")
Back to Journals » Journal of Inflammation Research » Volume 14
The 4G/5G Polymorphism of Plasminogen Activator Inhibitor Type 1 is a Predictor of Moderate-to-Severe Thyroid Eye Disease
Authors Katko M , Galgoczi E, Erdei A, Gazdag A, Berta E, Bodor M, Seres I, Hircsu I, Badics A, Ujhelyi B, Sira L, Bhattoa HP, Nagy EV
Received 17 February 2021
Accepted for publication 31 March 2021
Published 12 May 2021 Volume 2021:14 Pages 1883—1890
DOI https://doi.org/10.2147/JIR.S307046
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Monika Katko,1 Erika Galgoczi,1 Annamaria Erdei,1 Annamaria Gazdag,1 Eszter Berta,1 Miklos Bodor,1 Ildiko Seres,2 Ildiko Hircsu,1 Arpad Badics,1 Bernadett Ujhelyi,3 Livia Sira,1 Harjit Pal Bhattoa,4 Endre V Nagy1
1Division of Endocrinology, Department of Internal Medicine, Faculty of Medicine, University of Debrecen, Debrecen, Hungary; 2Division of Metabolic Diseases, Department of Internal Medicine, Faculty of Medicine, University of Debrecen, Debrecen, Hungary; 3Department of Ophthalmology, Faculty of Medicine, University of Debrecen, Debrecen, Hungary; 4Department of Laboratory Medicine, Faculty of Medicine, University of Debrecen, Debrecen, Hngary
Correspondence: Endre V Nagy
Division of Endocrinology, Department of Internal Medicine, Faculty of Medicine, University of Debrecen, 98 Nagyerdei krt, Debrecen, 4032, Hungary
Tel +36-30-337-1244
Email [email protected]
Introduction: Thyroid eye disease (TED) is an autoimmune disease of the orbits. Once developed, complete cure is rare. Plasminogen activator inhibitor type 1 (PAI-1) contributes to remodeling of connective tissue and has a central role in the pathogenesis of TED. We aimed to test if the 4G/5G polymorphism of PAI-1 is a predictor of the development of moderate-to-severe TED.
Methods: A total of 185 patients with Graves’ disease, 87 of them with TED, 98 without TED, as well as 201 healthy controls, were studied. Genomic DNA was isolated from peripheral blood samples. The 4G/5G polymorphism of the PAI-1 gene was analyzed by allele-specific PCR, and the distribution of genotypes was calculated in each group. Plasma PAI-1 and thyroid hormone levels were measured by ELISA and ECLIA, respectively.
Results: The 4G/4G genotype was associated with the development of moderate-to-severe TED (OR = 2.54; 95% CI: 1.26– 5.14; p < 0.01). The 4G/5G polymorphism of PAI-1 was not a predictor of plasma PAI-1 levels.
Conclusion: The 4G/4G genotype of PAI-1 is a risk factor for the development of moderate-to-severe TED. Patients with Graves’ disease who harbor this genotype may be candidates for special attention towards the development of TED.
Keywords: thyroid eye disease, Graves’ disease, PAI-1, 4G/5G polymorphism, risk factor
Introduction
Thyroid eye disease (TED) is an autoimmune disease of the orbital tissues which accompanies Graves’ disease (GD) in 30% of the cases. This disfiguring disease is affecting mostly women. The facial character change has a deep and long-continued impact on quality of life.1,2 Once developed, complete cure is rare. The distinction between mild and moderate-to-severe TED is substantiated as both the prognosis and the treatment modalities used are different in these stages.3 The most severe cases may result in the loss of vision. Up to now, only few genetic risk factors for TED have been identified.4
Recently, we have shown that plasminogen activator inhibitor-1 (PAI-1) driven remodeling in the orbital connective tissue has a central role in the pathogenesis of TED.5 PAI-1, a clade E member of the serine protease inhibitor (SERPIN) family, is the main regulator of fibrinolysis and plasmin dependent pericellular proteolysis by inhibiting tissue-type plasminogen activator (t-PA) and urokinase-type plasminogen activator (u-PA) [reviewed in6,7 and8]. Increased PAI-1 expression and/or activity stabilizes extracellular matrix and inhibits its clearance via inhibition of plasmin and subsequent matrix metalloproteinase activation. This maintains a supporting scaffold for proliferation and leads to connective tissue expansion, which has a key role during the course of TED.9 In addition, PAI-1 may be able to mediate the immune response independent of its influence on plasmin activity; PAI-1 contributes to innate immunity, prolongs the duration of inflammation by blocking the clearance of apoptotic neutrophils10 and by activation of macrophages through Toll-like receptor 4 (TLR4)11 acting as a damage-associated molecular pattern (DAMP).9 DAMPs could be held responsible for the unresolved chronic inflammation leading to autoimmune thyroid diseases like GD.12 Elevated plasma PAI-1 levels in GD due to hyperthyroidism were described and interpreted as a risk factor for hyperthyroidism induced endothelial dysfunction and hypercoagulation.13–15
A common 4G/5G single guanine insertion/deletion polymorphism in the promoter region of the PAI-1 gene is of functional importance in regulating PAI-1 expression.16 The presence of the 4G allele does not influence serum PAI-1 levels under normal conditions; however, it is linked to higher PAI-1 level and/or activity under the effect of certain factors, including interleukin-1α (IL-1α),16,17 angiotensin II,18 very low-density lipoprotein,19 and upstream stimulatory factor 1 (USF-1) transcription factor.20
We aimed to test if the 4G/5G polymorphism of PAI-1 is a predictor of the development and severity of TED in patients with GD.
