")
Back to Journals » Infection and Drug Resistance » Volume 15
The Comparison of Metagenomic Next-Generation Sequencing with Conventional Microbiological Tests for Identification of Pathogens and Antibiotic Resistance Genes in Infectious Diseases
Authors Lu H, Ma L, Zhang H, Feng L, Yu Y, Zhao Y, Li L, Zhou Y, Song L, Li W, Zhao J , Liu L
Received 15 April 2022
Accepted for publication 11 October 2022
Published 22 October 2022 Volume 2022:15 Pages 6115—6128
DOI https://doi.org/10.2147/IDR.S370964
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Hongzhi Lu,1 Li Ma,1 Hong Zhang,1 Li Feng,1 Ying Yu,1 Yihan Zhao,1 Li Li,1 Yujiao Zhou,1 Li Song,2,3 Wushuang Li,2,3 Jiangman Zhao,2,3 Lanxiang Liu4
1Department of Infectious Diseases, First Hospital of Qinhuangdao, Qinhuangdao, Hebei, 066000, People’s Republic of China; 2Shanghai Biotecan Pharmaceuticals Co., Ltd, Shanghai, 201204, People’s Republic of China; 3Shanghai Zhangjiang Institute of Medical Innovation, Shanghai, 201204, People’s Republic of China; 4Medical Imaging Center, First Hospital of Qinhuangdao, Qinhuangdao, Hebei, 066000, People’s Republic of China
Correspondence: Lanxiang Liu, Department of Medical Imaging Center, First Hospital of Qinhuangdao, 258 Wenhua Road, Qinhuangdao, 066000, Hebei, People’s Republic of China, Email [email protected]
Background: Metagenomic next-generation sequencing (mNGS) has been widely studied, due to its ability of detecting all the microbial genetic information unbiasedly in a sample at one time and not relying on traditional culture. However, the application of mNGS in the diagnosis of clinical pathogens remains challenging.
Methods: From December 2019 to March 2021, 134 specimens including Broncho alveolar lavage fluid (BAFL), blood, sputum, cerebrospinal fluid (CSF), bile, pleural fluid, pus, were continuously collected in The First Hospital of Qinhuangdao, and their retrospective diagnoses were classified into infectious disease (128, 95.5%) and noninfectious disease (6, 4.5%). The pathogen-detection performance of mNGS was compared with conventional microbiological tests (CMT) and culture method. In addition, the antibiotic resistance genes (ARGs) and evolutionary relationship of common drug-resistant A. baumannii were also analyzed.
Results: Compared with CMT and culture methods, mNGS showed higher sensitivity in pathogen detection (74.2% vs 57.8%; P < 0.001 and 66.3% vs 31.7%; P < 0.001, respectively). Importantly, for cases that mNGS-positive only, 18 (35%) cases result in diagnosis modification, and 7 (23%) cases confirmed the clinical diagnosis. In 17 cases that A. baumannii were both detected in mNGS and culture, ade genes were the most frequently detected ARGs (from 13 cases), followed by sul2 and APH(3”)-Ib (both from 12 cases). High consistency was observed among these ARGs and the related phenotype (100% for ade genes, 91.6% for sul2 and APH(3”)-Ib). A. baumannii strains were classified into three groups, and most were well-clustered. It suggested those strains may be the epidemic strains.
Conclusion: In our study, mNGS had a higher sensitivity than CMT and culture method. And the result of ARGs frequency and cluster analysis of A. baumannii was of great significance to the anti-infective therapy.
Keywords: metagenomic next-generation sequencing, sensitivity, antibiotic resistance genes, infection, conventional microbiological tests
Introduction
Infection is one of the main causes of death in critically ill patients. With the emergence of new pathogenic microorganisms, increasing number of drug-resistant pathogens and immunosuppressive hosts, the incidence and mortality rate of infectious diseases remain high.1–3
The traditional pathogen detection technology is mainly based on microbial culture which depends on the vitality of pathogens and media type, incubation temperature, oxygen levels, etc. The results of traditional culture method indicated that only 38% of patients with community-acquired pneumonia can be diagnosed by traditional detection method.4 Serological detection technology based on antigen or antibody is fast and easy to operate, but prior knowledge is necessary, while the detection sensitivity and specificity are also limited.5 With the development of molecular biology technology, nucleic acid detection has become the mainstream development trend of pathogen detection. It gets rid of the dependence of traditional detection on pathogen isolation and culture, and significantly improves the sensitivity of pathogen detection. In recent years, with the development and application of high-throughput sequencing technology in pathogen detection, mNGS has shown significant advantages compared to other detection methods.6,7
However, the etiological diagnosis of clinical infection remains challenging.8–10 Whether the detection results reflect the real infection status of patient needs to be combined with the clinical situation for comprehensive evaluation since mNGS only detects nucleic acids (including DNA and RNA) in samples. In order to establish the standard of clinical application and interpretation of these results, the efficacy of mNGS in the diagnosis of pathogens in clinical infectious diseases still needs more comprehensive studies. On the other hand, there is still little research on the consistency between antibiotic resistance genes (ARGs) detection and clinical practice. However, this consistency may play an important role when the results of routine culture-based antibiotic susceptibility test (AST) cannot be obtained.
Thus, to evaluate the pathogen- and ARGs-detecting performance of mNGS in infectious diseases, we retrospectively analyzed the detection results of mNGS and conventional microbiological tests (CMT) in 134 cases of infectious clinical specimens, for A. baumannii that detected by mNGS and traditional culture methods, the strain cluster analysis was carried out trying to obtain the dominant strains.
Materials and Methods
Study Patients
The comparative study between mNGS and traditional methodology is described in Figure 1. From December 2019 to March 2021, we reviewed 152 cases suspected of acute or chronic infection from respiratory and critical care medicine department, pneumology department, intensive care unit and department of hematopathology at The First Hospital of Qinhuangdao, China. Based on our inclusion/exclusion criteria, 134 samples were included for analysis. To study the sensitivity and specificity of pathogen detection methods, the 134 samples were categorized into 2 groups defined as infectious disease (ID) and noninfectious disease (NID) by clinicians according to the criteria of infection shown in Table S1. Specimens were analyzed by mNGS assay (Biotecan, China) as well as conventional microbiological testing (CMT) in a pairwise manner. The CMT used in this study were presented in Table S1, and they included blood culture, serological test, molecular diagnostic test, antigen detection and direct sputum smear method. ARGs were analyzed for specimens that A. baumannii been detected by both mNGS and CMTs.
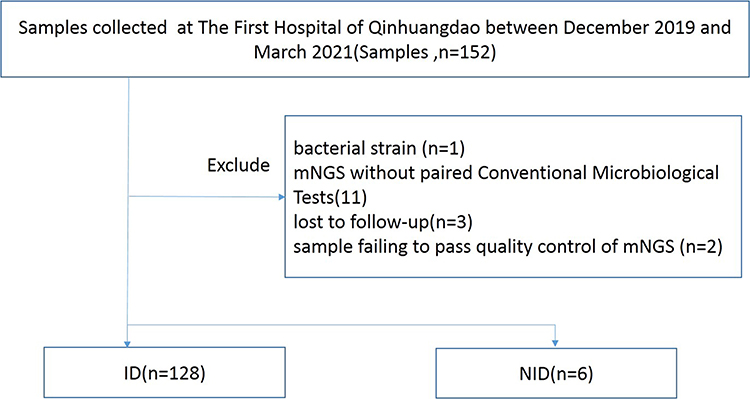 |
Figure 1 Flow chart of cases inclusion and exclusion. |
Metagenomic Next-Generation Sequencing and Analysis
In this research, nucleic acid detection and sequencing were performed based on NextSeq CN500 platform of Illumina (Illumina, Inc.). The experimental process is described in Figure 2. Patients with suspected infection were enrolled, and samples of infection sites or peripheral blood samples were collected according to standard protocol. Blood samples no less than 5mL were collected by cell-free DNA storage tube (CW2815M, CWBIO) which was anticoagulant blood collection vessels with Ethylene Diamine Tetraacetic Acid (EDTA) and special deoxyribonucleic acid (DNA) protective agent as described in the instructions, and were stored and transported at room temperature. Each sample from Broncho alveolar lavage fluid (BALF) or pleural fluid was at least 5 mL, and 1–3mL from sputum, cerebrospinal fluid (CSF) or other body fluids was collected in a dry sterile tube for cryopreservation and transported in drikold. Since the collected samples were suspected to be infectious, they were inactivated by 56°C water bath for 30 minutes before nucleic acid extraction.11–13 Different types of samples underwent different pretreatment. For nucleic acid extraction, blood samples were centrifuged at 1600 g for 10 minutes at 4°C to separate plasma. Then, transfer the plasma sample to new sterile tubes and cell-free DNA was extracted using HiPure circulating DNA MIDI kit (D3182-03B, Magen). Sputum samples were liquefied with 0.1% DTT at room temperature for 30 minutes. No pretreatment was required for BALF and other types of samples (including CSF, Pleural Fluid). DNA was extracted using a HostZEROTM Microbial DNA Kit (D4310, ZYMO RESEARCH) according to the protocol.
