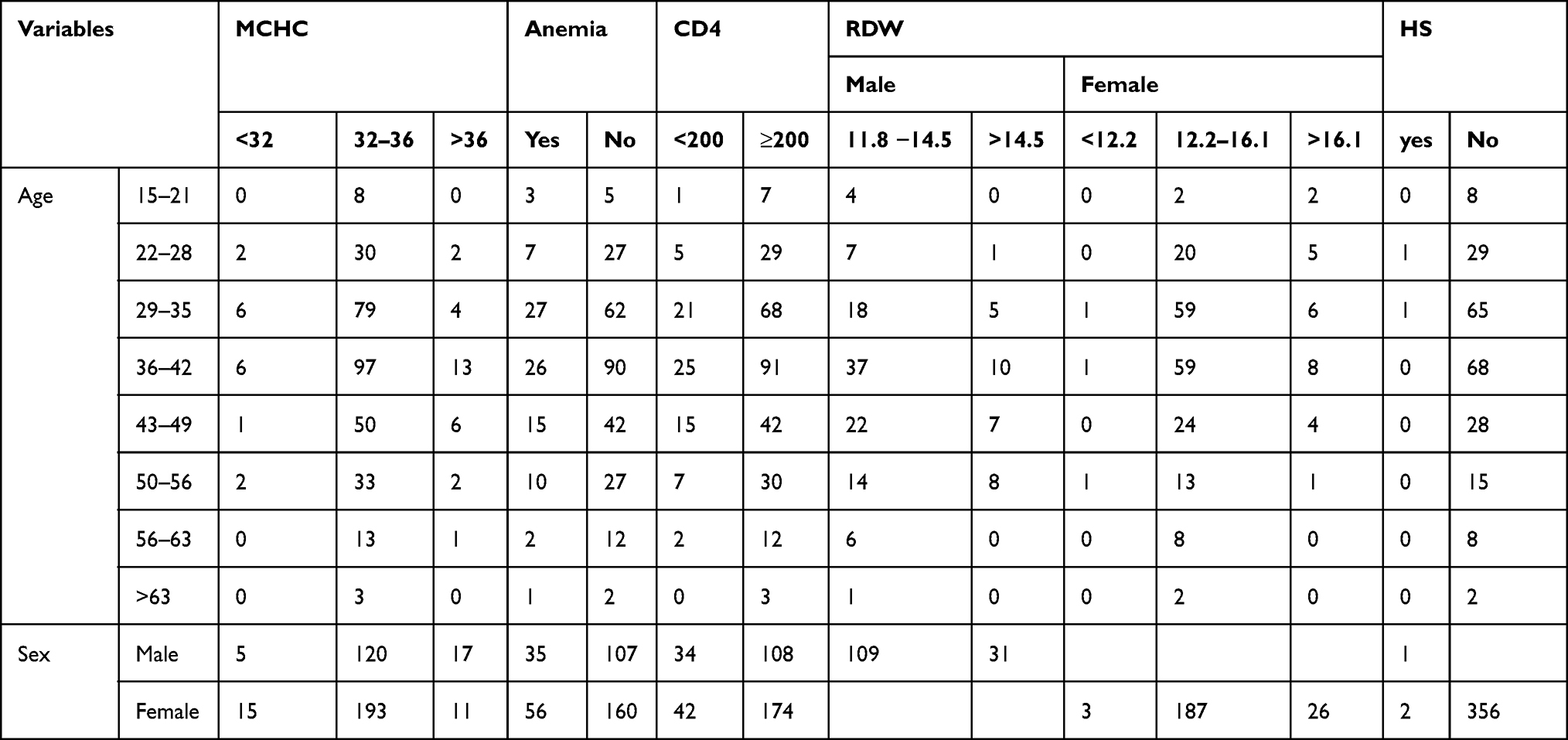 |
Table 2 Age and Sex Distribution of MCHC, Anemia, RDW, HS and CD4 of Study Participants |
The mean red cell distribution width was 13.99 with 1.35 SD. The distribution frequency for red cell distribution width (Figure 1). Of 91 anemic patients who were anemic, 14 (3.91%) of them had a reticulocyte count greater than 2.5. On the other hand, from these 91 previously mentioned patients, 72 (79.12%) of them were negative for DAT, while only 19 (20.88%) patients were positive for the DAT test. Among study participants, 75 (20.95%) had CD4 count less than 200/µL and 283 (79.05%) had CD4 count greater than 200/µL of blood (Figures 1 and 2).
