Back to Journals » Drug Design, Development and Therapy » Volume 15
The Effect of Nintedanib on T-Cell Activation, Subsets and Functions
Authors Ubieta K ![]() , Thomas MJ, Wollin L
, Thomas MJ, Wollin L
Received 31 October 2020
Accepted for publication 15 February 2021
Published 8 March 2021 Volume 2021:15 Pages 997—1011
DOI https://doi.org/10.2147/DDDT.S288369
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Kenia Ubieta, Matthew James Thomas, Lutz Wollin
Immunology & Respiratory Diseases Research, Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany
Correspondence: Lutz Wollin
Boehringer Ingelheim Pharma GmbH & Co. KG, Birkendorfer Str. 65, Biberach, 88397, Germany
Tel +49 7351 54-94993
Fax +49 7351 83-94993
Email [email protected]
Background: T cells are important regulators of inflammation and, via release of mediators, can contribute to pulmonary fibrosis. Nintedanib is approved for the treatment of idiopathic pulmonary fibrosis, systemic sclerosis-associated interstitial lung disease (ILD) and chronic fibrosing ILDs with a progressive phenotype. However, how nintedanib targets T cells has not been elucidated.
Materials and Methods: We investigated the immunomodulatory effects of nintedanib on T cells and peripheral blood mononuclear cells isolated from healthy donors. Cells were pre-incubated with different concentrations of nintedanib and then stimulated for 24 hours with anti-CD3 with or without anti-CD28 and with or without different cytokines. Levels of interferon gamma (IFN-γ), interleukin (IL)-2, IL-4, IL-5, IL-10, IL-12p70 and IL-13 were quantitated. Western blotting with primary antibodies against phospho-Lck-Y394, phospho-Lck-Y505, Lck-total and Cofilin examined the phosphorylation level of the Lck protein. In vitro T-cell proliferation, T-cell clustering and different T-cell populations were also assessed.
Results: Nintedanib blocked T-cell activation through inhibiting Lck-Y394 phosphorylation. Pretreatment of T cells with nintedanib reduced cluster formation as a marker of activation and inhibited the release of IFN-γ, IL-2, IL-4, IL-5, IL-10, IL-12p70 and IL-13 at clinically relevant concentrations ranging from 5– 77 nmol/L. Nintedanib did not alter T-cell proliferation or numbers of CD4+ and CD8+ T cells, but did increase stimulated Th17-like cells without increasing IL-17A levels.
Conclusion: These immunomodulatory effects may further explain how nintedanib slows the progression of pulmonary fibrosis in various ILDs.
Keywords: cytokines, fibrosis, inflammation, nintedanib, T cells, tyrosine kinase
Introduction
T cells are important regulators of the immune system and are central to controlling inflammation. They are present diffusely throughout the lung and are known to be involved in the pulmonary fibrosis seen in fibrosing interstitial lung diseases (ILDs), such as idiopathic pulmonary fibrosis (IPF), as well as in pulmonary arterial hypertension (PAH).1,2 T cells have also been identified in ectopic lymphoid tissues, contributing to sustained inflammation in patients with IPF and PAH.2,3 Pulmonary fibrosis can also manifest in several connective tissue diseases, including systemic sclerosis (SSc/scleroderma), rheumatoid arthritis (RA),4–6 and in patients with chronic hypersensitivity pneumonitis (cHP).7
Both the innate and adaptive immune systems are involved in the development of fibrosis.8 Accordingly, circulating peripheral blood mononuclear cells (PBMCs), including T cells, appear to play a prominent role in the pathogenesis of SSc, RA, and cHP.9–11 Fibrosis is characterized by the expansion of fibroblasts and excessive deposition of extracellular matrix (ECM) through signaling from various cytokines, chemokines, and other mediators. Pulmonary fibrosis is commonly preceded by inflammation due to T-cell infiltration, suggesting that these cells are important for the pathology of fibrosis. T cells are a major source of mediators that stimulate and transform fibroblasts,12 causing excessive deposition of ECM, which can lead to pulmonary fibrosis in patients with SSc-ILD, RA-ILD, and cHP,9,10,13 but which may also downregulate the fibrotic response (reviewed in Zhang et al).14
A broad range of different subsets of T cells is involved in the fibrogenic response, such as T helper cells (Th; including Th1, Th2, Th9, Th17, Th22), and T follicular helper cells, regulatory T (Treg) cells, natural killer T cells, γδ T cells, CD8+ cytotoxic T lymphocytes, and T follicular regulatory cells (reviewed in Heukels et al8 and Zhang et al14).
Depending on their activation status, interconnectivity and disease pathology, nearly all subsets of T cells are capable of releasing diverse mediators such as interleukin (IL)-2, IL-4, IL-9, IL-13, IL-17, IL-22 and interferon gamma (IFN-γ), to modulate the fibrotic response.14,18,19
Nintedanib is an oral, potent, small-molecule tyrosine kinase inhibitor targeting fibroblast growth factor receptor 1–3, platelet-derived growth factor receptor α and β, vascular endothelial growth factor receptor 1–3, and multiple non-receptor tyrosine kinases, including proto-oncogene tyrosine-protein kinase (Src), Lyn, lymphocyte-specific protein tyrosine kinase (Lck), Fms-like tyrosine kinase-3, colony-stimulating factor-1 receptor and several other tyrosine kinases. By binding to the intracellular adenosine triphosphate binding sites of these tyrosine kinases, nintedanib inhibits the activation of intracellular signal transduction pathways.15–17
Preclinical studies have demonstrated that nintedanib exerts antifibrotic and anti-inflammatory activities in models of lung fibrosis, whereas clinical trials have shown good efficacy and safety profiles in patients with IPF,18 SSc-ILD19 and, most recently, a range of fibrosing ILDs with a progressive phenotype.20 Nintedanib inhibits fibroblast-to-myofibroblast transformation and the proliferation of lung fibroblasts from patients with IPF.17,21,22 It also demonstrated a reduction in fibrosis and inflammation in different animal models of lung fibrosis.22–26
However, the underlying mechanisms by which nintedanib targets pulmonary fibrosis via T cells have not been explored. We know that T-cell activation is dependent on T-cell antigen receptor (TCR) signaling, which is initiated through the phosphorylation of several tyrosine kinases, such as Src and Lck,27,28 and dampened by the phosphorylation of Fyn.29,30 Phosphorylation leads to the activation of downstream signaling pathways that ultimately activate transcription factors responsible for T-cell proliferation, differentiation, or apoptosis.27 In particular, Lck is activated after T-cell stimulation and is required for T-cell proliferation and mediator release like IL-2.31 Inhibition of Lck can induce apoptosis in glucocorticoid-resistant T-acute lymphoblastic leukemia cells.32 The aim of this study was to explore the immunomodulatory effect of nintedanib on T-cell activation, subsets and function.
Materials and Methods
Determining the Inhibitory Potency of Nintedanib on Mediator Release in Human T Cells and PBMCs
Human whole blood was collected and PBMCs were isolated with a Ficoll density gradient; 1x105 cells were seeded in 100 μL X–Vivo™ 15 medium per well of a 96-well cell culture plate (Lonza, Basel, Switzerland). PBMCs were pre-incubated for 20 minutes with different concentrations of nintedanib (0.1–1000 nM) and then stimulated for 24 hours with either anti-CD3 (1 µg/mL) or anti-CD3 (1 µg/mL) in combination with anti-CD28 (0.5 µg/mL) at 37°C and 5% CO2 in the presence of nintedanib and dimethyl sulfoxide.
