Back to Journals » OncoTargets and Therapy » Volume 8
The human immunodeficiency virus protease inhibitor ritonavir is potentially active against urological malignancies
Authors Sato A
Received 23 December 2014
Accepted for publication 24 February 2015
Published 8 April 2015 Volume 2015:8 Pages 761—768
DOI https://doi.org/10.2147/OTT.S79776
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Faris Farassati
Akinori Sato
Department of Urology, National Defense Medical College, Tokorozawa, Japan
Abstract: The human immunodeficiency virus protease inhibitor ritonavir has recently been shown to have antineoplastic activity, and its use in urological malignancies is under investigation with an eye toward drug repositioning. Ritonavir is thought to exert its antineoplastic activity by inhibiting multiple signaling pathways, including the Akt and nuclear factor-kappaB pathways. It can increase the amount of unfolded proteins in the cell by inhibiting both the proteasome and heat shock protein 90. Combinations of ritonavir with agents that increase the amount of unfolded proteins, such as proteasome inhibitors, histone deacetylase inhibitors, or heat shock protein 90 inhibitors, therefore, induce endoplasmic reticulum stress cooperatively and thereby kill cancer cells effectively. Ritonavir is also a potent cytochrome P450 3A4 and P-glycoprotein inhibitor, increasing the intracellular concentration of combined drugs by inhibiting their degradation and efflux from cancer cells and thereby enhancing their antineoplastic activity. Furthermore, riotnavir’s antineoplastic activity includes modulation of immune system activity. Therapies using ritonavir are thus an attractive new approach to cancer treatment and, due to their novel mechanisms of action, are expected to be effective against malignancies that are refractory to current treatment strategies. Further investigations using ritonavir are expected to find new uses for clinically available drugs in the treatment of urological malignancies as well as many other types of cancer.
Keywords: drug repositioning, novel treatment
Introduction
New anticancer agents have been developed in an effort to improve treatment outcome in patients with advanced metastatic urological malignancies. Targeted therapies using tyrosine kinase inhibitors1–3 and inhibitors of the mammalian target of rapamycin4 have been replacing immunotherapy in the treatment of renal cancer, and the agents docetaxel,5 cabazitaxel,6 enzalutamide,7 and abiraterone8 have been used to treat castration-resistant prostate cancer. These treatments are innovative and have contributed to the improved survival of patients. In urothelial carcinoma, on the other hand, there have been no new therapeutic agents significantly improving survival; the cisplatin–gemcitabine combination is of limited efficacy but is still a mainstay in the treatment of metastatic disease.9 Because there is still no curative treatment for advanced urological malignancies, there is an urgent need for new agents or new combination therapies using agents currently available. Drug repositioning has recently emerged as an attractive strategy for finding candidate anticancer drugs among the existing drugs, and some noncancer drugs have been shown to be potent anticancer agents.10–12 Ritonavir is a human immunodeficiency virus (HIV) protease inhibitor approved by the US Food and Drug Administration (FDA)13 and widely used for the treatment of HIV infection. Its repositioning as an anticancer drug, however, has been suggested by the results of recent studies showing that ritonavir has antineoplastic effects such as induction of apoptosis and inhibition of inflammatory cytokine production, proteasome activity, and cell proliferation and survival.14 In this article, the anticancer activity of ritonavir and the underlying mechanism of action, as a single agent and in combination with other agents, are reviewed, with a focus on ritonavir’s possible use in treating urological malignancies.
Ritonavir’s mechanisms of action
Ritonavir’s mechanisms of action include inhibition of the proteasome; inhibition of heat shock protein 90 (HSP90), cytochrome P450 3A4 (CYP3A4), and P-glycoprotein; and modulation of immune system activity. Inhibition of the proteasome and HSP90 causes unfolded proteins to accumulate and thereby induces endoplasmic reticulum (ER) stress, whereas inhibition of CYP3A4 and P-glycoprotein increases the intracellular concentration of other drugs. Ritonavir may also act against malignancies by enhancing immune system activity (Figure 1).