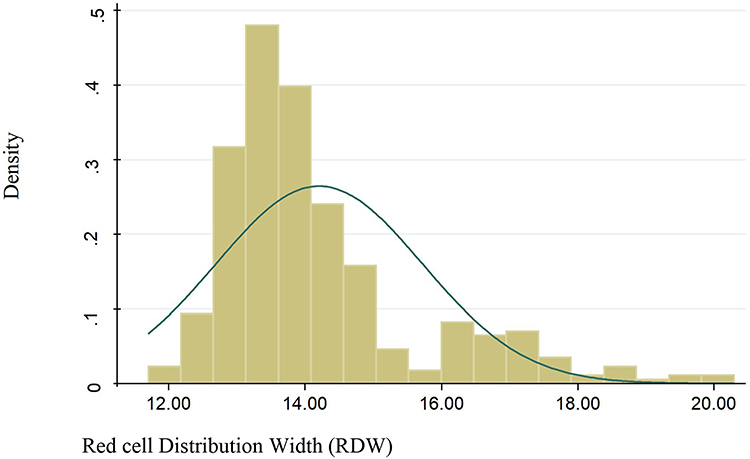 |
Figure 1 The normality distribution of red cell distribution width among HIV positive patients. |
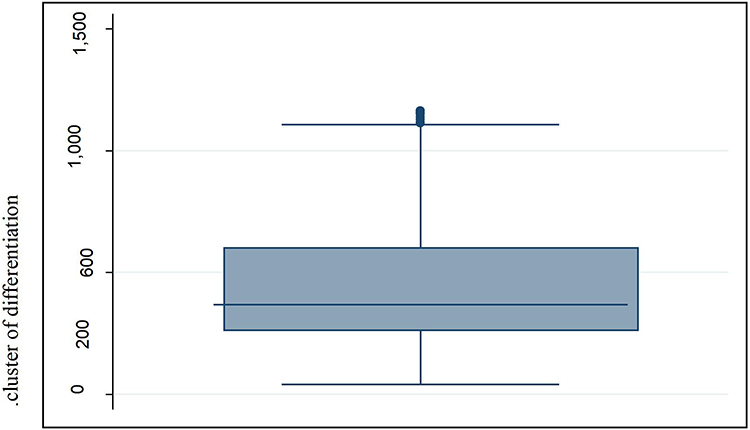 |
Figure 2 The box plot distribution of cluster of differentiation 4 (CD4). |
Discussion
Hereditary spherocytosis is one of the common inherited disorders that is characterized by decreased hemoglobin less than 12g/dl with jaundice, and splenomegaly.4 Patients with mild hereditary spherocytosis (HS) can maintain normal hemoglobin concentration, whereas typical and severe HS patients develop an anemic state.4,15
The prevalence of hereditary spherocytosis among study participants was 2 out of 358. However, the studies which indicate the global prevalence reported that the highest frequency of 1:5,000 is found in Northern European countries6 and 1:2000 in North America.
According to this study, the prevalence of HS is higher in females (2 out of 358), this study is supported by the study in china which is 1.49/100,000. In HS patients, mutations in ≥2 related genes can develop abnormality with heterogeneity.3 It can also cause by the synergistic or inhibitory effects of multiple mutations in related genes that can lead to the complex pathogenesis of HS. In combination with another disease, HS, combined with other diseases, can lead to inconsistent HS genotypes and phenotypes, causing high rates of missed diagnoses or even misdiagnoses in clinical HS cases.16
The reticulocytosis among the anemic study population was 15.38% which is consistent with the reticulocytosis amount on a study done in Black lion hospital, Ethiopia, which was 12.9%, and Lagos, Nigeria, which was 18%.16,17 Although reticulocytopenia occurs in some individuals with IHA, hemolysis either immune or non-immune enhances the index of bone marrow hemopoietic activity and causes increased reticulocyte count.18 Reticulocyte counts increased gradually with age. Analysis of the reticulocyte data showed an inverse relation between transfusion requirements and reticulocyte counts ≥200 × 109/L.