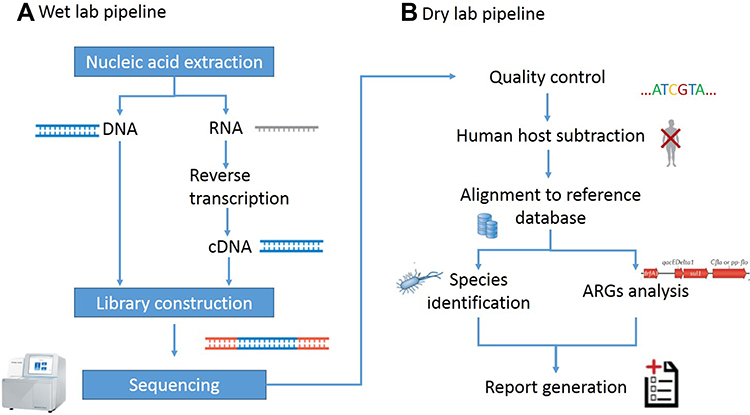 |
Figure 2 Schematic of metagenomic next-generation sequencing and analysis. (A) The wet lab pipeline including nucleic acid extraction, library construction and sequencing. (B) Dry lab pipeline including quality control, human host subtraction, alignment to reference database and report generation. |
The commercial library preparation kit for Illumina sequencing system was used to prepare the library. Library was constructed with Kapa hyper plus library preparation kit (kk8514, Kapa) according to the manufacturer’s protocol. Agilent 2100 Bioanalyzer was used to analyze the length of the inserted fragments in the library, and qubit dsDNA HS assay kit (Q32854, Thermo Fisher Scientific Inc.) was used to control the concentration of the library. Sequencing was performed on NextSeq CN500 platform (Illumina). Each test included internal control, negative control and positive control. Internal parameters were specific molecular tags placed in the sample before nucleic acid extraction to track the whole process and control the quality of the workflow. No-template water as a negative control enabled the detection of environment, reagent and cross-sample contamination. It might indicate a certain contamination when the negative control failed, and the test would be repeated. The positive control in this study were real clinical samples verified to contain known pathogens. If the targeted pathogens were not detected in positive controls, it could indicate there were failures in the workflow of the wet lab pipeline or the dry lab pipeline.
Quality Control
Due to the high homology of microbial sequences, the length of reads should be more than 50 bp. Considering the interference of human genome and the sensitivity of detection, the amount of effective sequencing data should not be less than 20 million without removing the components of human genome.
Data Filtering
In order to obtain high-quality sequence data, bioinformatics analysis method was used to further filter the qualified data. Fastp software (v0.23.1) is used to filter the original sequencing data to obtain high-quality sequencing data (clean data) to ensure the follow-up analysis. The specific steps include removing adapter sequence from reads and filtering out low complexity sequences. The processed effective data contain more than 90% of human hosts. In order to ensure the effective microbial data, the reads is compared to the version of grch38.p12 of human genome, so as to filter out the residual human source reads pollution which cannot be removed by experimental means, and only the microbial reads are reserved for subsequent classification.
Sequences Alignment
Two software were used to identify species of microorganisms based on marker sequence or whole genome alignment. According to the method provided by metaphlan2 software, the sequence information of the existing microbial database (mpa_v20_m200.fna) was analyzed to form a unique marker for each species, then the effective bacterial DNA reads were compared with the species marker sequence to identify the pathogen species. At the same time, Kraken software was used to customize the whole genome database of pathogenic microorganisms and classify pathogenic bacteria through whole genome alignment. The genomic data of pathogenic microorganisms were obtained from NCBI (ftp://ftp.ncbi.nlm.nih.gov/genomes/genbank/) and PATRIC (https://www.patricbrc.org/). According to the final pathogen alignment results, the parameters of all detected pathogens were calculated, including alignment sequence number, relative abundance, genome coverage.
Analysis of Antibiotic Resistance Genes and Phylogenetic on A. baumannii
Since A.baumannii was the bacterium most frequently detected by mNGS and CMT and in most cases had been found antibiotic resistance by AST, samples in which A. baumannii was both detected by mNGS and CMT had been analyzed for ARGs and phylogenetic tree. Based on the CARD (the Comprehensive Antimicrobial Resistance Database), the sequences annotated to pathogenic microorganisms were aligned to the established drug resistance gene database by blast (v 2.2.26). Only the sequences with similarity identity ≥90% and length more than 70 bp were retained. According to the starting position of the comparison, the coverage of the drug-resistant gene was calculated. The coverage ≥80% indicated that the drug-resistant gene was positive. Analysis of phylogenetic tree was based on the full genome sequences of the A. baumannii strains by RAxML (v2.0.1). The Codon Tree method selects single-copy PATRIC PGFams and analyzes aligned proteins and coding DNA from single-copy genes using the program RAxML (v2.0.1).
Criteria for a Positive mNGS Result
We established criteria for a potential mNGS result according to previous studies.14
- Bacteria (except mycobacteria), viruses and parasites: mNGS-found a microorganism (at the species level) with a coverage rate scored 10-fold higher than any other microorganism.15
- Fungi: mNGS-identified microorganism (species level) with a coverage scored 5-fold higher than any other fungus.16,17
- Mycobacteria: Mycobacterium tuberculosis (MTB) was considered positive when at least one specific sequence was mapped to the reference genome due to the difficulty of nucleic acid extraction and low possibility18 of environmental contamination.19 Nontuberculous mycobacterium (NTM) which commonly found in the environment was defined as positive when its relative abundance of bacteria was in the top 10.
Statistical Analysis
Comparative analysis was performed by Pearson χ2 test, Fisher exact test, or McNemar test for discrete variables. Spss22.0 software was used for data analysis. P values less than 0.05 were considered significant and all tests were two-tailed.
Results
Sample and Patient Characteristics
Demographic characteristics of patients are provided in Table 1. There were 87 males and 47 females with a median age of 65 years old participated in our research. The majority (66/134, 49.25%) of the samples were BALF. The rest include 41 samples (30.60%) from Blood, 11 samples (8.21%) from sputum, 11 samples (8.21%) from CSF, 5 samples (3.73%) from other types of samples including 2 from bile, and 2 samples from Pleural Fluid and 1 from pus (Table 2). In our research cohort, 128 cases (95.5%) were diagnosed as infectious diseases (ID) by clinicians and were divided into ID group, while 6 cases were diagnosed as non-infectious diseases (NID) by clinicians and were divided into NID group. Most patients were diagnosed with respiratory tract infection (95/134, 70.9%), followed by bloodstream infection (10/134, 7.5%), central nervous system infection (7/134, 5.2%) and intra-abdominal infection (6/134, 4.5%) as shown in Figure 3.