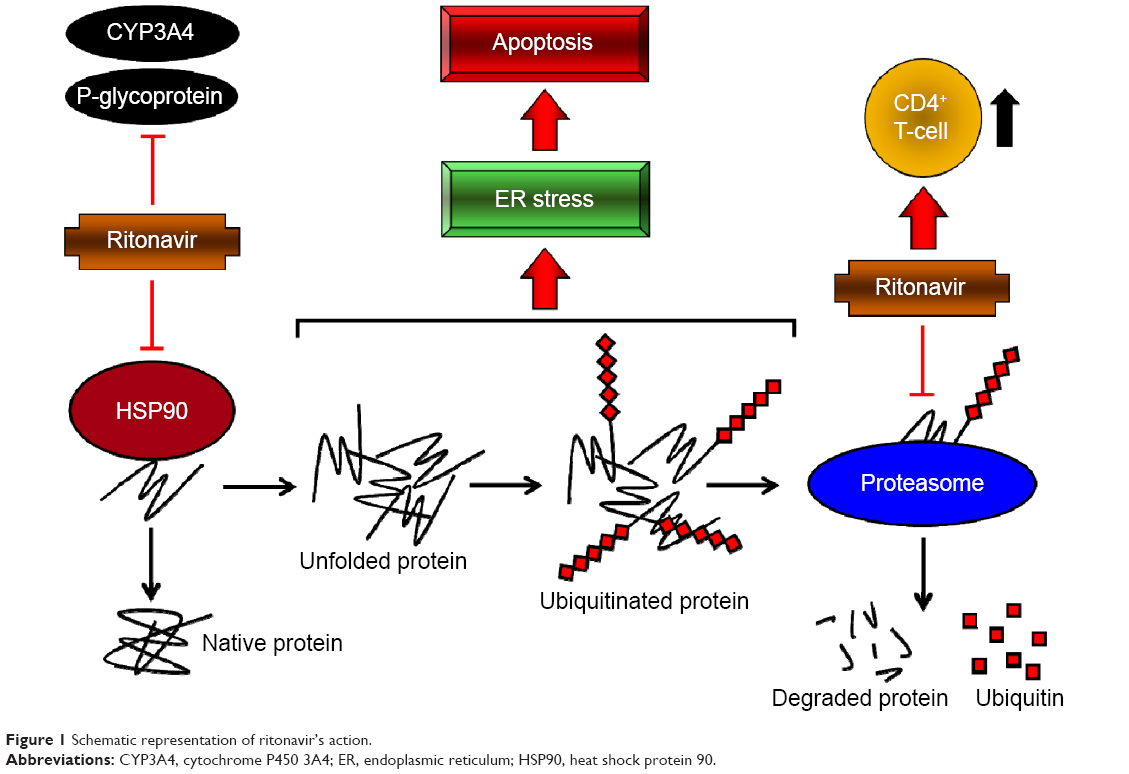 | Figure 1 Schematic representation of ritonavir’s action. |
Ritonavir acts as a proteasome inhibitor
Protein degradation by the ubiquitin–proteasome pathway affects the proliferation and survival of both normal and malignant cells,15 so proteasome inhibitors have been utilized in the treatment of malignancies. Bortezomib is widely used to treat patients with relapsed or refractory multiple myeloma,16,17 and carfilzomib is a new oral proteasome inhibitor that has been approved by the FDA for the treatment of multiple myeloma patients who have received at least two prior therapies including bortezomib.18 On the other hand, the efficacy of proteasome inhibitors is limited in patients with solid tumors.19–23 In an effort to ameliorate bortezomib’s efficacy in urological malignancies, combination therapies using bortezomib and a histone deacetylase (HDAC) inhibitor, either suberoylanilide hydroxamic acid (SAHA)24,25 or panobinostat,26 have been investigated. These studies demonstrated that the combinations induced robust ER stress and killed cancer cells synergistically.
Although ritonavir is an HIV protease inhibitor, it has been shown to also act as a proteasome inhibitor. Gaedicke et al27 focused on ritonavir’s ability to inhibit the chymotrypsin-like activity of isolated 20S proteasomes and showed that ritonavir inhibited the growth of murine lymphoma cells both in vitro and in vivo by acting like a proteasome inhibitor. Laurent et al28 showed that in glioma cells, ritonavir had cytostatic and cytotoxic effects due to inhibition of the chymotrypsin-like activity of the proteasome. In that study, however, ritonavir failed to inhibit the tumor growth in vivo because the therapeutic dose level was not reached in the tumor.
Inhibition of the transcription factor nuclear factor (NF)-kappaB is thought to be one of the important consequences of proteasome inhibition by ritonavir because proteasome inhibitors cause the NF-kappaB inhibitor IkappaB to accumulate in the cell by inhibiting its proteasome-dependent degradation.29,30 Pati et al31 reported the antineoplastic activity of ritonavir evaluated in experiments using Kaposi sarcoma cells both in vitro and in vivo. They found that ritonavir inhibited NF-kappaB activity and concluded that it had antineoplastic effects independent of its ability to inhibit the HIV protease. Ritonavir was also shown to inhibit transcriptional activation of NF-kappaB in adult T-cell leukemia cells, resulting in inhibition of the Bcl-xL, survivin, c-Myc, and cyclin D2 expression in them.32 Inhibition of NF-kappaB by ritonavir in a non-Hodgkin lymphoma was also reported.33
Ritonavir inhibits HSP90
Inhibition of HSP90 destabilizes the cancer cell’s aberrant protein subset34 and thereby increases unfolded proteins in the cell. This causes ER stress, and chronic or unresolved ER stress leads to apoptosis.35 Inhibition of HSP90 is therefore an attractive new strategy against cancer. The geldanamycin derivative 17-allylamino-17-demethoxygeldanamycin (17-AAG) was the first HSP90 inhibitor to be used clinically.36 Its efficacy has been investigated in patients with renal cancer,37 prostate cancer,38 melanoma,39 and breast cancer,40 but no objective responses were seen in those studies. One of the reasons for this low effectiveness of 17-AAG is that the unrepaired unfolded proteins could be degraded by the proteasome and not accumulated in the cell. Therefore, both HSP90 and the proteasome would have to be inhibited to produce enough ER stress to cause apoptosis. Ritonavir binds HSP90 and partially inhibits its chaperone function,41 but unlike 17-AAG, it also inhibits the proteasome.27,28 Ritonavir would therefore theoretically be the ideal agent to induce ER stress. Kraus el al42 showed that ritonavir increased the level of ER stress induced by bortezomib enough to cause apoptosis in bortezomib-resistant sarcoma cells, and Sato et al43 showed that a ritonavir–bortezomib combination enhanced ER stress and ubiquitinated unfolded protein accumulation synergistically in renal cancer cells. Interestingly, ritonavir was revealed to enhance 17-AAG activity by inhibiting the 17-AAG-induced expression of heat shock factor-1, an HSP transcription factor. Ritonavir thus enhances 17-AAG activity, leading to thorough HSP90 suppression.44
It has been reported that HSP90 interacts with Akt and that inhibition of Akt-HSP90 binding leads to the dephosphorylation and inactivation of Akt.45 Therefore, another important consequence of HSP90 inhibition by ritonavir would be inhibition of the Akt signaling pathway. Ritonavir reportedly inhibited the Akt signaling pathway in breast cancer cells,41 ovarian cancer cells,46 and pancreatic cancer cells.47
