Back to Journals » International Journal of Nanomedicine » Volume 20
Tetrahedral Framework Nucleic Acids Inhibit Oxidative Stress and Cardiomyocyte Apoptosis in Doxorubicin-Induced Cardiotoxicity
Authors Pan H, Ye Q, Li Y, Ma L ![]() , Ni Y
, Ni Y ![]()
Received 17 April 2025
Accepted for publication 16 July 2025
Published 21 July 2025 Volume 2025:20 Pages 9275—9289
DOI https://doi.org/10.2147/IJN.S529346
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Eng San Thian
Haizhou Pan,1,* Qianxi Ye,1,* Yifan Li,2 Liang Ma,1 Yiming Ni1
1Department of Cardiovascular Surgery, the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang Province, People’s Republic of China; 2Department of Orthopedics, the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yiming Ni, Department of Cardiovascular Surgery, the First Affiliated Hospital, Zhejiang University School of Medicine, No. 79 Qingchun Road, Hangzhou, People’s Republic of China, Tel +86-0571-87236348, Email [email protected]
Background: Doxorubicin-induced cardiotoxicity (DIC) is a major clinical limitation of doxorubicin therapy, driven by mitochondrial dysfunction and apoptosis. Tetrahedral framework nucleic acids (tFNAs), as novel 3D DNA nanostructures, exhibit antioxidative and anti-apoptotic properties, suggesting therapeutic potential for DIC.
Methods: The therapeutic efficacy of tFNAs was evaluated through in vivo (mouse model) and in vitro (cardiac cell lines) experiments. Apoptotic pathways were analyzed via AKT/p53 signaling inhibition assays, while cardiac function was assessed by histological examination and biochemical analysis.
Results: In vitro results demonstrated that tFNAs significantly attenuated DIC by suppressing AKT/p53-mediated apoptosis. In vivo studies confirmed functional improvement in cardiac tissue, validated by reduced biomarkers of cardiotoxicity and enhanced histological integrity.
Conclusion: tFNAs effectively mitigated DIC pathogenesis through dual mechanisms of mitochondrial protection and apoptosis inhibition. These findings position tFNAs as a promising therapeutic strategy for clinical DIC management, warranting further translational studies.
Keywords: doxorubicin-induced cardiotoxicity, tetrahedral framework nucleic acids, mitochondria, oxidative stress, apoptosis
Background
Doxorubicin (DOX), a powerful anthracycline antibiotic, is administered in a wide range of cancers.1 However, the application of DOX is limited due to its life-threatening cardiotoxicity.2 DOX causes bioenergetic failure and cell death in the heart and ultimately leads to cardiomyopathy.3 Emerging evidence highlights that the pathophysiological progression of doxorubicin-induced cardiotoxicity (DIC) is multifactorial, encompassing aberrations in genomic stability maintenance, perturbations in autophagic flux, and maladaptive alterations in cytosolic calcium dynamics.4–6 Although the underlying mechanism of DIC has been intensively studied,7,8 and our lack of feasible and useful methods still exists.
Mitochondrial dysfunction is one of the first events of cardiac injury upon DOX challenge.9 Beyond their primary role in adenosine triphosphate (ATP) generation, mitochondria critically regulate diverse cellular functions such as maintaining calcium ion balance, modulating inflammatory responses, and generating reactive oxygen species (ROS).10–12 Excessive reactive oxygen species induce oxidative damage to mitochondrial membrane integrity and function and ultimately cause cardiomyocyte fibrosis and apoptosis in DIC.2,3,13 DOX-induced oxidative stress directly elicits extensive cardiomyocyte apoptosis followed by severe cardiac dysfunction.14 Thus, it is worth investigating changes in mitochondrial function because of the pivotal role of mitochondria in DOX-induced cardiotoxicity.
Current preventative and therapeutic approaches for doxorubicin-induced cardiac injury, such as dexrazoxane, provide only limited cardioprotection and merely stabilize patients at a suboptimal level of cardiac function.14,15 This constraint is compounded by two critical clinical dilemmas: the absence of viable therapeutic alternatives to replace doxorubicin in chemotherapy regimens, which impedes actionable responses to cardiac injury biomarkers, and the inherent risk of compromising anticancer efficacy when individualizing dose reductions to mitigate cardiotoxicity. Consequently, the development of novel and effective strategies to prevent doxorubicin-induced cardiac injury represents an urgent unmet need in oncology and cardiology research.
Tetrahedral framework nucleic acids (tFNAs), self-assembled three-dimensional DNA nanostructures composed of four stoichiometrically equivalent single-stranded DNA (ssDNA) strands, demonstrate therapeutic potential in mitigating oxidative damage and inhibiting apoptosis across diverse pathological conditions.16,17 A previous study demonstrated the high stability, good biocompatibility and tunable physicochemical properties of tFNAs, indicating their potential as therapeutic biomaterials.18 tFNAs exhibit smooth endocytosis via the caveolin-mediated pathway and could inhibit Toll-like Receptor 2 signaling Pathway in acute ischemic stroke.19 While tFNAs have been applied in neural regeneration, osteoarthritis and acute kidney injury,8,20,21 their cardioprotective efficacy against DOX-induced toxicity remains unexplored since mitochondrial dysfunction and oxidative stress contribute greatly to DIC pathogenesis.17,20,21 No prior studies have investigated tFNA-mediated modulation of oxidative damage and apoptosis in cardiomyocytes under doxorubicin stress. Therefore, it is reasonable to consider the application of tFNAs to fight doxorubicin-induced cardiotoxicity. In this study, we established an in vitro DOX-treated AC16 model and an in vivo DOX insult mouse model to verify the therapeutic potential of tFNAs.
Materials and Methods
Materials, Cells and Animals
PCR sets were purchased from Abclonal, China. Doxorubicin were purchased from Selleck Chemicals. Single-stranded oligonucleotides were synthesized by Tsingke Biotech, China. Human AC16 cardiomyocyte cell line was purchased from PROCELL, China and was cultured in DMEM/F12 medium (Gibco, USA) supplemented with 1% penicillin-streptomycin (Gibco, USA) and 10% fetal bovine serum (FBS) (Gibco, USA), and maintained at 37°C in a 5% CO2 humidified incubator under standard aseptic protocols. C57BL/6J- mice were obtained from the Experimental Animal Center of Zhejiang University School of Medicine, the First Affiliated Hospital. All animal experiments were conducted in accordance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH). The experimental protocols were approved by the Institutional Animal Care and Use Committee of Zhejiang University School of Medicine, the First Affiliated Hospital.
