Back to Journals » International Journal of Nanomedicine » Volume 16
The Anti-Proliferative Activity of Coordination Compound-Based ZnO Nanoparticles as a Promising Agent Against Triple Negative Breast Cancer Cells
Authors Stepankova H ![]() , Swiatkowski M
, Swiatkowski M ![]() , Kruszynski R
, Kruszynski R ![]() , Svec P
, Svec P ![]() , Michalkova H, Smolikova V, Ridoskova A, Splichal Z, Michalek P, Richtera L, Kopel P
, Michalkova H, Smolikova V, Ridoskova A, Splichal Z, Michalek P, Richtera L, Kopel P ![]() , Adam V
, Adam V ![]() , Heger Z
, Heger Z ![]() , Rex S
, Rex S ![]()
Received 5 February 2021
Accepted for publication 27 May 2021
Published 1 July 2021 Volume 2021:16 Pages 4431—4449
DOI https://doi.org/10.2147/IJN.S304902
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Hana Stepankova,1,2 Marcin Swiatkowski,3 Rafal Kruszynski,3 Pavel Svec,1 Hana Michalkova,1 Vendula Smolikova,1,2 Andrea Ridoskova,1,4 Zbynek Splichal,1,2 Petr Michalek,1,2 Lukas Richtera,1,2 Pavel Kopel,5 Vojtech Adam,1,2 Zbynek Heger,1,2 Simona Rex1,2
1Department of Chemistry and Biochemistry, Mendel University in Brno, Brno, Czechia; 2Central European Institute of Technology, Brno University of Technology, Brno, Czechia; 3Institute of General and Ecological Chemistry, Lodz University of Technology, Lodz, Poland; 4Central European Institute of Technology, Mendel University in Brno, Brno, Czechia; 5Department of Inorganic Chemistry, Faculty of Science, Palacky University, Olomouc, Czechia
Correspondence: Simona Rex
Department of Chemistry and Biochemistry, Mendel University in Brno, Brno, Czechia
Tel +420-5-4513-3350
Fax +425-5-4521-2044
Email [email protected]
Purpose: The present study deals with the in vitro evaluation of the potential use of coordination compound-based zinc oxide (ZnO) nanoparticles (NPs) for the treatment of triple negative breast cancer cells (TNBrCa). As BrCa is one of the most prevalent cancer types and TNBrCa treatment is difficult due to poor prognosis and a high metastasis rate, finding a more reliable treatment option should be of the utmost interest.
Methods: Prepared by reacting zinc carboxylates (formate, acetate, propionate, butyrate, isobutyrate, valerate) and hexamethylenetetramine, 4 distinct coordination compounds were further subjected to two modes of conversion into ZnO NPs – ultrasonication with oleic acid or heating of pure precursors in an air atmosphere. After detailed characterization, the resulting ZnO NPs were subjected to in vitro testing of cytotoxicity toward TNBrCa and normal breast epithelial cells. Further, their biocompatibility was evaluated.
Results: The resulting ZnO NPs provide distinct morphological features, size, biocompatibility, and selective cytotoxicity toward TNBrCa cells. They internalize into two types of TNBrCa cells and imbalance their redox homeostasis, influencing their metabolism, morphology, and ultimately leading to their death via apoptosis or necrosis.
Conclusion: The crucial properties of ZnO NPs seem to be their morphology, size, and zinc content. The ZnO NPs with the most preferential values of all three properties show great promise for a future potential use in the therapy of TNBrCa.
Keywords: carboxylate, hexamethylenetetramine, nanoparticles, redox homeostasis imbalance, triple negative breast cancer, zinc oxide coordination compounds
Introduction
As breast cancer is the cancer type with the second highest prevalency, a huge attention is paid toward the development of novel drug modalities for its therapy.1 Based on its molecular subtypes, it is usually categorized into human epidermal growth factor receptor 2 (HER2) positive, estrogen receptor (ER) positive, progesterone receptor (PR) positive, luminal A, or luminal B,2 where ER and PR positive subtypes form nearly 70% of all breast cancer cases and 20% lack HER2, ER, and PR. Called triple negative breast cancer,3 these often possess an aggressive phenotype with an increased metastasis rate, poor prognosis,1 and unreliable treatability with numerous side effects due to conventional chemotherapy inhibiting both healthy and cancer cells.4
These side effects could be overcome by the employment of nano-scaled particles (NPs) with a specific affinity toward cancer cells.1 NPs have received increased attention thanks to the enhanced permeability and retention (EPR) effect, causing suitably sized particles to intrinsically accumulate in many types of solid tumors due to the presence of large pores in rapidly formed tumor neovasculature together with a lack of lymph vessels.5 However, the EPR effect is not present in all types of solid tumors and even where it is, there is only 2× increased delivery of NPs to tumors compared to critical off-target organs.6 For this reason, engineering of NPs to selectively bind to tumor cells rather than normal cells can greatly improve their therapeutic efficacy.7–9
Recently, zinc oxide (ZnO) NPs, generally known for their antibacterial properties,10 attracted much attention as a suitable platform for the therapy of various types of cancer. They provide different photo-oxidizing and photocatalytic effects, owning to their ultraviolet light absorption,11 semiconducting, and magnetic properties.12 They can induce selective inhibition of cancer cells,13 with additive or synergistic effects together with anti-cancer compounds.14 The induction of apoptosis by ZnO NPs was observed in breast cancer cells,15,16 including triple negative breast cancer.17,18 On the other hand, certain ZnO NPs can accumulate in various off-target organs, leading to toxicity for heart, brain, thymus, pancreas, liver, kidney, and others.19–22
This work is focused on the development of ZnO NPs converted from distinct coordination compounds providing selective cytotoxicity toward triple negative breast cancer cells of human origin, while maintaining biocompatibility, and avoiding the normal cells of breast epithelium.
Materials and Methods
Chemicals
Most of the listed chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) in an ACS purity. The Milli-Q® Water Purification System (Merck Millipore, Burlington, MA, USA) was employed to produce deionized water.
Synthesis of Coordination Compounds
Coordination compounds were synthesized according to literature procedure in direct reactions featuring Zn carboxylate and hexamethylenetetramine (HMTA) in aqueous medium23 (synthesis and analytical24 details are given in Supplementary Materials Paragraph S1.1). The reactions between Zn carboxylates (formate, acetate, propionate, butyrate, isobutyrate, valerate) and HMTA were carried out with metal to ligand (M:L) molar ratio of 2:1 (complementary to the previous research,23 in which stoichiometries of 1:1 and 1:2 were applied) and led to the formation of 4 coordination compounds (Supplementary Table S1). Similar to the previously reported data, in the synthesis with Zn valerate, both substrates crystallized separately and there was no evidence of a formation of coordination compounds (despite the used M:L molar ratio). The separate crystallization of salt and HMTA also occurred in the synthesis involving Zn butyrate with M:L molar ratio of 2:1.
Synthesis of ZnO NPs
Two strategies of a single precursor method were applied to the synthesis of ZnO NPs. In the first one (S1), samples of coordination precursors (containing 0.01 mol of Zn) were suspended in oleic acid (10 mL) with support of ultrasonic irradiation generated by the ultrasonic processor Hielscher 100UP (Hielscher Ultrasonics, Teltow, Germany) equipped with a sonotrode of diameter 10 mm (power 100 W, working frequency 30 kHz, maximum amplitude 70 µm) followed by heating at 220 °C for 2 h. After that the temperature was raised to 800 °C with a maximum heating range (~30 °C/min) in order to purify ZnO from oleic acid. In this approach, NPs A, B, C, and D were produced from precursors 1, 2, 3, and 5, respectively. In the second strategy (S2), precursor samples (containing 0.01 mol of Zn) were heated in an air atmosphere up to 500 °C (5 °C/min heating rate), and then left at this temperature for 1 h. In this approach, NPs E, F, G, and H were produced from precursors 1, 2, 3, and 5, respectively. Surface morphologies of NPs were studied using a scanning electron microscope (SEM, Hitachi S-4700 [Hitachi, Tokyo, Japan]) with an energy-dispersive X-ray spectrometer (EDS Thermo NORAN [Thermo Fisher Scientific, Waltham, MA, USA]).
Determination of Crystal Structure
The colorless prism crystals of compounds 3 and 5 were mounted on the Rigaku Synergy Dualflex automatic diffractometer equipped with Pilatus 300K detector (Rigaku, Tokyo, Japan). X-ray intensity data were collected using the monochromated CuKα mirror (λ=1.54184 Å, micro-focus sealed PhotonJet X-ray tube), radiation at 100.0(1) K, ω scan and the shuterless mode. The reflections inside an Ewald sphere were collected up to ϴ=79°. 61102 and 12308 strongest reflections were used for 3 and 5, respectively, to determine the unit cell parameters. The details are provided in Supplementary Table S2. No loss of intensity during the measurement occurred. Polarization, Lorentz, and numerical absorption corrections (using a Gaussian integration over a multifaceted crystal model) were used. A dual-space algorithm was used to solve the structure. The full-matrix, least-squares technique on F2 was used to anisotropically refine the non-hydrogen atoms, whereas the hydrogen atoms were found after four cycles of anisotropic refinement from difference Fourier synthesis, and refined as “riding” on the adjacent atom with geometric idealization after each cycle of the refinement. The individual isotropic displacement factors were 1.5× of parent methyl carbon and oxygen atoms and 1.2× of parent non-methyl carbon atoms. Rotation about local three-fold axes was allowed for the methyl groups. Programs SHELXT,25 SHELXL,26 and SHELXTL27 were used for all the calculations. International Tables for Crystallography were used to obtain atomic scattering factors.28 Supplementary Table S3, shows the interatomic bond distances and Supplementary Table S4, shows the intramolecular interactions. Further details are in the Supplementary Paragraph S1.2. The supplementary crystallographic data are in CCDC 1980739−1980740 and they can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, [email protected], or The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK.
Other Physical Measurements
The infrared (IR) spectra were recorded on the Jasco Fourier transform (FT)/IR 6200 spectrophotometer (Jasco Deutschland GmbH, Pfungstadt, Germany), in a form of KBr pellets. The spectral range was 4000−400 cm−1. The thermal analyses were performed using the TG/DTA-SETSYS−16/18 thermoanalyser coupled with the ThermoStar mass spectrometer (Balzers, Liechtenstein). Platinum crucibles were used to heat the samples up to 1000 °C (5 °C/min) in 1 L/h flow of synthetic air (composed of N2:O2 79:21). X-ray powder diffraction (XRPD) using the Powder Diffraction File and XPert PRO XRPD system (CENIMAT|i3N, Caparica, Portugal) equipped with CuKα1 radiation, Bragg-Brentano PW 3050/65 high-resolution goniometer, and PW 3011/20 proportional point detector were used to confirm the final products of decomposition.29
Zn Content Analysis
The stock ZnO NPs suspensions (1 mg/mL) were prepared by suspending the NPs in the appropriate solution (water in the case of Zn content determination), followed by ultrasonication for 2 h. To evaluate the Zn content, a standard solution of Zn (1 mg/mL; Merck Millipore) was used. Calibration solutions were acidified by HNO3 suprapure (1%, Penta Chemicals Unlimited, Prague, Czech Republic), obtained by sub-boiling distillation using the BSB-939IR (Berghof, Eningen unter Achalm, Germany). Suspensions of ZnO NPs were diluted with HNO3 (5%) for the selected calibration range (0.0–10.0 µg/mL). The atomic absorption spectrometer 240FS AA (Agilent Technologies, Santa Clara, CA, USA), with a deuterium background correction and flame atomization was used for Zn content analysis. The instrument operated under the oxygen flow of 13.5 L/min and acetylene flow of 2.0 L/min. The radiation source was the ultrasensitive hollow cathode lamp (Agilent Technologies) with the operational wavelength for Zn detection of 213.9 nm.
