Back to Journals » Journal of Inflammation Research » Volume 14
Targeting Neuroinflammation via Purinergic P2 Receptors for Disease Modification in Drug-Refractory Epilepsy
Authors Engel T ![]() , Smith J, Alves M
, Smith J, Alves M
Received 17 April 2021
Accepted for publication 12 June 2021
Published 18 July 2021 Volume 2021:14 Pages 3367—3392
DOI https://doi.org/10.2147/JIR.S287740
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Ning Quan
Tobias Engel,1,2,* Jonathon Smith,1,2,* Mariana Alves1
1Department of Physiology and Medical Physics, Royal College of Surgeons in Ireland, University of Medicine and Health Sciences, Dublin, D02 YN77, Ireland; 2FutureNeuro, Science Foundation Ireland Research Centre for Chronic and Rare Neurological Diseases, Royal College of Surgeons in Ireland, University of Medicine and Health Sciences, Dublin, D02 YN77, Ireland
*These authors contributed equally to this work
Correspondence: Tobias Engel
Department of Physiology and Medical Physics, Royal College of Surgeons in Ireland, University of Medicine and Health Sciences, 123 St Stephen’s Green, Dublin D02 YN77, Ireland
Tel +353-1-402-5199
Fax +35314022447
Email [email protected]
Abstract: Treatment of epilepsy remains a clinical challenge, with > 30% of patients not responding to current antiseizure drugs (ASDs). Moreover, currently available ASDs are merely symptomatic without altering significantly the progression of the disease. Inflammation is increasingly recognized as playing an important role during the generation of hyperexcitable networks in the brain. Accordingly, the suppression of chronic inflammation has been suggested as a promising therapeutic strategy to prevent epileptogenesis and to treat drug-refractory epilepsy. As a consequence, a strong focus of ongoing research is identification of the mechanisms that contribute to sustained inflammation in the brain during epilepsy and whether these can be targeted. ATP is released in response to several pathological stimuli, including increased neuronal activity within the central nervous system, where it functions as a neuro- and gliotransmitter. Once released, ATP activates purinergic P2 receptors, which are divided into metabotropic P2Y and ionotropic P2X receptors, driving inflammatory processes. Evidence from experimental models and patients demonstrates widespread expression changes of both P2Y and P2X receptors during epilepsy, and critically, drugs targeting both receptor subtypes, in particular the P2Y1 and P2X7 subtypes, have been shown to possess both anticonvulsive and antiepileptic potential. This review provides a detailed summary of the current evidence suggesting ATP-gated receptors as novel drug targets for epilepsy and discusses how P2 receptor–driven inflammation may contribute to the generation of seizures and the development of epilepsy.
Keywords: epilepsy, inflammation, purinergic signaling, ATP, P2 receptors, P2X7 receptor, P2Y1 receptor, disease modification, drug refractoriness
Introduction
Epilepsy encompasses a heterogeneous group of brain disorders which all share a predisposition to generate spontaneous, unprovoked seizures. With an incidence of ~1% and affecting up to 70 million persons worldwide, epilepsy is one of the most common chronic neurological diseases,1 and consequently is associated with a substantial economic burden.2 Adding significantly to the disease burden, epilepsy has a fourfold-increased risk of additional comorbidities (eg, mood, anxiety, and psychotic disorders).3,4 Epilepsy can result from genetic abnormalities (eg, de novo mutations, polymorphisms, copy-number variations) or insults to the brain, such as traumatic brain injury (TBI), stroke, prolonged seizures (eg, status epilepticus), or infection.5,6 Epileptogenesis, triggered following a precipitating insult to the brain, is the process of turning a normal healthy brain into a brain expressing epileptic seizures.7 Pathological changes occurring during epileptogenesis include ongoing neurodegeneration, structural and synaptic plasticity, increased permeability of the blood–brain barrier (BBB), aberrant neurogenesis, epigenetic changes, and inflammation.7,8 Temporal lobe epilepsy (TLE), which can be acquired following an insult to the brain, is the most common form of epilepsy in adults, is particularly prone to drug refractoriness, and involves structures within the limbic system, including the amygdala and hippocampus.1 Hippocampal sclerosis, characterized by a pattern of selective neuronal loss and reactive gliosis, is the most common pathological finding in brains of TLE patients.9 Of note, whereas some recent studies have investigated a potential role of ATP-gated P2 receptors in epilepsies with a genetic cause (eg, use of WAG/Rij rats, which is a model of genetic absence epilepsy10), most effort has been invested in studying these receptors in TLE,11 which is thus the main focus of the present review. While there are several options for the treatment of epilepsy, which include brain surgery, use of the ketogenic diet, or vagus nerve/deep-brain stimulation, antiseizure medication remains the first-choice treatment of epilepsy, with over 25 antiseizure drugs (ASDs) available.1,12 Current ASDs are, however, ineffective in >30% of patients, can cause severe side effects (eg, fatigue, irritability, and dizziness) and are thought to be mainly symptomatic.13 With the majority of ASDs targeting synaptic transmission (glutamate, γ-aminobutyric acid [GABA] and ion [Na+/Ca2+] channels) the focus is now on the identification of novel disease-modifying treatment strategies targeting underlying causes that provide long-lasting seizure control, either preventing the development of epilepsy or slowing down/reversing disease progression once epilepsy is established.14,15
It is now well established that besides glutamatergic- and GABAergic-dependent neurotransmission, other signaling systems contribute to increased brain hyperexcitability, possibly representing alternative treatment targets for epilepsy. In this regard, exciting evidence gathered over the last few decades has demonstrated a role for purinergic signaling in both the generation of seizures and the development of epilepsy. Purinergic signaling encompasses a complex system comprising purine-release mechanisms, specific purinergic receptors (adenosine-sensitive P1 and adenine and uracil nucleotide–sensitive P2 receptors), and enzymes metabolizing and thereby removing extracellularly released purines, such as ATP or adenosine.16,17 While the anticonvulsant function of the nucleoside adenosine via P1 receptors is well established,18 data generated over the last decade have also suggested an important role for P2 receptors during the generation of seizures and epilepsy.11 This includes evidence showing ATP release following seizures and during epilepsy, widespread expression changes of P2 receptors during epilepsy within the brain, and importantly drugs targeting P2-receptor subtypes have been shown to suppress seizures and even impact on the development of epilepsy. In the present review, we provide a detailed description of what is known regarding the expression and function of P2 receptors observed in both animal models of epilepsy and patients, and how targeting of P2-receptor subtypes impacts on seizures, epilepsy, and the underlying inflammatory processes.
Epilepsy and Inflammation
Because seizures are thought to be the result of an imbalance between excitatory and inhibitory activity of neural circuits in the brain, epilepsy has been considered primarily to be a disease of neurons, with the targeting of neuronal ion channels, both GABA and glutamate receptors, the main approach to stop/suppress seizures.13,19 However, this perception has changed considerably over recent decades, with mounting evidence from both experimental models and patients demonstrating a contribution of other cell types to increased brain hyperexcitability, including astrocytes and microglia.20–22
The role of astrocytes in epilepsy is well established. Astrocytes are key regulators of homeostasis in the central nervous system (CNS) during physiology and pathology, regulating crucial processes involved in gliotransmission, extracellular water and ion balance, maintenance of the BBB, regulation of arteriolar blood flow, energy supply, metabolism, oxidative stress, and neuroinflammation.23 Suggesting a proepileptogenic function, reactive astrocytes, which are characterized by hypertrophy of primary processes and a dramatic increase in the expression of GFAP, display several characteristics believed to contribute to increased brain hyperexcitability and seizures. This includes the release of proinflammatory molecules (eg, IL1β, IL6, or TNFα) and excitatory gliotransmitters (eg, ATP, glutamate), altered expression of ion channels (eg, inwardly rectifying potassium channel Kir4.1), leading to perturbation in spatial K+ buffering, changes in expression of Ca2+ and Cl– transporters, uncoupling of gap junctions, and promotion of Ca2+ waves, which in turn are thought to modulate the release of a number of gliotransmitters that may influence synaptic function.23–26 Moreover, astrocytes express the enzyme adenosine kinase (ADK), which catalyzes the conversion of adenosine into adenosine monophosphate, removing anticonvulsant adenosine from the extracellular space and thereby lowering seizure threshold.27 Further support for a proepileptic function of activated astrocytes stems from a study showing that the activation of astrocytes in the absence of other pathologies is sufficient to cause epileptic seizures.28
Microglia are the brain’s resident macrophages, and are crucial for physiological brain function. In their resting state, microglia have a ramified morphology with long protruding processes, and play a surveillance role by removing cellular debris and dead cells from the brain, synaptic pruning, and controlling the release of anti- and proinflammatory mediators. Following injury to the brain (eg, seizures), microglia are one of the first cell types to respond, and transition to an activated state.29 In contrast to astrocytes, microglia’s role during epilepsy is less clear, with both pro- and antiepileptic effects reported.30 However, these pro- or antiepileptic effects are largely dependent on the stage following the initial insult. While previously believed to have a mainly proconvulsant function via the secretion of proinflammatory cytokines (eg, IL1β),31 more recent research now has also suggested a protective function of microglia. In line with this, the depletion of microglia leads to an amplification and synchronization of neuronal activity, culminating in the generation of epileptic seizures. Interestingly, this effect is highly dependent on microglia’s ability to sense and catabolize extracellular ATP that is released by neurons and astrocytes following neuronal activation.32 This detrimental effect was further confirmed by two recent studies wherein microglia were depleted in a similar fashion.33,34 These conflicting reports can be attributed to the heterogeneity of activated microglia that is largely present following an insult. Resting microglia can either transition to the proinflammatory and neurotoxic M1 phenotype or the anti-inflammatory M2 phenotype.35 M1 microglia secrete proinflammatory cytokines (such as TNFa, IL1β, and IL6), produce reactive oxygen species to potentiate neuroinflammation and initiate phagocytosis essential for neuronal repair.36 However, prolonged M1 activation is neurotoxic, creating a positive-feedback loop of perpetuating inflammation and subsequent neurodegeneration.37 M2 microglia have an opposing function, secreting anti-inflammatory cytokines (such as IL4 and IL13) to promote neuronal protection, and are essential for repair mechanisms.38 Microglia are polarized into an M1 phenotype via classical proinflammatory cytokines, such as TNFα, IFNγ, and activation of TLR4-mediated NFκβ signaling, as well as induced by necrotic neurons, whereas the M2 phenotype is induced via anti-inflammatory cytokines, such as IL4 and IL13.39 Adding to the complexity, astrocyte microglia–dependent signaling occurs, with both cell types maintaining a tight relationship during seizures and epilepsy, impacting thereby on each other’s activation status and function.40 Microglia inflammatory action may precede astrocytic action. Microglia are known to stimulate the transition of astrocytes to both a neuroprotective41 and a neurotoxic phenotype,42 this stimulation being largely dependent on the proinflammatory cytokines released from presumably M1 microglia. IL6, TNFα, and IL1β induce a neuroprotective astrocyte phenotype,41 while IL1α stimulates the neurotoxic phenotype.42 Interestingly, the classical anti-inflammatory cytokines TGFβ or FGF released from M2 microglia can inhibit the formation of neurotoxic astrocytes.42 Furthermore, in cocultures of astrocytes and microglia, the proliferative capability of astrocytes is drastically reduced, suggesting that microglia tightly regulate astrocytic proliferation.43 Of note, neurogenesis is also regulated by microglia, with evidence suggesting that microglia may both promote and suppress aberrant neurogenesis seen in epilepsy models.21,44 While it is clear that microglia have a significant role in epilepsy and seizures, each phenotype’s role has not yet been fully characterized in disease progression. In rodent models, a mixed-microglia phenotype is expressed following status epilepticus and also in established epilepsy.45 However, less is known regarding the polarization state of microglia during epileptogenesis. Benson et al observed an elevated M1 response during epileptogenesis; however, this was much less pronounced than in the acute and chronic stages of epilepsy. An emerging theoretical strategy to reduce the establishment of epilepsy is to elevate the M2-activation profile following an insult whist simultaneously reducing the effect of M1 microglia.36
In addition to astrocytes and microglia, other cells of innate immunoresponse (eg, monocytes and macrophages) have been ascribed a proepileptogenic role.46 In animal models of seizures, microglia and neurons release chemoattractants to exacerbate BBB disruption and recruit monocytes, more specifically CCR2+ monocytes, across the BBB. Once infiltrated, they can release proconvulsant cytokines, such as IL1β, via STAT3 signaling. However, while monocyte infiltration may exacerbate inflammation and neuronal damage,47–49 no effect on seizure severity has been observed in a rodent model of status epilepticus.50
Suggesting proinflammatory mediators directly interfere with neuronal function and excitability, cytokines have been reported to modulate the function of glutamate and GABA receptors, eg, IL1β inhibits GABA-mediated inhibitory transmission and potentiates excitatory N-methyl-D-aspartate (NMDA)–receptor-dependent synaptic transmission51–53, alter voltage-gated ion-channel function and expression eg, IL6 increases activity of Nav1.7, thereby amplifying excitatory inputs on neurons,54–57 and modulate presynaptic exocytosis of both glutamate and GABA neurotransmitters.58,59 Cytokines increase the amplitude and network synchronicity of neuronal Ca2+ currents in neuronal culture via reducing GABAergic input.60 Interestingly, IL1β is able to reduce the anticonvulsant action of midazolam (a benzodiazepine that enhances endogenous GABA activity) in these cultures, suggesting IL1β may cause the drug resistance seen in patients.60 IL1β’s ability to reduce GABA-neurotransmitter release in hyperexcitable networks in the mouse hippocampus may underlie this pharmacoresistance.61 Reinforcing this notion, in tissue resected from drug-refractory epileptic patients, cytokines (IL1β) reduced GABAA-mediated currents.53 Furthermore, the IL1 type I receptor is closely associated with the NMDA-receptor subtype 2B (NR2B) subunit of the excitatory NMDA receptor,62 where IL1β induces phosphorylation of NR2B. Phosphorylated NR2B induces excitotoxicity, enhances NMDA inward currents by ~45%, and inhibits NMDA outward currents, all promoting the generation of seizure.52,63,64 TNFα is also known to reduce inhibitory drive via inducing internalization of GABAA receptors while concurrently promoting the expression of excitatory α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors.65
Demonstrating the therapeutic potential of targeting inflammation in epilepsy, drugs targeting specific inflammatory pathways (eg, TLR4, HMGB1, IL1β) have provided neuroprotection and disease modification, delaying the onset of epilepsy and suppressing epileptic seizures.66–68 Importantly, anti-inflammatory drugs have reached the stage of clinical trials,69 which should accelerate their future use in the clinic.
There is now compelling evidence of inflammation impacting on both seizure generation and the development of epilepsy. Consequently, drugs targeting inflammatory pathways represent a promising new therapeutic strategy to provide disease modification in epilepsy and treat patients not responding to current ASDs.
Animal Models of Epilepsy
The heterologous nature of epilepsy syndromes is reflected in the plethora of experimental models available to mimic both acute seizures (eg, models of status epilepticus) and epilepsy. This includes both in vitro and in vivo approaches, with each having their inherent advantages and limitations. Whereas in vitro models mainly allow the study of molecular mechanisms of neuronal excitability, they cannot completely replicate the complexity of epilepsy and its genesis in a living being, which includes inflammatory processes, changes in the BBB, and complex neuron–glia interactions, though advantages of in vitro models include a higher number of possible replicates within a shorter time frame, with less variability between samples. While a detailed description of animal models of epilepsy is outside of the scope of the present review, we give a short summary of the main in vivo models used to investigate purinergic signaling in epilepsy. For a more comprehensive list of in vitro and in vivo models of seizures and epilepsy please refer to previous reviews published on this subject.15,70,71
In vivo approaches to model seizures and epilepsy include the use of seizure-inducing chemoconvulsants (eg, KA, pilocarpine, or pentylenetetrazol [Ptz]), electrical stimulation (eg, perforant pathway, hippocampus, amygdala), genetic mutations, and injury models (eg, TBI, hyperthermia) with species spanning from flies to primates.71,72 To date, however, investigation of the impact of purinergic signaling on seizures and epilepsy has largely been restricted to the use of chemoconvulsants and electrical stimulation in rodent models using models of acute seizures and drug-refractory epilepsy. This primarily includes the glutamate agonist KA, the cholinergic agonist pilocarpine, and the GABAA-receptor inhibitor Ptz, which will be described in more detail.