Fabrication and Characterization of tFNAs
The tetrahedral DNA nanostructures (tFNAs) were programmatically assembled through a magnesium ion-dependent annealing process, wherein four sequence-engineered single-stranded oligonucleotides (Table 1) were stoichiometrically combined (1 μM each) in a 200 μL TM buffer system (10 mM Tris-HCl, 50 mM MgCl2, pH 8.0) to stabilize Holliday junction formation. Mg²+ neutralizes the negative charges on the DNA phosphate backbone, reducing electrostatic repulsion between double-stranded DNA and thereby promoting tighter binding between the DNA strands.22 Following thermal denaturation at 95°C for 10 minutes to dissociate secondary structures, the mixture underwent controlled cooling to 4°C over 20 minutes, enabling stepwise hybridization.17,18 Agarose gel electrophoresis (AGE) was performed to assess the synthesis of tFNAs. The structural morphology and dimensional characteristics of tFNAs were analyzed using atomic force microscopy (AFM) (Bruker Dimension Icon system, Germany), while their hydrodynamic diameter and zeta potential distribution were quantified via dynamic light scattering (DLS) (Zetasizer Nano ZS 90, Malvern Instruments, UK). To evaluate biostability, tFNAs were subjected to serum incubation (2% or 10% fetal bovine serum contained medium) across a physiological-relevant time course (0–12 h), followed by agarose gel electrophoresis (AGE) to monitor structural integrity. Electrophoretic banding patterns were documented using a Bio-Rad Gel Doc XR imaging system (USA), enabling temporal stability profiling under simulated in vivo conditions.
 |
Table 1 Sequence of Each ssDNA |
Cell Treatment
Before being treated with DOX (1μM) for 8/12 h, AC16 cells were preincubated with tFNAs (250 nM) or PBS for 12 h.
Cellular Uptake of tFNAs
The S1 ssDNA in the tFNAs was replaced with the Cy-5 fluorescent molecule-conjugated S1 ssDNA to verify the delivery of the tFNAs. Cultured AC16 cells were coincubated with Cy5-tFNA (250 nM) and incubated for 4 h, 8 h or 12 h. Following PBS rinsing (pH 7.4), cells underwent fixation in 4% paraformaldehyde (PFA)(Sigma-Aldrich) for 15 min at room temperature and subsequent nuclear counterstaining with 4′,6-diamidino-2-phenylindole (DAPI) (Beyotime Biotechnology, China; 1 μg/mL in PBS) for 10 min, with three PBS washes interspersed between each step to ensure removal of unbound reagents. Fluorescent imaging was performed using a confocal microscope (Olympus FV3000) equipped with a 405 nm excitation laser to visualize DAPI-stained nuclei.
Detection of Oxidative Stress
Cellular reactive oxygen species (ROS) levels were quantified using a fluorescent ROS Assay Kit (Beyotime, China) and MitoSOX™ Red Mitochondrial Superoxide Indicator (Thermo Fisher Scientific, USA), while mitochondrial membrane potential dynamics were assessed via a JC-1-based assay (Beyotime, China). Following staining protocols, fluorescence signals were captured by a confocal laser scanning microscope (Olympus FV-3000, Japan). Concurrently, oxidative stress biomarkers including malondialdehyde (MDA), glutathione (GSH), total superoxide dismutase (SOD), and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity were measured using assay kits (Nanjing Jiancheng, China), adhering to manufacturer-specified spectrophotometric protocols.
RT‒PCR Analysis
Gene expression analysis was conducted through quantitative real-time PCR (qPCR). Total RNA isolating from cell lysates was performed using TRIzol® Reagent (Thermo Fisher Scientific, USA; Cat. No. 15596018), followed by cDNA synthesis with the PrimeScript™ RT Reagent Kit (Takara, Japan; Cat. No. RR047Q). qPCR amplification was performed on a QuantStudio™ 5DX System (Thermo Fisher Scientific, USA) using PowerUp™ SYBR™ Green Master Mix (Thermo Fisher Scientific; Cat. No. A25742, USA). Relative gene expression levels were normalized to β-actin (internal control) and calculated via the comparative ΔΔCt method. Primer sequences targeting specific genes are listed in Table 2, with annealing temperatures optimized to ensure amplification efficiency (90–110%) and specificity, as validated by melt curve analysis.
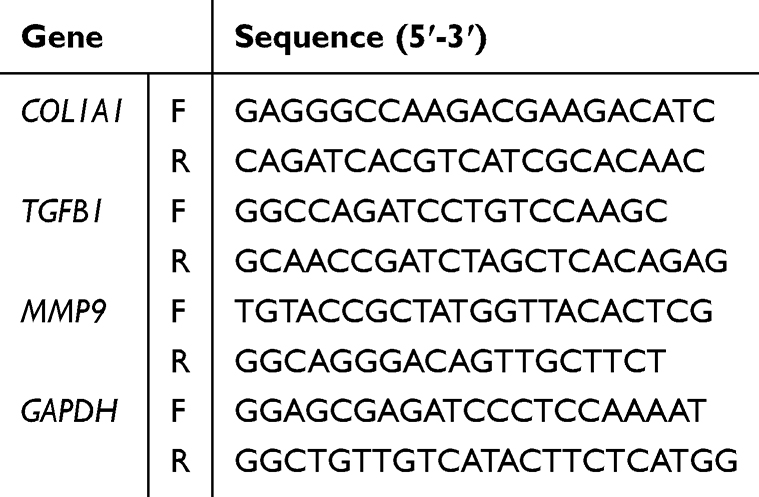 |
Table 2 Primer Sequence |
In vitro IF Staining
Immunofluorescence staining was performed on AC16 cells to visualize the target proteins. Cells were fixed in 4% paraformaldehyde (PFA) for 15 minutes, followed by permeabilization and blocking using QuickBlock™ Blocking Buffer for Immunolstaining (Beyotime, Shanghai, China). Subsequently, the cells underwent overnight incubation at 4°C with primary antibodies specific to collagen I (1:200 dilution; Proteintech, 14695-1-AP), TGF-β1 (1:200 dilution; Proteintech, 21898-1-AP), and MMP9 (1:200 dilution; Affinity, AF5228). On the following day, the cells were incubated in diluted Alexa Fluor™ 488 fluorescent secondary antibodies (1:500 dilution; Thermo Fisher Scientific, USA) at 37°C for 1 hour. Each incubation step was followed by washing with phosphate-buffered saline (PBS) (5 min for 3 times). Finally, nuclear staining was performed using DAPI (37°C, 10 minutes). Confocal laser scanning microscopy (Olympus FV-3000, Japan) was employed to acquire the images.
Cell Viability Detection and TdT-Mediated dUTP Nick End-Labeling (TUNEL) Staining
Cell viability was evaluated using a Calcein/PI Cell Viability/Cytotoxicity Assay Kit (Beyotime, Shanghai, China). In this assay, live cells are labeled with Calcein AM, which emits green fluorescence, while dead cells are stained with propidium iodide (PI), emitting red fluorescence. Additionally, cell apoptosis was assessed in vitro using a TUNEL staining kit (Beyotime, Shanghai, China).
Establishment of DIC Model in Mice
Male C57BL/6J mice aged 8 weeks were obtained from the Experimental Animal Center of Zhejiang University School of Medicine, the First Affiliated Hospital, and were allowed a 1-week adaptation period prior to the commencement of the study. Throughout the experiment, all mice were housed in individually ventilated cages under specific pathogen-free (SPF) conditions in an environmentally controlled facility. The animals received sterile food and water ad libitum, with environmental parameters maintained at a temperature of 20–25°C and humidity of 50 ± 5%.
