")
Back to Journals » OncoTargets and Therapy » Volume 14
The Association Between Inflammation and Immunosuppression: Implications for ICI Biomarker Development
Authors Sacdalan DB , Lucero JA
Received 14 December 2020
Accepted for publication 25 February 2021
Published 18 March 2021 Volume 2021:14 Pages 2053—2064
DOI https://doi.org/10.2147/OTT.S278089
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Gaetano Romano
Danielle Benedict Sacdalan,1,2 Josephine Anne Lucero3
1Department of Pharmacology and Toxicology, University of the Philippines Manila College of Medicine, Manila, Philippines; 2Division of Medical Oncology, Department of Medicine, Philippine General Hospital and University of the Philippines Manila, Manila, Philippines; 3Division of Hematology, Department of Medicine, Philippine General Hospital and University of the Philippines Manila, Manila, Philippines
Correspondence: Danielle Benedict Sacdalan Email [email protected]
Abstract: Evasion of immune destruction is considered one of the hallmarks of cancer. Chronic inflammation can enable immune escape by suppressing immune surveillance and permitting the development of tumors and creating a tumor microenvironment that sustains cancer. This includes generating mechanisms that prevent the effectiveness of anti-tumor treatment including immune checkpoint inhibitor therapy. In this review, we explore the interplay of inflammation and immunosuppression, their effects on the tumor microenvironment, and their implications for immune checkpoint inhibitor therapy particularly in the context of predictive biomarkers for their use.
Keywords: inflammation, immunosuppression, tumorigenesis, immune checkpoint inhibitor, biomarkers
Introduction
Disease may result from the failure of the body to detect noxious entities but also, from its inability to distinguish what is “self” from what is “non self”. In the case of cancer, the former is operative as developing tumors grow unchecked by the immune system to the detriment of the host. Evasion of immune destruction is considered one of the hallmarks of cancer.1
Tumors are immunogenic to varying degrees. Moreover, certain antigens are expressed almost exclusively by tumors and these cancer antigens are distinct from the host’s normal milieu.1,2 Immune surveillance describes the interplay between the host’s immune system and its response to the developing cancer. Rather than seeing this as a process that results in a dichotomous result – success or failure, tumor destruction or tumor growth – it is better to see that one of three outcomes are to be expected with the interaction of immune system and tumor: elimination, equilibrium, or escape, otherwise known as immunoediting. Host and tumor factors determine which of these come to fruition. Highly immunogenic tumors are readily dispatched by a competent immune system resulting in elimination of a nascent cancer. As a corollary to this, tumors developing in immunosuppressed hosts tend to be highly immunogenic due to failure of immunoediting.1,3 Less immunogenic tumors or immunogenic tumors pitted against a less robust immune system may only be partially eliminated resulting in an uneasy equilibrium manifesting as tumor dormancy. If this balance is maintained throughout a lifetime, the result is akin to elimination; however, if the immune system is somehow weakened, or the tumor acquires changes that allow it to circumvent the immune system, then immune escape occurs and cancer takes hold.2,3
Immunotherapy inhibits tumor cell growth by stoking the immune response against tumor cells, which stands in contrast to cytotoxic chemotherapy and driver gene targeted therapies that directly affect malignant cells themselves.4 Much progress has been made over the last century since William Coley first used a cocktail of live and inactivated bacteria in the hope of inducing an immune response against bone sarcomas.5 Greater understanding of the molecular biology and immunology of cancer has allowed the development of therapies that induce more reliable and effective immune reactions to tumors.
At the forefront of immuno-oncology are the immune checkpoint inhibitors. These monoclonal antibodies target programmed cell death protein ligand 1 (PD-L1), programmed cell death protein (PD-1), and cytotoxic T-lymphocyte antigen 4 (CTLA-4). PD-L1 ligand is found on tumor cells, while PD-1 receptors are selectively expressed on CD4+ and CD8+ T cells, natural killer (NK) T cells, B cells, monocytes, and dendritic cells. PD-1 is a member of the co-stimulatory receptor family CD28:7. Meanwhile, CTLA-4 is an inhibitor of T-cell function that interacts with its ligands CD80 and CD86. Tumors subvert these regulatory ligand–receptor interactions to facilitate immune escape. Blockade of these immune checkpoints reverses their negative regulatory effects and results in immune reactivation.6
This review will cover the interplay of inflammation, immunosuppression, and tumorigenesis. It will also discuss the implications of these on the development and performance of predictive biomarkers of immune checkpoint inhibitor therapy.
Inflammation and Cancer