In vivo approaches modeling acute seizures include the Ptz model, widely used in the screening of anticonvulsant drugs. Here, intraperitoneal injections of Ptz lead to a single, brief, nondamaging seizure.71 Other models used to determine the impact of interfering with purinergic signaling during acute seizures include the maximal electroshock model and the 6 Hz psychomotor-seizure test, which has also been described as a model of pharmacoresistant partial seizures. Serial administrations of subconvulsive doses of Ptz over a number of days (Ptz kindling model) are used to mimic the process of epileptogenesis and determine antiepileptic effects.71 Another widely used model of epileptogenesis is the amygdala kindling model, which consists in the repeated application of electrical stimuli via a depth electrode in the basolateral amygdala of rodents and induces permanently enhanced seizure susceptibility and other enduring brain alterations that are similar to those occurring in human TLE.71
KA- and pilocarpine-treated mice are usually the models of choice to study effects of drugs targeting the purinergic system on status epilepticus and TLE pathology. Systemically injected pilocarpine mimics certain aspects of TLE pathology, including a period of status epilepticus with subsequent development of spontaneous recurrent seizures and a pattern of hippocampal damage and sclerosis, particularly evident in the CA1 and hilar regions of the hippocampus.73 This model is, however, associated with high mortality, peripheral immunoresponses prior to the onset of status epilepticus, including white blood–cell activation with consequent increase in IL1β serum levels and BBB damage,74 and high interanimal variability in pathology and neuronal injury, which most likely reflects a mixture of an ischemic and excitotoxic insult.71,73 Similarly to systemic pilocarpine, systemic injections of KA also cause prolonged seizures and the development of epilepsy with high variability in hippocampal pathology in mice and variable epilepsy development.75 In contrast, intracerebral injections of KA (eg, intrahippocampal and intraamygdala [IA]), seem to be associated with less mortality, more consistent and reliable focal and unilateral hippocampal lesions, and spontaneous seizure onset and refractoriness to ASDs.75,76 Of note, a recent study demonstrated that intracerebral (ie, IA and intrahippocampal) KA causes similar changes in hippocampal gene expression to human TLE.77 However, each of these models replicates slightly different aspects of seizure-induced pathologies, which is important to keep in mind when interpreting data and potential conflicting results among studies.15,71
Purinergic Signaling via P2 Receptor — Overview
ATP Release
ATP acting as an intercellular signaling molecule was first described by Geoffrey Burnstock in 1972.78 It is well recognized that ATP functions as either a sole transmitter or co-transmitter in both the peripheral nervous system and the CNS.79 Here, ATP can act as a fast, excitatory neurotransmitter or as a neuromodulator, and is involved in a plethora of short- and long-term physiological and pathological processes, including inflammation, cellular survival, proliferation, cellular differentiation, and synaptic plasticity.17,80,81 Under physiological conditions, ATP is usually present at micromolar concentrations in the extracellular space; however, under pathological conditions (eg, inflammation, hyperexcitability, and cell death) extracellular ATP levels can reach the millimolar range.79,81,82 As such, ATP acts as an endogenous danger signal, known as a damage-associated molecular pattern (DAMP).83 ATP can enter the extracellular space passively by crossing the compromised membranes of damaged and dying cells79 or be actively released from different cell types, including neurons, astrocytes, microglia, and endothelial cells.79,84 ATP-release mechanisms include exocytosis of secretory granules, vesicular transport involving the vesicular nucleotide transporter (VNUT), and membrane channels such as ABC transporters, pannexins (eg, pannexin 1), connexins, and via purinergic receptors themselves (eg, P2X7 receptor).79,84 Once released into the extracellular space, ATP is rapidly metabolized by ectonucleotidases, including the ectonucleoside triphosphate diphosphohydrolase family, the ectonucleotide pyrophosphatase/phosphodiesterase family, and alkaline phosphatases, into such breakdown products as ADP and adenosine, which are also important neurotransmitters and neuromodulators in their own right.17,18,85
Purinergic P2 Receptors
Activated by adenine and uridine nucleotides, purinergic P2 receptors are further subdivided into two families according to their sequence homology, pharmacology, and mechanism of action. This includes the ionotropic P2X receptors and metabotropic P2Y receptors, both expressed throughout the CNS and functional on both neurons and glial cells.86
To date, seven mammalian subunits of the P2X-receptor family have been cloned (P2X1–7), ranging in length from 379 (P2X6) to 595 (P2X7) amino acids.87 Each subunit comprises two transmembrane domains: a large extracellular loop and an intracellular N- and C-terminus.88 P2X receptors can form either functional homo- or heterotrimers, which upon binding to their endogenous agonist ATP allow the passage of small cations, such as Na+, Ca2+, and K+, depolarizing the cell membrane. All P2X receptors are sensitive to ATP; however, with differing levels of affinity (P2X1 > P2X3 > P2X2 > P2X4, P2X5 > P2X6 > P2X7).89 P2X receptors are activated within milliseconds, and with the exception of P2X7, display fast desensitization (P2X3 > P2X1 > P2X2 > P2X4 > P2X788,90). While the contribution of P2X receptors to fast synaptic transmission is well-established,91 they have also been implicated in several other cellular processes, such as proliferation, differentiation, maturation, survival, cell adhesion, migration, and inflammation.92 Among the P2X receptors, due to its unique structural and functional characteristics, P2X7 has attracted particular attention as a target for brain diseases, including epilepsy.93 P2X7 has a relatively low affinity for ATP (EC50 ≥100 μM, activation threshold 0.3–0.5 mM, which has been reported decreased during inflammation [0.05–0.1 mM])94, suggesting that P2X7 activation occurs mainly under pathological conditions of high ATP release (which may occur during a seizure). It also has slow desensitization dynamics, the ability to permeabilize the cell membrane to molecules up to 900 Da in size, and is a key driver of inflammation via activation of the NLRP3 inflammasome.94 Moreover, suggesting a direct impact on neurotransmission, P2X7 has been shown to modulate both uptake and release of GABA and glutamate during epilepsy.95,96 P2X7 is highly expressed on cells of the immune system, including microglia. P2X7 expression has also been reported on oligodendrocytes and endothelial cells.97,98 Its expression and function with regard to astrocytes and neurons remains, however, controversial.99,100
The metabotropic P2Y-receptor family consists of eight G protein–coupled receptors — P2Y1,2,4,6,11–14.17,101 P2Y receptors can be activated by adenosine or uridine nucleotides: ATP (P2Y2, P2Y11), ADP (P2Y1, P2Y12, P2Y13), uridine-5′-triphosphate (UTP) (P2Y2, P2Y4), UDP (P2Y6, P2Y14), and UDP-glucose (P2Y14). P2Y receptors have an extracellular amino terminus, intracellular carboxyl terminus, and seven transmembrane-spanning motifs.102 P2Y receptors can also be grouped according to their coupling to specific G proteins: P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11 receptors are coupled with Gq/G11 proteins, stimulating phospholipase C and leading to the release of calcium from intracellular stores and the activation of PKC. P2Y11 can also couple with Gs, stimulating adenylate cyclase and increasing the production of cyclic adenosine monophosphate (cAMP).101 P2Y12, P2Y13, and P2Y14 receptors are coupled to Gi proteins, inhibiting adenylate cyclase and thereby decreasing cAMP production.101 Similarly to P2X receptors, P2Y receptors have been described to be expressed and functional on all brain cells and implicated in several cellular functions and pathological processes relevant to epileptogenesis and epilepsy, including synaptic reorganization, changes in neurotransmitter release, neurodegeneration, and neuroinflammation.103,104
Purinergic Receptors and Neuroinflammation
P2X Receptors and Neuroinflammation
With many inflammatory pathologies resulting in altered purinergic signaling and with ATP being one of the primary DAMPs to be released at sites of inflammation, the close association of P2 receptors with inflammatory signaling comes as no surprise.105 This positive-feedback loop created between inflammation and ATP release creates continuous neuroinflammation in the brain.81
All P2X-receptor subtypes have some degree of expression on immune-system cells of the body; however, within the brain, only P2rx1, P2rx4, P2rx6, and P2rx7 mRNA is predominantly expressed in mouse microglia.106,107 Microglia P2rx3 and P2rx5 expression is observed at much lower levels, with P2rx5 expression restricted to neonatal animals and no evidence of microglia P2rx2 expression at any age analyzed.106 All P2X receptors aside from P2X6 are expressed in astrocytes.108 Of all P2X receptors, P2X4 and P2X7 possess the largest body of evidence of a well-defined functional role in inflammation. P2X1 and P2X2 inflammatory function within the brain is currently unknown, and their association with inflammation stems from their expression changing at sites of inflammation observed in ischemia, axotomy, stab-wound injuries, and cerebellar lesions.109–112 Despite their expression in glial cells, an immunomodulatory function of P2X3, P2X5, and P2X6 within the brain is currently unknown.
Of P2X7’s various roles, its proinflammatory function has been widely explored, with many touting it as the gatekeeper of inflammation.113 P2X7 is known to be the most potent contributor to the release of proinflammatory cytokines,94 including but not limited to IL18,114 IL6,115 IL1β, and IL1α.116 With varying affinity of ATP required to activate P2X4 or P2X7, they work in tandem to respond to stimuli over a wide range of ATP concentrations.117 In many disease pathologies, including seizures, the proinflammatory cytokines IL1β, IL18, and TNFα are released. It is well known that pharmacological inhibition or genetic knockout of P2X4 or P2X7 can hinder the release of proinflammatory cytokines, such as IL1β.118–122 Proinflammatory and proconvulsive IL1β release is a two-signal process: priming (an inflammatory signal acts upon TLR4 to synthesise proIL1β) and activation (activation of the NLRP3 inflammasome and caspase1 is required to cleave pro-IL1β and for subsequent secretion of IL1β). P2X7 is important for NLRP3 activation, with P2X7’s pore-forming ability described as necessary to create a drop of local intracellular K+ concentration needed for activation of the inflammasome without compromising global cellular K+ homeostasis.123,124 P2X4 also possesses the ability to form a large conductance pore that can facilitate inflammasome activation, with P2X4-knockout mice showing impaired inflammasome activation and subsequent IL1β release following a spinal cord injury.118 However, P2X4 may act as the initial trigger, due to a higher affinity for ATP, and then via working with pannexin 1 to release ATP, with P2X7 amplifying the signal and elevating inflammation within the brain.92 P2X7 stimulation can also promote the activation of microglia and elevate their proliferation.125 Furthermore, P2X7 may contribute to further release of DAMPs, which act to potentiate inflammation via facilitating pyroptosis.126 Pyroptosis, a form of inflammatory programmed cell death, is downstream of the P2X7–NLRP3 axis, where the permeability of the cell membrane increases, inducing swelling and subsequent cell death. P2X7 activation to induce pore formation can also contribute to cell death and subsequent DAMP release via rearrangements of the cell cytoskeleton.127,128 Interestingly, P2X4 pore formation does not lead to cell death.129 The release of DAMPs is a fundamental component of inflammation, and can further act upon P2X7 to potentiate inflammation. Furthermore, P2X7 activation can stimulate production of cytokine precursors via upregulation of both the NFκB130 and NFAT131 proinflammatory transcription factors.
Aside from P2X7’s defined role in microglia, its astrocytic expression has also been reported. Functional astrocytic P2X7 has been observed in cortical brain slices132 and astrocytic cultures, where it induces cell death and the release of radical oxygen species and proinflammatory IL6.133 Furthermore, human primary astrocytes secrete IL1β mediated via the P2X7–NLRP2 signaling cascade in tandem with pannexin 1 in a manner similar to what is seen in microglia.134
With P2X4 higher ATP affinity than P2X7, P2X4 may have a more physiological role in inflammation. Upregulation of P2X4 is observed in many disease models that are associated with inflammation.135–137 P2X4 immunoreactivity has been observed in astrocytes;138 however, conflicting reports from electrophysiological experiments suggest no functional role of astrocytic P2X4.139 P2X4 is widely considered to drive microglia motility, process extension, activation, and recruitment to the site of damage, with ATP acting as a chemotactic factor for P2X4+ microglia.140,141 In fact, P2X4 activation drives microglia recruitment and motility via the PI3K–Akt pathway.140,142 P2X4 expression is mainly restricted to activated microglia, with P2X4 localized in intracellular lysosomal compartments of microglia and trafficked to the cell surface under inflammatory conditions.143 Microglia are shifted to an activated state in response to noxious stimuli and governed via the transcription factors IRF8 and IRF5 directly inducing expression of the P2rx4 gene.144 Furthermore, P2X4 also plays an anti-inflammatory role, which may occur following its acute proinflammatory function. P2X4 is associated with mediating microglia cell death as a physiological response, with suggestions that the repopulating microglia are regenerative.145 Following a spinal cord injury, microglia also release brain-derived neurotrophic factor146 mediated via P2X4 to promote regeneration.147 In multiple sclerosis models, activation of microglia P2X4 promotes an anti-inflammatory phenotype and facilitates remyelination.148
P2Y Receptors and Neuroinflammation
P2Y receptors have been repeatedly implicated in neuroinflammation; however, their exact role is not fully understood and seems to be dependent on downstream-signaling pathways.149 As with P2X receptors, many P2Y receptors are expressed on inflammatory cells. In isolated microglia, mRNA of all known rodent P2Y receptors can be detected.106 Among P2Y receptors, P2Y1 is one of the predominant targets of ATP in mediating danger signals in the brain during, eg, ischemia150 or trauma.151 As such, there is a plethora of evidence of P2Y1 inflammatory action. Glial P2Y1 expression is known to regulate astroglial proliferation and astrogliosis.112 In a model of cerebral ischemia, antagonism of P2Y1 reduced the astrocytic transcription of cytokines (eg, TNFα) and chemokines.152 Overexpression of P2Y1 in astrocytes can lead to the propagation of intracellular Ca2+ waves, which is responsible for the release of ATP and cytokines.152,153 Oxidative stress potentiates the activation of P2Y1 and the release of IL6.154 Interestingly, blockade of P2Y1 increases astrocytic response and is neuroprotective in TBI.41 Reinforcing tight cross-interaction of glial cells, microglia-derived cytokines have been demonstrated to downregulate P2Y1 expression and function on astrocytes, promoting formation of glial scars and increasing astrocyte process extension.41 Interestingly P2Y1 and P2Y12 activation can induce astrocyte proliferation in vitro.155 In the same study, activation of P2Y14 had no influence on proliferation. Though less explored, an anti-inflammatory role of P2Y1 through the release of IL6 has also been suggested.156
P2Y12 is heavily implicated in mediating inflammatory responses. In fact, P2Y12 immunoreactivity is widely accepted as a marker of resting microglia,157 ie, microglia in nonpathological conditions. Similarly to P2X4, the primary role of P2Y12 activation is to induce reactivity of microglia, elicit microglia-process outgrowth, and stimulate their migratory movement via PI3k/PLC signaling to sites of inflammation.140,158–160 As such, P2Y12 may promote neuronal damage in brain pathologies by guiding the inflammatory response to the site of injury.161,162 Interestingly, P2Y12 expression is downregulated during the phenotypic M1/M2 shift,163 reinforcing its role in resting microglia. Additionally, microglial P2Y12 and P2Y13 are involved in preventing astroglial proliferation mediated by ADPβS in cocultures of astrocytes and microglia.164 P2Y12 and P2Y13 are integrally linked, with P2Y13 acting to potentiate the chemotaxis response of P2Y12,165 and have been widely reported to be expressed in microglia and astrocytes.106,166–168 As reported for the other P2Y receptors, P2Y13 activation also increases intracellular Ca2+ in glial cells,167–169 contributing to the release of several proinflammatory cytokines (IL1β, IL6, and TNFα) from microglia cells.170 Furthermore, a recent study demonstrated that joint activation of P2Y1 and P2Y13 induce the retraction of microglial processes in epileptic and peritumoral human tissue.171
Much less is known about the remaining P2Y receptors in inflammation. P2Y6 has been implicated in microglia activation, leading to activation of inflammation signaling pathways.172 P2Y6 stimulation is also involved in inducing microglia phagocytosis of cell debris, which is beneficial to neuroregeneration.172 Interestingly, P2Y6 activation can inhibit P2X4 functioning in microglia, which results in reduced motility.173 P2Y2 plays a neuroprotective role via upregulation of antiapoptotic genes in astrocytes174 and promoting migration of astrocytes.175 Furthermore, P2Y2 mediates amyloid β–protein phagocytosis by microglia cells, suggesting a neuroprotective role in Alzheimer’s disease.176 Unfortunately, current understanding of P2Y4’s functional role in neuroinflammation is lacking, despite it being known to be expressed in microglia.106
Purinergic Signaling During Seizures and Epilepsy
ATP Release During Seizures and Epilepsy
Initial evidence suggesting a link between increased extracellular ATP levels and hyperexcitability was provided by two studies, one showing increased extracellular ATP concentrations in hippocampal brain slices of a seizure-prone strain of mice (inbred DBA/2 [D2] mice),177 and the other evidencing motor seizures caused by microinjections of ATP into the prepiriform cortex of rats.178 In vitro evidence suggesting that ATP can be released during increased neuronal activity stems from a study using depolarizing high K+ concentrations in slices of rat hippocampus.179 Further in vitro evidence was provided by Lopartar et al, showing increased extracellular ATP mediated via pannexin 1 in rat hippocampal slices treated with the glutamate agonist (S)-3,5-dihydroxyphenylglycine.180 In contrast to these findings, other methods, such as stimulation of the Schaffer collateral, did not increase extracellular ATP concentrations.181 In agreement with pannexin 1 activation contributing to extracellular ATP increases during seizures, a recent study using resected tissue from patients with epilepsy showed that extracellular ATP increased 80% during high K+–induced ictal discharge, which was blocked by pharmacological inhibition of pannexin 1.182 Of note, blocking pannexin 1 channels provides potent anticonvulsive effects in a mouse model of KA-induced seizures, suggesting a proconvulsant function of extracellular released ATP.182 In vivo evidence showing increased extracellular ATP during seizures/epilepsy was provided by Dona et al using a rat model of epilepsy induced by pilocarpine administration (360 mg/kg, IP). The authors showed that status epilepticus led to an increase of the ATP metabolites ADP, AMP, and adenosine, but not ATP. On the other hand, the same ATP metabolites were found to be decreased during epilepsy, though increased significantly following an epileptic seizure. This also included an approximately fourfold increase in extracellular ATP levels.183 Further suggesting a proconvulsant function of extracellular adenosine nucleotides, injection of ATP, the ATP analogue 2,3-O-(4-benzoylbenzoyl) ATP (BzATP) or ADP, into the lateral ventricle of mice caused high-amplitude, high-frequency polyspiking on electroencephalography (ATP)184 and increased seizure severity during KA (0.3 µg, IA)–induced status epilepticus (BzATP and ADP).119,185 In contrast to a proconvulsant function of extracellular ATP, injection of uridine-5′-triphosphate into the lateral ventricle of mice reduced seizure severity during IA KA-induced status epilepticus.185
P2-Receptor Expression Following Seizures and During Epilepsy
P2 receptors, including both P2X and P2Y receptors, are widely distributed throughout the brain, where they are expressed and functional on both neurons and glial cells, contributing to a plethora of physiological and pathological processes ranging from neurotransmission and neurogenesis to glial cell communication.93,104 While we still lack a detailed description of potential expression changes during epilepsy for most of the P2 receptors, this has changed in recent years, in particular for the P2X7 and P2Y1 subtypes.