The mice were allocated randomly into three experimental groups: Control group, DOX-treated group, and DOX and tFNA-treated group. Mice in DOX-treated group, and DOX and tFNA-treated group received DOX treatment to induce a model of DIC. Specifically, these mice were administered DOX (15 mg/kg, intraperitoneally) in three weekly doses, resulting in a cumulative total dose of 15 mg/kg. For those in DOX and tFNA-treated group, 100 μL of tFNA solution (250 nM) was administered via tail vein injection 12 hours prior to each DOX administration, over a 3-week period.
Echocardiographic Assessment
Following DOX challenge, M-mode echocardiographic assessments were conducted on the mice under isoflurane anesthesia (1–2%). Two-dimensional (2D)-guided M-mode echocardiography was performed using a 22–55 MHz FUJIFILM VisualSonics transducer (Toronto, ON, Canada) to evaluate cardiac function. Measurements were taken using a Vevo 2100 echocardiography system (FUJIFILM VisualSonics, Toronto, ON, Canada) according to established protocols. Echocardiographic parameters were calculated as the average of three to five consecutive cardiac cycles.23
Histological Analysis
After echocardiographic assessment, the mice were sacrificed. The hearts and other four organs, including the lungs, spleens, livers, and kidneys, were harvested from the mice. Hearts were cut in half, and half of the hearts with other organs were fixed with PFA. All the samples were subsequently dehydrated in a graded alcohol series, embedded in paraffin, and sheared into slices. Finally, all the samples were stained with H&E (hearts and other major organs) or Masson (hearts), scanned, and imaged for further analysis.
Transmission Electron Microscopy (TEM)
Some of the hearts collected from the previous step were sectioned into cubic fragments. These cubic heart tissue samples were fixed in 0.1 M PBS (pH 7.4) containing 2.5% glutaraldehyde for a minimum of 24 hours at 4°C. Following this, the samples underwent dehydration through a graded series of ethanol solutions before being embedded in Epon Araldite resin. Prior to fixation with 1% osmium tetroxide (OsO₄) for 1 hour, the dehydration process was completed. Ultrathin sections, ranging in thickness from 75 to 80 nm, were prepared using a Leica ultramicrotome (Wetzlar, Germany) fitted with a Diatome diamond knife. The sections were then sequentially stained with uranyl acetate for 10 minutes and lead citrate for an additional 5 minutes. Morphological examination of the sections was conducted using a Hitachi H600 transmission electron microscope (Hitachi, Japan) operated at an accelerating voltage of 40–120 kV. Digital images of the samples were captured and recorded using Digital Micrograph software.
Western Blot Analysis
Protein extraction and Western blotting analyses were conducted on both AC16 cells and heart tissue samples. After washing and harvesting, the samples were lysed using RIPA buffer (Beyotime, Shanghai, China). Protein concentrations of the lysates were quantified using a BCA protein assay kit (Beyotime, Shanghai, China).
For Western blot, the protein samples were diluted with NuPAGE™ LDS sample buffer (4×) (Invitrogen, USA), thoroughly mixed, and denatured by heating in a metal bath at 70°C for 10 minutes. The denatured proteins were then separated by SDS-PAGE according to their molecular weights. The separated proteins were transferred onto a 0.22nm or 0.45nm PVDF membrane, which was subsequently blocked using skim milk for 1 hour to reduce nonspecific binding. The PVDF membrane was then incubated at 4°C overnight in the following diluted primary antibodies, each diluted at a 1:1000 ratio:
Rabbit anti-cleaved-caspase-3 (Cell Signaling Technology, 9664)
Rabbit anti-caspase-3 (Cell Signaling Technology, 9662)
Rabbit anti-cleaved-PARP (Cell Signaling Technology, 94885)
Rabbit anti-PARP (Cell Signaling Technology, 9532)
Mouse anti-collagen I (Proteintech, 67288-1-Ig)
Rabbit anti-phospho-Akt (Ser473) (Cell Signaling Technology, 9271)
Rabbit anti-ANP (Proteintech, 27426-1-AP)
Rabbit anti-P53 (Proteintech, 10442-1-AP)
Mouse anti-GAPDH (Proteintech, 60004-1-Ig)
Following primary antibody incubation, the PVDF membrane was then given 1-hour incubation at room temperature in diluted HRP-conjugated goat anti-mouse IgG (H+L) or HRP-conjugated goat anti-rabbit IgG (H+L) secondary antibodies (ABclonal, Wuhan, China) (1:10,000). Immunoreactivity was subsequently visualized by Bio-Rad ChemiDoc MP detection system (Bio-Rad, Hercules, CA, USA).
Statistical Analysis
Every experimental analysis in this study was performed in triplicate, with data represented as the mean ± standard deviation. Statistical analysis was conducted using GraphPad Prism 9.5.0 software. Comparisons between two groups were analyzed using two-tailed Student’s t-tests. Results were considered statistically significant at *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Results
Synthesis and Characterization of the tFNAs
As previously described, tFNAs quickly self-assemble into four ssDNAs (S1, S2, S3, and S4, as shown in Table 1) according to the complementary base pairing principle (Figure 1A).18 The formation of tFNAs was validated through agarose gel electrophoresis analysis, as evidenced by the characteristic banding profile in Figure 1B. The atomic force microscopy (AFM) image revealed the uniform dispersion, uniform size, and tetrahedral structure of the tFNAs (Figure 1C). Physicochemical characterization via DLS indicated a particle size distribution of 11.08 ± 0.892 nm for tFNAs, while zeta potential analysis confirmed their electronegative surface charge (−6.997 ± 1.036 mV), as shown in Figure 1D. The stability of tFNAs in serum was then tested. tFNAs did not significantly degrade in 2% or 10% serum after 6 hours (Figure 1E). The cellular uptake of tFNAs in AC16 cells was subsequently observed via confocal microscopy. Twelve hours after being added to the cell culture medium, the tFNAs were still satisfactorily taken up by the AC16 cells (Figure 1F). All these results were consistent with those from previous studies.
