Back to Journals » Journal of Inflammation Research » Volume 19
Targeting Macrophage-Mediated Mechanisms in Sepsis-Induced Cardiomyopathy: From Pathophysiology to Therapeutic Strategies
Authors Wang J ![]() , Zhang J, Luo N
, Zhang J, Luo N ![]() , Zhou Z, Zhang Y, Guo K, Li R
, Zhou Z, Zhang Y, Guo K, Li R ![]() , Li C
, Li C
Received 23 March 2026
Accepted for publication 20 June 2026
Published 15 July 2026 Volume 2026:19 607880
DOI https://doi.org/10.2147/JIR.S607880
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Xin Du
Jun Wang,1,* Jingyi Zhang,1,* Nianfang Luo,1 Ziran Zhou,2 Yue Zhang,1 Kangyan Guo,1 Ruiting Li,3 Chang Li1
1Department of Cardiology, Jianghan University Affiliated Hubei Third People’s Hospital, Wuhan, Hubei, 430022, People’s Republic of China; 2The Second Clinical College, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, People’s Republic of China; 3Department of Critical Care Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, 430022, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Chang Li, Department of Cardiology, Jianghan University Affiliated Hubei Third People’s Hospital, Wuhan, Hubei, 430022, People’s Republic of China, Email [email protected] Ruiting Li, Department of Critical Care Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, 430022, People’s Republic of China, Email [email protected]
Abstract: Sepsis-induced cardiomyopathy (SICM) is a life-threatening complication of sepsis, characterized by dysregulated inflammation, metabolic disturbances, and complex cellular crosstalk. As central innate immune cells, macrophages exert a dual role in this pathology. This review focuses on the key mechanisms by which macrophages contribute to SICM. We discuss macrophage-driven pathogenesis across three interconnected themes. First, inflammatory activation. Macrophages amplify the inflammatory storm through dynamic M1/M2 polarization and activation of Toll-like receptor 4 (TLR4) / nuclear factor κB (NF-κB), as well as multiple inflammasome pathways such as NLR family pyrin domain containing 3 (NLRP3). Second, myocardial injury mechanisms. Activated macrophages promote cardiomyocyte damage via oxidative stress, suppression of mitophagy, direct cytotoxic effects, and indirect networks (such as MMP-9-mediated endothelial disruption). Third, regulatory layers. Immunometabolic reprogramming (enhanced glycolysis and succinate accumulation in M1 macrophages versus fatty acid oxidation in M2 macrophages) serves as an intrinsic driver linking immune activation to cardiomyocyte dysfunction. Additionally, macrophages engage in complex intercellular communication with neutrophils (NETs), T cells (PD-L1/PD-1), and via complement C5a/C5aR1 signaling, all of which amplify cardiac injury. Finally, based on these mechanisms, we discuss therapeutic strategies targeting macrophage polarization, metabolism, specific subsets (e.g. TREM2hi macrophages), and intercellular communication, aiming to offer a theoretical framework for developing precision immunotherapies for SICM.
Keywords: sepsis-induced cardiomyopathy, macrophage, immunometabolism, inflammasome, pyroptosis, therapeutic target
Introduction
Sepsis-induced cardiomyopathy (SICM) is clinically defined as an acute, reversible myocardial dysfunction occurring in a septic patient, characterized by left ventricular dilation, reduced ejection fraction (typically <50%), and impaired contractile response to volume infusion or catecholamines, in the absence of coronary artery disease or other known cardiac pathologies.1 Despite its reversibility, SICM doubles or triples mortality risk, yet no specific pharmacotherapy exists beyond supportive care and infection control. Macrophages are among the first cells to recognize pathogen-associated molecular patterns (PAMPs, eg, lipopolysaccharide (LPS)) and damage-associated molecular patterns (DAMPs, eg, high mobility group box 1 (HMGB1)). Through their phenotypic plasticity, secretory activity, and high degree of microenvironment interaction, macrophages critically shape the initiation, progression, and outcome of SICM.2
The need for this review arises from three critical gaps: First, while macrophages are recognized as central orchestrators of sepsis, their specific contribution to cardiomyocyte dysfunction—distinct from systemic inflammation—remains incompletely integrated. Second, recent breakthroughs in immunometabolism, single-cell omics, and intercellular communication (eg, NETosis, exosomal miRNAs) have generated a surge of mechanistic data that require synthesis into a coherent, SICM-focused framework. Third, and most importantly, the field lacks a critical appraisal of how much of our current knowledge derives from non-SICM models (eg, ischemia-reperfusion, atherosclerosis) versus genuine SICM-specific studies, leading to potential overgeneralization. Therefore, this review systematically examines macrophage-driven mechanisms in SICM with emphasis on (i) distinguishing direct evidence from indirect inference, (ii) integrating novel layers of regulation (metabolic, neuro-immune, complement), and (iii) identifying translatable therapeutic targets with acknowledgment of current limitations.
Macrophage Heterogeneity and Dynamic Evolution in SICM
Macrophages are not a homogeneous population. In the heart, they can be categorized based on origin into embryo-derived tissue-resident macrophages (eg, C-C chemokine receptor type 2 (CCR2)- subsets) and bone marrow-derived monocyte-derived macrophages (CCR2+) that are continuously replenished in adulthood.1,2 Under homeostasis, resident macrophages act as “guardians” of cardiac health by clearing apoptotic debris, promoting tissue repair, and even indirectly influencing cardiomyocyte electrophysiology through electrotonic coupling.3–5
Sepsis disrupts this equilibrium. Potent inflammatory signals (eg, LPS, interferon gamma (IFN-γ)) drive macrophage polarization towards the classically activated M1 phenotype. M1 macrophages are primary effectors of pro-inflammatory responses, producing high levels of tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), interleukin-6 (IL-6), inducible nitric oxide synthase (iNOS)-derived nitric oxide (NO), and reactive oxygen species (ROS) via pathways like TLR4/NF-κB and NLRP3 inflammasome activation.6–8 TNF-α and IL-1β directly impair cardiomyocyte contractility by disrupting calcium handling, while ROS and NO inhibit mitochondrial respiratory chain complexes, leading to ATP synthesis failure and a cellular “energy crisis”.6,7,9 Additionally, activated M1 macrophages release exosomes carrying miR-155-5p into the circulation. These exosomes are subsequently internalized by cardiomyocytes. MiR-155-5p may target MSK1 in cardiomyocytes, thereby activating the p38-MAPK signaling pathway. This activation triggers intrinsic inflammatory responses in cardiomyocytes, characterized by the production of pro-inflammatory cytokines such as IL-6 and TNF-α. Furthermore, it may promote the assembly of inflammasomes (eg, NLRP3), ultimately leading to pyroptosis of cardiomyocytes.6
As the disease progresses, microenvironmental cues shift. Cytokines like interleukin-4 (IL-4) and interleukin-13 (IL-13), signaling through the signal transducer and activator of transcription 6 (STAT6) pathway, promote alternative activation towards the M2 phenotype. M2 macrophages secrete anti-inflammatory cytokines (eg, interleukin-10 (IL-10), transforming growth factor beta (TGF-β)) and pro-repair factors (eg, vascular endothelial growth factor (VEGF), arginase-1 (Arg-1)), facilitating inflammation resolution and tissue healing.10–12 In the early phase of SICM, appropriate M2 polarization can be protective by clearing apoptotic cells and limiting excessive inflammation.13 However, persistent M2 polarization later in the course may drive excessive cardiac fibroblast activation via the TGF-β/Smad3 pathway, leading to aberrant collagen deposition, myocardial fibrosis, and subsequent diastolic dysfunction.14,15 Excessive M2 polarization might also suppress antimicrobial immunity, increasing the risk of secondary infections.16
Therefore, the progression and outcome of SICM hinge on the precise spatiotemporal balance between M1 and M2 macrophages. Therapeutic strategies should aim not simply to suppress or activate one pole, but to achieve precise spatiotemporal modulation of macrophage polarization according to the disease stage.
Core Mechanisms of Macrophage-Driven Myocardial Injury
Amplification of Key Inflammatory Signaling Pathways
The central role of macrophages in SICM begins with the recognition of danger signals. The TLR4 / Myeloid differentiation primary response 88 (MyD88) / NF-κB axis is a quintessential inflammatory pathway. LPS binding to TLR4 triggers a signaling cascade via the adaptor protein MyD88, culminating in the nuclear translocation of NF-κB, which initiates the transcription of a plethora of pro-inflammatory cytokines and chemokines, creating an “inflammatory storm”.17 These factors directly impair cardiomyocyte function and recruit more neutrophils and monocytes to the heart, amplifying local inflammation. Concurrently, NF-κB-induced iNOS generates excessive NO, suppressing mitochondrial function and reducing vascular reactivity.17 Cross-talk exists between the NF-κB pathway and NLRP3 inflammasome activation, with the former providing a priming signal for the latter (eg, upregulating NLRP3 and pro-IL-1β expression).18
Emerging optogenetics approaches offer precise control. For instance, blue light activation of transfected photoactivated adenylyl cyclase (bPAC) specifically elevates intracellular cyclic adenosine monophosphate (cAMP) levels in macrophages. cAMP activates protein kinase A (PKA), which suppresses pro-inflammatory signaling like NF-κB, reducing TNF-α and IL-1β secretion while promoting anti-inflammatory mediators like IL-10 and Arg-1,19 as illustrated in Figure 1.
