")
Back to Journals » Cancer Management and Research » Volume 11
The construction and analysis of ceRNA networks in invasive breast cancer: a study based on The Cancer Genome Atlas
Authors Gao C, Li H, Zhuang J, Zhang H, Wang K, Yang J, Liu C , Liu L, Zhou C, Sun C
Received 3 August 2018
Accepted for publication 24 October 2018
Published 17 December 2018 Volume 2019:11 Pages 1—11
DOI https://doi.org/10.2147/CMAR.S182521
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Beicheng Sun
Chundi Gao,1,* Huayao Li,1,* Jing Zhuang,2,3 HongXiu Zhang,4 Kejia Wang,5 Jing Yang,2 Cun Liu,6 Lijuan Liu,2,3 Chao Zhou,2,3 Changgang Sun2,3
1College of First Clinical Medicine, Shandong University of Traditional Chinese Medicine, Jinan 250014, People’s Republic of China; 2Department of Oncology, Weifang Traditional Chinese Hospital, Weifang 261041, People’s Republic of China; 3Department of Oncology, Affiliated Hospital of Weifang Medical University, Weifang 261031, People’s Republic of China; 4Institute of Virology, Jinan Center for Disease Control and Prevention, Jinan 250021, People’s Republic of China; 5College of Basic Medicine, Qingdao University, Qingdao 266071, People’s Republic of China; 6College of Traditional Chinese Medicine, Shandong University of Traditional Chinese Medicine, Jinan 250014, People’s Republic of China
*These authors contributed equally to this work
Background: Studies have shown that long noncoding RNAs (lncRNAs) make up the major proportion of the ceRNA network and can regulate gene expression by competitively binding to miRNAs. This reveals the existence of an RNA-miRNA regulatory pathway and is of great biological significance. CeRNAs, as competitive endogenous RNAs, have revealed a new mechanism of interaction between RNAs. Until now, the role of lncRNA-mediated ceRNAs in breast cancer and their regulatory mechanisms have been elucidated to some extent.
Purpose: In this study, comprehensive analysis of large-scale invasive breast cancer samples in TCGA were conducted to further explore the developmental mechanism of invasive breast cancer and the potential predictive markers for invasive breast cancer prognosis in the ceRNA network.
Methods: Abnormal expression profiles of invasive breast cancer associated mRNAs, lncRNAs and miRNAs were obtained from the TCGA database. Through further alignment and prediction of target genes, an abnormal lncRNA-miRNA-mRNA ceRNA network was constructed for invasive breast cancer. Through the overall survival analysis, Identification prognostic biomarkers for invasive breast cancer patients,In addition, we used Cytoscape plug-in BinGo for the different mRNA performance functional cluster analysis.
Results: Differential analysis revealed that 1059 lncRNAs, 86 miRNAs, and 2138 mRNAs were significantly different in invasive breast cancer samples versus normal samples. Then we construct an abnormal lncRNA-miRNA-mRNA ceRNA network for invasive breast cancer, consisting of 90 DElncRNAs, 18 DEmiRNAs and 26 DEmRNAs.Further, 4 out of 90 lncRNAs, 3 out of 26 mRNAs, and 2 out of 18 miRNAs were useful as prognostic biomarkers for invasive breast cancer patients (P value < 0.05). It is worth noting that based on the ceRNA network, we found that the LINC00466-Hsa-mir-204- NTRK2 LINC00466-hsa-mir-204-NTRK2 axis was present in 9 RNAs associated with the prognosis of invasive breast cancer.
Conclusion: This study provides an effective bioinformatics basis for further understanding of the molecular mechanism of invasive breast cancerand for predicting outcomes, which can guide the use of invasive breast cancerdrugs and subsequent related research.
Keywords: invasive breast cancer, cancer genome atlas, lncRNA–miRNA–mRNA ceRNA network, bioinformatics, diagnosis and prognosis biomarkers
Introduction
Breast cancer, as the most frequent cancer affecting women worldwide, has strong invasiveness and metastatic qualities,1 and the incidence and mortality rate of breast cancer continue to increase.2 Currently, chemotherapy and surgery are the main treatment methods for breast cancer.3 However, because of the high rate of tumor recurrence and resistance to chemotherapy, the therapeutic effect and prognosis of breast cancer have always failed to achieve satisfactory results.4 Therefore, an in-depth understanding of the molecular pathogenesis of breast cancer and the identification of new candidate therapeutic targets and biomarkers is urgently needed for breast cancer treatment.
This in-depth study on the molecular pathogenesis of tumors based on bioinformatics analysis exploits an important method for tumor research. It can not only explore the molecular pathogenesis of tumors in depth but also identify novel biomarkers for the onset and prognosis of tumors.5 The screening of biomarkers for the diagnosis and prognosis of breast cancer has a crucial role in the early diagnosis of breast cancer, in improving the prognosis of breast cancer and reducing breast cancer mortality.