 |
Figure 1 Synthesis and characterization of the tFNAs. (A) Schematic diagram of tFNAs synthesis. (B) Molecular weight detection by 2% AGE of each ssDNA and tFNAs. (C) Images acquired by AFM. (D) Sizes and zeta potentials of tFNAs measured by DLS. (E) Stability evaluation of tFNAs in fetal bovine serum by 2% AGE. (F) Immunofluorescence images for cellular uptake of Cy5-labeled tFNAs in AC16 cells (Cy5: red; nucleus: blue). Abbreviation: AGE, agarose gel electrophoresis. |
tFNAs Alleviated DOX-Induced Oxidative Stress in vitro
As established in prior studies, heightened oxidative stress constitutes a hallmark pathological mechanism underlying the progression of DIC.2 Therefore, preventing oxidative stress injury by scavenging ROS has become a priority in the treatment of DIC. To model oxidative stress, AC16 cells were first pretreated with tFNAs for 12 hours prior to 12-hour DOX challenge. Parallel control groups received either PBS (negative control) or DOX alone (positive control). We used DCFH-DA and MITOSOX fluorescent probes to test the ROS levels in vitro. The results revealed that pretreatment with tFNAs significantly reduced ROS levels in response to DOX insult (Figure 2A–D). The mitochondrial membrane is the target of oxidative stress, and JC-1 staining reflects the extent of damage to the integrity of the mitochondrial membrane. The change from aggregates (red) to monomers (green) in JC-1 staining indicates a significant decrease in the mitochondrial membrane potential, which is a marker of an impaired mitochondrial membrane. As shown in Figure 2E, tFNAs exerted marked effects against DOX insult in AC16 cells. Moreover, the production levels of several oxidative markers were measured. tFNAs had a profound effect against DOX stimulation, as evidenced by decreased MDA levels and NADPH oxidase activity, with preserved GSH levels and SOD activity (Figure 2F–I).
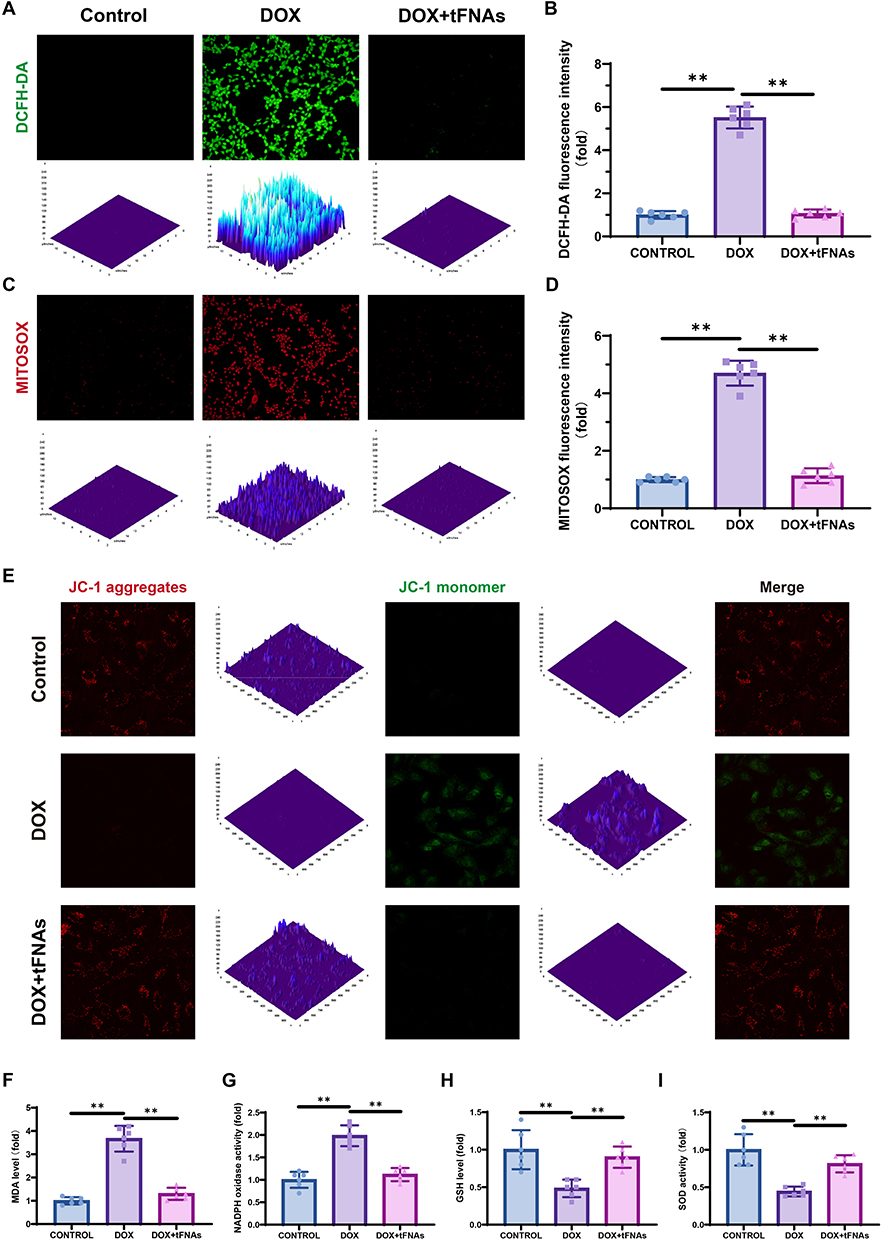 |
Figure 2 tFNAs alleviated DOX-induced oxidative stress and cardiomyocyte apoptosis in vitro. (A) Representative images of intracellular ROS measurement by DCFH assay. (B) Statistical analysis of DCFH assay, n=6. (C) Representative images of intracellular ROS measurement by MITOSOX assay. (D) Statistical analysis of MITOSOX assay, n=6. (E) Representative images of intracellular ROS measurement using JC-1 assay. (F–I) MDA, NADPH, GSH, SOD contents in AC16 cells, n=6. AC16 cells were preincubated with tFNAs (250 nM) or PBS for 12 h before being treated with DOX (1μM) for 12 h. The data are presented as the mean ± SD, **p < 0.01. |
Effects of tFNAs on DOX-Induced Fibrosis in vitro
Fibrosis a common feature of heart failure triggered by DOX insult.24 To explore whether tFNAs affect DOX-induced fibrosis, we tested the expression of fibrosis-related genes (COL1A1, TGFB1, and MMP9) in AC16 cells. Fibrotic remodeling, manifested through pathological accumulation of extracellular matrix components—most prominently type I collagen—serves as a conserved pathological signature across nearly all myocardial disorders.25 Fibrotic processes are driven by a complex interplay of mechanisms. For example, following myocardial infarction, the ischemic myocardium undergoes a marked upregulation of pro-inflammatory mediators and regenerative signaling molecules, notably including transforming growth factor-β1 (TGF-β1). TGF-β1 upregulation induces fibroblast activation through the canonical SMAD3-dependent pathway, which governs extracellular matrix (ECM) protein synthesis and pathological accumulation, thereby driving fibrotic progression.26,27 MMP9 exerts its proteolytic activity to disrupt extracellular matrix (ECM) homeostasis by skewing the equilibrium between matrix synthesis and degradation, thereby driving concurrent cardiac fibrotic remodeling and hypertrophic pathology.28,29 As shown in Figure 3A and B, COL1A1 and TGFB1 were upregulated, whereas MMP9 was downregulated in response to DOX challenge, and tFNA administration reversed these changes. Additionally, the protein expression of Collagen I, TGF-beta1 and MMP9 in AC16 cells was detected via IF staining. These results were consistent with the qPCR results (Figure 3C–E).
