Back to Journals » Drug Design, Development and Therapy » Volume 19
Targeting Lactylation Offers Therapy to Reverse Cell Death Resistance
Authors Wang Y ![]() , Chen J, Wang Y, Cao Y, Li Y, Zhu Y, Zhang Z, Wu S, Wang H
, Chen J, Wang Y, Cao Y, Li Y, Zhu Y, Zhang Z, Wu S, Wang H
Received 14 September 2025
Accepted for publication 11 December 2025
Published 20 December 2025 Volume 2025:19 Pages 11371—11394
DOI https://doi.org/10.2147/DDDT.S567488
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Yumin Wang,1,* Jinxia Chen,2,* Yan Wang,3,* Yuwei Cao,1 Yulin Li,1 Yonglin Zhu,1 Zhe Zhang,1 Shuang Wu,4 Hongquan Wang5– 7
1Department of Respiratory and Critical Care Medicine, Aerospace Center Hospital, Peking University Aerospace School of Clinical Medicine, Beijing, 100049, People’s Republic of China; 2Department of Blood Transfusion, The Fourth Hospital of Hebei Medical University, Shijiazhuang, 050000, People’s Republic of China; 3Hunan Provincial Key Laboratory of Hepatobiliary Disease Research & Division of Hepato-Biliary-Pancreatic Surgery, Department of Surgery, The Second Xiangya Hospital of Central South University, Changsha, 410011, People’s Republic of China; 4Department of Neurology, Zhongnan Hospital of Wuhan University, Wuhan, 430000, People’s Republic of China; 5Department of Geriatrics, Aerospace Center Hospital, Peking University Aerospace School of Clinical Medicine, Beijing, 100049, People’s Republic of China; 6Aerospace Medical Center, Aerospace Center Hospital, Beijing, 100049, People’s Republic of China; 7Inner Mongolia Key Laboratory of Allergic Diseases, Foundational and Translational Medical Research Center, Department of Allergy and General Surgery, Hohhot First Hospital, Hohhot, 010030, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shuang Wu, Email [email protected] Hongquan Wang, Email [email protected]
Abstract: Lactylation, a lactate-derived post-translational modification, emerges as a master metabolic regulator of resistance to regulated cell death (RCD) across pathologies including cancer, inflammatory disorders, and degenerative diseases. By dynamically modifying histones and non-histone proteins via lactyltransferases and delactylases, lactylation orchestrates convergent molecular pathways that suppress ferroptosis, cuproptosis, and apoptosis. This review synthesizes current understanding of lactylation as a central regulator of RCD resistance in diseases, especially in cancers. We dissect the molecular machinery through which lactylation subverts ferroptosis, cuproptosis, and apoptosis; evaluate its pathophysiological implications in diverse pathologies; and discuss emerging therapeutic strategies to disrupt lactylation-mediated cell death evasion. This metabolic-epigenetic crosstalk establishes a robust shield against RCD in disease microenvironments, promoting therapeutic resistance and pathological resilience. Targeting lactylation regulators (writers/erasers) or combining lactate modulation with RCD inducers represents a promising strategy to overcome treatment-refractory conditions in cancers.
Keywords: lactylation, regulated cell death, ferroptosis, cuproptosis, apoptosis
Introduction
Metabolic reprogramming is a fundamental hallmark of diverse pathological states, driving the accumulation of metabolites like lactate that serve as precursors for novel post-translational modifications. Among these, lysine lactylation (Kla)—a lactate-derived epigenetic mark initially identified in macrophages—has emerged as a critical regulator of gene expression beyond immunomodulation. Elevated lactate levels, characteristic of inflammatory microenvironments, ischemic tissues, and metabolic disorders, provide the substrate for enzymatic lactylation catalyzed by acyltransferases such as p300 and KAT7. This modification directly links cellular metabolism to epigenetic and non-epigenetic reprogramming, enabling context-dependent adaptations in disease progression. Crucially, lactylation’s influence extends to the regulation of fundamental cell death pathways, where it orchestrates resistance mechanisms that compromise tissue homeostasis and promote pathological resilience across cancer, neurodegenerative conditions, ischemic injuries, and autoimmune diseases.