In recent years, sufficient research evidence has shown that lncRNAs play key roles in the physiological and pathological processes of various organisms, such as embryonic development, organogenesis, and tumorigenesis.6 lncRNAs function as ceRNAs to communicate with mRNAs by competing for shared miRNAs and many lncRNAs serve as master regulators that affect the expression levels of dozens or even hundreds of target genes.7–9 A large number of studies have shown that the abnormal expression of ceRNAs is closely related to the occurrence, development, and prognosis of tumors, including breast cancer,10 For example, Zheng et al11 found that activation of the STARD13-associated ceRNA network attenuates YAP/TAZ nuclear accumulation and transcriptional activity in breast cancer by co-modulating Hippo and Rho-GTPase/F-actin signaling and proposed the possibility of promoting the inhibition of breast cancer stem cell traits via the STARD13-associated ceRNA network. Li et al found that growth arrest-specific transcript 5 (GAS5), as a ceRNA-antagonizing tumor promoter of miR-196a-5p-expressing triple-negative breast cancer (TNBC) cells, can suppress TNBC progression by competitively binding miR-196a-5p and, therefore, GAS5 may be a prognostic biomarker of TNBC.12
ceRNA networks can link the function of protein-coding mRNAs with the function of ncRNAs, such as miRNAs and lncRNAs.10 In this study, we first obtained meaningful differentially expressed lncRNAs, miRNAs, and mRNAs based on The Cancer Genome Atlas (TCGA) database (https://cancergenome.nih.gov/). Subsequently, by comparing, predicting, and integrating differentially expressed RNAs, we constructed a differentially expressed lncRNA–miRNA–mRNA ceRNA network for invasive breast cancer. Finally, to bring the results closer to the clinical treatment of invasive breast cancer, we performed an overall survival analysis of lncRNAs, miRNAs, and mRNAs in the ceRNA network to identify invasive breast cancer-related prognostic biomarkers. Through this study, we can not only further understand the molecular mechanism of invasive breast cancer but also provide potential lncRNA, miRNA and mRNA diagnostic biomarkers for invasive breast cancer, which will contribute to the early diagnosis, treatment, and prognosis of invasive breast cancer.
Methods
Data processing and differential expression analysis
TCGA database provides the analysis of high-throughput data for various genomic changes, including the expression data of mRNA, lncRNA, and miRNA. By combining bioinformatics analysis with the clinical information of patients, it lays the foundation for improving cancer prevention and the early detection and treatment of diseases. In this study, from TCGA database, we downloaded expression data on mRNA, lncRNA, and miRNA from invasive breast cancer. Among them, mRNA and lncRNA expression data from 1,208 samples, including 112 normal samples and 1,096 invasive breast cancer samples, and miRNA expression data from 1,193 samples, including 103 normal samples and 1,090 invasive breast cancer samples, were acquired. Based on the edger software package, the downloaded data were normalized and analyzed for differences to obtain differentially expressed lncRNA, miRNA, and mRNA molecules. The relevant data provided by TCGA are publicly available and open-ended and, therefore, do not require the approval of the local ethics committee.
Prediction of lncRNA–miRNA and miRNA–mRNA interactions
miRcode (http://www.mircode.org/)13 provides “whole transcriptome” human miRNA target predictions based on the comprehensive GENCODE gene annotation. In this study, miRcode online software was used to predict the interaction between lncRNAs and miRNAs and DElncRNAs and DEmiRNAs that could be compared successfully were obtained. In addition, the target mRNAs of miRNAs were predicted using TargetScan (http://www.targetscan.org/),14 miRDB (http://www.mirdb.org/miRDB/),15 and miRTarBase (http://mirtarbase.mbc.nctu.edu.tw)16 online analysis tools. To further improve the reliability of bioinformatics analysis, a Venny diagram was used to obtain the portion of the target mRNA that overlaps with the differentially expressed mRNA in invasive breast cancer and was further analyzed as DEmRNA. Finally, we established matched DElncRNA–DEmiRNA and DEmiRNA–DEmRNA pairs.
Construction of the DElncRNA–DEmiRNA–DEmRNA ceRNA network
Based on the relationships between DElncRNAs, DEmiRNAs, and DEmRNAs and the obtained DElncRNA–DEmiRNA and DEmiRNA–DEmRNA pairs, we used Cytoscape v3.017 to construct and visualize the DElncRNA–DEmiRNA–DEmRNA ceRNA network. Cytoscape is an open source software platform for visualizing molecular interaction networks and biological pathways and integrating these networks with annotations, gene expression profiles, and other state data. Cytoscape plug-in (BNGO)18 provides a comprehensive set of functional annotation tools for investigators to understand the biological meaning behind Gene Ontology (GO) and the Kyoto Encyclopedia list of genes. To further understand the function of these differentially expressed genes, functional enrichment analysis was performed using the Cytoscape plugin BiNGO, which involves the following three parts: molecular function (MF), biological process (BP), and cellular component (CC). P<0.05 was selected as the cutoff condition when GO conditions were selected.
Associations of DElncRNAs, DEmiRNAs, and DEmRNAs and patient prognosis
Each sample in TCGA was independent of each other, and it contained all sample data, such as gene expression, prognosis, and survival time. From TCGA database, we obtained clinical information about invasive breast cancer and combined the expression data of DElncRNAs, DEmiRNAs, and DEmRNAs with the clinical data. Cox regression analysis of survival packages and Kaplan–Meier curves was used to assess the relationship between the expression of DElncRNAs, DEmiRNAs, and DEmRNAs and invasive breast cancer patients’ overall survival. In addition, DElncRNAs, DEmiRNAs, and DEmRNAs that were significantly associated with overall survival were identified as prognostic biomarkers.
Results
TCGA-related data analysis and differential expression analysis
We calculated mRNA, miRNA, and lncRNA expression profiles between invasive breast cancer samples and nonmalignant samples obtained from TCGA. Based on the relevant limma software package, data on differential expression were extracted and analyzed. Using log 2-fold change ≥2.0 and P-value <0.01 as screening cutoff conditions, 2,138 differentially expressed mRNAs, 1,059 differentially expressed lncRNAs, and 86 differentially expressed miRNAs were screened out (Supplementary Tables 1–3). Among them, the 2,138 differentially expressed mRNAs included 1,375 upregulated mRNAs and 763 downregulated mRNAs and the 1,059 differentially expressed lncRNAs included 842 upregulated lncRNAs and 217 downregulated lncRNAs. In addition, 86 differentially expressed miRNAs contained 67 upregulated miRNAs and 19 downregulated miRNAs. Cluster analysis of RNA heat maps and volcano diagrams are shown in Figures 1–3.
