Back to Journals » ImmunoTargets and Therapy » Volume 10
The Cancer-Immunity Cycle in Multiple Myeloma
Authors Casey M, Nakamura K ![]()
Received 1 June 2021
Accepted for publication 10 July 2021
Published 16 July 2021 Volume 2021:10 Pages 247—260
DOI https://doi.org/10.2147/ITT.S305432
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Michael Shurin
Mika Casey, Kyohei Nakamura
Immune Targeting in Blood Cancers Laboratory, QIMR Berghofer Medical Research Institute, Herston, 4006, Australia
Correspondence: Kyohei Nakamura
Immune Targeting in Blood Cancers Laboratory, QIMR Berghofer Medical Research Institute, 300 Herston Road, Herston, 4006, Australia
Tel +61-7-3845-3907
Fax +61-7-3362-0111
Email [email protected]
Abstract: Multiple myeloma is a plasma cell malignancy that primarily affects the elderly. The global burden of multiple myeloma is increasing in many countries due to an aging population. Despite recent advances in therapy, myeloma remains an incurable disease, highlighting the pressing need for new therapies. Accumulating evidence supports that triggering the host immune system is a critical therapeutic mechanism of action by various anti-myeloma therapies. These anti-myeloma therapies include proteasome inhibitors, immunomodulatory drugs, monoclonal antibody drugs, and autologous stem cell transplantation. More recently, T cell-based immunotherapeutics (including chimeric antigen receptor T-cell therapies and bispecific T-cell engagers) have shown dramatic clinical benefits in patients with relapsed or refractory multiple myeloma. While immune-based therapeutic approaches are recognized as key modalities for improved clinical outcomes in myeloma patients, understanding the immune system in multiple myeloma patients remains elusive. The cancer-immunity cycle is a conceptual framework illustrating how immune cells recognize and eliminate tumor cells. Based on this framework, this review will provide an overview of the immune system in multiple myeloma patients and discuss potential therapeutic approaches to stimulate anti-tumor immunity.
Keywords: multiple myeloma, immune checkpoint, immunotherapy, immunosuppression
Introduction
Multiple myeloma (MM) is typically preceded by the asymptomatic precursor conditions known as monoclonal gammopathy of undetermined significance (MGUS) or smoldering multiple myeloma (SMM).1 This spectrum of plasma cell disorders, namely, plasma cell dyscrasias, is tightly associated with the clonal evolution of plasma cells.2,3 Chromosomal hyperdiploidy or a translocation involving the immunoglobulin heavy-chain gene locus is a key event triggering the expansion of plasma cells. The progression from MGUS to active MM requires the accumulation of several genomic alteration events, such as copy number abnormalities, secondary translocations, and somatic mutations.4 Intra-clonal heterogeneity is a key feature of plasma cell dyscrasias, which explains the mechanism underlying acquired chemo-resistance and disease relapse in MM.5 Growing evidence suggests that the immune system exerts selective pressure on malignant plasma cells, acting as a rheostat for regulating symptomatic progression in plasma cell dyscrasias.6 In addition to the importance of natural immunosurveillance, myeloma immunotherapy has emerged as a potential approach to eradicate chemotherapy-resistant plasma cell clones.7,8 For instance, chimeric antigen receptor (CAR) T-cell therapies and bispecific T-cell engager therapies are powerful modalities for immune-mediated destruction of malignant plasma cells. In this review, we will summarize the current understanding of the immune system in MM patients using the cancer-immunity cycle framework, and discuss immunological mechanisms of action in myeloma therapeutics.
The Cancer-Immunity Cycle in Multiple Myeloma
The cancer-immunity cycle is a series of stepwise functional events that describe the immune system recognizing and eliminating malignant cells.9 This framework consists of seven major steps: 1) release of tumor antigens, 2) antigen presentation, 3) priming and activation of effector T cells, 4) trafficking of T cells to tumors, 5) infiltration of T cells into tumors, 6) recognition of tumor cells by T cells, and 7) killing of tumor cells.9 This framework exquisitely depicts the mechanism of anti-tumor immunity against solid malignancies. However, disease-specific aspects should also be considered to understand the immune system in MM patients (Figure 1).
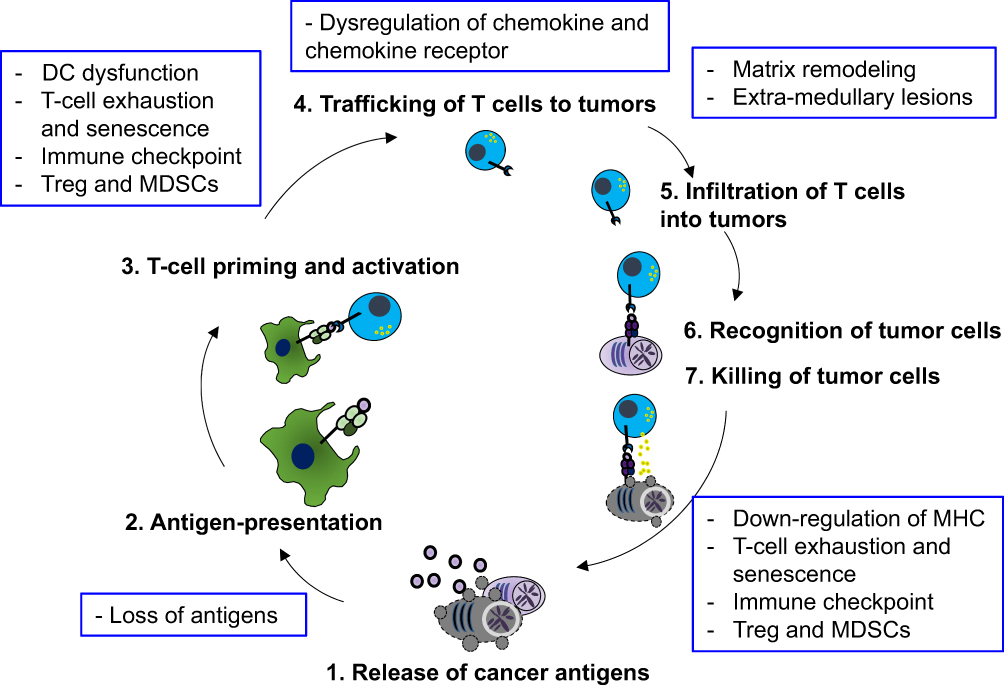 |
Figure 1 The cancer-immunity cycle in multiple myeloma and its negative regulation. A schematic illustrating seven steps of the cancer-immunity cycle and their negative regulatory mechanisms in multiple myeloma (blue boxes). Abbreviations: DC, dendritic cells; Tregs, regulatory T cells; MDSCs, myeloid-derived suppressor cells. |
Release of Tumor Antigens (Step 1)
Even in the absence of therapeutic interventions, cancer cells are constantly exposed to stressful stimuli, such as metabolic stress, hypoxia and immune-mediated destruction, all of which trigger the release of antigens from dying tumor cells. Tumor antigens are broadly classified as either tumor-associated antigens (TAAs) or neoantigens (also called tumor-specific antigens). TAAs are antigens that are aberrantly overexpressed in tumor tissues when compared to normal tissues. In contrast, neoantigens refer to antigens exclusively expressed by tumor cells, as these peptide fragments arise from somatically mutated self-proteins.
As seen in solid malignancies, myeloma cells express a wide variety of TAAs, such as cancer-testis antigens (MAGE-C1 and NY-ESO-1), WT-1, and MUC1. Additionally, the variable region of the clonal immunoglobulin, namely idiotype, has been recognized as an important TAA in MM.10 Historically, TAA-loaded dendritic cells (DCs) have been used as cancer vaccines for various malignancies including MM. Although DC-based cancer vaccine approaches have shown limited clinical efficacies as a single-agent therapy,11 they are actively being tested in combination with immune checkpoint inhibitors or other immunotherapeutics. Additionally, two immunogenic TAAs have been reported in MGUS patients: OFD1 (Oral-facial-digital syndrome 1 protein) and SOX2 (SRY-box transcription factor 2). OFD1 is a developmental antigen implicated in primary cilium formation and regulation of the Hedgehog and Wnt signaling pathways. Specific antibody responses against OFD1 protein were seen in 20% of MGUS patients, although not in newly diagnosed MM patients. OFD1-specific CD8+ T cells were detectable in both MGUS and MM patients. However, only MGUS patients-derived T cells underwent proliferation in response to OFD1-derived peptide,12 suggesting functional impairment of OFD1-specific T cells in active MM patients. Another MGUS-associated TAA, SOX2, is normally related to pluripotency and maintenance of stem cells. SOX2-specific T cells were observed in about 70% of MGUS patients, while absent in active MM patients. Notably, the absence of SOX2-specific T cells in MGUS patients was significantly associated with a higher risk of progression into active MM.13 While these results strongly suggest that these antigen-specific T cells play a critical role in myeloma immunosurveillance, it remains to be elucidated how malignant plasma cells evade the immune system, leading to symptomatic progression.