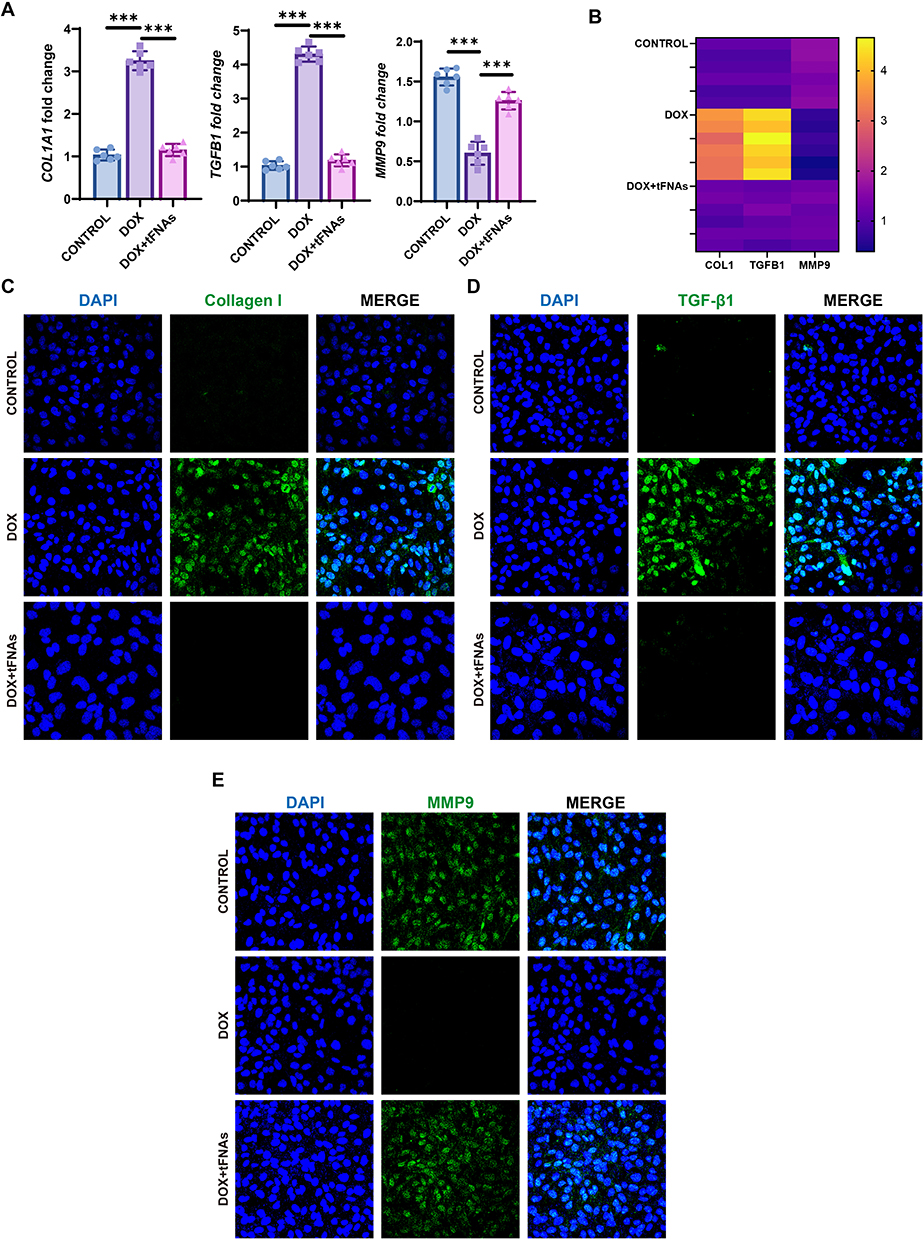 |
Figure 3 Effects of tFNAs on DOX-induced fibrosis in vitro. (A) qPCR results of AC16 cells with different treatment. (B) Heatmap of the previous qPCR results. (C–E) IF staining of Collagen I, TGF-β1 and MMP9 TGF-β1 of AC16 cells with different treatment. AC16 cells were preincubated with tFNAs (250 nM) or PBS for 12 h before being treated with DOX (1μM) for 12 h. ***p < 0.001. |
AKT/p53 Signaling May Be Responsible for the tFNA-Mediated Protective Effect on Cardiomyocyte Apoptosis Against DOX Insult
Apoptosis, a regulated cell death process, is usually triggered by DOX in cardiomyocytes.30,31 Live/dead dual fluorescence assays demonstrated marked cytotoxicity of DOX, evidenced by compromised cellular viability and substantial cell population depletion, whereas tFNA pretreatment effectively attenuated these detrimental effects (Figure 4A and B). TUNEL staining revealed that apoptosis was the main type of regulated cell death after DOX insult, and tFNAs had strong antiapoptotic effects on DIC (Figure 4C). Elevated cleaved caspase3 and cleaved PARP are the two typical hallmarks of enhanced activation of apoptosis.32,33 Activated caspase3 catalyzes the specific cleavage of PARP, causing it to lose its enzyme activity and ultimately leading to the apoptosis of cells. Western blot was performed to examine the expression of apoptosis-related signaling pathway proteins. Apparently, tFNAs reduced apoptosis in the context of DOX insult, as indicated by decreased cleaved caspase3 and cleaved PARP in 12-hour DOX insult (1μM) (Figure 4D and E). AKT itself can be activated under oxidative stress conditions.34 Acting as upstream of p53, phosphorylated AKT phosphorylates MDM2 at Serine 183 (Ser183), thereby stabilizing MDM2 in the nucleus and accelerating p53 degradation, which promotes cell survival.35 But after 12 hours of DOX treatment, we did not observe elevated pAKT or decreased p53 in the DOX+tFNAs group. We speculate that tFNAs treatment rapidly suppressed oxidative stress. Therefore, we reduced the DOX treatment duration to 8 hours and did another Western blot analysis, and we found that pAKT levels in the DOX+tFNAs group after 8-hour DOX treatment increased significantly compared to the DOX group, while p53 levels decreased significantly (Figure 4F and G). These results suggest that tFNAs may exert their effects by strongly activating the AKT/p53 pathway.