Ritonavir interferes with drug degradation mechanisms in cancer cells
Ritonavir inhibits CYP3A4 in liver microsomes. Ikezoe et al48 showed that ritonavir increased the antitumor activity of docetaxel in DU145 prostate cancer cells by protecting docetaxel from inactivation in the cells. Oostendorp et al49 clinically tried the combination of ritonavir and docetaxel in patients with solid tumor. On days 1 and 8 they gave the patients 10 mg or 100 mg oral docetaxel and 100 mg oral ritonavir, either simultaneously or 60 minutes before the docetaxel, and this was followed by 100 mg intravenous docetaxel on day 15 or 22. The combination was well tolerated, and ritonavir was shown to significantly enhance the bioavailability of docetaxel. Sato et al50 showed in renal cancer cells that ritonavir enhances the activity of the HDAC inhibitor panobinostat, which is a substrate of CYP3A4.51 Ritonavir also inhibits P-glycoprotein, a drug-efflux pump associated with multidrug resistance.52 Therefore, ritonavir could increase the concentrations of combined drugs, thus enhancing their activity, by inhibiting their degradation and efflux from the cell. A matter of great concern is that ritonavir could also inhibit CYP3A4 in the liver, increasing the serum concentrations of combined drugs and leading to adverse events. Hamberg et al51 conducted a clinical study in which panobinostat was administered in combination with the CYP3A4 inhibitor ketoconazole. They concluded that coadministration of panobinostat with CYP3A4 inhibitors is feasible because the observed increase in pharmacokinetics parameters (maximum serum concentration increased 1.6-fold and area under the curve increased 1.8-fold) was not clinically relevant. Inhibiting CYP3A4 activity thus does not necessarily cause severe adverse events. However, careful monitoring of the serum concentrations of combined drugs is mandatory for the patients’ safety.
Ritonavir’s antineoplastic activity includes modulation of immune system activity
Ritonavir acts against acquired immunodeficiency syndrome by decreasing plasma viremia and thereby increasing CD4+ T-cell counts.53 Interestingly, ritonavir has also been shown to increase lymphocyte levels itself. Weichold et al54 found that it increases the viability of peripheral blood mononuclear cells, decreases their susceptibility to apoptosis, and decreases their production of tumor necrosis factor. Similarly, Sloand et al55 showed that ritonavir decreases CD4+ T-cell apoptosis by inhibiting caspase 1 expression independent of HIV infection, which could increase the number of CD4+ T cells. CD4+ T cells are important in tumor immunity because they are needed to sustain the cytolytic CD8+ T cells, which can kill cancer cells, and because they recruit cells of the innate immune system, such as macrophages and mast cells.56,57 Ritonavir is thus thought to enhance tumor immunity and act against cancer by increasing the number of CD4+ T cells.
Experimental use of ritonavir to treat urological malignancies
In our laboratory at National Defense Medical College we have been investigating the antineoplastic activity of ritonavir in studies focusing on its possible use to treat urological malignancies such as renal cancer, prostate cancer, and bladder cancer. Although ritonavir alone is effective in some cancer cell lines, it exerts very strong anticancer activity when combined with agents such as proteasome inhibitors and HDAC inhibitors. These combinations are especially attractive because they generally induce drastic ER stress, which has recently been shown to be useful in the treatment of malignancies.58
Ritonavir as a single agent
Using human renal cancer cell lines, we have previously shown in vitro that ritonavir itself induces ER stress, evidenced by the increased expression of the ER stress markers such as glucose-regulated protein 78 and HSP70 and that it inhibits the growth of cancer cells in a dose-dependent fashion.59 This ER stress and growth inhibition are thought to be due to the ability of ritonavir to inhibit both HSP90 and the proteasome. In vivo, however, ritonavir as a single agent significantly suppressed tumor growth in a murine allograft tumor model using the mouse renal cancer cell line Renca (Figure 2) but not in xenograft models. These results, together with those of the in vivo study using glioma cells,28 indicate that in human solid tumors the effectiveness of ritonavir as a single agent is limited.