Compared to TAAs, neoantigens are less characterized in MM. In general, immune checkpoint inhibitors, such as anti-CTLA-4, anti-PD-1, and anti-PD-L1, have demonstrated promising clinical responses against tumors with high tumor mutational burden (TMB) and tumor neoantigen burden (TNB).14 This indicates that TMB and TNB are key determinants of tumor immunogenicity. Intriguingly, Miller et al reported that MM patients harboring high TMB and TNB were associated with decreased progression-free survival,15 although the mechanism remains elusive. High TMB and TNB conditions might reflect aggressive or refractory disease characteristics driven by genomic aberrations. Indeed, Perumal et al showed that relapsed MM patients have a higher TMB and TNB than newly diagnosed MM patients,16 supporting that the genomic complexity might be associated with high TMB/TNB status. These results also raise the possibility that natural immunosurveillance by myeloma-specific T cells might not effectively control disease progression under the current standard of care. Besides, given that anti-PD-1 mAb failed to improve clinical outcomes in MM patients,17,18 myeloma-specific T cell responses might not be predicted based on the TMB/TNB status. Recently, Perumal et al identified shared neoantigens across MM patients in major oncogenic driver genes, including KRAS, NRAS, and IRF4,16 providing an important implication for off-the-shelf neoantigen-based immunotherapy. For clinical applications of neoantigens, high-throughput profiling of myeloma neoantigens is warranted, such as clonality assessment and expression patterns of neoantigen peptides on the major histocompatibility complex (ie, the MHC ligandome).
Antigen Presentation and Priming of T Cells (Step 2 and 3)
Tumor antigens derived from dead tumor cells are captured by antigen-presenting cells (APCs) such as DCs. These antigens are subsequently exhibited on MHC-II via direct presentation or by MHC-I via a cross-presentation pathway. The activation of T cells is triggered by the recognition of peptide/MHC (pMHC) complexes by T-cell receptors (signal 1) and the engagement of the CD28 co-stimulatory receptor by its ligands CD80 and CD86 (signal 2). In solid malignancies, migratory DCs expressing CD141 (in humans) or CD103 (in mice) play a critical role in delivering antigens from tumor tissues to draining lymph nodes, where these DCs prime tumor-specific T cells.19 In contrast, it remains unknown how antigen-presentation occurs in MM. As the bone marrow (BM) acts as a major hematopoietic organ and a secondary lymphoid organ, the myeloma BM might harbor various APCs at different maturation stages. Indeed, Feuerer et al reported that mouse BM resident DCs could take up exogenous blood-borne antigens and prime T cells in the BM.20 Alternatively, another group demonstrated that circulating DCs carrying antigens migrate to the BM and stimulate BM-resident memory T cells.21 This suggests that both BM-resident DCs and circulating DCs might contribute to antigen-presentation in the myeloma BM. In addition to DCs, tumor-associated macrophages expressing CD169 also mediate cross-presentation in solid malignancies.22 Likewise, intra-vital imaging revealed that apoptotic myeloma cells are engulfed by CD169+ BM macrophages.23 The discrete roles of heterogeneous myeloid subsets for myeloma antigen presentation remain to be fully characterized.
Several studies have shown impaired antigen-presentation capacity in DCs from MM patients. This DC dysfunction could be due to aberrant cytokines released in the myeloma BM including IL-6, IL-10, and TGF-β.24 However, contradictory results have been reported regarding numerical and phenotypic alterations of DCs in the myeloma BM.24 Previous studies defined DCs based on the expression of CD11c and MHC-II; however, these markers are not sufficient to discriminate heterogeneous APC subsets in the BM.25 The comprehensive characterization of APCs at single-cell resolution might clarify how myeloma progression impairs antigen presentation.
In addition to DCs, T cell-intrinsic dysfunction has been reported in myeloma patients. While T cells from MGUS patients showed IFN-γ secretion upon stimulation by in vitro expanded DCs pulsed with autologous plasma cells, T cells from myeloma patients failed to show responses.26 Thus, T-cell hypo-responsiveness is induced during symptomatic progression. More recently, using single-cell RNA-sequencing (scRNA-seq) in combination with mass cytometry analysis, Bailur et al demonstrated that stem-like memory T cells expressing the transcription factor T-cell factor 1 (TCF1) were enriched in MGUS patients in comparison to age-matched healthy subjects.27 In contrast, active MM patients had decreased TCF1+ stem-like memory T cells, while terminal effector T cells were progressively increased.27 Another study on the immune landscape of SMM revealed loss of memory cytotoxic T cells expressing granzyme K and an increase in effector cytotoxic T cells expressing granzyme B.28 While disease progression from asymptomatic myeloma to active MM might be associated with T-cell dysfunction, further studies are warranted to understand whether T-cell dysfunction is explained by T-cell exhaustion, anergy or senescence.29
T-Cell Migration and Infiltration into Tumor Tissues (Step 4 and Step 5)
In solid malignancies, C-X-C chemokine receptor 3 (CXCR3) and CXCR4 have been recognized as major chemokine receptors for mobilization of T cells and NK cells.30 IFN-γ has been recognized as a key regulator for the CXCR3 chemokine system. Indeed, IFN-γ can induce CXCR3 on activated T cells and its ligands CXCL9/10/11 in tumor tissues. Recently, Chow et al showed that the interaction between CXCR3 and its ligands was not essential for T cell infiltration into tumor tissues but indispensable for the optimal CD8+ T cell responses elicited by anti-PD-1 blockade.31 Thus, the CXCR3 system has unique T-cell stimulatory functions, other than the mobilization of T cells. CXCR4 is critically implicated in the homing of lymphocytes into the BM, as BM stromal cells are major producers of its ligand, CXCL12. As discussed previously, the BM functions as a secondary lymphoid organ, where memory CD8 T cells undergo maintenance and proliferation.20,21,32 Recently, Khan et al showed that adoptive transfer of CD8+ T cells overexpressing CXCR4 exhibited a greater capacity for expansion with poly-functional phenotypes in the BM, leading to better control of subcutaneously transplanted tumors. Thus, temporal homing of T cells to the BM via the CXCR4-CXCL12 interaction might be a critical step for T cell activation and expansion.
In the myeloma BM, aberrant chemokine expression might be implicated in the mechanism of immune exclusion. Ponzetta et al showed that the symptomatic progression of myeloma was associated with increased levels of CXCL9/10 and decreased levels of CXCL12 in the BM.33 However, due to rapid down-modulation of CXCR3 expression on NK cells, NK cells were excluded from the myeloma BM.33 In contrast, the dynamics of migration and egress of T cells in the myeloma BM remain to be elucidated. It should be noted that malignant plasma cells also express multiple chemokine receptors, such as CXCR4 and CCR1 and that abnormal chemokine expression might foster extramedullary dissemination.34,35 Thus, the aberrant chemokine pattern might be implicated in both immune exclusion and metastatic dissemination of malignant plasma cells.
T-cell infiltration into tumor tissues is a crucial process for anti-tumor immunity, as supported by the fact that immune checkpoint inhibitors have shown limited efficacies against tumors with low T-cell infiltration, namely, immunologically “cold” tumors. As discussed later, various immunosuppressive cells critically limit T-cell infiltration and effector functions. T-cell infiltration into tumor tissues is also regulated by extracellular matrix components, such as glycoproteins, glucosaminoglycans, and proteoglycans.36 In the myeloma BM, a chondroitin sulfate proteoglycan, called versican, accumulates around myeloma lesions.37 Versican is known to undergo proteolytic cleavage by the ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) proteases. It was reported that higher levels of versican cleavage were associated with higher CD8 T cell-infiltration in MM patients after autologous stem-cell transplantation (ASCT).38 However, notably, higher infiltration of CD8 T cells was paradoxically associated with inferior outcomes in these patients.38 Given that ADAMTS activities are associated with inflammation, high levels of versican cleavage might reflect tumor-promoting BM environments.