PAN-T cells, CD4+ and CD8+ T cells were separated from other PBMCs by affinity chromatography using Miltenyi Biotec magnetic labeling kits for negative selection of the respective cells (Miltenyi Biotec, Bergisch Gladbach, Germany). The purity of isolated T cells was verified to be ≥88.1% (PAN-T cells), 84.7% (CD4+ T cells) and 89.7% (CD8+ T cells) by flow cytometry analysis using either FITC-conjugated anti-CD3, PE-conjugated anti-CD4, or APC-conjugated anti-CD8, respectively. The cells were seeded on a 96-well plate (1x105 cells/well) that was coated with 50 ng anti-CD3/well, pre-incubated for 20 minutes with different concentrations of nintedanib (0.1–1000 nM) and then stimulated for 24 hours with or without anti-CD28 (0.5 µg/mL). Pre-incubation was chosen because nintedanib is used as a chronic treatment in patients with the goal of achieving constant exposure.33 The cell culture supernatants were collected and the levels of IFN-γ, IL-2, IL-4, IL-5, IL-10, IL-12p70 and IL-13 were quantitated using a cytokine-specific detection antibody labeled with MSD SULFO-TAG™ (Meso Scale Discovery, Rockville, USA) reagent by a Meso Scale SECTOR imager. For each experiment, the half maximal inhibitory concentration (IC50) was calculated separately. The mean IC50 was calculated by non-linear fitting of results from experiments performed with blood from the different donors. The number of donors (1–8) for each experiment is given in the figures.
Cell Isolation and Culture
Primary human T cells were isolated from blood of healthy donors. Whole blood was incubated for 20 minutes with RosetteSep™ Human T cell Enrichment Cocktail (Stem Cell Technologies, Vancouver, Canada) and T cells were isolated by negative selection using a Ficoll density gradient; 1x105 cells were seeded in 100 μL per well of a 96-well plate in RPMI-1640 medium supplemented with 10% fetal calf serum (FCS), 1 mM pyruvate, 2 mM L-glutamine, 100 U/mL penicillin/streptomycin, 50 μM β-mercaptoethanol (RPMI/FCS) and used for experiments within 24 hours and 48 hours of isolation. Typically, 90% pure T-cell populations were obtained, measured by anti-CD3 staining and fluorescence-activated cell sorting (FACS) analysis.
Cell Stimulation
Sorted T-cell populations were cultured in RPMI/FCS and incubated for 20 minutes with nintedanib at 10, 30 and 100 nM. After 20 minutes of incubation, T cells were stimulated with anti-CD3 (10 µg/mL) in combination with anti-CD28 (2 µg/mL) and IL-2 (10 ng/mL) at 37°C in 5% CO2 for 24 or 48 hours in the presence of nintedanib and other cytokines, as shown in Table 1. After stimulation, cells were characterized either by flow cytometry or Western blot analysis. For flow cytometry analysis of intracellular cytokines, cells were stimulated for 4 hours with phorbol myristate acetate (5 ng/mL) and ionomycin (500 ng/mL).
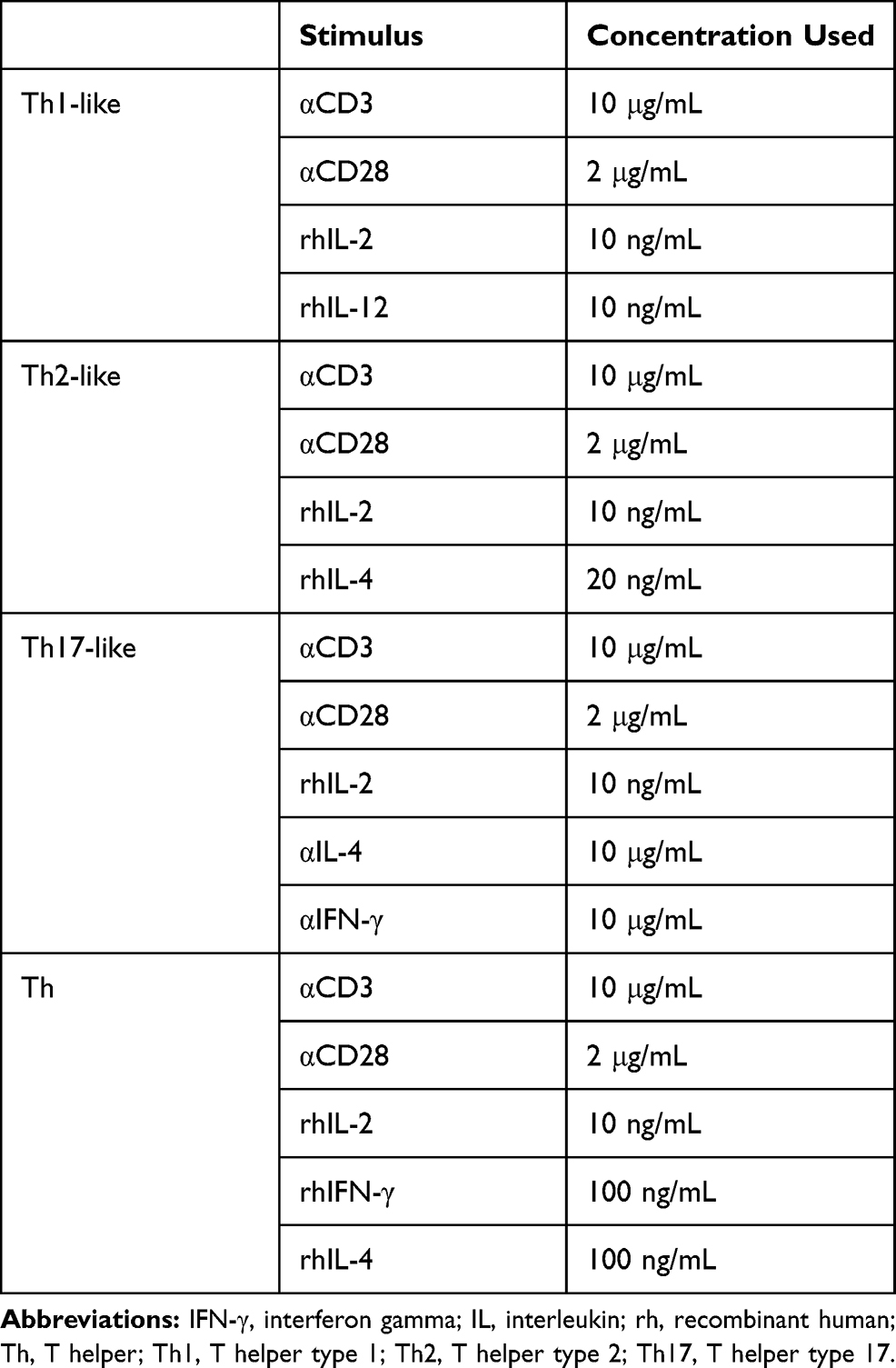 |
Table 1 Reagents Used for T-Cell Stimulation |
In vitro T-Cell Proliferation Assay
Isolated T cells from the blood of healthy donors were plated in 96-well plates; 1x105 cells were seeded in 100 μL per well in RPMI/FCS pretreated with nintedanib and stimulated as described above together with 10 µM bromodeoxyuridine (BrdU). The plates were placed in an incubator (37°C, 5% CO2) for 24 hours. The cell proliferation assay was performed according to the manufacturer’s instructions (Roche Applied Sciences, Penzberg, Germany). Control experiments included cells cultured without any stimuli. The experiment was done twice with triplicate samples.