Matching of DElncRNA–DEmiRNA and prediction of DEmiRNA–DEmRNA interactions
Based on the miRcode online software, we predicted and compared DElncRNA–DEmiRNA pairs and found successful comparisons of 90 DElncRNAs and 18 DEmiRNAs. The target genes of 18 DEmiRNAs were predicted using TargetScan, miRDB, and miRTarBase target gene prediction software. The target genes predicted by the three online software packages were simultaneously used as prediction results for target genes of DEmiRNAs, and finally, 506 target genes were obtained. To obtain a differentially expressed miRNA–mRNA interaction pair in invasive breast cancer, the predicted target gene was matched with differentially expressed mRNAs and the results showed that 26 DEmRNAs could be successfully matched with DEmiRNAs. Finally, 323 DElncRNA–DEmiRNA pairs and 28 DEmiRNA–DEmRNA interaction pairs were obtained.
Construction of the ceRNA network and functional enrichment analysis
To further explore the mechanism of invasive breast cancer development, a ceRNA network was constructed based on the interactions of 323 DElncRNA–DEmiRNA pairs and 28 DEmiRNA–DEmRNA pairs between 90 DElncRNAs, 18 DEmiRNAs, and 26 DEmRNAs. We used Cytoscape to visualize the network (Figure 4). To further understand the functional role of DEmRNAs in invasive breast cancer, we used the BiNGO plugin for functional enrichment analysis of these DEmRNAs. The results showed that the DEmRNAs were particularly rich in the MF, BP, and cell components’ classifications. In the BP group, these genes were mainly enriched in the regulation of DNA-dependent transcription, the RNA metabolic process, and gene expression, among others (Figure 5A). The MF was mainly enriched in multiple binding, such as chromatin DNA binding, DNA regulatory region binding, transcription regulator activity, and transcription factor binding (Figure 5B). In addition, CC terms were mainly involved in the cytoplasm and nucleoplasm (Figure 5C).