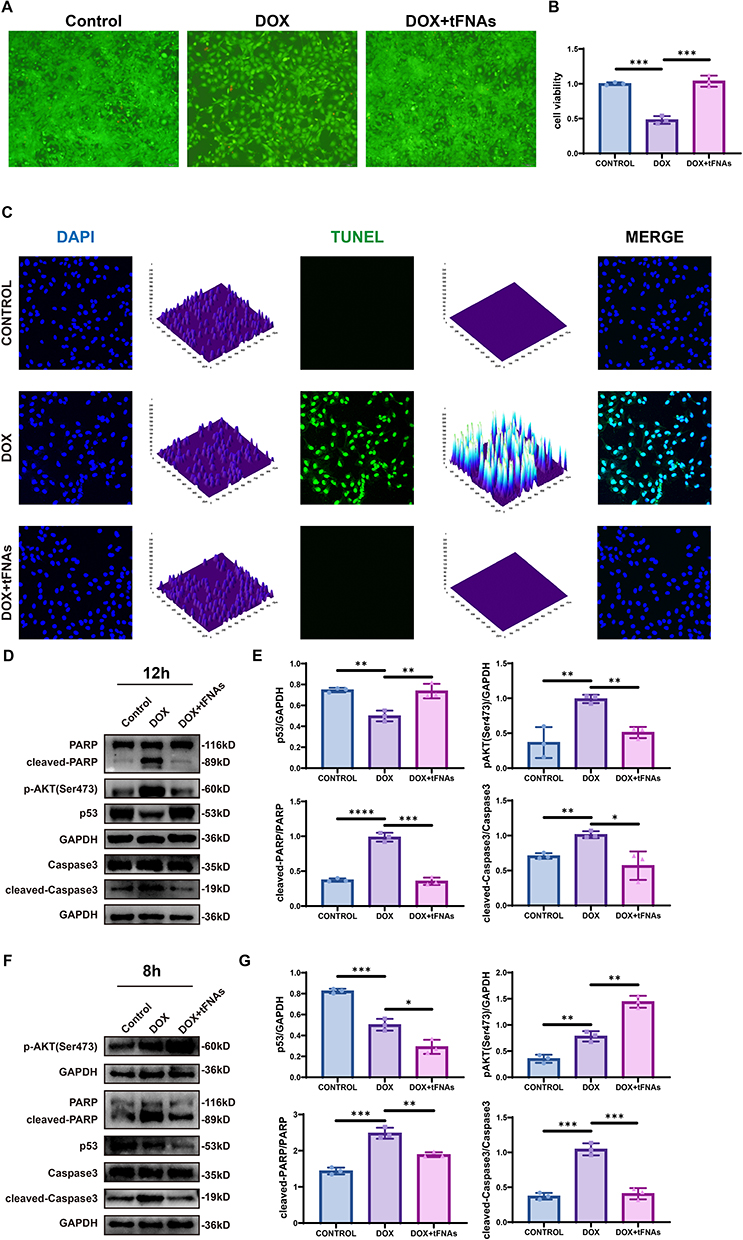 |
Figure 4 AKT/p53 signaling may be responsible for tFNAs-mediated protective role on cardiomyocyte apoptosis against DOX insult. (A and B). Representative images of live/dead staining of AC16 cells and quantification, n=3. (C) Representative images of TUNEL staining of AC16 cells of different groups. (D and E) Protein levels of cleaved-caspase-3, caspase-3, cleaved-PARP, PARP, phospho-AKT (Ser473), p53, and GAPDH detected by Western blot and their quantitative evaluation, n=3. AC16 cells were preincubated with tFNAs (250 nM) or PBS for 12 h before being treated with DOX (1μM) for 8/12 h. The data are presented as the mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
tFNAs Exerted Cardioprotective Effects on Mouse Hearts in Response to DOX Insult
We next used C57BL/6J mice aged 8 weeks to establish a DOX-induced murine model of cardiac injury. Before further experiments, we tested the in vivo biosafety of tFNAs in mice. Age-matched 8-week-old C57BL/6J mice (n=6/group) were allocated to: the control group, the DOX-treated group, and the DOX- and tFNA-treated groups. After the four-week experiment, echocardiographic analysis was carried out to measure mouse heart function. DOX challenge severely decreased the ejection fraction, fraction shortening, and heart rate, whereas tFNA treatment rescued the hearts from DOX insult, as indicated by increases in the ejection fraction, heart rate, and fraction shortening (Figure 5A–D). Additionally, Western blot analysis of heart tissue revealed elevated ANP levels in the DOX group and decreased ANP expression in the DOX + tFNAs group (Figure 5E). Also, DOX challenge t reduced mouse body weight and the ratio of heart weight (HW) to tibia length (TL), while tFNAs pretreatment partially rescued this phenotype (Figure 5F and G). These findings suggest that tFNAs protect mice from heart failure caused by DOX insult.
 |
Figure 5 tFNAs exerted cardioprotective effects to mice hearts in response to DOX insult. (A) Representative echocardiographic images of cardiac function. (B–D) Quantitative analysis of fraction shortening, ejection fraction and heart rate, n=6. (E) Protein levels of ANP and GAPDH detected by Western blot and their quantitative evaluation, n=3. (F) Quantitative analysis of mouse body weight, n=6. (G) Quantitative analysis of HW/TL. HW, heart weight; TL, tibia length. (H) Protein levels of collagen I and GAPDH detected by Western blot and their quantitative evaluation, n=3. (I) Representative images of Masson staining. (J) Representative TEM images of mice hearts in different groups. (K–N) MDA, NADPH, GSH, SOD contents in mice hearts, n=6. Mice were administrated with DOX (15 mg/kg, intraperitoneally) in three weekly doses, resulting in a cumulative total dose of 15 mg/kg. For those in DOX and tFNA-treated group, 100 μL of tFNA solution (250 nM) was administered via tail vein injection 12 hours prior to each DOX administration, over a 3-week period. The data are presented as the mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
Next, we aimed to explore the underlying mechanism by which tFNA treatment affects DOX-induced cardiac injury. The increase in the collagen I level in response to DOX insult was alleviated after tFNA treatment (Figure 5H). Compared with that in the DOX group, Masson staining revealed a decreased fibrotic area in the DOX + tFNA group (Figure 5I). TEM images of the myocardium revealed ultrastructural alterations inside the mitochondria. Ultrastructural analysis revealed extensive myocardial injury characterized by mitochondrial cristae fragmentation and sarcomeric disarray with myofilament dissolution. Furthermore, the tFNA treatment group presented an intact mitochondrial membrane and a regular mitochondrial ultrastructure (Figure 5J). The ELISA results for the myocardium shown in Figure 5I–L verified the protective effects of tFNAs against oxidative damage to mitochondria following DOX challenge.
Discussion
This study revealed for the first time the cardioprotective effects of tFNAs in a DIC model. Through both in vivo and in vitro experiments, we confirmed that tFNAs significantly alleviated myocardial injury through inhibiting mitochondrial oxidative stress and regulating apoptosis, and these effects may be achieved by activating AKT/p53 pathway (Figure 6).