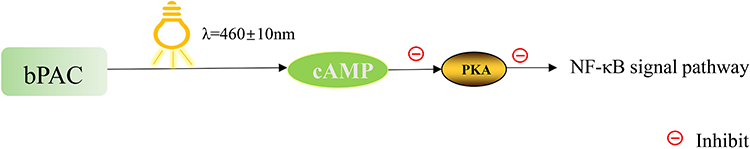 |
Figure 1 Optogenetic regulation of macrophage inflammatory response via the cAMP-PKA signaling pathway. Blue light (λ = 460 ± 10 nm) activates photoactivated adenylyl cyclase (bPAC) transfected into macrophages, leading to an increase in intracellular cyclic adenosine monophosphate (cAMP). Elevated cAMP activates protein kinase A (PKA), which subsequently suppresses the NF-κB signaling pathway, thereby reducing the expression of pro-inflammatory cytokines such as TNF-α and IL-1β. This optogenetic approach offers a precise spatiotemporal strategy to modulate macrophage-mediated inflammation in sepsis-induced cardiomyopathy (SICM). |
Inflammasome Activation
Macrophages in sepsis can be activated via multiple, often interconnected, inflammasome pathways. The NLRP3 inflammasome acts as a classic “danger signal integrator,” responding to various intracellular disturbances like K+ efflux, ROS burst, and lysosomal disruption. Its activation underpins macrophage pyroptosis and IL-1β release.20 The NLR family apoptosis inhibitory protein (NAIP) / NLR family CARD domain containing 4 (NLRC4) inflammasome specifically detects cytosolic bacterial components (eg, flagellin, type III secretion system proteins),21,22 inducing robust inflammation and, via caspase-1-mediated poly (ADP-ribose) polymerase 1 (PARP1) cleavage, epigenetically upregulating iNOS expression, leading to excessive NO production that directly inhibits cardiomyocyte mitochondrial respiration.23 The absent in melanoma 2 (AIM2) inflammasome serves as a cytosolic double-stranded DNA (dsDNA) “sensor.” In contexts like clonal hematopoiesis, metabolically hyperactive macrophages produce abundant ROS, causing DNA damage and replication stress, leading to activation by self or pathogen-derived cytosolic dsDNA, which exacerbates inflammation.24 The Pyrin inflammasome uniquely acts as an “indirect monitor” of bacterial toxin activity by sensing Rho GTPase inactivation. Notably, neutrophil extracellular traps (NETs) have been identified as a novel upstream activator of Pyrin in macrophages, amplifying inflammation.25,26 Despite distinct triggers, these inflammasomes converge on caspase-1 activation, which cleaves gasdermin D (GSDMD) to execute pyroptosis and processes pro-IL-1β and pro-interleukin-18 (pro-IL-18) into their active forms, forming a potent “inflammatory cell death” network that fuels the myocardial inflammatory storm.22,23,25–27 IL-1β and NO promote cardiomyocyte apoptosis and metabolic dysfunction, while IL-18 enhances IFN-γ secretion, increasing immune cell infiltration, as illustrated in Figure 2. Pyroptotic release of inflammatory mediators also damages coronary endothelium, promoting microthrombosis and worsening perfusion. The inflammasome adapter protein Caspase recruitment domain family member 8 (CARD8) acts as a critical “endogenous brake,” whose expression is finely tuned by the circular RNA (circRNA)-CARD8/miR-580-3p axis.28 In sepsis, dysregulation of this axis reduces CARD8 protein levels, lifting the restraint on pyroptosis and rendering macrophages more susceptible, thereby amplifying myocardial inflammation. NLR family pyrin domain containing 6 (NLRP6), conversely, is a negative regulator of type 2 immunity, and its deficiency enhances type 2 innate lymphoid cells (ILC2) and T helper 2 cells (Th2) activity, elevating type 2 cytokines (eg, IL-4, interleukin-5 (IL-5), IL-13) and skewing the type1/type2 balance, which may indirectly affect myocardial repair.29
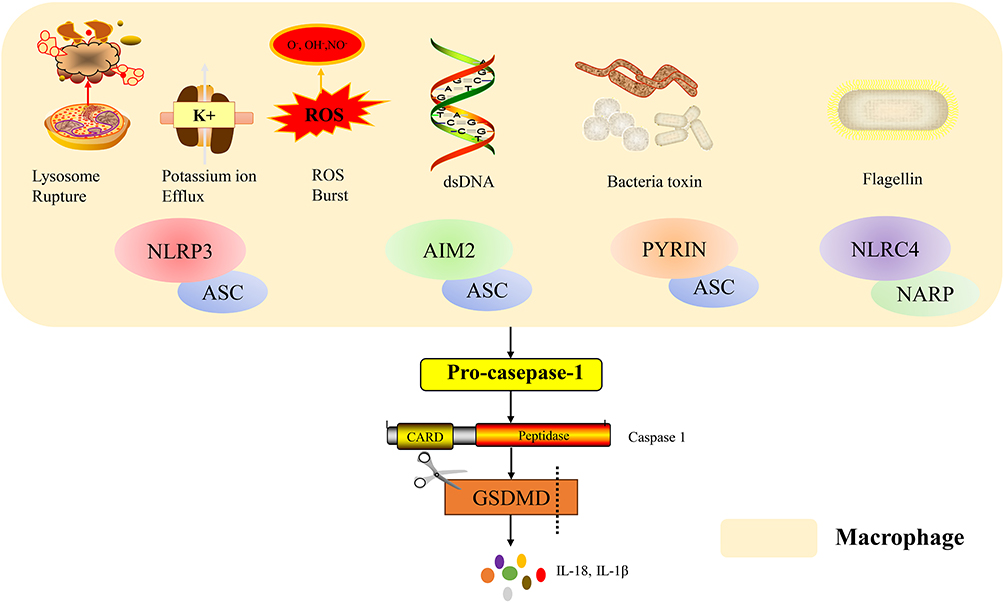 |
Figure 2 Mechanisms of inflammasome activation in macrophages. Different pathogen-associated molecular patterns (PAMPs) and danger signals activate specific inflammasome sensors. Lysosomal rupture, potassium ion efflux and ROS burst triggers the NLRP3 inflammasome. Cytosolic dsDNA is sensed by AIM2. Bacterial toxins activate the PYRIN inflammasome, while flagellin is recognized by NLRC4. NLRP3. AIM2 and PYRIN sensors recruit the adaptor protein ASC, while NLRC4 sensors recruit the adaptor protein NARP, leading to caspase-1 activation. Caspase-1 cleaves Gasdermin D (GSDMD), inducing pore formation and macrophage pyroptosis, accompanied by the release of IL-18 and IL-1β. The domain structure of pro-caspase-1 and its active form (Caspase-1, p20/p10) is shown. |
Oxidative Stress Injury
Activated macrophages generate excessive ROS and reactive nitrogen species (RNS) via nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NADPH oxidase 2 (NOX2)) and mitochondrial electron leak, directly damaging cardiomyocytes through lipid peroxidation, protein denaturation, and DNA damage, leading to structural disruption, contractile dysfunction, and apoptosis.30,31 In sepsis accompanied by hemolysis, macrophage phagocytosis of damaged erythrocytes causes iron overload, precipitating ferroptosis. Ferroptotic macrophages release oxidized lipids, inflammatory cytokines, and iron-loaded vesicles, which not only directly injure cardiomyocytes but also transmit ferroptotic signals via extracellular vesicles (EVs), inducing cardiomyocyte ferroptosis.30,31
Oxidative stress also alters the cargo of macrophage-derived small extracellular vesicles (sEVs), enriching them with pro-inflammatory miRNAs (eg, miR-155), oxidized lipids, and iron. Upon uptake by cardiomyocytes, these sEVs provoke intracellular inflammation, oxidative stress, and ferroptosis. Specifically, exosomes from IL-1β-stimulated macrophages (M-IL-exo) exhibit elevated miR-146a, which suppresses mitogen-activated protein kinase 4 (MAPK4) expression, affecting dynamin-related protein 1 (Drp-1) phosphorylation (Ser616), leading to excessive mitochondrial fission, loss of membrane potential, ROS burst, and reduced ATP synthesis, culminating in cardiomyocyte apoptosis or necrosis.32 Macrophage-derived EVs, rich in transferrin receptor (TfR) on their surface, can chelate protein-bound iron. TfR knockdown ablates the iron-chelating capacity and cardioprotective effects of EVs, underscoring their role in mitigating iron overload and oxidative stress.33 In myocardial infarction models, EV administration reduced cardiac iron content and oxidative markers (malondialdehyde (MDA), dihydroethidium (DHE)), enhanced antioxidant enzyme activity (glutathione (GSH), glutathione peroxidase 4 (GPX4)), limited infarct size, and improved cardiac function (left ventricular ejection fraction (LVEF), left ventricular fractional shortening (LVFS)).33
Collectively, these processes cause severe mitochondrial dysfunction, manifesting as fission/fusion imbalance (Drp1↑, mitofusin 2 (Mfn2)/optic atrophy 1 (OPA1)↓), impaired autophagy, and decreased respiratory chain complex activity, contributing to the release of cardiac enzymes and functional deterioration. Oxidatively stressed macrophages also secrete DAMPs like HMGB1 and S100 calcium-binding protein A8/A9 (S100A8/A9), which disrupt mitochondrial quality control in cardiomyocytes via TLR4/receptor for advanced glycation end products (RAGE) receptors, leading to insufficient ATP synthesis and myocardial stunning.
Suppression of Mitophagy
Impaired macrophage autophagy contributes to SICM. Oxidized low-density lipoprotein (ox-LDL)-treated macrophages exhibit reduced autophagosome formation, decreased microtubule-associated protein 1A/1B-light chain 3 (LC3)-II/I ratio, and downregulated Beclin1, alongside cellular senescence (elevated senescence-associated beta-galactosidase (SA-β-gal) activity, increased P16/P21 expression).34 These senescent macrophages exacerbate myocardial inflammation and injury via the senescence-associated secretory phenotype (SASP). Mechanistically, Notch1 signaling activation upregulates macrophage stimulating 1 (MST1), which phosphorylates Beclin1 (Thr108), enhancing its binding to B-cell lymphoma 2 (Bcl-2) and inhibiting the assembly of the autophagy initiation complex (autophagy-related 14-like protein (Atg14L)--Beclin1--vacuolar protein sorting 34 (Vps34)), thereby obstructing mitophagy.34 Autophagy inhibition leads to mitochondrial dysfunction (swelling, cristae disruption, elevated ROS), accelerates senescence, and activates the NLRP3 inflammasome, ultimately depressing cardiac function. Notch1 deficiency improves mitochondrial morphology and function, attenuating cardiac injury.34 Quercetin mitigates these effects by downregulating MST1, relieving autophagy inhibition, increasing the LC3-II/I ratio and Beclin1 expression, and restoring autophagic flux, thereby improving mitochondrial function, reducing ROS, and delaying senescence. This suggests that pharmacological (eg, rapamycin) or genetic strategies to enhance macrophage autophagy may be effective against SICM.34
Direct Cardiomyocyte Injury
Activated M1 macrophages highly express Fas ligand (FasL), which engages the death receptor Fas on cardiomyocytes, initiating caspase-8/caspase-3-mediated apoptosis and contributing to cardiomyocyte loss.35 Furthermore, macrophage-derived TNF-α and IL-1β can activate the N-methyl-D-aspartate receptor (NMDAR) on cardiomyocytes, causing calcium influx and intracellular calcium overload. Calcium overload triggers mitochondrial permeability transition pore (mPTP) opening, disrupts mitochondrial function, and activates Bcl-2-like protein 11 (Bim)/caspase-3-mediated apoptosis, ultimately leading to contractile dysfunction.36–38
Indirect Regulatory Networks
Endothelial Barrier Disruption and Microcirculatory Dysfunction
In sepsis, activated M1 macrophages secrete high levels of matrix metalloproteinase-9 (MMP-9). MMP-9 degrades key components of the myocardial capillary basement membrane (eg, collagen IV, laminin) and tight junction proteins (eg, zonula occludens-1 (ZO-1)), directly disrupting microvascular barrier integrity. This increases vascular permeability, leading to plasma leakage, myocardial edema (which increases oxygen diffusion distance and exacerbates hypoxia), and exposure of pro-coagulant subendothelial matrix, activating coagulation and promoting microthrombosis. Together, these events cause severe microcirculatory impairment.39
MMP-9 also degrades structural proteins within cardiomyocytes (eg, myosin light chain) and the extracellular matrix, directly impairing contractility; degrades connexin-43, disrupting electrical coupling and promoting arrhythmias; and disrupts the interstitial collagen network (types I and III), contributing to adverse ventricular remodeling.30 Moreover, MMP-9 cleaves latent pro-inflammatory factors and matrix-bound bioactive fragments from the extracellular matrix (ECM), recruiting more immune cells and creating a self-amplifying cycle of inflammation, as illustrated in Figure 3.