Inflammation is distinguished temporally as being either acute or chronic. In the former, physical, or chemical injury or exposure to an infectious agent is the antecedent event. Acute inflammation is intended to be a beneficial process that eliminates pathogens and necrotic cells. It subsequently initiates the healing process at the site of tissue injury. This inflammatory process is self-limiting and resolves after completion of tissue repair or elimination of pathogens.7,8 In the event that the body cannot abrogate the acute inflammatory response, the result is chronic inflammation. This persistent inflammation can serve as a trigger for the development of tumors.7 Epidemiological studies have demonstrated the association between solid tumors and inflammation.9 A quarter of all cancers are estimated to be attributable to chronic inflammation.10
Chronic inflammation can arise from intrinsic host factors as with the case of inflammatory bowel disease. In turn, extrinsic factors include viral infections such as with Hepatitis or EBV, certain parasitic infection, and environmental toxins. Tumor formation occurs through induction of free radicals such as reactive oxygen (ROS) and nitrogen species (RNS). ROS and RNS produced by inflammatory cells are associated with mutations in tumor suppressor and DNA repair genes that result in genomic instability. In addition, epigenetic events such as changes in DNA methylation as in the case of smoking may also alter gene expression resulting in tumorigenesis.8 Long-standing inflammation is generally thought to result in tumor initiation, progression, and metastasis by providing a tumor-promoting microenvironment.8,11
An important driver of this change is the generation of pro-inflammatory cytokines that in turn recruit immune cells characteristic of the chronic inflammatory state. These immune cells depending on their functional phenotype may prevent or enable the subsequent development of tumors. Macrophages are of the monocytic lineage and are attributed to persistent inflammation. These phagocytes functionally exist within a spectrum. On one end, there is the classically activated M1 (CAM) endotype while on the other is the alternatively activated M2 (AAM) endotype.12,13 Neutrophils, another component of the innate immune system, functionally mirrors the polarized macrophages with N1 and N2 analogues.14 The M1 and N1 phenotypes have an anti-tumor function while the M2 and N2 phenotypes have the opposite.12–14 Myeloid-derived suppressor cells (MDSCs) are important inhibitory cells in the tumor microenvironment that play a role in suppressing tumor-directed immune response. Monocytic MDSC (M-MDSC) are akin to monocytes, while polymorphonuclear MDSC (PMN-MDSC) are morphologically and phenotypically similar to neutrophils.15 Dendritic cells are a central component of the microenvironment which plays a crucial role in immune activation and the induction of tolerance.11,16
Cytokines play a vital role in the control of inflammation and the immune system. These signaling molecules are synthesized in response to changes in the environment and take part in a myriad of cellular functions. Cytokines are usually classified as either being pro- and anti-inflammatory.7,11 Proinflammatory cytokines implicated in carcinogenesis include IL-1, IL-6, IL-15, TNF-a, colony stimulating factors (CSF), and the macrophage migration inhibitory factor (MIF). Biological processes ascribed to MIF function include the regulation of the p53 tumor suppressor gene, angiogenesis, cell cycle, and senescence. Due to its role in these events, it is a strong candidate linking inflammation and cancer. Specifically, MIF initiates the phosphorylation of ERK1/ERK2 pathways leading to the increase in COX2 and NOS2. MIF also facilitates the inhibition of apoptosis, both p53-dependent and independent, and growth arrest and the activation of NF-ΚB. Another cytokine, IL-8, can have angiogenic activity in some cancers, for example non-small cell lung cancer (NSCLC), and can function as a positive autocrine growth factor.10,17
Several molecular signaling pathways have been identified in inflammation-mediated cancer and immune activation. Epithelial to mesenchymal transition (EMT), which is vital in the metastatic process is governed by various cytokines such as transforming growth factor-β (TGF-β), IL-1β, and TNF-α, which consequently stimulate NF-κB as well as the signal transduction and activation of transcription (STAT3) inflammatory cascades. NF-κB activation is central to both inflammation and oncogenesis, this transcription factor influences the expression of various genes related to malignancy and inflammation.7,10 In the setting of continuous inflammation, NF-κB is activated and results in the transcription of target inflammatory genes such as the tumor necrosis factor α, which is a pro-inflammatory cytokine with pleiotropic functions.17 In addition, activation of NF-κB can enhance Wnt signaling leading to the dedifferentiation of non-stem tumor epithelial cells into tumor-initiating cells in mouse intestine.8,18 Mitogen-activated protein kinase (MAPK) activation influences cell growth, cell differentiation, and cell survival. It also plays a role in managing immune and stress responses. Three MAPK-mediated pathways have been well studied – ERK1/2, P38 MAPK, and JNK – and these govern cell proliferation, apoptosis, and tumorigenesis.7 Other signaling pathways associated with inflammation-mediated cancer include: JAK/STAT, Phosphoinositide-3-Kinase (PI3K) pathway, and the CREB signaling pathway.7,19