Enhancing T-cell migration and infiltration into tumor tissues is an important consideration for an immunotherapeutic approach against extramedullary MM (EMM). Many clinical studies have concluded that EMM was associated with an inferior prognosis in patients who received current standard therapies, including ASCT or daratumumab (anti-CD38 mAb) therapies.39,40 Although CAR T-cell therapy against B-cell maturation antigen (BCMA) has shown good responses in some patients with EMM,41 it remains challenging to control extramedullary lesions. The immune microenvironment of EMM has not been fully characterized. Given that malignant plasma cells in extramedullary lesions frequently show high Ki-67 expression,42 highly proliferative myeloma cells might overwhelm T-cell infiltration, leading to the generation of immunologically “cold” EMM. Genetically modified CAR-T cell therapies with enhanced migration capacities are being developed to treat solid malignancies,43 which might provide a clue to improve control of EMM.
Recognition and Elimination of Tumor Cells (Step 6 and Step 7)
Cytotoxic T cells recognize pMHC-I derived from neoantigens and TAAs in tumor cells. The TCR-pMHC-I interaction leads to the formation of an immunological synapse, a highly organized subcellular structure at which adhesion and co-signaling molecules accumulate. The immunological synapse acts as a molecular platform for cytotoxic granule exocytosis and release of IFN-γ to eliminate tumor cells.44 Cytotoxic T cells fail to recognize and eliminate tumor cells by multiple mechanisms, including 1) down-regulation of pMHC-I expression on tumor cells, 2) negative regulation of T cells through the interaction between immune checkpoint receptors and their ligands, 3) functional exhaustion of T cells, and 4) resistance to immune-mediated killing of tumor cells.
Several lines of evidence suggest that proteasome inhibitors down-regulate expression levels of MHC-I on malignant plasma cells.45,46 As NK cells preferentially recognize tumor cells with low levels of MHC-I, bortezomib-treated myeloma cells show an increased sensitivity to NK cell-mediated cytotoxicity.45 Recently, Kowalewski et al performed MHC ligandome profiling of myeloma cells treated with carfilzomib (a second-generation proteasome inhibitor) to assess qualitative and quantitative changes of pMHC. Intriguingly, carfilzomib specifically downregulated pMHC expression in peptides with aromatic C-termini,46 indicating that proteasome inhibitors modulate pMHC expression in a peptide motif-specific manner. Still, it remains to be understood how therapy-induced changes of MHC-I expression differentially affects T cell-dependent and NK cell-dependent control of myeloma. It is also possible that the dynamic changes in expression levels of MHC-restricted myeloma antigens might contribute to immune escape during clonal evolution of malignant plasma cells.
Immune checkpoint inhibitors against PD-1 and PD-L1 are known to reinvigorate functionally exhausted effector T cells.47 In an initial study, pembrolizumab (anti-PD-1 mAb) in combination with pomalidomide and dexamethasone showed an objective response rate of 60% in heavily treated MM patients.48 However, the combination approach did not show clinical benefits in Phase III clinical trials.17,18 Clinicopathological and immunological characteristics of anti-PD-1 responders might be warranted to utilize PD-1/PD-L1 blockade in MM. Although PD-1/PD-L1 blockade has been reported to enhance the expansion of neoantigen-specific T cells in a subset of MM patients,16 it failed to rescue proliferation and cytokine production of BM CD8 T cells in the majority of MM patients.49 Of note, in the presence of TGF-β inhibitors, anti-PD-1 mAb improved functional responses in ex vivo isolated BM CD8 T cells,49 suggesting that extrinsic control might be implicated in T-cell hypo-responsiveness.
One of the key pathways regulating cytotoxic lymphocyte activity at the immunological synapse is the nectin family molecules: the co-activating receptor CD226 (also known as DNAM-1), the inhibitor receptor TIGIT (T-cell immunoreceptor with immunoglobulin and ITIM domains), and their shared ligand, CD155. CD155 is broadly expressed on tumor cells, including malignant plasma cells, and the interaction between CD226 and CD155 allows cytotoxic lymphocytes to recognize and eliminate tumor cells. Indeed, in the preclinical de novo Vk*MYC myeloma model, deficiency of CD226 accelerated disease progression with shortened survival,50 highlighting the prominent role of CD226 for immunosurveillance. Recently, two groups have provided mechanistic insights into how tumor cells can evade CD226-mediated recognition and elimination. Weulersse et al showed that CD226 underwent transcriptional repression by Eomes (Eomesodermin, a key transcriptional factor upregulated in exhausted T cells), and thus, CD226 expression was downregulated in exhausted T cells.51 Braun et al showed that CD155 on tumor cells triggered ligand-induced internalization and subsequent proteasomal degradation of CD226 in cytotoxic lymphocytes.52 Together, CD226 undergoes both transcriptional and post-transcriptional regulation in the tumor microenvironment. Notably, both studies concluded that CD8+ T cells with reduced expression levels of CD226 poorly responded to PD-1 blockade.51,52
The CD226-dependent functions also undergo negative regulation by TIGIT, the inhibitory checkpoint molecule upregulated on activated T cells and NK cells. Due to its higher binding affinity to CD155, TIGIT competitively dampens CD226 functions.53 TIGIT is the most frequently expressed inhibitory immune checkpoint molecule in MM BM T cells, and TIGIT-expressing T cells represent functionally exhausted phenotypes.54 Importantly, anti-TIGIT mAb reinvigorated myeloma antigen-specific T cells in MM patients, and improved disease control either by monotherapy or post-ASCT maintenance therapy in preclinical models.54,55 TIGIT has multifaceted inhibitor mechanisms, including 1) functional competition with CD226, 2) T/NK-cell intrinsic regulation, 3) enhancing immunosuppressive activities in regulatory T (Treg) cells, and 4) negative regulation of DCs.53 Thus, the imbalance between CD226 and TIGIT provides critical implications for myeloma immunotherapy.
In addition to the imbalance between CD226 and TIGIT expression, multiple factors might be implicated in T-cell dysfunction including transcriptional and epigenetic reprograming of T cells, metabolic alterations, and extrinsic regulation by immunosuppressive subsets. Age-associated immune senescence and infection might also sculpt T-cell phenotypes in MM patients. Besides, due to spatial heterogeneity of MM, T cells isolated from BM aspirates might not reflect bona-fide tumor-infiltrating T cells.6 Thus, in-depth profiling of intra-tumoral T cells might provide a clearer insight into T-cell dysfunction in MM patients.
Immune Subsets Positively Regulating the Cancer-Immunity Cycle
The framework of cancer-immunity cycle was originally designed to understand anti-tumor T cells immunity. However, NK cells are also recognized as a key subset of the cancer-immunity cycle, either by stimulating DCs and adaptive immunity or by direct recognition and elimination of tumor cells.56 Several contradictory results have been reported regarding the numbers and functions of NK cells in MM patients.24 Recently, Barberi et al reported an increase in NK cells in MM patients, compared to healthy subjects. Intriguingly, they showed that a subset of NK cells with CD94lowCD56dim expression possessed a high proliferative potential and cytotoxic activity against malignant plasma cells.57 Thus, heterogeneity of NK-cell subsets might be an important aspect of the efficacies of immunomodulatory drugs and mAbs that elicit antibody-dependent cellular cytotoxicity (ADCC). Notably, immunomodulatory drugs also stimulate BM type 1 innate lymphoid cells (ILC1) to produce IFN-γ,58 suggesting that ILC1 might also contribute to anti-myeloma immunity. NKT cells are also recognized as a key subset bridging innate and adaptive immunity. As malignant plasma cells express CD1d, CD1d-restricted NKT cells might exert direct cytotoxicity against MM. It is reported that NKT cells isolated from active MM showed a marked deficiency of ligand-dependent IFN-γ production.59 Still, DCs pulsed with α-galactosylceramide (α-GalCer) could restore NKT cell functions in active MM patients, indicating that NKT cell hypo-responsiveness is reversible.59 It is possible that NK cells, ILC1 cells and NKT cells play non-redundant roles during myeloma immunosurveillance and immunotherapy, though further studies are necessary to understand their discrete functions.