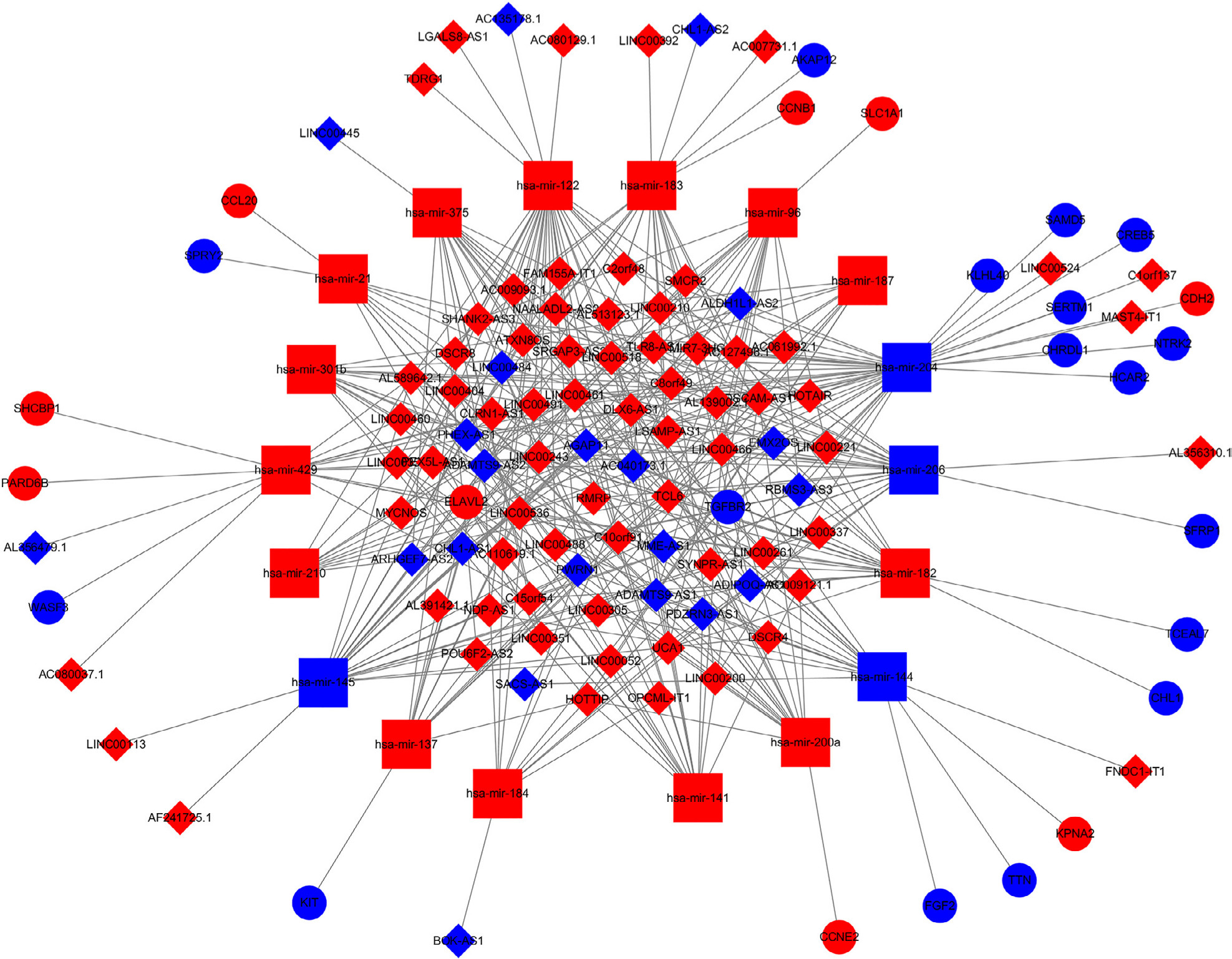 | Figure 4 The dysregulated lncRNA–mRNA–miRNA ceRNA network. Notes: Diamond denotes lncRNA, square represents miRNA, and round rectangle represents mRNA. All shapes in red and blue colors stand for upregulation and downregulation, respectively. |
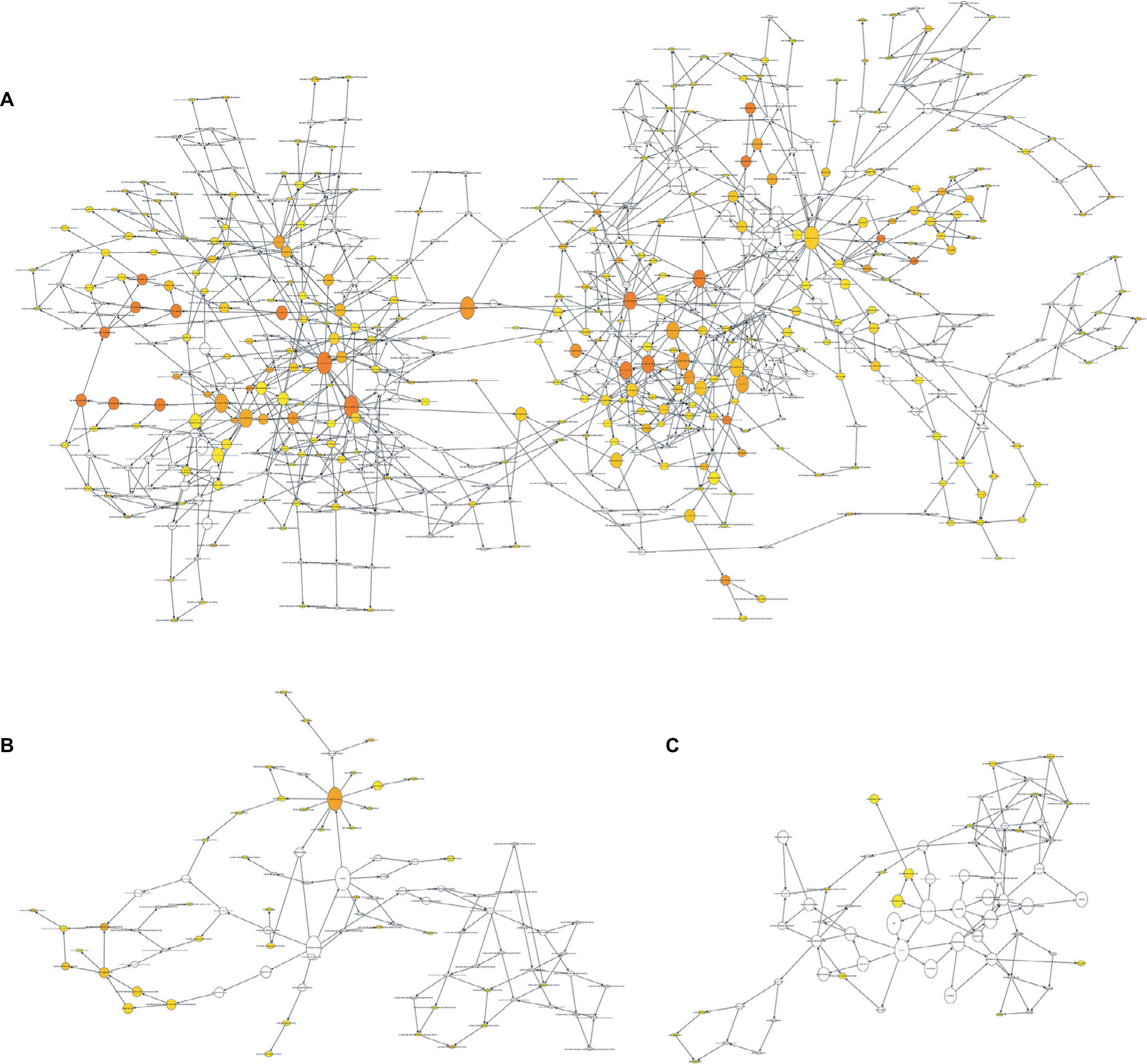 | Figure 5 Gene Ontology analysis of differentially expressed mRNAs associated with breast cancer. Note: (A) The enrichment analysis of biological processes; (B) enrichment analysis of molecular function; and (C) enrichment analysis of cellular component. |
Prognostic overall survival assessment of lncRNAs, miRNAs, and mRNAs
Survival analysis based on a survival package found that using P<0.05 as the screening criteria, 4 lncRNAs (AC007731.1, AL356479.1, LINC00460, and LINC00466) among 90 DElncRNAs were closely associated with the overall survival of invasive breast cancer patients. High expression of LINC00460 and LINC00466 was associated with high survival rates. For AC007731.1, however, low expression was associated with a high survival rate. The effect of AL356479.1 on the survival rate of patients changed over time. High expression during the early stage was associated with a higher survival rate. Two of the 18 DEmiRNAs (hsa-mir-204 and hsa-mir-301b) were associated with prognosis. High expression of hsa-mir-204 suggested a long survival time, while high expression of hsa-mir-301b suggested that the survival time was relatively short. In addition, among the 26 DEmRNAs, 3 mRNAs (KPNA2, NTRK2, and SFRP1) were associated with overall survival. High expression of NTRK2 and SFRP1 and low expression of KPNA2 showed an association with a higher survival rate (Figure 6). It is worth noting that based on the ceRNA network, we found that the LINC00466–hsa-mir-204–NTRK2 axis was linked to nine RNAs associated with the prognosis of invasive breast cancer.