In rare situations, measurements of erythrocyte membrane proteins may be required to determine the nature of the membrane abnormality, and in the absence of family history, molecular genetic analysis may be used to assess whether inheritance is recessive or non-dominant. It is especially vital to rule out stomatocytosis if a splenectomy is not an option due to the risk of thrombosis. Mild HS does not require splenectomy and can be handled without the use of folate supplements.18
In general, this study is the first study that revealed the current prevalence of HS because there was no study concerning HS among HIV patients as well as non-infected patients. This might be due to the lack of hematology analyzers that measure MRV and reticulocyte count which are key to diagnose HS.
Strength and Limitation
Strength of the Study
In this study, hematological analyses, such as reticulocyte count, mean reticulocyte volume, and immature reticulocyte fraction, were performed by automation which was not common in developing countries.
Limitation
The first limitation of this study was that the only prevalence of HS was studied; it did not allow us to observe causality in the relationship between HS and its associated factors. The serum lactate dehydrogenase, haptoglobin, and unconjugated bilirubin were not tested for additional evidence of hemolysis.
Conclusion and Recommendation
Conclusion
According to the findings of this cross-sectional study, HS in HIV patient was rare public health problem.
Recommendations
The ART clinicians were recommended to screen HS for individuals who are anemic with increased reticulocyte count. Additionally, we recommend that additional study to be done by using sensitive and specific advanced technology products like flow cytometry and advanced molecular tests which also help to quantify the amount of RBC having membrane abnormality that is known to be the probability for hemolysis. It is better for researchers in the hematology area to give attention to set the reference interval of immature reticulocyte fraction and mean reticulocyte volume. Even though it is less frequent, HS diagnosis needs prior identification to minimize the severity and burden of the disease because it may result in fatal condition. We suggest policy makers to develop guideline for HIV patient by considering HS and work to make screening tests for HS to be available in every ART clinic across the country.
Abbreviation
AIDS, Acquired Immune Deficiency Syndrome; ART, Anti-Retroviral Therapy; CD, Cluster of Differentiation; CI, Confidence Interval; DAT, Direct Antihuman Globulin Test; DIHA, Drug Induced Hemolytic Anemia; EDTA, Ethylene diamine tetra acetic acid; FC, Fragment of Crystallization; HAART, Highly Active Anti-Retroviral Therapy; Hb, Hemoglobin; HIV, Human Immune deficiency Virus; IHA, Immune Hemolytic Anemia; MCH, Mean Cell Hemoglobin; MCHC, Mean Cell Hemoglobin concentration; MCV, Mean Cell Volume; PLWHIV, Peoples Living with Human Immune deficiency Virus; RBC, Red Blood Cell; UOGCSH, University of Gondar comprehensive specialized hospital; WHO, World Health Organization.
Data Sharing Statement
All relevant data are available within the manuscript. In case of need, the data that support the findings of this study are available from the corresponding author on reasonable request.
Ethical Approval and Consent to Participate
The study was carried out after receiving ethical approval from the University of Gondar college of medicine and health sciences (CMHS), school of biomedical and laboratory science research, and ethical review committee (Reference number SBLS/2750). All activities in this research work were based on Helsinki declaration. Furthermore, supports and permission letter were secured from UOGSCH. In addition, following an explanation of the purpose, the benefits and the possible risks of the study, written informed consent was taken from a parent/legal guardian, and assent was sought from children before commencement of the study. It was made clear that participation in the study was purely on a voluntary basis and refusal was possible. To ensure confidentiality of data, study participants were coded by using unique codes, and only authorized persons were accessing the collected data. The study participants with abnormal findings were linked to the physicians who are working at the ART clinic for proper patient care.
Acknowledgments
The authors are grateful to the study participants for their voluntary participation. We would like to thank data collectors for their collaboration. The authors would also like to thank the University of Gondar for providing ethical clearance.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research received post graduate fee grant from the University of Gondar.
Disclosure
The authors have declared that they have no competing interests.
References
1. Rodwell C, Aymé S. Rare disease policies to improve care for patients in Europe. Biochim Biophys Acta Mol Basis Dis. 2015;1852(10):2329–2335. doi:10.1016/j.bbadis.2015.02.008
2. Barcellini W, Mariani M, Vercellati C, et al. Clinical and haematologic features of 300 patients affected by hereditary spherocytosis as a function of the type of the membrane protein defect. American Society of Hematology; 2007.