Expression changes for P2X receptors following seizures (eg, status epilepticus) or during epilepsy have been reported for P2X2, P2X4, and P2X7. No changes have been found so far in expression of the remaining P2X receptors (ie, P2X1, P2X3, P2X5, and P2X6).119 While P2X2 has been shown to be downregulated following KA (3 µg, IA),119 P2X4 expression increases in the hippocampus following systemic KA injections (8–22 mg/kg, IP),142,186 but not IA KA- or pilocarpine (360 mg/kg, IP)-induced status epilepticus.119,187 In contrast, P2X4 expression is decreased in the hippocampi of pilocarpine (360 mg/kg, IP)-treated epileptic rats.187 As mentioned previously, among the P2X-receptor family, most data are available for P2X7. P2X7 expression has been reported to be upregulated in mice following intraperitoneal KA (hippocampus),186 IA KA-induced status epilepticus in mice (hippocampus, cortex, striatum, thalamus, and cerebellum),119,188,189 and pilocarpine-induced status epilepticus in rats (hippocampus).187 Studies have suggested P2X7 to be upregulated on neurons post–status epilepticus using immunohistochemical approaches, detecting P2X7 at glutamatergic nerve terminals,187 and a soluble EGFP BAC transgenic P2X7 mouse model, where EGFP is under the transcriptional control of the P2rx7 promoter.119,188 Others, however, have failed to detect neuronal expression of P2X7 using P2X7 reporter mice where P2X7 is fused to EGFP.189,190 Of note, a recent study comparing both transgenic P2X7 reporter mouse lines observed aberrant P2X7 expression and increased P2X4 expression in the soluble EGFP BAC transgenic P2X7 mouse model, questioning the validity of this reporter mouse.191 P2X7 expression has also been shown on oligodendrocytes post–status epilepticus; however, its expression was not detected on astrocytes.119,189 During epilepsy, P2X7 expression has been shown to be increased in the hippocampus and cortex in both experimental rodent models and patients with TLE.95,187,188,192 Regarding its cell type–specific expression using immunohistochemistry, P2X7 expression has been shown to increase on microglia, mossy fibers, and glutamatergic nerve terminals of epileptic pilocarpine (360–380 mg/kg, IP)-treated rats.187,193 A mainly microglial and neuronal P2X7 induction during epilepsy was confirmed by using the soluble EGFP BAC transgenic P2X7 mouse model.188,192 Regarding neuronal expression, Barros-Barbosa found P2X7 to be increased in neocortical nerve terminals of patients with epilepsy.95 However, similarly to status epilepticus, neuronal expression of P2X7 receptors during epilepsy could not be confirmed in a more recent study using the P2X7 reporter mouse where P2X7 is fused to EGFP.189 P2X7 expression has also been found on oligodendrocytes, while absent on astrocytes during epilepsy in mice.189 Based on these results, whereas there is broad consensus on P2X7 receptors being increased on microglia following status epilepticus and during epilepsy, whether P2X7 receptors increase on neurons is still a matter of debate.
Changes in the transcription and expression of the P2Y receptors have been reported in different brain structures, including the hippocampus and cortex, using different mouse models, including KA (3 μg, IA and 18-22 mg/kg, IP)185, 186 and pilocarpine (340 mg/kg, IP),185 pilocarpine (340 mg/kg, IP),185 and KA (18–22 mg/kg, IP)-induced status epilepticus mouse model.186 In the IP KA-induced status epilepticus mouse model, P2ry6, P2ry12, and P2ry13 mRNA transcripts were increased in the hippocampus.186 A more detailed study using the IA KA-induced status epilepticus mouse model, identified an interesting pattern between changes in P2Y-receptor transcript levels and the two main agonists of these receptors (adenosine and uridine nucleotides). While transcript levels of adenosine-sensitive receptors were decreased (P2ry1, P2ry12, and P2ry13), transcript levels of uridine-sensitive receptors were increased (P2ry2, P2ry4, and P2ry6) in the hippocampus. At the protein level, hippocampal P2Y receptors coupled with Gq were increased (P2Y1, P2Y2, P2Y4, and P2Y6) and P2Y receptors coupled with Gi downregulated or unchanged (P2Y12, P2Y13, P2Y14).185 In contrast, P2Y-receptor expression was mainly upregulated in the cortex post–status epilepticus194 and in the hippocampi of epileptic mice and patients with drug-refractory epilepsy.185,195 Regarding cell type–specific expression, while P2Y1 has been shown to be expressed in microglia following IA KA-induced status epilepticus,196 P2Y2 and P2Y4 have been shown to be mainly expressed in astrocytes in brain tissue from patients with intractable epilepsy.195
Targeting of P2X Receptors During Seizures and Epilepsy
As stated before, among the P2X receptors, though not the only one investigated, P2X7 has attracted most attention as a potential therapeutic target for seizure suppression and treatment of epilepsy.93
Treatment with P2X7 antagonists has been shown to provide anticonvulsive effects in several experimental models of status epilepticus and acute seizures. P2X7 antagonists (A438079 and Brilliant Blue G [BBG]) reduce both seizure severity during status epilepticus and resulting neurodegeneration in the KA (3 µg, IA) mouse model (pre- and posttreatment regime).119,188 Using the same IA KA mouse model, seizure suppression is also evident in P2X7 Knockout mice and via P2X7-targeting antibodies delivered into the lateral ventricle.119 Interestingly, P2X7 antagonists potentiated effects of the anticonvulsant lorazepam, suggesting their potential as adjunctive treatment for pharmacoresistant status epilepticus.119 Confirming an anticonvulsive effect of P2X7 antagonism, a more recent study using three P2X7 antagonists (BBG, A438079, and A740003) showed reduced seizure severity during coriaria lactone (40 mg/kg, IP)-induced status epilepticus in rats.197 Suggesting anticonvulsive effects of P2X7 antagonism not being dependent on developmental stage, P2X7 antagonism also reduces seizures in a KA (2 µg, ICV) rat model of early-life seizures using the P2X7 antagonist A438079198 and in a model of hypoxia-induced seizures (5% O2 for 15 minutes) in 7-day-old mouse pups using two P2X7 antagonists (A438079 and JNJ47965567).121 The effects of P2X7 antagonism on neonatal seizures are even more exciting, as commonly used ASDs, such as phenobarbital, show only limited effects in suppressing seizures in infants.199,200 Moreover, the use of commonly used ASDs is undesirable at a time when the brain is developing rapidly and particularly vulnerable to any toxic effects. Therefore, the targeting of P2X7, thought to be mainly activated during pathological release of ATP,201 may represent a promising novel treatment strategy for neonates.202 Other studies, however, have shown only a moderate or no anticonvulsant effect of P2X7 antagonism. Using the maximal electroshock seizure–threshold test and the Ptz (87 mg/kg, IP) seizure-threshold test in mice, Fischer et al showed no anticonvulsant effects provided by P2X7 antagonism using four antagonists (JNJ47965567, AFC5128, BBG, and tanshinone IIA sulfonate). However, further strengthening the potential of P2X7 antagonism as adjunctive treatment, P2X7 antagonists potentiated anticonvulsant effects of carbamazepine in the maximal electroshock-seizure test.203 In line with this, a subsequent study by Nieczym et al204 found only a weak anticonvulsant effect of the P2X7 antagonist BBG on Ptz (1% Ptz 2 mL/min, IV) seizure threshold, maximal electroshock-seizure threshold, and 6 Hz psychomotor seizure-threshold tests. Moreover, no effect of P2X7 antagonism on seizures was observed in WAG/Rij rats, a model of genetic absence epilepsy.10 Suggesting a proconvulsive function of the P2X7 receptor, Kim and Kang reported exacerbated seizure severity in P2X7-KO mice using a pilocarpine (150, 175, 200, 225, or 250 mg/kg, IP) mouse model.205 In addition, research by the same group using the pilocarpine (380 mg/kg, IP) rat model showed that P2X7 antagonism (oxidized ATP [OxATP] and BBG) protected against astroglial cell death,206 reduced the infiltration of neutrophils into the frontoparietal cortex,207 and increased status epilepticus–induced hippocampal neurodegeneration.208 Suggesting, however, a model-specific effect, research by the same group showed that P2X7 deficiency did not alter seizure severity in the systemic KA (25 mg/kg, IP) mouse model or the picrotoxin (5 mg/kg, IP) mouse model.205 As such, while P2X7 antagonism impacts on seizure severity in models of status epilepticus, these effects seem to be minor or absent during acute nondamaging seizures.
Suggesting antiepileptogenic potential of P2X7 antagonism, Soni et al showed that the P2X7 antagonist BBG decreased mean kindling score and restored cognitive deficits and motor coordination using the Ptz (30 mg/kg, IP) kindling model in rats.209 Similar results were obtained in a more recent study by Fischer et al using the P2X7 antagonists JNJ47965567, AFC5128, and BBG.203 In the same vein, Amorim et al showed that injection of a P2X7-targeting siRNA post–status epilepticus delayed the emergence of the first epileptic seizure and reduced the frequency and severity of seizures in the pilocarpine (370 mg/kg, IP) rat model.210 Interestingly, a recent study by Jamali-Raeufy et al showed that the P2X7 antagonist BBG and linagliptin, which blocks the enzyme dipeptidyl peptidase4 and is used for the treatment of diabetes, reduced seizure severity and neuronal cell death more efficiently when given in combination in a KA (4 μg, intrahippocampal) rat model. Epileptic rats treated with both inhibitors also showed improved spatial learning at a later stage, probably caused by fewer seizures during the initial insult.211 In agreement with P2X7 antagonism impacting on epilepsy-related comorbidities, treatment of pilocarpine-induced epileptic rats with the P2X7 antagonist BBG promoted antidepressive and anti-anxiety effects. BBG did not, however, affect the development of spontaneous recurrent seizures, but inhibited microglia activation following pilocarpine (single dose of 30 mg/kg and repeated doses of 10 mg/kg, IP)-induced status epilepticus in rats.212 However, similarly to status epilepticus, treatment with the P2X7 antagonists AZ10606120 and BBG following pilocarpine (300 mg/kg, IP)-induced status epilepticus in mice resulted in the development of a more severe epileptic phenotype.213
Suggesting P2X7 antagonism as treatment for established epilepsy, Amhaoul et al showed that treatment with the P2X7 antagonist JNJ47965567 during epilepsy reduced seizure severity, though without affecting the total number of seizures. In this study, epileptic rats were treated for 1 week 3 months after KA (22.2±2.02 mg/kg, IP)-induced status epilepticus.214 The antiepileptic potential of P2X7 antagonism was further demonstrated in a study carried out by Jimenez-Pacheco et al. In this study, mice subjected to IA KA (0.3 µg) were treated with the P2X7 antagonist JNJ47965567 for 5 days, starting at 10 days post–status epilepticus. In contrast to the previous study, P2X7 antagonism reduced the total number of seizures during treatment, and remarkably, during an additional recording period following treatment withdrawal, suggesting disease-modifying potential.192 Therefore, in contrast to acute seizures, P2X7 seems to play a more prominent role during epilepsy once pathological processes have been initiated.