Hydrodynamic Diameter and ζ-Potential Measurement
The ZnO NPs were suspended in water (1 mg/mL) and diluted 200× for hydrodynamic diameter (HDD) measurement and 50× for ζ-potential determination. Quasielastic dynamic light scattering and Doppler microelectrophoresis were used, respectively, both employing the Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). The refractive index of 2.000 was used for the dispersive phase and 1.333 for the dispersive environment. Equilibration lasted for 120 s. To determine the HDD and polydispersive index (PdI), the cuvettes ZEN0040 (Brand GmbH, Wertheim, Germany) were used with the measurement of each suspension in hexaplicates. For ζ-potential determination, the cells DTS1070 (Brand GmbH, Wertheim, Germany) were used with the measurement of each suspension in triplicates. The measurements were performed for three separate suspensions of each ZnO NPs. The calculations were performed as in.30
Stability of ZnO NPs in Water
ZnO NPs were suspended in deionized water and imaged using a SEM Tescan MAIA 3 (Tescan Ltd., Brno, Czech Republic) with a field-emission gun. To obtain 768×858 pixel images, the In-lens SE detector was used (working distance of 2.41–3.06 mm, acceleration voltage 5 kV, magnification 100,000×, sample area 2.08 µm). UH resolution mode was employed for full frame capture with the enabled image shift correction for image accumulation (~0.32 µs/pixel dwell time, 2.4 nm spot size).
Stability of ZnO NPs Size in Ringer’s Solution
ZnO NPs suspended in Ringer’s solution pH 7.4 (containing 6.50 g NaCl, 0.42 g KCl, 0.25 g CaCl2, and 0.20 g NaHCO3 in 1 L of deionized water) were kept at 37 °C with the evaluation of HDD at different time points.
Cell Lines
Normal breast cell line HBL-100 and triple negative breast cancer cell lines MDA-MB-231 and MDA-MB-468 were purchased from American Type Culture Collection (Manassas, VA, USA). HBL-100 were cultured in DMEM (Dulbecco’s Modified Eagle’s Medium) and both cancer cell lines were cultured in RPMI (Roswell Park Memorial institute) 1640 medium, both supplemented with penicillin (100 U/mL), streptomycin (0.1 mg/mL), and fetal bovine serum (FBS, 10% v/v). Humidified incubator (Galaxy 170 R, Eppendorf, Hamburg, Germany) with 5% CO2 was employed to maintain the cells.
24hIC50 Determination
The cell viability was assayed by measuring mitochondrial activity using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as in.31 Incubation in 1:1 ratio with the varying concentrations of ZnO NPs in medium (maximum of 1 mg/mL) lasted for 24 h. The 24hIC50 concentration was determined from the viability data.
Wound Healing Assay
An artificial wound was formed in a cell monolayer as in.31 The influence of the treatment with ZnO NPs (24hIC50) in medium was captured at the beginning and at each time point (0; 2; 4, 8; 12; 24; 48; and 72 h [only 24, 48 and 72 h are shown in this work]).
Hard Protein Corona Formation
ZnO NPs were exposed to human plasma or FBS. Human plasma was obtained as in31 and kept at –80 °C until use. Plasma pooled from multiple donors and FBS were defrosted and spun at 21,000 rcf and 4 °C for 30 min to remove aggregates formed during the defrosting. Next, the supernatant was incubated at 37 °C and 350 rpm for 35 min and again spun to remove aggregates. ZnO NPs in water (1 mg/mL) were mixed in 1:1 ratio (v/v) with FBS or plasma and the mixture was incubated at 37 °C and 350 rpm for 35 min. As a control of the follow-up cleaning process from unbound proteins, phosphate buffered saline (PBS) mixed with FBS or plasma was used. The rest of the assay was performed as in31 with some notable changes. The pellets were washed 5× with PBS by centrifugation at 15,000 rcf and 4 °C for 15 min. Finally, the pellets were resuspended in deionized water (100 µL) and mixed (10 µL) with loading buffer (5 µL; 50 mM Tris/HCl, 20% glycerol, 0.1% bromophenol blue, and 2% sodium dodecyl sulphate [SDS]). NEB protein ladder 10–250 kDa (5 µL, New England Biolabs, Ipswich, MA, USA) was used as a molar mass marker. The protein bands were imaged using near infrared irradiation at 700 nm on Azure c600 (Azure Biosystems, Dublin, CA, USA).
Opsonization with Complement C3b Element
ZnO NPs in water were mixed with human serum from male AB clotted whole blood (H6914, cleaned from aggregates in the same way as plasma and FBS) in 1:1 ratio and incubated at 37 °C and 350 rpm for 35 min. After the incubation, ZnO NPs with bound proteins were 3× centrifuged at 15,000 rcf and 4 °C for 15 min with the washing of the pellets containing NPs and bound proteins by PBS. Finally, pellets were resuspended in deionized water (100 µL), mixed in 2:1 ratio with loading buffer and resolved on 6% SDS PAGE at 200 V and 4 °C for 30 min. After the electrotransfer of the proteins onto the Immobilon®-FL PVDF membrane (Merck Millipore), the membrane was blocked with skim milk for 1 h at 20 °C. Primary complement C3 monoclonal antibody (LF-MA0132, Thermo Fisher Scientific, 1:1000) was used, with incubation at 20 °C for 1 h. Secondary antibody labelled with horseradish peroxidase (HRP, p0260, Dako, Santa Clara, CA, USA, 1:5000) was incubated with the membrane at 20 °C for 1 h. Chemiluminescent signal was developed with Clarity Western ECL Blotting Substrate (Bio-Rad, Hercules, CA, USA) and membranes were imaged using Azure c600 (Azure Biosystems).
Hemocompatibility
To determine the hemocompatibility of ZnO NPs in water, fresh human red blood cells (RBCs) were used as in31 with the ZnO NPs diluted in PBS (pH 7.4; 500, 250, 125, and 62.5 µg/mL).
Ethidium Bromide (EtBr) Displacement
The ability of ZnO NPs to displace EtBr was studied as in31 with some notable changes. Plasmid MP px459 was used and ZnO NPs in water (1000; 500; 250; 125 µg/mL, diluted in PBS with 10 mM NaCl) were added (final volume 25 µL). As a control, plasmid (1 µg) with EtBr (6 µM), and ZnO NPs-free PBS with NaCl (10 mM) was used. The rate of displacement was calculated as a percentage decrease of EtBr fluorescence.
Plasmid Cleaving Assay
ZnO NPs in water were diluted (to 500 µg/mL) with Tris-HCl buffer (50 mM) containing NaCl (50 mM, pH 7.2). After that, plasmid MP px330 (50 µg/mL) was incubated with the ZnO NPs (final volume 10 µL), according to.31 Loading buffer (5 µL, 30% glycerol with 0.1% bromophenol blue) was used, with 2-Log DNA ladder as a molar mass marker.
Reactive Oxygen Species (ROS) Formation
10,000 cells seeded in a 24-well plate were incubated with ZnO F-H in medium (55 µg/mL) for 6 h, stained with CellROX® Deep Red (Thermo Fisher Scientific, 1:500 in PBS), detached by accutase, and analyzed on flow cytometer BD Accuri C6 Plus (BD Biosciences, Franklin Lakes, NJ, USA), with the 35 µL/min flow rate and at least 10,000 cells analyzed in each group.
Lipid Peroxidation
The analysis was performed as in32 with the cells treated with ZnO F-H in medium (55 µg/mL) for 12 h. Cumene hydroperoxide (CH, 150 µM) was administered as a positive control. Fiji ImageJ software (National Institute of Health, Bethesda, MD, USA) was used to calculate the fluorescence intensity.
F-Actin Disorganization, Nuclei-to-Cytoplasm (N/C) Ratio, and Cell Morphology
~40,000 cells/coverslip seeded were treated with ZnO F-H in medium (55 µg/mL) for 24 h, followed by fixation, labeling of F-actin filaments, imaging, and analysis as in32 with the following exceptions. To visualize ZnO NPs, confocal reflectance microscopy was employed, with irradiation by a solid state 633 nm laser and detection window at 631–636 nm. To evaluate morphometric parameters, a minimum of 20 cells in each group were analyzed. Orientation J plugin with a cubic-spline gradient and 7 ROI in each picture was used to analyze coherency.33 Cell morphology upon treatment with ZnO F-H in medium (55 µg/mL) for 6 h was evaluated by Cryo-SEM as in.32
Apoptosis Induction
~600,000 cells seeded into each well of a 6-well plate were treated with ZnO F-H in medium (55 µg/mL) for 6 or 24 h. Cells detached by accutase, as well as spontaneously detached cells were dual stained for translocated phosphatidylserine and cell permeability using the PE Annexin V Apoptosis Detection Kit I (BD Biosciences). The analysis was performed using flow cytometry as in22 with at least 100,000 cells analyzed in each group. Further, cytopathological May-Grünwald-Giemsa staining was performed upon treatment with ZnO F-H (55 µg/mL) for 24 h according to.30
Descriptive Statistics
The main tendency was measured using the mean and the dispersion using the standard deviation (SD). To analyze the differences between groups, an unpaired t-test in software GraphPad (GraphPad Software, San Diego, CA, USA) was used. Figures were processed using Microsoft Office PowerPoint software (Redmond, WA, USA) and images using Fiji Image J (National Institute of Health), unless otherwise mentioned.