Immune Subsets Negatively Regulating the Cancer-Immunity Cycle
Tumor tissues contain heterogeneous immunosuppressive myeloid subsets, including immature monocytes and neutrophils (also known as myeloid-derived suppressor cells: MDSCs) and tumor-associated macrophages (TAM).60,61 Tumor progression triggers disturbance of hematopoiesis in the BM, and myeloid subsets acquire immunosuppressive activities in response to growth factors and pro-inflammatory cytokines derived from tumor tissues.61 These myeloid cells potently suppress T cell activation in both antigen-specific and non-specific manners by sequestration of amino acids, reactive oxygen and nitrogen species, and production of immunosuppressive metabolites.61 Thus, MDSCs and TAMs negatively regulate multiple steps of the cancer-immunity cycle, including T-cell priming and activation, infiltration, and T cell-mediated cytotoxicity against tumor cells. As myeloma primary grows in the BM, where myeloid cells and their precursor abundantly exist, various myeloid subsets with different maturation stages are known to hamper T-cell activation.28,62,63
Treg cells play a critical role in maintaining self-tolerance and the prevention of autoimmunity. CTLA-4 expressed in Treg cells is recognized as a key regulator of T-cell priming by its higher binding affinity and avidity for CD80/86, compared to CD28.64 Additionally, Treg cells can suppress effector lymphocyte functions by consumption of IL-2, production of cytokines (TGF-β and IL-10), and adenosine generation via ectoenzymes (CD38/CD39/CD73).65 Like tumor-infiltrating effector lymphocytes, activated Treg cells in the tumor microenvironment also express immune checkpoint molecules. Several lines of evidence suggest that PD-1 negatively regulates Treg functions.65–67 Indeed, an expansion of activated Treg cells was reported in gastric cancer patients experiencing an exacerbation of tumor progression after PD-1 blockade therapy,66 suggesting that anti-PD-1 triggered activation of Treg cells. A subsequent study showed that the balance of PD-1 expression between CD8+ T cells and Treg cells in the tumor microenvironment was a key determinant for predicting therapeutic responsiveness to PD-1 blockade.67 This finding may partially explain why PD-1 blockade led to inferior outcomes in myeloma patients,17,18 although validation is highly warranted. It should be noted that BM Treg cells have unique features and functions compared to Treg cells in the blood and lymphoid organs.68,69 The phenotype of BM Treg cells is further sculpted by myeloma progression, such as acquisition of type 1 interferon signature genes.28,70
Regulatory B (Breg) cells are also recognized as a key immunosuppressive subset by their ability to produce anti-inflammatory cytokines such IL-10, IL-35, and TGF-β. Zhang et al reported the expansion of IL-10-producing CD19+CD24highCD38high Breg cells in the BM from newly diagnosed MM patients.71 Indeed, ex vivo isolated BM Breg cells could suppress NK cell-mediated ADCC against autologous MM cells coated with anti-SLAMF7 mAb (elotuzumab),71 supporting the negative impact on anti-tumor immunity. Yet, it remains largely unknown how myeloma progression triggers expansion of Breg cells in the BM.
Therapeutic Activation of Cancer-Immunity Cycle
Various myeloma therapeutics can fuel the cancer-immunity cycle. For example, cytotoxic agents can trigger the release of tumor antigens (step 1) and stimulate APCs (step 2). Blocking a negative regulator of phagocytosis has emerged as a new modality to promote engulfment of tumor cells and subsequent antigen-presentation (step 2). Immune checkpoint inhibitors act at the T cell-APC interface (step 3) and at the T cell-cancer cell interface (step 6 and 7). Clinical benefits of anti-CD38 mAbs and ASCT might be explained by their pleiotropic impacts on multiple steps. Lastly, CAR-T cell and bispecific T-cell engager therapies have emerged as powerful approaches to recognize and eliminate malignant plasma cells irrespective of TCR specificity (step 6 and step 7). Here, we discuss their key immunological mechanisms of myeloma therapeutics (Figure 2).
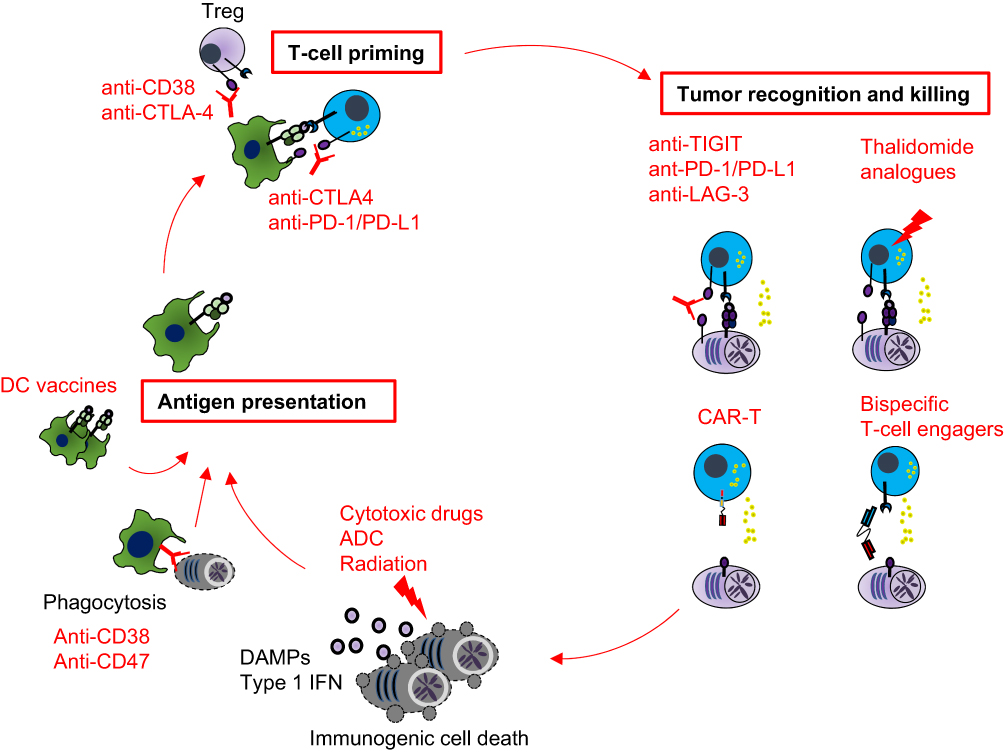 |
Figure 2 Therapeutic approaches to stimulate the cancer-immunity cycle in multiple myeloma. A schematic illustrating how anti-myeloma therapies differentially stimulate the cancer-immunity cycle. Myeloma antigen-presentation can be augmented by immunogenic cell death inducers, enhancing phagocytosis or DC-based vaccine. Regulatory T (Treg) cells are critical regulators for T-cell priming, and thus mAbs against CTLA-4 or CD38 stimulate T-cell priming. Immunomodulatory drugs and immune checkpoint inhibitors improve recognition and killing of myeloma cells by cytotoxic lymphocytes. Chimeric antigen receptor (CAR) T-cell therapies and bispecific T-cell engager antibodies allow T cells to recognize and eliminate tumor cells in a MHC-independent fashion. Autologous stem cell transplant has pleiotropic impacts on the cancer-immunity cycle. |
Induction of Immunogenic Cell Death
Although apoptosis is classified as an immunologically silent form of cell death, certain anti-cancer chemotherapy drugs can induce an immunostimulatory form of cell death, called immunogenic cell death (ICD). Indeed, various anti-myeloma drugs, such as cyclophosphamide, doxorubicin, and proteasome inhibitors, are reported to trigger ICD.72,73 Mechanistically, these drugs trigger the exposure and release of immunostimulatory damage-associated molecular patterns (DAMPs) from dying tumor cells. These DAMPs include adenosine triphosphate (ATP), nuclear proteins, and exposure of calreticulin (an ER-localized protein).72 Together with other tumor antigens released from dying tumor cells, these DAMPs promote the maturation of DCs, providing vaccine effects in the cancer-immunity cycle (step 1 and step 2). In preclinical myeloma models, adaptive immunity is indispensable for the optimal efficacies by bortezomib, cyclophosphamide, and LCL161, an inhibitor for IAP (inhibitors of apoptosis protein).23,50,73 More recently, Gulla et al showed that bortezomib stimulated the cGAS/STING pathway and production of type I IFNs, and that a STING agonist, ADU-S100, could potentiate bortezomib-mediated anti-myeloma immunity.73 These results highlight that ICD induced by anti-myeloma agents fuels the immune-mediated control of myeloma. However, detrimental impacts of these chemotherapies should be considered, such as lymphodepletion (by alkylating agents) and down-regulation of MHC-I (by proteasome inhibitors).46 In this context, tumor-specific killing by antibody-drug conjugates might be a potential approach to maximize the therapeutic benefits of ICD while preserving viability and functions of non-malignant lymphocytes.74,75
The ascorbic effect by radiation therapy is also explained by immunogenic tumor cell death and subsequent stimulation of anti-tumor immunity. Growing preclinical and clinical evidence supports that radiation therapy augments the efficacies of immune checkpoint inhibitors in solid malignancies.76 Recently, Kazandjian et al reported only modest clinical benefits of anti-PD-L1 and concomitant hypofractionated radiotherapy in patients with relapsed and refractory MM.77 Nevertheless, radiation therapy might act as an optimal partner for other immunotherapeutics due to its ability to modulate the tumor microenvironment.