The dysregulation of regulated cell death (RCD) pathways—including apoptosis, ferroptosis, and copper-dependent cuproptosis—represents a pivotal mechanism underlying treatment resistance and disease exacerbation. Ferroptosis, driven by iron-dependent lipid peroxidation, contributes to neurodegeneration, ischemia-reperfusion injury, and organ dysfunction; cuproptosis, mediated by mitochondrial copper toxicity, influences metabolic diseases and metal homeostasis disorders; while defective apoptosis underlies autoimmune pathologies and developmental abnormalities. Resistance to these RCD modalities enables pathological cell survival, fostering chronic inflammation, irreversible tissue damage, and therapeutic failure across multiple disease contexts.1–6 Emerging evidence reveals that lactylation subverts RCD execution through convergent molecular strategies: it epigenetically silences pro-death genes, stabilizes antioxidant transcripts via RNA methylation, enhances radical-trapping capacity, and disrupts metal ion homeostasis. By hijacking these effector mechanisms, lactylation establishes a robust shield against cellular demise in disease-perturbed environments.5,7,8
Specifically, lactylation universally enables RCD evasion by rewiring cellular machinery: In ferroptosis, it upregulates glutathione synthesis via GCLM/GCLC lactylation,9,10 activates NRF2 through PRDX1 modification,11 and reduces iron availability via histone-driven NFS1 expression.12 For cuproptosis, lactylation of IGF-1R stabilizes the suppressor CDKN2A,13 while NUDT21 lactylation destabilizes FDX1 mRNA to disable copper toxicity.14 In apoptosis, caspase-3 lactylation directly impedes proteolytic activation.15 These findings position lactylation as a master metabolic switch that rewires epigenetic landscapes, metabolic fluxes, and stress-response networks to sustain pathological cell survival. Despite these advances, key knowledge gaps persist regarding tissue-specific “writer” enzymes, lactylation crosstalk with other PTMs, and context-dependent target selectivity dictating RCD sensitivity.16,17