Results and Discussion
Synthesis Stoichiometry Affects the Composition of the Coordination Compounds Formed by Zn Propionate and Isobutyrate but Not Formate or Acetate
The reactions between Zn carboxylates and HMTA led to the formation of two previously described (1 and 2)23 and two new (3 and 5) coordination compounds (Supplementary Table S3). The stoichiometry used in the synthesis (metal: HMTA equal to 2:1 [this work], 1:1, and 1:2)23 does not affect the composition of formed compounds 1 and 2, i. e. M:L is always 1:2 in 1 and 1:1 in 2. Additionally, both these compounds (in opposition to the other discussed) are mononuclear molecular systems. The synthesis stoichiometries used previously (1:2 and 1:1)23 were not reflected in compounds containing propionate and butyrate ions (3A and 4A with M:L of 2:1). Repetition of these syntheses with resulting product M:L ratio (i. e. 2:1) led to the formation of compound 3 (M:L of 3:1) and separate crystallization of pure Zn butyrate [polymeric solventless Zn(n-C3H7COO)2] form34 and pure HMTA. It proves that excess of a neutral ligand is necessary for the formation of the 3, 3A, and 4A because in these cases the final compound possesses a smaller amount of ligand than was used in the synthesis. Hypothetical coordination compound of Zn butyrate with M:L of 3:1 was not formed in the solid state probably because of a lower dissociation constant (and higher stability) of polymeric Zn butyrate34 than Zn propionate35,36 (Zn butyrate crystallized as a first, and HMTA later, after precipitation of the whole salt). The synthesis stoichiometry affects the composition of the coordination compounds formed by Zn isobutyrate but it does not affect the M:L ratio in the product. In both cases, it is 1:1, but the compound formed in reactions with 1:2 and 1:1 stoichiometries of substrates is zig-zag one-dimensional polymer (5A),23 while a 2:1 reaction stoichiometry leads to the formation of a dinuclear molecular compound (5). In this reaction, the competition between anions and HMTA in an inner coordination sphere leads to the formation of four carboxylate bridges between two neighboring Zn cations in case of HMTA deficiency (compound 5) and one carboxylate bridge between two subsequent atoms in chains in case of a stoichiometric amount of HMTA (compound 5A). In general, a lengthening of anions with non-branched aliphatic chains leads to a lower number of neutral molecules present in the resulting compounds. This effect is not observed for anions with branched aliphatic chains.
The Four Compounds Have Distinct Crystal and Molecular Structure
The appliance of carboxylate anions differing slightly in the length of the aliphatic chain allows constructing diverse coordination moieties containing the same neutral ligand (HMTA). Coordination numbers of Zn cations, as well as coordination modes of ligands also vary in the studied compounds. All studied compounds are dissimilar, the 1 and 2 are mononuclear compounds with HMTA molecules located in outer and inner coordination sphere, respectively, the 3 (Figure 1Aa) is a two-dimensional coordination polymer with a hexagonal tiling (Figure 1B), and 5 is a dinuclear molecular compound (Figure 1Ab). The structures of 1 and 2 were described in,23 thus the respective data are not discussed in the current work. All atoms of 3 (Figure 1Aa) are located at general positions but the presence of the glide planes going through the asymmetric unit moiety expands it into a polymeric net. Each asymmetric unit contains six structurally different Zn cations divided into two distinguishable coordination systems composed of Zn3(C2H5COO)5(OH) heteroionic units interlinked by the neutral HMTA molecules. Each unit possesses cations in two different coordination environments. Two Zn cations of each unit (Zn1/Zn2 and Zn4/Zn5) adopt a slightly distorted trigonal bipyramidal geometry37 with apexes occupied by HMTA N atom and O atom of OH− ion, and three base corners created by the O atoms of bridging carboxylate ions. One Zn cation of each unit (Zn3 and Zn6) exists in an almost ideal tetragonal bipyramidal geometry38 with apexes inhabited also by HMTA N atom and O atom of OH− ion, and four O atoms of bridging carboxylate ions creating polyhedron base. Consequently, all carboxylate anions bridge two Zn2+ ions; however, one pair of cations is bridged by one carboxylate anion, while two other pairs of cations are bridged by two carboxylate anions. The central OH− ion links all three cations forming one heteroionic unit. The Zn3(C2H5COO)5(OH) moieties are connected to three tridentate HMTA molecules via Zn–N coordination bonds (Supplementary Table S2). The Zn···Zn distances within Zn3(C2H5COO)5(OH) units vary in the range of 3.32–3.73 Å. Compound 3 is a very rare example of a polymer composed of trinuclear Zn-carboxylate moieties assembled further by bridging N ligands. The two-dimensional polymeric net of 3 extends along the crystallographic (0 1 0) plane and is composed of rings with nodes occupied by Zn cations and HMTA molecules. The whole polymeric system creates a 3,3,4-c 3-nodal net with stoichiometry (3-c)(3-c)3(4-c)3 and is described by the {3.122}3{33.122.13}3{33} Schlafli symbol. Considering the simplified net with nodes located only at Zn cations, the polymer adopts a kagome pattern (kgm topological type; Shubnikov plane net (3.6.3.6)), i. e. 4-c unimodal net described by the {32.62.72} Schlafli symbol (Supplementary Figure S9). 39 This is distinctly different from the simple 3-c unimodal net ({4.82} Schlafli symbol) existing in compounds 3A and 4A.
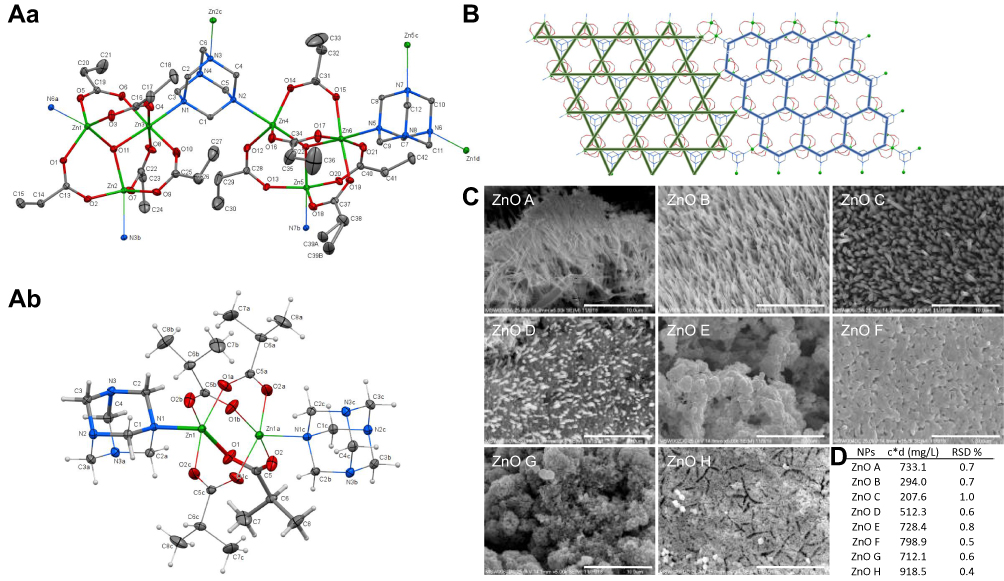 |
Figure 1 Distinct morphology of the produced nanoparticles originates in the molecular and crystal structure of the coordination compounds and their conversion method. (A) The molecular structure of the compounds 3 (a) and 5 (b), with atom numbering scheme, plotted with 50% probability of displacement ellipsoids of non-hydrogen atoms. The hydrogen atoms are omitted to improve clarity in 3, and they are plotted as spheres of arbitrary radii in 5. The symmetry generated atoms, indicated by the d letters, were generated via the following symmetry transformations: 3 (a) x+1, y, z; (b) x, -y+1.5, z-0.5; (c) x, -y+1.5, z+0.5; (d) x-1, y, z; 5 (a) x, -y+1, z; (b) -x+1, -y+1, -z; (c) -x+1, y, -z. (B) The part of a polymeric net of compound 3. The kagome pattern (left) and the general hexagonal motif (right). (C) The SEM micrographs showing morphology of the produced NPs. Scale bar, 3 µm for ZnO F and 10 µm for the rest. (D) Zinc content in the NPs determined by atomic absorption spectroscopy. Abbreviations: NPs, nanoparticles; RSD, relative standard deviation; SEM, scanning electron microscope; ZnO, zinc oxide. |
The asymmetric unit of 5 (Figure 1Ab) contains one fourth of the molecular compound. Consequently, one molecule occupies four asymmetric units and the unit cell contains two complex molecules. The Zn1, N1, N2, C1, and C4 atoms occupy a special position i of C2/m space group with site symmetry m and multiplicity 4, thus half of each of them resides in the asymmetric unit. The central atom in 5 is surrounded by four O atoms (each belonging to one bridging carboxylate ion) creating the tetragonal pyramid37 base and one N atom of HMTA molecule located at polyhedron apex. Therefore, two Zn atoms existing in the molecule are bridged by four carboxylate ions and HMTA molecules acting as monodentate ligands. The Zn cations within one molecule are separated by about 2.892(1) Å which is larger than the doubled Van der Waals radius of Zn (2.78 Å).
Carboxylate anions bridging five-coordinated central atoms of 3 exhibit a very slight asymmetricity of formed coordination bonds, and all the other carboxylate anions (of 3 and 4) display a strong asymmetricity of Zn-O bonds (Supplementary Table S3). The computed bond valences40,41 where Rij is the bond-valence parameter for i-j bond42–44 equal to 1.7588 Å for RZn-N and 1.6950 Å for RZn-O,45 b is the constant equal to 0.37 Å,46,47 show that in the compound 3 the Zn(pentacoordinated)–O(carboxylate) bonds are about 2× stronger than Zn(pentacoordinated)–N bonds and the strength of Zn(pentacoordinated)–O(OH−) is in the middle of the previous ones (Supplementary Table S3). The coordination bonds formed by hexacoordinated Zn ions possess similar strength, which agrees with the considerable regularity of coordination polyhedra. Compound 5 possesses the reverse strength of coordination bonds, i. e. the Zn–N bonds are stronger than Zn–O ones. This originates from constraints imposed by a rigid polymeric net existing in 3, which prevents the formation of coordination Zn–O of the most privileged strength (with bond valence equal to 0.4 v.u. for pentacoordinated divalent cations and 0.33 v.u. for hexacoordinated divalent cations). The computed total valences of Zn cations are close to the expected value +2 (Supplementary Table S3), which proves attainment of an optimal coordination sphere geometry for used building block ligands and lack of significant strains in the coordination moieties.
The absence of classical hydrogen bond donors (except one) leads to the presence of only weak hydrogen bonds in the studied compounds. The OH– ion (present in compound 3) is formally a classical hydrogen bond donor; however, it shares its electron density with three Zn2+ cations (via three Zn–O coordination bonds); thus, a partial negative charge on the O atom is diminished, which subsequently reduces a polarization of O–H bond and a partial positive charge of H atom. This results in a decreasing propensity of possible O–H···A interactions. The polymeric net of compound 3 contains multiple intermolecular weak hydrogen bonds connecting the C-H(HMTA) donors and O(carboxylate) acceptors (Supplementary Table S4). The subsequent nets are relatively well separated, and only H···H distances close to the doubled Van der Waals radius exist. In compound 5 (beside intramolecular C–H···O), one intermolecular C–H···N hydrogen bond exists, and it links the molecules to the piles extending along the crystallographic [0 0 1] axis (Supplementary Table S4).