Materials and Methods
Two hundred and twenty unrelated patients with Graves’ disease (GD) in the database of the Endocrine Clinic of the University of Debrecen were studied. The inclusion criterion was a history of GD that started more than 2 years ago; the median time since the onset of GD was 10 years (minimum 2 years and maximum 53 years). The presence/absence of TED was verified. Severity was defined as the most severe stage detected during the course of TED according to the ETA-EUGOGO severity scale;3 if this could not be verified retrospectively, the patient was excluded from the analysis. Another exclusion criterion was the initiation of interferon or insulin therapy between the diagnosis of GD and the onset of TED. The study population of this case–control study included 185 GD patients: 87 with TED and 98 without TED, as well as 201 sex-matched controls who were unrelated healthy volunteers living in the same area as the patients.
Blood was drawn from patients and controls and the resulting whole blood, serum and plasma samples were stored at −70°C until use. Genomic DNA was isolated from peripheral blood samples by silica adsorption method using the NucleoSpin Blood kit (Macherey-Nagel GmbH, Düren, Germany) according to the manufacturer’s instructions. The concentration and purity of the isolated genomic DNA were measured by a Nanodrop 2000 (Thermo Fisher Scientific Inc, Waltham, MA, USA) spectrophotometer. To detect the PAI-1 4G/5G polymorphism, an allele-specific polymerase chain reaction (PCR) method was used as described by Prabhudesai et al.21 Two PCR reactions were prepared per sample, each containing a common oligonucleotide primer and an internal standard primer (which is not specific for either allele but serves as a control for the PCR reaction) and a primer specific for the 4G or 5G allele per reaction. The oligonucleotide sequences of the primers were as follows: forward primer (internal control): 5ʹ-AAGCTTTTACCATGGTAACCCCTGGT-3ʹ, 4G allele primer: 5ʹ-GTCTGGACACGTGGGGA-3ʹ, 5G allele primer: 5ʹ-GTCTGGACACGTGGGGGA-3ʹ, and the common downstream primer: 5ʹ-TGCAGCCAGCCACGTGATTGTCTA-3ʹ. The primers used were manufactured by Eurofins Genomics (Ebersberg, Germany). PCR reactions were performed in a volume of 20 µL with the following composition: 100 ng DNA sample, 2 µL 10x buffer, 1 µL allele-specific primer (10 pmol), 1 µL common primer (10 pmol), 0.2 µL internal standard primer (10 pmol), 1 µL of 25 mM MgCl2, 0.2 µL of 25 mM deoxyribonucleotide triphosphate (dNTP) mix, 0.2 µL of 5U/µL Taq polymerase (Thermo Fisher Scientific Inc, Waltham, MA, USA) in nuclease free water in a Bio-Rad T100™ Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, USA). The setup was 94°C for 5 minutes, 30 cycles: 94°C for 1 minute, 60°C for 50 seconds and 72 °C for 1 minute 15 seconds, then 72°C for 5 minutes and storage at 4°C. PCR products were separated by gel electrophoresis (2% agarose gel, 100 V, 50 minutes) using a Wide Mini-Sub Cell GT System (Bio-Rad Laboratories, Hercules, CA, USA), stained with GelRed (Biotium Inc, Fremont, CA, USA) and visualized using a UV transilluminator (Analytik Jena, Jena, Germany).
The concentration of PAI-1 protein in plasma samples was measured by ELISA (PAI-1 DuoSet ELISA kit, R&D Systems, Minneapolis, USA). Serum thyrotropin (TSH), free thyroxine (fT4) and free triiodothyronine (fT3) were measured by ECLIA (Roche Diagnostics GmbH, Mannheim, Germany). The reference ranges are: TSH: 0.3–4.2 mU/L, fT4: 12–22 pmol/L and fT3: 2.4–6.3 pmol/L.
GraphPad Prism (GraphPad Software, San Diego, CA, USA) and STATISTICA (StatSoft Inc., Tulsa, OK, USA) software were used for statistical analyses. Genotype and allele frequencies were expressed in absolute numbers and percentages. Plasma PAI-1 and serum TSH, fT4 and fT3 concentrations were expressed as median and interquartile range. In controls, Hardy–Weinberg equilibrium was checked by Chi-square test. Comparison of genotype and allele frequencies was also performed by Chi-square for trend test in an additive (5G/5G vs 4G/5G vs 4G/4G), and Chi-square test in a multiplicative (4G vs 5G), a dominant (4G/4G + 4G/5G vs 5G/5G), and a recessive (4G/4G vs 4G/5G + 5G/5G) model. The additive model indicates that the risk of disease is increased r-fold (factor r > 1) for genotype 4G/5G and by 2r-fold for genotype 4G/4G relative to the genotype 5G/5G. The multiplicative model indicates that the risk of disease is increased r-fold with each additional 4G allele. The dominant model indicates that either one or two copies of 4G allele are required for an r-fold increase in disease risk, while the recessive model indicates that two copies of 4G allele are required for an r-fold increase in disease risk.22 Risk assessment was performed by calculating odds ratio (OR) with 95% confidence interval (CI). Chi-square test and Kruskal–Wallis H-test were used to compare demographic and biochemical parameters. A value of p <0.05 was considered statistically significant. The Bonferroni correction was applied for comparisons with multiple genetic models.
Results
The genotype distribution and the allele frequency of the 4G/5G polymorphism of PAI-1 did not differ between patients with or without TED, if all TED patients were included in the analysis irrespective of disease severity (after the Bonferroni correction, p > 0.0125). Details are provided in Table 1.