IFN-γ-secreting effector T cells play a central role in tumor immune surveillance. Tumors escape immune surveillance and maintain an immunosuppressive TME via the earlier described mechanisms specifically the recruitment of regulatory cells that include M2, N2, and MDSCs. Regulatory T cells (Treg) are also associated with suppression of immune surveillance and the maintenance of the exhausted T cell phenotype.20,21 Tumors likewise produce suppressor molecules against anti-tumor T cell responses such as IL-10 and TGF-β. Tumor growth is also associated with immunomodulation of T cell response through enhancement of co-inhibitory molecules or immune checkpoints, such as cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) or programmed cell death-1 (PD-1), on T cells.22 Following successful checkpoint blockade, there is restoration of the ability of CD8+ T cells within the tumor to generate IL-2 and to proliferate when analyzed ex vivo. Current evidence shows that clinical response with active ICI therapy is mediated through restored function of pre-existing TIL more than recruitment of new effector CD8+ T cells into the TME.23
Chronic Inflammation and Immunosuppression
Early studies done on melanoma show that T cells harvested from tumors exhibit tumor-specificity, but have functional deficiency. This dysfunction of tumor antigen-specific T cells has been described prior in in vitro studies.24 These results suggest that tumor progression in the face of specific adaptive immunity likely arises from immune suppressive mechanisms acting at the level of the tumor microenvironment (TME).23 The TME is dominated by the tumor but its survival is dependent on an interplay of other cellular players including marrow-derived immune cells, lymphocytes, blood vessels, fibroblasts, and extracellular matrix.20,21,25 The composition of the TME places the tumor into one of three possible immune-phenotypes: immune inflamed, immune-excluded, or immune desert. These latter two can both be considered non-inflamed tumors (Table 1).5,23,26
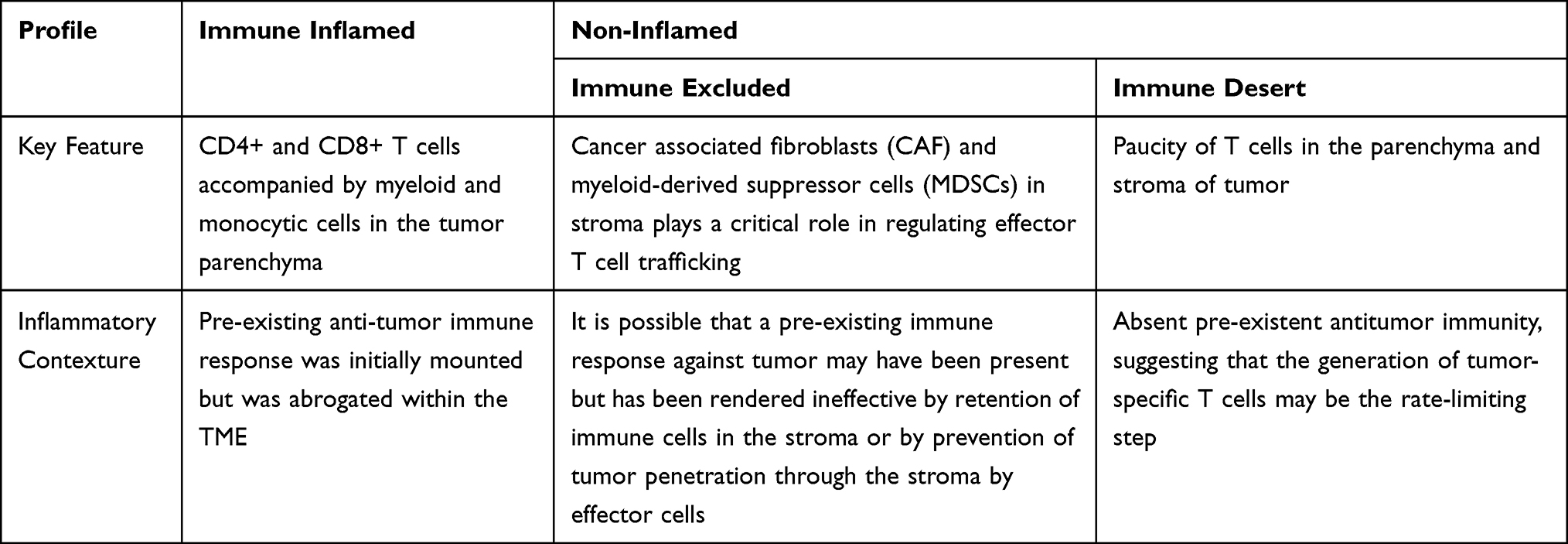 |
Table 1 Summary of TME Phenotypes |
In immune inflamed tumors, immune cells are found in proximity to tumor cells.5 These are characterized by the presence of CD4+ and CD8+ T cells accompanied by myeloid and monocytic cells in the tumor parenchyma. The T cell-inflamed subset of tumors are characterized by expression of T cell markers and chemokines that mediate recruitment of effector lymphocytes into the TME. These include CCL-2, CCL-3, CCL-4, and CCL-5. In vitro studies have demonstrated that expression of these chemokines can recruit CD8+ effector T cells to the TME.23 The CXCR3-CXCL9/CXCL10 axis is a requirement for CD8+ T-cell trafficking into the TME. Thus, CXCL9 and CXCL10 in tumor tissues are associated with elevated intratumoral T-cell infiltration.27 Fragments taken from these tumors may stain positive for PD-L1 on infiltrating immune cells or even in tumor cells themselves. It is possible that in this phenotype, a pre-existing anti-tumor immune response was initially mounted but was abrogated within the TME. Reversal of immune suppression and consequently clinical response results from anti-PD-L1/PD-1 therapy in patients with inflamed tumors. However, this may not be the case in every instance.20,26
In immune-excluded tumors, the stroma plays a critical role in regulating effector T cell trafficking into the tumor. Specifically, cancer-associated fibroblasts (CAFs) act through extracellular matrix (ECM)–mediated T cell trapping and CXCL12-regulated T cell exclusion. Another important component of the TME, myeloid-derived suppressor cells (MDSCs), also play a role in restricting accumulation of T cells into the vicinity of the cancer cells.28 It is possible that a pre-existing immune response against tumor may have been present but has been rendered ineffective by retention of immune cells in the stroma or by prevention of tumor penetration through the stroma by effector cells. T cell migration through the tumor stroma functions as the rate-limiting step in the operative immune response in this phenotype. After treatment with anti-PD-L1/PD-1 agents, stroma-associated T cells can show evidence of activation and proliferation but not infiltration, and clinical responses are uncommon.20,26
In the parenchyma and stroma of immune-desert tumors there exists a paucity of T cells.20 Although cells of the myeloid lineage may be present, the predominant feature of this profile is the presence of a non-inflamed TME with little to no CD8+ T cells. It comes as little surprise that these tumors rarely respond to anti-PD-L1/PD-1 therapy.5,26 In contrast to the previous two phenotypes, “immune-deserts” reflect an absent pre-existent antitumor immunity, which suggests that the generation of tumor-specific T cells may be the rate-limiting step.26
Challenges in Immune Checkpoint Inhibition
As promising as immunotherapy can be, not all patients exhibit a dramatic response. Frequency of rapid tumor shrinkage from single-agent anti-PD-L1/PD-1 or CTLA4 antibodies is <40% depending on the individual’s indication.4,26 In addition to this, the immunologic and financial toxicity associated with ICI therapy warrants the identification of biomarkers that will reliably predict which patients will benefit from this type of therapy.5,6,29 Predictive biomarkers (Table 2 and Figure 1) are measurable biological quantities or identifiable phenotypes that presage response to a specific therapy.20
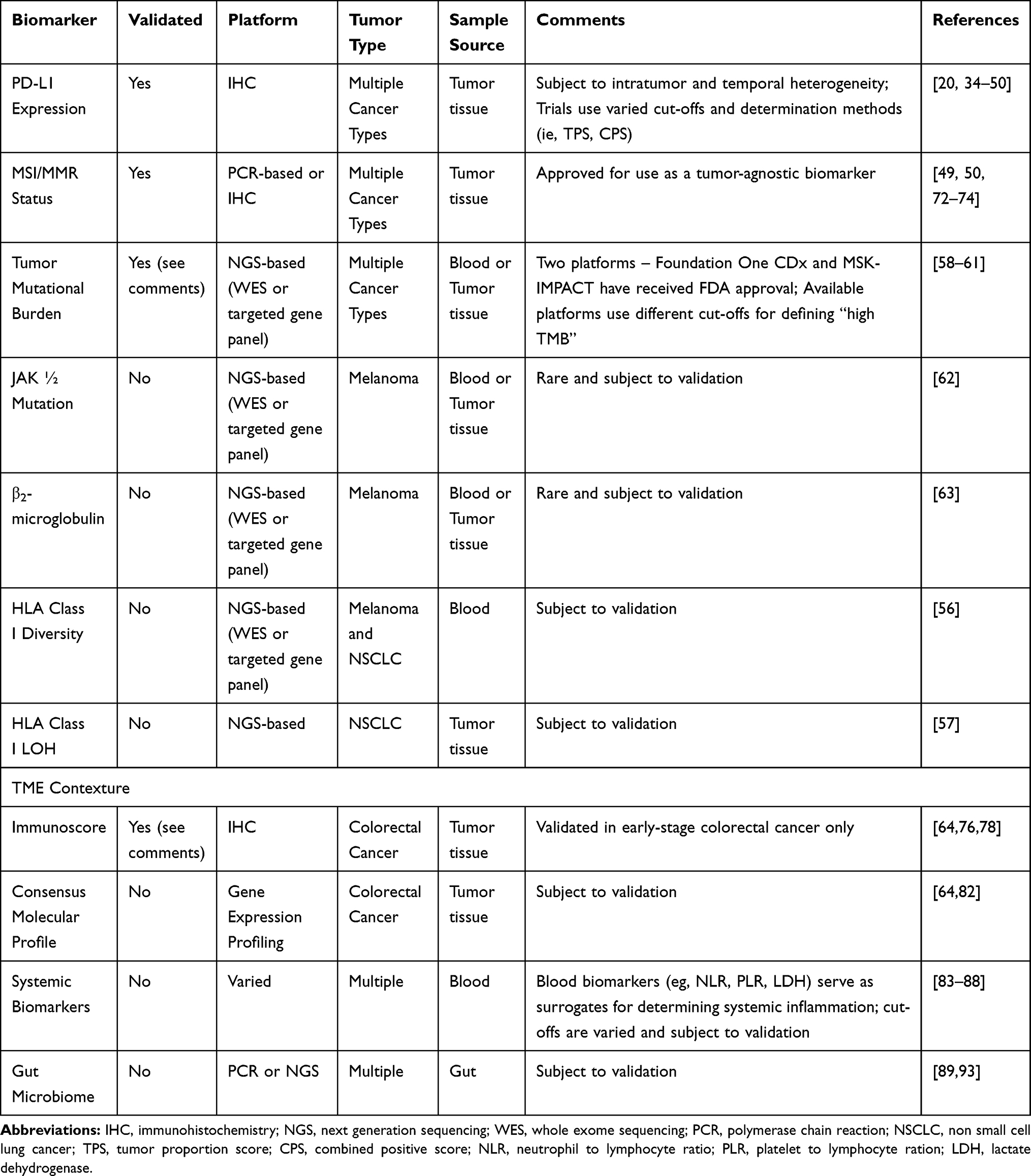 |
Table 2 Summary of Included Predictive Biomarkers in ICI Therapy |
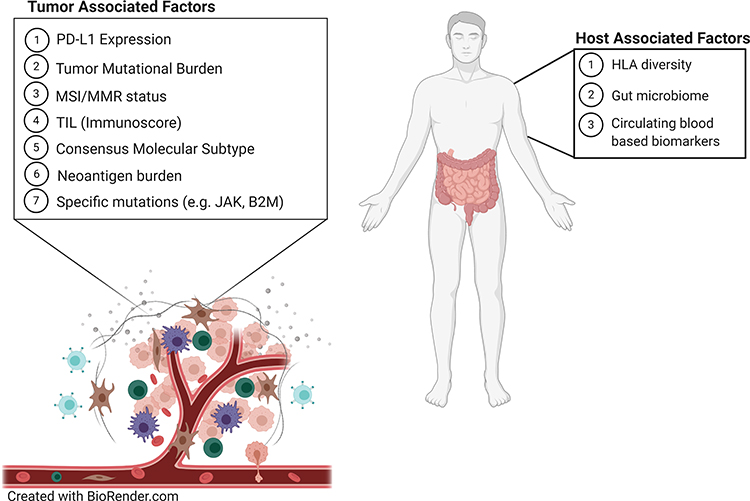 |
Figure 1 Predictive biomarkers that affect the tumor microenvironment and immune checkpoint inhibitor therapy |