T-Cell Clustering
Images of activated T cells were taken with an IncuCyte ZOOM® imager (Essen Bioscience, Ann Arbor, USA). Images were taken with a 310 objective lens every 2 hours for 48 hours. IncuCyte ZOOM software was used to automatically score and quantify cluster formation. Clusters were defined as cell aggregates occupying an area of at least 2000 µm2 and an eccentricity (non-circularity) of 0.8, and were expressed as the number of clusters per square millimeter.34 Five different locations were imaged in each well over the entire time course, and clustering analysis was dependent on the quantification of at least 8–16 total time-lapse videos.
Antibodies and Flow Cytometry
The following antibodies were used for flow cytometry: 7-AAD for viability, FITC-conjugated anti-CD3, BUV395-conjugated anti-CD4, APC-conjugated anti-CD8, BV421-conjugated anti-IL-4, PECy7-conjugated anti-IFN-γ (all from BD Biosciences, New Jersey, USA) and BV605-conjugated anti-IL-17A (BioLegend, San Diego, USA). Analyses of expression of cell surface molecules at a single-cell level were performed by flow cytometry with a FACS LSR-Fortessa flow cytometer (BD Biosciences, New Jersey, USA). Cells from tissue culture were stained with a sufficient amount of FACS antibodies for 20 minutes at 4°C in the dark. Cells were washed with 200 µL FACS buffer (2% FCS and 2 mM ethylenediaminetetraacetic acid in phosphate-buffered saline) and suspended in 200 µL FACS buffer. For analysis of intracellular proteins, cells were fixed and permeabilized with the Fix&Perm kit (BD Biosciences, New Jersey, USA) according to the manufacturer’s instructions, stained, and analyzed as described above.
The cell number was calculated using AccuCheck Counting Beads (Thermo Fisher Scientific, Waltham, USA). The final absolute count was performed according to the manufacturer’s instructions. 100 µL of counting beads was added to 100 µL of cell suspension for staining with FACS antibodies and measured by flow cytometry. The final absolute count was determined by dividing the number of cells counted with the total number of beads counted multiplied by the number of AccuCheck Counting Beads per µL (cells/µL).
Measurement of IL-17A Concentration in Supernatants
The concentration of IL-17A in the supernatant from T-cell cultures was measured with an IL-17A enzyme-linked immunosorbent assay kit (eBioscience, California, USA). Briefly, aliquots from the supernatant together with a biotin-conjugated anti-human IL-17A antibody were incubated overnight onto anti-human IL-17A-coated microwells. Following incubation with peroxidase-conjugated antibodies to IL-17A, color was developed with hydrogen peroxide and tetramethylbenzidine peroxidase, and the absorbance at 450 nM was measured. Wavelength correction was performed by absorbance at 620 nM. The concentration of IL-17A in each sample was determined by interpolation from a standard curve.
Protein Isolation
T cells from cell culture were washed twice with cold Dulbecco’s phosphate-buffered saline. Afterwards, cells were incubated with 100 μL of radioimmunoprecipitation assay buffer (Thermo Fisher Scientific, Waltham, USA) containing a protease-inhibitor cocktail (Sigma-Aldrich) and a phosphatase-inhibitor cocktail (Thermo Fisher Scientific, Waltham, USA) for 30 minutes at 4°C and centrifuged at 13,000 rpm for 20 minutes at 4°C. Supernatant was stored at –80°C until immunoblotting analyses.
Western Blotting
Cells were seeded onto 96-well plates with 1×105 cells/well, pre-incubated for 20 minutes with nintedanib (10, 30 and 100 nM) and subsequently stimulated with anti-CD3 (10 µg/mL) in combination with anti-CD28 (2 µg/mL) for 5, 10 and 15 minutes. To examine the phosphorylation level of the Lck protein, cells were lysed in cell lysis buffer. Primary antibodies against phospho-Lck-Y394, phospho-Lck-Y505, Lck-total (Life Technologies, Carlsbad, USA) and Cofilin (Cell Signaling Technology, Danvers, USA) were used. The detection was done by a capillary electrophoresis-based protein analysis system (ProteinSimple, San Jose, USA).
Statistical Analyses
All experiments were repeated at least three times (if not stated otherwise) and done in technical triplicates. Data are shown as mean ± standard error of mean or mean ± standard deviation. Unless otherwise noted, the statistical differences between groups were analyzed by one-way analysis of variance with subsequent Dunnett’s multiple comparison test for all parametric data, and Kruskal–Wallis test followed by Dunn’s multiple comparison test for non-parametric data. Statistical significance was accepted at P<0.05. The tests were performed using GraphPad Prism version 7 for Windows (GraphPad Software, La Jolla, USA).
Ethical Statement
Blood was donated by internal donors at the center for occupational health at Boehringer Ingelheim in Biberach; the donors provided signed informed consent that allows use for scientific purposes. All research was performed in accordance with the principles stated in the Declaration of Helsinki. Ethical approval was provided by the Federal State Medical Association of Baden-Württemberg, reference no. F-2016-121.
Results
Nintedanib Inhibits Lck Phosphorylation in T Cells
To explore whether nintedanib inhibits Y394 phosphorylation in our T-cell system, we performed a Western blot analysis of protein isolated from a primary culture of T cells pretreated with nintedanib at 10, 30 and 100 nM and stimulated with anti-CD3/CD28 at different time points. After 15 minutes of stimulation with anti-CD3/CD28, Y394 phosphorylation was significantly increased and this was inhibited by nintedanib in a concentration-dependent manner (Figure 1A and C). Total Lck was not reduced by nintedanib (Figure 1). However, no phosphorylation or differences between untreated cells or those treated with nintedanib were observed after 5 and 10 minutes of stimulation with anti-CD3/CD28 (data not shown). As expected, no Y505 phosphorylation was observed after stimulation with anti-CD3/CD28 (data not shown). These results confirmed that nintedanib blocks Lck phosphorylation in T cells.