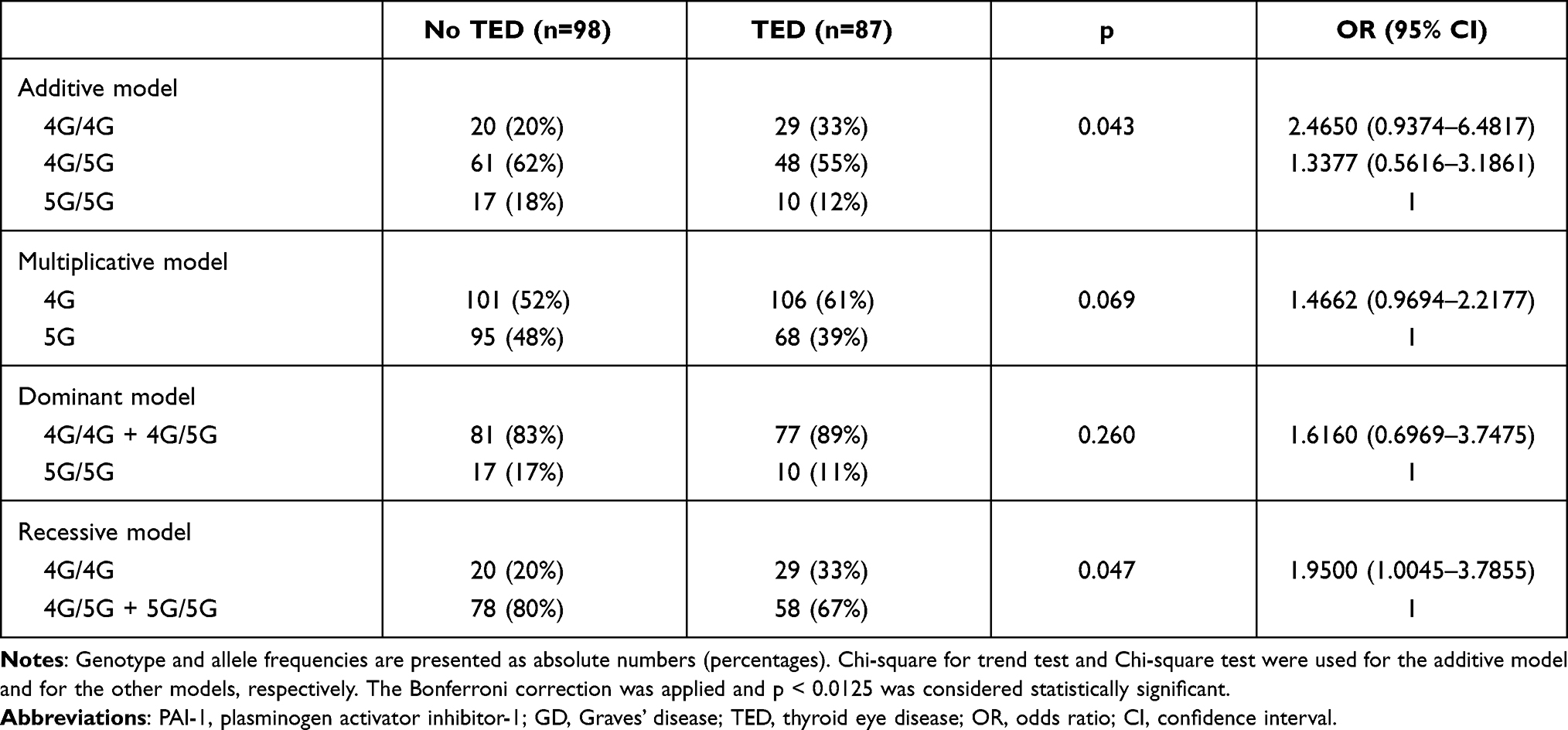 |
Table 1 Comparison of Genotype and Allele Frequencies of the PAI-1 4G/5G Polymorphism in GD Patients without TED vs GD Patients with TED Including All Severities |
Statistical analysis was also performed by comparing patients with moderate-to-severe TED according to the ETA-EUGOGO criteria3 as one group (n=49), with patients with no or only mild TED (n=136) as the other group. In the recessive model, the 4G/4G genotype was more common in patients with moderate-to-severe TED (p = 0.008, Table 2). In this comparison, ie, moderate-to-severe TED vs no or mild TED, with these sample sizes the highest statistical power is offered also by the recessive model (76%) with medium effect size (OR = 2.54). These results show that two copies of 4G allele are required for increased risk of moderate-to-severe TED in patients with GD.
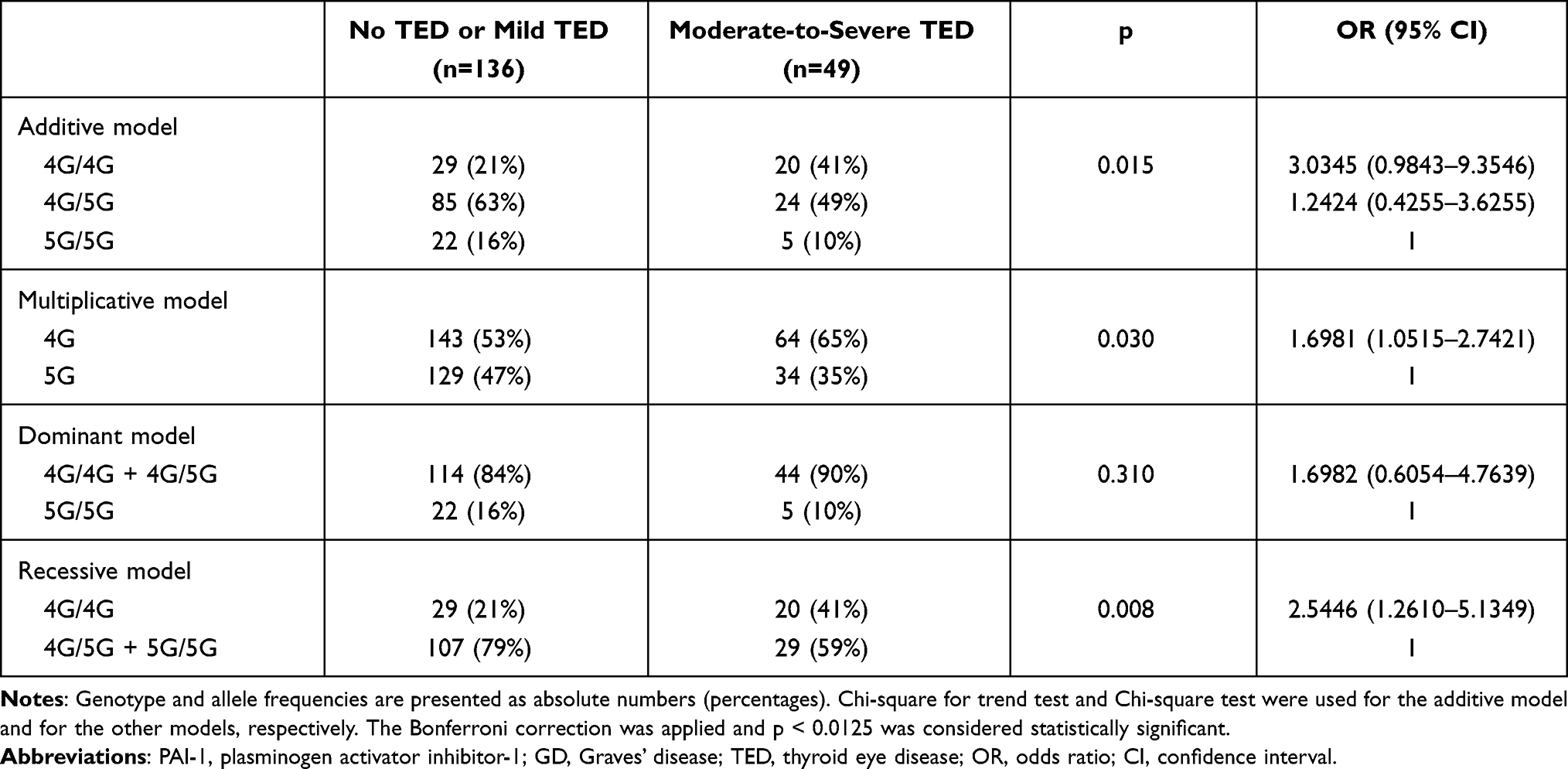 |
Table 2 Comparison of Genotype and Allele Frequencies of the PAI-1 4G/5G Polymorphism in GD Patients with No or Mild TED vs GD Patients with Moderate-to-Severe TED |