Enhancing Phagocytosis
Signal regulatory protein α (SIRPα) is known to negatively regulate macrophage phagocytosis through the recognition of CD47, a ligand frequently overexpressed on hematological tumor cells. Thus, blocking the interaction between CD47 and SIRPα can stimulate macrophage phagocytosis.61 Indeed, anti-CD47 in combination with rituximab (anti-CD20 mAb) has shown promising clinical responses in patients with non-Hodgkin’s lymphoma.78 Importantly, CD47 blockade can also trigger CD8 T cell-dependent control of tumors by stimulating cross-priming by DCs,79 supporting that targeting phagocytosis checkpoint augments step 1 and step 2 in the cancer-immunity cycle. In MM patients, several CD47 inhibitors are being tested as a monotherapy or in combination with bortezomib and dexamethasone (NCT04445701 and NCT03530683).
Immunomodulatory Drugs
Together with immunomodulatory functions, thalidomide analogues (such as lenalidomide and pomalidomide) have multifaceted anti-tumor effects by inhibiting tumor proliferation, anti-angiogenesis, and inhibiting pro-inflammatory cytokine production.80 The key molecular mechanism of immunomodulatory drugs is explained by their direct binding to cereblon (a substrate receptor for the E3 ubiquitin ligase), followed by proteasome degradation of the zinc finger transcription factors of Ikaros (IKZF1) and Aiolos (IKZF3). The direct anti-myeloma activity of thalidomide analogues is explained by the fact that proteasomal degradation of IKZF1 and IKZF3 leads to transcriptional repression of MYC and IRF4 (interferon regulatory factor 4), both of which are key players for myeloma survival and proliferation. As IKZF1 and IKZF3 act as transcriptional repressors of the IL2 gene, thalidomide analogues can augment IL-2 production from T cells.81 Additionally, they can directly augment NK cell-mediated cytotoxicity against tumor cells.82 The activation of NK cells triggers the formation of actin mesh-like structure, through which lytic granules and vesicles containing IFN-γ are released toward tumor cells. Lenalidomide increases the opening of the actin mesh-like structure, promoting granule exocytosis in NK cells.83 The positive impact on cytotoxic activity is also supported by clinical efficacies of thalidomide analogues in combination with anti-SLAMF7 mAb (elotuzumab) that elicits ADCC by NK cells.84,85 Lenalidomide also augments the efficacy of CAR T-cell therapy in a preclinical model.86 These results indicate that immunomodulatory drugs critically support the final step of the cancer-immunity cycle (step 7). Moreover, thalidomide analogues hamper induction of MDSCs and Treg cells,87,88 highlighting multifaceted impacts on the cancer-immunity cycle.
Autologous Stem-Cell Transplantation (ASCT)
Growing evidence supports that the clinical benefits of ASCT are not simply explained by the cytoreductive effects of high-dose chemotherapy. ASCT pleiotropically enhances the cancer-immunity cycle by inducing ICD, increasing the generation of antigen-specific T cells, and resulting in T cell-dependent control of myeloma.89,90 Using mass cytometry-based immune profiling, Kourelis et al showed that the early post-ASCT period was associated with the immunosuppressive status characterized by an increase in senescence or exhausted T-cell subsets and activated Treg cells.91 Among T-cell subsets, CD8 T cells undergo rapid homeostatic proliferation after ASCT, as supported by the fact that an inverted CD4/CD8 ratio is observed for nearly one year after ASCT.92 Importantly, the emergence of T cells with an exhausted/senescent phenotype predicts disease relapse after ASCT,92 suggesting that T cells might be actively implicated in post-ASCT immunosurveillance. More recently, Lee et al reported an expansion of effector and memory T cells subsets post-ASCT, which was associated with a skewed TCR repertoire.93 Together, cytoreduction and immune-reconstitution by ASCT might favor the generation and expansion of myeloma antigen-specific T cells. In this context, immunomodulatory drugs are widely used as post-ASCT consolidation and maintenance therapies to help antigen-specific T cells responses to eliminate malignant plasma cells. To accelerate T cell-mediated control of residual MM cells, CAR-T therapies and bispecific T-cell engagers are being tested as post-ASCT therapies.89,94
Anti-CD38 Monoclonal Antibodies (mAbs)
Anti-CD38 monoclonal antibodies (mAbs) (daratumumab and isatuximab) have multiple anti-tumor mechanisms, including direct apoptosis by cross-linking stimulation, antibody-dependent cellular phagocytosis (ADCP) by macrophages, ADCC by NK cells, and complement-dependent cytotoxicity.95,96 Additionally, anti-CD38 mAbs deplete CD38-expressing immunosuppressive subsets, including Treg cells.97 Compared to daratumumab, isatuximab is known to potently suppress the enzymatic activity of CD38, reducing the generation of immunosuppressive adenosine.98 Given that adenosine inhibits effector lymphocyte functions as well as ADCC and ADCP,99 preventing adenosine generation might provide additional therapeutic benefits. The immunostimulatory effects of anti-CD38 mAbs have been supported by an increase in cytotoxic lymphocytes expressing high levels of granzymeB100 and by an increase of TCR clonality after daratumumab therapy.97 Thus, anti-CD38 mAbs pleiotropically modulate the cancer-immunity cycle in MM.
Immune Checkpoint Inhibitors (ICIs)
It is appreciated that anti-CTLA-4 blockade mainly acts on T-cell priming (step 3), while anti-PD-1/PD-L1 blockade can target effector lymphocytes in peripheral tissues (step 6 and step 7).64 As described previously, these ICIs have shown limited clinical efficacies in patients with MM. Still, there is a possibility that these ICIs bring some clinical benefits in combination with other modalities. The balance between TIGIT and DNAM-1 provides important implications for myeloma immunotherapies. In addition to tumor recognition and cytotoxicity, blocking TIGIT might also improve T-cell priming by modulating Treg cell activities and tolerogenic DCs functions.53 LAG-3 (lymphocyte-activation gene 3) is recognized as a major immune checkpoint molecule upregulated on BM T cells in relapsed MM patients after ASCT.93,101 Due to its high affinity to pMHC-II complexes, LAG-3 inhibits the activation of CD4 T cells at the interface between T cells and APC.47 More recently, a liver-secreted protein called fibrinogen-like protein 1 (FGL1) was identified as a ligand of LAG-3,102 providing a mechanistic insight into how LAG3 blockade can reinvigorate exhausted CD8 T cells in combination with other immune checkpoint inhibitors. It remains to be clarified whether the FGL1-mediated immune regulation contributes to T-cell dysfunction in MM. Currently, anti-TIGIT mAb and anti-LAG-3 mAb are being tested in clinical trials in patients with relapsed/refractory MM (NCT04150965).
CAR T-Cell Therapies and Bispecific T-Cell Engagers
BCMA-directed CAR T-cell therapies and bispecific T-cell engagers are designed to recognize BCMA on malignant plasma cells by single-chain variable domain fragments (scFv) derived from anti-BCMA mAb. CARs deliver signal 1 (eg, via CD3ζ chain domain) and signal 2 (via CD28 and/or 4-1BB domain), leading to activation of T cells. In bispecific T-cell engagers, bispecific antibodies bind to malignant plasma cells by one arm containing anti-BCMA scFv, and trigger the activation of cytotoxic T cells by another arm containing anti-CD3 scFv.103 Both CAR T-cell therapies and bispecific T-cell engagers can stimulate T cells in an MHC-independent manner, bypassing the multiple steps of the cancer-immunity cycle. The first FDA-approved anti-BCMA CAR T-cell therapy (idecabtagene vicleucel) has demonstrated clinical benefits with an overall response rate of ~73% in patients with relapsed/refractory MM.104 Other BCMA-directed CAR T-cell therapies and bispecific T-cell engagers also demonstrated promising clinical responses in clinical trials.103 However, relapse remains a major issue, which might be explained by multiple mechanisms including loss of BCMA antigen, selection of BCMA-negative clones, T-cell dysfunction or the immunosuppressive microenvironment.105–107 In this context, other myeloma antigens have been identified as potential targets, such as activated integrin β7 and GPRC5D (G-protein-coupled receptor family C group 5 member D).108,109 Additionally, dual targeting approaches are actively being tested, such as APRIL-based CAR-T (targeting BCMA and TACI),110 combined infusion of anti-CD19 and anti-BCMA CAR-T,111 and co-targeting BCMA with SLAMF7 or CD38.111,112 In addition to down-regulation of antigens, overcoming T-cell intrinsic dysfunction remains a key for better clinical efficacies.6,112 In this context, off-the-shelf allogeneic CAR-T cell therapies are actively being tested in clinical trials (NCT04093596 and NCT04142619). More recently, allogeneic cord blood-derived CAR NK cell therapy has shown impressive responses in CD19+ B-cell malignancies.113 These approaches will not only broaden the utility of CAR-based therapies but also reveal new combination strategies for better outcomes.