 |
Figure 1 Inhibition of Lck phosphorylation by nintedanib. Automated capillary electrophoresis Western blot was performed using antibodies against Lck-Y394 (A) and total Lck (B). Cells were pretreated for 20 min with nintedanib and stimulated for 15 min with αCD3/αCD28. The area under the curve of the intensity of the chemiluminescence signal is shown as mean ± standard deviation of 3 donors. Panel (C) shows example Western blot with Lck-Y394 and cofilin. Panel (D) shows total Lck control Western blot. Abbreviation: Lck, lymphocyte-specific protein tyrosine kinase. |
Nintedanib Inhibits the Release of Mediators by PBMCs and T Cells
To determine the inhibitory potency of nintedanib on the release of mediators by human PBMCs and T cells, these cells were stimulated with anti-CD3 or anti-CD3 in combination with anti-CD28. In PBMCs, nintedanib blocked the release of anti-CD3- or anti-CD3/CD28-stimulated IFN-γ, IL-2, IL-4, IL-5, IL-10, IL-12p70 and IL-13 in a concentration-dependent manner, with IC50 values ranging from 17 to 59 nmol/L (Figure 2 and Supplementary Figures 1–4). The same effect was observed in PAN-T cells with IC50 values ranging from 5 to 77 nmol/L; however, anti-CD3-stimulated IL-12p70 was below the detection limit. In CD4+ and CD8+ T cells, only IL-2 levels were determined, as cell yields were too low to determine all other mediators (Figure 2 and Supplementary Figures 1–4). Nintedanib also attenuated the release of anti-CD3- or anti-CD3/CD28-stimulated IL-2 in a concentration-dependent manner, with IC50 values ranging from 23 to 83 nmol/L. These results are summarized in Table 2.
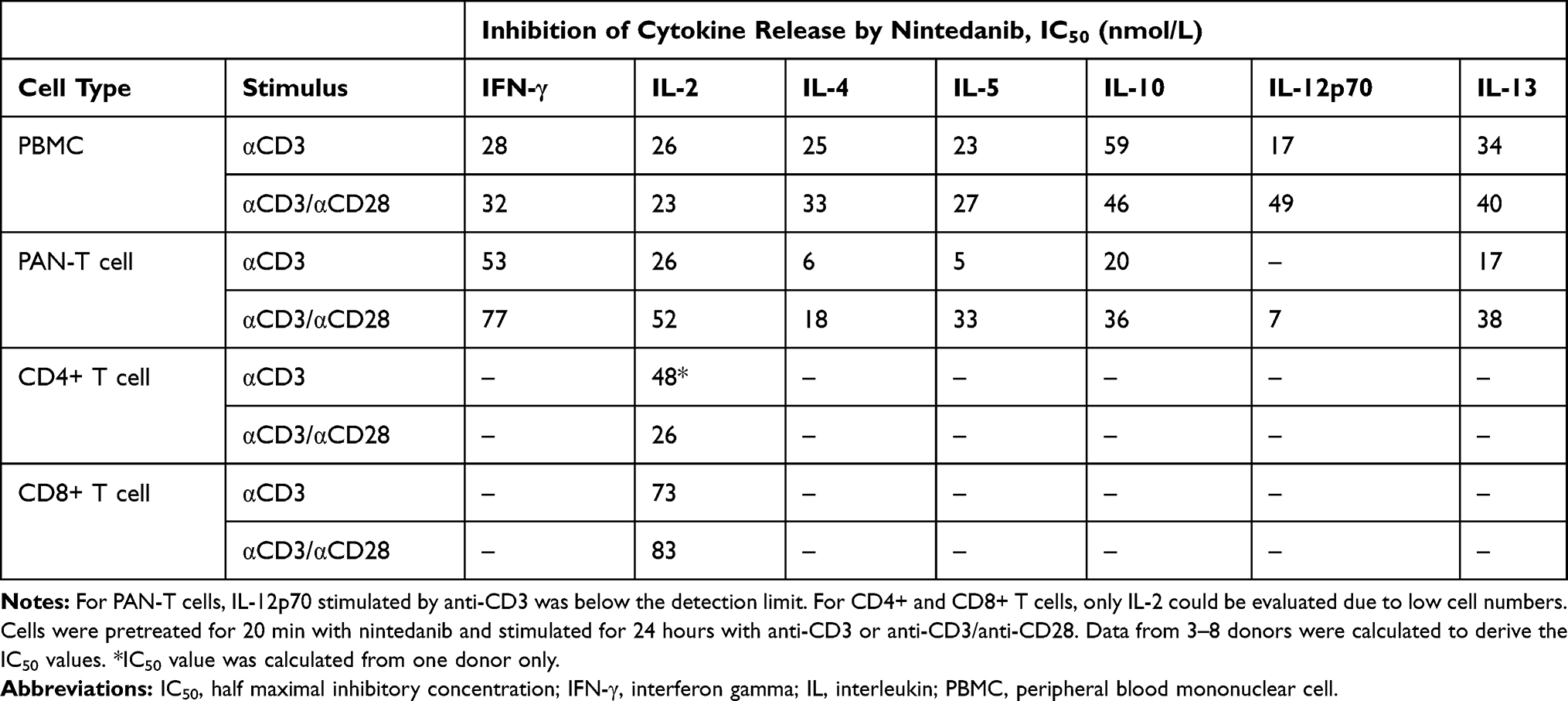 |
Table 2 Nintedanib Inhibits the Release of Cytokines |
 |
Figure 2 Nintedanib blocks the release of anti-CD3 or anti-CD3/anti-CD28-stimulated IL-2 from PBMCs, T cells, CD4+ and CD8+ specific T cells. IL-2 levels were determined in the cell culture supernatants from primary PBMCs, T cells and CD4+ and CD8+ specific T cells from healthy donors. Cells were pretreated for 20 min with nintedanib and stimulated for 24 hours with anti-CD3 or anti-CD3/anti-CD28. All data are presented as mean ± standard deviation. The number of donors is indicated in the figure. Abbreviations: IC50, half maximal inhibitory concentration; IL, interleukin; PBMC, peripheral blood mononuclear cell. |
Normal T-Cell Proliferation Upon Nintedanib Treatment
To investigate the inhibitory potency of nintedanib on T-cell proliferation and activation, human PAN-T cells were isolated from healthy donors, treated with nintedanib, and stimulated with anti-CD3/CD28, IL-2, anti-IL-4 and anti-IFN-γ to examine cluster formation and proliferation. T cells pretreated with nintedanib at 30 and 100 nM failed to cluster following stimulation with the above cocktail (Figure 3A and B). Proliferation of the PAN-T cells was also slightly enhanced following stimulation with the above cocktail. Although nintedanib demonstrated reduced cluster formation, it did not alter the proliferation of stimulated T cells, as evidenced by similar absorbance values of BrdU between cells that were pretreated with nintedanib and cells that were not (Supplementary Figure 5).
 |
Figure 3 Nintedanib decreases T-cell clustering. (A) Images were taken using an IncuCyte ZOOM live imager of highly purified T cells from healthy donors after 48 hours of activation with αCD3/αCD28, αIL-4, αIFN-γ and IL-2 and pretreated with or without nintedanib at 10, 30 and 100 nM. Scale bar (0–200 µm). (B) The number of clusters per square millimeter is shown for n=5–18 time-lapse videos over 48 hours of n=4–6 donors. Data presented as mean ± standard error of mean, *P<0.05. Abbreviations: IFN-γ, interferon gamma; IL, interleukin. |
The Number of CD4+ and CD8+ T Cells Does Not Change with Nintedanib Treatment
To investigate the effect of nintedanib on T-cell subsets, we performed flow cytometry analyses on CD4+ and CD8+ T-cell populations after pretreatment with nintedanib and stimulation with anti-CD3/CD28, IL-2, anti-IL-4 and anti-IFN-γ. T cells pretreated with nintedanib did not show significant differences in CD4+ and CD8+ cell numbers compared with non-pretreated cells (Supplementary Figure 6).