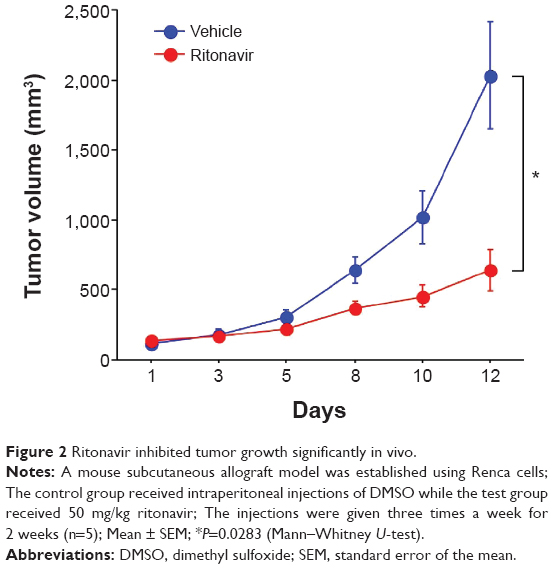 | Figure 2 Ritonavir inhibited tumor growth significantly in vivo. |
Ritonavir in combination with proteasome inhibitors
Proteasome inhibitors inhibit the degradation of unfolded proteins and therefore cause them to accumulate and induce ER stress. Because ritonavir inhibits HSP9041 and increases the amount of unfolded proteins in the cell, the combination of ritonavir and a proteasome inhibitor is expected to cause drastic accumulation of unfolded proteins and, thereby, drastic ER stress. Figure 3 shows photomicrographs of PC-3 human prostate cancer cells treated with the combination of ritonavir and the proteasome inhibitor bortezomib. The combination killed PC-3 cells drastically (most of the cells are floating), whereas each individual agent had a minimal effect at the indicated concentrations. The ritonavir–bortezomib combination was shown in mice xenograft tumor models to suppress tumor growth significantly and shown in vitro to kill renal cancer cells synergistically.43 It also induced ER stress and ubiquitinated unfolded protein accumulation synergistically. A beneficial effect of combining ritonavir and bortezomib was also found in an in vitro study using human bladder cancer cells,60 where the combination decreased the expression of cyclin D1 and cyclin-dependent kinase (CDK)4 and increased the expression of p21, inhibiting both the expression and function of the cyclin D1/CDK4 complex. In that study, the combination induced ER stress, caused ubiquitinated proteins to accumulate, and enhanced histone acetylation synergistically by decreasing the expression of HDACs. We have shown in vitro that ritonavir combined with carfilzomib, a novel proteasome inhibitor that inhibits proteasomal activity irreversibly, inhibits human renal cancer growth synergistically by inducing ER stress and autophagy,61 and studies using ritonavir and the novel proteasome inhibitor delanzomib are currently underway at our laboratory.
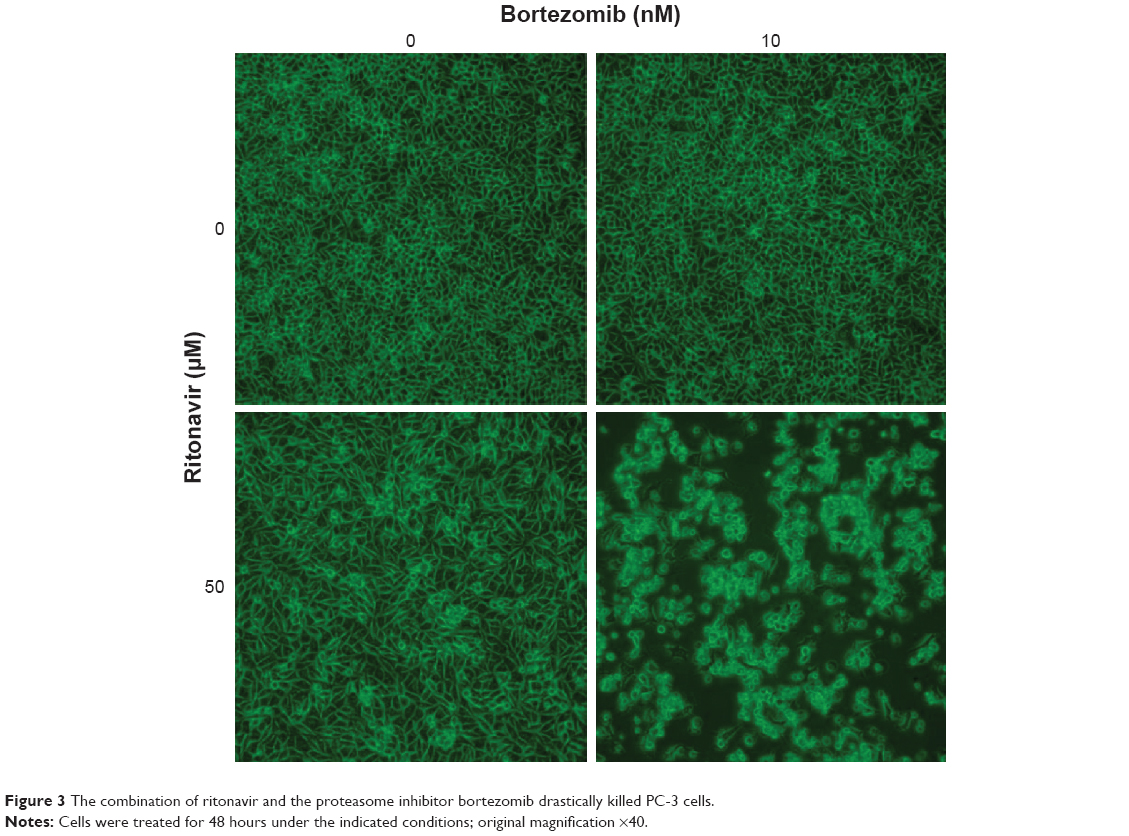 | Figure 3 The combination of ritonavir and the proteasome inhibitor bortezomib drastically killed PC-3 cells. |
Ritonavir in combination with HDAC inhibitors
The combination of ritonavir and HDAC inhibitors is also an attractive approach to killing cancer cells effectively. Levels of histone acetylation reflect the balance between the activities of histone acetyltransferases and HDACs,62 and the acetylation and deacetylation of histones play important roles in the regulation of gene transcription.63 Their deacetylation, for example, tightens their interaction with DNA, resulting in a closed chromatin structure and inhibiting gene transcription.64 HDACs are associated with a number of cellular oncogenes and tumor suppressor genes leading to development of malignancy,65,66 so compounds that target HDACs have generated great interest as anticancer drugs.67 HDAC inhibitors basically exert their antineoplastic activity by inducing histone acetylation, but they have another important mechanism of action: inhibition of HSP90. Many HDAC inhibitors inhibit HDAC6, and ablation of HDAC6 induces hyperacetylation of HSP90, disrupting its chaperone function.68,69 Ritonavir acts not only as an HSP90 inhibitor and a proteasome inhibitor but also as a CYP3A4 inhibitor and P-glycoprotein inhibitor. It is therefore expected to further increase the amount of unfolded proteins due to HDAC inhibitors and also to enhance the activity