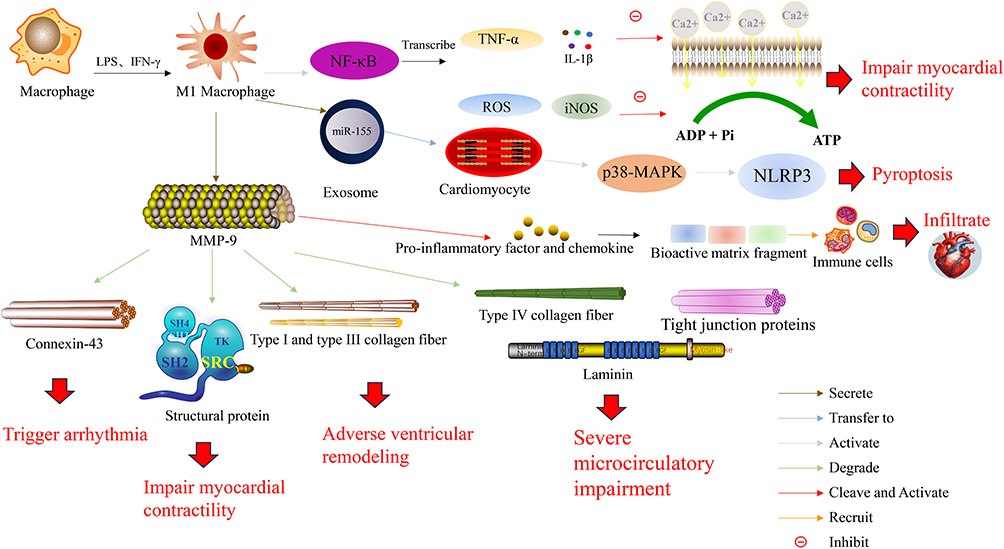 |
Figure 3 Pro-inflammatory role of M1 macrophages in cardiac injury and remodeling. Upon stimulation with LPS and IFN-γ, macrophages polarize into the M1 phenotype, leading to NF-κB activation. Activated NF-κB drives the production of TNF-α and IL-1β, which impair myocardial contractility by suppressing calcium (Ca2⁺) influx. Concurrently, NF-κB activation promotes ROS generation and iNOS expression, which attenuate myocardial contractility through inhibition of ADP/ATP metabolism. M1 macrophages also secrete exosomes containing miR-155, which are transferred into cardiomyocytes to activate the p38-MAPK–NLRP3 axis and promote pyroptosis. In parallel, M1 macrophage-derived MMP-9 exerts multifaceted detrimental effects: it cleaves and activates pro-inflammatory factors and chemokines into bioactive matrix fragments, thereby recruiting immune cells and promoting infiltration; it degrades connexin-43 expression, triggering arrhythmia; it degrades type I and III collagen fibers, leading to adverse ventricular remodeling; it decreases structural proteins, resulting in impaired myocardial contractility; and it degrades type IV collagen fibers, tight junction proteins, and laminin, causing severe microcirculatory impairment. |
In addition to MMP-9-mediated proteolytic injury, emerging evidence identifies macrophage extracellular traps (METs) as a critical contributor to microvascular compromise in SICM.40 METs are web-like structures composed of DNA, histones, and granule proteins released by activated macrophages in response to LPS, IL-1β, or complement C5a stimulation.41,42 Structurally analogous to neutrophil extracellular traps (NETs), METs exert multifaceted deleterious effects on the myocardial microenvironment. First, METs directly compromise endothelial integrity by degrading adherens junction proteins (eg, vascular endothelial cadherin) and tight junction components (eg, ZO-1, occludin) via histone-driven cytotoxicity and associated proteases, thereby exacerbating vascular leakage and interstitial edema.43 Second, METs serve as a scaffold for platelet adhesion and fibrin deposition, promoting microthrombosis and regional ischemia.44 Third, METs function as potent danger signals: their DNA backbone activates the AIM2 inflammasome in neighboring macrophages and cardiomyocytes, while associated HMGB1 and IL-1β sustain TLR4/NF-κB signaling, creating a self-amplifying inflammatory loop.45 Notably, pyroptotic macrophage-derived microvesicles can induce NET formation in neutrophils, establishing a bidirectional MET-NET axis that further amplifies cardiac injury (Figure 3).44,46 The convergence of MMP-9-mediated matrix degradation and MET-driven endothelial disruption underscores the central role of macrophage-derived extracellular effectors in SICM-associated microcirculatory failure.
Autonomic Nervous System Dysregulation
Macrophage-derived IL-1β and other inflammatory mediators act on central nervous system regions like the subfornical organ (SFO) and paraventricular nucleus (PVN), triggering intense sympathetic nervous system (SNS) activation.47 Locally in the heart, macrophages exposed to high norepinephrine (NE) levels may see their beta-2 adrenergic receptor (ADRB2) signaling shift towards pro-inflammatory non-canonical pathways like MAPK. Parasympathetic vagal activity is often suppressed in sepsis, impairing the endogenous “cholinergic anti-inflammatory pathway (CAIP)”.47 Consequently, alpha-7 nicotinic acetylcholine receptor (α7nAChR) on macrophages receive insufficient acetylcholine signals, leading to disinhibition of NF-κB and uncontrolled release of TNF-α and IL-1β, as illustrated in Figure 4. Excessive inflammation suppresses myocardial contraction via iNOS/NO overproduction, inhibition of β-adrenergic receptor signaling, and disruption of calcium homeostasis. SNS activation also mobilizes monocytes/macrophages from bone marrow and spleen. Systemic inflammatory signals can access the brain via circumventricular organs, activating microglia and releasing mediators like prostaglandin E2 (PGE2) that further drive sympathetic excitation, creating a vicious cycle of “peripheral inflammation → central activation → enhanced sympathetic output → worsened peripheral inflammation,” significantly increasing the risk of malignant ventricular arrhythmias.47
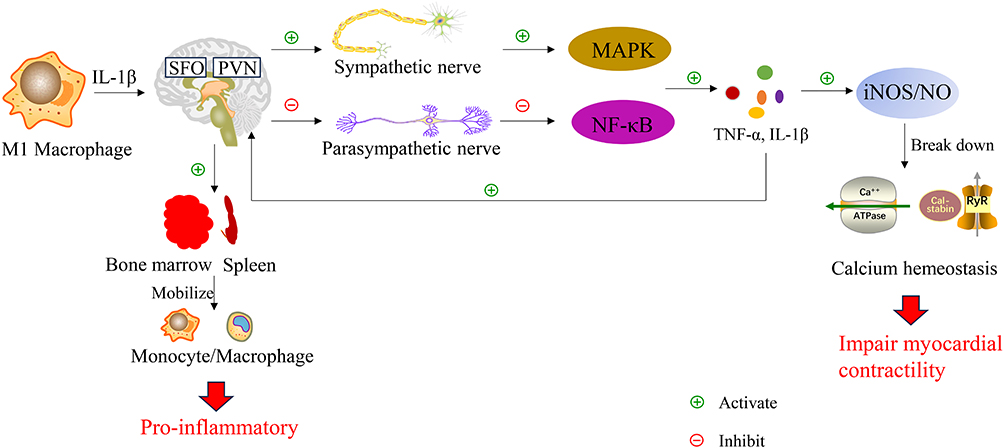 |
Figure 4 Neuro-immune crosstalk in cardiac inflammation. Pro-inflammatory cytokines such as IL-1 act on central nervous system regions including the subfornical organ (SFO) and paraventricular nucleus (PVN), modulating autonomic outflow. Sympathetic activation promotes MAPK and NF-κB pathways, enhancing TNF-α and IL-1 production. Excessive inflammation leads to overproduction of iNOS/NO, which disrupts calcium homeostasis and suppresses cardiac contractility. Parasympathetic activity may exert anti-inflammatory effects, highlighting bidirectional neuro-immune interactions in cardiovascular pathology. |
Regulation of Repair and Pathological Fibrosis
During the intermediate phase, Smad3 activation promotes beneficial polarization, enhances phagocytic capacity (eg, via milk fat globule-EGF factor 8 protein (Mfge8)), and stimulates anti-inflammatory cytokine (IL-10, TGF-β1) secretion to curb excessive inflammation; indeed, Smad3 deficiency increases cardiomyocyte apoptosis.48 However, persistent transforming growth factor beta 1 (TGF-β1) signaling, via Smad2/3 activation, potently drives cardiac fibroblast-to-myofibroblast transition (expressing α-smooth muscle actin (α-SMA)) and stimulates synthesis and deposition of collagen types I and III, promoting fibrosis.49 Notably, macrophages (particularly M2) can themselves undergo TGF-β/Smad3-driven transdifferentiation into myofibroblasts (see Figure 5), directly contributing to excessive ECM accumulation.50,51 Thus, uncontrolled M2-driven repair can shift from adaptive healing to pathological fibrosis, severely compromising long-term cardiac function.