Nintedanib Increases the Number of Th17-Like Cells
To determine whether the reduced cluster formation by nintedanib also decreases the numbers of Th1-, Th2- or Th17-like cells, T cells were pretreated with nintedanib and stimulated for 24 hours with different stimuli (Table 1), and flow cytometry of the intracellular levels of IL-4, IFN-γ and IL-17A was performed. Cells pretreated with nintedanib and stimulated with anti-CD3/CD28, IL-2, anti-IL-4 and anti-IFN-γ (Th17-like stimuli) demonstrated an increase in the percentage and number of Th17-like cells (Figure 4A–C) and a reduction in the percentage and number of Th1- and Th2-like cells measured by intracellular staining of IFN-γ and IL-4, respectively (Figure 5A). However, cells pretreated with nintedanib and stimulated with Th1 and Th2 stimuli did not show any differences in the number of Th1- and Th2-like cells in comparison with non-pretreated cells (Supplementary Figure 7). Also, the number of Tregs did not change (data not shown).
 |
Figure 4 Increased Th17-like polarized CD4+ T-cell number after pretreatment with nintedanib. Flow cytometry analyses of CD4+ T cells in CD3+ T-cell culture from healthy donors pretreated for 20 min with or without nintedanib at 10, 30 and 100 nM and stimulated for 24 hours with αCD3/αCD28, αIL-4, αIFN-γ and IL-2. (A) FACS dot plots show one representative experiment of CD4+ gated, IL-17+ cells. (B) Percentage and (C) absolute counts of the total CD4+/IL-17+ T cells in total CD3+ T-cell culture (n=4–6). (D) IL-17A in the cell culture supernatant was determined by ELISA. Bars represent mean values ± standard deviation, *P<0.05. Abbreviations: ELISA, enzyme-linked immunosorbent assay; FACS, fluorescence-activated cell sorting; IFN-γ, interferon gamma; IL, interleukin; SSC, side-scatter. |
 |
Figure 5 Decreased intracellular levels of IFN-γ and IL-4 in cells pretreated with nintedanib and stimulated with Th17 stimuli. Flow cytometry analyses of CD4+ T cells in CD3+ T-cell culture from healthy donors pretreated for 20 min with or without nintedanib at 10, 30 and 100 nM and stimulated for 24 hours with αCD3/αCD28, αIL-4, αIFN-γ and IL-2. (A) FACS dot plots show one representative experiment of CD4+ gated, IFN-γ+ and IL-4+ T cells. (B) Percentage of the total CD4+/IFN-γ+ T cells and (C) CD4+/IL-4+ T cells in total CD3+ T-cell culture (n=4–6). Bars represent mean values ± standard deviation, *P<0.05. Abbreviations: FACS, fluorescence-activated cell sorting; IFN-γ, interferon gamma; IL, interleukin. |
The increased numbers of Th17-like cells occurred in a concentration-dependent manner with nintedanib pretreatment, with the highest numbers of cells pretreated with 100 nM of nintedanib. Surprisingly, nintedanib did not affect the levels of IL-17A release in these cells (Figure 4D). In accordance with the high intracellular levels of IL-17A, pretreatment with nintedanib and Th17 stimuli showed a decreased percentage of intracellular levels of IFN-γ and IL-4 (Figure 5), with no change in the absolute cell number between cells with or without pretreatment with nintedanib (Supplementary Figure 8). These data indicate that decreased cluster formation following nintedanib treatment is not accompanied by a reduction in Th1, Th2 or Th17 cells.
Excess IFN-γ and IL-4 Prevent the Increase of Th17-Like Cells Induced by Nintedanib
Naïve T cells differentiate into Th1, Th2, Th17 or Treg cells, depending on the cytokine profile. IFN-γ or IL-4 can inhibit the differentiation from naïve T cells to Th17 cells.35 To determine whether the increased number of Th17-like cells was due to the inhibition of nintedanib of the IFN-γ and IL-4 release, stimulation of cells with anti-CD3/CD28, IL-2 and recombinant IFN-γ and IL-4 after nintedanib pretreatment was performed in vitro. Indeed, the stimulation with recombinant IFN-γ and IL-4 prevented the increase in the number of Th17-like cells in cells pretreated with nintedanib (Figure 6). These data indicate that the activity of nintedanib to inhibit IFN-γ and IL-4, as shown in Table 2, drives the differentiation of T cells to Th17 cells, but without stimulating IL-17 release.
 |
Figure 6 Excess IL-4 and IFN-γ prevent the increase of Th17-like cells induced by nintedanib. Flow cytometry analyses of CD4+ T cells in CD3+ T-cell culture from healthy donors pretreated for 20 min with or without nintedanib at 10, 30 and 100 nM and stimulated for 24 hours with αCD3/αCD28, rhIL-4, with (rescued) or without (unrescued) rhIFN-γ and IL-2. (A) FACS dot plots show one representative experiment of CD4+ gated, IL-17+ cells. (B) Percentage and (C) total number of CD4+IL-17+ T cells in total CD3+ T-cell culture (n=4-6). Bars represent mean values ± standard deviation, *P<0.05. + = unrescued, R = rescued. Abbreviations: FACS, fluorescence-activated cell sorting; IFN-γ, interferon gamma; IL, interleukin; rh, recombinant human; SSC, side-scatter. |
Discussion
Nintedanib Elicits Immunomodulatory Effects That Dampen T-Cell Activity
Historically, the role of T cells in pulmonary fibrosis has been controversial, especially with regard to subsets of CD4+ T cells, which possess both profibrotic and antifibrotic activities in pulmonary fibrosis.36 Lck is required for the activation of mature T cells through the TCR and is also possibly required for survival.27,28,37 In T cells, Lck activity is enhanced by Y394 phosphorylation, which promotes T-cell activation.38 In our assays, pretreatment with nintedanib blocked the activation of T cells (stimulated by anti-CD3/anti-CD28) through the inhibition of Lck-Y394 phosphorylation in a concentration-dependent manner, which was similar to a previous report.15 Because cluster formation of T cells is considered a hallmark of T-cell activation,39–41 the inhibition of T-cell activation was evidenced by reduced cluster formation in T cells exposed to nintedanib at concentrations in the range of the exposure in patients treated with 150 mg twice daily, leading to maximum plasma concentration at steady state of 39–74 nmol/L.33,42,43 Interestingly, T-cell proliferation was not altered upon nintedanib treatment. A decrease in T-cell clustering without altered T-cell proliferation has been described before by Zumwalde et al.34 As CD28-mediated T-cell activation leads to the production of various cytokines and chemokines in the lungs,44 inhibiting TCR cluster formation is one of the immunomodulatory effects of nintedanib identified in our study.