IR Spectra Show Multiplication of Most of HMTA Bands in Compounds 3 and 5
The IR spectra of compounds 1 and 2 were described in23 thus respective data are not discussed in the current work. Most of HMTA bands present in the spectra of compounds 3 and 5 (Supplementary Figure S1, Supplementary Table S5) are multiplicated (in comparison to the spectrum of pure HMTA)48 due to the formation of the coordination bonds by some (but not all) nitrogen atoms.49 The bands corresponding to ρCH2 (1240 cm−1) and νCN (1007 cm−1) vibrations split up into quartet and doublet, respectively, in the spectrum of 3, and into doublet and doublet, respectively, in the spectrum of 5. The multiplication to a larger number of bands in the case of 3 in comparison to 5 is a consequence of the tridentate function of HMTA in 3 in contrast to monodentate HMTA in 5. The separation parameter Δν, concerning bands of stretching vibrations of the carboxylate group, is 179 cm−1 (3) and 197 cm−1 (5). These values are in the range characteristic for bridging anions23 which agrees with the structural data. The absence of the considerable shifts of bands involving non-coordination N atoms of HMTA (e. g. bending NCN vibrations) is caused by a lack of strong or medium strength D–H···N hydrogen bonds.50,51 Blue (3) and red (5) shifts of bands assigned to stretching symmetric and asymmetric vibrations of carboxylate anions methyl groups originates from a different environment of each anion in a crystal net and consequently different vibration freedom of these groups. The hydroxide anion is represented in a spectrum of 3 by a typical broadened band with a maximum at about 3447 cm−1, caused by the stretching OH vibrations.
Thermogravimetric Analysis Shows Decomposition of Compounds 3 and 5 at Similar Temperatures
The thermal decompositions of compounds 1 and 2 were described in23 thus respective data are not discussed in the current work. The thermal decompositions of 3 and 5 are a one stage processes composed of multiple (7 and 6, respectively) overwrapping substages (Supplementary Figure S2). Due to the absence of the water molecules in the structures of these compounds, both are stable above 100 °C. The presence of some analogy between coordination moieties (occurrence of the bridging carboxylate ions) causes the start of decomposition of 3 and 5 at similar temperatures (142 °C and 155 °C, respectively). The first substage is connected to the decomposition of coordination moieties accompanied by sublimation of the HMTA molecules (endothermic process, Supplementary Figure S2), which, upon elevation of the temperature, is changed to exothermic combustion of HMTA.52 Upon further increase of the temperature, the simultaneous oxidation of the anions occurs, accompanied by the formation of carbon deposit, which is oxidized much slower in comparison to ligands (it is visible on the TG curves as a mild weight loss at the end of the decomposition process, Supplementary Figure S2). The temperature needed for the formation of pure ZnO slightly differs for the studied compounds, and it is equal to 428 °C and 450 °C for compounds 3 and 5, respectively. Total losses (found/calculated: 67.3/66.0% for 3 and 78.1/78.6% for 5) correspond to the registered (by XRPD) final product, i. e. ZnO.
Morphology of ZnO NPs is Bound to the Structure of Coordination Compounds and the Conversion Method
The structure of the coordination compounds and the conversion conditions are crucial for controlling the morphology of the resulting ZnO NPs (Figure 1C). In all cases, a hexagonal form of ZnO was formed (wurtzite structure, a = b = 3.249 Å, c = 5.203 Å, Z = 2, space group = P63mc). The NPs synthesized with the usage of oleic acid (S1) are straight nanopoles, while those produced from pure precursors (S2) create complex spherical and polyhedral agglomerates. The mononuclear precursors 1 and 2 allow the production of cylindrical nanowires, polymeric 3 lead to production of hexagonal rods, and dinuclear 5 to a mixture of hexagonal rods and spherical/polyhedral particles. It must be outlined that the currently used alteration of the precursor conversion procedure described previously23 allows change of the shape of the particles (obtained from 1 and 2) from nanoplates to nanopoles and nanospheres.
Furthermore, the used conversion method also influences the Zn content in the produced NPs (Figure 1D). In general, NPs synthesized from pure precursors contain significantly more Zn than NPs synthesized with the usage of oleic acid. No correlation between the structure of the coordination compounds and the Zn content in the NPs was found.
ZnO NPs Show Favorable Stability for Future in vivo Use
The HDD is one of the most crucial parameters when considering NPs for therapy. NPs need to be >5.5 nm to avoid renal clearance and >10 nm to avoid rapid clearance to healthy tissues through the pores in normal blood vessels.53 At the same time, size <200 nm is required to exploit the EPR effect and escape tumor vasculature54 and NPs >100 nm can be cleared in the liver, kidneys, and spleen by phagocyting cells of the reticuloendothelial system (RES).55 Therefore, the size impairment of the ZnO NPs synthesized in this work needs to be addressed before commencing in vivo testing. The differences in dimensions of solid NPs result mainly from precursor structures (Figure 2A top). A comparison of NPs prepared from mononuclear precursors via both strategies demonstrate that the larger NPs are created from 1 (375 and 385 nm for ZnO A and E vs 280 and 195 nm for ZnO B and F, respectively), due to its larger outer coordination sphere and presence of the easily degradable [Zn(H2O)6]2+ coordination moieties.23 The dimensions of NPs produced from polynuclear precursors are larger for 3 (450 and 325 nm for ZnO C and G, respectively) because the polymeric net of 3 is more stable and cations are packed more closely than in the supramolecular net of dinuclear 5 (375 and 275 nm for ZnO D and H, respectively). The usage of additional dispersing agent and ultrasonication, beside the shape of NPs, also changes their size and uniformity at some degree. For compounds without an outer coordination sphere (2, 3, and 5), the application of S2 (without additional dispersing factors) leads to NPs more uniform in size. Such effect is not observed for 1 as the coordination units are “dispersed” (well separated) by the outer coordination sphere species also in S2.
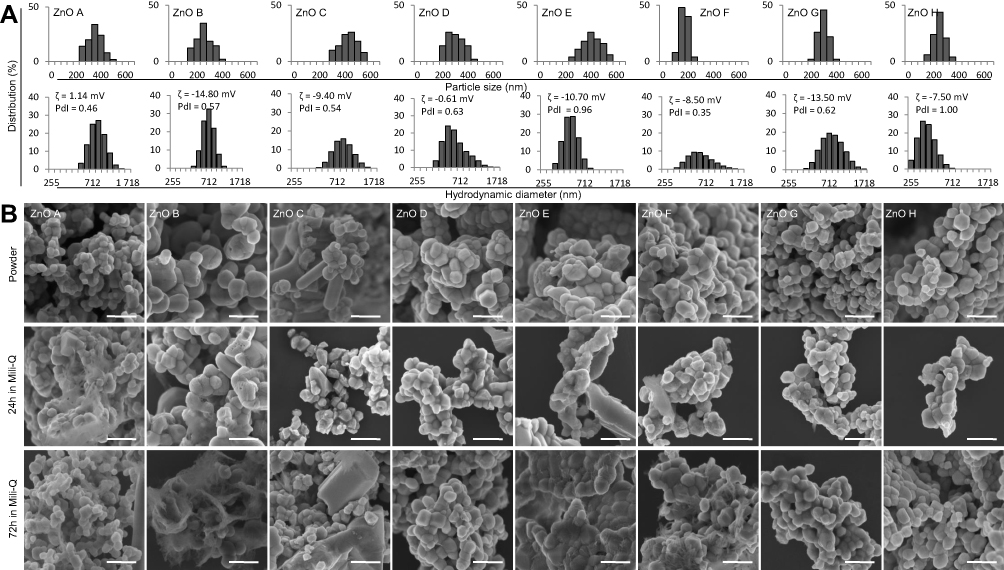 |
Figure 2 The size distribution of the ZnO NPs is influenced by their precursor structures and conversion method. ((A) top) The ZnO NPs size distribution based on SEM measurements. ((A) bottom) HDD distributions, PdI, and ζ-potential values of the ZnO NPs suspended in water. (B) The SEM micrographs showing the morphology changes of ZnO NPs suspended in water for up to 72 h. Scale bar, 500 nm. Abbreviations: HDD, hydrodynamic diameter; NPs, nanoparticles; PdI, polydispersive index; SEM, scanning electron microscope; ZnO, zinc oxide. |
A similar trend was obtained for the HDD of the produced ZnO NPs suspended in water (Figure 2A bottom). Interestingly, NPs with larger HDD were prepared using the synthesis with the usage of oleic acid and ultrasonication (ZnO A = 825 nm; B = 712 nm; C = 825 nm; D = 531 nm), rather than those prepared from pure precursors (ZnO E = 459 nm; F = 531 nm; G = 712 nm; H = 342 nm). The HDD in water was also quite heterogeneous, with the PdI ranging from 0.35 for ZnO F to 1.00 for ZnO H. Except for ZnO F, the NPs were more homogeneous when synthesized with the usage of oleic acid and ultrasonication. The HDD did not significantly change upon suspension of the NPs in Ringer’s solution at 37 °C (Supplementary Figure S3), mimicking human plasma environment56 and it changed only slightly during further incubation.
All ZnO NPs showed slightly negative ζ-potential upon suspension in water (Figure 2A bottom, inset). For ZnO A and D, the ζ-potential was close to the neutral value (−1.14 and −0.61 mV, respectively). The other NPs possessed ζ-potential in the range of −7.5 through −14.8 mV without dependence on the synthesis method. Further, the ζ-potential was stable for up to 72 h. For potential in vivo use, the ζ-potential needs to be large enough to prevent aggregation of the NPs (at least |3−8| mV)53 but at the same time small enough not to provoke rapid clearance into neighboring tissues and RES through electrostatic means.57 Also, slightly negative ζ-potential prevents interactions between the NPs and negatively charged membranes of cells forming the endothelial layer of blood vessels and hinders clearance from the organism.54 It has been shown that the ζ-potential for larger NPs (above 250 nm) does not differ between blood plasma, interstitial fluid, and intracellular fluid,53 so measuring in one can serve as a good approximation of the other environments. Taken together, the ζ-potential of most of these ZnO NPs is favorable for future in vivo use.
Further, the stability of the ZnO NPs morphology upon suspending in a liquid was tested (Figure 2B). It was shown that suspension of ZnO NPs leading to a sustained release of Zn2+ ions can facilitate their pronounced internalization into cells and enable the anticancer activity58 by causing mitochondrial damage and disrupting cellular Zn homeostasis, leading to a disequilibrium in protein activity.59 Most of the ZnO NPs did not show any morphological changes even after 72 h incubation in water, suggesting that the potential release of the Zn2+ ions is very gradual. The notable exceptions were ZnO B and E, both made from coordination compound 2, which showed morphological changes after 72 h.
ZnO NPs Prepared from Pure Precursors Exhibit Selective Cytotoxicity for Triple Negative Breast Cancer Cells
In general, the ZnO NPs prepared from pure precursors showed more selective cytotoxicity toward triple negative breast cancer cells, whereas the ZnO NPs prepared using oleic acid and ultrasonication showed similar cytotoxicity toward both normal and cancer breast cells or they were even less cytotoxic for cancer cells (Figure 3A). The only exceptions were ZnO E, which did not show selective cytotoxicity, and ZnO D, which were selectively cytotoxic toward cancer cells. However, the cancer cell cytotoxicity of ZnO H, made from the same precursor as ZnO D, was much higher. There is no single parameter differentiating NPs, which can be directly responsible for differences in their cytotoxicity. Such differences result from a combination of morphology, size, and Zn content. Based on the current studies, the first requirement for the selective cytotoxicity is a spherical morphology of NPs. The second one is their size, with the upper limit between 385 nm (ZnO E non-selective cytotoxicity) and 325 nm (ZnO F selective cytotoxicity). Finally, Zn content should be as high as possible. The obligatory existence of these three features together explains high selective cytotoxicity of ZnO F-H, and a lack of selectivity in the case of ZnO E (too large) and ZnO A-C (inappropriate morphology: nanowires/hexagonal rods). The moderate selectivity of ZnO D is a result of mixed morphology (spherical NPs and hexagonal rods) and lower Zn content in comparison to ZnO F-H.