While compelling evidence has now demonstrated a role for P2X7 mediating hyperexcitability in the brain, the mechanisms by which it modulates seizure severity are still to be determined. While affecting numerous pathways in the CNS, the most likely explanation is that P2X7 effects are mediated, at least in part, via their prominent role in driving inflammatory processes. P2X7 is an important driver of microglial activation and proliferation,125 and has been shown to be highly expressed under physiological conditions and during epilepsy.189,190 Suggesting a functional role of P2X7 regulating inflammatory processes during seizures and epilepsy, blocking of P2X7 during status epilepticus leads to decreased hippocampal IL1β levels.119 Moreover, targeting of P2X7 receptors during epilepsy reduces both microgliosis and astrogliosis.192 Although not shown to be increased on astrocytes following seizures or during epilepsy, astrocytes can also be activated via P2X7.215 As outlined before, astrocytes can reduce seizure thresholds via various mechanisms (eg, dysregulation of extracellular ionic balance, impaired neurotransmitter reuptake, release of proinflammatory cytokines and purines, eg, ATP, and removal of extracellular adenosine via ADK).27,28,216,217 Interestingly, P2X7 antagonism has been shown to protect against astrocyte death following pilocarpine (380 mg/kg, IP)-induced status epilepticus, possibly contributing to its proconvulsive function in this model.206 Further in line with P2X7 impacting on astrocytes, P2X7-deficient mice subjected to KA (25 mg/kg, IP) show decreased astroglial autophagy through the regulation of FAK- and PHLPP1/2-mediated Akt-S473 phosphorylation.218 P2X7 has also been shown to promote microglial activation and astroglial apoptosis via PDI-mediated redox/S-nitrosylation following pilocarpine (380 mg/kg, IP)-induced status epilepticus in rats.219 It is, however, important to keep in mind that while suppressing inflammatory pathways is the most likely explanation, P2X7 is involved in a myriad of different effects in pathological conditions, including BBB disruption, changes in neurotransmitter levels, synaptic reorganization, and neurogenesis, to name just a few.220 Of note, suggesting P2X7 activation modulating extracellular levels of the neurotransmitters GABA and glutamate, Barros-Barbosa et al demonstrated that by using isolated nerve terminals of the neocortex from patients with epilepsy, P2X7 antagonism prevented the uptake of GABA more efficiently in tissue from epileptic patients, thereby potentially increasing GABAergic rundown.95 Further supporting a role of P2X7 receptors in the control of neurotransmitter levels in epilepsy, a more recent study observed that P2X7 modulated both GABA and glutamate release depending on the availability of extracellular Ca2+.96
The only other P2X receptors for which a functional role has been investigated in the setting of seizures and epilepsy are P2X3 and P2X4. In the case of P2X4, Ulmann et al showed that despite experiencing no changes in seizure severity during status epilepticus, P2X4-deficient mice were partially protected from seizure-induced neurodegeneration following KA (8–22 mg/kg, IP)-induced status epilepticus, possibly via regulating the activation of microglia.142 As for P2X3, Xia et al showed that treatment with the P2X3 NF110 reduced mean kindling scores and improved other pathological parameters, such as memory deficits, motor activity, neuronal damage, and hippocampal inflammation, in the Ptz (30 mg/kg, IP)-induced kindling rat model.221
Targeting of P2Y Receptors During Seizures and Epilepsy
While P2X receptors have attracted most attention as possible drug targets for epilepsy, increasing evidence also suggests a role of the metabotropic P2Y receptors,11,222 with P2Y1 and P2Y12 being the most studied.222 Evidence suggesting a role for P2Y12 during status epilepticus stems from a study using P2Y12-knock-out mice. Using the KA (18–22 mg/kg, IP) mouse model of status epilepticus, Eyo et al found that P2Y12-deficient mice showed a more severe seizure phenotype and reduced microglial process extension in the hippocampus.160 They suggested that neuronal NMDA-receptor activation contributes to an influx of Ca2+, which increased ATP release from neurons to activate microglial P2Y12, potentiating the extension of microglia processes.160 In line with P2Y12 regulating microglial processes, Avignone et al also showed an increase in microglia motility following treatment with the P2Y12 agonist 2-Me-ADP in a mouse model of KA (15 and 5 mg/kg, IP)-induced status epilepticus.223 In a more recent study, using resected hippocampal tissue from TLE patients, Milior et al showed that while low doses of ADP induced microglial process extension via blocking of P2Y12, high doses of ADP caused microglial process retraction and membrane ruffling, which was blocked by the coapplication of antagonists against P2Y1 and P2Y13.171 Moreover, microglial P2Y12 has been suggested to contribute to status epilepticus–induced aberrant neurogenesis and increased immature neuronal projections.224
Initial evidence suggesting a role for P2Y1 during seizures showed that P2Y1 antagonism (MRS2179) reduced neuronal cell death following KA (10 mg/kg, IP)-induced status epilepticus in rats, though without affecting seizure severity.225 In contrast, a more recent study published by our group showed that P2Y1-deficient mice subjected to IA KA (3 µg) presented a lower seizure threshold, experiencing more severe seizures during status epilepticus and increased seizure-induced neurodegeneration in the hippocampus.196 In line with genetic deletion of P2Y1, treatment with the P2Y1 antagonist MRS2500 prior to IA KA exacerbated seizure severity during status epilepticus and resulting neurodegeneration. Moreover, mice pretreated with the P2Y1 agonist MRS2365 had less severe seizures during status epilepticus and less seizure-induced neurodegeneration.196 However, suggesting a context-specific role for P2Y1 during status epilepticus, treatment with the P2Y1 agonist MRS2365 or P2Y1 antagonist MRS2500 10 minutes following IA KA injection when mice had presented their first seizure burst showed the opposite results when compared to the pretreatment regime. Here, the P2Y1 antagonist reduced seizure severity and neurodegeneration, while treatment with the P2Y1 receptor agonist increased seizure severity. These results suggest that P2Y1-based treatment is highly dependent on the timing of intervention. Why P2Y1 behaves differently according to the timing of treatment initiation remains elusive. However, immunohistochemical analysis shows that while P2Y1 receptors are mainly expressed in neurons during physiological conditions, they become rapidly upregulated in microglia following IA KA. Therefore, a likely explanation may be a cell type–specific contribution of P2Y1, depending on where it is increased, with predominant neuronal P2Y1 expression reducing hyperexcitability and predominant P2Y1 microglial expression contributing to increased hyperexcitability via driving inflammatory processes.196 To fully address a possible cell type–specific contribution of P2Y1 to seizures requires, however, the use of a cell type–specific deletion of P2Y1 (ie, in neurons and microglia). The role of P2Y1 is, however, not restricted to microglia, and P2Y1 has also been shown to be functional on astrocytes.112 In a rapid amygdala kindling model, P2Y1 has been proposed to contribute to seizure generation and epilepsy via the mediation of astrocytic Ca2+ oscillations226 and to restore normal excitatory synaptic activity in the hippocampus by blocking TNFα-induced Ca2+ release from astrocytes.227 Recently and in line with these results, Martorell et al, using a rapid kindling model in rats consisting of ten daily trains of biphasic rectangular current pulses at subthreshold postdischarge intensity applied for 3 consecutive days, showed that P2Y1 inhibition rescued both the abnormal pattern of astroglial Ca2+ activity and plastic properties of CA3–CA1 synapses in the epileptic hippocampus.228
In contrast to studies analyzing the impact of targeting P2Y receptors during status epilepticus, only one study has investigated a possible contribution of P2Y receptors to epileptogenesis and epilepsy.196 Here, the authors showed that treatment with the P2Y1 antagonist MRS2500 post-IA KA (3 µg)-induced status epilepticus, delayed the onset of epilepsy, and when applied during epilepsy, suppressed epileptic seizures, though this effect did not persist during treatment withdrawal. Changes in inflammation during treatment were not analyzed.196
Targeting of P2Y receptors has shown promising results during acute seizures and also during epilepsy (Table 1). Again, as with P2X receptors, the exact mechanisms of how these receptors impact on seizures or epilepsy remains to be established; however, results so far suggest a prominent role of P2Y receptor–driven inflammation. While P2Y12 seems to be more important for the activation of microglia,160 effects mediated via P2Y1 have been suggested for both microglia and astrocytes.196 Of note, in the case of P2Y1, neuronal expression seems to be associated with protective mechanisms,226 and thus targeting of P2Y1 in specific cell types may represent a better and possibly more efficient pharmacological strategy to treat seizures and epilepsy. Whether a cell type–specific function is also the case for other P2Y receptors remains to be established.
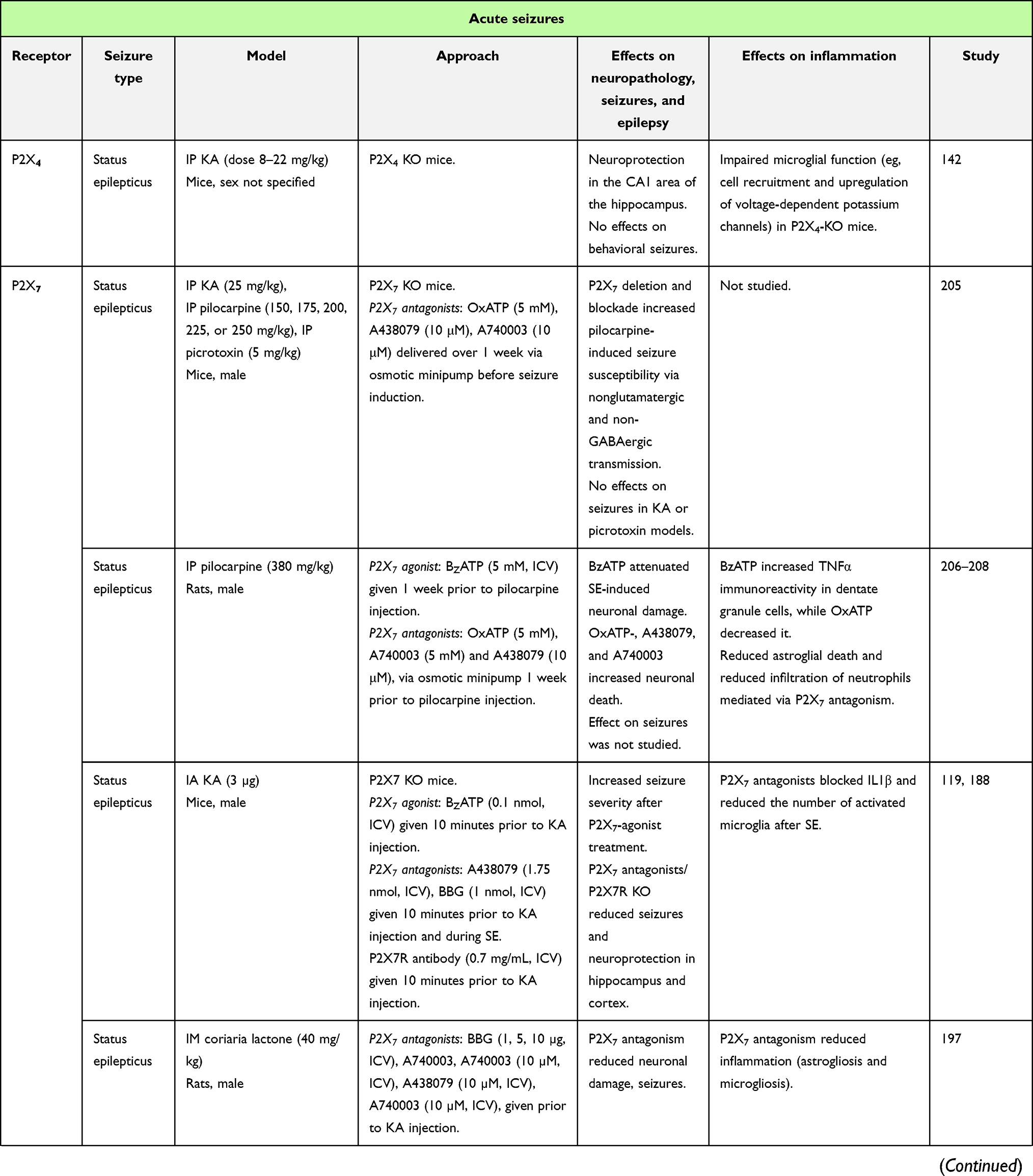 | 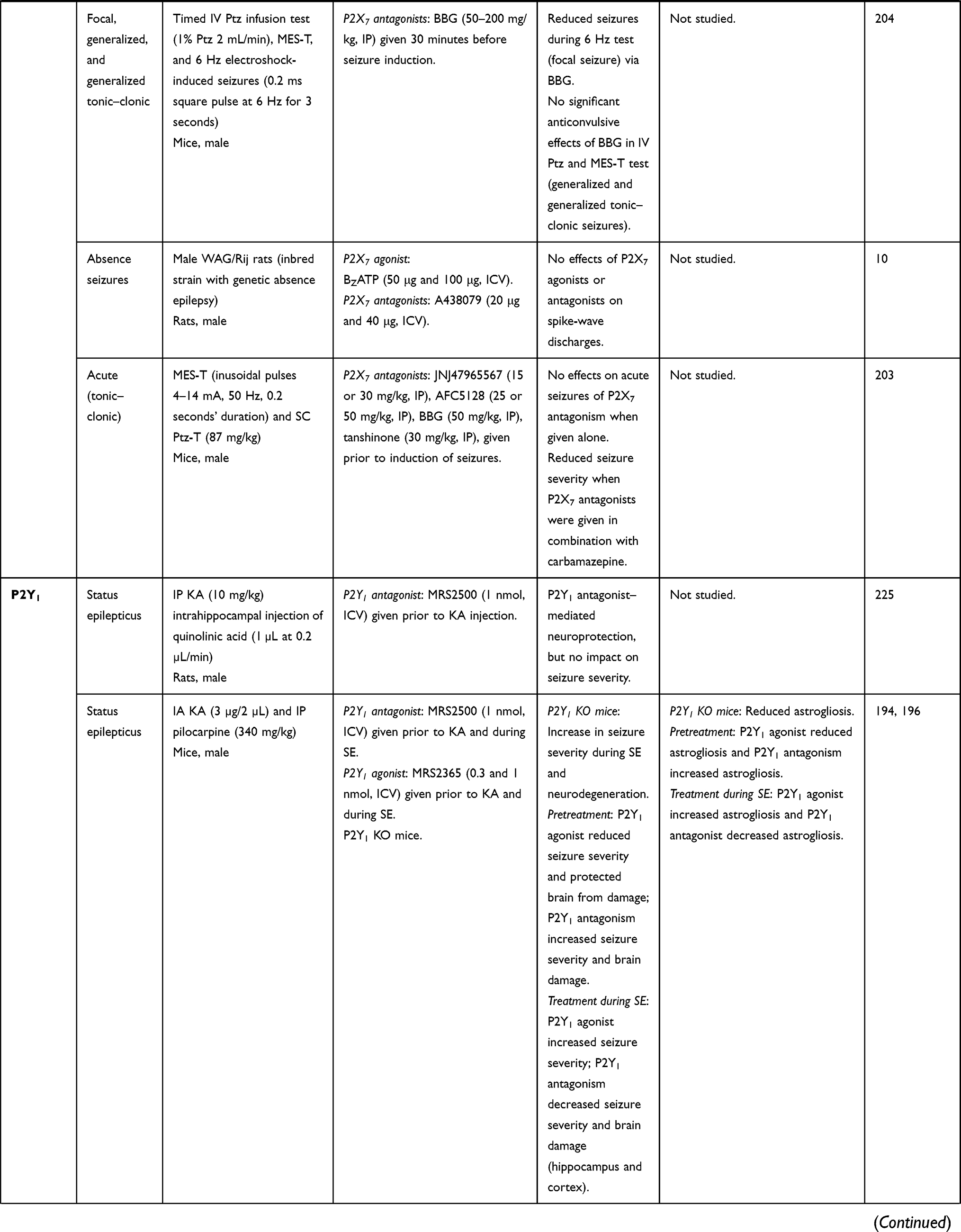 | 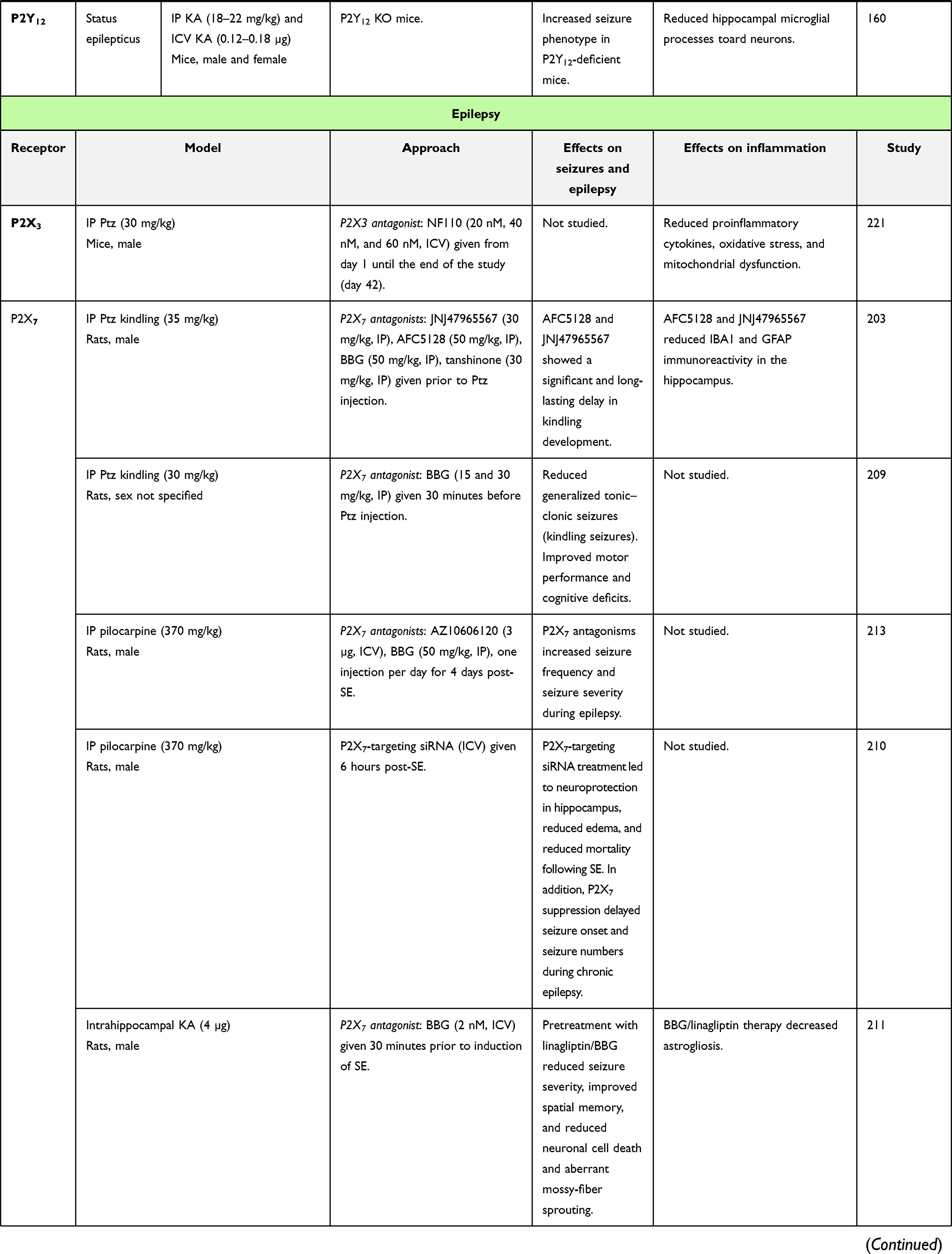 | 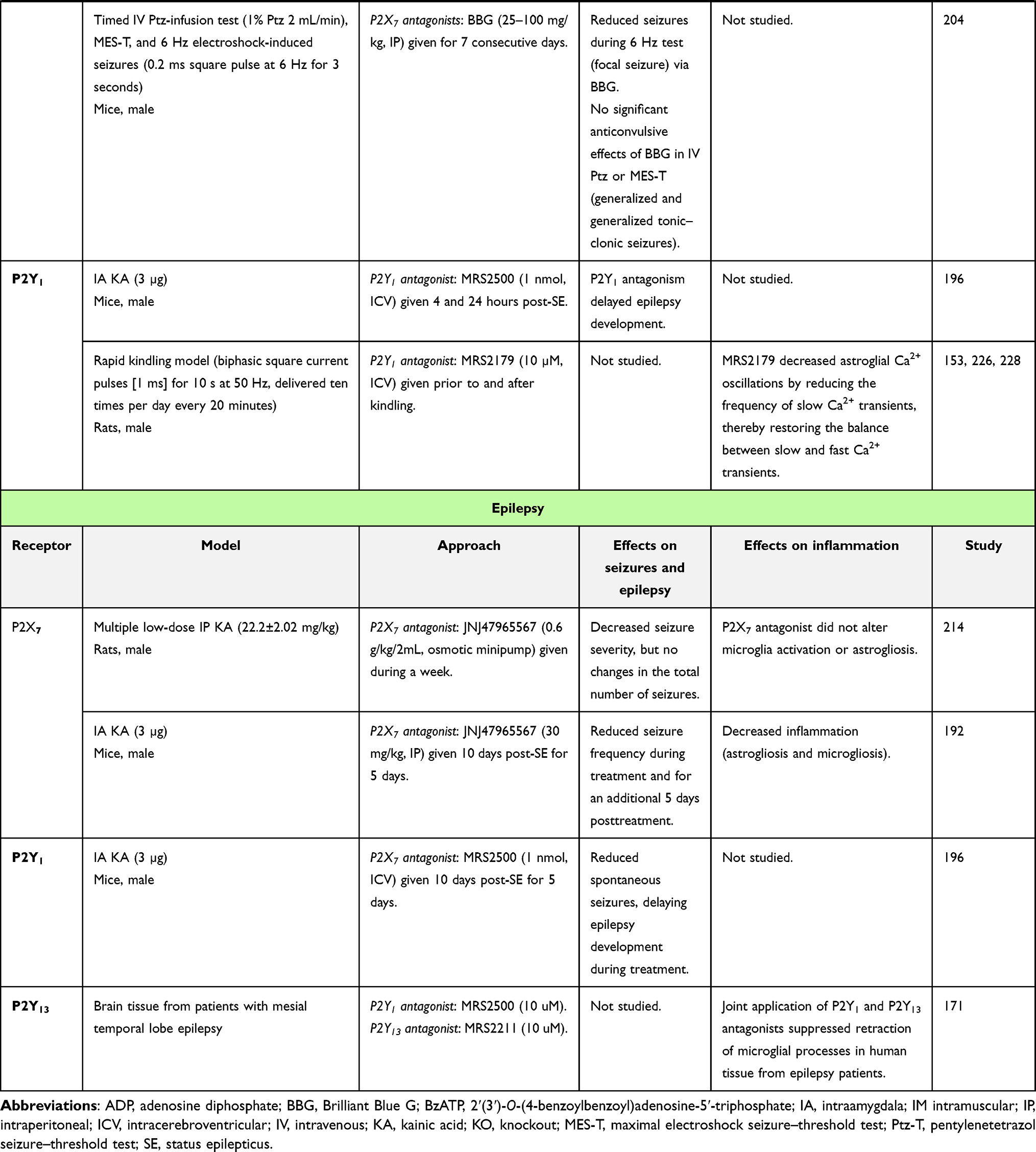 |
Table 1 In vivo studies demonstrating the impact of P2-receptor targeting in seizures and epilepsy (selected examples) |
Conclusion
Over the last decade, compelling evidence suggesting an important role of P2 receptors during pathological brain hyperexcitability has been steadily increasing, with encouraging data showing that targeting of these receptors not only suppresses neuroinflammatory processes but also provides seizure-suppressive and disease-modifying effects (Figure 1). However, to advance therapeutic strategies based on P2-receptor targeting further toward clinical application, several issues should be addressed.