of HDAC inhibitors by inhibiting their degradation or efflux from the cell. The combination of ritonavir and the HDAC inhibitor SAHA was shown to inhibit human renal cancer growth synergistically in vitro.59 It inhibited the expression of the inhibitor of apoptosis family members X-linked inhibitor of apoptosis protein and survivin and increased the expression of active caspase 3, thus inducing apoptosis. It also promoted histone acetylation, suggesting that ritonavir enhanced the activity of SAHA. The efficacy of ritonavir in combination with the HDAC inhibitor panobinostat was also investigated.50 That combination induced caspase-dependent apoptosis and inhibited human renal cancer growth both in vitro and in vivo by enhancing histone acetylation. This enhancement was thought to be in part due to CYP3A4 inhibition by ritonavir because panobinostat has been shown to be a CYP3A4 substrate.51 Interestingly, the combination decreased the expression of HDACs, which would be another mechanism enhancing histone acetylation.
Ritonavir in combination with 17-AAG
Combining the HSP90 inhibitor 17-AAG with ritonavir is a reasonable approach because the combination could inhibit HSP90 function cooperatively. In fact, the combination of 17-AAG and ritonavir caused apoptosis and inhibited the growth of human renal cancer cells effectively in vitro.44 It inhibited the expression of cyclin D1, CDK4, survivin, and X-linked inhibitor of apoptosis protein. Although 17-AAG inhibits HSP90, it generally increases the expression of HSPs,70 and this could attenuate the effect of 17-AAG. Ritonavir was found in that study to suppress 17-AAG-induced HSP expression by inhibiting the expression of heat shock factor-1, an HSP transcription factor. Thus ritonavir enhanced 17-AAG activity, leading to thorough HSP90 suppression.
Future perspectives
Ritonavir has been shown to be potentially active against cancer. Because its antineoplastic activity is much stronger when ritonavir is combined with specific agents, future investigations should be devoted to finding the drug combination with the highest efficacy. Combining ritonavir with agents that increase the amount of unfolded proteins in the cell (eg, proteasome inhibitors, HDAC inhibitors, and HSP90 inhibitors) is a promising approach because the resulting combinations would be expected to increase the amount of unfolded proteins and thereby induce ER stress leading to apoptosis. Agents that are substrates of CYP3A4 and/or P-glycoprotein are also good candidates because their intracellular concentration can be increased by ritonavir. Most importantly, ritonavir’s mechanisms do not act separately. Bortezomib, for example, is thought not only to enhance ER stress in combination with ritonavir but at its increased concentration due to ritonavir’s inhibition of CYP3A4 further to enhance the ER stress. Clinical trials using ritonavir-containing regimens are also important. Because ritonavir could affect CYP3A4 in the liver as well as in cancer cells, Phase I study with careful drug monitoring is mandatory. Furthermore, we have shown that ritonavir-containing regimens are effective against cancers with very different characteristics: renal cancer, prostate cancer, and bladder cancer. This suggests that ritonavir-containing regimens work irrespective of cancer type and that investigating the effects of ritonavir-containing combinations on other types of cancer would be a promising next step.