This review synthesizes current understanding of lactylation as a central regulator of RCD resistance across disease states. We dissect the molecular machinery through which lactylation subverts ferroptosis, cuproptosis, and apoptosis; evaluate its pathophysiological implications in diverse pathologies; and discuss emerging therapeutic strategies to disrupt lactylation-mediated cell death evasion. By integrating advances in lactylome profiling, RCD modulation, and metabolic targeting, we aim to establish a unified framework for overcoming lactylation-driven resistance in refractory diseases.
Core Mechanisms of Lysine Lactylation
Similar to other PTMs, the protein lactylation process is a dynamic and reversible modification regulated by three types of enzymes: lactyltransferases (“writers”), delactylases (“erasers”), and lactylation binding proteins (“readers”)10,17–22 (Figure 1 and Table 1). We define the enzymes mediating protein lactylation as lactylation regulators. Writers catalyze the addition of lactoyl groups (or lactyl moiety) to lysine residues on both histone and non-histone target proteins, while erasers remove these lactoyl modifications. Readers recognize lactylated proteins and translate this modification into diverse cellular functions by modulating downstream signaling pathways.23 Collectively, these enzymatic systems establish a sophisticated mechanism for the precise regulation of Kla, functioning as critical epigenetic regulators.10
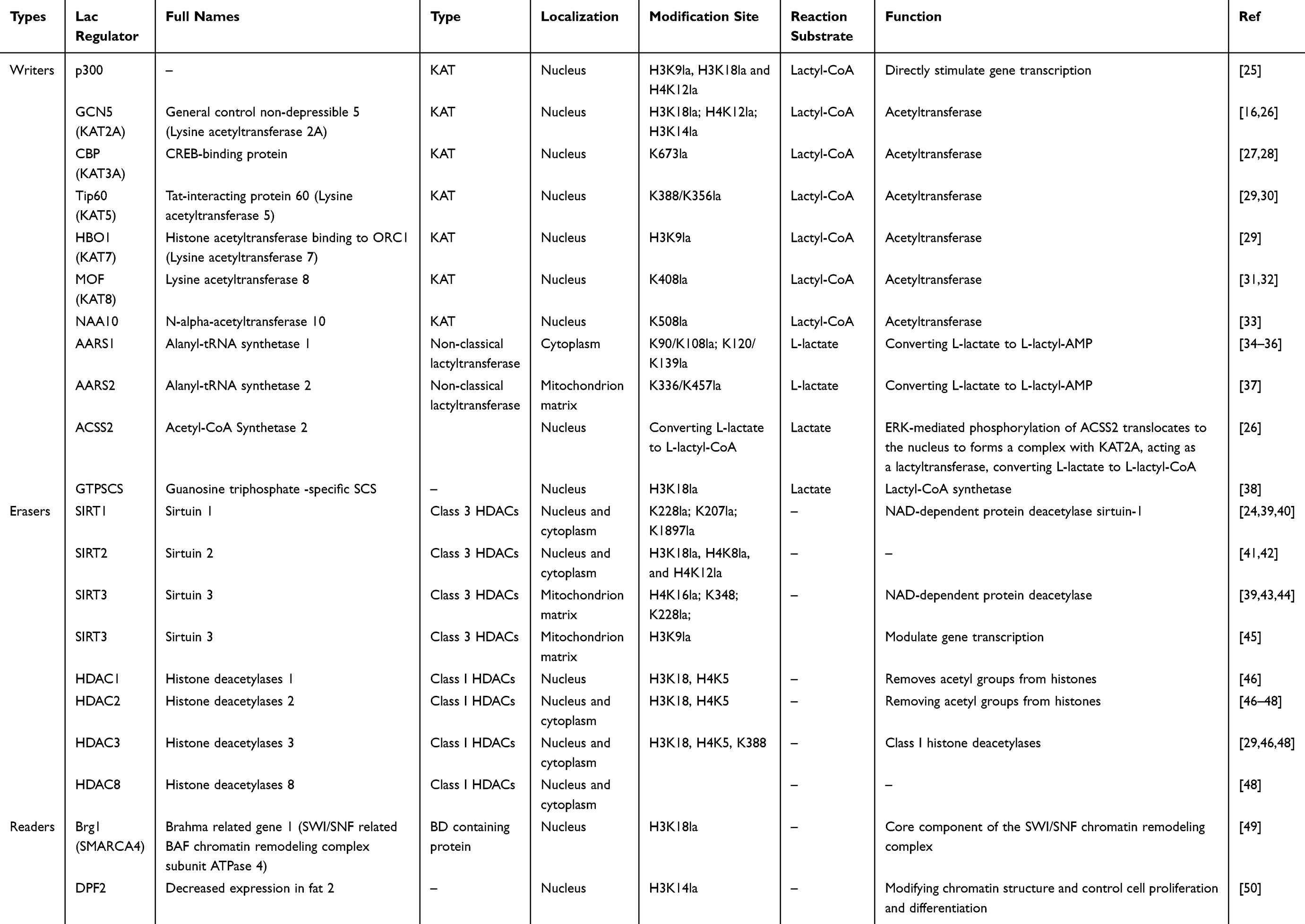 |
Table 1 The Function of Lysine Lactylation Regulator Proteins24 |
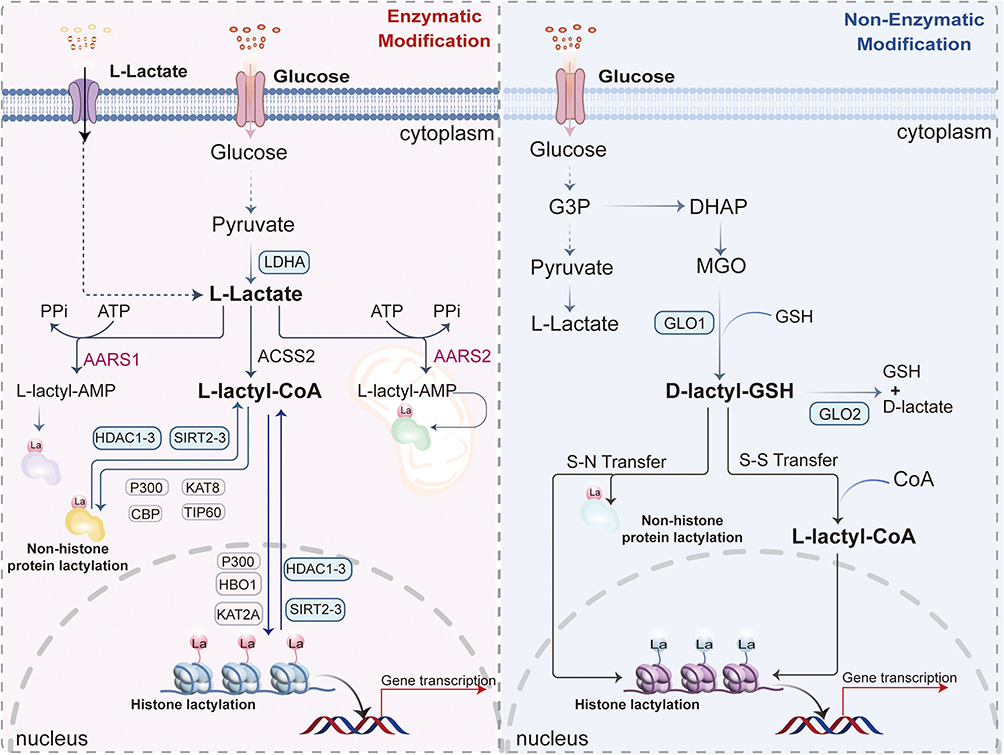 |
Figure 1 Core mechanisms of lactylation modification on histones and nonhistone proteins, including lactyl moiety sources and involved enzymes. Lactylation occurs via enzymatic reactions—mediated by diverse enzymes within the nucleus, cytoplasm, and mitochondrial matrix—or non-enzymatic reactions in the cytoplasm. Endogenous/exogenous L-lactate converts to L-lactyl-CoA, providing the L-lactyl moiety (La) for lysine lactylation (Kla) on target proteins. Identified Kla writers include acetyltransferases (p300, CBP, KAT8, KAT2A, TIP60, KAT7), while erasers comprise deacetylases (HDAC1-3, SIRT2-3). Alternatively, the glyoxalase intermediate S-D-lactylglutathione (D-lactyl-GSH) serves as a source for non-enzymatic D-lactyl group transfer to lysine residues. Parallel mechanisms include: AARS1/2 lactyltransferases catalyzing La transfer via L-lactyl-AMP complexes; and glyceraldehyde-3-phosphate (G3P) accumulation (following PKM2 inhibition) driving non-enzymatic S-lactylation of KEAP1 cysteine residues, thereby activating NRF2. |