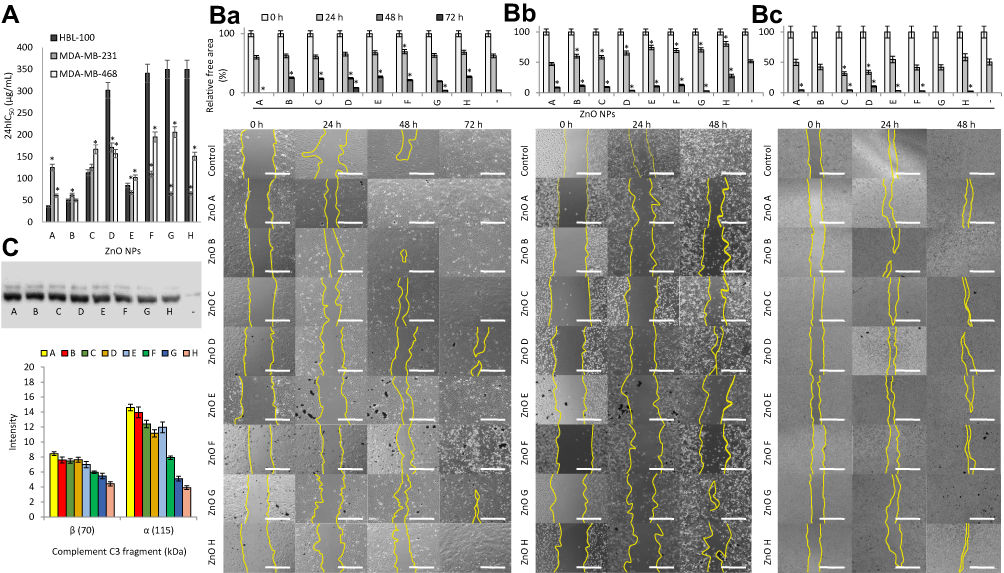 |
Figure 3 ZnO NPs F, G, and H are selectively cytotoxic for triple negative breast cancer cells. (A) The 24hIC50 values of the ZnO NPs for normal breast epithelial cells HBL-100 and triple negative breast cancer cells MDA-MB-231 and MDA-MB-468 as revealed by MTT assay. *Shows significantly (p<0.05) altered 24hIC50 values compared to 24hIC50 values in HBL-100 cells. (B) Micrographs and the relative free area measurements showing the artificial wound healing ability of cells (a, HBL-100; b, MDA-MB-231; c, MDA-MB-468) treated with 24hIC50 concentrations of the ZnO NPs. Scale bar, 400 µm. *Shows significantly (p<0.05) different values of relative free area compared to untreated cells. (C) Opsonization of ZnO NPs with complement component C3 detected by immunoblotting (top) and densitometric quantitation (bottom). Abbreviations: MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NPs, nanoparticles; ZnO, zinc oxide. |
The 24hIC50 values were determined in the range of 65−110 µg/mL for MDA-MB-231, 150−210 µg/mL for MDA-MB-468, and 340−350 µg/mL for HBL-100. This agrees with the results obtained for other cancer cell lines, as 50 nm ZnO nanospheres showed 24hIC50 of 25 µg/mL for tongue squamous cell carcinoma CAL-27.60 Moreover, the viability of the cells declined with a dose-dependent trend (Supplementary Figure S4), indicating that ZnO NPs influence cell metabolism before triggering cell death.61
This selective cytotoxicity toward cancer cells has been observed for other ZnO NPs as well.62 These include ZnO thin film-coated chips selective for lymphocyte Raji cells in comparison to normal peripheral blood mononuclear cells.58 It was proven that this cytotoxicity was facilitated by a release of Zn2+ ions,58 however, no such release was observed in this work. Further, 20 nm ZnO nanospheres showed selective cytotoxicity toward small-cell lung cancer cells, where more than 4× higher concentration of ZnO NPs was required to inhibit the proliferation of normal lung-derived cells. The same effect was also observed upon intravenous application of ZnO NPs into orthotopic mouse models, where no observable adverse effects were elicited.61
Next, as ZnO NPs have been shown to hinder the attachment of adherent cancer cells, as well as cause apoptosis in suspended cancer cells,58 the influence of 24hIC50 of ZnO NPs on cell migration and attachment was studied via a wound healing assay (Figure 3B). In general, while the migration and attachment of normal breast cells and breast cancer cells MDA-MB-468 was hindered by most ZnO NPs only after 48 h of treatment, in the case of breast cancer cells MDA-MB-231, the inhibition was observable after 24 h of treatment with most ZnO NPs. The highest inhibition was observed in MDA-MB-231 cells treated with ZnO H.
ZnO NPs Show Good Biocompatibility
Before further testing, the in vitro prediction of ZnO NPs biocompatibility was performed. First, their biological identity upon encountering a biological environment was tested (Supplementary Figure S5). As shown by the absence of proteins from FBS or plasma, the biological identity of most ZnO NPs was retained in both FBS, used during in vitro cellular experiments, and human plasma, predicting their behavior in vivo. The only notable exceptions were ZnO A and B that interacted with low-molecular proteins from FBS.
Inorganic NPs often rapidly interact with complex biomolecules in biological fluids, forming a biomolecular corona around these NPs and changing their biological identity. This influences their behavior on the NPs-cell level, and resulting in their altered immunocompatibility, biodistribution, pharmacokinetics, and cell interactions.63–66 With regards to the ZnO NPs, their reactivity with phosphate ions67,68 and different plasma proteins69 has been demonstrated, affecting their cytotoxicity, biodistribution, and immune response.59 Also, they were shown to interact with immunocompetent cells in the hemolymph of Bombyx mori, resulting in a declined viability of hemocytes, generation of ROS, alterations of morphology, and apoptosis of these hemocytes.12
Next, the opsonization of ZnO NPs by a complement component C3 contained in human serum was studied, as complement proteins are an important part of the innate immune system, and opsonization by them can lead to rapid clearance of the NPs from the blood.70 All of the studied ZnO NPs were opsonized by complement component C3 (Figure 3C), although the opsonization was significantly lowered for ZnO NPs prepared from pure precursors. This was especially notable for ZnO F, G, and H.
Finally, to understand the hemocompatibility of ZnO NPs, their ability to lyse RBCs was tested (Supplementary Figure S6). All ZnO NPs showed a high rate of hemolysis at 500 µg/mL with the highest by ZnO A. The lowest hemolysis was caused by ZnO H. The hemocompatibility rapidly increased with the decreased concentration of ZnO NPs, as most did not cause any hemolysis at 250 µg/mL. Only ZnO A caused slight hemolysis at 125 µg/mL. As an acceptable rate of hemolysis is ~5%,71 ZnO C-H were hemocompatible at a concentration as high as 250 µg/mL and ZnO A and B at 125 µg/mL.
Overall, even though the size of the tested ZnO NPs was quite large, they showed good biocompatibility, especially ZnO NPs produced from pure precursors. For this reason, they were deemed suitable for further testing.
ZnO NPs Exposure Distinctly Imbalances Redox Homeostasis in Triple Negative Breast Cancer Cells
To further elucidate the mechanism of their selective cytotoxicity, ZnO NPs were subjected to testing of their interaction with DNA. For this purpose, their ability to replace EtBr, a conventional double stranded DNA intercalator, was investigated. EtBr has a low binding affinity of 105 M−1.72 which is why substances with higher binding affinity to DNA can easily replace EtBr and decrease its fluorescence.31 For all ZnO NPs, their dose-dependent displacement of EtBr intercalated into double stranded DNA was proven (Figure 4A), which might be facilitated by the free Zn2+ ions.73 The highest displacement was shown for ZnO A and the lowest for ZnO C.
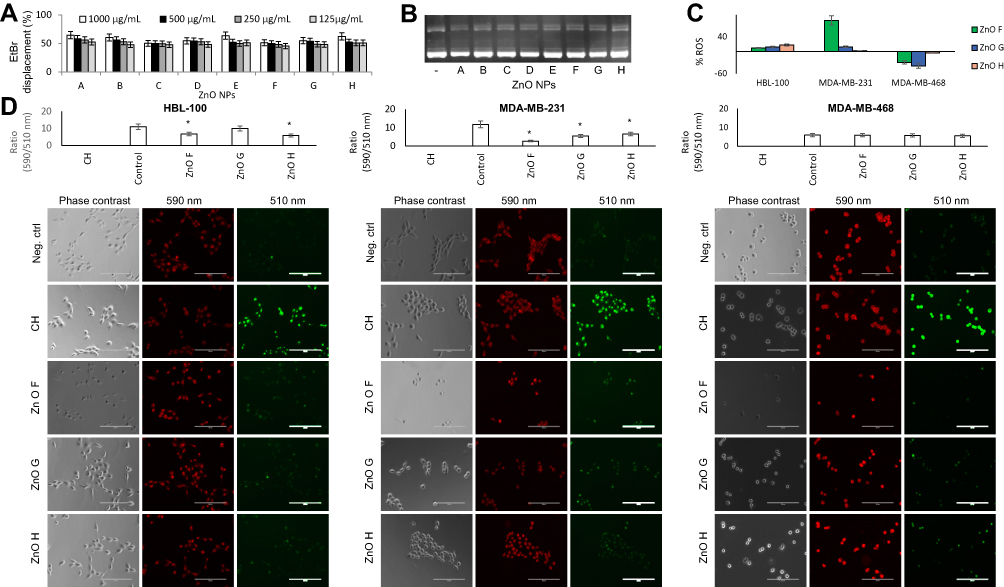 |
Figure 4 Redox homeostasis is imbalanced in the triple negative breast cancer cells. (A) EtBr displacement caused by interaction of DNA with various concentrations of ZnO NPs. (B) Agarose gel showing the stability of EtBr-stained plasmid DNA exposed to ZnO NPs. (C) The quantitative determination of ROS production in cells exposed to ZnO F, G, and H compared to the untreated cells. (D) Quantitative evaluation and representative micrographs showing lipid peroxidation in cells exposed to ZnO F, G, and H. Quantitation is represented as a ratio of fluorescence intensity upon 590 nm irradiation compared with the fluorescence intensity upon 510 nm irradiation. Scale bar, 200 µm. *Shows significantly (p<0.05) altered lipid peroxidation compared to the untreated cells. Abbreviations: CH, cumene hydroperoxide; EtBr, ethidium bromide; NPs, nanoparticles; ROS, reactive oxygen species; ZnO, zinc oxide. |
For small, 20 nm ZnO NPs, intercalation has been proven to cause genotoxicity and DNA leakage from nuclei, not only in immortalized cell lines but also in situ in mice orthotopic xenograft models.61 However, the ZnO NPs prepared in this work did not cause any fragmentation of the plasmid DNA (Figure 4B).