- While mounting data suggest anticonvulsive and antiepileptogenic effects via targeting of P2 receptors, the mechanisms of action of these receptors during seizures and epilepsy are still poorly understood. P2 receptors are described as one of the gatekeepers of inflammation, with continuous P2-receptor activation believed to contribute to sustained pathological neuroinflammation in the brain, possibly contributing to seizures and the development of epilepsy. However, how exactly P2 receptors contribute to inflammation during seizures and epilepsy is yet not fully understood.
- P2 receptors have been shown to change their expression in different brain regions following seizures and during epilepsy; however, we still lack reliable data regarding their cell type–specific expression or their subcellular localization, which is probably due to a lack of reliable detection tools for most of the purinergic receptors. For example, while it is well accepted that P2X7 is expressed in microglia during epilepsy, its expression on neurons is still a matter of debate.99 As such, better detection tools (eg, nanobodies, P2-receptor reporter mice190) are needed to establish the exact expression profile of these receptors during epilepsy.
- New data suggest a cell type–specific function of P2 receptors during seizures. For example, neuronal P2Y1 expression seems to be anticonvulsive, whereas P2Y1 expressed on microglia may be proconvulsive.196 Therefore, further investigation is needed to clarify the exact cell-specific contribution of these receptors to disease progression, requiring the use of cell type–specific knockout mice (eg, Cre-LoxP).
- To date, the majority of studies investigating a role for P2 receptors in epilepsy have been performed in rodent models of seizures, including acute seizures, such as Ptz or maximal electroshock, and models mimicking TLE, such as KA- or pilocarpine-induced status epilepticus. Taking in consideration that epilepsy is a heterogeneous group of epilepsy syndromes with many etiologies, the analysis of a possible role of P2 receptors should be extended to other disease models such as models of acquired epilepsy (eg, TBI) or genetic models.
- None of the P2 receptor–based treatments have resulted in complete cessation of seizures; however, promising data have suggested P2 receptor–based treatments as adjunctive treatment for drug-refractory epilepsy potentiating effects of anticonvulsants (eg, P2X7 antagonism boosting effects of lorazepam in the IA KA mouse model119). Importantly, new treatments are unlikely to be applied as monotherapies, and are most likely used as adjunctive therapy in combination with ASDs already in clinical use. Therefore, future studies should target P2 receptors in combination with ASDs to identify better options for seizure control during pharmacoresistant epilepsy.
- Studies to date have analyzed P2 receptors separately; however, this receptor family is constituted of P2X and P2Y receptors, which can have synergistic effects on each other. Multitargeting approaches should be carried out in future studies. For example, inhibition of both P2Y1 and P2X7 at the same time may improve their effectiveness against seizures and delay or stop the progression of epilepsy, possibly improving previously reported disease-modifying effects.192
- The lack of reliable biomarkers to predict seizures and/or the development of epilepsy remains an important gap in the field. Blood-based inflammation markers have been suggested as strong candidates for the diagnosis of epilepsy.22 Of note, a recent study showed that blood-purine levels correlated with seizure severity in mice and were elevated in humans with epilepsy, demonstrating their diagnostic potential.229 Future studies should be designed to establish the diagnostic and prognostic potential of purinergic signaling for epilepsy and whether we can stratify patients according to the need for P2 receptor–based treatment.
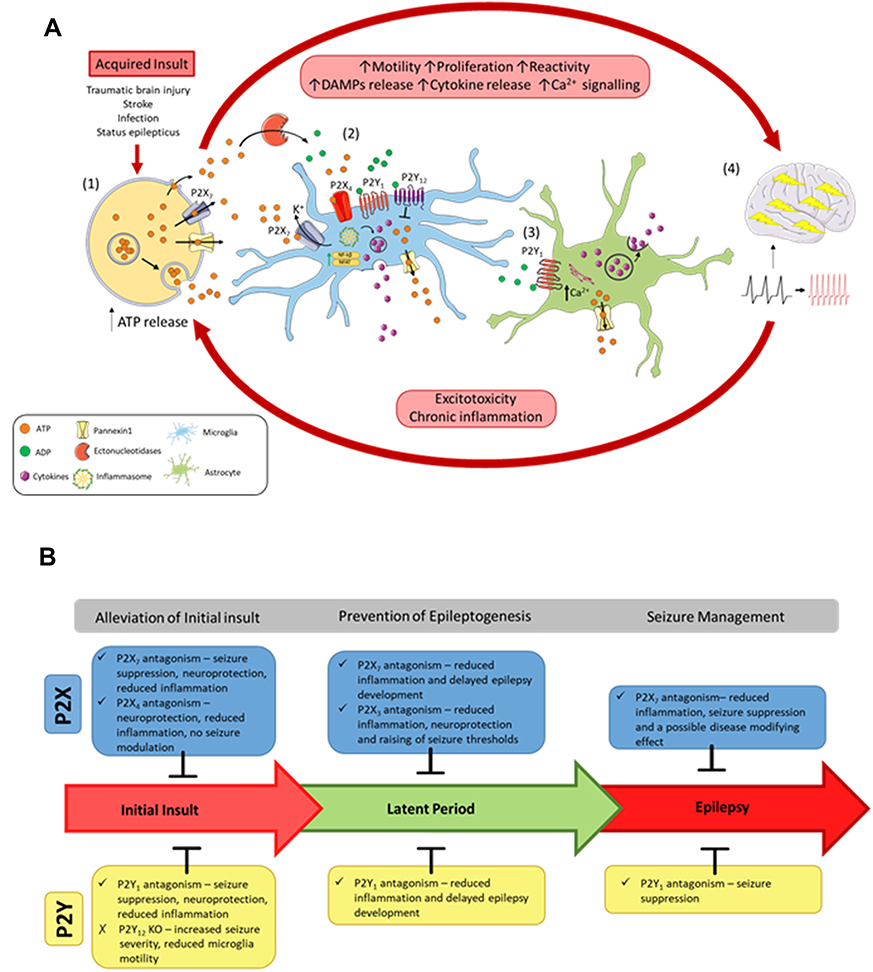 |
Figure 1 The vicious cycle of inflammation and purinergic signaling underlying epilepsy progression. (A) Following an acquired initial insult to the brain, (1) ATP is released from cells via exocytotic mechanisms, through leakage across damaged membranes, and through purinergic channels, such as P2X7 receptors and pannexin 1. Once released, ATP activates P2 receptors or is metabolized by ectonucleotidases into different breakdown products, such as ADP. ATP and ADP act on the P2 receptors to induce motility, proliferation, and reactivity of microglia and astrocytes. (2) Activation of P2X receptors on microglia via ATP induces inflammasome activation and upregulation of inflammatory transcription factors (NFκB and NFAT), leading to cytokine release. Activation of P2Y1 also induces cytokine release. However, the P2Y12 receptor acts to oppose these proinflammatory cascades. P2 receptor activation also stimulates release of ATP from microglia. (3) Activation of P2Y1 on astrocytes releases Ca2+ from the endoplasmic reticulum, leading to further cytokine release and ATP release. Acting as a paracrine-signaling molecule, ATP can potentiate these inflammatory cascades. (4) An increase in inflammation of the brain and large amounts of ATP release increases the excitability of the brain, resulting in seizures and epilepsy progression. Chronic inflammation and excitotoxicity induced by seizures leads to further ATP release and epilepsy progression. (B) Effects of blocking P2 receptors at the different stages of acquired epilepsy progression following an initial insult to the brain (eg, status epilepticus). |
Abbreviations
ADK, adenosine kinase; ADP, adenosine-5ʹ-diphosphate; AMP, adenosine monophosphate; ASDs, antiseizure drugs; ATP, adenosine 5ʹ-triphosphate; BBB, blood–brain barrier; BBG, Brilliant Blue G; BzATP, 2,3-O-(4-benzoylbenzoyl) ATP; cAMP, cyclic AMP; CNS, central nervous system; DAMPs, damage-associated molecular patterns; GABA, γ-aminobutyric acid; GFAP, glial fibrillary acidic protein; HMGB1, high-mobility group box 1; ILAE, International League Against Epilepsy; IL, interleukinIRF5, interferon regulatory factor; IA, intraamygdala; IP, intraperitoneal; IV, intravenous; NFAT, nuclear factor of activated T cells; NFκB, Nuclear factor κ light–chain enhancer of activated B cells; NMDA, N-methyl-D-aspartate; NR2B, N-methyl-D-aspartate receptor subtype 2B; NLRP3, NLR family, pyrin domain containing 3; OxATP, oxidized ATP; P2XRs, P2X receptors; P2YRs, P2Y receptors; PDI, protein disulfide isomerase; PI3K, phosphatidylinositol-3-kinase; Ptz, pentylenetetrazol; STAT3, signal transducer and activator of transcription 3; TLE, temporal lobe epilepsy; TLR4, Toll-like receptor 4; TNF, tumor necrosis factor; TBI, traumatic brain injury; VNUT, vesicular nucleotide transporter.
Funding
This work was supported by funding from Science Foundation Ireland (17/CDA/4708 and 16/RC/3948, cofunded under the European Regional Development Fund and by FutureNeuro industry partners) and the Irish Research Council (Government of Ireland Postdoctoral Fellowship Programme, GOIPD/2020/865).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Thijs RD, Surges R, O’Brien TJ, Sander JW. Epilepsy in adults. Lancet. 2019;393(10172):689–701. doi:10.1016/S0140-6736(18)32596-0
2. Allers K, Essue BM, Hackett ML, et al. The economic impact of epilepsy: a systematic review. BMC Neurol. 2015;15(1):245. doi:10.1186/s12883-015-0494-y
3. Lin JJ, Mula M, Hermann BP. Uncovering the neurobehavioural comorbidities of epilepsy over the lifespan. Lancet. 2012;380(9848):1180–1192.
4. Keezer MR, Sisodiya SM, Sander JW. Comorbidities of epilepsy: current concepts and future perspectives. Lancet Neurol. 2016;15(1):106–115. doi:10.1016/S1474-4422(15)00225-2
5. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58(4):512–521. doi:10.1111/epi.13709
6. Klein P, Dingledine R, Aronica E, et al. Commonalities in epileptogenic processes from different acute brain insults: do they translate? Epilepsia. 2018;59(1):37–66.
7. Pitkanen A, Lukasiuk K, Dudek FE, Staley KJ. Epileptogenesis. Cold Spring Harb Perspect Med. 2015;5(10):a022822. doi:10.1101/cshperspect.a022822
8. Henshall DC, Kobow K. Epigenetics and epilepsy. Cold Spring Harb Perspect Med. 2015;5(12). doi:10.1101/cshperspect.a022731
9. Thom M. Review: hippocampal sclerosis in epilepsy: a neuropathology review. Neuropathol Appl Neurobiol. 2014;40(5):520–543. doi:10.1111/nan.12150
10. Dogan E, Aygun H, Arslan G, et al. The role of NMDA receptors in the effect of purinergic P2X7 receptor on spontaneous seizure activity in WAG/Rij rats with genetic absence epilepsy. Front Neurosci. 2020;14:414. doi:10.3389/fnins.2020.00414
11. Engel T, Alves M, Sheedy C, Henshall DC. ATPergic signalling during seizures and epilepsy. Neuropharmacology. 2016;104:140–153. doi:10.1016/j.neuropharm.2015.11.001
12. Moshe SL, Perucca E, Ryvlin P, Tomson T. Epilepsy: new advances. Lancet. 2015;385(9971):884–898. doi:10.1016/S0140-6736(14)60456-6
13. Bialer M, White HS. Key factors in the discovery and development of new antiepileptic drugs. Nat Rev Drug Discov. 2010;9(1):68–82. doi:10.1038/nrd2997
14. Cross JH, Lagae L. The concept of disease modification. Eur J Paediatr Neurol. 2020;24:43–46. doi:10.1016/j.ejpn.2019.12.005
15. Loscher W. The holy grail of epilepsy prevention: preclinical approaches to antiepileptogenic treatments. Neuropharmacology. 2020;167:107605. doi:10.1016/j.neuropharm.2019.04.011
16. Burnstock G. Purinergic signalling and neurological diseases: an update. CNS Neurol Disord Drug Targets. 2017;16(3):257–265. doi:10.2174/1871527315666160922104848
17. Burnstock G. Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev. 2007;87(2):659–797.
18. Boison D. Adenosinergic signaling in epilepsy. Neuropharmacology. 2016;104:131–139. doi:10.1016/j.neuropharm.2015.08.046
19. Rho JM, White HS. Brief history of anti-seizure drug development. Epilepsia Open. 2018;3(Suppl 2):114–119. doi:10.1002/epi4.12268
20. Coulter DA, Steinhauser C. Role of astrocytes in epilepsy. Cold Spring Harb Perspect Med. 2015;5(3):a022434. doi:10.1101/cshperspect.a022434
21. Hiragi T, Ikegaya Y, Koyama R. Microglia after seizures and in epilepsy. Cells. 2018;7(4):26. doi:10.3390/cells7040026
22. Vezzani A, Balosso S, Ravizza T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat Rev Neurol. 2019;15(8):459–472. doi:10.1038/s41582-019-0217-x
23. Verhoog QP, Holtman L, Aronica E, van Vliet EA. Astrocytes as guardians of neuronal excitability: mechanisms underlying epileptogenesis. Front Neurol. 2020;11:591690.
24. Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci. 2013;36(3):174–184. doi:10.1016/j.tins.2012.11.008
25. Xu S, Sun Q, Fan J, et al. Role of astrocytes in post-traumatic epilepsy. Front Neurol. 2019;10:1149. doi:10.3389/fneur.2019.01149
26. Walrave L, Vinken M, Leybaert L, Smolders I. Astrocytic connexin43 channels as candidate targets in epilepsy treatment. Biomolecules. 2020;10(11):1578. doi:10.3390/biom10111578
27. Boison D. The adenosine kinase hypothesis of epileptogenesis. Prog Neurobiol. 2008;84(3):249–262. doi:10.1016/j.pneurobio.2007.12.002
28. Robel S, Buckingham SC, Boni JL, et al. Reactive astrogliosis causes the development of spontaneous seizures. J Neurosci. 2015;35(8):3330–3345. doi:10.1523/JNEUROSCI.1574-14.2015
29. Bilbo S, Stevens B. Microglia: the brain’s first responders. Cerebrum. 2017;2017.
30. Kinoshita S, Koyama R. Pro- and anti-epileptic roles of microglia. Neural Regen Res. 2021;16(7):1369–1371. doi:10.4103/1673-5374.300976
31. Vezzani A, Conti M, De Luigi A, et al. Interleukin-1beta immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. J Neurosci. 1999;19(12):5054–5065. doi:10.1523/JNEUROSCI.19-12-05054.1999
32. Badimon A, Strasburger HJ, Ayata P, et al. Negative feedback control of neuronal activity by microglia. Nature. 2020;586(7829):417–423. doi:10.1038/s41586-020-2777-8
33. Zhao XF, Liao Y, Alam MM, et al. Microglial mTOR is neuronal protective and antiepileptogenic in the pilocarpine model of temporal lobe epilepsy. J Neurosci. 2020;40(40):7593–7608. doi:10.1523/JNEUROSCI.2754-19.2020
34. Liu M, Jiang L, Wen M, et al. Microglia depletion exacerbates acute seizures and hippocampal neuronal degeneration in mouse models of epilepsy. Am J Physiol Cell Physiol. 2020;319(3):C605–C610. doi:10.1152/ajpcell.00205.2020
35. Fernandes A, Miller-Fleming L, Pais TF. Microglia and inflammation: conspiracy, controversy or control? Cell Mol Life Sci. 2014;71(20):3969–3985. doi:10.1007/s00018-014-1670-8
36. Therajaran P, Hamilton JA, O’Brien TJ, Jones NC, Ali I. Microglial polarization in posttraumatic epilepsy: potential mechanism and treatment opportunity. Epilepsia. 2020;61(2):203–215. doi:10.1111/epi.16424
37. Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. 2016;53(2):1181–1194. doi:10.1007/s12035-014-9070-5
38. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27(1):451–483. doi:10.1146/annurev.immunol.021908.132532
39. Huang M, Li Y, Wu K, et al. Paraquat modulates microglia M1/M2 polarization via activation of TLR4-mediated NF-kappaB signaling pathway. Chem Biol Interact. 2019;310:108743. doi:10.1016/j.cbi.2019.108743
40. Sano F, Shigetomi E, Shinozaki Y, et al. Reactive astrocyte-driven epileptogenesis is induced by microglia initially activated following status epilepticus. JCI Insight. 2021;6(9). doi:10.1172/jci.insight.135391
41. Shinozaki Y, Shibata K, Yoshida K, et al. Transformation of astrocytes to a neuroprotective phenotype by microglia via P2Y1 receptor downregulation. Cell Rep. 2017;19(6):1151–1164. doi:10.1016/j.celrep.2017.04.047
42. Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. doi:10.1038/nature21029
43. Quintas C, Fraga S, Goncalves J, Queiroz G. P2Y receptors on astrocytes and microglia mediate opposite effects in astroglial proliferation. Purinergic Signal. 2011;7(2):251–263. doi:10.1007/s11302-011-9235-x
44. Cho KO, Lybrand ZR, Ito N, et al. Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat Commun. 2015;6:6606. doi:10.1038/ncomms7606
45. Benson MJ, Manzanero S, Borges K. Complex alterations in microglial M1/M2 markers during the development of epilepsy in two mouse models. Epilepsia. 2015;56(6):895–905. doi:10.1111/epi.12960
46. Broekaart DWM, Anink JJ, Baayen JC, et al. Activation of the innate immune system is evident throughout epileptogenesis and is associated with blood-brain barrier dysfunction and seizure progression. Epilepsia. 2018;59(10):1931–1944. doi:10.1111/epi.14550
47. Tian DS, Peng J, Murugan M, et al. Chemokine CCL2-CCR2 signaling induces neuronal cell death via STAT3 activation and IL-1beta production after status epilepticus. J Neurosci. 2017;37(33):7878–7892. doi:10.1523/JNEUROSCI.0315-17.2017
48. Feng L, Murugan M, Bosco DB, et al. Microglial proliferation and monocyte infiltration contribute to microgliosis following status epilepticus. Glia. 2019;67(8):1434–1448. doi:10.1002/glia.23616
49. Bosco DB, Tian DS, Wu LJ. Neuroimmune interaction in seizures and epilepsy: focusing on monocyte infiltration. FEBS J. 2020;287(22):4822–4837. doi:10.1111/febs.15428
50. Varvel NH, Neher JJ, Bosch A, et al. Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc Natl Acad Sci U S A. 2016;113(38):E5665–5674. doi:10.1073/pnas.1604263113
51. Wang S, Cheng Q, Malik S, Yang J. Interleukin-1beta inhibits gamma-aminobutyric acid type A (GABA(A)) receptor current in cultured hippocampal neurons. J Pharmacol Exp Ther. 2000;292(2):497–504.
52. Yang S, Liu ZW, Wen L, Qiao HF, Zhou WX, Zhang YX. Interleukin-1beta enhances NMDA receptor-mediated current but inhibits excitatory synaptic transmission. Brain Res. 2005;1034(1–2):172–179. doi:10.1016/j.brainres.2004.11.018
53. Roseti C, van Vliet EA, Cifelli P, et al. GABAA currents are decreased by IL-1beta in epileptogenic tissue of patients with temporal lobe epilepsy: implications for ictogenesis. Neurobiol Dis. 2015;82:311–320. doi:10.1016/j.nbd.2015.07.003
54. Viviani B, Gardoni F, Marinovich M. Cytokines and neuronal ion channels in health and disease. Int Rev Neurobiol. 2007;82:247–263.
55. Yan J, Melemedjian OK, Price TJ, Dussor G. Sensitization of dural afferents underlies migraine-related behavior following meningeal application of interleukin-6 (IL-6). Mol Pain. 2012;8:6. doi:10.1186/1744-8069-8-6
56. Wu Z, Wang S, Gruber S, Mata M, Fink DJ. Full-length membrane-bound tumor necrosis factor-alpha acts through tumor necrosis factor receptor 2 to modify phenotype of sensory neurons. Pain. 2013;154(9):1778–1782. doi:10.1016/j.pain.2013.05.038
57. Vezzani A, Viviani B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology. 2015;96(Pt A):70–82. doi:10.1016/j.neuropharm.2014.10.027
58. Huang KF, Huang WT, Lin KC, Lin MT, Chang CP. Interleukin-1 receptor antagonist inhibits the release of glutamate, hydroxyl radicals, and prostaglandin E(2) in the hypothalamus during pyrogen-induced fever in rabbits. Eur J Pharmacol. 2010;629(1–3):125–131. doi:10.1016/j.ejphar.2009.11.060
59. Rossi S, Furlan R, De Chiara V, et al. Interleukin-1beta causes synaptic hyperexcitability in multiple sclerosis. Ann Neurol. 2012;71(1):76–83. doi:10.1002/ana.22512
60. Clarkson BDS, Kahoud RJ, McCarthy CB, Howe CL. Inflammatory cytokine-induced changes in neural network activity measured by waveform analysis of high-content calcium imaging in murine cortical neurons. Sci Rep. 2017;7(1):9037. doi:10.1038/s41598-017-09182-5
61. Zhu G, Okada M, Yoshida S, et al. Effects of interleukin-1beta on hippocampal glutamate and GABA releases associated with Ca2+-induced Ca2+ releasing systems. Epilepsy Res. 2006;71(2–3):107–116. doi:10.1016/j.eplepsyres.2006.05.017
62. Gardoni F, Boraso M, Zianni E, et al. Distribution of interleukin-1 receptor complex at the synaptic membrane driven by interleukin-1beta and NMDA stimulation. J Neuroinflammation. 2011;8(1):14. doi:10.1186/1742-2094-8-14
63. Viviani B, Bartesaghi S, Gardoni F, et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23(25):8692–8700. doi:10.1523/JNEUROSCI.23-25-08692.2003
64. Balosso S, Maroso M, Sanchez-Alavez M, et al. A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1beta. Brain. 2008;131(Pt 12):3256–3265. doi:10.1093/brain/awn271
65. Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. 2005;25(12):3219–3228. doi:10.1523/JNEUROSCI.4486-04.2005
66. Maroso M, Balosso S, Ravizza T, et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16(4):413–419. doi:10.1038/nm.2127
67. Iori V, Iyer AM, Ravizza T, et al. Blockade of the IL-1R1/TLR4 pathway mediates disease-modification therapeutic effects in a model of acquired epilepsy. Neurobiol Dis. 2017;99:12–23. doi:10.1016/j.nbd.2016.12.007
68. Noe FM, Polascheck N, Frigerio F, et al. Pharmacological blockade of IL-1beta/IL-1 receptor type 1 axis during epileptogenesis provides neuroprotection in two rat models of temporal lobe epilepsy. Neurobiol Dis. 2013;59:183–193. doi:10.1016/j.nbd.2013.07.015
69. French JA, Koepp M, Naegelin Y, et al. Clinical studies and anti-inflammatory mechanisms of treatments. Epilepsia. 2017;58(Suppl 3):69–82. doi:10.1111/epi.13779
70. Campos G, Fortuna A, Falcao A, Alves G. In vitro and in vivo experimental models employed in the discovery and development of antiepileptic drugs for pharmacoresistant epilepsy. Epilepsy Res. 2018;146:63–86.
71. Loscher W. Animal models of seizures and epilepsy: past, present, and future role for the discovery of antiseizure drugs. Neurochem Res. 2017;42(7):1873–1888. doi:10.1007/s11064-017-2222-z
72. Loscher W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure. 2011;20(5):359–368. doi:10.1016/j.seizure.2011.01.003
73. Curia G, Longo D, Biagini G, Jones RS, Avoli M. The pilocarpine model of temporal lobe epilepsy. J Neurosci Methods. 2008;172(2):143–157. doi:10.1016/j.jneumeth.2008.04.019
74. Marchi N, Oby E, Batra A, et al. In vivo and in vitro effects of pilocarpine: relevance to ictogenesis. Epilepsia. 2007;48(10):1934–1946. doi:10.1111/j.1528-1167.2007.01185.x
75. Levesque M, Avoli M. The kainic acid model of temporal lobe epilepsy. Neurosci Biobehav Rev. 2013;37(10 Pt 2):2887–2899.
76. Welzel L, Schidlitzki A, Twele F, Anjum M, Loscher W. A face-to-face comparison of the intra-amygdala and intrahippocampal kainate mouse models of mesial temporal lobe epilepsy and their utility for testing novel therapies. Epilepsia. 2020;61(1):157–170. doi:10.1111/epi.16406
77. Conte G, Parras A, Alves M, et al. High concordance between hippocampal transcriptome of the mouse intra-amygdala kainic acid model and human temporal lobe epilepsy. Epilepsia. 2020;61(12):2795–2810. doi:10.1111/epi.16714
78. Burnstock G. Purinergic nerves. Pharmacol Rev. 1972;24(3):509–581.
79. Rodrigues RJ, Tome AR, Cunha RA. ATP as a multi-target danger signal in the brain. Front Neurosci. 2015;9:148. doi:10.3389/fnins.2015.00148
80. Khakh BS, North RA. Neuromodulation by extracellular ATP and P2X receptors in the CNS. Neuron. 2012;76(1):51–69. doi:10.1016/j.neuron.2012.09.024
81. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. 2014;509(7500):310–317. doi:10.1038/nature13085
82. Dale N, Frenguelli BG. Release of adenosine and ATP during ischemia and epilepsy. Curr Neuropharmacol. 2009;7(3):160–179. doi:10.2174/157015909789152146
83. Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10(12):826–837. doi:10.1038/nri2873
84. Lecca D, Ceruti S. Uracil nucleotides: from metabolic intermediates to neuroprotection and neuroinflammation. Biochem Pharmacol. 2008;75(10):1869–1881. doi:10.1016/j.bcp.2007.12.009
85. Zimmermann H. Ectonucleotidases in the nervous system. Novartis Found Symp. 2006;276:113–128;discussion 128–130, 233-117, 275-181.
86. Burnstock G. An introduction to the roles of purinergic signalling in neurodegeneration, neuroprotection and neuroregeneration. Neuropharmacology. 2016;104:4–17.