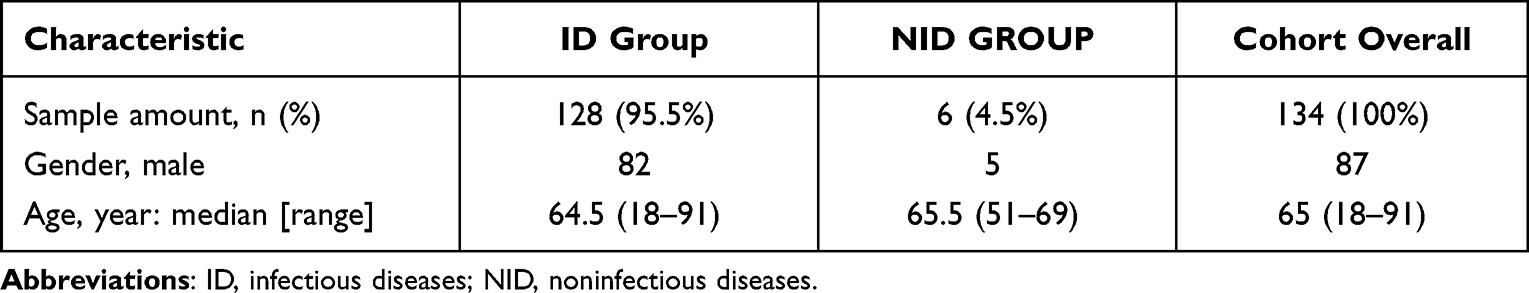 |
Table 1 Demographics Characteristics of Samples |
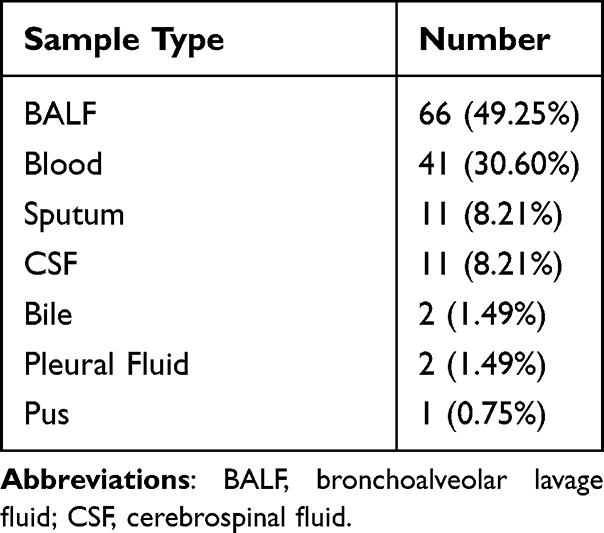 |
Table 2 Distribution of Sample Types |
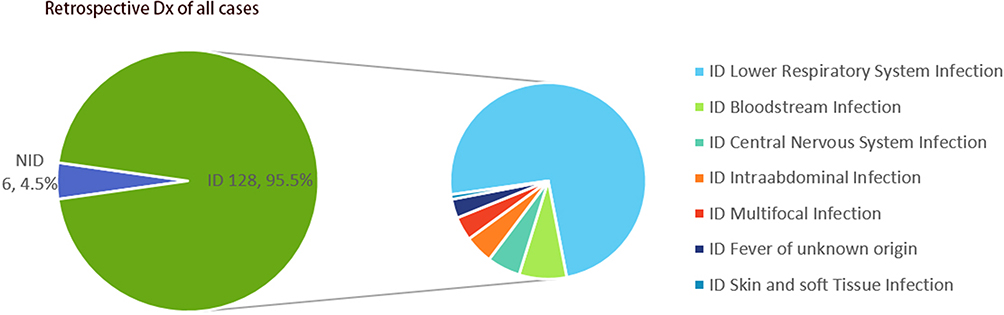 |
Figure 3 Distribution of the infectious diseases of clinical specimens. The pie chart demonstrated the cause of the retrospective diagnosis in ID (95.5%) and NID (4.5%) groups. Lower respiratory tract infection accounted for 70.9% of cases in ID groups, followed by bloodstream infection (7.5%), central nervous system infections (5.2%), intra-abdominal infection (4.5%) multifocal infections (3.9%), fever of unknown origin (3.1%), and skin and soft tissue infections (0.8%). Abbreviations: ID, infectious disease; NID, noninfectious disease; BALF, bronchoalveolar lavage fluid; CSF, cerebrospinal fluid; Dx, diagnosis. |
Diagnostic Performance Comparison of mNGS and CMT
Comparison of the Diagnostic Performance of mNGS and CMT
The comparison of the diagnostic results of NGS with the CMT method for all 134 samples is shown in Figure 4A. The positivity rates of mNGS and CMT method for the ID and NID groups are illustrated in Figure 3. In Chi square test, the positive rates of mNGS and CMT were significantly different within ID group (70.9% vs 55.2%, P<0.01), but there was no difference within NID group. The positive predictive value was 99%, and negative predictive value of mNGS was 13.2%. As expected, mNGS increased the sensitivity rate from 57.8% to 74.2% compared with CMT, while the difference of specificity was not significant (83.3% vs 83.3%, P>0.05) (Figure 4B).
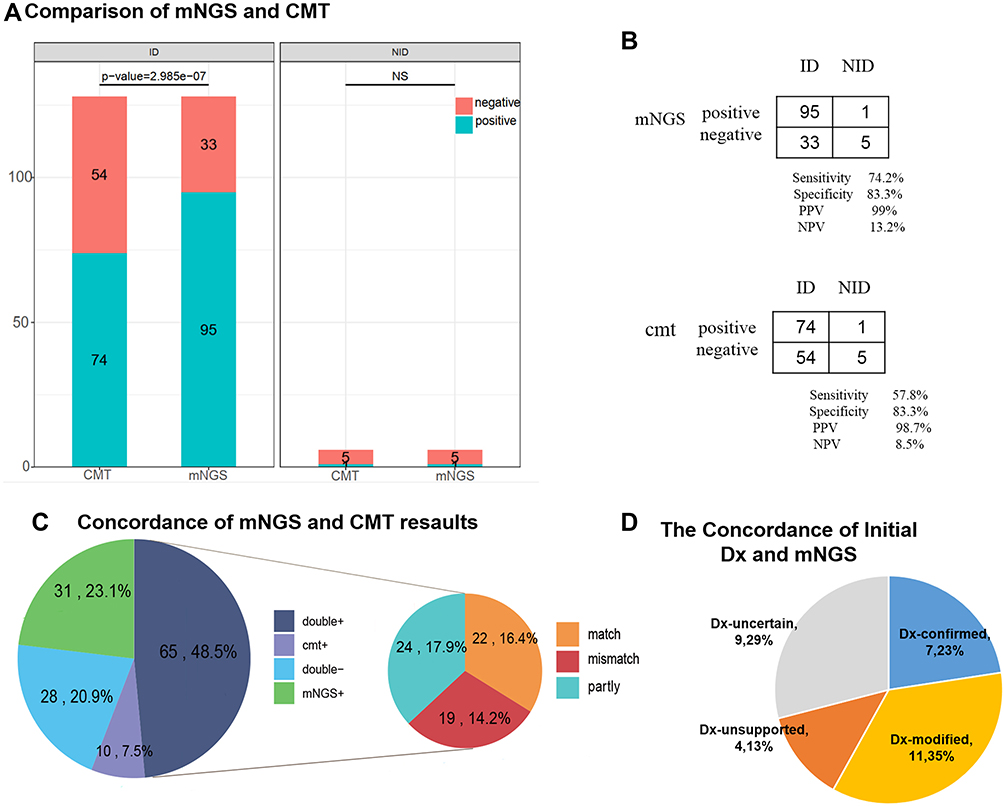 |
Figure 4 Diagnostic performance comparison of mNGS and CMT. (A) Positive and negative samples (y-axis) for pairwise mNGS and CMT is plotted against ID, NID group (x-axis). (B) Contingency tables showed the sensitivity and specificity of mNGS and CMT respectively. mNGS increased the sensitivity in comparison with that of culture (P < 0.001) while there were no differences in specificity between them (P > 0.05). (C) Pie chart demonstrated the positivity distribution of mNGS and CMT results from two groups. For the double-positive subset, a proportion of complete matching (22/65) and partial matching (at least 1 pathogen identified in the test was confirmed by the other) (24/65) was observed, with 19 conflicts between mNGS and CMT. (D) For microbes only identified in mNGS, the primary empirical diagnosis was confirmed (23%) or modified (35%) according to mNGS, whereas 13% of the samples were considered unreliable (diagnosis unsupported) and 29% were uncertain. Abbreviations: CMT, conventional microbiological tests. NPV, negative predictive values; PPV, positive predictive values; NS, not significant; ID, infectious disease; NID, noninfectious disease; e mNGS+, positive only by mNGS; double+ match, both positive by CMT and mNGS and pathogens completely overlapped; double+ mismatch, both positive by CMT and mNGS and no pathogens overlapped; double+ partly, both positive by CMT and mNGS and partly microorganism was overlapped. |
Concordance Between mNGS and CMT for Pathogen Detection
In our results, mNGS and CMT were both positive in 65 of 134 (48.5%) cases and were both negative in 28 of 134 (20.9%) cases. Thirty-one cases were only positive in mNGS (23.1%), and 10 cases were only positive in CMT (7.5%) (Figure 4C). Total 65 cases were both positive in mNGS and CMT, 22 of 65 cases were completely matched (pathogens completely overlapped between mNGS and CMT) and 19 cases were totally mismatched (no pathogens overlapped between mNGS and CMT), the remaining 24 cases were partially matched (at least one microorganism was overlapped between mNGS and CMT). For cases only positive in mNGS, 11 (35%) cases result in diagnosis modification, and 7 (23%) cases confirmed the clinical diagnosis (Figure 4D).