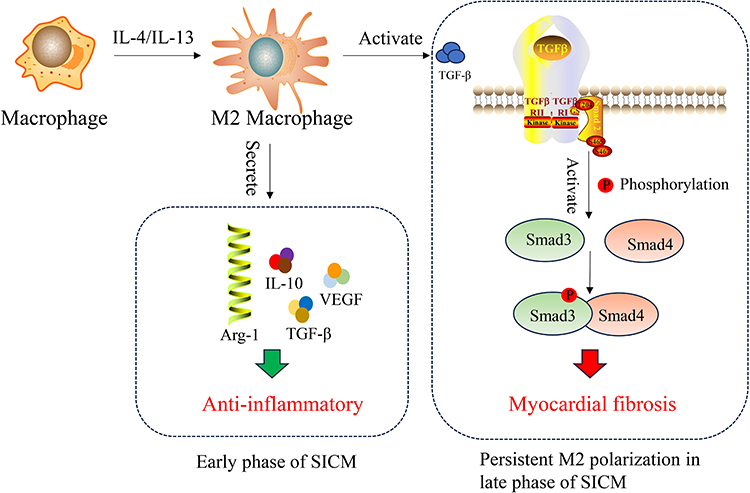 |
Figure 5 Protective and fibrotic roles of M2 macrophages in myocardial repair. In the early stage after injury, M2 macrophages exhibit anti-inflammatory properties and promote tissue repair. However, persistent M2 activation, driven by IL-4/IL-13, can lead to excessive collagen deposition (type I and III fibers), resulting in myocardial fibrosis and adverse structural remodeling. |
Metabolic Reprogramming: Bridging Immune Activation and Cardiac Dysfunction
Recent research highlights the tight coupling of macrophage immune function with their intrinsic metabolic state. In sepsis, activated M1 macrophages exhibit a pronounced increase in glycolysis, akin to the “Warburg effect”.52 This process, largely driven by hypoxia-inducible factor-1 alpha (HIF-1α), upregulates glucose transporter 1 (GLUT1) and key glycolytic enzymes to rapidly generate ATP and biosynthetic precursors supporting massive cytokine production.31,53 Critically, the glycolytic intermediate succinate accumulates. Oxidation of succinate drives mitochondrial ROS (mtROS) production, which stabilizes HIF-1α and activates the NLRP3 inflammasome, creating a positive feedback loop.54,55 Circulating succinate can also act directly on cardiomyocytes via its receptor G protein-coupled receptor 91 (GPR91), inducing ROS and inhibiting oxidative phosphorylation.56
M2 macrophages rely more on fatty acid oxidation (FAO) for energy. Activation of the peroxisome proliferator-activated receptor gamma (PPARγ) pathway promotes fatty acid uptake and mitochondrial β-oxidation, supporting the long-term survival and repair functions of M2 cells.57–59 However, excessive FAO can lead to lipid accumulation and excess acetyl-coenzyme A (acetyl-CoA), potentially activating the NLRP3 inflammasome under certain conditions, illustrating the complexity of metabolic regulation.57 Therefore, targeting macrophage metabolic reprogramming, eg, using glycolytic inhibitors (2-deoxyglucose (2-DG)) or succinate dehydrogenase (SDH) modulators, represents a promising therapeutic avenue.
Complex Intercellular Communication Network
Macrophages do not act in isolation but engage in complex crosstalk with other cardiac cells.
Synergy with Other Immune Cells: Microvesicles from pyroptotic macrophages can be engulfed by neutrophils, inducing mitochondrial dysfunction and mtROS burst in the latter, which activates GSDMD and promotes NET formation.44 NETs directly damage cardiomyocytes and endothelial cells and promote microthrombosis, contributing to organ dysfunction.60 During sepsis, cardiac endothelial cells, cardiomyocytes, and fibroblasts secrete chemokines like C-C motif chemokine ligand 2 (CCL2), which recruits CCR2-expressing monocytes from the circulation into the heart, where they differentiate into pro-inflammatory M1 macrophages, amplifying local injury.61,62 Combined CCR2/C-C chemokine receptor type 5 (CCR5) inhibition appears more effective than single targeting, suggesting potential for multi-target intervention in SICM.61 Furthermore, macrophage-expressed programmed death-ligand 1 (PD-L1) binding to programmed cell death protein 1 (PD-1) on T cells delivers inhibitory signals, causing T cell exhaustion and potentially enhancing regulatory T cell (Treg)-mediated immunosuppression, shaping an immune microenvironment unfavorable for myocardial repair.63–65 The cyclooxygenase-2 (COX2)/PGE2 pathway may contribute to this suppression by upregulating PD-L1 and promoting Treg function.63
Complement System Interaction: The complement fragment complement component 5a (C5a), binding to C5a receptor 1 (C5aR1) on macrophages, enhances their inflammatory response via G protein-coupled receptor (GPCR) signaling, MAPK/extracellular signal-regulated kinase (ERK), NF-κB, and phosphoinositide 3-kinase / protein kinase B (PI3K/Akt) pathways, driving M1 polarization, cytokine release, and ROS production. C5a is also a potent neutrophil chemoattractant, promoting NETosis. Additionally, C5a acting on cardiomyocyte C5a receptor (C5aR) can cause calcium overload and apoptosis. Thus, C5a exerts a dual “immune-cardiomyocyte” hit, amplifying inflammation, oxidative stress, apoptosis, and dysfunction in SICM.66 However, simply blocking C5aR1 may be detrimental in specific contexts (eg, transfusion-related acute lung injury (TRALI) models67), suggesting that direct C5 targeting might be a safer strategy.
Macrophage Migration Inhibitory Factor (MIF): MIF plays a complex role. It enhances immune recognition by upregulating TLR4 expression but exacerbates myocardial depression when overproduced.68 MIF promotes monocyte migration/infiltration via receptors like CCR2, C-X-C motif chemokine receptor 4 (CXCR4), and angiotensin II type 1 receptor (AT-1R), amplifies systemic and local inflammation (IL-6, monocyte chemoattractant protein-1 (MCP-1)), directly activates the NLRP3 inflammasome, modulates MMP activity, promotes ROS generation, and antagonizes glucocorticoid actions.61–63 MIF deficiency attenuates endoplasmic reticulum (ER) stress and calcium handling abnormalities, improving contractile function.68–70 MIF’s effects are cell source-dependent, with cardiac-derived MIF potentially being protective and immune-cell-derived MIF being predominantly pro-inflammatory.68 MIF also influences cell survival/apoptosis via NF-κB and c-Jun N-terminal kinase (JNK)/mitochondrial pathways.70–72 Thus, MIF is a multi-faceted player and a potential therapeutic target.
Novel Therapeutic Strategies Targeting Macrophages
Regarding the novel therapeutic strategies targeting macrophages in SICM, we have organized and summarized the relevant information in this section, and presented some treatment options along with their mechanism, evidence level, safety risks and other indicators in Table 1.
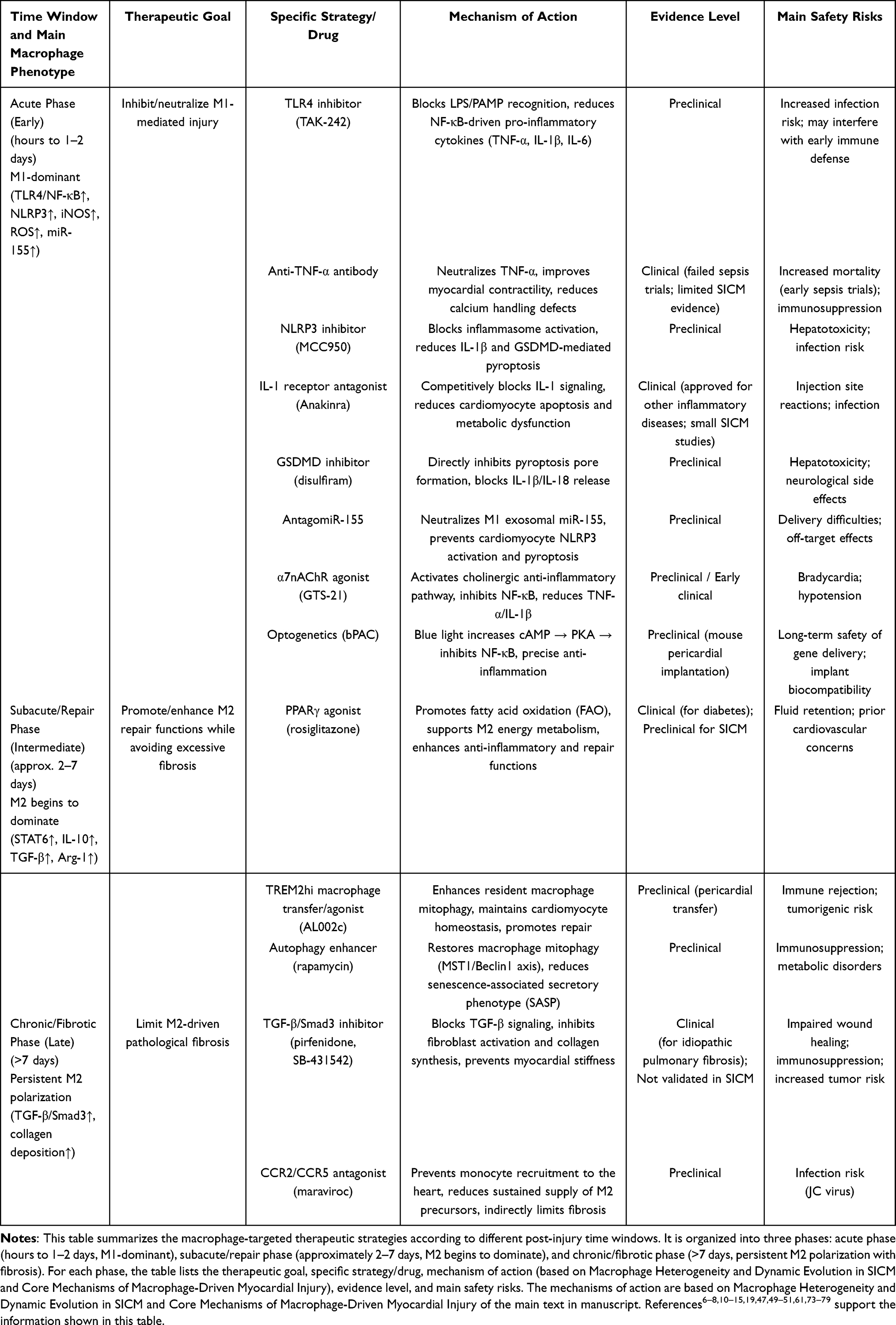 |
Table 1 Therapeutic Strategies Targeting Macrophages Across Different Time Windows of SICM |
Polarization
miR-133a Pathway
The miR-133a pathway exerts its protective effect by directly targeting and suppressing suppressor of cytokine signaling 3 (SOCS3), thereby blocking the activation of the JAK/STAT3 signaling cascade. Inhibition of this pathway reduces the polarization of pro-inflammatory M1 macrophages while promoting a shift toward the reparative M2 phenotype.80 The evidence supporting this pathway is the most complete among the five, including dual-luciferase reporter assays, macrophage-cardiomyocyte co-culture, a cecal ligation and puncture (CLP) rat model, and validation using a nanoparticle-based delivery system. Regarding safety, nanoparticle delivery systems carry a moderate risk of off-target effects, and long-term miR-133a overexpression may affect other SOCS family members, potentially disrupting immune homeostasis.