Concluding Remarks
Despite the recent success of immunotherapies, it is still challenging to achieve cure by myeloma immunotherapies. One of the major advantages of CAR-T cell therapies and bispecific T-cell engager antibodies is their ability to recognize tumor cells in an MHC-independent fashion; however, eliciting antigen-specific T cells might also be required for preventing relapse after cessation of these therapies. In this context, combining different therapeutic modalities is a rational approach. Alternatively, given that T-cell dysfunction critically limits adaptive immunity against MM, harnessing innate anti-tumor immunity by NK cells and macrophages might overcome immunotherapeutic resistance in MM. Overall, an in-depth understanding of the cancer immunity cycle will provide a clue for better clinical efficacies of myeloma therapeutics.
Acknowledgment
The authors appreciate Bianca Nowlan for critical reading and helpful suggestions. K.N. is supported by the Naito Foundation and NHMRC Project Grant (1159593). K.N. is a recipient of recipients of a Leukemia Foundation of Australia SERP grant. This project was supported by grant 2000538 awarded through the 2020 Priority-driven Collaborative Cancer Research Scheme and funded by the Leukaemia Foundation with the support of Cancer Australia.
Disclosure
The authors have no conflicts of interest to declare.
References
1. van de Donk N, Pawlyn C, Yong KL. Multiple myeloma. Lancet. 2021;397(10272):410–427. doi:10.1016/S0140-6736(21)00135-5
2. Ledergor G, Weiner A, Zada M, et al. Single cell dissection of plasma cell heterogeneity in symptomatic and asymptomatic myeloma. Nat Med. 2018;24(12):1867–1876. doi:10.1038/s41591-018-0269-2
3. Rustad EH, Yellapantula V, Leongamornlert D, et al. Timing the initiation of multiple myeloma. Nat Commun. 2020;11(1):1917. doi:10.1038/s41467-020-15740-9
4. van Nieuwenhuijzen N, Spaan I, Raymakers R, Peperzak V. From MGUS to multiple myeloma, a paradigm for clonal evolution of premalignant cells. Cancer Res. 2018;78(10):2449–2456. doi:10.1158/0008-5472.CAN-17-3115
5. Schürch CM, Rasche L, Frauenfeld L, Weinhold N, Fend F. A review on tumor heterogeneity and evolution in multiple myeloma: pathological, radiological, molecular genetics, and clinical integration. Virchows Arch. 2020;476(3):337–351. doi:10.1007/s00428-019-02725-3
6. Nakamura K, Smyth MJ, Martinet L. Cancer immunoediting and immune dysregulation in multiple myeloma. Blood. 2020;136(24):2731–2740. doi:10.1182/blood.2020006540
7. Minnie SA, Hill GR. Immunotherapy of multiple myeloma. J Clin Invest. 2020;130(4):1565–1575. doi:10.1172/JCI129205
8. Nadeem O, Tai YT, Anderson KC. Immunotherapeutic and targeted approaches in multiple myeloma. Immunotargets Ther. 2020;9:201–215. doi:10.2147/ITT.S240886
9. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. doi:10.1016/j.immuni.2013.07.012
10. Nguyen-Pham TN, Lee YK, Kim HJ, Lee JJ. Immunotherapy using dendritic cells against multiple myeloma: how to improve? Clin Dev Immunol. 2012;2012:397648. doi:10.1155/2012/397648
11. Hoang MD, Jung SH, Lee HJ, et al. Dendritic cell-based cancer immunotherapy against multiple myeloma: from bench to clinic. Chonnam Med J. 2015;51(1):1–7. doi:10.4068/cmj.2015.51.1.1
12. Blotta S, Tassone P, Prabhala RH, et al. Identification of novel antigens with induced immune response in monoclonal gammopathy of undetermined significance. Blood. 2009;114(15):3276–3284. doi:10.1182/blood-2009-04-219436
13. Dhodapkar MV, Sexton R, Das R, et al. Prospective analysis of antigen-specific immunity, stem-cell antigens, and immune checkpoints in monoclonal gammopathy. Blood. 2015;126(22):2475–2478. doi:10.1182/blood-2015-03-632919
14. Jiang T, Shi T, Zhang H, et al. Tumor neoantigens: from basic research to clinical applications. J Hematol Oncol. 2019;12(1):93. doi:10.1186/s13045-019-0787-5
15. Miller A, Asmann Y, Cattaneo L, et al. High somatic mutation and neoantigen burden are correlated with decreased progression-free survival in multiple myeloma. Blood Cancer J. 2017;7(9):e612. doi:10.1038/bcj.2017.94
16. Perumal D, Imai N, Lagana A, et al. Mutation-derived neoantigen-specific T-cell responses in multiple myeloma. Clin Cancer Res. 2020;26(2):450–464. doi:10.1158/1078-0432.CCR-19-2309
17. Usmani SZ, Schjesvold F, Oriol A, et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): a randomised, open-label, Phase 3 trial. Lancet Haematol. 2019;6(9):e448–e458. doi:10.1016/S2352-3026(19)30109-7
18. Mateos MV, Blacklock H, Schjesvold F, et al. Pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): a randomised, open-label, phase 3 trial. Lancet Haematol. 2019;6(9):e459–e469. doi:10.1016/S2352-3026(19)30110-3
19. Roberts EW, Broz ML, Binnewies M, et al. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell. 2016;30(2):324–336. doi:10.1016/j.ccell.2016.06.003
20. Feuerer M, Beckhove P, Garbi N, et al. Bone marrow as a priming site for T-cell responses to blood-borne antigen. Nat Med. 2003;9(9):1151–1157. doi:10.1038/nm914
21. Cavanagh LL, Bonasio R, Mazo IB, et al. Activation of bone marrow-resident memory T cells by circulating, antigen-bearing dendritic cells. Nat Immunol. 2005;6(10):1029–1037. doi:10.1038/ni1249
22. Asano K, Nabeyama A, Miyake Y, et al. CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity. 2011;34(1):85–95. doi:10.1016/j.immuni.2010.12.011
23. Chesi M, Mirza NN, Garbitt VM, et al. IAP antagonists induce anti-tumor immunity in multiple myeloma. Nat Med. 2016;22(12):1411–1420. doi:10.1038/nm.4229
24. Guillerey C, Nakamura K, Vuckovic S, Hill GR, Smyth MJ. Immune responses in multiple myeloma: role of the natural immune surveillance and potential of immunotherapies. Cell Mol Life Sci. 2016;73(8):1569–1589. doi:10.1007/s00018-016-2135-z
25. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. doi:10.1146/annurev-immunol-020711-074950
26. Dhodapkar MV, Krasovsky J, Osman K, Geller MD. Vigorous premalignancy-specific effector T cell response in the bone marrow of patients with monoclonal gammopathy. J Exp Med. 2003;198(11):1753–1757. doi:10.1084/jem.20031030
27. Bailur JK, McCachren SS, Doxie DB, et al. Early alterations in stem-like/resident T cells, innate and myeloid cells in the bone marrow in preneoplastic gammopathy. JCI Insight. 2019;5:e127807. doi:10.1172/jci.insight.127807
28. Zavidij O, Haradhvala NJ, Mouhieddine TH, et al. Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat Cancer. 2020;1(5):493–506. doi:10.1038/s43018-020-0053-3
29. Cohen AD, Raje N, Fowler JA, Mezzi K, Scott EC, Dhodapkar MV. How to train your T cells: overcoming immune dysfunction in multiple myeloma. Clin Cancer Res. 2020;26(7):1541–1554. doi:10.1158/1078-0432.CCR-19-2111
30. Kohli K, Pillarisetty VG, Kim TS. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 2021. doi:10.1038/s41417-021-00303-x
31. Chow MT, Ozga AJ, Servis RL, et al. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity. 2019;50(6):1498–1512.e1495. doi:10.1016/j.immuni.2019.04.010
32. Di Rosa F, Pabst R. The bone marrow: a nest for migratory memory T cells. Trends Immunol. 2005;26(7):360–366. doi:10.1016/j.it.2005.04.011
33. Ponzetta A, Benigni G, Antonangeli F, et al. Multiple myeloma impairs bone marrow localization of effector natural killer cells by altering the chemokine microenvironment. Cancer Res. 2015;75(22):4766–4777. doi:10.1158/0008-5472.CAN-15-1320