Writers
Current evidence indicates that typical lactylation writers, or lactyltransferases, are derived from lysine acetyltransferase (KAT) family enzymes. While these enzymes primarily catalyze lysine acetylation, recent research demonstrates their capacity to also mediate lysine lactylation (Kla). To date, the identified lactylation writers include, including p300 (lysine acetyltransferase 3B, KAT3B),25 general control non-depressible 5 (GCN5; also named as lysine acetyltransferase 2A[KAT2A]),16 CREB-binding protein (CBP; also named as lysine acetyltransferase 3A[KAT3A]),27 Tat-interacting protein 60 (Tip60; also named as lysine acetyltransferase 5[KAT5]),29 histone acetyltransferase binding to ORC1 (HBO1; also named as lysine acetyltransferase 7[KAT7]),29 males absent on the first (MOF; also named as lysine acetyltransferase 8[KAT8]),32 and N-alpha-acetyltransferase 10 (NAA10).33 Recent studies have been identified as a non-classical lactyltransferase, ie alanyl-tRNA synthetase 1/2 (AARS1/2), which directly utilize lactate as a lactyl group donor to catalyze Kla.34–36 The detailed role of these lactylation writers refer to recent excellent review.23
Erasers
The histone deacetylases (HDACs) and sirtuins (SIRTs) were originally associated with lysine residues deacetylation of lysine residues and have been revealed to implicate in regulating protein deacylases.23 Several isozymes of histone deacetylases (HDACs) and sirtuins (SIRTs) were identified as erasers, which catalyze the removal of other lysine acylations.23 The erasers from HDACs include HDAC1,46 HDAC2,46–48 HDAC3,29,46,48 and HDAC8.48 The erasers from SIRTs include SIRT1,24,39,40 SIRT241,42 and SIRT3.39,43,44 The detailed role of these lactylation writers refer to recent excellent review.23
Readers
Research on Kla-specific “readers” remains an emerging field with limited progress. To date, only a few readers have been identified, including brahma-related gene-1 (BRG1)49 and decreased expression in fat 2 (DPF2).50 BRG1 functions as a reader of H3K18la[31], while DPF2 recognizes H3K14la to modify chromatin structure and control cell proliferation and differentiation.50 Currently, protein lactylation readers have not been unambiguously identified, and distinguishing their specific roles in lysine acetylation versus lactylation constitutes an ongoing research challenge.