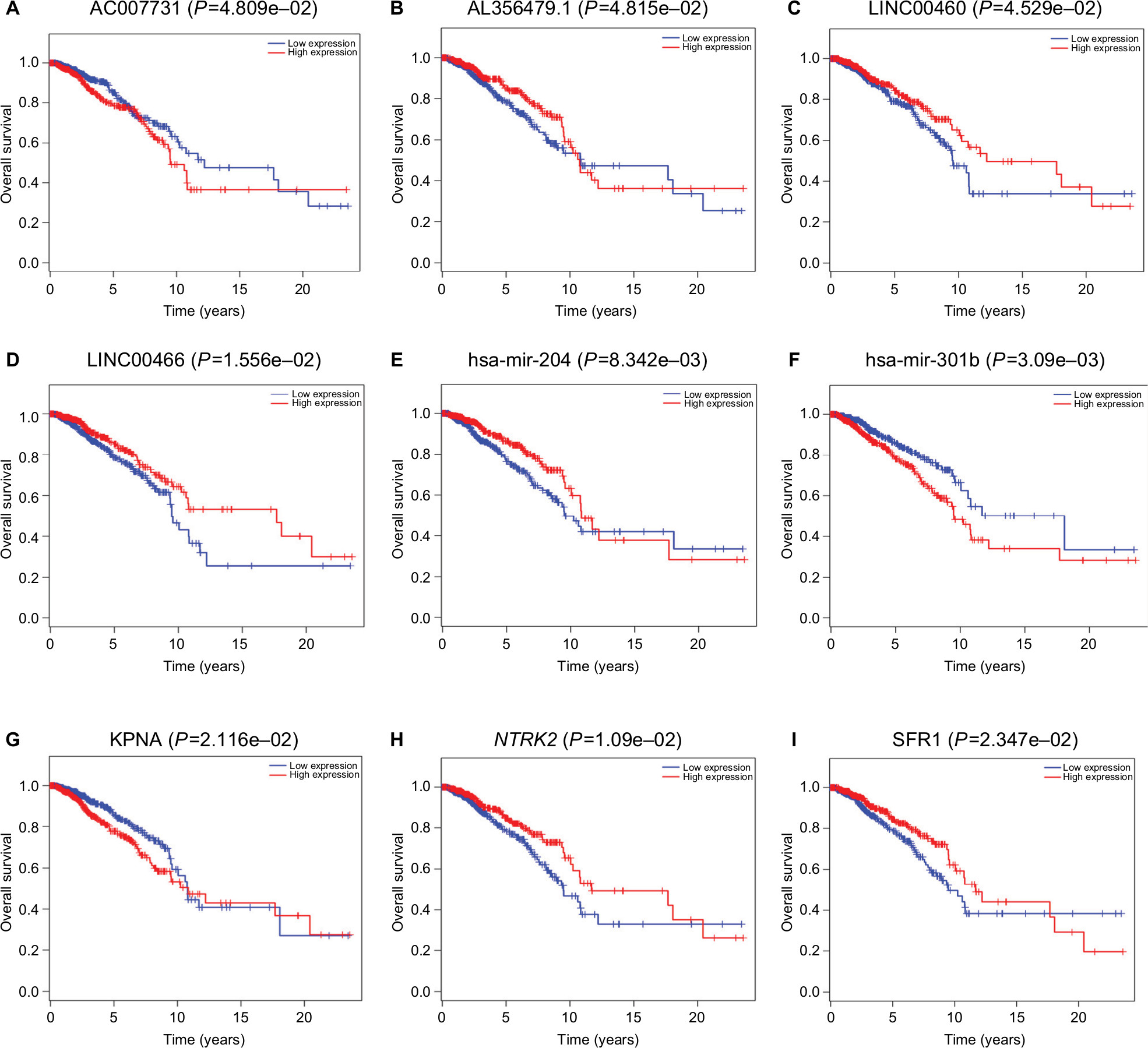 | Figure 6 Kaplan–Meier survival curves of four DElncRNAs (A–D), two DEmiRNAs (E–F), and three DEmRNAs (G–I) associated with the overall survival in breast cancer. |
Discussion
As one of the most invasive malignant tumors in women, it is very urgent and meaningful to deeply explore the molecular pathogenesis of breast cancer. In this study, we used bioinformatics analysis of the invasive breast cancer-associated lncRNA–miRNA–mRNA ceRNA network to study the molecular pathogenesis of invasive breast cancer. We obtained a total of 90 DElncRNAs, 18 DEmiRNAs, and 26 DEmRNAs, and by analyzing overall survival, 4 lncRNAs (AC007731, AL356479, LINC00460, and LINC00466), 2 miRNAs (hsa-mir-204 and hsa-mir-301b), and 3 mRNAs (KPNA2, NTRK2, and SFRP1) were found to be associated with overall survival in invasive breast cancer patients.
To further investigate the cellular mechanisms involved in invasive breast cancer, GO analysis of 26 DEmRNAs was performed. GO analysis indicated that DEmRNAs were mainly enriched in single-cell proliferation and apoptosis, including DNA-dependent transcription, the processes of RNA metabolism, chromatin DNA binding, DNA regulatory region binding, transcriptional regulation activity, and transcription factor binding, and cytoplasm and nucleoplasm were associated with gene mutations and the malignant proliferation of invasive breast cancer cells, which are consistent with the findings of current invasive breast cancer research.