Generally, one of the main mechanisms behind NPs cytotoxicity is their effect on redox homeostasis,32 which can cause damage to various cellular structures,74 such as mitochondrial oxidative damage60 and activation of signaling cascades leading to apoptosis.74 For ZnO NPs, increased cellular ROS was found in lymphocyte cancer cells,58 hemocytes,12 lung small-cell carcinoma,61 tongue squamous cell carcinoma,60 and colorectal adenocarcinoma.61 The ROS generation can be caused by ZnO semiconductor properties,62 or it can be the result of proinflammatory cell response against the NPs.59 The increased ROS caused by ZnO NPs was proven to be of both peroxide and superoxide species, where the peroxide levels were increased even after 5 days of exposure.12 It was also found that although ROS was increased in cancer and normal cells, normal cells were less liable to it.61
Interestingly, the influence of ZnO NPs prepared in this work (55 µg/mL in all groups) varied based on the exposed cell line (Figure 4C). Normal epithelial cells HBL-100 showed only a slight increase in intracellular ROS generation, with the highest increase for ZnO H. In MDA-MB-231 cells ZnO F caused an increase of ROS by 85% compared to the untreated control and ZnO G and H caused only a slight increase of ROS. On the other hand, ROS generation in MDA-MB-468 was significantly decreased by both ZnO F and G and slightly decreased by ZnO H, suggesting that the cytotoxicity mechanism is different for each of the triple negative breast cancer cells. As cancer cells require very precisely tuned ROS levels,75 decreased ROS generation can misbalance cellular signaling in cancer cells and hinder cell growth, proliferation, and protein synthesis.76
Often, increased intracellular ROS levels can lead to oxidative degradation of lipids in cell membrane, resulting in cell damage.77 In line with ROS induction, increased lipid peroxidation was observed in MDA-MB-231 cells treated with all three ZnO NPs (Figure 4D), whereas no significant changes were observed in MDA-MB-468 cells. The results further support the distinct mode of action in the two triple negative breast cancer cells.
ZnO NPs Exposure Leads to Morphological Changes in Cells
The ZnO NPs internalized more in both types of triple negative breast cancer cells than in the normal epithelial cells (Figure 5, Supplementary Figure S7, and Supplementary Figure S8). Moreover, in cancer cells, the ZnO NPs were evenly localized throughout the cytoplasm, whereas in HBL-100, they formed small aggregates near the cytoplasmic perinuclear area. This propensity to accumulate in the perinuclear area has been observed in the past for ZnO nanocrystals internalized into papilloma cells.59
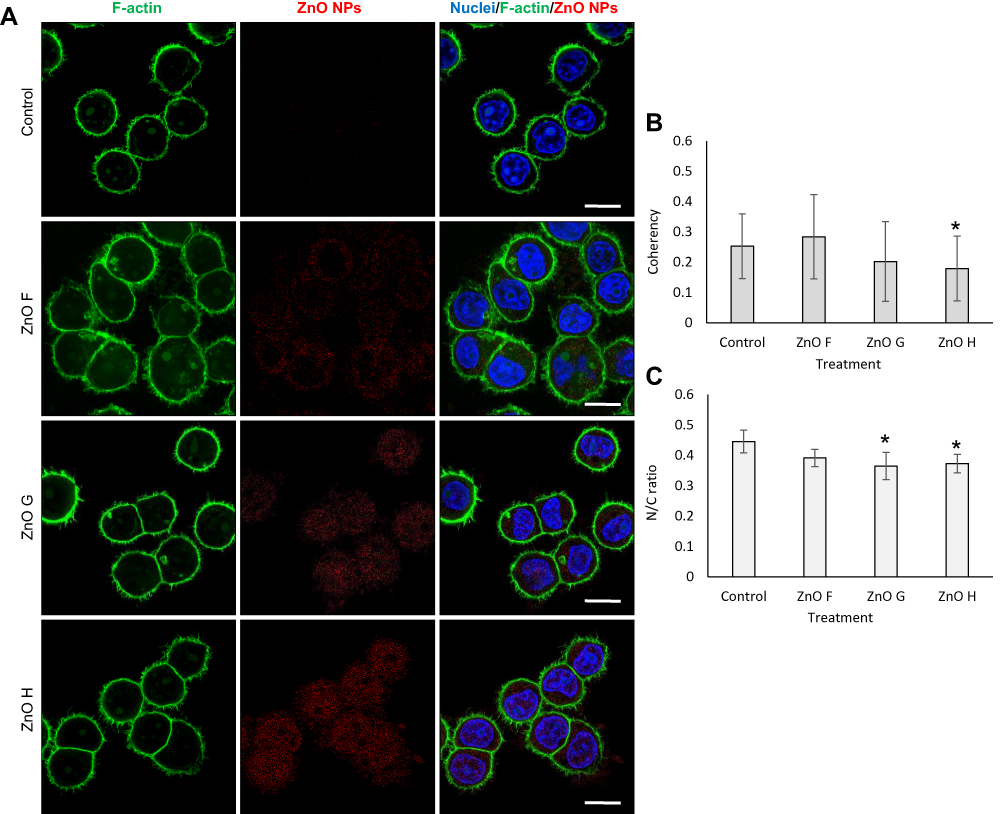 |
Figure 5 ZnO NPs distribute evenly in the cytoplasm of cancer cells. (A) Representative confocal micrographs showing MDA-MB-468 cells treated with ZnO NPs for 24 h. F-actin was stained with phalloidin and ZnO NPs were imaged using their reflectance. Scale bar, 10 µm. (B) The coherency of F-actin filaments. *Shows significantly (p<0.05) altered coherency compared to the untreated cells. (C) N/C ratio of cells. *Shows significantly (p<0.05) altered N/C ratio compared to untreated cells. Abbreviations: N/C, nuclei-to-cytoplasm; NPs, nanoparticles; ZnO, zinc oxide. |
Upon internalization, marked changes to cytoskeleton coherency were found in the MDA-MB-468 cells exposed to ZnO H and changes to N/C ratio in cells exposed to both ZnO G and H (Figure 5), suggesting that this treatment led to an increase in the cellular area through cytoplasm extension and reorganization of the cytoskeletal network.32 We believe that these morphological changes, together with the decreased ROS could explain the distinct cytotoxicity of the ZnO NPs in MDA-MB-468.32 In HBL-100, a decrease in cytoskeleton coherency was found upon exposure to ZnO H and an increase in cytoskeleton coherency upon exposure to ZnO F (Supplementary Figure S7). On the other hand, no significant influence on either the N/C ratio or cytoskeleton coherency was found for MDA-MB-231 (Supplementary Figure S8).
Furthermore, shrinkage of the whole cells was observed for HBL-100 exposed to all three ZnO NPs, whereas apoptotic protrusions of cell membrane were observed in both triple negative breast cancer cells (Figure 6). Also, for MDA-MB-231, their overall elongated shape markedly changed into spherical shape upon exposure to ZnO H.
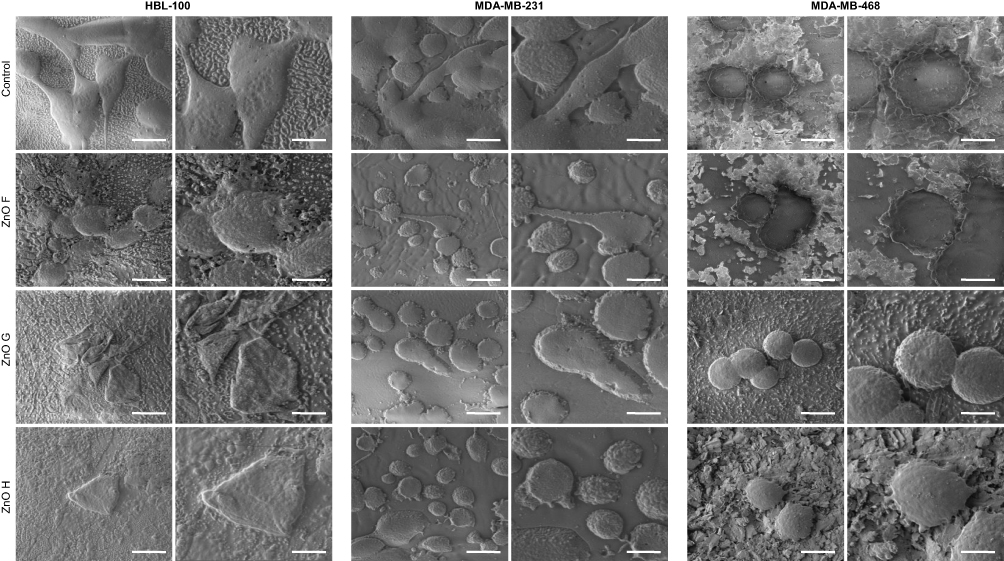 |
Figure 6 Normal epithelial cells shrink in response to ZnO NPs exposure and cancer cells form apoptotic blebs. Representative cryo-SEM micrographs indicating changes in the morphology of cells exposed to ZnO NPs for 6 h. Scale bar, 20 µm (left) and 10 µm (right). Abbreviations: NPs, nanoparticles; SEM, scanning electron microscope; ZnO, zinc oxide. |
The changes in the cell morphology in response to exposure to ZnO NPs are in accordance with the works of others. Multiple structural abnormalities, including blebbing, cell membrane disruption, hypervacuolation, and altered morphology were caused by 40 nm ZnO NPs in hemocytes upon their phagocytosis and diffusion through the plasma membrane.12 Membrane disruption leading to cytoplasmic release was also observed in small-cell lung cancer cells exposed to 20 nm ZnO NPs61 and swollen mitochondria were observed in tongue squamous cell carcinoma cells exposed to 50 nm ZnO NPs.60 Even though morphological changes were visible in cells exposed to ZnO NPs prepared in this work, no membrane disruption was observed.
Distinct Mode of Cell Death Shown for the Triple Negative Breast Cancer Cells
Flow cytometry of cell permeability/translocated phosphatidyl serine dual-stained cells together with May-Grünwald-Giemsa staining (Figure 7) revealed that prolonged ZnO NPs exposure led to slightly increased apoptosis and necrosis in HBL-100 cells and significantly increased number of apoptotic MDA-MB-231 cells. Very large induction of necrosis was observed in MDA-MB-468 cells exposed to ZnO H. The results confirm that whereas in HBL-100 and MDA-MB-231, increased intracellular ROS can lead to induction of apoptosis, the mechanism of action differs for MDA-MB-468 cells.