87. Khakh BS, North RA. P2X receptors as cell-surface ATP sensors in health and disease. Nature. 2006;442(7102):527–532. doi:10.1038/nature04886
88. North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82(4):1013–1067. doi:10.1152/physrev.00015.2002
89. Coddou C, Stojilkovic SS, Huidobro-Toro JP. Allosteric modulation of ATP-gated P2X receptor channels. Rev Neurosci. 2011;22(3):335–354. doi:10.1515/rns.2011.014
90. North RA, Jarvis MF. P2X receptors as drug targets. Mol Pharmacol. 2013;83(4):759–769. doi:10.1124/mol.112.083758
91. Pankratov Y, Lalo U, Krishtal OA, Verkhratsky A. P2X receptors and synaptic plasticity. Neuroscience. 2009;158(1):137–148. doi:10.1016/j.neuroscience.2008.03.076
92. Burnstock G. P2X ion channel receptors and inflammation. Purinergic Signal. 2016;12(1):59–67. doi:10.1007/s11302-015-9493-0
93. Beamer E, Fischer W, Engel T. The ATP-gated P2X7 receptor as a target for the treatment of drug-resistant epilepsy. Front Neurosci. 2017;11:21. doi:10.3389/fnins.2017.00021
94. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 receptor in infection and inflammation. Immunity. 2017;47(1):15–31. doi:10.1016/j.immuni.2017.06.020
95. Barros-Barbosa AR, Fonseca AL, Guerra-Gomes S, et al. Up-regulation of P2X7 receptor-mediated inhibition of GABA uptake by nerve terminals of the human epileptic neocortex. Epilepsia. 2016;57(1):99–110. doi:10.1111/epi.13263
96. Barros-Barbosa AR, Oliveira A, Lobo MG, Cordeiro JM, Correia-de-sa P. Under stressful conditions activation of the ionotropic P2X7 receptor differentially regulates GABA and glutamate release from nerve terminals of the rat cerebral cortex. Neurochem Int. 2018;112:81–95. doi:10.1016/j.neuint.2017.11.005
97. Oliveira SD, Coutinho-Silva R, Silva CL. Endothelial P2X7 receptors’ expression is reduced by schistosomiasis. Purinergic Signal. 2013;9(1):81–89. doi:10.1007/s11302-012-9332-5
98. Zhao YF, Tang Y, Illes P. Astrocytic and oligodendrocytic P2X7 receptors determine neuronal functions in the CNS. Front Mol Neurosci. 2021;14:641570. doi:10.3389/fnmol.2021.641570
99. Illes P, Khan TM, Rubini P. Neuronal P2X7 receptors revisited: do they really exist? J Neurosci. 2017;37(30):7049–7062. doi:10.1523/JNEUROSCI.3103-16.2017
100. Miras-Portugal MT, Sebastian-Serrano A, de Diego Garcia L, Diaz-Hernandez M. Neuronal P2X7 receptor: involvement in neuronal physiology and pathology. J Neurosci. 2017;37(30):7063–7072. doi:10.1523/JNEUROSCI.3104-16.2017
101. von Kugelgen I. Pharmacological profiles of cloned mammalian P2Y-receptor subtypes. Pharmacol Ther. 2006;110(3):415–432. doi:10.1016/j.pharmthera.2005.08.014
102. Jacobson KA, Paoletta S, Katritch V, et al. Nucleotides acting at P2Y receptors: connecting structure and function. Mol Pharmacol. 2015;88(2):220–230. doi:10.1124/mol.114.095711
103. Jacobson KA, Boeynaems JM. P2Y nucleotide receptors: promise of therapeutic applications. Drug Discov Today. 2010;15(13–14):570–578. doi:10.1016/j.drudis.2010.05.011
104. Guzman SJ, Gerevich Z. P2Y receptors in synaptic transmission and plasticity: therapeutic potential in cognitive dysfunction. Neural Plast. 2016;2016:1207393. doi:10.1155/2016/1207393
105. Inoue K. Purinergic systems in microglia. Cell Mol Life Sci. 2008;65(19):3074–3080. doi:10.1007/s00018-008-8210-3
106. Crain JM, Nikodemova M, Watters JJ. Expression of P2 nucleotide receptors varies with age and sex in murine brain microglia. J Neuroinflammation. 2009;6:24. doi:10.1186/1742-2094-6-24
107. Crain JM, Watters JJ. Microglial P2 purinergic receptor and immunomodulatory gene transcripts vary by region, sex, and age in the healthy mouse CNS. Transcr Open Access. 2015;3(2). doi:10.4172/2329-8936.1000124
108. Abbracchio MP, Ceruti S. Roles of P2 receptors in glial cells: focus on astrocytes. Purinergic Signal. 2006;2(4):595–604. doi:10.1007/s11302-006-9016-0
109. Florenzano F, Viscomi MT, Cavaliere F, Volonte C, Molinari M. Cerebellar lesion up-regulates P2X1 and P2X2 purinergic receptors in precerebellar nuclei. Neuroscience. 2002;115(2):425–434. doi:10.1016/S0306-4522(02)00397-4
110. Cavaliere F, Florenzano F, Amadio S, et al. Up-regulation of P2X2, P2X4 receptor and ischemic cell death: prevention by P2 antagonists. Neuroscience. 2003;120(1):85–98. doi:10.1016/S0306-4522(03)00228-8
111. Viscomi MT, Florenzano F, Conversi D, Bernardi G, Molinari M. Axotomy dependent purinergic and nitrergic co-expression. Neuroscience. 2004;123(2):393–404. doi:10.1016/j.neuroscience.2003.09.030
112. Franke H, Krugel U, Grosche J, et al. P2Y receptor expression on astrocytes in the nucleus accumbens of rats. Neuroscience. 2004;127(2):431–441. doi:10.1016/j.neuroscience.2004.05.003
113. Adinolfi E, Giuliani AL, De Marchi E, Pegoraro A, Orioli E, Di Virgilio F. The P2X7 receptor: a main player in inflammation. Biochem Pharmacol. 2018;151:234–244. doi:10.1016/j.bcp.2017.12.021
114. Mehta VB, Hart J, Wewers MD. ATP-stimulated release of interleukin (IL)-1beta and IL-18 requires priming by lipopolysaccharide and is independent of caspase-1 cleavage. J Biol Chem. 2001;276(6):3820–3826. doi:10.1074/jbc.M006814200
115. Lu W, Albalawi F, Beckel JM, Lim JC, Laties AM, Mitchell CH. The P2X7 receptor links mechanical strain to cytokine IL-6 up-regulation and release in neurons and astrocytes. J Neurochem. 2017;141(3):436–448. doi:10.1111/jnc.13998
116. He Y, Taylor N, Fourgeaud L, Bhattacharya A. The role of microglial P2X7: modulation of cell death and cytokine release. J Neuroinflammation. 2017;14(1):135. doi:10.1186/s12974-017-0904-8
117. Illes P, Verkhratsky A, Tang Y. Pathological ATPergic signaling in major depression and bipolar disorder. Front Mol Neurosci. 2019;12:331. doi:10.3389/fnmol.2019.00331
118. de Rivero Vaccari JP, Bastien D, Yurcisin G, et al. P2X4 receptors influence inflammasome activation after spinal cord injury. J Neurosci. 2012;32(9):3058–3066. doi:10.1523/JNEUROSCI.4930-11.2012
119. Engel T, Gomez‐Villafuertes R, Tanaka K, et al. Seizure suppression and neuroprotection by targeting the purinergic P2X7 receptor during status epilepticus in mice. FASEB J. 2012;26(4):1616–1628. doi:10.1096/fj.11-196089
120. Adinolfi E, Capece M, Franceschini A, et al. Accelerated tumor progression in mice lacking the ATP receptor P2X7. Cancer Res. 2015;75(4):635–644. doi:10.1158/0008-5472.CAN-14-1259
121. Rodriguez-Alvarez N, Jimenez-Mateos EM, Engel T, et al. Effects of P2X7 receptor antagonists on hypoxia-induced neonatal seizures in mice. Neuropharmacology. 2017;116:351–363. doi:10.1016/j.neuropharm.2017.01.005
122. Albalawi F, Lu W, Beckel JM, Lim JC, McCaughey SA, Mitchell CH. The P2X7 receptor primes IL-1beta and the NLRP3 inflammasome in astrocytes exposed to mechanical strain. Front Cell Neurosci. 2017;11:227. doi:10.3389/fncel.2017.00227
123. Franceschini A, Capece M, Chiozzi P, et al. The P2X7 receptor directly interacts with the NLRP3 inflammasome scaffold protein. FASEB J. 2015;29(6):2450–2461. doi:10.1096/fj.14-268714
124. Abe T, Lee A, Sitharam R, Kesner J, Rabadan R, Shapira SD. Germ-cell-specific inflammasome component NLRP14 negatively regulates cytosolic nucleic acid sensing to promote fertilization. Immunity. 2017;46(4):621–634. doi:10.1016/j.immuni.2017.03.020
125. Monif M, Reid CA, Powell KL, Smart ML, Williams DA. The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J Neurosci. 2009;29(12):3781–3791. doi:10.1523/JNEUROSCI.5512-08.2009
126. Zhao H, Chen Y, Feng H. P2X7 receptor-associated programmed cell death in the pathophysiology of hemorrhagic stroke. Curr Neuropharmacol. 2018;16(9):1282–1295. doi:10.2174/1570159X16666180516094500
127. Turola E, Furlan R, Bianco F, Matteoli M, Verderio C. Microglial microvesicle secretion and intercellular signaling. Front Physiol. 2012;3:149. doi:10.3389/fphys.2012.00149
128. Karasawa A, Michalski K, Mikhelzon P, Kawate T. The P2X7 receptor forms a dye-permeable pore independent of its intracellular domain but dependent on membrane lipid composition. Elife. 2017;6. doi:10.7554/eLife.31186
129. Bernier LP, Ase AR, Boue-Grabot E, Seguela P. P2X4 receptor channels form large noncytolytic pores in resting and activated microglia. Glia. 2012;60(5):728–737. doi:10.1002/glia.22301
130. Ferrari D, Wesselborg S, Bauer MK, Schulze-Osthoff K. Extracellular ATP activates transcription factor NF-kappaB through the P2Z purinoreceptor by selectively targeting NF-kappaB p65. J Cell Biol. 1997;139(7):1635–1643. doi:10.1083/jcb.139.7.1635
131. Ferrari D, Stroh C, Schulze-Osthoff K. P2X7/P2Z purinoreceptor-mediated activation of transcription factor NFAT in microglial cells. J Biol Chem. 1999;274(19):13205–13210. doi:10.1074/jbc.274.19.13205
132. Kamatsuka Y, Fukagawa M, Furuta T, Ohishi A, Nishida K, Nagasawa K. Astrocytes, but not neurons, exhibit constitutive activation of P2X7 receptors in mouse acute cortical slices under non-stimulated resting conditions. Biol Pharm Bull. 2014;37(12):1958–1962. doi:10.1248/bpb.b14-00000
133. Munoz FM, Patel PA, Gao X, et al. Reactive oxygen species play a role in P2X7 receptor-mediated IL-6 production in spinal astrocytes. Purinergic Signal. 2020;16(1):97–107. doi:10.1007/s11302-020-09691-5
134. Minkiewicz J, de Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome. Glia. 2013;61(7):1113–1121. doi:10.1002/glia.22499
135. Ulmann L, Hatcher JP, Hughes JP, et al. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci. 2008;28(44):11263–11268. doi:10.1523/JNEUROSCI.2308-08.2008
136. Wixey JA, Reinebrant HE, Carty ML, Buller KM. Delayed P2X4R expression after hypoxia-ischemia is associated with microglia in the immature rat brain. J Neuroimmunol. 2009;212(1–2):35–43. doi:10.1016/j.jneuroim.2009.04.016
137. Vazquez-Villoldo N, Domercq M, Martin A, Llop J, Gomez-Vallejo V, Matute C. P2X4 receptors control the fate and survival of activated microglia. Glia. 2014;62(2):171–184. doi:10.1002/glia.22596
138. Kukley M, Barden JA, Steinhauser C, Jabs R. Distribution of P2X receptors on astrocytes in juvenile rat hippocampus. Glia. 2001;36(1):11–21. doi:10.1002/glia.1091
139. Jabs R, Matthias K, Grote A, Grauer M, Seifert G, Steinhauser C. Lack of P2X receptor mediated currents in astrocytes and GluR type glial cells of the hippocampal CA1 region. Glia. 2007;55(16):1648–1655. doi:10.1002/glia.20580
140. Ohsawa K, Irino Y, Nakamura Y, Akazawa C, Inoue K, Kohsaka S. Involvement of P2X4 and P2Y12 receptors in ATP-induced microglial chemotaxis. Glia. 2007;55(6):604–616. doi:10.1002/glia.20489
141. Montilla A, Mata GP, Matute C, Domercq M. Contribution of P2X4 receptors to CNS function and pathophysiology. Int J Mol Sci. 2020;21(15). doi:10.3390/ijms21155562
142. Ulmann L, Levavasseur F, Avignone E, et al. Involvement of P2X4 receptors in hippocampal microglial activation after status epilepticus. Glia. 2013;61(8):1306–1319. doi:10.1002/glia.22516
143. Raouf R, Chabot-Dore AJ, Ase AR, Blais D, Seguela P. Differential regulation of microglial P2X4 and P2X7 ATP receptors following LPS-induced activation. Neuropharmacology. 2007;53(4):496–504. doi:10.1016/j.neuropharm.2007.06.010
144. Masuda T, Iwamoto S, Yoshinaga R, et al. Transcription factor IRF5 drives P2X4R+-reactive microglia gating neuropathic pain. Nat Commun. 2014;5(1):3771. doi:10.1038/ncomms4771
145. Kawai K, Baba S. Studies on drug metabolism by use of isotopes. XV. Stability of deuterium-label in p-hydroxylation of l-ephedrine. Chem Pharm Bull (Tokyo). 1975;23(4):920–922. doi:10.1248/cpb.23.920
146. Trang T, Beggs S, Wan X, Salter MW. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J Neurosci. 2009;29(11):3518–3528. doi:10.1523/JNEUROSCI.5714-08.2009
147. Su WF, Wu F, Jin ZH, et al. Overexpression of P2X4 receptor in Schwann cells promotes motor and sensory functional recovery and remyelination via BDNF secretion after nerve injury. Glia. 2019;67(1):78–90. doi:10.1002/glia.23527
148. Zabala A, Vazquez-Villoldo N, Rissiek B, et al. P2X4 receptor controls microglia activation and favors remyelination in autoimmune encephalitis. EMBO Mol Med. 2018;10(8). doi:10.15252/emmm.201708743
149. Forster D, Reiser G. Supportive or detrimental roles of P2Y receptors in brain pathology?--The two faces of P2Y receptors in stroke and neurodegeneration detected in neural cell and in animal model studies. Purinergic Signal. 2015;11(4):441–454. doi:10.1007/s11302-015-9471-6
150. Carmo MR, Simoes AP, Fonteles AA, Souza CM, Cunha RA, Andrade GM. ATP P2Y1 receptors control cognitive deficits and neurotoxicity but not glial modifications induced by brain ischemia in mice. Eur J Neurosci. 2014;39(4):614–622. doi:10.1111/ejn.12435
151. Choo AM, Miller WJ, Chen YC, et al. Antagonism of purinergic signalling improves recovery from traumatic brain injury. Brain. 2013;136(Pt 1):65–80. doi:10.1093/brain/aws286
152. Kuboyama K, Harada H, Tozaki-Saitoh H, Tsuda M, Ushijima K, Inoue K. Astrocytic P2Y(1) receptor is involved in the regulation of cytokine/chemokine transcription and cerebral damage in a rat model of cerebral ischemia. J Cereb Blood Flow Metab. 2011;31(9):1930–1941. doi:10.1038/jcbfm.2011.49
153. Wellmann M, Alvarez-Ferradas C, Maturana CJ, Saez JC, Bonansco C. Astroglial Ca(2+)-dependent hyperexcitability requires P2Y1 purinergic receptors and pannexin-1 channel activation in a chronic model of epilepsy. Front Cell Neurosci. 2018;12:446. doi:10.3389/fncel.2018.00446
154. Fujita T, Tozaki-Saitoh H, Inoue K. P2Y1 receptor signaling enhances neuroprotection by astrocytes against oxidative stress via IL-6 release in hippocampal cultures. Glia. 2009;57(3):244–257. doi:10.1002/glia.20749
155. Quintas C, Fraga S, Goncalves J, Queiroz G. Opposite modulation of astroglial proliferation by adenosine 5ʹ-O-(2-thio)-diphosphate and 2-methylthioadenosine-5ʹ-diphosphate: mechanisms involved. Neuroscience. 2011;182:32–42. doi:10.1016/j.neuroscience.2011.03.009
156. Rothaug M, Becker-Pauly C, Rose-John S. The role of interleukin-6 signaling in nervous tissue. Biochim Biophys Acta. 2016;1863(6 Pt A):1218–1227. doi:10.1016/j.bbamcr.2016.03.018
157. Mildner A, Huang H, Radke J, Stenzel W, Priller J. P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia. 2017;65(2):375–387. doi:10.1002/glia.23097
158. Irino Y, Nakamura Y, Inoue K, Kohsaka S, Ohsawa K. Akt activation is involved in P2Y12 receptor-mediated chemotaxis of microglia. J Neurosci Res. 2008;86(7):1511–1519. doi:10.1002/jnr.21610
159. Dissing-Olesen L, LeDue JM, Rungta RL, Hefendehl JK, Choi HB, MacVicar BA. Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J Neurosci. 2014;34(32):10511–10527. doi:10.1523/JNEUROSCI.0405-14.2014
160. Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, Wu LJ. Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J Neurosci. 2014;34(32):10528–10540. doi:10.1523/JNEUROSCI.0416-14.2014
161. Amadio S, Montilli C, Magliozzi R, Bernardi G, Reynolds R, Volonte C. P2Y12 receptor protein in cortical gray matter lesions in multiple sclerosis. Cereb Cortex. 2010;20(6):1263–1273. doi:10.1093/cercor/bhp193