“False Positives” and “False Negatives” of mNGS
In the ID group, up to 27 pathogens detected by CMT were lost in mNGS. Among these “mNGS false-negative” cases, 19 CMT results were paradoxical with clinical diagnosis, and 4 were detected by mNGS without meeting the positive criteria, while the other 4 were completely unidentified by mNGS. For the only one case of “mNGS false-positive” in the NID group, the possible explanation could be the micro-colonization of virus in blood (Table 3).
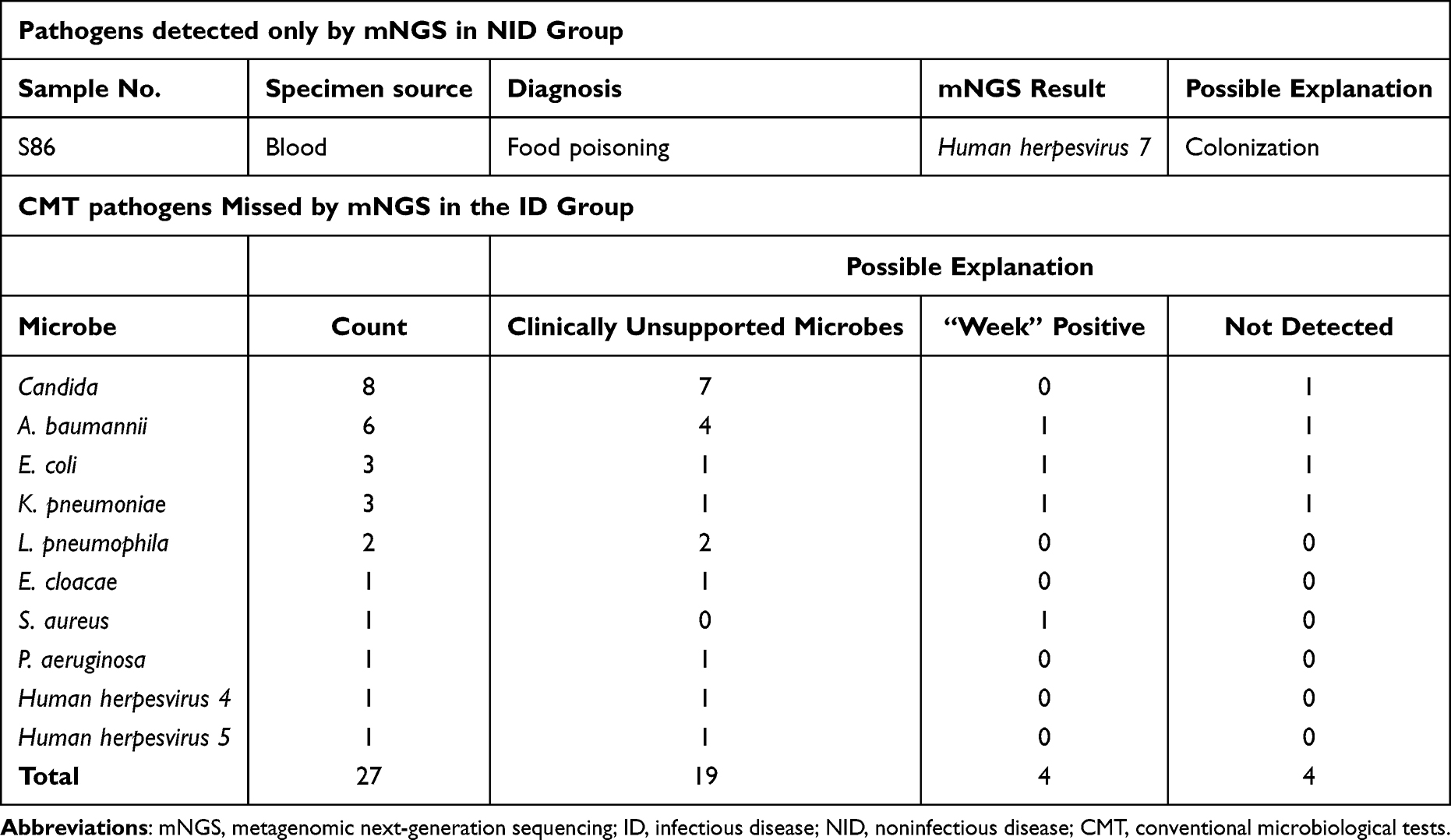 |
Table 3 “False Positive” and “False Negative”Results of mNGS |
Diagnostic Performance Comparison of mNGS and Culture
For the 104 cases tested by both culture and mNGS, mNGS had higher sensitivity than culture method, as shown in Figure 5A. Compared with culture, mNGS increased the overall sensitivity by 34.6% (66.3% vs 31.7%). In the BALF and blood samples, mNGS detection had significantly higher sensitivity than the culture method (P = 0.034 for BALF, P < 0.01 for blood). The positive rate of BALF was higher than blood samples in both mNGS and traditional culture method (71.8% vs 58.9% of BALF, 41.0% vs 10.2% of blood). The high concordance between mNGS and culture for pathogen detection of BALF is shown in Figure 5B. To be specific, in 16 double-positive cases of mMGS and culture, only one case was mismatch, while 10 cases of 16 were total match.