3. Hassoun H, Palek J. Hereditary spherocytosis: a review of the clinical and molecular aspects of the disease. Blood Rev. 1996;10(3):129–147. doi:10.1016/S0268-960X(96)90021-1
4. Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372(9647):1411–1426. doi:10.1016/S0140-6736(08)61588-3
5. Panigrahi I, Phadke S, Agarwal A, Gambhir S, Agarwal S. Clinical profile of hereditary spherocytosis in North India. J Assoc Physicians India. 2002;50:1360–1367.
6. Eber S, Lux SE. Hereditary spherocytosis—defects in proteins that connect the membrane skeleton to the lipid bilayer. In: Seminars in Hematology. Elsevier; 2004:118–141.
7. Rocha S, Costa E, Catarino C, et al. Erythropoietin levels in the different clinical forms of hereditary spherocytosis. Br J Haematol. 2005;131(4):534–542. doi:10.1111/j.1365-2141.2005.05802.x
8. Iolascon A, Del Giudice EM, Perrotta S, Alloisio N, Morlé L, Delaunay J. Hereditary spherocytosis: from clinical to molecular defects. Haematologica. 1998;83(3):240–257.
9. Remus R, Zeschnigk M, Zuther I, et al. The state of DNA methylation in the promoter regions of the human red cell membrane protein (band 3, protein 4.2, and β‐spectrin) genes. Gene Funct Dis. 2001;2(4):171–184. doi:10.1002/1438-826X(200112)2:4<171::AID-GNFD171>3.0.CO;2-O
10. Mariani M, Barcellini W, Vercellati C, et al. Clinical and hematologic features of 300 patients affected by hereditary spherocytosis grouped according to the type of the membrane protein defect. Haematologica. 2008;93(9):1310–1317. doi:10.3324/haematol.12546
11. Saif MW. HIV-associated autoimmune hemolytic anemia: an update. AIDS Patient Care STDS. 2001;15(4):217–224. doi:10.1089/10872910151133783
12. Seedat F, Patel M, Waja F, Sigauke F, Variava E. Hereditary spherocytosis and human immunodeficiency virus (HIV) infection: is there an association?; 2015.
13. Carr J, Geesaman S, Czader M. Performance evaluation of the new UniCel DxH800 coulter cellular analysis system in a large hospital setting. Lab Med. 2012;43(5):157–163. doi:10.1309/LMEJRJLI8L5ZCDWP
14. Chang C-C, Kass L. Clinical significance of immature reticulocyte fraction determined by automated reticulocyte counting. Am J Clin Pathol. 1997;108(1):69–73. doi:10.1093/ajcp/108.1.69
15. Delhommeau F, Cynober T, Schischmanoff P, et al. Natural history of hereditary spherocytosis during the first year of life. J Am Society Hematol. 2000;95(2):393–397.
16. Ibrahim J, Taye M, Defar M. Prevalence of autoimmune hemolytic anemia in human immunodeficiency virus–infected anemic adults: a cross-sectional study at Tikur Anbessa specialized teaching hospital from June 5, 2015, to September 10, 2015, Addis Ababa, Ethiopia. Am J Clin Pathol. 2018;150:S108. doi:10.1093/ajcp/aqy097.261
17. Adewumi AA, Titilope AA, Osamuedemen VA, et al. Prevalence of HIV-related autoimmune haemolytic anaemia in Lagos, Nigeria. Niger Med J. 2014;55(1):63. doi:10.4103/0300-1652.128175
18. Barcellini W, Fattizzo B. Clinical applications of hemolytic markers in the differential diagnosis and management of hemolytic anemia. Dis Markers. 2015;2015. doi:10.1155/2015/635670
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Anemia in HIV Patients Attending Highly Active Antiretroviral Therapy Clinic at Hoima Regional Referral Hospital: Prevalence, Morphological Classification, and Associated Factors
Kaudha R, Amanya R, Kakuru D, Muhumuza Atwooki R, Mutebi Muyoozi R, Wagubi R, Muwanguzi E, Okongo B
HIV/AIDS - Research and Palliative Care 2023, 15:621-632
Published Date: 12 October 2023