PARP1/NLRP3 Pathway
Activation of PARP1 promotes the assembly and activation of the NLRP3 inflammasome, which in turn induces M1 polarization of macrophages and the release of pro-inflammatory cytokines such as IL-1β. Inhibition of PARP1 (eg, by atractylenolide I) blocks NLRP3 inflammasome signaling and suppresses M1 polarization.81 However, the evidence supporting this pathway remains preliminary, as it is derived exclusively from LPS-stimulated RAW 264.7 macrophage and H9c2 cardiomyocyte co-culture systems, without validation in established in vivo sepsis models such as cecal ligation and puncture (CLP) or LPS-challenged rodents. Consequently, the safety profile of this approach remains unknown, although broad-spectrum PARP inhibitors are known to interfere with DNA repair and may carry potential genotoxicity with long-term use.
DANCR/IGF2BP2/HK2 Pathway
The long non-coding RNA DANCR interacts with the RNA-binding protein IGF2BP2 to stabilize HK2 (hexokinase 2) mRNA, thereby promoting glycolytic metabolic reprogramming in macrophages and providing energy and substrates for M1 polarization. Knockdown of DANCR disrupts the DANCR/IGF2BP2/HK2 axis and suppresses aerobic glycolysis required for M1 polarization.82 Mechanistically, this pathway is well characterized (including RIP and RNA stability assays) and has been validated in a CLP mouse model, but it lacks macrophage-specific knockout studies and has received limited citations to date. From a safety perspective, antisense oligonucleotides targeting lncRNAs pose moderate risks of liver accumulation and immunogenicity, and inhibiting HK2 may interfere with normal glucose metabolism in other cell types such as neurons and immune cells.
Piezo1/PI3K/AKT Pathway
Piezo1 is a mechanosensitive ion channel that, when overexpressed in septic cardiomyopathy, suppresses the activity of the PI3K/AKT signaling pathway. Knockdown or inhibition of Piezo1 restores PI3K/AKT signaling, thereby driving macrophage polarization toward the anti-inflammatory, reparative M2 phenotype while suppressing M1 polarization.83 Strong support for this pathway comes from myeloid-specific Piezo1 knockout mice and an LPS-induced model, although it lacks validation using the CLP model and direct molecular binding data. In terms of safety, because Piezo1 is widely expressed in vascular endothelium and erythrocytes, systemic inhibition may affect blood pressure regulation and red blood cell volume; however, myeloid-specific targeting as used in the knockout models could mitigate such risks.
STAT3/FoxO3a/Sirt1 Pathway
This pathway involves the transcription factors STAT3 and FoxO3a together with the deacetylase Sirt1. Under septic conditions, activation of the STAT3/FoxO3a axis promotes M1 polarization of macrophages and suppresses Sirt1 expression. Therapeutic intervention (eg, with ginsenoside Rc) inhibits STAT3/FoxO3a signaling while upregulating Sirt1, which not only suppresses M1 polarization but also directly improves mitochondrial function in cardiomyocytes.84 The evidence is based on network pharmacology, in vitro reverse validation (using a STAT3 agonist), and a CLP model, but it lacks independent genetic confirmation such as macrophage-specific knockout. Regarding safety, ginsenoside Rc is a natural product with relatively low toxicity, although long-term high-dose use may cause estrogen-like effects or drug interactions, and excessive Sirt1 activation has been linked to potential tumor promotion.
Enhancing the Function of Protective Macrophage Subsets
Activation of TREM2 enhances mitophagy and maintains cardiomyocyte homeostasis, improving cardiac function in septic mice;73 direct delivery of TREM2hi macrophages to the pericardial space also reduces inflammation and increases survival, representing a promising cell-based therapy. Currently, these strategies are supported only by preclinical evidence in septic mouse models, and their safety concerns include potential immune rejection and tumorigenic risk associated with cell transplantation, as well as possible off-target effects of TREM2 agonists that remain to be evaluated.
Inflammatory Signaling Pathways
The TLR4 inhibitor TAK-242 blocks LPS binding to TLR4, thereby inhibiting the MyD88/NF-κB axis and reducing production of pro-inflammatory cytokines such as TNF-α and IL-1β;74 however, evidence is limited to preclinical models, and broad suppression of innate immunity may increase infection risk and interfere with early host defense.
The anti-TNF-α antibody infliximab neutralizes TNF-α, improving cardiomyocyte contractility and calcium handling;75 although it has reached clinical use for autoimmune diseases, sepsis trials have shown no survival benefit and even potential harm, with safety concerns including increased mortality, immunosuppression, and tuberculosis reactivation.
Inflammasome Activation
The NLRP3 inhibitor MCC950 reduces caspase-1-mediated pyroptosis and production of IL-1β and IL-18;76 available data derive from preclinical inflammatory models, and safety concerns include hepatotoxicity (which led to termination of clinical trials for other indications) and potentially increased susceptibility to bacterial infection.
The GSDMD inhibitor disulfiram blocks gasdermin D pore formation, preventing pyroptosis and release of IL-1β/IL-18 from macrophages;77 this strategy is still preclinical, and its safety is challenged by hepatotoxicity, neurological side effects (eg, peripheral neuropathy), and drug–drug interactions.
Oxidative Stress Injury
AntagomiR-155 neutralizes miR-155 derived from M1 macrophage exosomes, thereby preventing cardiomyocyte NLRP3 inflammasome activation and subsequent pyroptosis;6 this approach remains at the preclinical stage in cardiovascular disease models, with safety concerns such as difficulties in oligonucleotide delivery, off-target effects, and possible hepatotoxicity.
M-IL-exo (IL-1β-stimulated macrophage-derived exosomes) carry elevated miR-146a, which suppresses MAPK4 and leads to excessive mitochondrial fission, ROS burst, and cardiomyocyte apoptosis;32 although mechanistically promising, this strategy is still preclinical, and its safety risks include heterogeneity of exosome preparations, potential pro-tumorigenic effects of miR-146a, and the lack of standardized production protocols.
Suppression of Mitophagy
Autophagy enhancers (eg, rapamycin and quercetin) have been shown to restore autophagic flux by suppressing MST1-mediated Beclin1 inhibition, leading to improved mitochondrial function, reduced ROS levels, and delayed macrophage senescence. Specifically, rapamycin was tested in an LPS-induced septic rat model,78 while quercetin was assessed using clinical serum samples from 20 SIC patients compared with 20 healthy controls, together with a CLP-induced septic rat model.85
Direct Cardiomyocyte Injury
IL-1 receptor antagonist (Anakinra) competitively blocks IL-1 signaling, reducing cardiomyocyte apoptosis, calcium overload, and metabolic dysfunction induced by macrophage-derived IL-1β.6,7 Among septic patients with concurrent liver dysfunction and disseminated intravascular coagulation (DIC) (a pattern indicative of macrophage activation syndrome), the IL-1 receptor antagonist Anakinra improved 28-day survival, but showed no significant effect in other septic patients.79
Indirect Regulatory Networks
The α7nAChR agonist (GTS-21) activates the cholinergic anti-inflammatory pathway, thereby inhibiting NF-κB in macrophages and reducing TNF-α/IL-1β release;47 accordingly, it has demonstrated anti-inflammatory effects in preclinical and early clinical studies using sepsis and inflammation models.86,87
MMP-9 inhibitors (eg, doxycycline, SB-3CT) have been shown to prevent degradation of tight junction proteins (eg, ZO-1) and basement membrane components, thereby preserving microvascular barrier integrity and reducing myocardial edema in myocarditis and aneurysm models.39 However, evidence from SICM models is lacking.
Clopidogrel51 and pioglitazone49,50 have clinical evidence supporting their anti-fibrotic effects; however, these data are derived from fibrotic models, and only mechanistic inferences are available for SICM.
Metabolic Reprogramming
The PPARγ agonist rosiglitazone promotes fatty acid oxidation (FAO) and supports M2 polarization, thereby enhancing anti-inflammatory and repair functions;12 although this mechanism is well established in metabolic research, direct evidence in SICM remains preclinical, and safety concerns include fluid retention and prior controversies over cardiovascular risks.
The SGLT2 inhibitor downregulates PFKFB3, suppresses glycolysis, and shifts macrophage polarization away from the pro-inflammatory M1 phenotype;53 this class of drugs has already entered clinical use for diabetes and heart failure, but their application in SICM is still mechanistic, with known safety issues such as ketoacidosis and genitourinary infections.
The glycolysis inhibitor 2-DG inhibits the Warburg effect in activated M1 macrophages, reducing HIF-1α stabilization and pro-inflammatory cytokine production;53 evidence is limited to preclinical models, and its safety is challenged by hyperglycemia, neurotoxicity, and QT interval prolongation.
The SDH modulator dimethyl malonate (DMM) blocks succinate oxidation, thereby reducing mitochondrial ROS and NLRP3 inflammasome activation in macrophages;54,55 this strategy is at a preclinical stage, and potential risks include interference with the tricarboxylic acid cycle and general cellular energy metabolism.
Intercellular Communication Network
The CCR2/CCR5 antagonist maraviroc blocks monocyte recruitment to the heart by inhibiting CCL2-CCR2/CCR5 signaling, thereby reducing local M1 macrophage accumulation;61 this approach has been tested in preclinical models of pulmonary hypertension and myocardial infarction, but direct evidence in SICM is lacking, and safety concerns include increased risk of infection (eg, JC virus reactivation) and potential hepatotoxicity.
By dismantling the DNA scaffold of both NETs and METs, DNase I not only attenuates direct cytotoxicity but also disrupts the platelet-activating and inflammasome-amplifying functions of these extracellular traps;44,62 available data are preclinical, and safety risks include possible impairment of local NET-mediated antimicrobial defense and bleeding tendency.88
The C5aR1 antagonist avacopan blocks complement C5a receptor 1 on macrophages, reducing M1 polarization, cytokine release, and ROS production;44,60 this drug has reached clinical approval for other immune-mediated diseases, but its efficacy in SICM has not been validated, and safety concerns include an increased risk of encapsulated bacterial infections.
MIF inhibitor ISO-1 inhibits macrophage migration inhibitory factor (MIF), thereby reducing TLR4 expression, NLRP3 inflammasome activation, and myocardial depression;68–72,89 evidence remains preclinical, and potential risks include disruption of normal immune regulation and altered glucocorticoid sensitivity.
Limitations and Perspectives
While the above sections delineate plausible mechanistic pathways linking macrophage dysfunction to myocardial injury, it is imperative to critically assess the translational relevance of the cited evidence. A substantial portion of the mechanistic insights—particularly regarding ferroptosis,30,44 exosomal miR-146a,32 and metabolic reprogramming31,53–55—derives from in vitro studies or animal models of non-septic conditions such as myocardial infarction, atherosclerosis, or osteoarthritis. Although these models provide valuable mechanistic hypotheses, they may not fully recapitulate the unique, poly-microbial, and dynamically evolving inflammatory milieu of human sepsis-induced cardiomyopathy (SICM). Direct evidence from SICM-specific models, especially large animal or human tissue studies, remains sparse. For instance, the role of succinate accumulation as a key inflammatory driver has been elegantly demonstrated in LPS-stimulated macrophages,53,55 but circulating succinate levels and their direct cardiodepressant effects in septic patients have yet to be validated. Furthermore, macrophage-targeted interventions showing promise in murine endotoxemia models (eg, optogenetics19) face significant hurdles in clinical translation, including species differences in immune signaling and the complexity of human sepsis with variable co-morbidities. Therefore, while these mechanistic pathways represent promising therapeutic frontiers, their direct causative role in human SICM requires confirmation through dedicated translational studies, including analysis of patient myocardial samples and longitudinal biomarker validation.