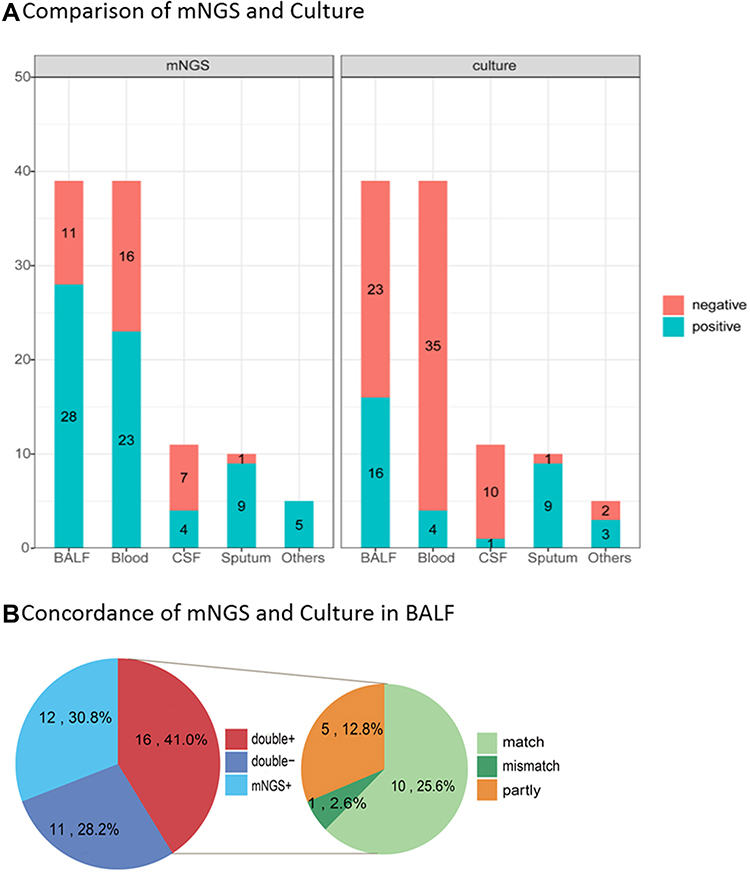 |
Figure 5 The positivity rate between mNGS and culture for different sample types. (A) Compared with the culture, mNGS increased the overall sensitivity by 34.6% (66.3% vs 31.7%). In cases of BALF and blood samples, mNGS detection had significantly higher sensitivity than the culture method (P = 0.034 for BALF, P < 0.01 for blood). (B) In all the 39 cases of BALF sample performed both mNGS and culture, high concordance for pathogen detection of BALF was shown in 16 double-positive cases of mMGS and culture. Only one case of mismatch was observed (2.6%), while 10 cases were total matched (25.6%). |
Comparison Analysis at the Pathogen-Type Level
In the 106 positive cases in mNGS or CMT, 40 kinds of suspected pathogenic microorganism were detected (Figure 6). The most commonly detected bacterium was Acinetobacter baumannii (26/106), followed by Klebsiella pneumonia (20/106) and Pseudomonas aeruginosa (11/106). For fungi, Candida albicans was the most commonly detected pathogen (17/106), followed by Pneumocystis jirovecii (8/106) and Aspergillus fumigatus (4/106). Human herpesvirus 4 was the most commonly detected virus (14/106). The proportion of mNGS-positive samples was significantly higher than CMT-positive samples in terms of MTB, NTM, virus, and fungus, especially NTM such as Mycobacterium abscessus and Mycobacterium chelonae. Fungus such as Pneumocystis jirovecii and Aspergillus fumigatus were mNGS-positive only. For cases that virus were detected, most cases (33/36) were mNGS-positive only, except one case of Human herpesvirus 4 was positive in both two methods, and two cases of Human herpesvirus 4 or Human herpesvirus 5 was CMT-positive only, showing “false positive” by mNGS but explained as clinically unsupported microbes.
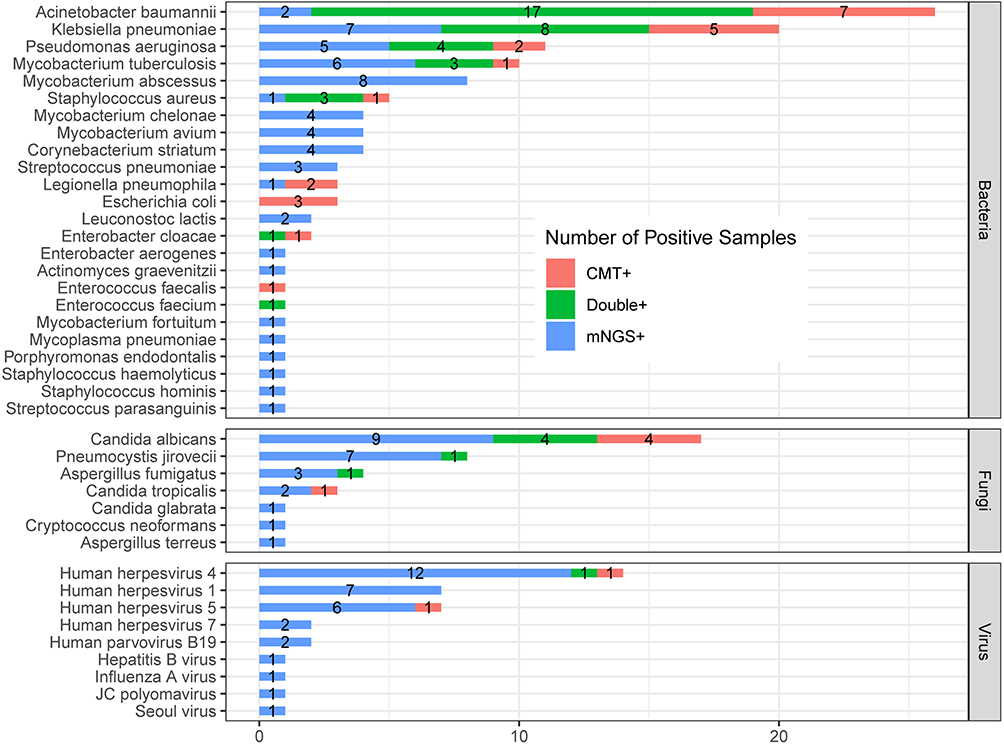 |
Figure 6 The overlap of positivity in pathogen between mNGS and culture. A total of 40 different pathogens were detected in the infectious disease group with their corresponding frequencies plotted in histograms. The proportion of mNGS positive samples was significantly higher than that of CMT positive samples in terms of MTB, NTM, virus, and fungus (P < 0.05). |
Comparison Analysis of Drug Resistant Bacteria
A. baumannii was the most commonly detected bacteria in our study shown in Figure 6. For the 17 cases that A. baumannii both detected in mNGS and CMT we performed ARGs analysis since A. baumannii often showed multiple drug resistance clinically. A total of 33 ARGs were detected in 14 cases (Figure 7) and 3 cases did not detect ARGs (Table 4), detail result of ARGs was shown in Table S2 Resistance-nodulation-cell division (RND) antibiotic efflux pump gene (ade genes) were detected in 13 of 14 cases and adeL was the most frequent ARG detected (13 cases of 14, detection frequency was 92.8%), followed by adeG, adeI, adeH, adeM, adeN (12 cases of 14, detection frequency was 85.7%) and adeF, adeC, adeK, adeS (12 cases of 14, detection frequency was 78.5%). Besides, sul2 and APH(3”)-Ib both showed high detection frequency. There was a high consistency between these ARGs and phenotypes (Table 4). In addition, AST was also conducted in other 16 cases except for S26 (Table 4). It was observed that 11 of 12 cases that detected sul_2 resistance gene showed resistance of sulfonamides, and resistance of aminoglycosides was detected in 11 cases contained APH(3”)-Ib resistance gene. All the 12 ade-positive cases showed multidrug resistance.