As discussed earlier, tumor-induced immunosuppression operates in two main ways. The first involves expression of immunosuppressive molecules or their receptors, including programmed death-ligand 1/programmed death-1 (PD-L1/PD-1), galectin-9/TIM-3, IDO1, LAG-3, and CTLA4 – immune checkpoints that inhibit the activation of effector T cells, ultimately leading to immune escape by the tumor. The second occurs by inducing immunosuppressive cells to accumulate around the tumor and secrete immunosuppressive factors, which inactivate cytolytic T lymphocytes (CTL).30,31
PD-1 receptor downregulates T cells, reducing the activity of T lymphocytes when binding to PD-L1 ligand. PD-L1 is a receptor found on the cell membranes of malignant cells. Expression of PD-L1 can favor tumor cell survival by activating the PI3K/AKT pathway, facilitating the Warburg effect to favor metabolism via anaerobic glycolysis.32 In addition, binding of PD-1 to its ligand will inhibit kinase signaling pathways, leading to inactivation of the T cell. This mechanism of inhibiting T lymphocytes allows tumor cells to escape the immune system and thus survive and develop further.30,33 In relation to this, PD-L1 expression serves as a predictive biomarker for response to anti-PD-L1 or anti-PD1 therapy. For this purpose, immunohistochemistry assays for PD-L1 protein expression have been developed for clinical use as companion diagnostics.20,34,35 Currently, the FDA has approved PD-L1 IHC as a companion diagnostic for anti- PD1 therapy for several solid tumors including: NSCLC, Gastric Cancer and GE Junction Cancer, Head and Neck SCC, Cervical Cancer, and Urothelial Carcinoma. Data in support of the use of PD-L1 as a predictive biomarker in these indications have come from Phase II and III trials.36–50 Interestingly, while the earlier mentioned trials have found a positive association between PD-L1 expression levels and response to PD1-directed ICI therapy, other studies have failed to do so. Even when PD-L1 expression is correlated with response, patients with low to no detectable PD-L1 expression have been seen to experience durable clinical benefit.51–53 On balance therefore, PD-L1 expression remains a useful yet imperfect predictor of ICI response. The absence of a standardized criteria and cut-offs for assessing positivity across trials is an issue across trials. Other potential reasons behind the incongruent results of PD-L1 biomarker studies include the use of different detection assays, heterogeneity in the expression of PD-L1 by tumors, and variations in the temporal expression of this ligand.20 A recent meta-analysis investigated the utility of other checkpoint proteins, specifically IDO1 and LAG3 as prognostic biomarkers in NSCLC. This study showed a potential use for these proteins, but their value is based on limited evidence.54
Nucleated cells express major histocompatibility complex (MHC) proteins on their cell surface. These are encoded by the human leukocyte antigen (HLA) gene complex. HLA genes are the most polymorphic genes in the human genome and encode key components of immunogenicity as these are the lynchpin of antigen presentation and therefore of the adaptive immune system.20 Specifically, HLA-I diversity is characterized by significant sequence variation in peptide-binding regions.20,55 The presence of a more diverse array of HLA-I molecules was associated with increased survival based on an analysis of 1535 patients with ICI-treated tumors.56 Conversely, HLA-I LOH is an immune escape mechanism under strong selection pressures that can result in resistance to immunotherapy.57
Another biomarker is tumor mutational burden (TMB), which is a measure of the number of mutations in a cancer. The number of mutations correlates with the number neo-antigens present in a tumor which can be immunogenic and thereupon trigger a T cell response against it.58 Traditional TMB detection is performed by whole-exome sequencing (WES) as the gold standard. This method however can be costly and cumbersome and therefore next generating sequencing (NGS) targeted gene platforms are increasingly being favored. In relation to this, the FDA has approved two such platforms – Foundation One CDx and MSK-IMPACT – for use as companion diagnostics to anti-PD-1 ICIs.58–60 A direct relationship between ICI responsiveness and TMB has been described.58–61 Despite this observation challenges remain with the use of TMB as a biomarker predictive biomarker for response with ICI therapy. First, there is currently no consensus in the definition of TMB cutoffs for patient stratification.55,58 Secondly, specific genomic alterations influence tumor response to ICI regardless of TMB status. For example, PD-L1 amplification results in response to ICI regardless of TMB levels. Loss-of-function mutations in JAK1/2 which play a role in chronic inflammation result in decreased sensitivity to IFN-γ have been seen to result in acquired resistance to immunotherapy first in mouse models and subsequently with melanoma. Alterations in β2-microglobulin have likewise been found to correlate with poor outcomes in melanoma.7,20,58,62,63 Third, TMB has been widely associated with solid tumors, but it is critical to keep in mind that the total number of mutations per coding area of genome is substantially different among tumor types (as well as among individuals with the same types of tumors).58 Fourth, at least some assumptions on the utility of TMB are based on results from retrospective studies which need to be validated in prospective studies.58 All these taken together highlight limitations of TMB as a predictive biomarker, particularly when used in isolation.