Mitochondrial Influence on Lactylation and Cell Death Regulation
Beyond its well-established role as a product of cytoplasmic glycolysis, lactate production and its subsequent utilization for lactylation are intimately linked to mitochondrial function, creating a bidirectional regulatory axis that profoundly influences cell death susceptibility. Mitochondria themselves can contribute to the lactate pool. The mitochondrial lactate dehydrogenase (mLDHA) complex facilitates the conversion of pyruvate to lactate within the organelle, particularly under conditions of impaired electron transport chain (ETC) function or hypoxia.51,52 This mitochondrial-derived lactate can then contribute to both intra-mitochondrial and nuclear lactylation events. Furthermore, mitochondrial metabolism dictates the availability of key metabolites that influence lactylation. For instance, inhibition of the mitochondrial pyruvate carrier (MPC) reduces mitochondrial pyruvate import, potentially shunting pyruvate towards lactate production in the cytoplasm, thereby increasing the substrate for lactylation.53
Crucially, lactylation directly targets a repertoire of mitochondrial proteins, thereby directly intervening in cell death pathways. As detailed in Non-Histone Lactylation Regulates Resistance to Cuproptosis, the core executor of cuproptosis, FDX1, is a mitochondrial protein, and its regulation via NUDT21 lactylation indirectly impacts mitochondrial copper toxicity.14 More directly, several mitochondrial enzymes and regulators are subject to lactylation, which can alter their function. For example, lactylation of the complex I subunit NDUFV1 has been shown to suppress its activity, leading to ETC dysfunction, increased ROS production, and altered apoptotic sensitivity.28 Similarly, lactylation of the mitochondrial sirtuin SIRT3, a key deacetylase and delactylase, can impair its activity, leading to hyper-acetylation and hyper-lactylation of mitochondrial substrates, which disrupts oxidative metabolism and promotes a metabolic state resistant to certain cell death inducers.39,43 In the context of ferroptosis, mitochondrial lipid peroxidation is a key event, and lactylation-driven upregulation of NFS1, an iron-sulfur cluster biogenesis enzyme located in mitochondria, depletes the free iron necessary for propagating lipid peroxidation, thereby conferring resistance.12
This mitochondrial-lactylation axis creates a sophisticated feedback loop. Mitochondrial dysfunction (eg, ETC impairment, ROS overproduction) can promote lactate generation and subsequent lactylation. This lactylation, in turn, can either exacerbate mitochondrial dysfunction (eg, via NDUFV1 lactylation) or initiate adaptive survival programs (eg, via NFS1 upregulation or SIRT3 inhibition) that ultimately dictate cellular fate by modulating the threshold for ferroptosis, cuproptosis, and apoptosis. Therefore, the mitochondrial influence on lactylation represents a fundamental layer of metabolic-epigenetic crosstalk that fine-tunes cellular stress responses and death signaling, emphasizing the need to consider the organelle’s role for a fully integrated understanding of lactylation in pathophysiology.