For the control population, we found that there is a Hardy–Weinberg equilibrium for the 4G/5G polymorphism of PAI-1, ie, the observed genotype distribution did not differ from the expected genotype distribution (p = 0.29); the distribution of the PAI-1 4G/5G genotype and allele frequency in patients with GD (if all GD patients irrespective of the presence or absence of TED were analyzed) were similar to that of the control population in any of the models studied (Table 3). However, the latter comparison did not have sufficient statistical power to detect a true difference.
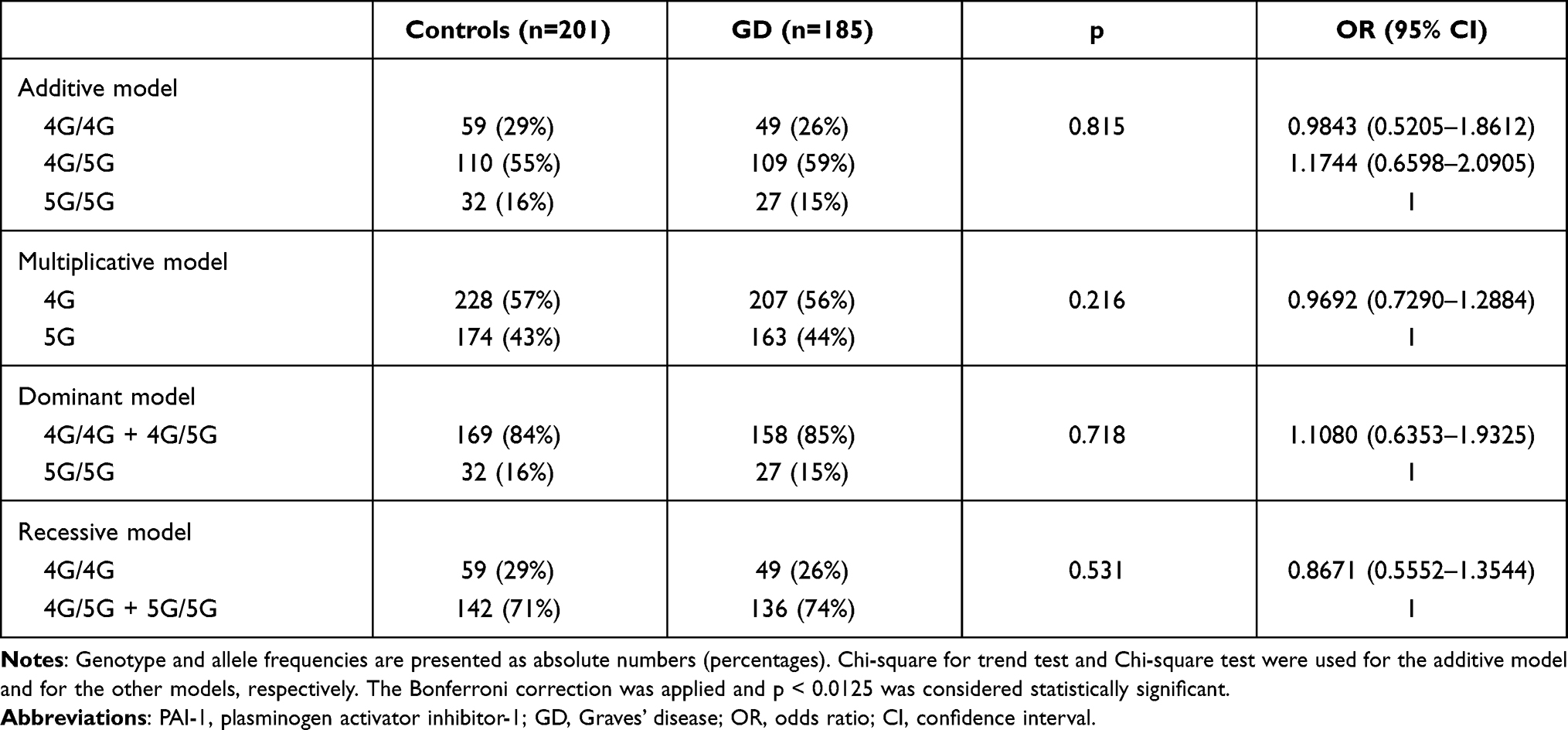 |
Table 3 Comparison of Genotype and Allele Frequencies of the PAI-1 4G/5G Polymorphism in Controls vs Graves’ Disease Irrespective of the Presence/Absence of Thyroid Eye Disease |
Laboratory parameters of the thyroid function, plasma PAI-1 levels, male-to-female ratio, age at inclusion, age at diagnosis of GD, and GD duration did not differ in GD patients with different 4G/5G genotypes (Table 4).