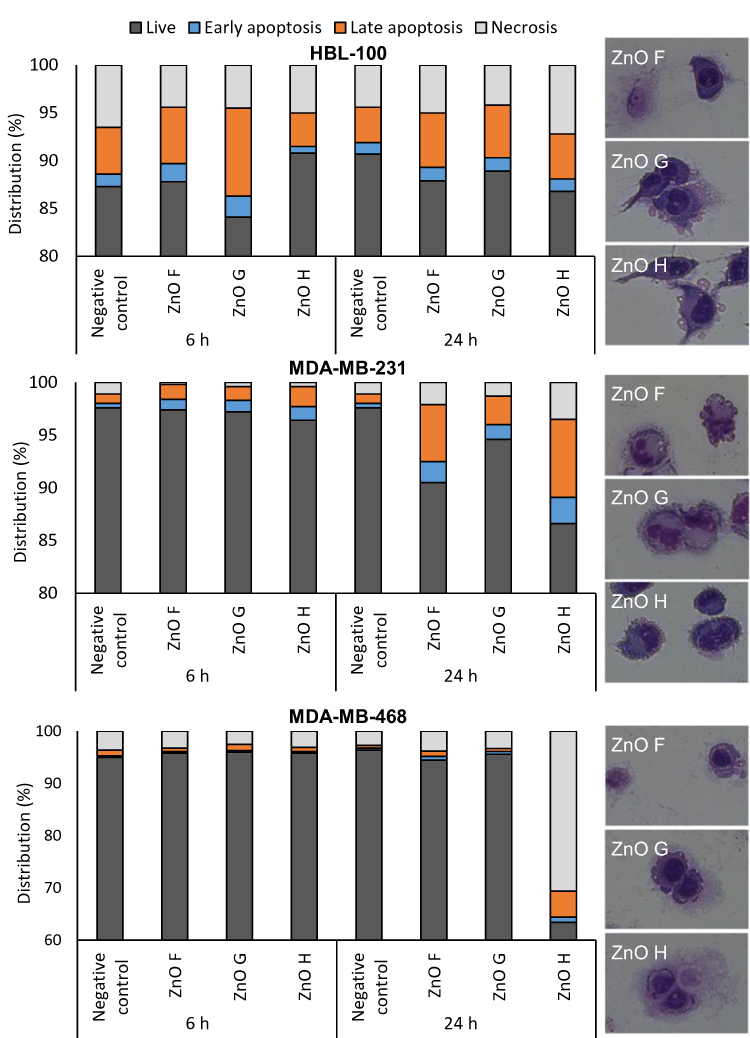 |
Figure 7 ZnO NPs lead to distinct modes of cell death in the exposed cells. Induction of early and late apoptosis and necrosis in cells dual-stained for translocated phosphatidylserine and cell permeability. Insets show representative micrographs of cells stained for cytopathological features according to May-Grünwald-Giemsa. Abbreviations: NPs, nanoparticles; ZnO, zinc oxide. |
In the past, the cytotoxicity of ZnO NPs has been linked to the release of Zn2+ ions inducing apoptosis,58 as well as direct cell-NPs contact,78 leading to necrosis.61 Also, autophagy and mitophagy have been suggested as the mechanisms responsible for cell death upon exposure to ZnO NPs.60 The apoptosis was found to be strongly dependent on caspase induction58 and PARP cleavage.61
Conclusion
This study describes the mechanisms underlying the cytotoxic activity of coordination compound-based ZnO NPs in breast human cells in vitro. Throughout the study, it is shown that even though the current nanoparticles still offer obstacles due to their size, the ones prepared from pure precursors with a combination of a suitable size, morphology, and Zn content intrinsically target hard-to-heal triple negative breast cancer cells with the ability to misbalance their redox homeostasis and morphology, ultimately leading to apoptosis and/or necrosis. We also show their very good biocompatibility and propensity to retain their biological identity in the complex in vivo-mimicking environments. The high Zn content, together with the spherical morphology of the ZnO H shows to possess the highest cytotoxicity for the breast cancer cells, probably due to the influence on cell morphology, migration and adhesion. Small size, such as in ZnO F, leads to the highest increase of intracellular ROS but also toxicity for normal cells. Further fine-tuning of these properties can therefore lead to synthesis of NPs with distinct mode of cytotoxicity. To the best of our knowledge, this is the first study taking a complex look at the intrinsic triple negative breast cancer cell targeting ability of coordination compound-based ZnO NPs. It is clear that this description offers great opportunities for future synthesis of cancer nanomedicines naturally exploiting the responses of triple negative breast cancer cells to ZnO NPs.
Acknowledgments
The authors wish to express their thanks to Lucie Markova for perfect technical assistance.
Funding
The synthesis and characterization of coordination compounds and ZnO nanoparticles, as well as crystallographic part of this work, was financed by funds allocated by the Ministry of Science and Higher Education (Poland) to the Institute of General and Ecological Chemistry, Lodz University of Technology. For testing of the in vitro effects of these nanoparticles, the financial support from ERDF “Multidisciplinary research to increase application potential of nanomaterials in agricultural practice” (No. CZ.02.1.01/0.0/0.0/16_025/0007314), ERC Starting Grant 759585 and the Czech Science Foundation (project no. 19-13766J) are gratefully acknowledged.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jain V, Kumar H, Anod HV, et al. A review of nanotechnology-based approaches for breast cancer and triple-negative breast cancer. J Control Release. 2020;326:628–647. doi:10.1016/j.jconrel.2020.07.003
2. Greish K, Mathur A, Al Zahrani R, et al. Synthetic cannabinoids nano-micelles for the management of triple negative breast cancer. J Control Release. 2018;291:184–195. doi:10.1016/j.jconrel.2018.10.030
3. Saraiva DP, Cabral MG, Jacinto A, Braga S. How many diseases is triple negative breast cancer: the protagonism of the immune microenvironment. ESMO Open. 2017;2(4):11. doi:10.1136/esmoopen-2017-000208
4. Sinha R, Kim GJ, Nie SM, Shin DM. Nanotechnology in cancer therapeutics: bioconjugated nanoparticles for drug delivery. Mol Cancer Ther. 2006;5(8):1909–1917. doi:10.1158/1535-7163.MCT-06-0141
5. Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. doi:10.1016/S0168-3659(99)00248-5
6. Nakamura Y, Mochida A, Choyke PL, Kobayashi H. Nanodrug delivery: is the enhanced permeability and retention effect sufficient for curing cancer? Bioconjugate Chem. 2016;27(10):2225–2238. doi:10.1021/acs.bioconjchem.6b00437
7. Fang KJ, Wang LF, Huang HY, Dong SW, Guo YL. Therapeutic efficacy and cardioprotection of nucleolin-targeted doxorubicin-loaded ultrasound nanobubbles in treating triple-negative breast cancer. Nanotechnology. 2021;32(24):13. doi:10.1088/1361-6528/abed03
8. Grewal IK, Singh S, Arora S, Sharma N. Polymeric nanoparticles for breast cancer therapy: a comprehensive review. Biointerface Res Appl Chem. 2021;11(4):11151–11171.
9. Mohamed H, Asker M, Kotb N, Abdelwahab H. Preparation and characterization of folate decorated 5-fluro uracil loaded albumin nanoparticles and in vitro evaluation of its cytotoxic effect. Biointerface Res Appl Chem. 2021;11(3):10412–10428.
10. Sirelkhatim A, Mahmud S, Seeni A, et al. Review on zinc oxide nanoparticles: antibacterial activity and toxicity mechanism. Nano Micro Letters. 2015;7(3):219–242. doi:10.1007/s40820-015-0040-x
11. Buzea C, Pacheco II, Robbie K. Nanomaterials and nanoparticles: sources and toxicity. Biointerphases. 2007;2(4):MR17–71. doi:10.1116/1.2815690
12. Mir AH, Qamar A, Qadir I, Naqvi AH, Begum R. Accumulation and trafficking of zinc oxide nanoparticles in an invertebrate model, Bombyx mori, with insights on their effects on immuno-competent cells. Sci Rep. 2020;10(1):14. doi:10.1038/s41598-020-58526-1
13. Akhtar MJ, Ahamed M, Kumar S, Khan MAM, Ahmad J, Alrokayan SA. Zinc oxide nanoparticles selectively induce apoptosis in human cancer cells through reactive oxygen species. Int J Nanomed. 2012;7:845–857.
14. Deng YX, Zhang HJ. The synergistic effect and mechanism of doxorubicin-ZnO nanocomplexes as a multimodal agent integrating diverse anticancer therapeutics. Int J Nanomed. 2013;8:1835–1841.
15. Kollur SP, Prasad SK, Pradeep S, et al. Luteolin-fabricated ZnO nanostructures showed PLK-1 mediated anti-breast cancer activity. Biomolecules. 2021;11(3):21. doi:10.3390/biom11030385
16. Shamasi Z, Es-haghi A, Yazdi MET, Amiri MS, Homayouni-Tabrizi M. Role of Rubia tinctorum in the synthesis of zinc oxide nanoparticles and apoptosis induction in breast cancer cell line. Nanomed J. 2021;8(1):65–72.
17. D’Souza JN, Prabhu A, Nagaraja GK, Navada KM, Kouser S, Manasa DJ. Unravelling the human triple negative breast cancer suppressive activity of biocompatible zinc oxide nanostructures influenced by Vateria indica (L.) fruit phytochemicals. Mater Sci Eng C Mater Biol Appl. 2021;122:17. doi:10.1016/j.msec.2021.111887
18. Pouresmaeil V, Haghighi S, Raeisalsadati AS, Neamati A, Homayouni-Tabrizi M. The anti-breast cancer effects of green-synthesized zinc oxide nanoparticles using carob extracts. Anti Cancer Agents Med Chem. 2021;21(3):316–326. doi:10.2174/1871520620666200721132522
19. Cho WS, Duffin R, Howie SEM, et al. Progressive severe lung injury by zinc oxide nanoparticles; the role of Zn2+ dissolution inside lysosomes. Part Fibre Toxicol. 2011;8:16. doi:10.1186/1743-8977-8-27
20. Cho WS, Duffin R, Thielbeer F, et al. Zeta potential and solubility to toxic ions as mechanisms of lung inflammation caused by metal/metal oxide nanoparticles. Toxicol Sci. 2012;126(2):469–477. doi:10.1093/toxsci/kfs006
21. Li CH, Shen CC, Cheng YW, et al. Organ biodistribution, clearance, and genotoxicity of orally administered zinc oxide nanoparticles in mice. Nanotoxicology. 2012;6(7):746–756. doi:10.3109/17435390.2011.620717
22. Wang B, Feng WY, Wang M, et al. Acute toxicological impact of nano- and submicro-scaled zinc oxide powder on healthy adult mice. J Nanopart Res. 2008;10(2):263–276. doi:10.1007/s11051-007-9245-3
23. Swiatkowski M, Kruszynski R. Structurally diverse coordination compounds of zinc as effective precursors of zinc oxide nanoparticles with various morphologies. Appl Organomet Chem. 2019;33(4):e4812. doi:10.1002/aoc.4812
24. Welcher FJ. The Analytical Uses of Ethylenediamine Tetraacetic Acid. Van Nostrand; 1965.
25. Sheldrick GM. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr Sect A. 2015;71:3–8. doi:10.1107/S2053273314026370
26. Sheldrick GM. Crystal structure refinement with SHELXL. Acta Crystallogr Sect C Struct Chem. 2015;71:3–8. doi:10.1107/S2053229614024218
27. Sheldrick GM. A short history of SHELX. Acta Crystallogr Sect A. 2008;64:112–122. doi:10.1107/S0108767307043930
28. Wilson AJ, Geist V. International Tables for Crystallography, Volume C: Mathematical, Physical and Chemical Tables.
29. International Center of Diffraction Data. Powder diffraction file. 2003.
30. Tesarova B, Dostalova S, Smidova V, et al. Surface-PASylation of ferritin to form stealth nanovehicles enhances in vivo therapeutic performance of encapsulated ellipticine. Appl Mater Today. 2020;18:11.