Conclusion
The HIV protease inhibitor ritonavir is potentially an anticancer agent. Its mechanisms of action include inhibition of the proteasome, inhibition of HSP90, inhibition of CYP3A4 and P-glycoprotein, and modulation of immune system activity. In urological cancer cells, ritonavir exerts strong anticancer activity when combined with proteasome inhibitors, HDAC inhibitors, or an HSP90 inhibitor. Further studies exploring more effective drugs to be combined with ritonavir are underway. Ritonavir-containing regimens should also be clinically tried in patients with advanced urological malignancies because they act by mechanisms completely different from those of the currently available noncurative treatments. Ritonavir is thus a promising anticancer agent, and investigation of its use in urological malignancies as well as other types of cancer is strongly encouraged.
Disclosure
The author reports no conflicts of interest in this work.
References
Escudier B, Eisen T, Stadler WM, et al; TARGET Study Group. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356(2):125–134. | ||
Motzer RJ, Rini BI, Bukowski RM, et al. Sunitinib in patients with metastatic renal cell carcinoma. JAMA. 2006;295(21):2516–2524. | ||
Rixe O, Bukowski RM, Michaelson MD, et al. Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: a phase II study. Lancet Oncol. 2007;8(11):975–984. | ||
Motzer RJ, Escudier B, Oudard S, et al; RECORD-1 Study Group. Phase 3 trial of everolimus for metastatic renal cell carcinoma: final results and analysis of prognostic factors. Cancer. 2010;116(18):4256–4265. | ||
Tannock IF, de Wit R, Berry WR, et al; TAX 327 Investigators. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351(15):1502–1512. | ||
de Bono JS, Oudard S, Ozguroglu M, et al; TROPIC Investigators. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376(9747):1147–1154. | ||
Scher HI, Beer TM, Higano CS, et al; Prostate Cancer Foundation/Department of Defense Prostate Cancer Clinical Trials Consortium. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet. 2010;375(9724):1437–1446. | ||
Reid AH, Attard G, Danila DC, et al. Significant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. J Clin Oncol. 2010;28(9):1489–1495. | ||
Teply BA, Kim JJ. Systemic therapy for bladder cancer – a medical oncologist’s perspective. J Solid Tumors. 2014;4(2):25–35. | ||
Shim JS, Liu JO. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int J Biol Sci. 2014;10(7):654–663. | ||
Vlahopoulos S, Critselis E, Voutsas IF, et al. New use for old drugs? Prospective targets of chloroquines in cancer therapy. Curr Drug Targets. 2014;15(9):843–851. | ||
Aldea M, Craciun L, Tomuleasa C, et al. Repositioning metformin in cancer: genetics, drug targets, and new ways of delivery. Tumour Biol. 2014;35(6):5101–5110. | ||
Schouten JT. FDA approves 2 new protease inhibitors: ritonavir (Norvir) and Crixivan (Indinavir sulfate). Food and Drug Administration. STEP Perspect. 1996;8(1):7–8. | ||
Chow WA, Jiang C, Guan M. Anti-HIV drugs for cancer therapeutics: back to the future? Lancet Oncol. 2009;10(1):61–71. | ||
Naujokat C, Hoffmann S. Role and function of the 26S proteasome in proliferation and apoptosis. Lab Invest. 2002;82(8):965–980. | ||
Richardson PG, Barlogie B, Berenson J, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348(26):2609–2617. | ||
Berenson JR, Jagannath S, Barlogie B, et al. Safety of prolonged therapy with bortezomib in relapsed or refractory multiple myeloma. Cancer. 2005;104(10):2141–2148. | ||
Herndon TM, Deisseroth A, Kaminskas E, et al. U.S. Food and Drug Administration approval: carfilzomib for the treatment of multiple myeloma. Clin Cancer Res. 2013;19(17):4559–4563. | ||
Morris MJ, Kelly WK, Slovin S, et al. A phase II trial of bortezomib and prednisone for castration resistant metastatic prostate cancer. J Urol. 2007;178(6):2378–2383; discussion 2383–2384. | ||
Kondagunta GV, Drucker B, Schwartz L, et al. Phase II trial of bortezomib for patients with advanced renal cell carcinoma. J Clin Oncol. 2004;22(18):3720–3725. | ||
Besse B, Planchard D, Veillard AS, et al. Phase 2 study of frontline bortezomib in patients with advanced non-small cell lung cancer. Lung Cancer. 2012;76(1):78–83. | ||
Mackay H, Hedley D, Major P, et al. A phase II trial with pharmacodynamic endpoints of the proteasome inhibitor bortezomib in patients with metastatic colorectal cancer. Clin Cancer Res. 2005;11(15):5526–5533. | ||