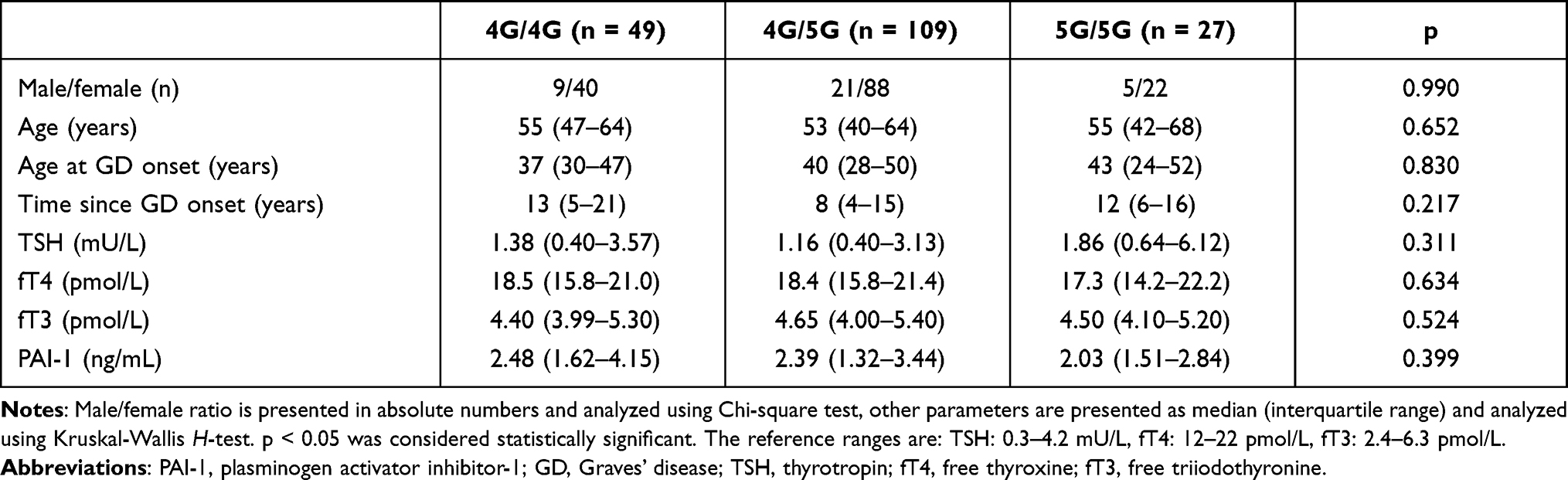 |
Table 4 Lack of Correlation Between Clinical Parameters and PAI-1 4G/5G Genotypes in Patients with Graves’ Disease |
Discussion
PAI-1 is the major inhibitor of the plasminogen-plasmin system. In addition to its role in fibrinolytic processes, it contributes to tissue homeostasis and regulates immunity by controlling immune cell migration and regulating clearance of apoptotic and necrotic cells by macrophages.23 Furthermore, PAI-1 can influence the duration and magnitude of immune responses.10,11 Presumably, PAI-1 level or activity may be potential factors in the development of autoimmune diseases. The 4G/5G insertion/deletion polymorphism of the PAI-1 gene influences PAI-1 expression in response to several stimuli, including IL-1α.17–20 For its role in autoimmune diseases, systemic lupus erythematosus (SLE) and rheumatoid arthritis have been studied,24 and an association has been found between the 4G allele and the development of SLE.25,26
Data on PAI-1 polymorphisms in TED, up till now, have not been published, and only few candidate other genes have been studied in this respect. CTLA-4,27 vitamin D receptor,28 TNF, IL-1029 and deiodinase-2 polymorphisms30 were recently described in patient cohorts of comparable size. However, unlike these genes which are immune response related, PAI-1 acts on the other side of the orbital process, stabilizing the extracellular matrix and facilitating its expansion, which explains nearly all signs and symptoms of TED. This may link the 4G/4G genotype to clinically more severe disease.
In Graves’ disease, we found that the 4G/4G genotype of PAI-1 was more frequent in patients with moderate-to-severe TED compared to patients with mild TED or without TED, representing a risk factor for moderate-to-severe TED (OR = 2.54, p < 0.01). It is not known which immune mediators may exploit the fact that the presence of the 4G allele in homozygote form in an individual makes him/her prone to more severe TED. The main candidate is IL-1α, a cytokine with emerging importance in the initiation and maintenance of inflammation,31 which is known to upregulate PAI-1 expression with higher efficacy in cells carrying the 4G allele of the PAI-1 gene.16,17 IL-1α may have a role in the pathogenesis of TED, as positive association has been found between the T allele of IL-1α −889 C/T polymorphism, characterized by higher promoter activity of IL-1α gene, and the susceptibility to TED.32 Serum IL-1α levels were higher in TED patients than in controls,33 and in vitro activation of human orbital fibroblasts upregulates the synthesis of IL-1α.34 The level of IL-1α in the tear samples of GD patients, even without TED, was higher compared to controls.35 Our previous finding, that PAI-1 concentration in the tear of TED patients is higher than in patients without TED36 further supports the potential role of an IL-1α – PAI-1 interaction. The locally elevated cytokine level in the orbital connective tissue during TED development may lead to increased PAI-1 synthesis by orbital fibroblasts, and this process may be more marked in the presence of the 4G allele. The consequent high local PAI-1 level may have an adverse effect on the development and course of TED by increasing the amount of extracellular matrix, stabilizing it, and promoting immune cell migration.9 On the other hand, increase of local PAI-1 level or activity in the orbits is not necessarily accompanied by a similar change of plasma PAI-1 levels.