31. Tesarova B, Charousova M, Dostalova S, et al. Folic acid-mediated re-shuttling of ferritin receptor specificity towards a selective delivery of highly cytotoxic nickel(II) coordination compounds. Int J Biol Macromol. 2019;126:1099–1111. doi:10.1016/j.ijbiomac.2018.12.128
32. Michalkova H, Skubalova Z, Sopha H, et al. Complex cytotoxicity mechanism of bundles formed from self-organised 1-D anodic TiO2 nanotubes layers. J Hazard Mater. 2020;388:12. doi:10.1016/j.jhazmat.2020.122054
33. Clemons TD, Bradshaw M, Toshniwal P, et al. Coherency image analysis to quantify collagen architecture: implications in scar assessment. RSC Adv. 2018;8(18):9661–9669. doi:10.1039/C7RA12693J
34. Blair J, Howie RA, Wardell JL. Structure of monoclinic zinc N-butanoate. Acta Crystallogr Sect C Cryst Struct Commun. 1993;49:219–221. doi:10.1107/S0108270192007108
35. Clegg W, Little IR, Straughan BP. Orthorhombic andhydrous zinc(II) propionate. Acta Crystallogr Sect C Cryst Struct Commun. 1987;43:456–457. doi:10.1107/S0108270187095398
36. Goldschmied E, Rae AD, Stephenson NC. The crystal structure of ZnII propionate (C6H10O4Zn)n. Acta Crystallogr Sect B Struct Crystallogr Cryst Chem. 1977;33:2117–2120. doi:10.1107/S0567740877007857
37. Favas MC, Kepert DL. Aspects of the stereochemistry of eight-coordination. In: Progress in Inorganic Chemistry. Wiley Online Libraray; 1978:179–249.
38. Kepert DL. Aspects of the Stereochemistry of Six-Coordination. Vol. 23. New York, USA: John Wiley; 1977.
39. Blatov VA, Shevchenko AP, Proserpio DM. Applied topological analysis of crystal structures with the program package ToposPro. Cryst Growth Des. 2014;14(7):3576–3586. doi:10.1021/cg500498k
40. Brown ID. Influence of chemical and spatial constraints on the structures of inorganic compounds. Acta Crystallogr Sect B Struct Commun. 1997;53:381–393. doi:10.1107/S0108768197002474
41. Zachariasen WH. Bond lengths in oxygen and halogen compounds of D-element and F-element. J Less-Common Metals. 1978;62:1–7. doi:10.1016/0022-5088(78)90010-3
42. Trzesowska A, Kruszynski R, Bartczak TJ. New bond-valence parameters for lanthanides. Acta Crystallogr Sect B Struct Sci Cryst Eng Mat. 2004;60:174–178. doi:10.1107/S0108768104002678
43. Trzesowska A, Kruszynski R, Bartczak TJ. New lanthanide-nitrogen bond-valence parameters. Acta Crystallogr Sect B Struct Sci Cryst Eng Mat. 2005;61:429–434. doi:10.1107/S0108768105016083
44. Trzesowska A, Kruszynski R, Bartczak TJ. Bond-valence parameters of lanthanides. Acta Crystallogr Sect B Struct Commun. 2006;62:745–753. doi:10.1107/S0108768106016429
45. Kruszynski R. Synthesis of coordination compounds via dehalogenation of zinc bromoacetate in presence of some amines. Inorg Chim Acta. 2011;371(1):111–123. doi:10.1016/j.ica.2011.03.049
46. Brese NE, Okeeffe M. Bond-valence parameters for solids. Acta Crystallogr Sect B Struct Commun. 1991;47:192–197. doi:10.1107/S0108768190011041
47. Brown ID. Chemical and steric constraints in inorganic solids. Acta Crystallogr Sect B Struct Commun. 1992;48:553–572. doi:10.1107/S0108768192002453
48. Jensen JO. Vibrational frequencies and structural determinations of hexamethylenetetraamine. Spectroc Acta Pt A Molec Biomolec Spectr. 2002;58(7):1347–1364. doi:10.1016/S1386-1425(01)00585-6
49. Kruszynski R, Sieranski T, Bilinska A, Bernat T, Czubacka E. Alkali metal halogenides coordination compounds with hexamethylenetetramine. Struct Chem. 2012;23(5):1643–1656. doi:10.1007/s11224-012-9961-x
50. Kruszynski R, Sieranski T, Swiatkowski M, et al. On the coordination behaviour of the hmta toward alkali metal cations in presence of perchlorate anions. J Chem Crystallogr. 2015;45(10–12):484–494. doi:10.1007/s10870-015-0618-7
51. Sieranski T, Kruszynski R. On the governing of alkaline earth metal nitrate coordination spheres by hexamethylenetetramine. J Coord Chem. 2013;66(1):42–55. doi:10.1080/00958972.2012.744835
52. Sieranski T, Kruszynski R. Magnesium sulphate complexes with hexamethylenetetramine and 1,10-phenanthroline Thermal, structural and spectroscopic properties. J Therm Anal Calorim. 2012;109(1):141–152. doi:10.1007/s10973-011-1693-4
53. Taylor Z, Marucho M. The self-adaptation ability of zinc oxide nanoparticles enables reliable cancer treatments. Nanomaterials. 2020;10(2):18. doi:10.3390/nano10020269
54. Kobayashi H, Watanabe R, Choyke PL. Improving conventional enhanced permeability and retention (EPR) effects; What is the appropriate target? Theranostics. 2014;4(1):81–89. doi:10.7150/thno.7193
55. Li SD, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5(4):496–504. doi:10.1021/mp800049w
56. Corazzari I, Gilardino A, Dalmazzo S, Fubini B, Lovisolo D. Localization of CdSe/ZnS quantum dots in the lysosomal acidic compartment of cultured neurons and its impact on viability: potential role of ion release. Toxicol Vitro. 2013;27(2):752–759. doi:10.1016/j.tiv.2012.12.016
57. Hunley C, Marucho M. Electrical double layer properties of spherical oxide nanoparticles. Phys Chem Chem Phys. 2017;19(7):5396–5404. doi:10.1039/C6CP08174F
58. Moon SH, Choi WJ, Choi SW, et al. Anti-cancer activity of ZnO chips by sustained zinc ion release. Toxicol Rep. 2016;3:430–438. doi:10.1016/j.toxrep.2016.03.008
59. Dumontel B, Susa F, Limongi T, et al. ZnO nanocrystals shuttled by extracellular vesicles as effective Trojan nano-horses against cancer cells. Nanomedicine. 2019;14(21):2815–2833. doi:10.2217/nnm-2019-0231
60. Wang JF, Gao ST, Wang SY, Xu ZN, Wei LM. Zinc oxide nanoparticles induce toxicity in CAL 27 oral cancer cell lines by activating PINK1/Parkin-mediated mitophagy. Int J Nanomed. 2018;13:3441–3450. doi:10.2147/IJN.S165699
61. Tanino R, Amano Y, Tong XX, et al. Anticancer activity of ZnO nanoparticles against human small-cell lung cancer in an orthotopic mouse model. Mol Cancer Ther. 2020;19(2):502–512. doi:10.1158/1535-7163.MCT-19-0018
62. Hanley C, Layne J, Punnoose A, et al. Preferential killing of cancer cells and activated human T cells using ZnO nanoparticles. Nanotechnology. 2008;19(29):10. doi:10.1088/0957-4484/19/29/295103
63. Cedervall T, Lynch I, Lindman S, et al. Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc Natl Acad Sci U S A. 2007;104(7):2050–2055. doi:10.1073/pnas.0608582104
64. Lynch I, Dawson KA. Protein-nanoparticle interactions. Nano Today. 2008;3(1–2):40–47. doi:10.1016/S1748-0132(08)70014-8
65. Monopoli MP, Aberg C, Salvati A, Dawson KA. Biomolecular coronas provide the biological identity of nanosized materials. Nat Nanotechnol. 2012;7(12):779–786. doi:10.1038/nnano.2012.207
66. Nel AE, Madler L, Velegol D, et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8(7):543–557. doi:10.1038/nmat2442
67. Laurenti M, Lamberti A, Genchi GG, et al. Graphene oxide finely tunes the bioactivity and drug delivery of mesoporous ZnO scaffolds. ACS Appl Mater Interfaces. 2019;11(1):449–456. doi:10.1021/acsami.8b20728
68. Reed RB, Ladner DA, Higgins CP, Westerhoff P, Ranville JF. Solubility of nano-zinc oxide in environmentally and biologically important matrices. Environ Toxicol Chem. 2012;31(1):93–99. doi:10.1002/etc.708
69. Deng ZJ, Mortimer G, Schiller T, Musumeci A, Martin D, Minchin RF. Differential plasma protein binding to metal oxide nanoparticles. Nanotechnology. 2009;20(45):9. doi:10.1088/0957-4484/20/45/455101
70. Noris M, Remuzzi G. Overview of complement activation and regulation. Semin Nephrol. 2013;33(6):479–492. doi:10.1016/j.semnephrol.2013.08.001
71. Dobrovoiskaia MA, Clogston JD, Neun BW, Hall JB, Patri AK, McNeil SE. Method for analysis of nanoparticle hemolytic properties in vitro. Nano Lett. 2008;8(8):2180–2187. doi:10.1021/nl0805615
72. Luedtke NW, Hwang JS, Nava E, Gut D, Kol M, Tor Y. The DNA and RNA specificity of eilatin Ru(II) complexes as compared to eilatin and ethidium bromide. Nucleic Acids Res. 2003;31(19):5732–5740. doi:10.1093/nar/gkg758
73. Pastre D, Pietrement O, Zozime A, Le Cam E. Study of the DNA/ethidium bromide interactions on mica surface by atomic force microscope: influence of the surface friction. Biopolymers. 2005;77(1):53–62. doi:10.1002/bip.20185
74. Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta Mol Cell Res. 2016;1863(12):2977–2992. doi:10.1016/j.bbamcr.2016.09.012
75. Aggarwal V, Tuli HS, Varol A, et al. Role of reactive oxygen species in cancer progression: molecular mechanisms and recent advancements. Biomolecules. 2019;9(11):26. doi:10.3390/biom9110735
76. Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010;44(5):479–496. doi:10.3109/10715761003667554
77. Ayala A, Munoz MF, Arguelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med Cell Longev. 2014;2014:31. doi:10.1155/2014/360438
78. Moos PJ, Chung K, Woessner D, Honeggar M, Cutler NS, Veranth JM. ZnO particulate matter requires cell contact for toxicity in human colon cancer cells. Chem Res Toxicol. 2010;23(4):733–739. doi:10.1021/tx900203v
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.