162. Webster CM, Hokari M, McManus A, et al. Microglial P2Y12 deficiency/inhibition protects against brain ischemia. PLoS One. 2013;8(8):e70927. doi:10.1371/journal.pone.0070927
163. Gomez Morillas A, Besson VC, Lerouet D. Microglia and neuroinflammation: what place for P2RY12? Int J Mol Sci. 2021;22(4). doi:10.3390/ijms22041636
164. Quintas C, Pinho D, Pereira C, Saraiva L, Goncalves J, Queiroz G. Microglia P2Y(6) receptors mediate nitric oxide release and astrocyte apoptosis. J Neuroinflammation. 2014;11(1):141. doi:10.1186/s12974-014-0141-3
165. Kyrargyri V, Madry C, Rifat A, et al. P2Y13 receptors regulate microglial morphology, surveillance, and resting levels of interleukin 1beta release. Glia. 2020;68(2):328–344. doi:10.1002/glia.23719
166. Bianco F, Fumagalli M, Pravettoni E, et al. Pathophysiological roles of extracellular nucleotides in glial cells: differential expression of purinergic receptors in resting and activated microglia. Brain Res Brain Res Rev. 2005;48(2):144–156. doi:10.1016/j.brainresrev.2004.12.004
167. Carrasquero LM, Delicado EG, Bustillo D, Gutierrez-Martin Y, Artalejo AR, Miras-Portugal MT. P2X7 and P2Y13 purinergic receptors mediate intracellular calcium responses to BzATP in rat cerebellar astrocytes. J Neurochem. 2009;110(3):879–889. doi:10.1111/j.1471-4159.2009.06179.x
168. Fischer W, Norenberg W, Franke H, Schaefer M, Illes P. Increase of intracellular Ca2+ by P2Y but not P2X receptors in cultured cortical multipolar neurons of the rat. J Comp Neurol. 2009;516(5):343–359. doi:10.1002/cne.22079
169. Zheng W, Talley Watts L, Holstein DM, Wewer J, Lechleiter JD. P2Y1R-initiated, IP3R-dependent stimulation of astrocyte mitochondrial metabolism reduces and partially reverses ischemic neuronal damage in mouse. J Cereb Blood Flow Metab. 2013;33(4):600–611. doi:10.1038/jcbfm.2012.214
170. Liu PW, Yue MX, Zhou R, et al. P2Y12 and P2Y13 receptors involved in ADPbetas induced the release of IL-1beta, IL-6 and TNF-alpha from cultured dorsal horn microglia. J Pain Res. 2017;10:1755–1767. doi:10.2147/JPR.S137131
171. Milior G, Morin-Brureau M, Chali F, et al. Distinct P2Y receptors mediate extension and retraction of microglial processes in epileptic and peritumoral human tissue. J Neurosci. 2020;40(7):1373–1388. doi:10.1523/JNEUROSCI.0218-19.2019
172. Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, et al. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature. 2007;446(7139):1091–1095. doi:10.1038/nature05704
173. Bernier LP, Ase AR, Boue-Grabot E, Seguela P. Inhibition of P2X4 function by P2Y6 UDP receptors in microglia. Glia. 2013;61(12):2038–2049. doi:10.1002/glia.22574
174. Chorna NE, Santiago-Perez LI, Erb L, et al. P2Y receptors activate neuroprotective mechanisms in astrocytic cells. J Neurochem. 2004;91(1):119–132. doi:10.1111/j.1471-4159.2004.02699.x
175. Wang M, Kong Q, Gonzalez FA, et al. P2Y nucleotide receptor interaction with alpha integrin mediates astrocyte migration. J Neurochem. 2005;95(3):630–640. doi:10.1111/j.1471-4159.2005.03408.x
176. Kim HJ, Ajit D, Peterson TS, et al. Nucleotides released from Abeta(1)(-)(4)(2) -treated microglial cells increase cell migration and Abeta(1)(-)(4)(2) uptake through P2Y(2) receptor activation. J Neurochem. 2012;121(2):228–238. doi:10.1111/j.1471-4159.2012.07700.x
177. Wieraszko A, Seyfried TN. ATP-induced synaptic potentiation in hippocampal slices. Brain Res. 1989;491(2):356–359. doi:10.1016/0006-8993(89)90070-X
178. Wu PH, Phillis JW. Distribution and release of adenosine triphosphate in rat brain. Neurochem Res. 1978;3(5):563–571. doi:10.1007/BF00963759
179. Heinrich A, Ando RD, Turi G, Rozsa B, Sperlagh B. K+ depolarization evokes ATP, adenosine and glutamate release from glia in rat hippocampus: a microelectrode biosensor study. Br J Pharmacol. 2012;167(5):1003–1020. doi:10.1111/j.1476-5381.2012.01932.x
180. Lopatar J, Dale N, Frenguelli BG. Pannexin-1-mediated ATP release from area CA3 drives mGlu5-dependent neuronal oscillations. Neuropharmacology. 2015;93:219–228. doi:10.1016/j.neuropharm.2015.01.014
181. Lopatar J, Dale N, Frenguelli BG. Minor contribution of ATP P2 receptors to electrically-evoked electrographic seizure activity in hippocampal slices: evidence from purine biosensors and P2 receptor agonists and antagonists. Neuropharmacology. 2011;61(1–2):25–34. doi:10.1016/j.neuropharm.2011.02.011
182. Dossi E, Blauwblomme T, Moulard J, et al. Pannexin-1 channels contribute to seizure generation in human epileptic brain tissue and in a mouse model of epilepsy. Sci Transl Med. 2018;10(443):443. doi:10.1126/scitranslmed.aar3796
183. Dona F, Conceicao IM, Ulrich H, et al. Variations of ATP and its metabolites in the hippocampus of rats subjected to pilocarpine-induced temporal lobe epilepsy. Purinergic Signal. 2016;12(2):295–302. doi:10.1007/s11302-016-9504-9
184. Sebastian-Serrano A, Engel T, de Diego-garcia L, et al. Neurodevelopmental alterations and seizures developed by mouse model of infantile hypophosphatasia are associated with purinergic signalling deregulation. Hum Mol Genet. 2016;25(19):4143–4156. doi:10.1093/hmg/ddw248
185. Alves M, Gomez-Villafuertes R, Delanty N, et al. Expression and function of the metabotropic purinergic P2Y receptor family in experimental seizure models and patients with drug-refractory epilepsy. Epilepsia. 2017;58(9):1603–1614. doi:10.1111/epi.13850
186. Avignone E, Ulmann L, Levavasseur F, Rassendren F, Audinat E. Status epilepticus induces a particular microglial activation state characterized by enhanced purinergic signaling. J Neurosci. 2008;28(37):9133–9144. doi:10.1523/JNEUROSCI.1820-08.2008
187. Dona F, Ulrich H, Persike DS, et al. Alteration of purinergic P2X4 and P2X7 receptor expression in rats with temporal-lobe epilepsy induced by pilocarpine. Epilepsy Res. 2009;83(2–3):157–167. doi:10.1016/j.eplepsyres.2008.10.008
188. Jimenez-Pacheco A, Mesuret G, Sanz-Rodriguez A, et al. Increased neocortical expression of the P2X7 receptor after status epilepticus and anticonvulsant effect of P2X7 receptor antagonist A-438079. Epilepsia. 2013;54(9):1551–1561. doi:10.1111/epi.12257
189. Morgan J, Alves M, Conte G, et al. Characterization of the expression of the ATP-gated P2X7 receptor following status epilepticus and during epilepsy using a P2X7-EGFP reporter mouse. Neurosci Bull. 2020;36(11):1242–1258. doi:10.1007/s12264-020-00573-9
190. Kaczmarek-Hajek K, Zhang J, Kopp R, et al. Re-evaluation of neuronal P2X7 expression using novel mouse models and a P2X7-specific nanobody. Elife. 2018;7. doi:10.7554/eLife.36217
191. Ramirez-Fernandez A, Urbina-Trevino L, Conte G, et al. Deviant reporter expression and P2X4 passenger gene overexpression in the soluble EGFP BAC transgenic P2X7 reporter mouse model. Sci Rep. 2020;10(1):19876. doi:10.1038/s41598-020-76428-0
192. Jimenez-Pacheco A, Diaz-Hernandez M, Arribas-Blazquez M, et al. Transient P2X7 receptor antagonism produces lasting reductions in spontaneous seizures and gliosis in experimental temporal lobe epilepsy. J Neurosci. 2016;36(22):5920–5932. doi:10.1523/JNEUROSCI.4009-15.2016
193. Vianna EP, Ferreira AT, Naffah-Mazzacoratti MG, et al. Evidence that ATP participates in the pathophysiology of pilocarpine-induced temporal lobe epilepsy: fluorimetric, immunohistochemical, and Western blot studies. Epilepsia. 2002;43(Suppl 5):227–229. doi:10.1046/j.1528-1157.43.s.5.26.x
194. Alves M, Smith J, Engel T. Differential expression of the metabotropic P2Y receptor family in the cortex following status epilepticus and neuroprotection via P2Y1 antagonism in mice. Front Pharmacol. 2019;10:1558. doi:10.3389/fphar.2019.01558
195. Sukigara S, Dai H, Nabatame S, et al. Expression of astrocyte-related receptors in cortical dysplasia with intractable epilepsy. J Neuropathol Exp Neurol. 2014;73(8):798–806. doi:10.1097/NEN.0000000000000099
196. Alves M, De Diego Garcia L, Conte G, et al. Context-specific switch from anti- to pro-epileptogenic function of the P2Y1 receptor in experimental epilepsy. J Neurosci. 2019;39(27):5377–5392. doi:10.1523/JNEUROSCI.0089-19.2019
197. Huang C, Chi XS, Li R, et al. Inhibition of P2X7 receptor ameliorates nuclear factor-kappa B mediated neuroinflammation induced by status epilepticus in rat hippocampus. J Mol Neurosci. 2017;63(2):173–184. doi:10.1007/s12031-017-0968-z
198. Mesuret G, Engel T, Hessel EV, et al. P2X7 receptor inhibition interrupts the progression of seizures in immature rats and reduces hippocampal damage. CNS Neurosci Ther. 2014;20(6):556–564. doi:10.1111/cns.12272
199. Glass HC, Shellhaas RA, Wusthoff CJ, et al. Contemporary profile of seizures in neonates: a Prospective Cohort Study. J Pediatr. 2016;174:98–103e101. doi:10.1016/j.jpeds.2016.03.035
200. Soul JS. Acute symptomatic seizures in term neonates: etiologies and treatments. Semin Fetal Neonatal Med. 2018;23(3):183–190. doi:10.1016/j.siny.2018.02.002
201. Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science. 1996;272(5262):735–738. doi:10.1126/science.272.5262.735
202. Menendez Mendez A, Smith J, Engel T. Neonatal seizures and purinergic signalling. Int J Mol Sci. 2020;21(21):7832. doi:10.3390/ijms21217832
203. Fischer W, Franke H, Krugel U, et al. Critical evaluation of P2X7 receptor antagonists in selected seizure models. PLoS One. 2016;11(6):e0156468. doi:10.1371/journal.pone.0156468
204. Nieoczym D, Socala K, Wlaz P. Evaluation of the anticonvulsant effect of brilliant blue G, a selective P2X7 receptor antagonist, in the iv PTZ-, maximal electroshock-, and 6 Hz-induced seizure tests in mice. Neurochem Res. 2017;42(11):3114–3124. doi:10.1007/s11064-017-2348-z
205. Kim JE, Kang TC. The P2X7 receptor-pannexin-1 complex decreases muscarinic acetylcholine receptor-mediated seizure susceptibility in mice. J Clin Invest. 2011;121(5):2037–2047. doi:10.1172/JCI44818
206. Kim JE, Kwak SE, Jo SM, Kang TC. Blockade of P2X receptor prevents astroglial death in the dentate gyrus following pilocarpine-induced status epilepticus. Neurol Res. 2009;31(9):982–988. doi:10.1179/174313209X389811
207. Kim JE, Ryu HJ, Yeo SI, Kang TC. P2X7 receptor regulates leukocyte infiltrations in rat frontoparietal cortex following status epilepticus. J Neuroinflammation. 2010;7(1):65. doi:10.1186/1742-2094-7-65
208. Kim JE, Ryu HJ, Kang TC. P2X7 receptor activation ameliorates CA3 neuronal damage via a tumor necrosis factor-alpha-mediated pathway in the rat hippocampus following status epilepticus. J Neuroinflammation. 2011;8(1):62. doi:10.1186/1742-2094-8-62
209. Soni N, Koushal P, Reddy BV, Deshmukh R, Kumar P. Effect of GLT-1 modulator and P2X7 antagonists alone and in combination in the kindling model of epilepsy in rats. Epilepsy Behav. 2015;48:4–14. doi:10.1016/j.yebeh.2015.04.056
210. Amorim RP, Araujo MGL, Valero J, et al. Silencing of P2X7R by RNA interference in the hippocampus can attenuate morphological and behavioral impact of pilocarpine-induced epilepsy. Purinergic Signal. 2017;13(4):467–478. doi:10.1007/s11302-017-9573-4
211. Jamali-Raeufy N, Barati H, Baluchnejadmojarad T, Roghani M, Goudarzi M. Combination therapy with dipeptidyl peptidase-4 and P2X7 purinoceptor inhibitors gives rise to antiepileptic effects in rats. J Chem Neuroanat. 2020;110:101855. doi:10.1016/j.jchemneu.2020.101855
212. Hong S, Xin Y, JiaWen W, et al. The P2X7 receptor in activated microglia promotes depression- and anxiety-like behaviors in lithium -pilocarpine induced epileptic rats. Neurochem Int. 2020;138:104773. doi:10.1016/j.neuint.2020.104773
213. Rozmer K, Gao P, Araujo MGL, et al. Pilocarpine-induced status epilepticus increases the sensitivity of P2X7 and P2Y1 receptors to nucleotides at neural progenitor cells of the juvenile rodent hippocampus. Cereb Cortex. 2017;27(7):3568–3585. doi:10.1093/cercor/bhw178
214. Amhaoul H, Ali I, Mola M, et al. P2X7 receptor antagonism reduces the severity of spontaneous seizures in a chronic model of temporal lobe epilepsy. Neuropharmacology. 2016;105:175–185. doi:10.1016/j.neuropharm.2016.01.018
215. Khan MT, Deussing J, Tang Y, Illes P. Astrocytic rather than neuronal P2X7 receptors modulate the function of the tri-synaptic network in the rodent hippocampus. Brain Res Bull. 2019;151:164–173. doi:10.1016/j.brainresbull.2018.07.016
216. Bedner P, Dupper A, Huttmann K, et al. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain. 2015;138(Pt 5):1208–1222. doi:10.1093/brain/awv067
217. Illes P, Burnstock G, Tang Y. Astroglia-derived ATP modulates CNS neuronal circuits. Trends Neurosci. 2019;42(12):885–898. doi:10.1016/j.tins.2019.09.006
218. Lee DS, Kim JE. P2 x 7 receptor inhibits astroglial autophagy via regulating FAK- and PHLPP1/2-mediated AKT-S473 phosphorylation following kainic acid-induced seizures. Int J Mol Sci. 2020;21(18).
219. Lee DS, Kim JE. Protein disulfide isomerase-mediated S-nitrosylation facilitates surface expression of P2X7 receptor following status epilepticus. J Neuroinflammation. 2021;18(1):14. doi:10.1186/s12974-020-02058-y
220. Sperlagh B, Illes P. P2X7 receptor: an emerging target in central nervous system diseases. Trends Pharmacol Sci. 2014;35(10):537–547. doi:10.1016/j.tips.2014.08.002
221. Xia J, Wang H, Zhang Q, Han Z. Modulation of P2X purinoceptor 3 (P2X3) in pentylenetetrazole-induced kindling epilepsy in rats. Med Sci Monit. 2018;24:6165–6177. doi:10.12659/MSM.910352
222. Alves M, Beamer E, Engel T. The metabotropic purinergic P2Y receptor family as novel drug target in epilepsy. Front Pharmacol. 2018;9:193. doi:10.3389/fphar.2018.00193
223. Avignone E, Lepleux M, Angibaud J, Nagerl UV. Altered morphological dynamics of activated microglia after induction of status epilepticus. J Neuroinflammation. 2015;12(1):202. doi:10.1186/s12974-015-0421-6
224. Mo M, Eyo UB, Xie M, et al. Microglial P2Y12 receptor regulates seizure-induced neurogenesis and immature neuronal projections. J Neurosci. 2019;39(47):9453–9464. doi:10.1523/JNEUROSCI.0487-19.2019
225. Simoes AP, Silva CG, Marques JM, et al. Glutamate-induced and NMDA receptor-mediated neurodegeneration entails P2Y1 receptor activation. Cell Death Dis. 2018;9(3):297. doi:10.1038/s41419-018-0351-1
226. Alvarez-Ferradas C, Morales JC, Wellmann M, et al. Enhanced astroglial Ca2+ signaling increases excitatory synaptic strength in the epileptic brain. Glia. 2015;63(9):1507–1521. doi:10.1002/glia.22817
227. Nikolic L, Shen W, Nobili P, Virenque A, Ulmann L, Audinat E. Blocking TNFalpha-driven astrocyte purinergic signaling restores normal synaptic activity during epileptogenesis. Glia. 2018;66(12):2673–2683. doi:10.1002/glia.23519
228. Martorell A, Wellmann M, Guiffa F, Fuenzalida M, Bonansco C. P2Y1 receptor inhibition rescues impaired synaptic plasticity and astroglial Ca(2+)-dependent activity in the epileptic hippocampus. Neurobiol Dis. 2020;146:105132. doi:10.1016/j.nbd.2020.105132
229. Beamer E, Lacey A, Alves M, et al. Elevated blood purine levels as a biomarker of seizures and epilepsy. Epilepsia. 2021;62(3):817–828. doi:10.1111/epi.16839
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.