The lncRNAs are ncRNA molecules with lengths >200 nucleotides. They do not participate in the coding of proteins, but they play an important role in the physiological and pathological processes of the human body.19 Studies have found that the main functions of lncRNAs occur through their interactions with miRNAs, mainly through miRNA binding to silence the miRNA, thereby indirectly regulating mRNA, DNA, and protein expressions. In humans, lncRNAs are involved in chromosomal rearrangements, gene expression, cell development, proliferation, apoptosis, and other physiological and pathological processes.20 In recent years, many studies have found that lncRNAs play a crucial role in the molecular pathogenesis of cancer and that the molecular mechanisms of action of lncRNA in breast cancer and other cancers, chromatin modification, and DNA methylation are key epigenetic events that are fundamentally disturbed during the development of cancer.21,22
In this study, we analyzed the overall survival rate of patients associated with DElncRNAs in the ceRNA network. We screened four lncRNAs that correlated with the prognosis of patients with invasive breast cancer. It is noteworthy that among the four lncRNAs, LINC00460 has been confirmed to be involved in the molecular pathogenesis of many kinds of tumors and has a strong relationship with the occurrence, development, proliferation, metastasis, and prognosis of cancer. Xing et al,23 using luciferase reporter gene assays and Western blot analysis, revealed that LINC00460 promotes MMP-9 expression by targeting miR-539, thereby acting as an oncogenic RNA in malignant meningiomas and accelerating the proliferation and metastasis of meningiomas. Using real-time quantitative polymerase chain reactions, Transwell assays, Western blots, and immunofluorescence assays, the researchers found that the interference of LINC00460 expression could inhibit the proliferation, invasion, and migration of non-small-cell cancers and interference with the expression of LINC00460 could affect epithelial–mesenchymal transition (EMT)-related proteins. The expression changes were used to confirm that LINC00460 could affect tumor invasion and migration by affecting EMT, so LINC00460’s expression had potential in the prognosis and treatment of non-small-cell lung cancer patients.24,25 In addition, LINC00460 has been proven to have roles in esophageal squamous cell carcinoma, laryngeal squamous cell carcinoma, nasopharyngeal carcinoma, and other malignant tumors. It can be used as a new biomarker for diagnosis and prognosis and may have great value in guiding clinical treatment.26–28 However, although we identified four lncRNAs that were closely related to the diagnosis and prognosis of invasive breast cancers using the bioinformatics analysis, the complex molecular regulation mechanisms among them require further clinical trials.
The important role of miRNAs in posttranscriptional gene regulation reveals another fine-tuning step in the expression of genetic information. The cellular pathways involved in the regulation of miRNAs play an important role in the physiological and pathological processes of the body’s development. At the molecular level, miRNAs inhibit protein production by affecting the stability of their target mRNAs and/or by downregulating their translation.29 Although the number of miRNAs is very small, miRNAs can participate in the regulation of cell development, proliferation, apoptosis, differentiation, and metabolism.30 Increasing numbers of studies have shown that abnormal miRNA expression can participate in many types of cancer.31,32 Breast cancer is one of these cancers, and miRNA can either act as an oncogene or tumor suppressor gene in breast cancer.33 Therefore, as a key factor in signal cascades, miRNAs are potentially useful biomarkers in clinical diagnosis and may become targets and tools for cancer treatment development.34
In this study, we have identified two miRNAs related to the diagnosis and prognosis of invasive breast cancer, including hsa-mir-204 and hsa-mir-301b. Shen et al,35 using real-time qPCR, MTT assay, flow cytometry, and Western blot analysis, found that miR-204 regulates the biological behavior of breast cancer cells through direct targeting of FOXA1, including cell proliferation, invasion, metastasis, and apoptosis. In addition, hsa-mir-204 can be used to predict the outcomes of patients with clear cell renal cell carcinoma, lung adenocarcinoma, and other cancers.32,36 Fort et al37 found that the correlation between hsa-miR-301b and candidate target genes supports it having a role in the inhibition of tumor suppressor genes in prostate cancer, indicating the oncogenic role of miR-301b in prostate cancer. In addition, these researchers also found that hsa-miR-301b is involved in the pathogenesis of cancer in both pancreatic cancer and thyroid papillary carcinoma.38,39
In this study, the specific molecular pathogenesis of hsa-miR-301b’s involvement in breast cancer needs further investigation. miRNAs regulate gene expression mainly by inducing targeted mRNA degradation. In this study, we identified a total of three mRNAs (KPNA2, NTRK2, and SFRP1) that have been shown to be associated with the prognosis of invasive breast cancer. KPNA2 has been found to play a role in altering cell phenotype and carcinogenesis. Through immunohistochemical evaluation and long-term follow-up of 1,494 invasive breast cancer patients, it was found that KPNA2 expression was associated with the negative estrogen receptor and triple-negative phenotype. Survival analysis showed that KPNA2 expression was associated with a poor prognosis (P<0.0001).40 Studies have found that miR-200c is essential for maintaining the characteristics, behavior, and sensitivity to chemotherapy in invasive breast cancer epithelial cells.
NTRK2, a direct target of miR-200c, codes for proteins expressed in mesenchymal or neuron-derived cells and may contribute to the relative risk for symptom burden during and shortly after the period of chemotherapy in women with early-stage breast cancer.41–43 Li et al,44 using the gene microarray analysis, found that miR-105/93-3p activates Wnt/β-catenin signaling by the downregulation of SFRP1, thereby promoting stem cells, chemoresistance, and TNBC cell metastasis. The has-miR-542-5p is a predictive biomarker and potential target for the treatment of breast cancer patients. Studies have confirmed that has-miR-542-5p functions through a mechanism involving its target gene SFRP1.45 Therefore, KPNA2, NTRK2, and SFRP1 are involved in the pathogenesis, infiltration, and metastasis of invasive breast cancer.