TED is more common in younger women, but more severe in men and patients older than 50 years,37 suggesting that sex hormones may affect the course of the disease. Both estrogen and testosterone can regulate PAI-1 expression in endothelial cells;38,39 however, the potential role of sex steroids in the orbital connective tissue remodeling of TED patients remains to be elucidated.
Smoking is one of the major risk factors for the development and worsening of TED.40 Elevated PAI-1 expression was found in endothelial cells and in alveolar epithelial cells in vitro after treatment with cigarette smoke extract.41,42 Moreover, in smokers, significantly higher plasma levels of PAI-1 were found in patients with untreated essential hypertension carrying 4G/4G genotype as compared with 4G/5G and 5G/5G genotypes.43 Based on these studies, one may assume, that smoking can potentiate the effect of 4G/4G genotype on PAI-1 level in tissues of GD patients, leading to increased risk for TED. Unfortunately, insufficient data available on smoking status at the time of GD onset prevented us from analyzing the interaction between PAI-1 polymorphism and smoking.
One limitation of our study is the small sample size. The incidence of moderate-to-severe TED is 1 per 100 000 population44,45 which explains the limited access to these patients. Second, in the GD vs control comparison, due to the lack of power, the similarity of genotype and allele frequencies of the 4G/5G polymorphism cannot be considered conclusive. Further, the potential presence of an as of now unknown linkage disequilibrium might lead to overestimation of the effect size and misinterpretation of the results.
Plasma PAI-1 levels in GD patients did not differ significantly between groups with different 4G/5G genotypes. Of the patients included in this study, the active phase of GD and TED had ended in the past, and we have no information on how patients’ plasma and/or tissue PAI-1 levels behaved when GD and TED developed. Further, the major source of PAI-1 in the circulation are endothelial cells,46 therefore, plasma level may not reflect the PAI-1 level in tissues, including orbital connective tissue. Hyperthyroidism per se results in higher PAI-1 levels,13–15 and theoretically, this may be exaggerated in GD patients carrying the 4G allele, contributing to TED development. T3 induces PAI-1 expression at transcription level, but the thyroid hormone response element in the promoter region of the PAI-1 gene is localized at positions −327/-312,47 which does not overlap with the 4G/5G polymorphism at position −675.16 USF-1 can regulate the 4G/5G polymorphism-dependent PAI-1 expression in mast cells,20 and is involved in T3 responsiveness of hepatocytes.48 The potential interaction between USF-1 and T3 in the 4G/5G polymorphism-dependent regulation of PAI-1 expression remains to be elucidated in cell types relevant in TED.
Identification of genetic susceptibility may identify individual patients who are prone to the development of TED during the course of GD. According to our data, the 4G/4G genotype of PAI-1 is a risk factor for the development of moderate-to-severe TED. Patients with Graves’ disease who harbor this genotype may be candidates for special attention towards TED during the course of their thyroid disease. Preventive measures, and treatment modalities of Graves’ hyperthyroidism which do not facilitate the development or worsen the course of TED, may be required for these patients.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics Approval
The study was approved by the Regional and Institutional Ethics Committee of the University of Debrecen. The study was carried out in accordance with the Declaration of Helsinki.
Consent to Participate
Consent has been obtained from each patient after full explanation of the purpose and nature of all procedures used.
Acknowledgment
The authors thank Zsanett Molnar for technical assistance.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Hungarian National Research, Development and Innovation Office – NKFIH (grant number: K116419).
Disclosure
The authors have no conflicts of interest to declare.