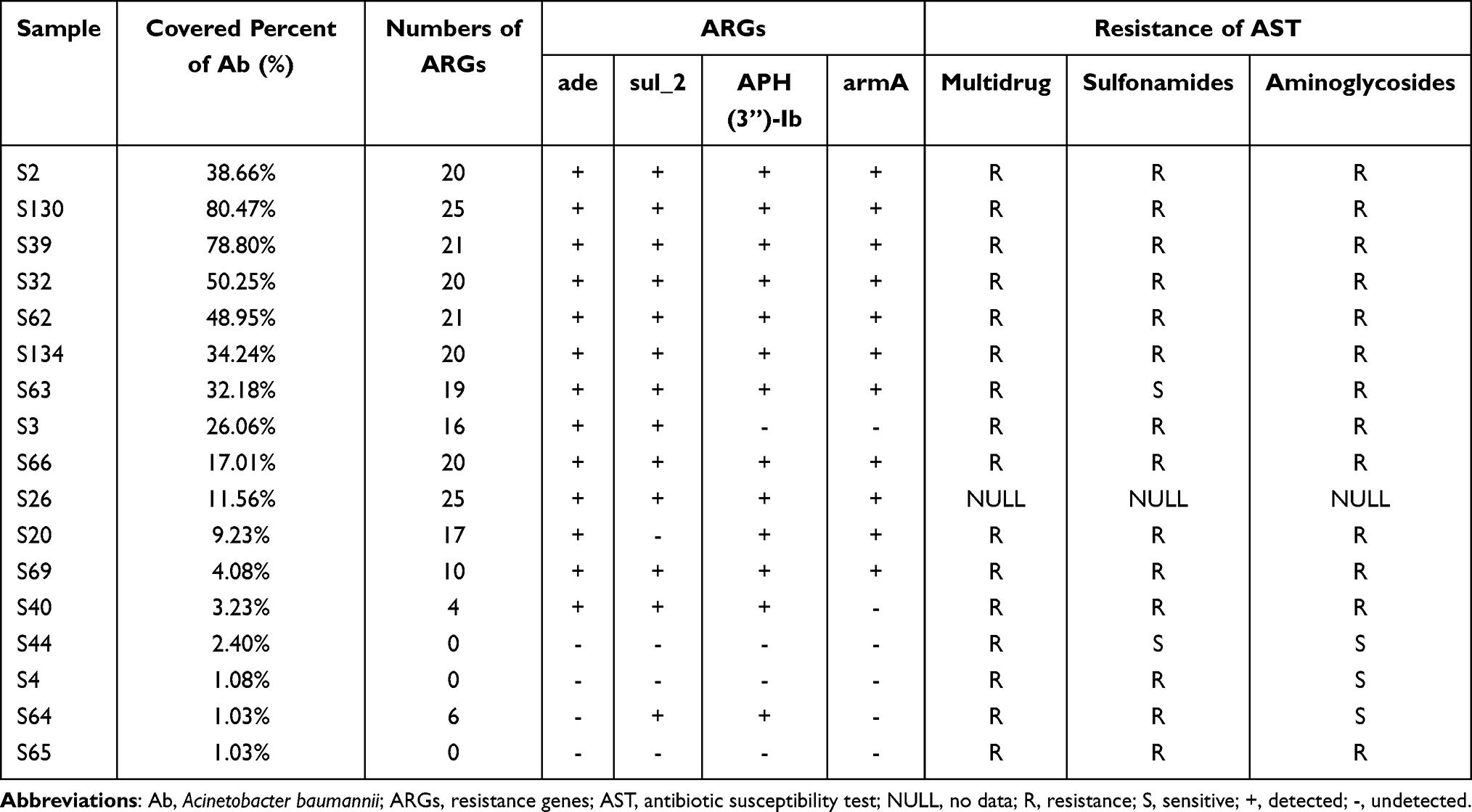 |
Table 4 Resistance Genes Found by mNGS in Relation to Pathogens |
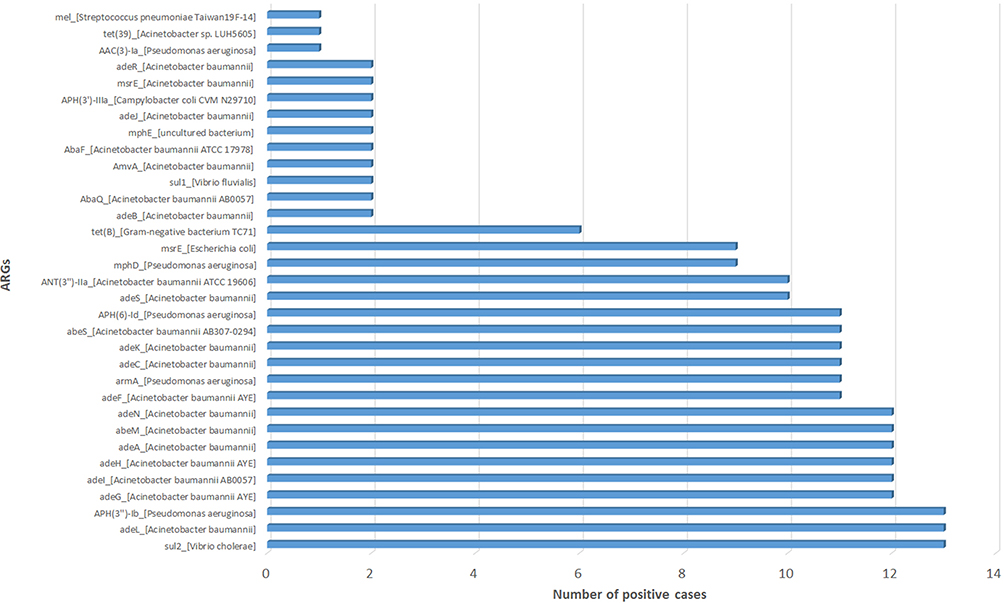 |
Figure 7 Frequency of ARGs detected from 17 cases that A. baumannii was both positive in mNGS and CMT. A total of 33 antiARGs were detected in 14 cases except 3 negative cases; of which adeL, sul2, and APH(3”)-Ib showed high detection frequency (13/14). |
Besides that, we selected the dominant strains of A. baumannii in each of 17 cases by evolutionary analysis, the result showed that 17 strains of A. baumannii were mainly classified into three groups (Figure 8). There were 15 cases of 17 clustered in group B or group C, furthermore, strain XH731 (7 cases of 17, 41.18%) and strain AB07 (5 cases of 17, 29.41%) were dominant strains between all A. baumannii strains analyzed.
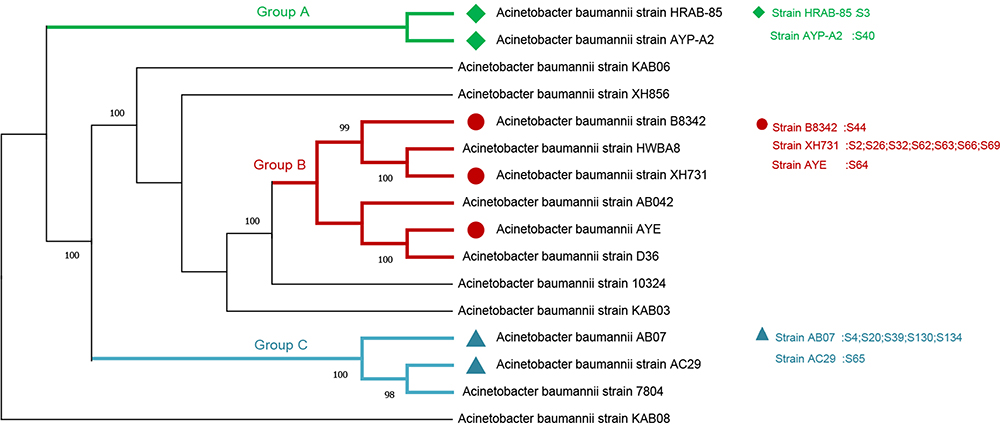 |
Figure 8 Evolutionary analysis of A. baumannii strains both positive in mNGS and CMT. Among the 17 cases, 15 were clustered in group B or group C, strain XH731 (7 cases of 17, 41.18%) and AB07 (5 cases of 17, 29.41%) were dominant strains. |
Discussion
Rapid and accurate microbial identification technology is always the focus of clinical microbiology. Traditional microbial test, such as morphology, culture, antigen antibody and targeted nucleic acid detection, has limitations in sensitivity, specificity, timeliness, information content, and unable to quickly identify unknown or rare pathogens. Since mNGS can analyze all the microorganisms in the infected samples of patients, it has been widely used in the etiological detection of acute and critical patients.20 In this study, 134 suspected infection samples were collected, including BALF, sputum, blood, CSF and pleural fluid, bile, etc. We compared the clinical characteristics and the detection results of mNGS with traditional methods. In addition, for cases that A. baumannii were detected both by mNGS and CMT, we analyzed the ARGs and evolutionary relationship of common drug-resistant A. baumannii, expecting to study the ARGs and dominant strains of A. baumannii.