Deeper insights into the functions of specific macrophage subsets, particularly resident populations like TREM2hi macrophages, are revealing novel therapeutic targets. Future research should leverage single-cell multi-omics and spatial transcriptomics to map the dynamic landscape of macrophage subsets throughout SICM progression. However, several critical limitations must be explicitly acknowledged.
First, the overwhelming majority of mechanistic evidence—including polarization dynamics, inflammasome activation, and metabolic reprogramming—derives from murine models (eg, cecal ligation and puncture or LPS injection) or in vitro systems using immortalized macrophage cell lines. Human data are strikingly sparse: only a handful of studies have characterized cardiac macrophages from septic patients, and no prospective clinical trial has specifically targeted macrophages in SICM. Second, significant species differences exist in macrophage subset markers (eg, TREM2, CCR2), Toll-like receptor signaling thresholds, and inflammasome regulation, which profoundly impact translational predictability.90 Third, current experimental interventions (eg, optogenetics, TREM2 agonists, GSDMD inhibitors) face formidable clinical hurdles, including delivery specificity to cardiac macrophages, off-target immunosuppression (eg, increased secondary infection risk).91 There is no clear solution for patients with pre-existing immunosuppression.92
Finally, most studies focus on early hyperinflammation, whereas the prolonged immunosuppressive phase—where macrophages also contribute to chronic cardiac dysfunction and fibrosis—remains understudied. Therefore, while macrophage-targeted strategies hold promise, future efforts must prioritize (i) validation in human samples (post-mortem cardiac tissue, circulating exosomes/monocytes from septic patients), (ii) large animal models with clinically relevant endpoints, and (iii) safety-focused trial designs addressing infection risk. Without such rigorous translational scrutiny, the leap from bench to bedside for macrophage-directed therapies in SICM will remain perilously wide.
Developing tools for spatiotemporally precise targeting of specific subsets or functions (eg, inhibiting pro-inflammatory output without compromising phagocytosis) is crucial for efficacy and safety. Identifying reliable biomarkers reflecting macrophage activation status (eg, specific EV miRNAs, metabolites like succinate) will aid in early diagnosis, risk stratification, and treatment monitoring. Ultimately, integrating macrophage-targeted strategies with antimicrobial therapy, hemodynamic support, and direct cardiomyocyte protection into a multi-targeted, personalized treatment framework holds the key to overcoming the challenge of SICM.
Abbreviations
SICM, Sepsis-Induced Cardiomyopathy; PAMPs, Pathogen-Associated Molecular Patterns; DAMPs, Damage-Associated Molecular Patterns; LPS, Lipopolysaccharide; HMGB1, High Mobility Group Box 1; CCR2, C-C Chemokine Receptor Type 2; IFN-γ, Interferon Gamma; TNF-α, Tumor Necrosis Factor Alpha; IL-1β, Interleukin-1 Beta; IL-6, Interleukin-6; iNOS, Inducible Nitric Oxide Synthase; NO, Nitric Oxide; ROS, Reactive Oxygen Species; VEGF, Vascular Endothelial Growth Factor; Arg-1, Arginase-1; TGF-β, Transforming Growth Factor Beta; TLR4, Toll-Like Receptor 4; NF-κB, Nuclear Factor Kappa B; NLRP3, NLR Family Pyrin Domain Containing 3; MyD88, Myeloid Differentiation Primary Response 88; bPAC, Blue Light-Activated Adenylyl Cyclase; cAMP, Cyclic Adenosine Monophosphate; PKA, Protein Kinase A; NAIP, NLR Family Apoptosis Inhibitory Protein; NLRC4, NLR Family CARD Domain Containing 4; PARP1, Poly (ADP-Ribose) Polymerase 1; AIM2, Absent in Melanoma 2; dsDNA, Double-Stranded DNA; NETs, Neutrophil Extracellular Traps; GSDMD, Gasdermin D; CARD8, Caspase Recruitment Domain Family Member 8; circRNA, Circular RNA; miRNA, MicroRNA; NLRP6, NLR Family Pyrin Domain Containing 6; ILC2, Type 2 Innate Lymphoid Cells; Th2, T Helper 2 Cells; NADPH, Nicotinamide Adenine Dinucleotide Phosphate; NOX2, NADPH Oxidase 2; RNS, Reactive Nitrogen Species; EVs, Extracellular Vesicles; sEVs, Small Extracellular Vesicles; M-IL-exo, Interleukin-1β-Stimulated Macrophage-Derived Exosomes; MAPK, Mitogen-Activated Protein Kinase; Drp1, Dynamin-Related Protein 1; TfR, Transferrin Receptor; MDA, Malondialdehyde; DHE, Dihydroethidium; GSH, Glutathione; GPX4, Glutathione Peroxidase 4; LVEF, Left Ventricular Ejection Fraction; LVFS, Left Ventricular Fractional Shortening; Mfn2, Mitofusin 2; OPA1, Optic Atrophy 1; ox-LDL, Oxidized Low-Density Lipoprotein; LC3, Microtubule-Associated Protein 1A/1B-Light Chain 3; SA-β-gal, Senescence-Associated Beta-Galactosidase; MST1, Macrophage Stimulating 1; SASP, Senescence-Associated Secretory Phenotype; FasL, Fas Ligand; NMDAR, N-Methyl-D-Aspartate Receptor; mPTP, Mitochondrial Permeability Transition Pore; MMP-9, Matrix Metalloproteinase-9; ZO-1, Zonula Occludens-1; SFO, Subfornical Organ; PVN, Paraventricular Nucleus; SNS, Sympathetic Nervous System; NE, Norepinephrine; ADRB2, Beta-2 Adrenergic Receptor; CAIP, Cholinergic Anti-Inflammatory Pathway; α7nAChR, Alpha-7 Nicotinic Acetylcholine Receptor; PGE2, Prostaglandin E2; FAO, Fatty Acid Oxidation; PPARγ, Peroxisome Proliferator-Activated Receptor Gamma; HIF-1α, Hypoxia-Inducible Factor-1 Alpha; GLUT1, Glucose Transporter 1; GPR91, G Protein-Coupled Receptor 91; CCL2, C-C Motif Chemokine Ligand 2; PD-L1, Programmed Death-Ligand 1; PD-1, Programmed Cell Death Protein 1; Treg, Regulatory T Cell; COX2, Cyclooxygenase-2; C5a, Complement Component 5a; C5aR1, C5a Receptor 1; GPCR, G Protein-Coupled Receptor; PI3K/Akt, Phosphoinositide 3-Kinase / Protein Kinase B; TRALI, Transfusion-Related Acute Lung Injury; MIF, Macrophage Migration Inhibitory Factor; AT-1R, Angiotensin II Type 1 Receptor; ER, Endoplasmic Reticulum; JNK, c-Jun N-Terminal Kinase; SGLT2is, Sodium-Glucose Cotransporter-2 Inhibitors; PFKFB3, 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3; SDH, Succinate Dehydrogenase; TREM2, Triggering Receptor Expressed on Myeloid Cells 2; METs, Macrophage Extracellular Traps; 2-DG, 2-Deoxyglucose; SOCS3, Suppressor of cytokine signaling 3; DIC, Disseminated intravascular coagulation.
Data Sharing Statement
Data sharing is not applicable to this article as no data were created or analysed in this study.
Author Contributions
Jun Wang; Writing - original draft, Writing - review & editing
Jingyi Zhang; Supervision, Project administration, Formal analysis, Writing - review & editing
Nianfang Luo; Methodology, Software, Writing - review & editing
Ziran Zhou; Methodology, Writing – review & editing
Yue Zhang; Data curation, Writing - review & editing
Kangyan Guo; Data curation, Writing - review & editing
Ruiting Li; Conceptualization, Supervision, Visualization, Writing - review & editing
Chang Li; Conceptualization, Supervision, Funding acquisition, Project administration, Formal analysis, Validation, Writing – original draft, Writing – review & editing
All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and to be accountable for all aspects of the work.
Funding
The study was supported by the National Natural Science Foundation of China (grant number 82472224).
Disclosure
The authors declare that they have no conflict of interests.