34. Roccaro AM, Mishima Y, Sacco A, et al. CXCR4 regulates extra-medullary myeloma through epithelial-mesenchymal-transition-like transcriptional activation. Cell Rep. 2015;12(4):622–635. doi:10.1016/j.celrep.2015.06.059
35. Vandyke K, Zeissig MN, Hewett DR, et al. HIF-2α promotes dissemination of plasma cells in multiple myeloma by regulating CXCL12/CXCR4 and CCR1. Cancer Res. 2017;77(20):5452–5463. doi:10.1158/0008-5472.CAN-17-0115
36. Mushtaq MU, Papadas A, Pagenkopf A, et al. Tumor matrix remodeling and novel immunotherapies: the promise of matrix-derived immune biomarkers. J Immunother Cancer. 2018;6(1):65. doi:10.1186/s40425-018-0376-0
37. Hope C, Ollar SJ, Heninger E, et al. TPL2 kinase regulates the inflammatory milieu of the myeloma niche. Blood. 2014;123(21):3305–3315. doi:10.1182/blood-2014-02-554071
38. Dhakal B, Pagenkopf A, Mushtaq MU, et al. Versican proteolysis predicts immune effector infiltration and post-transplant survival in myeloma. Leuk Lymphoma. 2019;60(10):2558–2562. doi:10.1080/10428194.2019.1585836
39. Bhutani M, Foureau DM, Atrash S, Voorhees PM, Usmani SZ. Extramedullary multiple myeloma. Leukemia. 2020;34(1):1–20. doi:10.1038/s41375-019-0660-0
40. Touzeau C, Moreau P. How I treat extramedullary myeloma. Blood. 2016;127(8):971–976. doi:10.1182/blood-2015-07-635383
41. Xu J, Chen LJ, Yang SS, et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc Natl Acad Sci U S A. 2019;116(19):9543–9551. doi:10.1073/pnas.1819745116
42. Firsova MV, Mendeleeva LP, Kovrigina AM, Solovev MV, Savchenko VG. Plasmacytoma in patients with multiple myeloma: morphology and immunohistochemistry. BMC Cancer. 2020;20(1):346. doi:10.1186/s12885-020-06870-w
43. Tian Y, Li Y, Shao Y, Zhang Y. Gene modification strategies for next-generation CAR T cells against solid cancers. J Hematol Oncol. 2020;13(1):54. doi:10.1186/s13045-020-00890-6
44. Dieckmann NM, Frazer GL, Asano Y, Stinchcombe JC, Griffiths GM. The cytotoxic T lymphocyte immune synapse at a glance. J Cell Sci. 2016;129(15):2881–2886. doi:10.1242/jcs.186205
45. Shi J, Tricot GJ, Garg TK, et al. Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell-mediated lysis of myeloma. Blood. 2008;111(3):1309–1317. doi:10.1182/blood-2007-03-078535
46. Kowalewski DJ, Walz S, Backert L, et al. Carfilzomib alters the HLA-presented peptidome of myeloma cells and impairs presentation of peptides with aromatic C-termini. Blood Cancer J. 2016;6(4):e411. doi:10.1038/bcj.2016.14
47. Salik B, Smyth MJ, Nakamura K. Targeting immune checkpoints in hematological malignancies. J Hematol Oncol. 2020;13(1):111. doi:10.1186/s13045-020-00947-6
48. Badros A, Hyjek E, Ma N, et al. Pembrolizumab, pomalidomide, and low-dose dexamethasone for relapsed/refractory multiple myeloma. Blood. 2017;130(10):1189–1197. doi:10.1182/blood-2017-03-775122
49. Kwon M, Kim CG, Lee H, et al. PD-1 blockade reinvigorates bone marrow CD8(+) T cells from patients with multiple myeloma in the presence of TGFβ inhibitors. Clin Cancer Res. 2020;26(7):1644–1655. doi:10.1158/1078-0432.CCR-19-0267
50. Guillerey C, Ferrari de Andrade L, Vuckovic S, et al. Immunosurveillance and therapy of multiple myeloma are CD226 dependent. J Clin Invest. 2015;125(5):2077–2089. doi:10.1172/JCI77181
51. Weulersse M, Asrir A, Pichler AC, et al. Eomes-dependent loss of the co-activating receptor CD226 restrains CD8(+) T cell anti-tumor functions and limits the efficacy of cancer immunotherapy. Immunity. 2020;53(4):824–839.e810. doi:10.1016/j.immuni.2020.09.006
52. Braun M, Aguilera AR, Sundarrajan A, et al. CD155 on tumor cells drives resistance to immunotherapy by inducing the degradation of the activating receptor CD226 in CD8(+) T cells. Immunity. 2020;53(4):805–823.e815. doi:10.1016/j.immuni.2020.09.010
53. Manieri NA, Chiang EY, Grogan JL. TIGIT: a key inhibitor of the cancer immunity cycle. Trends Immunol. 2017;38(1):20–28. doi:10.1016/j.it.2016.10.002
54. Guillerey C, Harjunpaa H, Carrie N, et al. TIGIT immune checkpoint blockade restores CD8(+) T-cell immunity against multiple myeloma. Blood. 2018;132(16):1689–1694. doi:10.1182/blood-2018-01-825265
55. Minnie SA, Kuns RD, Gartlan KH, et al. Myeloma escape after stem cell transplantation is a consequence of T-cell exhaustion and is prevented by TIGIT blockade. Blood. 2018;132(16):1675–1688. doi:10.1182/blood-2018-01-825240
56. Bald T, Krummel MF, Smyth MJ, Barry KC. The NK cell-cancer cycle: advances and new challenges in NK cell-based immunotherapies. Nat Immunol. 2020;21(8):835–847. doi:10.1038/s41590-020-0728-z
57. Barberi C, De Pasquale C, Allegra A, et al. Myeloma cells induce the accumulation of activated CD94 low NK cells by cell-to-cell contacts involving CD56 molecules. Blood Adv. 2020;4(10):2297–2307. doi:10.1182/bloodadvances.2019000953
58. Kini Bailur J, Mehta S, Zhang L, et al. Changes in bone marrow innate lymphoid cell subsets in monoclonal gammopathy: target for IMiD therapy. Blood Adv. 2017;1(25):2343–2347. doi:10.1182/bloodadvances.2017012732
59. Dhodapkar MV, Geller MD, Chang DH, et al. A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J Exp Med. 2003;197(12):1667–1676. doi:10.1084/jem.20021650
60. Nakamura K, Smyth MJ. Targeting cancer-related inflammation in the era of immunotherapy. Immunol Cell Biol. 2017;95(4):325–332. doi:10.1038/icb.2016.126
61. Nakamura K, Smyth MJ. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol Immunol. 2020;17(1):1–12. doi:10.1038/s41423-019-0306-1
62. Nakamura K, Kassem S, Cleynen A, et al. Dysregulated IL-18 is a key driver of immunosuppression and a possible therapeutic target in the multiple myeloma microenvironment. Cancer Cell. 2018;33(4):634–648.e635. doi:10.1016/j.ccell.2018.02.007
63. Perez C, Botta C, Zabaleta A, et al. Immunogenomic identification and characterization of granulocytic myeloid-derived suppressor cells in multiple myeloma. Blood. 2020;136(2):199–209. doi:10.1182/blood.2019004537
64. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069–1086. doi:10.1158/2159-8290.CD-18-0367
65. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol. 2019;16(6):356–371. doi:10.1038/s41571-019-0175-7
66. Kamada T, Togashi Y, Tay C, et al. PD-1(+) regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U S A. 2019;116(20):9999–10008. doi:10.1073/pnas.1822001116
67. Kumagai S, Togashi Y, Kamada T, et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol. 2020;21(11):1346–1358. doi:10.1038/s41590-020-0769-3
68. Zhao E, Wang L, Dai J, et al. Regulatory T cells in the bone marrow microenvironment in patients with prostate cancer. Oncoimmunology. 2012;1(2):152–161. doi:10.4161/onci.1.2.18480
69. Glatman Zaretsky A, Konradt C, Depis F, et al. T regulatory cells support plasma cell populations in the bone marrow. Cell Rep. 2017;18(8):1906–1916. doi:10.1016/j.celrep.2017.01.067
70. Kawano Y, Zavidij O, Park J, et al. Blocking IFNAR1 inhibits multiple myeloma-driven Treg expansion and immunosuppression. J Clin Invest. 2018;128(6):2487–2499. doi:10.1172/JCI88169
71. Zhang L, Tai YT, Ho M, et al. Regulatory B cell-myeloma cell interaction confers immunosuppression and promotes their survival in the bone marrow milieu. Blood Cancer J. 2017;7(3):e547. doi:10.1038/bcj.2017.24
72. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. doi:10.1146/annurev-immunol-032712-100008
73. Gulla A, Morelli E, Samur MK, et al. Bortezomib induces anti-multiple myeloma immune response mediated by cGAS/STING pathway activation. Blood Cancer Discov. 2021. doi:10.1158/2643-3230.BCD-21-0047