Mechanistically, MSI-H/dMMR tumors are a specific type of high TMB tumor that generates a high mutation associated neo-antigen (MANA) load due to defective genomic repair machinery.64 DNA mismatch repair (MMR) mechanisms maintain genomic integrity. The main function of MMR proteins is the correction of single base nucleotide mismatches that arise during DNA replication and recombination. Mutations arising from these errors include indels that cause frameshifts that produce neoantigens that are more immunogenic because of their greater sequence divergence from self-peptides. It is therefore not surprising that MSI-H/dMMR status is associated with improved ICI response due to its increased TMB.20,58,65,66 Determination of microsatellite/mismatch repair status can be made using sequencing or IHC-based methods.67,68 Studies on colorectal cancer have demonstrated that T cell infiltration into the tumor bed is associated with positive outcomes. Approximately 15% of all CRCs are MSI-H/dMMR. MSI-H/dMMR disease is prognostic with stage II MSI-H/dMMR tumors carrying a lower risk of recurrence than stage II MSI-L/pMMR tumors with a 35% reduction in the risk of death associated with MSI-H status on pooled analysis.69 Conversely, in the case of stage 4 MSI-H/dMMR disease (~3% of all mCRCs), the opposite is the case. Patients with MSI-H/dMMR tumors that metastasize have a dismal prognosis. Notably, expression of the immune checkpoint proteins PD-L1/PD-1 and CTLA4 are upregulated in these tumors.70 These observations suggest that MSI-H/dMMR status of CRCs might respond well to immune checkpoint blockade.71 Phase II trial data have shown the benefit of PD-1-directed ICI therapy in the treatment of MSI-H/dMMR tumors regardless of primary site leading to FDA approval of Pembrolizumab and Nivolumab for this tumor agnostic indication.49,50,72–74 Notably, approximately 50% of all CRC exhibit dysfunction of the MMR protein MSH3 in the context of inflammation and release of IL-6 from infiltrating immune cells. Isolated MSH3 dysfunction allows cancer cells to accumulate non-mononucleotide microsatellite frameshift mutations that are likely not very immunogenic, as well as DNA double strand breaks. The latter being a result of the MSH3s contribution to homologous recombination repair. The effect of the loss of MSH3 function in this setting on the effectiveness of immune checkpoint blockade has not been tested to date.75
For over a century, immune infiltration of cancers has been thought to be a positive prognostic factor for patient outcomes; but these insights have not directly informed clinical decision making. With the emergence of newer imaging, sequencing, and computational tools it has become possible to demonstrate the prognostic importance of the immune contexture more clearly.76 Gene expression profiling (GEP) and IHC staining have been used to characterize the immune infiltrate in colorectal cancer and to subsequently correlate this with patient outcomes.77 Refinement of this technique has allowed the development of an Immunoscore that accounts for the density and location of CD3+ and CD8+ T cells within the tumor. This scoring system is based on the numeration of lymphocyte populations at the core and invasive margins of tumors. What results is a total ranging from an Immunoscore 0 (I0) when low densities of T cells are found in both regions to an Immunoscore 4 (I4) when high densities are found. This has recently been validated in a cohort of 2681 early-stage colon cancer patients from 14 centers across 13 countries. Interestingly, the authors showed that patients with a high Immunoscore had similar tumor relapse and survival independent of their MSI-H/dMMR status. Specifically, higher Immunoscores correlated with better clinical outcomes in patients with stage I–III CRC.64,78 Therefore, it is possible to conclude that the favorable prognosis observed in MSI-H/dMMR localized CRC is related to high immune infiltration, which gives credence to the notion that TILs are important in disease control by preventing local and distant tumor spread.64,76 Interestingly, the heterogeneity of infiltrating T cell density appears to be greater in MSI-H/dMMR compared to MSI-L/pMMR tumors.79,80 A small cohort study of MSI-H/dMMR colon cancer showed that higher CD3+ and CD8+ T cell densities on cancer cells are associated with a higher ORR and duration of disease control following treatment with Pembrolizumab.80 Additional validation of immunophenotyping as a predictor of response to immunotherapy in metastatic disease is necessary and prospective trials for this purpose such as NCT03608046 are underway.81
The consensus molecular subtype (CMS) classification is potentially a new biomarker of response to immune therapies that has likewise been developed for CRC. This system classifies tumors into one of four subtypes based on GEP. CMS1 and CMS4 are immune-reactive, so-called “hot” tumors which are and highly invested with immune cells in contrast to CMS2 and CMS3, which are “cold” tumors.64,82 It needs mentioning that care is to be taken when selecting biopsies and resection specimens for CMS classification because this system has been developed from samples of primary non-metastatic CRC and it is not totally reproducible on metastatic samples or validated in the metastatic setting. Moreover, spatial- and temporal-tumor heterogeneity, seen widely in the metastatic setting, can muddy CMS status. Therefore, the source of the sample as well as prior treatments before collection have to be taken into account when applying the CMS classification.64
Blood- and serum-derived biomarkers allow assessment of potential ICI response in a minimally invasive manner. These biomarkers also serve as surrogates that estimate underlying systemic inflammation, which in turn has been associated with more dismal outcomes in treatment, including ICI therapy.83–87 Peripheral blood neutrophil to lymphocyte ratio (NLR) values greater than five were associated with decreased PFS and OS in multiple studies of anti-CTLA4 and anti-PD1 across a wide range of cancer types.20,83,85,88 Several other features of peripheral blood composition have been associated with ICI response such as platelet to lymphocyte ratio (PLR), total lymphocyte count (TLC), monocyte count, relative eosinophil count, T cell clonality, circulating Treg cell levels, cytokine levels (for example, IL-6, IL-8 and IL-10), circulating monocytes or MDSCs, and lactate dehydrogenase (LDH) activity. The key challenge with these blood-based biomarkers is the need to standardize cut-off values and for validation in large prospective studies.20,84,85