Regulated Cell Death
Core Mechanism of Ferroptosis
Ferroptosis is a non-apoptotic, regulated cell death mechanism characterized by iron-dependent lipid peroxidation (LPO) and compromised antioxidant defenses1,3,6,54–56 (Figure 2). This process culminates in plasma membrane rupture driven by iron accumulation and lethal LPO,57 with its induction precisely regulated through the balance of pro-ferroptotic factors and cellular defense systems mediated by specific inducers and inhibitors.2 Core triggers include inhibition of the SLC7A11-GSH-GPX4 axis and intracellular free iron overload.58–60 Iron catalyzes LPO through both enzymatic and non-enzymatic pathways: non-enzymatically via Fe2⁺-mediated Fenton reactions that convert H2O2 to hydroxyl radicals, initiating peroxidation of polyunsaturated fatty acid phospholipids (PUFA-PLs); enzymatically through Fe2⁺ acting as a cofactor for ALOXs/POR, oxidizing ACSL4-esterified and LPCAT3-incorporated PUFA-PLs into peroxidized derivatives (PUFA-PL-OOH).2,59,61–68 Critically, iron’s redox activity enables radical propagation, as Fe2⁺ reacts with PUFA-PL-OOH to generate new radicals that perpetuate chain reactions.69
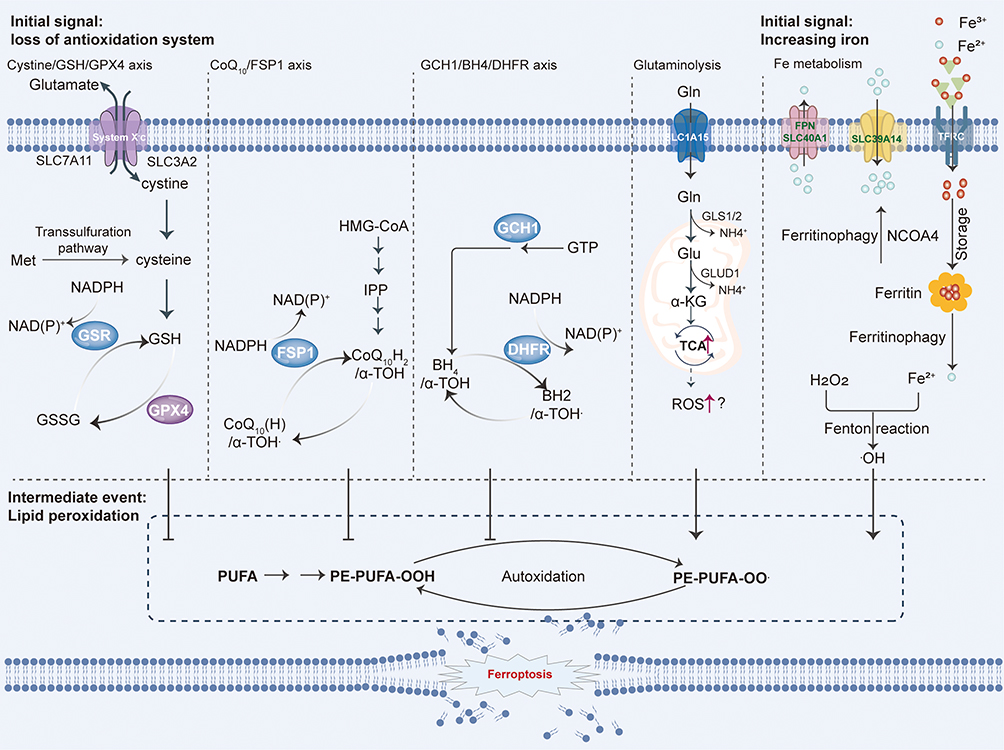 |
Figure 2 Core mechanisms of ferroptosis. Ferroptosis is initiated by iron-dependent peroxidation of polyunsaturated fatty acid (PUFA)-containing phospholipids (PUFA-PLs). When pro-ferroptotic drivers—including PUFA-PL synthesis/peroxidation and dysregulated iron metabolism—overwhelm cellular antioxidant defenses, lethal accumulation of lipid peroxides ruptures cellular membranes, triggering ferroptotic cell death. Cells counteract ferroptosis through four major defense systems that detoxify lipid peroxides: the GPX4/xCT system (cytosolic and mitochondrial GPX4 isoforms); the FSP1/CoQH2 system (plasma membrane); the DHODH/CoQH2 system (mitochondria); the GCH1/BH4 system. Mechanistically, ACSL4 and LPCAT3 mediate PUFA-PL synthesis, rendering membranes susceptible to peroxidation via enzymatic (eg, ALOXs, POR) and non-enzymatic (iron-dependent Fenton reaction) pathways. Iron further amplifies peroxidation as a cofactor for lipoxygenases. Ferroptosis ensues when pro-ferroptotic forces exceed the detoxification capacity of these defense systems, culminating in membrane-disrupting lipid peroxide accumulation and cell death. |