 |
Figure 6 Schematic illustration of the antioxidant, anti-fibrosis, anti-apoptosis and cardioprotective effects of tFNAs in DOX-stimulated cardiomyocyte and mice model of DIC. |
Multiple factors are substantiated in the pathogenesis of DIC, encompassing inhibition of DNA/RNA/protein synthesis, dysregulation of autophagy, and disruption of intracellular calcium homeostasis.5,13,36 However, emerging evidence highlights the indispensable roles of ROS overproduction and cardiomyocyte apoptosis in DOX-induced myocardial damage. DOX-elicited oxidative stress activates both extrinsic and intrinsic apoptotic cascades, provoking extensive cardiomyocyte death and consequent severe impairment of cardiac function.14 The heart constitutes the primary target organ for DOX-induced toxicity. Under physiological conditions, the quinone group within the DOX molecule undergoes enzymatic reduction via NADH-dependent reductases, forming a semiquinone radical intermediate. This semiquinone moiety rapidly reacts with molecular oxygen (O2), generating superoxide anion radicals (O2−) while concurrently regenerating DOX’s original quinone structure. This cyclic redox reaction perpetuates continuous production of superoxide radicals, culminating in a massive “supernova” of ROS.37 Therefore, maintaining redox homeostasis may represent an effective strategy for ameliorating doxorubicin-induced cardiotoxicity.38
Current preventative and therapeutic approaches for doxorubicin-induced cardiac injury, such as dexrazoxane or dose-limiting strategies, provide only limited cardioprotection in DIC or significantly reduces anti-cancer efficacy. Dexrazoxane, as a catalytic inhibitor of topoisomerase II, protects cardiac cells by binding to the ATPase domain of TOP2β, stabilizing its closed conformation and thereby preventing DNA unwinding and religation.39 The mechanism by which dexrazoxane prevents DNA damage through degradation of TOP2B protein may compromise the antitumor efficacy of doxorubicin and potentially increase the risk of secondary tumors.40 Despite these limitations, emerging therapeutic strategies aim to circumvent the cardioprotection-chemotherapy efficacy trade-off by targeting downstream pathways of oxidative stress and mitochondrial dysfunction.
In sarcopenia, tFNAs were found to inhibit Drp1-mediated excessive mitochondrial fission, upregulate the expression of the mitochondrial fusion protein Mfn1, restore mitochondrial network integrity, and reduce ROS production. By balancing the expression of the pro-apoptotic protein BIM and the anti-apoptotic protein BCL, tFNAs block the mitochondrial apoptosis pathway and decrease myotube apoptosis.16 tFNAs restore mitochondrial membrane potential, reduce cytochrome c (Cyto C) release and caspase activation, and decrease the apoptosis rate of renal tubular epithelial cells in acute kidney injury.21 In our research, we demonstrate that tFNAs not only counteract oxidative stress and reduce cell apoptosis, but also alleviate fibrosis and suppress the expression of related genes, thereby partially restoring cardiac function in DIC. These results position tFNAs as a multi-organ protective agent targeting the mitochondrial-ROS-apoptosis-fibrosis axis.
Conclusion
This study revealed that tFNAs efficiently penetrate myocardial cells, exhibiting significant therapeutic benefits for DIC. Both in vitro and in vivo experiments demonstrated that tFNA administration effectively mitigated oxidative stress-induced damage and resultant apoptosis, mediated through suppression of the p53/AKT signaling pathway. These findings highlight tFNAs as a potential nanotherapeutic strategy for managing DIC through targeted modulation of critical cellular stress-response mechanisms.
Acknowledgments
This work was sponsored by the Science Technology Department of Zhejiang Province, China (Grant No. 2023C03087).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Volkova M, Russell R. Anthracycline cardiotoxicity: prevalence, pathogenesis and treatment. Curr Cardiol Rev. 2012;7(4):214–220. doi:10.2174/157340311799960645
2. Silber JH, Barber G. Doxorubicin-induced cardiotoxicity. N Engl J Med. 1995;333(20):1359–1360.
3. Carvalho FS, Burgeiro A, Garcia R, Moreno AJ, Carvalho RA, Oliveira PJ. Doxorubicin-induced cardiotoxicity: from bioenergetic failure and cell death to cardiomyopathy. Med Res Rev. 2014;34(1):106–135. doi:10.1002/med.21280
4. Wang Y, Lu X, Wang X, et al. atg7-based autophagy activation reverses doxorubicin-induced cardiotoxicity. Circ Res. 2021;129(8):e166–e182. doi:10.1161/CIRCRESAHA.121.319104
5. Zhang S, Liu X, Bawa-Khalfe T, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18(11):1639–1642. doi:10.1038/nm.2919
6. Huang KM, Zavorka Thomas M, Magdy T, et al. Targeting OCT3 attenuates doxorubicin-induced cardiac injury. Proc Natl Acad Sci U S A. 2021;118(5):e2020168118. doi:10.1073/pnas.2020168118
7. Zhang X, Hu C, Kong CY, et al. FNDC5 alleviates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via activating AKT. Cell Death Differ. 2020;27(2):540–555. doi:10.1038/s41418-019-0372-z
8. Fang X, Wang H, Han D, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116(7):2672–2680. doi:10.1073/pnas.1821022116
9. Detmer FJ, Alpert NM, Moon SH, et al. PET imaging of mitochondrial function in acute doxorubicin-induced cardiotoxicity: a proof-of-principle study. Sci Rep. 2022;12(1):6122. doi:10.1038/s41598-022-10004-6
10. Patron M, Tarasenko D, Nolte H, et al. Regulation of mitochondrial proteostasis by the proton gradient. EMBO J. 2022;41(16):e110476. doi:10.15252/embj.2021110476
11. Vringer E, Tait SWG. Mitochondria and cell death-associated inflammation. Cell Death Differ. 2023;30(2):304–312. doi:10.1038/s41418-022-01094-w
12. Matsuyama S, Reed JC. Mitochondria-dependent apoptosis and cellular pH regulation. Cell Death Differ. 2000;7(12):1155–1165. doi:10.1038/sj.cdd.4400779
13. Takemura G, Fujiwara H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog Cardiovasc Dis. 2007;49(5):330–352. doi:10.1016/j.pcad.2006.10.002
14. Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. 2012;52(6):1213–1225. doi:10.1016/j.yjmcc.2012.03.006
15. Renu K, A VG, TP PB, Arunachalam S. Molecular mechanism of doxorubicin-induced cardiomyopathy – an update. Eur J Pharmacol. 2018;818:241–253. doi:10.1016/j.ejphar.2017.10.043
16. Yu X, Wang Y, Ran L, et al. Tetrahedral framework nucleic acids inhibit muscular mitochondria-mediated apoptosis and ameliorate muscle atrophy in sarcopenia. Nano Lett. 2023;23(18):8816–8826. doi:10.1021/acs.nanolett.3c01502