We also determined the inhibitory potency of nintedanib in stimulated T-cell subsets on IFN-γ, IL-2, IL-4, IL-5, IL-10, IL-12p70 and IL-13. Reduced T-cell activation subsequently led to a reduced release of mediators. Nintedanib was able to inhibit the release of all mediators tested in PBMCs, and also in CD3+ PAN-T cells, with the exception of IL-12p70 when stimulated with only anti-CD3. Similarly, in stimulated CD4+ and CD8+ T cells, IL-2 release was inhibited with nintedanib. By downregulating T-cell-secreted mediators, it can be assumed that the cellular interactions between T cells and fibroblasts are modulated. Reducing secretion of several detrimental mediators may elicit a clinical benefit in the fibrotic milieu of ILDs.
There are contradicting views as to whether CD4+ and CD8+ T-cell subsets contribute to fibrosis. Some studies have demonstrated a reduction in the intensity of lung fibrosis after neutralizing these T-cell subsets,45,46 whereas others have failed to improve the extent of lung fibrosis.47 This may be attributable to the degree of T-cell depletion achieved.1 Nonetheless, in our experiments, nintedanib had no effect on the numbers of CD4+ and CD8+ T cells. Altogether, nintedanib attenuates the activity of different T-cell subsets and reduces the release of IFN-γ, IL-2, IL-4, IL-5, IL-10, IL-12p70 and IL-13 without affecting total T-cell numbers.
Nintedanib Elevated the Numbers of Th17-Like Cells without Promoting IL-17 Release
Due to the reduced cluster formation, we wanted to evaluate whether there were differences in the numbers of T-cell subsets upon nintedanib pretreatment. There was an increase in the number of Th17-like cells upon nintedanib treatment, which was also concentration-dependent, but numbers of Th1- and Th2-like cells were not affected.
In our assays, nintedanib appeared to drive the differentiation of T cells into Th17 cells through the inhibition of IFN-γ and IL-4. For patients with IPF, where levels of both Th17 and natural killer cells are reduced compared with controls,48 this may explain some of the clinical efficacy of nintedanib in IPF, and potentially in ILD as well. In contrast, an increase in Th17 cells (which shift the balance of the Th17:Treg cell ratio) has been demonstrated in autoimmune diseases, like RA, contributing to its pathology.35 Interestingly, the increase in Th17-like cells was not associated with an increase in the production of IL-17A. This could be reassuring, as IL-17 can induce fibroblast proliferation and collagen secretion,49 although its role in IPF in humans is not well understood. The role of Th17 cells and IL-17 in SSc is even more controversial (reviewed in Chizzolini et al50). Long-term studies of T cells cultured under Th17 stimuli and in the presence of nintedanib to fully differentiate T cells into Th17 are necessary to confirm this result. Studies to investigate whether or not nintedanib modulates the secretion of IL-17 after long-term exposure will be needed as well. Currently, we assume that the increase of Th17-like cells without increased IL-17A production will not affect the anti-inflammatory and antifibrotic activity of nintedanib and will have no major impact on the clinical efficacy.
Effects of Nintedanib on T Cells and Its Potential in Treating (Pulmonary) Fibrosis
There has been an increasing interest in the role of T cells in pulmonary fibrosis, due to their capacity to secrete profibrotic mediators.36 In our study, nintedanib inhibited the release of IFN-γ, IL-2, IL-4, IL-5, IL-10, IL-12p70 and IL-13, which may elicit a clinical benefit in pulmonary fibrosis in ILDs.
IL-2 has been shown to bind to lung fibroblasts, and addition of IL-2 to fibroblast cultures led to enhanced expression of the JE gene coding for the monocyte chemoattractant protein-1.51 IL-2 is also capable of inducing fibroblast proliferation.52 Hence, IL-2 may integrate monocytes and fibroblasts into a coordinated response. IL-4 and IL-13 have been shown to directly act on lung fibroblast to induce a fibrogenic response53 and to sustain the profibrotic effect of macrophages.54 IL-13 is also known to regulate fibroblast activation.55,56 IL-5-stimulated eosinophil-conditioned medium drives fibroblasts to produce several novel pro-inflammatory and pro-remodeling mediators.57
However, IFN-γ, which is also downregulated by nintedanib, exhibits potent antifibrotic activity by suppressing collagen synthesis in fibroblasts and by promoting the activation of inflammatory M1 macrophages that support ECM degradation (reviewed in Wynn55). IL-10 has been shown to attenuate the inhibitory effects of IFN-γ on lung fibroblasts.58 In summary, an effect of the reduced mediator levels on the fibrotic response seems likely.
Interestingly, nintedanib did not influence the percentage and cell numbers of various T-cell subsets, including CD4+, CD8+, Th1, Th2, and Treg.
Preclinical studies have also shown that nintedanib interferes with essential fibrotic processes, including fibroblast proliferation and migration; fibroblast-to-myofibroblast transformation; ECM secretion and deposition;21,22 and induction of apoptosis.15
Furthermore, nintedanib has consistently been shown to slow the annual rate of decline in lung function across a range of fibrosing ILDs, including SSc-ILD,19 IPF18 and other fibrosing ILDs with a progressive phenotype.20 Consequently, nintedanib has been approved for the treatment of IPF in more than 70 countries, and more recently for the treatment of SSc-ILD in the US, Europe and Japan and for chronic fibrosing ILDs with a progressive phenotype in the US.
However, not all T-cell-targeted treatments have demonstrated clinical benefits in fibrosing diseases. Abatacept, a fusion protein that inhibits T-cell co-stimulation-associated inflammation in autoimmune diseases, recently failed to demonstrate improvements in skin fibrosis in patients with diffuse cutaneous SSc.59
This study may have some limitations. For example, PBMCs and T cells in this study were derived from the blood of healthy donors and artificially stimulated to mimic activation. However, in patients with fibrotic ILDs like IPF, SSc-ILD, RA-ILD or cHP, T cells might have different features, and the milieu in fibrotic lungs might elicit a gain of function, which is not reflected in our experimental settings. The purity of isolated T cells was 80–90%, and we cannot exclude the possibility that some observed effects may be due to other cell populations. Additional experiments are warranted, with prolonged drug exposure and to confirm the current results in cells derived from patients with fibrotic ILDs.
Conclusion
Nintedanib elicits immunomodulatory effects that dampen T-cell activity by reducing the secretion of mediators capable of modulating the inflammatory and fibrotic response. Reducing secretion of these mediators may elicit a clinical benefit in fibrosing ILDs. These results support the clinical findings that nintedanib can slow disease progression in several ILDs and provide insight into the mechanism of fibrotic diseases where there is a strong T-cell involvement.
Abbreviations
BrdU, bromodeoxyuridine; cHP, chronic hypersensitivity pneumonitis; ECM, extracellular matrix; FACS, fluorescence-activated cell sorting; FCS, fetal calf serum; IC50, half maximal inhibitory concentration; IFN-γ, interferon gamma; IL, interleukin; ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis; PAH, pulmonary arterial hypertension; PBMC, peripheral blood mononuclear cell; RA, rheumatoid arthritis; SSc, systemic sclerosis; TCR, T-cell antigen receptor; Th, T helper cells; Treg, regulatory T cells.
Ethics Approval and Informed Consent
Blood was donated by internal donors at the center for occupational health at Boehringer Ingelheim in Biberach; the donors provided signed informed consent that allows use for scientific purposes. All research was performed in accordance with the principles stated in the Declaration of Helsinki. Ethical approval was provided by the Federal State Medical Association of Baden-Württemberg, reference no. F-2016-121.