74. Tai YT, Mayes PA, Acharya C, et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. 2014;123(20):3128–3138. doi:10.1182/blood-2013-10-535088
75. Richardson PG, Lee HC, Abdallah AO, et al. Single-agent belantamab mafodotin for relapsed/refractory multiple myeloma: analysis of the lyophilised presentation cohort from the pivotal DREAMM-2 study. Blood Cancer J. 2020;10(10):106. doi:10.1038/s41408-020-00369-0
76. Gong J, Le TQ, Massarelli E, Hendifar AE, Tuli R. Radiation therapy and PD-1/PD-L1 blockade: the clinical development of an evolving anticancer combination. J Immunother Cancer. 2018;6(1):46. doi:10.1186/s40425-018-0361-7
77. Kazandjian D, Dew A, Hill E, et al. Avelumab, a PD-L1 inhibitor, in combination with hypofractionated radiotherapy and the abscopal effect in relapsed refractory multiple myeloma. Oncologist. 2021;26(4):288–e541. doi:10.1002/onco.13712
78. Advani R, Flinn I, Popplewell L, et al. CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N Engl J Med. 2018;379(18):1711–1721. doi:10.1056/NEJMoa1807315
79. Liu X, Pu Y, Cron K, et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med. 2015;21(10):1209–1215. doi:10.1038/nm.3931
80. Quach H, Ritchie D, Stewart AK, et al. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia. 2010;24(1):22–32. doi:10.1038/leu.2009.236
81. Gandhi AK, Kang J, Havens CG, et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN.). Br J Haematol. 2014;164(6):811–821. doi:10.1111/bjh.12708
82. Davies FE, Raje N, Hideshima T, et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood. 2001;98(1):210–216. doi:10.1182/blood.V98.1.210
83. Lagrue K, Carisey A, Morgan DJ, Chopra R, Davis DM. Lenalidomide augments actin remodeling and lowers NK-cell activation thresholds. Blood. 2015;126(1):50–60. doi:10.1182/blood-2015-01-625004
84. Dimopoulos MA, Dytfeld D, Grosicki S, et al. Elotuzumab plus pomalidomide and dexamethasone for multiple myeloma. N Engl J Med. 2018;379(19):1811–1822. doi:10.1056/NEJMoa1805762
85. Dimopoulos MA, Lonial S, White D, et al. Elotuzumab, lenalidomide, and dexamethasone in RRMM: final overall survival results from the phase 3 randomized ELOQUENT-2 study. Blood Cancer J. 2020;10(9):91. doi:10.1038/s41408-020-00357-4
86. Wang X, Walter M, Urak R, et al. Lenalidomide enhances the function of CS1 chimeric antigen receptor-redirected T cells against multiple myeloma. Clin Cancer Res. 2018;24(1):106–119. doi:10.1158/1078-0432.CCR-17-0344
87. Kuwahara-Ota S, Shimura Y, Steinebach C, et al. Lenalidomide and pomalidomide potently interfere with induction of myeloid-derived suppressor cells in multiple myeloma. Br J Haematol. 2020;191(5):784–795. doi:10.1111/bjh.16881
88. Galustian C, Meyer B, Labarthe MC, et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol Immunother. 2009;58(7):1033–1045. doi:10.1007/s00262-008-0620-4
89. Minnie SA, Hill GR. Autologous stem cell transplantation for myeloma: cytoreduction or an Immunotherapy? Front Immunol. 2021;12:651288. doi:10.3389/fimmu.2021.651288
90. Vuckovic S, Minnie SA, Smith D, et al. Bone marrow transplantation generates T cell-dependent control of myeloma in mice. J Clin Invest. 2019;129(1):106–121. doi:10.1172/JCI98888
91. Kourelis TV, Villasboas JC, Jessen E, et al. Mass cytometry dissects T cell heterogeneity in the immune tumor microenvironment of common dysproteinemias at diagnosis and after first line therapies. Blood Cancer J. 2019;9(9):72. doi:10.1038/s41408-019-0234-4
92. Chung DJ, Pronschinske KB, Shyer JA, et al. T-cell exhaustion in multiple myeloma relapse after autotransplant: optimal timing of immunotherapy. Cancer Immunol Res. 2016;4(1):61–71. doi:10.1158/2326-6066.CIR-15-0055
93. Lee L, Alrasheed N, Khandelwal G, et al. Increased immune-regulatory receptor expression on effector T cells as early indicators of relapse following autologous stem cell transplantation for multiple myeloma. Front Immunol. 2021;12:618610. doi:10.3389/fimmu.2021.618610
94. Parrondo RD, Ailawadhi S, Sher T, Chanan-Khan AA, Roy V. Autologous stem-cell transplantation for multiple myeloma in the era of novel therapies. JCO Oncol Pract. 2020;16(2):56–66. doi:10.1200/JOP.19.00335
95. Morandi F, Horenstein AL, Costa F, Giuliani N, Pistoia V, Malavasi F. CD38: a target for immunotherapeutic approaches in multiple myeloma. Front Immunol. 2018;9:2722. doi:10.3389/fimmu.2018.02722
96. Hashmi H, Husnain M, Khan A, Usmani SZ. CD38-directed therapies for management of multiple myeloma. Immunotargets Ther. 2021;10:201–211. doi:10.2147/ITT.S259122
97. Krejcik J, Casneuf T, Nijhof IS, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood. 2016;128(3):384–394. doi:10.1182/blood-2015-12-687749
98. Deckert J, Wetzel MC, Bartle LM, et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin Cancer Res. 2014;20(17):4574–4583. doi:10.1158/1078-0432.CCR-14-0695
99. Nakamura K, Casey M, Oey H, et al. Targeting an adenosine-mediated “don’t eat me signal” augments anti-lymphoma immunity by anti-CD20 monoclonal antibody. Leukemia. 2020;34(10):2708–2721. doi:10.1038/s41375-020-0811-3
100. Adams HC
101. Lucas F, Pennell M, Huang Y, et al. T cell transcriptional profiling and immunophenotyping uncover LAG3 as a potential significant target of immune modulation in multiple myeloma. Biol Blood Marrow Transplant. 2020;26(1):7–15. doi:10.1016/j.bbmt.2019.08.009
102. Wang J, Sanmamed MF, Datar I, et al. Fibrinogen-like Protein 1 is a major immune inhibitory ligand of LAG-3. Cell. 2019;176(1–2):334–347.e312. doi:10.1016/j.cell.2018.11.010
103. Yu B, Jiang T, Liu D. BCMA-targeted immunotherapy for multiple myeloma. J Hematol Oncol. 2020;13(1):125. doi:10.1186/s13045-020-00962-7
104. Munshi NC, Anderson LD
105. Samur MK, Fulciniti M, Aktas samur A, et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat Commun. 2021;12(1):868. doi:10.1038/s41467-021-21177-5
106. Brudno JN, Maric I, Hartman SD, et al. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. 2018;36(22):2267–2280. doi:10.1200/JCO.2018.77.8084
107. Ahn S, Leblay N, Neri P. Understanding the mechanisms of resistance to T cell-based immunotherapies to develop more favorable strategies in multiple myeloma. Hemasphere. 2021;5(6):e575. doi:10.1097/HS9.0000000000000575
108. Hosen N, Matsunaga Y, Hasegawa K, et al. The activated conformation of integrin β(7) is a novel multiple myeloma-specific target for CAR T cell therapy. Nat Med. 2017;23(12):1436–1443. doi:10.1038/nm.4431
109. Smith EL, Harrington K, Staehr M, et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci Transl Med. 2019;11(485):eaau7746. doi:10.1126/scitranslmed.aau7746
110. Lee L, Draper B, Chaplin N, et al. An APRIL-based chimeric antigen receptor for dual targeting of BCMA and TACI in multiple myeloma. Blood. 2018;131(7):746–758. doi:10.1182/blood-2017-05-781351
111. Yan Z, Cao J, Cheng H, et al. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: a single-arm, Phase 2 trial. Lancet Haematol. 2019;6(10):e521–e529. doi:10.1016/S2352-3026(19)30115-2
112. D’Agostino M, Raje N. Anti-BCMA CAR T-cell therapy in multiple myeloma: can we do better? Leukemia. 2020;34(1):21–34. doi:10.1038/s41375-019-0669-4
113. Liu E, Marin D, Banerjee P, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545–553. doi:10.1056/NEJMoa1910607
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.