The microbiome is emerging as an important regulator of immune checkpoint inhibitor efficacy and toxicity. It has also been shown to promote carcinogenesis in gastrointestinal, colon, liver, and gallbladder cancers.5 The mammalian immune system uses pattern recognition receptors (PRRs), which include Toll-like receptors and NOD-like receptors to recognize microbes. They then activate specific pathways that activate inflammatory stimuli causing further tumor proliferation or resistance to cell death.89 Tumors themselves harbor microbiomes that have been shown to be like that of normal adjacent tissue. Furthermore, as tumors develop, circulating bacteria may colonize the TME allowing it to interact with tumor cells, consequently affecting tumor immunity, and tumor response to therapy.90 It has been postulated that these tumor microbiomes are the result of chronic inflammation, vascular permeability, and the immunosuppressive status of the tumor.91 Multiple studies have shown that the composition of the gut microbiome affects immune function and the efficacy of immune checkpoint inhibitor therapy. Increased intake of dietary fiber, fermented foods, and plant protein have been shown to maintain a healthy gut microbiome.20 Conversely, drugs such as antibiotics and proton pump inhibitors may lead to a less healthy gut microbiome and consideration has to be given about the judicious use of these.92,93 Maintaining a diverse gut microbiota results in diminished inflammatory biomarkers that contributes to better responses to immunotherapy.93 Apart from response to therapy, a healthy host microbiome has also been shown to be protective against drug-induced adverse effects, particularly for CTLA-4 blockade-induced colitis. Table 3 summarizes bacteria that have been found to be beneficial as studied in humans. However, it is more likely that those that respond well to immunotherapy have a combination of various microbiota and not just the presence or absence of a specific species.5,93
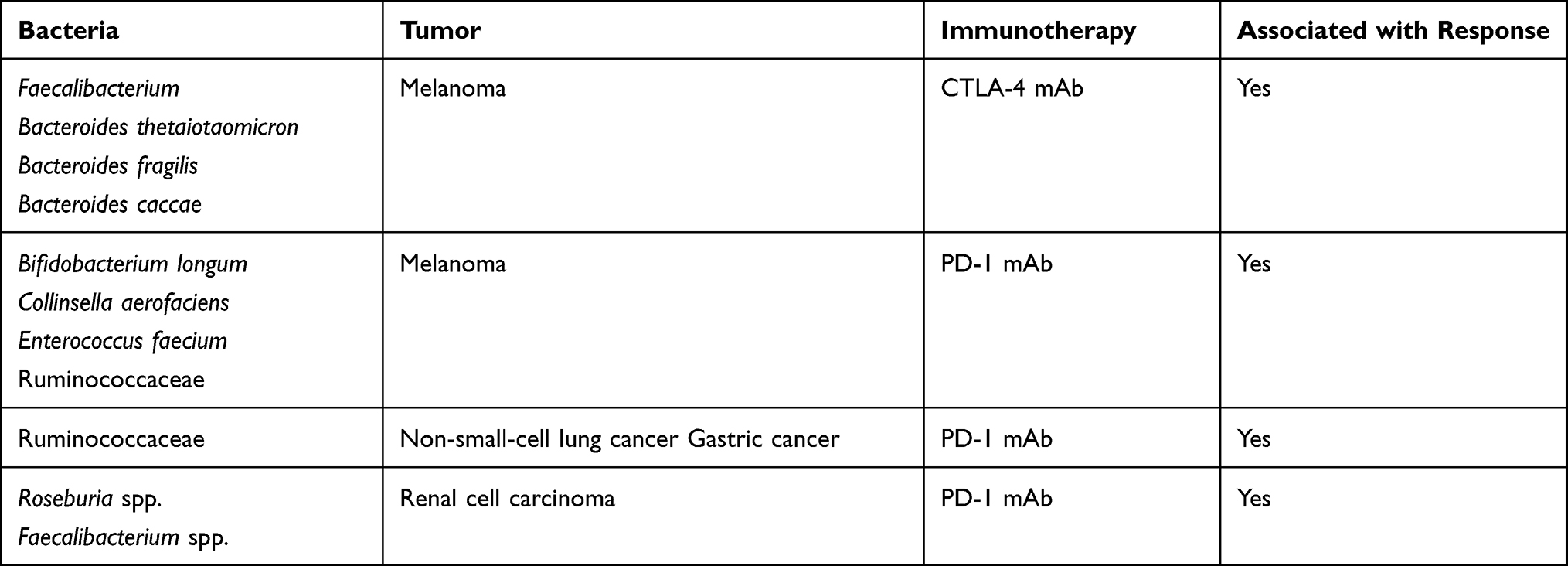 |
Table 3 Association Between Specific Bacteria as Predictive Biomarker for ICI Response |
Single biomarkers are inadequate to capture the immune status of a patient. Multiple studies have shown that a combination of multiple factors yields a higher positive predictive value for overall response rate, improved overall survival, and longer progression-free survival. Models that include tumor mutational burden, PD-L1 expression, tumor infiltrating lymphocytes, and T cell-inflamed gene expression profiling in various combinations.55 Predictive models are currently being styled using computational methods to analyze both the number and type of tumor antigens to determine survival.94,95 More studies are needed to investigate applications to various cancer types.55
Conclusion
Chronic inflammation and immunosuppression can drive development of cancer. The interplay of these with tumor and host factors can be used to predict response to immune checkpoint blockade. While some of these biomarkers have received regulatory approval as companion diagnostics, others remain in development. Given the disparate strengths and limitations of each biomarker, on balance it is likely that comprehensive tools that incorporate several biomarkers may more accurately predict treatment response to immune checkpoint inhibitor therapy.
Ethics Statement
This review does not involve human participants, identifiable human tissue, biological samples, or data. It is exempted from ethical review based on the National Ethical Guidelines for Health and Health-Related Research of the Republic of the Philippines published in 2017.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674.
2. Finn OJ. Immuno-oncology: understanding the function and dysfunction of the immune system in cancer. Ann Oncol. 2012;23(SUPPL.8):8–11. doi:10.1093/annonc/mds256
3. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271. doi:10.1146/annurev-immunol-031210-101324
4. Duffy MJ, Crown J. Biomarkers for predicting response to immunotherapy with immune checkpoint inhibitors in cancer patients. Clin Chem. 2019;65(10):1228–1238. doi:10.1373/clinchem.2019.303644
5. Esfahani K, Roudaia L, Buhlaiga N, Del Rincon SV, Papneja N, Miller WH. A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol. 2020;27(S2):87–97. doi:10.3747/co.27.5223
6. Arora S, Velichinskii R, Lesh RW, et al. Existing and emerging biomarkers for immune checkpoint immunotherapy in solid tumors. Adv Ther. 2019;36(10):2638–2678. doi:10.1007/s12325-019-01051-z
7. Qu X, Tang Y, Hua S. Immunological approaches towards cancer and inflammation: a cross talk. Front Immunol. 2018;9. doi:10.3389/fimmu.2018.00563
8. Wang D, Du Bois RN. Immunosuppression associated with chronic inflammation in the tumor microenvironment. Carcinogenesis. 2015;36(10):1085–1093. doi:10.1093/carcin/bgv123
9. Michels N, van Aart C, Morisse J, Huybrechts I. Chronic inflammation toward cancer incidence: an epidemiological systematic review. J Glob Oncol. 2018;4(Supplement 2):24s–24s. doi:10.1200/jgo.18.56200
10. Perwez Hussain S, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2007;121(11):2373–2380. doi:10.1002/ijc.23173
11. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. doi:10.1016/j.cell.2010.01.025
12. Poh AR, Ernst M. Targeting macrophages in cancer: from bench to bedside. Front Oncol. 2018;8:1–16. doi:10.3389/fonc.2018.00049
13. Wang J, Li D, Cang H, Guo B. Crosstalk between cancer and immune cells: role of tumor-associated macrophages in the tumor microenvironment. Cancer Med. 2019;8(10):4709–4721. doi:10.1002/cam4.2327
14. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer. 2020;22(Box1)
15. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37(3):208–220.