References
1. Aissaoui N, Boissier F, Chew M, Singer M, Vignon P. Sepsis-induced cardiomyopathy. Eur Heart J. 2025;46(34):3339–18. doi:10.1093/eurheartj/ehaf340
2. Epelman S, Lavine KJ, Beaudin AE, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40(1):91–104. doi:10.1016/j.immuni.2013.11.019
3. Epelman S, Liu PP, Mann DL. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol. 2015;15(2):117–129. doi:10.1038/nri3800
4. Aurora AB, Porrello ER, Tan W, et al. Macrophages are required for neonatal heart regeneration. J Clin Invest. 2014;124(3):1382–1392. doi:10.1172/JCI72181
5. Hulsmans M, Clauss S, Xiao L, et al. Macrophages facilitate electrical conduction in the heart. Cell. 2017;169(3):510–522.e20. doi:10.1016/j.cell.2017.03.050
6. Xu Y, Zhang C, Cai D, Zhu R, Cao Y. Exosomal miR-155-5p drives widespread macrophage M1 polarization in hypervirulent Klebsiella pneumoniae-induced acute lung injury via the MSK1/p38-MAPK axis. Cell Mol Biol Lett. 2023;28(1):92. doi:10.1186/s11658-023-00505-1
7. Kawaguchi M, Takahashi M, Hata T, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123(6):594–604. doi:10.1161/CIRCULATIONAHA.110.982777
8. Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. doi:10.1016/j.immuni.2014.06.008
9. Koentges C, Cimolai MC, Pfeil K, et al. Impaired SIRT3 activity mediates cardiac dysfunction in endotoxemia by calpain-dependent disruption of ATP synthesis. J Mol Cell Cardiol. 2019;133:138–147. doi:10.1016/j.yjmcc.2019.06.008
10. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44(3):450–462. doi:10.1016/j.immuni.2016.02.015
11. Jenkins SJ, Ruckerl D, Thomas GD, et al. IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J Exp Med. 2013;210(11):2477–2491. doi:10.1084/jem.20121999
12. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447(7148):1116–1120. doi:10.1038/nature05894
13. Nahrendorf M, Swirski FK, Aikawa E, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204(12):3037–3047. doi:10.1084/jem.20070885
14. Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 2004;4(8):583–594. doi:10.1038/nri1412
15. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014;11(5):255–265. doi:10.1038/nrcardio.2014.28
16. Anderson CF, Mosser DM. A novel phenotype for an activated macrophage: the type 2 activated macrophage. J Leukoc Biol. 2002;72(1):101–106. doi:10.1189/jlb.72.1.101
17. Lin YH, Wang YH, Peng YJ, et al. Interleukin 26 skews macrophage polarization towards M1 phenotype by activating cJUN and the NF-κB pathway. Cells. 2020;9(4):938. doi:10.3390/cells9040938
18. Huang X, Yao Y, Hou X, et al. Macrophage SCAP contributes to metaflammation and lean NAFLD by activating STING-NF-κB signaling pathway. Cell Mol Gastroenterol Hepatol. 2022;14(1):1–26. doi:10.1016/j.jcmgh.2022.03.006
19. Xia G, Shi H, Su Y, et al. Photoactivated adenylyl cyclases attenuate sepsis-induced cardiomyopathy by suppressing macrophage-mediated inflammation. Front Immunol. 2022;13:1008702. doi:10.3389/fimmu.2022.1008702
20. Zhang J, Liu X, Wan C, et al. NLRP3 inflammasome mediates M1 macrophage polarization and IL-1β production in inflammatory root resorption. J Clin Periodontol. 2020;47(4):451–460. doi:10.1111/jcpe.13258
21. Kay C, Wang R, Kirkby M, et al. Molecular mechanisms activating the NAIP-NLRC4 inflammasome: implications in infectious disease, autoinflammation, and cancer. Immunol Rev. 2020;297(1):67–82. doi:10.1111/imr.12906
22. Amaral MP, Cardoso FD, de Farias IS, et al. NAIP/NLRC4 inflammasome participates in macrophage responses to Trypanosoma cruzi by a mechanism that relies on cathepsin-dependent caspase-1 cleavage. Front Immunol. 2023;14:1282856. doi:10.3389/fimmu.2023.1282856
23. Vance RE. The NAIP/NLRC4 inflammasomes. Curr Opin Immunol. 2015;32:84–89. doi:10.1016/j.coi.2015.01.010
24. Fidler TP, Xue C, Yalcinkaya M, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. 2021;592(7853):296–301. doi:10.1038/s41586-021-03341-5
25. Liu C, Zhou Y, Tu Q, et al. Alpha-linolenic acid pretreatment alleviates NETs-induced alveolar macrophage pyroptosis by inhibiting pyrin inflammasome activation in a mouse model of sepsis-induced ALI/ARDS. Front Immunol. 2023;14:1146612. doi:10.3389/fimmu.2023.1146612
26. Li H, Li Y, Song C, et al. Neutrophil extracellular traps augmented alveolar macrophage pyroptosis via AIM2 inflammasome activation in LPS-Induced ALI/ARDS. J Inflamm Res. 2021;14:4839–4858. doi:10.2147/JIR.S321513
27. Sun L, Ma W, Gao W, et al. Propofol directly induces caspase-1-dependent macrophage pyroptosis through the NLRP3-ASC inflammasome. Cell Death Dis. 2019;10(8):542. doi:10.1038/s41419-019-1761-4
28. Chen S, Wen L, Wu Y, et al. Circular RNA circ-CARD8 regulates alveolar macrophage pyroptosis through the miR-580-3p/CARD8 pathway in acute lung injury. PLoS One. 2024;19(12):e0314936. doi:10.1371/journal.pone.0314936
29. Chenuet P, Marquant Q, Fauconnier L, et al. NLRP6 negatively regulates type 2 immune responses in mice. Allergy. 2022;77(11):3320–3336. doi:10.1111/all.15388
30. Huo S, Wang M, Du M, et al. Macrophage-derived S100A9 promotes diabetic cardiomyopathy by disturbing mitochondrial quality control via STAT3 activation. Int J Biol Sci. 2025;21(7):3061–3080. doi:10.7150/ijbs.111128
31. Liu B, Xian Y, Chen X, et al. Inflammatory fibroblast-like synoviocyte-derived exosomes aggravate osteoarthritis via enhancing macrophage glycolysis. Adv Sci. 2024;11(14):e2307338. doi:10.1002/advs.202307338
32. Ma C, Yang Z, Wang J, et al. Interleukin-1β-stimulated macrophage-derived exosomes improve myocardial injury in sepsis via regulation of mitochondrial homeostasis: experimental research. Int J Surg. 2025;111(1):283–301. doi:10.1097/JS9.0000000000001915
33. Guo D, Yang X, Yu R, et al. Macrophage-derived extracellular vesicles represent a promising endogenous iron-chelating therapy for iron overload and cardiac injury in myocardial infarction. J Nanobiotechnology. 2024;22(1):527. doi:10.1186/s12951-024-02800-1
34. Cao H, Jia Q, Yan L, et al. Quercetin suppresses the progression of atherosclerosis by regulating MST1-Mediated autophagy in ox-LDL-Induced RAW264.7 macrophage foam cells. Int J Mol Sci. 2019;20(23):6093. doi:10.3390/ijms20236093
35. Pan JJ, Shi HM, Luo XP, et al. Recombinant TFPI-2 enhances macrophage apoptosis through upregulation of Fas/FasL. Eur J Pharmacol. 2011;654(2):135–141. doi:10.1016/j.ejphar.2010.12.015
36. Hu F, Zhang S, Chen X, et al. MiR-219a-2 relieves myocardial ischemia-reperfusion injury by reducing calcium overload and cell apoptosis through HIF1α/ NMDAR pathway. Exp Cell Res. 2020;395(1):112172. doi:10.1016/j.yexcr.2020.112172
37. Ramachandra CJA, Hernandez-Resendiz S, Crespo-Avilan GE, et al. Hausenloy DJ. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine. 2020;57:102884. doi:10.1016/j.ebiom.2020.102884
38. Lei X, Tan G, Wang Y, et al. Mitochondrial calcium nanoregulators reverse the macrophage proinflammatory phenotype through restoring mitochondrial calcium homeostasis for the treatment of osteoarthritis. Int J Nanomed. 2023;18:1469–1489. doi:10.2147/IJN.S402170
39. Bendeck MP. Macrophage matrix metalloproteinase-9 regulates angiogenesis in ischemic muscle. Circ Res. 2004;94(2):138–139. doi:10.1161/01.RES.0000117525.23089.1A
40. Deng J, Qu Y, Jiang Y, et al. Dendritic cells derived MMP9 fuels sepsis-induced cardiac dysfunction through JNK/AP1 driven pro-inflammatory cytokine and nitric oxide storm. Inflammation. 2026. doi:10.1007/s10753-025-02374-6
41. Chen Z, Gao F. The dual role of macrophage extracellular traps in host defense and disease: mechanisms and therapeutic implications. Biomolecules. 2025;15(9):1220. doi:10.3390/biom15091220
42. Baz AA, Hao H, Lan S, et al. Emerging insights into macrophage extracellular traps in bacterial infections. FASEB J. 2024;38(13):e23767. doi:10.1096/fj.202400739R
43. Zhang J, Huang J, Qi T, et al. SHP2 protects endothelial cell barrier through suppressing VE-cadherin internalization regulated by MET-ARF1. FASEB J. 2019;33(1):1124–1137. doi:10.1096/fj.201800284R
44. Kuang L, Wu Y, Shu J, Yang J, Zhou H, Huang X. Pyroptotic macrophage-derived microvesicles accelerate formation of neutrophil extracellular traps via GSDMD-N-expressing mitochondrial transfer during sepsis. Int J Biol Sci. 2024;20(2):733–750. doi:10.7150/ijbs.87646
45. Wang J, Li R, Peng Z, Hu B, Rao X, Li J. HMGB1 participates in LPS‑induced acute lung injury by activating the AIM2 inflammasome in macrophages and inducing polarization of M1 macrophages via TLR2, TLR4, and RAGE/NF‑κB signaling pathways. Int J Mol Med. 2020;45(1):61–80. doi:10.3892/ijmm.2019.4402
46. Chen L, Zhao Y, Lai D, et al. Neutrophil extracellular traps promote macrophage pyroptosis in sepsis. Cell Death Dis. 2018;9(6):597. doi:10.1038/s41419-018-0538-5
47. Fujiu K, Manabe I. Nerve-macrophage interactions in cardiovascular disease. Int Immunol. 2022;34(2):81–95. doi:10.1093/intimm/dxab036
48. Chen B, Huang S, Su Y, et al. Macrophage Smad3 protects the infarcted heart, stimulating phagocytosis and regulating inflammation. Circ Res. 2019;125(1):55–70. doi:10.1161/CIRCRESAHA.119.315069
49. Jia Y, Qin Y, Yuan FL, et al. Macrophage-to-Myofibroblast transition contributes to cutaneous scarring formation through the TGF-β/Smad3 signaling pathways. Cell Biol Int. 2025;49(5):494–507. doi:10.1002/cbin.70002
50. Wang S, Meng XM, Ng YY, et al. TGF-β/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget. 2016;7(8):8809–8822. doi:10.18632/oncotarget.6604
51. Chen J, Tang Y, Zhong Y, et al. P2Y12 inhibitor clopidogrel inhibits renal fibrosis by blocking macrophage-to-myofibroblast transition. Mol Ther. 2022;30(9):3017–3033. doi:10.1016/j.ymthe.2022.06.019