Phuphanich S, Supko JG, Carson KA, et al. Phase 1 clinical trial of bortezomib in adults with recurrent malignant glioma. J Neurooncol. 2010;100(1):95–103. | ||
Sato A, Asano T, Ito K, Asano T. Vorinostat and bortezomib synergistically cause ubiquitinated protein accumulation in prostate cancer cells. J Urol. 2012;188(6):2410–2418. | ||
Sato A, Asano T, Ito K, Sumitomo M, Asano T. Suberoylanilide hydroxamic acid (SAHA) combined with bortezomib inhibits renal cancer growth by enhancing histone acetylation and protein ubiquitination synergistically. BJU Int. 2012;109(8):1258–1268. | ||
Sato A, Asano T, Isono M, Ito K, Asano T. Panobinostat synergizes with bortezomib to induce endoplasmic reticulum stress and ubiquitinated protein accumulation in renal cancer cells. BMC Urol. 2014;14:71. | ||
Gaedicke S, Firat-Geier E, Constantiniu O, et al. Antitumor effect of the human immunodeficiency virus protease inhibitor ritonavir: induction of tumor-cell apoptosis associated with perturbation of proteasomal proteolysis. Cancer Res. 2002;62(23):6901–6908. | ||
Laurent N, de Boüard S, Guillamo JS, et al. Effects of the proteasome inhibitor ritonavir on glioma growth in vitro and in vivo. Mol Cancer Ther. 2004;3(2):129–136. | ||
Hideshima T, Richardson P, Chauhan D, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61(7):3071–3076. | ||
Mitsiades N, Mitsiades CS, Poulaki V, et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc Natl Acad Sci U S A. 2002;99(22):14374–14379. | ||
Pati S, Pelser CB, Dufraine J, Bryant JL, Reitz MS, Weichold FF. Antitumorigenic effects of HIV protease inhibitor ritonavir: inhibition of Kaposi sarcoma. Blood. 2002;99(10):3771–3779. | ||
Dewan MZ, Uchihara JN, Terashima K, et al. Efficient intervention of growth and infiltration of primary adult T-cell leukemia cells by an HIV protease inhibitor, ritonavir. Blood. 2006;107(2):716–724. | ||
Kariya R, Taura M, Suzu S, Kai H, Katano H, Okada S. HIV protease inhibitor Lopinavir induces apoptosis of primary effusion lymphoma cells via suppression of NF-κB pathway. Cancer Lett. 2014;342(1):52–59. | ||
Sepp-Lorenzino L, Ma Z, Lebwohl DE, Vinitsky A, Rosen N. Herbimycin A induces the 20 S proteasome- and ubiquitin-dependent degradation of receptor tyrosine kinases. J Biol Chem. 1995;270(28):16580–16587. | ||
Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13(3):184–190. | ||
Taldone T, Gozman A, Maharaj R, Chiosis G. Targeting Hsp90: small-molecule inhibitors and their clinical development. Curr Opin Pharmacol. 2008;8(4):370–374. | ||
Ronnen EA, Kondagunta GV, Ishill N, et al. A phase II trial of 17-(Allylamino)-17-demethoxygeldanamycin in patients with papillary and clear cell renal cell carcinoma. Invest New Drugs. 2006;24(6):543–546. | ||
Heath EI, Hillman DW, Vaishampayan U, et al. A phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin Cancer Res. 2008;14(23):7940–7946. | ||
Solit DB, Osman I, Polsky D, et al. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin Cancer Res. 2008;14(24):8302–8307. | ||
Gartner EM, Silverman P, Simon M, et al. A phase II study of 17-allylamino-17-demethoxygeldanamycin in metastatic or locally advanced, unresectable breast cancer. Breast Cancer Res Treat. 2012;131(3):933–937. | ||
Srirangam A, Mitra R, Wang M, et al. Effects of HIV protease inhibitor ritonavir on Akt-regulated cell proliferation in breast cancer. Clin Cancer Res. 2006;12(6):1883–1896. | ||
Kraus M, Malenke E, Gogel J, et al. Ritonavir induces endoplasmic reticulum stress and sensitizes sarcoma cells toward bortezomib-induced apoptosis. Mol Cancer Ther. 2008;7(7):1940–1948. | ||
Sato A, Asano T, Ito K, Asano T. Ritonavir interacts with bortezomib to enhance protein ubiquitination and histone acetylation synergistically in renal cancer cells. Urology. 2012;79(4):966.e13–966.e21. | ||
Sato A, Asano T, Ito K, Asano T. 17-Allylamino-17-demethoxygeldanamycin and ritonavir inhibit renal cancer growth by inhibiting the expression of heat shock factor-1. Int J Oncol. 2012;41(1):46–52. | ||
Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci U S A. 2000;97(20):10832–10837. | ||
Kumar S, Bryant CS, Chamala S, et al. Ritonavir blocks AKT signaling, activates apoptosis and inhibits migration and invasion in ovarian cancer cells. Mol Cancer. 2009;8:26. | ||