References
1. Terwee C, Wakelkamp I, Tan S, Dekker F, Prummel MF, Wiersinga W. Long-term effects of Graves’ ophthalmopathy on health-related quality of life. Eur J Endocrinol. 2002;146:751–757. doi:10.1530/eje.0.1460751
2. Kahaly GJ, Petrak F, Hardt J, Pitz S, Egle UT. Psychosocial morbidity of Graves’ orbitopathy. Clin Endocrinol (Oxf). 2005;63:395–402. doi:10.1111/j.1365-2265.2005.02352.x
3. Bartalena L, Baldeschi L, Boboridis K, et al.; (EUGOGO) EGoGO. The 2016 European Thyroid Association/European Group on Graves’ Orbitopathy Guidelines for the Management of Graves’ Orbitopathy. Eur Thyroid J. 2016;5:9–26. doi:10.1159/000443828
4. Stan MN, Bahn RS. Risk factors for development or deterioration of Graves’ ophthalmopathy. Thyroid. 2010;20:777–783. doi:10.1089/thy.2010.1634
5. Galgoczi E, Jeney F, Gazdag A, et al. Cell density-dependent stimulation of PAI-1 and hyaluronan synthesis by TGF-β in orbital fibroblasts. J Endocrinol. 2016;229:187–196. doi:10.1530/JOE-15-0524
6. Declerck PJ, Gils A. Three decades of research on plasminogen activator inhibitor-1: a multifaceted serpin. Semin Thromb Hemost. 2013;39:356–364. doi:10.1055/s-0033-1334487
7. Rabieian R, Boshtam M, Zareei M, Kouhpayeh S, Masoudifar A, Mirzaei H. Plasminogen activator inhibitor type-1 as a regulator of fibrosis. J Cell Biochem. 2018;119:17–27. doi:10.1002/jcb.26146
8. Milenkovic J, Milojkovic M, Jevtovic Stoimenov T, Djindjic B, Miljkovic E. Mechanisms of plasminogen activator inhibitor 1 action in stromal remodeling and related diseases. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2017;161:339–347. doi:10.5507/bp.2017.046
9. Longo CM, Higgins PJ. Molecular biomarkers of Graves’ ophthalmopathy. Exp Mol Pathol. 2019;106:1–6. doi:10.1016/j.yexmp.2018.11.004
10. Park YJ, Liu G, Lorne EF, et al. PAI-1 inhibits neutrophil efferocytosis. Proc Natl Acad Sci U S A. 2008;105:11784–11789. doi:10.1073/pnas.0801394105
11. Gupta KK, Xu Z, Castellino FJ, Ploplis VA. Plasminogen activator inhibitor-1 stimulates macrophage activation through toll-like receptor-4. Biochem Biophys Res Commun. 2016;477:503–508. doi:10.1016/j.bbrc.2016.06.065
12. Kawashima A, Yamazaki K, Hara T, et al. Demonstration of innate immune responses in the thyroid gland: potential to sense danger and a possible trigger for autoimmune reactions. Thyroid. 2013;23:477–487. doi:10.1089/thy.2011.0480
13. Erem C, Ersoz HO, Karti SS, et al. Blood coagulation and fibrinolysis in patients with hyperthyroidism. J Endocrinol Invest. 2002;25:345–350. doi:10.1007/BF03344016
14. Li Y, Chen H, Tan J, Wang X, Liang H, Sun X. Impaired release of tissue plasminogen activator from the endothelium in Graves’ disease - indicator of endothelial dysfunction and reduced fibrinolytic capacity. Eur J Clin Invest. 1998;28:1050–1054. doi:10.1046/j.1365-2362.1998.00381.x
15. Popławska-Kita A, Szelachowska M, Modzelewska A, et al. Endothelial dysfunction in Graves’ disease. Adv Med Sci. 2013;58:31–37. doi:10.2478/v10039-012-0047-1
16. Dawson SJ, Wiman B, Hamsten A, Green F, Humphries S, Henney AM. The two allele sequences of a common polymorphism in the promoter of the plasminogen activator inhibitor-1 (PAI-1) gene respond differently to interleukin-1 in HepG2 cells. J Biol Chem. 1993;268:10739–10745. doi:10.1016/S0021-9258(18)82047-6
17. Roncal C, Orbe J, Belzunce M, Rodríguez JA, Páramo JA. The 4G/5G PAI-1 polymorphism influences the endothelial response to IL-1 and the modulatory effect of pravastatin. J Thromb Haemost. 2006;4:1798–1803. doi:10.1111/j.1538-7836.2006.02031.x
18. Roncal C, Orbe J, Rodriguez JA, et al. Influence of the 4G/5G PAI-1 genotype on angiotensin II-stimulated human endothelial cells and in patients with hypertension. Cardiovasc Res. 2004;63:176–185. doi:10.1016/j.cardiores.2004.03.023
19. Eriksson P, Nilsson L, Karpe F, Hamsten A. Very-low-density lipoprotein response element in the promoter region of the human plasminogen activator inhibitor-1 gene implicated in the impaired fibrinolysis of hypertriglyceridemia. Arterioscler Thromb Vasc Biol. 1998;18:20–26. doi:10.1161/01.ATV.18.1.20
20. Ma Z, Jhun B, Jung SY, Oh CK. Binding of upstream stimulatory factor 1 to the E-box regulates the 4G/5G polymorphism-dependent plasminogen activator inhibitor 1 expression in mast cells. J Allergy Clin Immunol. 2008;121:1006–1012.e1002. doi:10.1016/j.jaci.2007.11.015
21. Prabhudesai A, Shetty S, Ghosh K, Kulkarni B. Investigation of plasminogen activator inhibitor-1 (PAI-1) 4G/5G promoter polymorphism in Indian venous thrombosis patients: a case-control study. Eur J Haematol. 2017;99:249–254. doi:10.1111/ejh.12912
22. Clarke GM, Anderson CA, Pettersson FH, Cardon LR, Morris AP, Zondervan KT. Basic statistical analysis in genetic case-control studies. Nat Protoc. 2011;6:121–133. doi:10.1038/nprot.2010.182
23. Draxler DF, Sashindranath M, Medcalf RL. Plasmin: a modulator of immune function. Semin Thromb Hemost. 2017;43:143–153. doi:10.1055/s-0036-1586227
24. Bae SC, Lee YH. Association between plasminogen activator inhibitor-1 (PAI-1) 4G/5G polymorphism and circulating PAI-1 level in systemic lupus erythematosus and rheumatoid arthritis: a meta-analysis. Z Rheumatol. 2020;79:312–318. doi:10.1007/s00393-019-00689-y
25. Anaya-Macias BU, De la Cruz-mosso U, Palafox-Sánchez CA, et al. The −675 4G/5G PAI-1 polymorphism confers genetic susceptibility to systemic lupus erythematosus, its clinical manifestations, and comorbidities in Mexican-Mestizo population. Autoimmunity. 2020;53:71–77. doi:10.1080/08916934.2019.1700957