Compared with the CMT and culture methods, mNGS showed higher sensitivity in pathogen detection (74.2% vs 57.8%, and 66.3% vs 31.7%, respectively), which is consistent with the previous research results.14,21 Because of the small sample size in NID group, there was no statistical difference between the specificity of mNGS and traditional methods. In different sample types, mNGS showed different positive rates. Due to the fact that respiratory tract was a complex organ system that was inhabited by niche-specific communities of microorganisms,22,23 more microbial sequences and higher positive rate were detected in respiratory tract samples such as BALF and sputum than blood and CSF, however, communities of microorganisms also made it more difficult to explain the results of respiratory tract samples. Hence, more research is needed to further distinguish pathogens infection or communities of microorganisms colonization in mNGS results. The positivity criteria in our study might lead to lower sensitivity, there were 27 cases of false-negative by mNGS, while 19 of 27 CMT results were paradoxical with clinical diagnosis, and 4 of 27 “false-negative” microbes were detected by mNGS but discarded because of failing to meet the criteria. Based on the comprehensive analysis of clinical diagnosis and the original sequence number of microbes, finally only 4 cases were real “false-negative” that pathogens did not been detected by mNGS.
In our study, mNGS shows higher sensitivity than traditional methods in pathogen detection of fungi, viruses, and mycobacteria, which is consistent with the results of previous studies.14,24 Clinical detection of fungal infection mainly relies on conventional microscopy or traditional culture method. However, the diagnosis is often delayed or missed due to the tedious detection process, long fungal culture cycle and other reasons, leading to the aggravation of infection and even endangering the life of patients. The characteristics of high efficiency, high speed, and high accuracy of mNGS make it an effective supplement to traditional fungal detection methods.25 In recent years, the incidence of NTM is increasing rapidly, and it has become one of the important public health problems threatening human health.26,27 With the increasing popularity of NGS technology and the reduction of cost, it will play an increasingly important role in the diagnosis of NTM disease.28,29 Virus is a common pathogen of acute respiratory tract infection and central nervous system infection, which has a wide variety and variability that can cause the outbreak of infectious diseases.30,31 In our results, mNGS showed obvious advantages over CMT methods for virus detection, most virus were detected by mNGS only. Consistent with the results of previous studies, the application of mNGS improves the sensitivity of virus detection.32 While, with the increasing application of mNGS, many studies have found that there may be a small amount of virus colonization in the blood, such as Torque teno virus (TTV), Epstein-Barr virus (EBV) also called Human herpesvirus 4, Cytomegalovirus (CMV) also called Human herpesvirus 5, Human herpesvirus 7, which may be related to the host’s immune status rather than causing infection.33–35 So that, the one case of “mNGS false-positive” in the NID group Human herpesvirus 7 detected in our study was explained might be the micro-colonization of virus in blood.36
Timely and effective drug treatment is very important to resist infection. Traditional method AST was used to screen suitable anti-infective drugs. However, this method not only time-consuming, but also unable to detect the drug sensitivity of un-cultural microorganism.37 In this study, we explored the possible application value of mNGS technology based on shotgun sequencing in the detection of ARGs from clinical specimens. The results showed that ARGs we detected were highly consistent with the drug resistance phenotype of A. baumannii. This finding suggested that the detection of ARGs by mNGS method can predict drug resistance, which may help clinicians choose sensitive antibiotics. However, it should be noted that the low genome coverage of drug-resistant bacteria detected by mNGS would affect the detection of ARGs, resulting in false negative. To be specific, in our results, we found that the genome coverage of A. baumannii in 3 cases was less than 2.4%, and the ARGs could not be detected, thus leading to false negative. As predominantly causes of infections,38 A. baumannii strains detected in our study were classified into three groups, and most cases were well clustered. The data of this study with small size of samples suggested that strains of A. baumannii may have high infection rate. The result of ARGs frequency and cluster analysis would be helpful to the control infections caused by A. baumannii.
There were some limitations in this study. Firstly, the sample size was relatively small and diagnosis was unbalanced in ID and NID groups based on a single-center retrospective study. Secondly, due to the lack of standards for interpreting mNGS results, the criteria for the positive mNGS result in this study referenced to previous researches might not be the best one, which would lead false positive or false negative. In the future, more prospective and multicenter studies with large samples size would be conducted, and the interpretation criteria of mNGS results would be further studied. Thirdly, it is difficult to accurately distinguish from which bacteria that the ARGs were detected in the samples based on mNGS dataset. However, among these samples, A. baumannii was the main dominant bacteria, other bacteria were not detected or the number of detected reads was small. It was indirectly supported by the fact that the ARGs detected were probably carried by A. baumannii. This is also a limitation of ARG analysis by mNGS-based shotgun sequencing in our study, further study about this is needed in future research. Finally, our mNGS tests were delivered to the centralized laboratory rather than an in-house microbiology laboratory, which may sacrifice sensitivity rate because of reduced viability due to increased turnaround time from bedside to bench. Antibiotic usage might also infect detection of pathogens in this study, and we will include these factors in our future studies.
Conclusions
In summary, mNGS had a higher sensitivity than CMT and culture method. In the BALF and blood samples, it showed a trend of higher sensitivity of fungi, viruses and NTM. The ARGs detected by mNGS may guide the usages of antibiotics in case of the genome coverage of A. baumannii no less than 2.4%. Through evolutionary analysis, the clustering results of A. baumannii strains may suggest the dominant strains of infection. Therefore, based on the above findings and other advantages of mNGS, such as short time-consuming for diagnosis of the pathogen, not affected by previous antibiotic exposure and so on, we suggest that mNGS should be used more often in early diagnosis of pathogen. In the absence of the results of antibiotic susceptibility test, the detection of ARGs by mNGS has a certain guiding significance for the usage of antibiotics. However, the interpretation of mNGS results is still a challenge for clinicians to guide the clinical treatment of infectious diseases.
Abbreviations
mNGS, Metagenomic next-generation sequencing; CMT, conventional microbiological tests; AST, antibiotic susceptibility test; BALF, Bronchoalveolar lavage fluid; CSF, Cerebrospinal fluid; Dx, Diagnosis; ID, Infectious disease; NID, Noninfectious disease; PPV, Positive predictive value; NPV, Negative predictive value; EDTA, Ethylene Diamine Tetraacetic Acid; ARG, antibiotic resistance gene; NTM, Non-tuberculous Mycobacteria; EBV, Epstein-Barr virus; CMV, Cytomegalovirus; TTV, Torque teno virus.
Data Sharing Statement
The datasets generated for this study can be found in the NCBI BioProject database (BioProject ID: PRJNA750848).
Ethics Approval and Informed Consent
The study was conducted in accordance with the Declaration of Helsinki. All procedures were performed in accordance with the ethical standards of the Clinical Research Ethics Committee of the First Hospital of Qinhuangdao, and informed consent was obtained from all individuals included in the study.
Acknowledgments
This study was funded by National Key Research & Development Plan (2018YFE0102400) and Shanghai Special Project for Artificial Intelligence Innovation and Development (2020-RGZN-02039).
Disclosure
The authors declare that they have no competing interests.