Acknowledgments
The technical assistance of Angela Ostermann and Verena Brauchle in conducting the study is gratefully acknowledged. Under the authors’ conceptual direction and feedback, editorial support was provided by Henry Chung (PhD) of MediTech Media, and funded by Boehringer Ingelheim International GmbH, in accordance with Good Publication Practice (GPP3) guidelines. This paper was presented as a poster at the American Thoracic Society 2019 International Conference. The poster’s abstract was published in the American Journal of Respiratory and Critical Care Medicine 2019;199 Abstract Issue: Abstract A4600.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was conducted at Boehringer Ingelheim, Biberach, Germany.
Disclosure
Matthew James Thomas and Lutz Wollin are employees of Boehringer Ingelheim, the manufacturer of nintedanib. Kenia Ubieta was an employee of Boehringer Ingelheim at the time the study was conducted. His current affiliation is at Siemens Healthineers, Siemens Healthcare GmbH, Henkestr. 127, 91052 Erlangen, Germany. The authors report no other conflicts of interest in this work.
References
1. Luzina IG, Todd NW, Iacono AT, Atamas SP. Roles of T lymphocytes in pulmonary fibrosis. J Leukoc Biol. 2008;83(2):237–244. doi:10.1189/jlb.0707504
2. Perros F, Dorfmuller P, Montani D, et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185(3):311–321. doi:10.1164/rccm.201105-0927OC
3. Marchal-Sommé J, Uzunhan Y, Marchand-Adam S, et al. Cutting edge: nonproliferating mature immune cells form a novel type of organized lymphoid structure in idiopathic pulmonary fibrosis. J Immunol. 2006;176(10):5735–5739. doi:10.4049/jimmunol.176.10.5735
4. Wells AU, Lorimer S, Majumdar S, et al. Fibrosing alveolitis in systemic sclerosis: increase in memory T-cells in lung interstitium. Eur Respir J. 1995;8(2):266–271. doi:10.1183/09031936.95.08020266
5. Popp W, Ritschka L, Scherak O, et al. Bronchoalveolar lavage in rheumatoid arthritis and secondary Sjögren’s syndrome. Lung. 1990;168(4):221–231. doi:10.1007/BF02719696
6. Fischer A, Du Bois R. du Bois R. Interstitial lung disease in connective tissue disorders. Lancet. 2012;380(9842):689–698. doi:10.1016/S0140-6736(12)61079-4
7. Fernández Pérez ER, Brown KK. Fibrotic hypersensitivity pneumonitis. Curr Respir Care Rep. 2014;3(4):170–178. doi:10.1007/s13665-014-0094-0
8. Heukels P, Moor CC, von der Thusen JH, Wijsenbeek MS, Kool M. Inflammation and immunity in IPF pathogenesis and treatment. Respir Med. 2019;25:79–91. doi:10.1016/j.rmed.2018.12.015
9. O’Reilly S, Hugle T, van Laar JM. T cells in systemic sclerosis: a reappraisal. Rheumatology. 2012;51(9):1540–1549. doi:10.1093/rheumatology/kes090
10. Cope AP, Schulze-Koops H, Aringer M. The central role of T cells in rheumatoid arthritis. Clin Exp Rheumatol. 2007;25(5 Suppl 46):S4–11.
11. Barrera L, Mendoza F, Zuniga J, et al. Functional diversity of T-cell subpopulations in subacute and chronic hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2008;177(1):44–55. doi:10.1164/rccm.200701-093OC
12. Tanjore H, Xu XC, Polosukhin VV, et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. 2009;180(7):657–665. doi:10.1164/rccm.200903-0322OC
13. Spagnolo P, Rossi G, Cavazza A, et al. Hypersensitivity pneumonitis: a comprehensive review. J Investig Allergology Clin Immunol. 2015;25(4):237–250.
14. Zhang M, Zhang S. T cells in fibrosis and fibrotic diseases. Front Immunol. 2020;11:1142. doi:10.3389/fimmu.2020.01142
15. Hilberg F, Roth GJ, Krssak M, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68(12):4774–4782. doi:10.1158/0008-5472.CAN-07-6307
16. Hilberg F, Tontsch-Grunt U, Baum A, et al. Triple angiokinase inhibitor nintedanib directly inhibits tumor cell growth and induces tumor shrinkage via blocking oncogenic receptor tyrosine kinases. J Pharmacol Exp Ther. 2018;364(3):494–503. doi:10.1124/jpet.117.244129
17. Wollin L, Wex E, Pautsch A, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45(5):1434–1445. doi:10.1183/09031936.00174914
18. Richeldi L. du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082. doi:10.1056/NEJMoa1402584
19. Distler O, Highland KB, Gahlemann M, et al. Nintedanib for systemic sclerosis–associated interstitial lung disease. N Engl J Med. 2019;380(26):2518–2528. doi:10.1056/NEJMoa1903076
20. Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381(18):1718–1727. doi:10.1056/NEJMoa1908681
21. Hostettler KE, Zhong J, Papakonstantinou E, et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir Res. 2014;15(1):157. doi:10.1186/s12931-014-0157-3
22. Wollin L, Maillet I, Quesniaux V, Holweg A, Ryffel B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther. 2014;349(2):209–220. doi:10.1124/jpet.113.208223
23. Ackermann M, Kim YO, Wagner WL, et al. Effects of nintedanib on the microvascular architecture in a lung fibrosis model. Angiogenesis. 2017;20(3):359–372. doi:10.1007/s10456-017-9543-z
24. Huang J, Maier C, Zhang Y, et al. Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Ann Rheum Dis. 2017;76(11):1941–1948. doi:10.1136/annrheumdis-2016-210823
25. Lee HY, Hur J, Kim IK, et al. Effect of nintedanib on airway inflammation and remodeling in a murine chronic asthma model. Exp Lung Res. 2017;43(4–5):187–196. doi:10.1080/01902148.2017.1339141
26. Wollin L-S, Tomsic C, Erb KJ. The effect of nintedanib in a mouse model of severe allergic lung inflammation and remodeling. Am J Respir Crit Care Med. 2018;197:A1062.