Batchu RB, Gruzdyn OV, Bryant CS, et al. Ritonavir-mediated induction of apoptosis in pancreatic cancer occurs via the RB/E2F-1 and AKT pathways. Pharmaceuticals (Basel). 2014;7(1):46–57. | ||
Ikezoe T, Hisatake Y, Takeuchi T, et al. HIV-1 protease inhibitor, ritonavir: a potent inhibitor of CYP3A4, enhanced the anticancer effects of docetaxel in androgen-independent prostate cancer cells in vitro and in vivo. Cancer Res. 2004;64(20):7426–7431. | ||
Oostendorp RL, Huitema A, Rosing H, et al. Coadministration of ritonavir strongly enhances the apparent oral bioavailability of docetaxel in patients with solid tumors. Clin Cancer Res. 2009;15(12):4228–4233. | ||
Sato A, Asano T, Isono M, Ito K, Asano T. Ritonavir acts synergistically with panobinostat to enhance histone acetylation and inhibit renal cancer growth. Mol Clin Oncol. 2014;2(6):1016–1022. | ||
Hamberg P, Woo MM, Chen LC, et al. Effect of ketoconazole-mediated CYP3A4 inhibition on clinical pharmacokinetics of panobinostat (LBH589), an orally active histone deacetylase inhibitor. Cancer Chemother Pharmacol. 2011;68(3):805–813. | ||
Washington CB, Duran GE, Man MC, Sikic BI, Blaschke TF. Interaction of anti-HIV protease inhibitors with the multidrug transporter P-glycoprotein (P-gp) in human cultured cells. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;19(3):203–209. | ||
Markowitz M, Saag M, Powderly WG, et al. A preliminary study of ritonavir, an inhibitor of HIV-1 protease, to treat HIV-1 infection. N Engl J Med. 1995;333(23):1534–1539. | ||
Weichold FF, Bryant JL, Pati S, Barabitskaya O, Gallo RC, Reitz MS. HIV-1 protease inhibitor ritonavir modulates susceptibility to apoptosis of uninfected T cells. J Hum Virol. 1999;2(5):261–269. | ||
Sloand EM, Kumar PN, Kim S, Chaudhuri A, Weichold FF, Young NS. Human immunodeficiency virus type 1 protease inhibitor modulates activation of peripheral blood CD4(+) T cells and decreases their susceptibility to apoptosis in vitro and in vivo. Blood. 1999;94(3):1021–1027. | ||
Knutson KL, Disis ML. Augmenting T helper cell immunity in cancer. Curr Drug Targets Immune Endocr Metabol Disord. 2005;5(4):365–371. | ||
Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54(8):721–728. | ||
Liu Y, Ye Y. Proteostasis regulation at the endoplasmic reticulum: a new perturbation site for targeted cancer therapy. Cell Res. 2011;21(6):867–883. | ||
Sato A, Asano T, Horiguchi A, Ito K, Sumitomo M, Asano T. Combination of suberoylanilide hydroxamic acid and ritonavir is effective against renal cancer cells. Urology. 2010;76(3):764.e7–764.e13. | ||
Sato A, Asano T, Ito K, Asano T. Ritonavir combined with bortezomib synergistically induces endoplasmic reticulum stress and ubiquitinated protein accumulation in bladder cancer cells. J Urol. 2013;189(4):e242–e243. | ||
Sato A, Asano T, Isono M, Ito K, Asano T. Ritonavir synergizes with carfilzomib to induce endoplasmic reticulum stress and autophagy in renal cancer cells. J Urol. 2014;191(4):e374. | ||
Wade PA. Transcriptional control at regulatory checkpoints by histone deacetylases: molecular connections between cancer and chromatin. Hum Mol Genet. 2001;10(7):693–698. | ||
Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1(3):194–202. | ||
Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389(6649):349–352. | ||
Hess-Stumpp H. Histone deacetylase inhibitors and cancer: from cell biology to the clinic. Eur J Cell Biol. 2005;84(2–3):109–121. | ||
Fukuda H, Sano N, Muto S, Horikoshi M. Simple histone acetylation plays a complex role in the regulation of gene expression. Brief Funct Genomic Proteomic. 2006;5(3):190–208. | ||
Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006;5(1):37–50. | ||
Bali P, Pranpat M, Bradner J, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280(29):26729–26734. | ||
Fiskus W, Ren Y, Mohapatra A, et al. Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor-alpha levels and transcriptional activity: a result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin Cancer Res. 2007;13(16):4882–4890. | ||
Georgakis GV, Li Y, Younes A. The heat shock protein 90 inhibitor 17-AAG induces cell cycle arrest and apoptosis in mantle cell lymphoma cell lines by depleting cyclin D1, Akt, Bid and activating caspase 9. Br J Haematol. 2006;135(1):68–71. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.