It is worth noting that based on the ceRNA network, we compared and identified nine biomarkers related to the prognosis of invasive breast cancer. We obtained a meaningful lncRNA–miRNA–mRNA regulatory axis, namely, LINC00466–hsa-mir-204–NTRK2. This suggests that LINC00466, hsa-mir-204, and NTRK2 are involved in the molecular mechanism of invasive breast cancer, while hsa-mir-204 and NTRK2 are involved in or closely related to the molecular mechanism of invasive breast cancer. Thus, LINC00466 is a new lncRNA that can be used in the prediction of invasive breast cancer and may become a new target for the treatment of invasive breast cancer. The discovery of this axis provides new insights into the molecular mechanisms of invasive breast cancer and may provide new approaches to its treatment.
Conclusion
In this study, we first constructed an invasive breast cancer-associated ceRNA network consisting of 90 DElncRNAs, 18 DEmiRNAs, and 26 DEmRNAs. Based on the total survival analysis, we obtained a total of nine prognostic biomarkers associated with invasive breast cancer, including AC007731, AL356479, LINC00460, LINC00466, hsa-mir-204, hsa-mir-301b, KPNA2, NTRK2, and SFRP1. We obtained a LINC00466–hsa-mir-204–NTRK2 axis that correlates with the treatment and prognosis of invasive breast cancer through comparative integration. This further demonstrates the contribution of lncRNA, miRNA, and mRNA interactions to the pathogenesis of invasive breast cancer. This study further investigated the molecular pathogenesis of invasive breast cancer and provides new diagnostic and prognostic biomarkers that can be used to guide the treatment and in-depth study of invasive breast cancer.
Acknowledgment
This work was supported by grants from the National Natural Science Foundation of China (81673799) and the National Natural Science Foundation of China Youth Fund (81703915).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
Kroenke CH, Michael YL, Poole EM, et al. Postdiagnosis social networks and breast cancer mortality in the after breast cancer pooling project. Cancer. 2017;123(7):1228–1237. | ||
Gradishar WJ, Anderson BO, Balassanian R, et al. Invasive breast cancer version 1.2016, nccn clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2016;14(3):324–354. | ||
Zhang N, Cao C, Zhu Y, et al. Primary breast lymphoma: a single center study. Oncol Lett. 2017;13(2):1014–1018. | ||
Song J, Ye A, Jiang E, et al. Reconstruction and analysis of the aberrant lncRNA-miRNA-mRNA network based on competitive endogenous RNA in CESC. J Cell Biochem. 2018;119(8):6665–6673. | ||
Jariwala N, Sarkar D. Emerging role of lncRNA in cancer: a potential avenue in molecular medicine. Ann Transl Med. 2016;4(15):286. | ||
Caley DP, Pink RC, Trujillano D, Carter DR. Long noncoding RNAs, chromatin, and development. ScientificWorldJournal. 2010;10:90–102. | ||
Ergun S, Oztuzcu S. Oncocers: ceRNA-mediated cross-talk by sponging miRNAs in oncogenic pathways. Tumour Biol. 2015;36(5):3129–3136. | ||
Yang L, Duff MO, Graveley BR, Carmichael GG, Chen LL. Genomewide characterization of non-polyadenylated RNAs. Genome Biol. 2011;12(2):R16. | ||
Qi X, Zhang DH, Wu N, Xiao JH, Wang X, Ma W. ceRNA in cancer: possible functions and clinical implications. J Med Genet. 2015;52(10):710–718. | ||
Zheng L, Xiang C, Li X, et al. STARD13-correlated ceRNA network-directed inhibition on YAP/TAZ activity suppresses stemness of breast cancer via co-regulating Hippo and Rho-GTPase/F-actin signaling. J Hematol Oncol. 2018;11(1):72. | ||
Li S, Zhou J, Wang Z, Wang P, Gao X, Wang Y. Long noncoding RNA GAS5 suppresses triple negative breast cancer progression through inhibition of proliferation and invasion by competitively binding miR-196a-5p. Biomed Pharmacother. 2018;104:451–457. | ||
Jeggari A, Marks DS, Larsson E. miRcode: a map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics. 2012;28(15):2062–2063. | ||
Park K, Kim KB. miRTar Hunter: a prediction system for identifying human microRNA target sites. Mol Cells. 2013;35(3):195–201. | ||
Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43(Database issue):D146–D152. | ||
Hsu SD, Tseng YT, Shrestha S, et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014;42(Database issue):D78–D85. | ||
Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. | ||
Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448–3449. | ||
Gibb EA, Brown CJ, Lam WL. The functional role of long non-coding RNA in human carcinomas. Mol Cancer. 2011;10:38. | ||
Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15(1):7–21. | ||
Gibb EA, Vucic EA, Enfield KS, et al. Human cancer long non-coding RNA transcriptomes. PLoS One. 2011;6(10):e25915. | ||
Vikram R, Ramachandran R, Abdul KS. Functional significance of long non-coding RNAs in breast cancer. Breast Cancer. 2014;21(5):515–521. | ||
Xing H, Wang S, Li Q, Ma Y, Sun P. Long noncoding RNA LINC00460 targets miR-539/MMP-9 to promote meningioma progression and metastasis. Biomed Pharmacother. 2018;105:677–682. | ||
Li K, Sun D, Gou Q, et al. Long non-coding RNA linc00460 promotes epithelial-mesenchymal transition and cell migration in lung cancer cells. Cancer Lett. 2018;420:80–90. | ||