27. Nel AE. T-cell activation through the antigen receptor. Part 1: signaling components, signaling pathways, and signal integration at the T-cell antigen receptor synapse. J Allergy Clin Immunol. 2002;109(5):758–770. doi:10.1067/mai.2002.124259
28. Straus DB, Weiss A. Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell. 1992;70(4):585–593. doi:10.1016/0092-8674(92)90428-F
29. Lovatt M, Filby A, Parravicini V, Werlen G, Palmer E, Zamoyska R. Lck regulates the threshold of activation in primary T cells, while both Lck and Fyn contribute to the magnitude of the extracellular signal-related kinase response. Mol Cell Biol. 2006;26(22):8655. doi:10.1128/MCB.00168-06
30. Yasuda K, Nagafuku M, Shima T, et al. Cutting edge: Fyn is essential for tyrosine phosphorylation of Csk-binding protein/phosphoprotein associated with glycolipid-enriched microdomains in lipid rafts in resting T cells. J Immunol. 2002;169(6):2813–2817. doi:10.4049/jimmunol.169.6.2813
31. Molina TJ, Kishihara K, Siderovski DP, et al. Profound block in thymocyte development in mice lacking p56lck. Nature. 1992;357(6374):161–164. doi:10.1038/357161a0
32. Serafin V, Capuzzo G, Milani G, et al. Glucocorticoid resistance is reverted by LCK inhibition in pediatric T-cell acute lymphoblastic leukemia. Blood. 2017;130(25):2750–2761. doi:10.1182/blood-2017-05-784603
33. Wind S, Schmid U, Freiwald M, et al. Clinical pharmacokinetics and pharmacodynamics of nintedanib. Clin Pharmacokinet. 2019;58(9):1131–1147. doi:10.1007/s40262-019-00766-0
34. Zumwalde NA, Domae E, Mescher MF, Shimizu Y. ICAM-1-dependent homotypic aggregates regulate CD8 T cell effector function and differentiation during T cell activation. J Immunol. 2013;191(7):3681–3693. doi:10.4049/jimmunol.1201954
35. Figueiredo AS, Schumacher A. The T helper type 17/regulatory T cell paradigm in pregnancy. Immunology. 2016;148(1):13–21. doi:10.1111/imm.12595
36. Re S L, Lison D, Huaux F. CD4+ T lymphocytes in lung fibrosis: diverse subsets, diverse functions. J Leukoc Biol. 2013;93(4):499–510. doi:10.1189/jlb.0512261
37. Seddon B, Legname G, Tomlinson P, Zamoyska R. Long-term survival but impaired homeostatic proliferation of naive T cells in the absence of p56lck. Science. 2000;290(5489):127–131. doi:10.1126/science.290.5489.127
38. Rossy J, Williamson DJ, Gaus K. How does the kinase Lck phosphorylate the T cell receptor? Spatial organization as a regulatory mechanism. Front Immunol. 2012;3:167. doi:10.3389/fimmu.2012.00167
39. Lanzavecchia A, Iezzi G, Viola A. From TCR engagement to T cell activation: a kinetic view of T cell behavior. Cell. 1999;96(1):1–4. doi:10.1016/S0092-8674(00)80952-6
40. Lee K-H, Dinner AR, Tu C, et al. The immunological synapse balances T cell receptor signaling and degradation. Science. 2003;302(5648):1218–1222. doi:10.1126/science.1086507
41. Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity. 2006;25(1):117–127. doi:10.1016/j.immuni.2006.04.010
42. Eisen T, Shparyk Y, Macleod N, et al. Effect of small angiokinase inhibitor nintedanib (BIBF 1120) on QT interval in patients with previously untreated, advanced renal cell cancer in an open-label, Phase II study. Invest New Drugs. 2013;31(5):1283–1293. doi:10.1007/s10637-013-9962-7
43. Mross K, Stefanic M, Gmehling D, et al. Phase I study of the angiogenesis inhibitor BIBF 1120 in patients with advanced solid tumors. Clin Cancer Res. 2010;16(1):311–319. doi:10.1158/1078-0432.CCR-09-0694
44. Okazaki T, Nakao A, Nakano H, et al. Impairment of bleomycin-induced lung fibrosis in CD28-deficient mice. J Immunol. 2001;167(4):1977–1981. doi:10.4049/jimmunol.167.4.1977
45. Huaux F, Liu T, McGarry B, Ullenbruch M, Xing Z, Phan SH. Eosinophils and T lymphocytes possess distinct roles in bleomycin-induced lung injury and fibrosis. J Immunol. 2003;171(10):5470–5481. doi:10.4049/jimmunol.171.10.5470
46. Barbarin V, Arras M, Misson P, et al. Characterization of the effect of interleukin-10 on silica-induced lung fibrosis in mice. Am J Respir Cell Mol Biol. 2004;31(1):78–85. doi:10.1165/rcmb.2003-0299OC
47. Christensen PJ, Goodman RE, Pastoriza L, Moore B, Toews GB. Induction of lung fibrosis in the mouse by intratracheal instillation of fluorescein isothiocyanate is not T-cell-dependent. Am J Pathol. 1999;155(5):1773–1779. doi:10.1016/S0002-9440(10)65493-4
48. Galati D, De Martino M, Trotta A, et al. Peripheral depletion of NK cells and imbalance of the Treg/Th17 axis in idiopathic pulmonary fibrosis patients. Cytokine. 2014;66(2):119–126. doi:10.1016/j.cyto.2013.12.003
49. Nakashima T, Jinnin M, Yamane K, et al. Impaired IL-17 signaling pathway contributes to the increased collagen expression in scleroderma fibroblasts. J Immunol. 2012;188(8):3573–3583. doi:10.4049/jimmunol.1100591
50. Chizzolini C, Dufour AM, Brembilla NC. Is there a role for IL-17 in the pathogenesis of systemic sclerosis? Immunol Lett. 2018;195:61–67. doi:10.1016/j.imlet.2017.09.007
51. Gruss HJ, Scott C, Rollins BJ, Brach MA, Herrmann F. Human fibroblasts express functional IL-2 receptors formed by the IL-2R alpha- and beta-chain subunits: association of IL-2 binding with secretion of the monocyte chemoattractant protein-1. J Immunol. 1996;157(2):851–857.
52. Kang R, Tang D, Lotze MT, Zeh III HJ. Autophagy is required for IL-2-mediated fibroblast growth. Exp Cell Res. 2013;319(4):556–565. doi:10.1016/j.yexcr.2012.11.012
53. Saito A, Okazaki H, Sugawara I, Yamamoto K, Takizawa H. Potential action of IL-4 and IL-13 as fibrogenic factors on lung fibroblasts in vitro. Int Arch Allergy Immunol. 2003;132(2):168–176. doi:10.1159/000073718
54. Su S, Zhao Q, He C, et al. miR-142-5p and miR-130a-3p are regulated by IL-4 and IL-13 and control profibrogenic macrophage program. Nat Commun. 2015;6(1):8523. doi:10.1038/ncomms9523
55. Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208(7):1339–1350. doi:10.1084/jem.20110551
56. Guo J, Yao H, Lin X, et al. IL-13 induces YY1 through the AKT pathway in lung fibroblasts. PLoS One. 2015;10(3):e0119039. doi:10.1371/journal.pone.0119039
57. Esnault S, Bernau K, Torr EE, Bochkov YA, Jarjour NN, Sandbo N. RNA-sequencing analysis of lung primary fibroblast response to eosinophil-degranulation products predicts downstream effects on inflammation, tissue remodeling and lipid metabolism. Respir Res. 2017;18(1):188. doi:10.1186/s12931-017-0669-8
58. Hashimoto T, Nakamura M, Oshika Y, et al. Interleukin-10 relieves the inhibitory effects of interferon-γ on normal human lung fibroblasts. Int J Mol Med. 2001;7(2):149–154.
59. Khanna D, Spino C, Johnson S, et al. Abatacept in early diffuse cutaneous systemic sclerosis - results of a Phase 2 investigator-initiated, multicenter, double-blind randomized placebo-controlled trial. Arthritis Rheumatol. 2020;72(1):125–136. doi:10.1002/art.41055
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.