26. Yousef AA, Mohamed FY, Boraey NF, et al. Association of plasminogen activator inhibitor 1 (PAI-1) 4G/5G polymorphism and susceptibility to SLE in Egyptian Children and Adolescents: a Multicenter Study. J Inflamm Res. 2020;13:1103–1111. doi:10.2147/JIR.S277373
27. Chen DP, Chu YC, Wen YH, Lin WT, Hour AL, Wang WT. Investigation of the correlation between Graves’ Ophthalmopathy and CTLA4 gene polymorphism. J Clin Med. 2019;8(11):1842. doi:10.3390/jcm8111842
28. Maciejewski A, Kowalczyk MJ, Gasińska T, et al. The role of vitamin D receptor gene polymorphisms in thyroid-associated orbitopathy. Ocul Immunol Inflamm. 2020;28:354–361. doi:10.1080/09273948.2019.1629605
29. Petunina NA, Martirosian NS, Trukhina LV, et al. Association between polymorphic markers in candidate genes and the risk of manifestation of endocrine ophthalmopathy in patients with Graves’ disease. Ter Arkh. 2018;90:35–39. doi:10.26442/terarkh2018901035-39
30. Shahida B, Planck T, Åsman P, Lantz M. Study of deiodinase type 2 polymorphisms in Graves’ disease and ophthalmopathy in a Swedish population. Eur Thyroid J. 2018;7:289–293. doi:10.1159/000490892
31. Di Paolo NC, Shayakhmetov DM. Interleukin 1α and the inflammatory process. Nat Immunol. 2016;17:906–913. doi:10.1038/ni.3503
32. Khalilzadeh O, Anvari M, Esteghamati A, et al. Graves’ ophthalmopathy and gene polymorphisms in interleukin-1alpha, interleukin-1beta, interleukin-1 receptor and interleukin-1 receptor antagonist. Clin Exp Ophthalmol. 2009;37:614–619. doi:10.1111/j.1442-9071.2009.02093.x
33. Laban-Guceva N, Bogoev M, Antova M. Serum concentrations of interleukin (IL-)1alpha, 1beta, 6 and tumor necrosis factor (TNF-) alpha in patients with thyroid eye disease (TED). Med Arh. 2007;61:203–206.
34. Cao HJ, Wang HS, Zhang Y, Lin HY, Phipps RP, Smith TJ. Activation of human orbital fibroblasts through CD40 engagement results in a dramatic induction of hyaluronan synthesis and prostaglandin endoperoxide H synthase-2 expression. Insights into potential pathogenic mechanisms of thyroid-associated ophthalmopathy. J Biol Chem. 1998;273:29615–29625. doi:10.1074/jbc.273.45.29615
35. Mandić JJ, Kozmar A, Kusačić-Kuna S, et al. The levels of 12 cytokines and growth factors in tears: hyperthyreosis vs euthyreosis. Graefes Arch Clin Exp Ophthalmol. 2018;256:845–852. doi:10.1007/s00417-017-3892-6
36. Ujhelyi B, Gogolak P, Erdei A, et al. Graves’ orbitopathy results in profound changes in tear composition: a study of plasminogen activator inhibitor-1 and seven cytokines. Thyroid. 2012;22:407–414. doi:10.1089/thy.2011.0248
37. Kendler DL, Lippa J, Rootman J. The initial clinical characteristics of Graves’ orbitopathy vary with age and sex. Arch Ophthalmol. 1993;111:197–201. doi:10.1001/archopht.1993.01090020051022
38. Smith LH, Coats SR, Qin H, et al. Differential and opposing regulation of PAI-1 promoter activity by estrogen receptor alpha and estrogen receptor beta in endothelial cells. Circ Res. 2004;95:269–275. doi:10.1161/01.RES.0000136521.70093.f1
39. Jin H, Lin J, Fu L, et al. Physiological testosterone stimulates tissue plasminogen activator and tissue factor pathway inhibitor and inhibits plasminogen activator inhibitor type 1 release in endothelial cells. Biochem Cell Biol. 2007;85:246–251. doi:10.1139/O07-011
40. Wiersinga WM. Smoking and thyroid. Clin Endocrinol (Oxf). 2013;79:145–151. doi:10.1111/cen.12222
41. Hu XY, Ma YH, Wang C, Yang YH. Effects of simvastatin on cigarette smoke extract induced tissue-type plasminogen activator and plasminogen activator inhibitor-1 expression in human umbilical vein endothelial cells. Chin Med J (Engl). 2009;122:2380–2385.
42. Xu X, Wang H, Wang Z, Xiao W. Plasminogen activator inhibitor-1 promotes inflammatory process induced by cigarette smoke extraction or lipopolysaccharides in alveolar epithelial cells. Exp Lung Res. 2009;35:795–805. doi:10.3109/01902140902912519
43. Jastrzebska M, Goracy I, Naruszewicz M. Relationships between fibrinogen, plasminogen activator inhibitor-1, and their gene polymorphisms in current smokers with essential hypertension. Thromb Res. 2003;110:339–344. doi:10.1016/j.thromres.2003.08.002
44. Bahn RS. Graves’ ophthalmopathy. N Engl J Med. 2010;362:726–738. doi:10.1056/NEJMra0905750
45. Laurberg P, Berman DC, Bülow Pedersen I, Andersen S, Carlé A. Incidence and clinical presentation of moderate to severe graves’ orbitopathy in a Danish population before and after iodine fortification of salt. J Clin Endocrinol Metab. 2012;97:2325–2332. doi:10.1210/jc.2012-1275
46. Loskutoff DJ, Sawdey M, Mimuro J. Type 1 plasminogen activator inhibitor. Prog Hemost Thromb. 1989;9:87–115.
47. Chen CY, Chi LM, Chi HC, et al. Stable isotope labeling with amino acids in cell culture (SILAC)-based quantitative proteomics study of a thyroid hormone-regulated secretome in human hepatoma cells. Mol Cell Proteomics. 2012;11:
48. Jackson-Hayes L, Song S, Lavrentyev EN, et al. A thyroid hormone response unit formed between the promoter and first intron of the carnitine palmitoyltransferase-Ialpha gene mediates the liver-specific induction by thyroid hormone. J Biol Chem. 2003;278:7964–7972. doi:10.1074/jbc.M211062200
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.