Yue QY, Zhang Y. Effects of Linc00460 on cell migration and invasion through regulating epithelial-mesenchymal transition (EMT) in non-small cell lung cancer. Eur Rev Med Pharmacol Sci. 2018;22(4):1003–1010. | ||
Ge SS, Wu YY, Gao W, et al. [Expression of long non-coding RNA LINC00460 in laryngeal squamous cell carcinoma tissue and its clinical significance]. Lin Chung Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2018;32(1):18–22. | ||
Huang GW, Xue YJ, Wu ZY, et al. A three-lncRNA signature predicts overall survival and disease-free survival in patients with esophageal squamous cell carcinoma. BMC Cancer. 2018;18(1):147. | ||
Kong YG, Cui M, Chen SM, Xu Y, Xu Y, Tao ZZ. lncRNA-LINC00460 facilitates nasopharyngeal carcinoma tumorigenesis through sponging miR-149-5p to up-regulate IL6. Gene. 2018;639:77–84. | ||
Shukla GC, Singh J, Barik S. MicroRNAs: processing, maturation, target recognition and regulatory functions. Mol Cell Pharmacol. 2011;3(3):83–92. | ||
Wahid F, Shehzad A, Khan T, Kim YY. MicroRNAs: synthesis, mechanism, function, and recent clinical trials. Biochim Biophys Acta. 2010;1803(11):1231–1243. | ||
Heyn H, Engelmann M, Schreek S, et al. MicroRNA miR-335 is crucial for the BRCA1 regulatory cascade in breast cancer development. Int J Cancer. 2011;129(12):2797–2806. | ||
Zhao J, Cheng W, He X, et al. Construction of a specific SVM classifier and identification of molecular markers for lung adenocarcinoma based on lncRNA-miRNA-mRNA network. Onco Targets Ther. 2018;11:3129–3140. | ||
Tavazoie SF, Alarcón C, Oskarsson T, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451(7175):147–152. | ||
Xue J, Chen LZ, Li ZZ, Hu YY, Yan SP, Liu LY. MicroRNA-433 inhibits cell proliferation in hepatocellular carcinoma by targeting p21 activated kinase (PAK4). Mol Cell Biochem. 2015;399(1–2):77–86. | ||
Shen SQ, Huang LS, Xiao XL, et al. miR-204 regulates the biological behavior of breast cancer MCF-7 cells by directly targeting FOXA1. Oncol Rep. 2017;38(1):368–376. | ||
Wang Z, Zhang Z, Zhang C, Xu Y. Identification of potential pathogenic biomarkers in clear cell renal cell carcinoma. Oncol Lett. 2018;15(6):8491–8499. | ||
Fort RS, Mathó C, Oliveira-Rizzo C, Garat B, Sotelo-Silveira JR, Duhagon MA. An integrated view of the role of miR-130b/301b miRNA cluster in prostate cancer. Exp Hematol Oncol. 2018;7:10. | ||
Ab Mutalib NS, Othman SN, Mohamad Yusof A, Abdullah Suhaimi SN, Muhammad R, Jamal R. Integrated microRNA, gene expression and transcription factors signature in papillary thyroid cancer with lymph node metastasis. PeerJ. 2016;4:e2119. | ||
Ye S, Yang L, Zhao X, Song W, Wang W, Zheng S. Bioinformatics method to predict two regulation mechanism: TF-miRNA-mRNA and lncRNA-miRNA-mRNA in pancreatic cancer. Cell Biochem Biophys. 2014;70(3):1849–1858. | ||
Alshareeda AT, Negm OH, Green AR, et al. KPNA2 is a nuclear export protein that contributes to aberrant localisation of key proteins and poor prognosis of breast cancer. Br J Cancer. 2015;112(12):1929–1937. | ||
Cochrane DR, Spoelstra NS, Howe EN, Nordeen SK, Richer JK. MicroRNA-200c mitigates invasiveness and restores sensitivity to microtubule-targeting chemotherapeutic agents. Mol Cancer Ther. 2009;8(5):1055–1066. | ||
Howe EN, Cochrane DR, Richer JK. Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res. 2011;13(2):R45. | ||
Young EE, Kelly DL, Shim I, Baumbauer KM, Starkweather A, Lyon DE. Variations in COMT and NTRK2 influence symptom burden in women undergoing breast cancer treatment. Biol Res Nurs. 2017;19(3):318–328. | ||
Li HY, Liang JL, Kuo YL, et al. miR-105/93-3p promotes chemoresistance and circulating miR-105/93-3p acts as a diagnostic biomarker for triple negative breast cancer. Breast Cancer Res. 2017;19(1):133. | ||
Zhu QN, Renaud H, Guo Y. Bioinformatics-based identification of miR-542-5p as a predictive biomarker in breast cancer therapy. Hereditas. 2018;155:17. |
Figure 1 The differentially expressed mRNAs of invasive breast cancer.
Notes: (A) Heat maps of breast cancer-related differentially expressed mRNAs. The color from green to red shows a trend from low expression to high expression. (B) Volcano diagrams of breast cancer-related differentially expressed mRNAs. The red dot represents upregulated mRNA, and the green dot represents downregulated mRNA.
Figure 2 The differentially expressed lncRNAs of invasive breast cancer.
Notes: (A) Heat maps of breast cancer-related differentially expressed lncRNAs. The color from green to red shows a trend from low expression to high expression. (B) Volcano diagrams of breast cancer-related differentially expressed lncRNAs. The red dot represents upregulated lncRNA, and the green dot represents downregulated lncRNA.
Figure 3 The differentially expressed miRNAs of invasive breast cancer.
Notes: (A) Heat maps of breast cancer-related differentially expressed miRNAs. The color from green to red shows a trend from low expression to high expression. (B) Volcano diagrams of breast cancer-related differentially expressed mRNAs. The red dot represents upregulated miRNA, and the green dot represents downregulated miRNA
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.