17. Shi S, Chen T, Lu W, Chen Y, Xiao D, Lin Y. Amelioration of osteoarthritis via tetrahedral framework nucleic acids delivering microRNA‐124 for cartilage regeneration. Adv Funct Mater. 2023;33(46):2305558. doi:10.1002/adfm.202305558
18. Zhang T, Tian T, Zhou R, et al. Design, fabrication and applications of tetrahedral DNA nanostructure-based multifunctional complexes in drug delivery and biomedical treatment. Nat Protoc. 2020;15(8):2728–2757. doi:10.1038/s41596-020-0355-z
19. Zhou M, Zhang T, Zhang B, et al. A DNA nanostructure-based neuroprotectant against neuronal apoptosis via inhibiting toll-like receptor 2 signaling pathway in acute ischemic stroke. ACS Nano. 2022;16(1):1456–1470. doi:10.1021/acsnano.1c09626
20. Li J, Yao Y, Wang Y, et al. Modulation of the crosstalk between Schwann cells and macrophages for nerve regeneration: a therapeutic strategy based on a multifunctional tetrahedral framework nucleic acids system. Adv Mater. 2022;34(46):2202513. doi:10.1002/adma.202202513
21. Yan R, Cui W, Ma W, Li J, Liu Z, Lin Y. Typhaneoside-tetrahedral framework nucleic acids system: mitochondrial recovery and antioxidation for acute kidney injury treatment. ACS Nano. 2023;17(9):8767–8781. doi:10.1021/acsnano.3c02102
22. Steitz TA. A mechanism for all polymerases. Nature. 1998;391(6664):231–232. doi:10.1038/34542
23. Liang X, Wang S, Wang L, Ceylan AF, Ren J, Zhang Y. Mitophagy inhibitor liensinine suppresses doxorubicin-induced cardiotoxicity through inhibition of Drp1-mediated maladaptive mitochondrial fission. Pharmacol Res. 2020;157:104846. doi:10.1016/j.phrs.2020.104846
24. González A, Schelbert EB, Díez J, Butler J. Myocardial interstitial fibrosis in heart failure: biological and translational perspectives. J Am Coll Cardiol. 2018;71(15):1696–1706. doi:10.1016/j.jacc.2018.02.021
25. de Haas HJ, Arbustini E, Fuster V, Kramer CM, Narula J. Molecular imaging of the cardiac extracellular matrix. Circ Res. 2014;114(5):903–915. doi:10.1161/CIRCRESAHA.113.302680
26. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71(4):549–574. doi:10.1007/s00018-013-1349-6
27. Bujak M, Ren G, Kweon HJ, et al. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. 2007;116(19):2127–2138. doi:10.1161/CIRCULATIONAHA.107.704197
28. Piccoli MT, Gupta SK, Viereck J, et al. Inhibition of the cardiac fibroblast-enriched lncRNA Meg3 prevents cardiac fibrosis and diastolic dysfunction. Circ Res. 2017;121(5):575–583. doi:10.1161/CIRCRESAHA.117.310624
29. Duarte S, Baber J, Fujii T, Coito AJ. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol J Int Soc Matrix Biol. 2015;44-46:147–156. doi:10.1016/j.matbio.2015.01.004
30. Liu D, Ma Z, Di S, et al. AMPK/PGC1α activation by melatonin attenuates acute doxorubicin cardiotoxicity via alleviating mitochondrial oxidative damage and apoptosis. Free Radic Biol Med. 2018;129:59–72. doi:10.1016/j.freeradbiomed.2018.08.032
31. Pan JA, Tang Y, Yu JY, et al. miR-146a attenuates apoptosis and modulates autophagy by targeting TAF9b/P53 pathway in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2019;10(9):668. doi:10.1038/s41419-019-1901-x
32. Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. 2017;7(1):20–37. doi:10.1158/2159-8290.CD-16-0860
33. Porter AG, Jänicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6(2):99–104. doi:10.1038/sj.cdd.4400476
34. Ryu KY, Lee H, Woo H, et al. Dasatinib regulates LPS-induced microglial and astrocytic neuroinflammatory responses by inhibiting AKT/STAT3 signaling. J Neuroinflammation. 2019;16(1):190. doi:10.1186/s12974-019-1561-x
35. Chibaya L, Karim B, Zhang H, Jones SN. Mdm2 phosphorylation by Akt regulates the p53 response to oxidative stress to promote cell proliferation and tumorigenesis. Proc Natl Acad Sci. 2021;118(4):e2003193118. doi:10.1073/pnas.2003193118
36. Mukhopadhyay P, Rajesh M, Bátkai S, et al. CB1 cannabinoid receptors promote oxidative stress and cell death in murine models of doxorubicin-induced cardiomyopathy and in human cardiomyocytes. Cardiovasc Res. 2010;85(4):773–784. doi:10.1093/cvr/cvp369
37. Sarfraz M, Afzal A, Raza SM, et al. Liposomal co-delivered oleanolic acid attenuates doxorubicin-induced multi-organ toxicity in hepatocellular carcinoma. Oncotarget. 2017;8(29):47136–47153. doi:10.18632/oncotarget.17559
38. Tai P, Chen X, Jia G, et al. WGX50 mitigates doxorubicin-induced cardiotoxicity through inhibition of mitochondrial ROS and ferroptosis. J Transl Med. 2023;21(1):823. doi:10.1186/s12967-023-04715-1
39. Vavrova A, Jansova H, Mackova E, et al. Catalytic inhibitors of topoisomerase II differently modulate the toxicity of anthracyclines in cardiac and cancer cells. PLoS One. 2013;8(10):e76676. doi:10.1371/journal.pone.0076676
40. Liu C, Shen M, Liu Y, et al. CRISPRi/a screens in human iPSC-cardiomyocytes identify glycolytic activation as a druggable target for doxorubicin-induced cardiotoxicity. Cell Stem Cell. 2024;31(12):1760–1776.e9. doi:10.1016/j.stem.2024.10.007
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Vitamin D Ameliorates Apoptosis and Inflammation by Targeting the Mitochondrial and MEK1/2-ERK1/2 Pathways in Hyperoxia-Induced Bronchopulmonary Dysplasia
Hu J, Wu Z, Wang H, Geng H, Huo J, Zhu X, Zhu X
Journal of Inflammation Research 2022, 15:4891-4906
Published Date: 25 August 2022
Eriochloa villosa Alleviates Progression of Benign Prostatic Hyperplasia in vitro and in vivo
Baek EB, Hwang YH, Park S, Hong EJ, Won YS, Kwun HJ
Research and Reports in Urology 2022, 14:313-326
Published Date: 24 September 2022
Zinc Oxide Particles Can Cause Ovarian Toxicity by Oxidative Stress in Female Mice Model

Xu Y, Zhao Y, Liu S, Lv S, Chen L, Wang W, Feng Y, Fu F, Xu H
International Journal of Nanomedicine 2022, 17:4947-4960
Published Date: 19 October 2022
Pomegranate Seeds and Peel Ethanolic Extracts Anticancer Potentials and Related Genetic, Histological, Immunohistochemical, Apoptotic and Oxidative Stress Profiles: In vitro Study
Nasr M, Naeem SA, El-Shenbaby I, Mohamed FMA, Mahmoud SM, Abuamara TMM, Abd-Elhay WM, Elbayoumy FMAE, Elkot A, Shikhon T, Abo-akrab M, Doma MA, Hasan A
Journal of Experimental Pharmacology 2023, 15:191-205
Published Date: 15 April 2023
Remimazolam Suppresses Oxidative Stress and Apoptosis in Cerebral Ischemia/Reperfusion Injury by Regulating AKT/GSK-3β/NRF2 Pathway
Duan M, Yu N, Liu J, Zhao Y, Zhang J, Song S, Wang S
Drug Design, Development and Therapy 2025, 19:111-128
Published Date: 8 January 2025