52. Yang J, Li S, Li Z, et al. Targeting YAP1-regulated glycolysis in fibroblast-like synoviocytes impairs macrophage infiltration to ameliorate diabetic osteoarthritis progression. Adv Sci. 2024;11(5):e2304617. doi:10.1002/advs.202304617
53. Lin XF, Cui XN, Yang J, et al. SGLT2 inhibitors ameliorate NAFLD in mice via downregulating PFKFB3, suppressing glycolysis and modulating macrophage polarization. Acta Pharmacol Sin. 2024;45(12):2579–2597. doi:10.1038/s41401-024-01389-3
54. Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. doi:10.1038/nature11986
55. Mills EL, Kelly B, Logan A, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 2016;167(2):457–470.e13. doi:10.1016/j.cell.2016.08.064
56. Fu J, Han Z, Wu Z, et al. GABA regulates IL-1β production in macrophages. Cell Rep. 2022;41(10):111770. doi:10.1016/j.celrep.2022.111770
57. Hou Y, Wei D, Zhang Z, et al. FABP5 controls macrophage alternative activation and allergic asthma by selectively programming long-chain unsaturated fatty acid metabolism. Cell Rep. 2022;41(7):111668. doi:10.1016/j.celrep.2022.111668
58. Xiao S, Qi M, Zhou Q, et al. Macrophage fatty acid oxidation in atherosclerosis. Biomed Pharmacother. 2024;170:116092. doi:10.1016/j.biopha.2023.116092
59. Liu S, Zhang H, Li Y, et al. S100A4 enhances protumor macrophage polarization by control of PPAR-γ-dependent induction of fatty acid oxidation. J Immunother Cancer. 2021;9(6):e002548. doi:10.1136/jitc-2021-002548
60. Mihlan M, Wissmann S, Gavrilov A, et al. Neutrophil trapping and nexocytosis, mast cell-mediated processes for inflammatory signal relay. Cell. 2024;187(19):5316–5335.e28. doi:10.1016/j.cell.2024.07.014
61. Abid S, Marcos E, Parpaleix A, et al. CCR2/CCR5-mediated macrophage-smooth muscle cell crosstalk in pulmonary hypertension. Eur Respir J. 2019;54(4):1802308. doi:10.1183/13993003.02308-2018
62. Pozzi S, Satchi-Fainaro R. The role of CCL2/CCR2 axis in cancer and inflammation: the next frontier in nanomedicine. Adv Drug Deliv Rev. 2024;209:115318. doi:10.1016/j.addr.2024.115318
63. Kumar S, Tailor D, Dheeraj A, et al. Uncovering therapeutic targets for macrophage-mediated T cell suppression and PD-L1 therapy sensitization. Cell Rep Med. 2024;5(9):101698. doi:10.1016/j.xcrm.2024.101698
64. Pu Y, Ji Q. Tumor-Associated macrophages regulate PD-1/PD-L1 Immunosuppression. Front Immunol. 2022;13:874589. doi:10.3389/fimmu.2022.874589
65. Ortega MA, Boaru DL, De Leon-Oliva D, et al. PD-1/PD-L1 axis: implications in immune regulation, cancer progression, and translational applications. J Mol Med. 2024;102(8):987–1000. doi:10.1007/s00109-024-02463-3
66. Jiang K, Lu S, Li D, et al. Blockade of C5aR1 alleviates liver inflammation and fibrosis in a mouse model of NASH by regulating TLR4 signaling and macrophage polarization. J Gastroenterol. 2023;58(9):894–907. doi:10.1007/s00535-023-02002-w
67. Van der Velden S, Van Osch TLJ, Seghier A, et al. Complement activation drives antibody-mediated transfusion-related acute lung injury via macrophage trafficking and formation of NETs. Blood. 2024;143(1):79–91. doi:10.1182/blood.2023020484
68. Zhao L, Du GL, Ruze A, et al. Novel function of macrophage migration inhibitory factor in regulating post-infarct inflammation and the therapeutic significance. J Adv Res. 2026;80:905–923. doi:10.1016/j.jare.2025.05.030
69. Xu X, Ren J. Macrophage migration inhibitory factor (MIF) knockout preserves cardiac homeostasis through alleviating Akt-mediated myocardial autophagy suppression in high-fat diet-induced obesity. Int J Obes. 2015;39(3):387–396. doi:10.1038/ijo.2014.174
70. Zhao L, Zhao BH, Ruze A, et al. Distinct roles of MIF in the pathogenesis of ischemic heart disease. Cytokine Growth Factor Rev. 2024;80:121–137. doi:10.1016/j.cytogfr.2024.10.005
71. Rassaf T, Weber C, Bernhagen J. Macrophage migration inhibitory factor in myocardial ischaemia/reperfusion injury. Cardiovasc Res. 2014;102(2):321–328. doi:10.1093/cvr/cvu071
72. Chagnon F, Metz CN, Bucala R, et al. Endotoxin-induced myocardial dysfunction: effects of macrophage migration inhibitory factor neutralization. Circ Res. 2005;96(10):1095–1102. doi:10.1161/01.RES.0000168327.22888.4d
73. Zhang K, Wang Y, Chen S, et al. TREM2hi resident macrophages protect the septic heart by maintaining cardiomyocyte homeostasis. Nat Metab. 2023;5(1):129–146. doi:10.1038/s42255-022-00715-5
74. Zhao X, Zhang J, Xu F, Shang L, Liu Q, Shen C. TAK-242 alleviates diabetic cardiomyopathy via inhibiting pyroptosis and TLR4/CaMKII/NLRP3 pathway. Open Life Sci. 2024;19(1):20220957. doi:10.1515/biol-2022-0957
75. Ozer EK, Goktas MT, Kilinc I, et al. Infliximab alleviates the mortality, mesenteric hypoperfusion, aortic dysfunction, and multiple organ damage in septic rats. Can J Physiol Pharmacol. 2017;95(7):866–872. doi:10.1139/cjpp-2016-0628
76. Li S, Guo Z, Zhang ZY. Protective effects of NLRP3 inhibitor MCC950 on sepsis-induced myocardial dysfunction. J Biol Regul Homeost Agents. 2021;35(1):141–150. doi:10.23812/20-662-A
77. Yang L, Lyu L, Ming J, Che C. Effect of co-treatment with disulfiram and resatorvid on the pyroptosis of monocytes in sepsis. Biochim Biophys Acta Mol Basis Dis. 2025;1871(3):167704. doi:10.1016/j.bbadis.2025.167704
78. Walley KR. Sepsis-Induced myocardial dysfunction and mammalian target of rapamycin signalling pathways. Can J Cardiol. 2019;35(7):809–812. doi:10.1016/j.cjca.2019.04.003
79. Shakoory B, Carcillo JA, Chatham WW, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial. Crit Care Med. 2016;44(2):275–281. doi:10.1097/CCM.0000000000001402
80. Zheng C, Gao Y, Wei X, Huang X, Wu D, Tian J. MiR-133a encapsulated in hyaluronic acid nanoparticles alleviates sepsis-induced cardiomyopathy by decreasing M1 macrophage polarization via the SOCS3/JAK/STAT3 pathway. Arch Biochem Biophys. 2026;775:110673. doi:10.1016/j.abb.2025.110673
81. Wang D, Lin Z, Zhou Y, et al. Atractylenolide I ameliorates sepsis-induced cardiomyocyte injury by inhibiting macrophage polarization through the modulation of the PARP1/NLRP3 signaling pathway. Tissue Cell. 2024;89:102424. doi:10.1016/j.tice.2024.102424
82. Ma C, Liu S, Zhao Y, Liu H, Zhang X. DANCR promotes septic cardiomyopathy by enhancing macrophage glycolytic reprogramming via the IGF2BP2/HK2 axis. Front Cell Dev Biol. 2025;13:1628915. doi:10.3389/fcell.2025.1628915
83. Liu X, Du S, Jiang Y, Chen Y. Piezo1 exacerbates psoriasis by promoting macrophage M1 polarization and inhibits autophagy via activating PI3K/AKT signaling pathway. Inflamm Res. 2025;74(1):149. doi:10.1007/s00011-025-02111-7
84. Jinzhong Wang MS, Jian Fu MS. STAT3/FoxO3a/Sirt1 pathway inhibition by ginsenoside Rc ameliorates cardiomyocyte damage in septic cardiomyopathy by altering macrophage polarization. J Mol Histol. 2025;56(3):148. doi:10.1007/s10735-025-10417-3
85. Zhao H, Lin X, Chen Q, Wang X, Wu Y, Zhao X. Quercetin inhibits the NOX2/ROS-mediated NF-κB/TXNIP signaling pathway to ameliorate pyroptosis of cardiomyocytes to relieve sepsis-induced cardiomyopathy. Toxicol Appl Pharmacol. 2023;477:116672. doi:10.1016/j.taap.2023.116672
86. Zhou J, Wu K, Ma Y, et al. GTS-21 alleviates sepsis-induced atrial fibrillation susceptibility by modulating macrophage polarization and Neuregulin-1 secretion. Int Immunopharmacol. 2025;154:114561. doi:10.1016/j.intimp.2025.114561
87. Kong W, Kang K, Gao Y, et al. GTS-21 protected against LPS-Induced sepsis myocardial injury in mice through α7nAChR. Inflammation. 2018;41(3):1073–1083. doi:10.1007/s10753-018-0759-x
88. Xue J, Ren Y, Chen Y, Zhu H, Wang Y. Inhibition of neutrophil infiltration and NETosis ameliorates sepsis-induced cardiac injury and reduces myocardial inflammation and apoptosis. Int Immunopharmacol. 2026;168(Pt 1):115862. doi:10.1016/j.intimp.2025.115862
89. Yu XY, Chen HM, Liang JL, et al. Hyperglycemic myocardial damage is mediated by proinflammatory cytokine: macrophage migration inhibitory factor. PLoS One. 2011;6(1):e16239. doi:10.1371/journal.pone.0016239
90. Seok J, Warren HS, Cuenca AG, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110(9):3507–3512. doi:10.1073/pnas.1222878110
91. Marshall JC, Leligdowicz A. Gaps and opportunities in sepsis translational research. EBioMedicine. 2022;86:104387. doi:10.1016/j.ebiom.2022.104387
92. Deinhardt-Emmer S, Chousterman BG, Schefold JC, et al. Sepsis in patients who are immunocompromised: diagnostic challenges and future therapies. Lancet Respir Med. 2025;13(7):623–637. doi:10.1016/S2213-2600(25)00124-9
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Extracellular Histones Activate Endothelial NLRP3 Inflammasome and are Associated with a Severe Sepsis Phenotype

Beltrán-García J, Osca-Verdegal R, Pérez-Cremades D, Novella S, Hermenegildo C, Pallardó FV, García-Giménez JL
Journal of Inflammation Research 2022, 15:4217-4238
Published Date: 25 July 2022
Characterization of Cathepsin B in Mediating Silica Nanoparticle-Induced Macrophage Pyroptosis via an NLRP3-Dependent Manner
Ma L, Han Z, Yin H, Tian J, Zhang J, Li N, Ding C, Zhang L
Journal of Inflammation Research 2022, 15:4537-4545
Published Date: 8 August 2022
Pyroptosis and Intervertebral Disc Degeneration: Mechanistic Insights and Therapeutic Implications
Ge Y, Chen Y, Guo C, Luo H, Fu F, Ji W, Wu C, Ruan H
Journal of Inflammation Research 2022, 15:5857-5871
Published Date: 17 October 2022
Pyroptosis and Inflammasome-Related Genes-NLRP3, NLRC4 and NLRP7 Polymorphisms Were Associated with Risk of Lung Cancer
Jing X, Yun Y, Ji X, Yang E, Li P
Pharmacogenomics and Personalized Medicine 2023, 16:795-804
Published Date: 25 August 2023
The Mechanism of Pyroptosis and Its Application Prospect in Diabetic Wound Healing
Al Mamun A, Shao C, Geng P, Wang S, Xiao J
Journal of Inflammation Research 2024, 17:1481-1501
Published Date: 6 March 2024