16. Zhu S, Yang N, Wu J, et al. Tumor microenvironment-related dendritic cell deficiency: a target to enhance tumor immunotherapy. Pharmacol Res. 2020;159:519. doi:10.1016/j.phrs.2020.104980
17. Kanterman J, Sade-Feldman M, Baniyash M. New insights into chronic inflammation-induced immunosuppression. Semin Cancer Biol. 2012;22(4):307–318. doi:10.1016/j.semcancer.2012.02.008
18. Schwitalla S, Fingerle AA, Cammareri P, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152(1–2):25–38. doi:10.1016/j.cell.2012.12.012
19. Qian S, Golubnitschaja O, Zhan X. Chronic inflammation: key player and biomarker-set to predict and prevent cancer development and progression based on individualized patient profiles. EPMA J. 2019;10(4):365–381. doi:10.1007/s13167-019-00194-x
20. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19(3):133–150. doi:10.1038/s41568-019-0116-x
21. Shen M, Kang Y. Complex interplay between tumor microenvironment and cancer therapy. Front Med. 2018;12(4):426–439.
22. Santarpia M, Karachaliou N. Tumor immune microenvironment characterization and response to anti-PD-1 therapy. Cancer Biol Med. 2015;12(2):74–78. doi:10.7497/j.issn.2095-3941.2015.0022
23. Gajewski TF, Corrales L, Williams J, Horton B, Sivan A, Spranger S. Cancer immunotherapy targets based on understanding the t cell-inflamed versus non-t cell-inflamed tumor microenvironment. Adv Exp Med Biol. 2017;1036:19–31.
24. Zippelius A, Batard P, Rubio-Godoy V, et al. Effector function of human tumor-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res. 2004;64(8):2865–2873.
25. Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell. 2017;31(3):326–341.
26. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–330. doi:10.1038/nature21349
27. Mikucki ME, Fisher DT, Matsuzaki J, et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun. 2015;6. doi:10.1038/ncomms8458
28. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348(6230):74-80.
29. Lapuente-Santana O, Eduati F. Toward systems biomarkers of response to immune checkpoint blockers. Front Oncol. 2020;10:1–9. doi:10.3389/fonc.2020.00001
30. Jiang X, Wang J, Deng X, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. 2019;18(1):1–17. doi:10.1186/s12943-018-0930-x
31. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi:10.1146/annurev.immunol.26.021607.090331
32. Guo Q, Huang F, Goncalves C, Del Rincón SV, Miller WH Translation of cancer immunotherapy from the bench to the bedside. 143 Adv Cancer ResElsevier Inc 2019 1–62 p
33. Vajaitu C, Draghici CC, Solomon I, et al. The central role of inflammation associated with checkpoint inhibitor treatments. J Immunol Res. 2018;2018:2018. doi:10.1155/2018/4625472
34. PDL1 F 2. pd-l1-ihc-22c3-pharmdx-p150013s014. Available from: https://www.fda.gov/medical-devices/recently-approved-devices/pd-l1-ihc-22c3-pharmdx-p150013s014.
35. FDA2. pd-l1-ihc-28-8-pharmdx-p150025s013. Available from: https://www.fda.gov/medical-devices/recently-approved-devices/pd-l1-ihc-28-8-pharmdx-p150025s013.
36. Checkmate 275. NCT02387996. Available from: https://clinicaltrials.gov/ct2/show/NCT02387996.
37. Keynote059. NCT02335411. Available from: https://clinicaltrials.gov/ct2/show/NCT02335411.
38. Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. doi:10.1056/NEJMoa1507643
39. Hellmann MD, Paz-Ares L, Bernabe Caro R, et al. Nivolumab plus ipilimumab in advanced non–small-cell lung cancer. N Engl J Med. 2019;381(21):2020–2031. doi:10.1056/NEJMoa1910231
40. Ferris RL, Blumenschein G, Fayette J, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375(19):1856–1867. doi:10.1056/NEJMoa1602252
41. Sharma P, Retz M, Siefker-Radtke A, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, Phase 2 trial. Lancet Oncol. 2017;18(3):312–322. doi:10.1016/S1470-2045(17)30065-7
42. Schmid P, Adams S, Rugo HS, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379(22):2108–2121. doi:10.1056/NEJMoa1809615
43. Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N Engl J Med. 2016;375(19):1823–1833. doi:10.1056/NEJMoa1606774
44. Mok TSK, Wu YL, Kudaba I, et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, Phase 3 trial. Lancet. 2019;393(10183):1819–1830. doi:10.1016/S0140-6736(18)32409-7
45. Escudier B, Sharma P, McDermott DF, et al. CheckMate 025 randomized phase 3 study: outcomes by key baseline factors and prior therapy for nivolumab versus everolimus in advanced renal cell carcinoma. Eur Urol. 2017;72:962–971. doi:10.1016/j.eururo.2017.02.010
46. Fuchs CS, Doi T, Jang RW, et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 clinical KEYNOTE-059 trial. JAMA Oncol. 2018;4(5):1–8.
47. Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387(10027):1540–1550. doi:10.1016/S0140-6736(15)01281-7
48. Vuky J, Balar AV, Castellano D, et al. Long-term outcomes in KEYNOTE-052: phase II study investigating first-line pembrolizumab in cisplatin-ineligible patients with locally advanced or metastatic urothelial cancer. J Clin Oncol. 2020;38(23):2658–2666. doi:10.1200/JCO.19.01213
49. Le DT, Kim TW, van Cutsem E, et al. Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability–high/mismatch repair–deficient metastatic colorectal cancer: KEYNOTE-164. J Clin Oncol. 2020;38(1):11–19. doi:10.1200/JCO.19.02107
50. Marabelle A, Le DT, Ascierto PA, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair–deficient cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol. 2020;38(1):1–10. doi:10.1200/JCO.19.02105
51. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer. N Engl J Med. 2015;373(2):123–135. doi:10.1056/NEJMoa1504627
52. Carbone DP, Reck M, Paz-Ares L, et al. First-line nivolumab in stage iv or recurrent non–small-cell lung cancer. N Engl J Med. 2017;376(25):2415–2426. doi:10.1056/NEJMoa1613493
53. Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803–1813. doi:10.1056/NEJMoa1510665
54. Tan HN, Rodriguez CC, Monte KA, Tan PG, Cornelio GDH. 106P Immune checkpoint proteins as a prognostic biomarker of overall survival in non-small cell lung cancer: a meta-analysis and systematic review. Ann Oncol. 2020;31:S283. doi:10.1016/j.annonc.2020.08.227
55. Bai R, Lv Z, Xu D, Cui J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark Res. 2020;8(1):1–17. doi:10.1186/s40364-020-00209-0
56. Chowell D, Morris LGT, Grigg CM, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science (80-). 2018;359(6375):582–587. doi:10.1126/science.aao4572
57. McGranahan N, Rosenthal R, Hiley CT, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171(6):1259–1271.e11. doi:10.1016/j.cell.2017.10.001
58. Jardim DL, Goodman A, Gagliato DDM, Kurzrock R. ll review the challenges of tumor mutational burden as an immunotherapy biomarker; 2020. 2021.
59. Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–1031. doi:10.1038/nbt.2696
60. Cheng DT, Mitchell TN, Zehir A, et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagno. 2015;17(3):251–264. doi:10.1016/j.jmoldx.2014.12.006
61. Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598–2608. doi:10.1158/1535-7163.MCT-17-0386
62. Shin DS, Zaretsky JM, Escuin-Ordinas H, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7(2):188–201. doi:10.1158/2159-8290.CD-16-1223
63. Zaretsky JM, Garcia-Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–829. doi:10.1056/NEJMoa1604958
64. Huyghe N, Baldin P, Van Den Eynde M. Immunotherapy with immune checkpoint inhibitors in colorectal cancer: what is the future beyond deficient mismatch-repair tumours? Gastroenterol Rep. 2020;8(1):11–24. doi:10.1093/gastro/goz061
65. Modrich P. Mechanisms in eukaryotic mismatch repair. J Biol Chem. 2006;281(41):30305–30309. doi:10.1074/jbc.R600022200
66. Viale G, Trapani D, Curigliano G. Mismatch repair deficiency as a predictive biomarker for immunotherapy efficacy. Biomed Res Int. 2017;2017:1–8. doi:10.1155/2017/4719194
67. Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018;7(3):746–756. doi:10.1002/cam4.1372
68. Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome: part I. The utility of immunohistochemistry. J Mol Diagno. 2008;10(4):293–300. doi:10.2353/jmoldx.2008.080031
69. Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23(3):609–618. doi:10.1200/JCO.2005.01.086
70. Llosa NJ, Cruise M, Tam A, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5(1):43–51. doi:10.1158/2159-8290.CD-14-0863
71. Ganesh K, Stadler ZK, Cercek A, et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol. 2019;16(6):361–375. doi:10.1038/s41575-019-0126-x
72. Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–1191. doi:10.1016/S1470-2045(17)30422-9
73. FDA FDA-approves-first-cancer-treatment-any-solid-tumor-specific-genetic-feature. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-cancer-treatment-any-solid-tumor-specific-genetic-feature.
74. FDA-grants-nivolumab-accelerated-approval-msi-h-or-dmmr-colorectal-cancer. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-nivolumab-accelerated-approval-msi-h-or-dmmr-colorectal-cancer.
75. Carethers JM. Abstract IA4: the role of inflammation and DNA mismatch repair in colorectal cancer. Cancer Res. 2017;77(22Supplement):IA4LP–IA4.
76. Pagès F, Mlecnik B, Marliot F, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;391(10135):2128–2139. doi:10.1016/S0140-6736(18)30789-X
77. Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (80-). 2006;313(5795):1960–1964. doi:10.1126/science.1129139
78. Marliot F, Chen X, Kirilovsky A, et al. Analytical validation of the Immunoscore and its associated prognostic value in patients with colon cancer. J Immunother Cancer. 2020;8(1):13–15. doi:10.1136/jitc-2019-000272
79. Yoon HH, Shi Q, Heying EN, et al. Intertumoral heterogeneity of CD3þ and CD8þ T-cell densities in the microenvironment of DNA mismatch-repair–deficient colon cancers: implications for prognosis. Clin Cancer Res. 2019;25(1):125–133. doi:10.1158/1078-0432.CCR-18-1984
80. Chakrabarti S, Huebner LJ, Finnes HD, et al. Intratumoral CD3 + and CD8 + T-Cell densities in patients with DNA mismatch repair–deficient metastatic colorectal cancer receiving programmed cell death-1 blockade. JCO Precis Oncol. 2019;3:1–7.
81. NCT03608046. Available from: https://clinicaltrials.gov/ct2/show/NCT03608046.
82. Aderka D, Stintzing S, Heinemann V. Explaining the unexplainable: discrepancies in results from the CALGB/SWOG 80405 and FIRE-3 studies. Lancet Oncol. 2019;20(5):e274–83. doi:10.1016/S1470-2045(19)30172-X
83. Sacdalan DB, Lucero JA, Sacdalan DL. Prognostic utility of baseline neutrophil-to-lymphocyte ratio in patients receiving immune checkpoint inhibitors: a review and meta-analysis. Onco Targets Ther. 2018;11:11. doi:10.2147/OTT.S153290
84. Dolan RD, McSorley ST, Horgan PG, Laird B, McMillan DC. The role of the systemic inflammatory response in predicting outcomes in patients with advanced inoperable cancer: systematic review and meta-analysis. Crit Rev Oncol Hematol. 2017;116:134–146. doi:10.1016/j.critrevonc.2017.06.002
85. Khoja L, Atenafu EG, Templeton A, et al. The full blood count as a biomarker of outcome and toxicity in ipilimumab-treated cutaneous metastatic melanoma. Cancer Med. 2016;5(10):2792–2799. doi:10.1002/cam4.878
86. Buisan O, Orsola A, Oliveira M, et al. Role of inflammation in the perioperative management of urothelial bladder cancer with squamous-cell features: impact of neutrophil-to-lymphocyte ratio on outcomes and response to neoadjuvant chemotherapy. Clin Genitourin Cancer. 2017; 15:e697–e706. doi: 10.1016/j.clgc.2017.01.024
87. Zaragoza J, Caille A, Beneton N, et al. High neutrophil to lymphocyte ratio measured before starting ipilimumab treatment is associated with reduced overall survival in patients with melanoma. Br J Dermatol. 2016;174(1):146–151. doi:10.1111/bjd.14155
88. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer. 2020;20(9):485–503. doi:10.1038/s41568-020-0281-y
89. Kovács T, Mikó E, Ujlaki G, Sári Z, Bai P. The microbiome as a component of the tumor microenvironment. Adv Exp Med Biol. 2020;1225:137–153.
90. Nejman D, Livyatan I, Fuks G, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science (80-). 2020;368(6494):973–980. doi:10.1126/science.aay9189
91. Zhao K, Hu Y. Microbiome harbored within tumors: a new chance to revisit our understanding of cancer pathogenesis and treatment. Signal Transduct Target Ther. 2020;5(1):2–4. doi:10.1038/s41392-020-00244-1
92. Lee KA, Shaw HM, Bataille V, Nathan P, Spector TD. Role of the gut microbiome for cancer patients receiving immunotherapy: dietary and treatment implications. Eur J Cancer. 2020;138:149–155. doi:10.1016/j.ejca.2020.07.026
93. Wang J, Yang HR, Wang DJ, Wang XX. Association between the gut microbiota and patient responses to cancer immune checkpoint inhibitors (Review). Oncol Lett. 2020;20(6):1–9. doi:10.3892/ol.2020.11862
94. Balachandran VP, Łuksza M, Zhao JN, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551(7681):S12–6. doi:10.1038/nature24462
95. Jiang P, Gu S, Pan D, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24(10):1550–1558. doi:10.1038/s41591-018-0136-1
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.