References
1. Chalmers J, Campling J, Ellsbury G, Hawkey PM, Madhava H, Slack M. Community-acquired pneumonia in the United Kingdom: a call to action. Pneumonia. 2017;9:15. doi:10.1186/s41479-017-0039-9
2. Troeger C, Blacker B, Khalil IA; Collaborators GBDLRI. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory infections in 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect Dis. 2018;18(11):1191–1210. doi:10.1016/S1473-3099(18)30310-4
3. Machhi J, Herskovitz J, Senan AM, et al. The natural history, pathobiology, and clinical manifestations of SARS-CoV-2 Infections. J Neuroimmune Pharmacol. 2020;15(3):359–386. doi:10.1007/s11481-020-09944-5
4. Jain S, Self WH, Wunderink RG, et al. Community-acquired pneumonia requiring hospitalization among US adults. N Engl J Med. 2015;373(5):415–427. doi:10.1056/NEJMoa1500245
5. Krishna NK, Cunnion KM. Role of molecular diagnostics in the management of infectious disease emergencies. Med Clin North Am. 2012;96(6):1067–1078. doi:10.1016/j.mcna.2012.08.005
6. Chiu CY, Miller SA. Clinical metagenomics. Nat Rev Genet. 2019;20(6):341–355. doi:10.1038/s41576-019-0113-7
7. Gu W, Miller S, Chiu CY. Clinical metagenomic next-generation sequencing for pathogen detection. Annu Rev Pathol. 2019;14:319–338. doi:10.1146/annurev-pathmechdis-012418-012751
8. Wilson MR, O’Donovan BD, Gelfand JM, et al. Chronic meningitis investigated via metagenomic next-generation sequencing. JAMA Neurol. 2018;75(8):947–955. doi:10.1001/jamaneurol.2018.0463
9. Lee RA, Al Dhaheri F, Pollock NR, Sharma TS, Dekker JP. Assessment of the clinical utility of plasma metagenomic next-generation sequencing in a pediatric hospital population. J Clin Microbiol. 2020;58(7). doi:10.1128/JCM.00419-20
10. Han D, Li Z, Li R, Tan P, Zhang R, Li J. mNGS in clinical microbiology laboratories: on the road to maturity. Crit Rev Microbiol. 2019;45(5–6):668–685. doi:10.1080/1040841X.2019.1681933
11. Woldesemayat B, Gebremicael G, Zealiyas K, et al. Effect of heat inactivation for the detection of severe acute respiratory syndrome-Corona virus-2 (SARS-CoV-2) with reverse transcription real time polymerase chain reaction (rRT-PCR): evidence from Ethiopian study. BMC Infect Dis. 2022;22(1):163. doi:10.1186/s12879-022-07134-7
12. Pastorino B, Touret F, Gilles M, de Lamballerie X, Charrel RN. Heat Inactivation of different types of SARS-CoV-2 samples: what protocols for biosafety, molecular detection and serological diagnostics? Viruses. 2020;12(7):735. doi:10.3390/v12070735
13. Kampf G, Voss A, Scheithauer S. Inactivation of coronaviruses by heat. J Hosp Infect. 2020;105(2):348–349. doi:10.1016/j.jhin.2020.03.025
14. Miao Q, Ma Y, Wang Q, et al. Microbiological diagnostic performance of metagenomic next-generation sequencing when applied to clinical practice. Clin Infect Dis. 2018;67(suppl_2):S231–S240. doi:10.1093/cid/ciy693
15. Langelier C, Zinter MS, Kalantar K, et al. Metagenomic sequencing detects respiratory pathogens in hematopoietic cellular transplant patients. Am J Respir Crit Care Med. 2018;197(4):524–528. doi:10.1164/rccm.201706-1097LE
16. Bittinger K, Charlson ES, Loy E, et al. Improved characterization of medically relevant fungi in the human respiratory tract using next-generation sequencing. Genome Biol. 2014;15(10):487. doi:10.1186/s13059-014-0487-y
17. Schlaberg R, Chiu CY, Miller S, et al. Validation of metagenomic next-generation sequencing tests for universal pathogen detection. Arch Pathol Lab Med. 2017;141(6):776–786. doi:10.5858/arpa.2016-0539-RA
18. Ozcolpan OO, Surucuoglu S, Ozkutuk N, Cavusoglu C. Klinik Örneklerden Soyutlanan ve DNA Dizi Analizi ile Tanımlanan Tüberküloz Dışı Mikobakterilerin Dağılımı*[Distribution of nontuberculous mycobacteria isolated from clinical specimens and identified with DNA sequence analysis]. Mikrobiyol Bul. 2015;49(4):484–493. Turkish. doi:10.5578/mb.9698
19. van Ingen J, Kohl TA, Kranzer K, et al. Global outbreak of severe Mycobacterium chimaera disease after cardiac surgery: a molecular epidemiological study. Lancet Infect Dis. 2017;17(10):1033–1041. doi:10.1016/S1473-3099(17)30324-9
20. Li N, Cai Q, Miao Q, Song Z, Fang Y, Hu B. High-throughput metagenomics for identification of pathogens in the clinical settings. Small Methods. 2021;5(1):2000792. doi:10.1002/smtd.202000792
21. Wang S, Ai J, Cui P, Zhu Y, Wu H, Zhang W. Diagnostic value and clinical application of next-generation sequencing for infections in immunosuppressed patients with corticosteroid therapy. Ann Transl Med. 2020;8(5):227. doi:10.21037/atm.2020.01.30
22. Man WH, de Steenhuijsen Piters WA, Bogaert D. The microbiota of the respiratory tract: gatekeeper to respiratory health. Nat Rev Microbiol. 2017;15(5):259–270. doi:10.1038/nrmicro.2017.14
23. Gao L, Xu T, Huang G, Jiang S, Gu Y, Chen F. Oral microbiomes: more and more importance in oral cavity and whole body. Protein Cell. 2018;9(5):488–500. doi:10.1007/s13238-018-0548-1
24. Li H, Gao H, Meng H, et al. Detection of pulmonary infectious pathogens from lung biopsy tissues by metagenomic next-generation sequencing. Front Cell Infect Microbiol. 2018;8:205. doi:10.3389/fcimb.2018.00205
25. Wang J, Han Y, Feng J. Metagenomic next-generation sequencing for mixed pulmonary infection diagnosis. BMC Pulm Med. 2019;19(1):252. doi:10.1186/s12890-019-1022-4
26. Prevots DR, Marras TK. Epidemiology of human pulmonary infection with nontuberculous mycobacteria: a review. Clin Chest Med. 2015;36(1):13–34. doi:10.1016/j.ccm.2014.10.002
27. Sharma SK, Upadhyay V. Epidemiology, diagnosis & treatment of non-tuberculous mycobacterial diseases. Indian J Med Res. 2020;152(3):185–226. doi:10.4103/ijmr.IJMR_902_20
28. Huang Z, Zhang C, Fang X, et al. Identification of musculoskeletal infection with non-tuberculous mycobacterium using metagenomic sequencing. J Infect. 2019;78(2):158–169. doi:10.1016/j.jinf.2018.10.002
29. Appak O, Turkel S, Esen N, Ozkutuk AA. Comparison of polymerase chain reaction-restriction enzyme analysis method and DNA sequence analysis results in the identification of non-tuberculous mycobacteria. Acta Microbiol Immunol Hung. 2018;65(4):515–527. doi:10.1556/030.65.2018.027
30. Tan L, Yao J, Yang Y, et al. Current status and challenge of pseudorabies virus infection in China. Virol Sin. 2021;36(4):588–607. doi:10.1007/s12250-020-00340-0
31. Mourya DT, Yadav PD, Ullas PT, et al. Emerging/re-emerging viral diseases & new viruses on the Indian horizon. Indian J Med Res. 2019;149(4):447–467. doi:10.4103/ijmr.IJMR_1239_18
32. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. doi:10.1038/s41586-020-2008-3
33. Han D, Li R, Shi J, Tan P, Zhang R, Li J. Liquid biopsy for infectious diseases: a focus on microbial cell-free DNA sequencing. Theranostics. 2020;10(12):5501–5513. doi:10.7150/thno.45554
34. Chen X, Ding S, Lei C, et al. Blood and bronchoalveolar lavage fluid metagenomic next-generation sequencing in pneumonia. Can J Infect Dis Med Microbiol. 2020;2020:6839103. doi:10.1155/2020/6839103
35. Mouton W, Conrad A, Bal A, et al. Torque teno virus viral load as a marker of immune function in allogeneic haematopoietic stem cell transplantation recipients. Viruses. 2020;12(11):1292. doi:10.3390/v12111292
36. Ongradi J, Kovesdi V, Kovats E. Az emberi 7-es herpeszvírus[Human herpesvirus 7]. Orv Hetil. 2010;151(16):645–651. Hungarian. doi:10.1556/OH.2010.28856
37. Forbes JD, Knox NC, Ronholm J, Pagotto F, Reimer A. Metagenomics: the next culture-independent game changer. Front Microbiol. 2017;8:1069. doi:10.3389/fmicb.2017.01069
38. Kyriakidis I, Vasileiou E, Pana ZD, Tragiannidis A. Acinetobacter baumannii antibiotic resistance mechanisms. Pathogens. 2021;10(3). doi:10.3390/pathogens10030373
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.