Expansion of the labile iron pool is further driven by mechanisms such as ferritinophagy—a selective autophagic process that degrades ferritin to release stored iron—and enhanced mitochondrial Fe2⁺ transport, which amplifies iron availability for peroxidation reactions within mitochondria.66,70–75 Cellular susceptibility to ferroptosis is heightened by ferroportin suppression, ferritinophagy, increased transferrin uptake, or dysregulated mitochondrial iron handling. Cellular defenses against ferroptosis encompass both GPX4-dependent (GSH-reliant) and GPX4-independent systems, including the FSP1–CoQ10, GCH1–BH4, DHODH–CoQH2, MBOAT1/2–MUFA, and SC5D–7-DHC axes that neutralize lipid peroxides. Given its mechanistic complexity and significant disease implications, ferroptosis represents a promising therapeutic target.24,76–85
Cuproptosis
Tsvetkov et al in 2022 introduced the concept of cuproptosis.4 Prior research had established several key findings regarding copper’s role in cell death: disulfiram (DSF) exhibits anticancer activity;86 copper itself can induce cell death87 and accelerate cancer cell death;88 copper-based compounds induce cell death;86 copper triggers non-apoptotic programmed cancer cell death;86 elesclomol induces cancer cell apoptosis86 by selectively transporting copper to mitochondria to kill cells;89 copper enhances the anti-tumor activity of disulfiram;86 and ferredoxin 1 (FDX1) and lipoyl synthase (LIAS) were identified as key regulators of copper toxicity.90 These major findings on copper-induced cell death provided the foundation for understanding cuproptosis.
Cuproptosis represents a distinct form of cell death, differing mechanistically from necroptosis, apoptosis, pyroptosis, and ferroptosis (Figure 3). Copper, an essential micronutrient and trace metal,91 functions as a crucial structural and catalytic cofactor for numerous enzymes. It is indispensable for diverse biological processes—including biocompound synthesis, oxidative phosphorylation (OXPHOS), iron homeostasis, connective tissue cross-linking, reactive oxygen species (ROS) detoxification, and signal transmission—across nearly all organisms.92,93 However, copper can also be cytotoxic. Copper overload stimulates damaging ROS generation via copper-mediated Fenton reactions and disrupts iron-sulfur cofactors. Chronic copper exposure leads to toxicity, and elevated intracellular copper levels are implicated in various diseases, including cancer. Copper is also recognized as a crucial component for tumor growth and metastasis.94
 |
Figure 3 Core mechanisms of cuproptosis. Cuproptosis is initiated by the cellular uptake of Cu2⁺, mediated by either the copper transporter SLC31A1 or copper ionophores. Intracellular Cu2⁺ selectively binds to lipoylated proteins within the tricarboxylic acid (TCA) cycle. This binding induces mitochondrial proteotoxic stress through two key events: copper-dependent oligomerization of the lipoylated proteins, and destabilization of iron-sulfur (Fe-S) cluster proteins. Together, these events lead to a toxic gain of function, ultimately resulting in cell death. Abbreviations: ATP7B, ATPase copper transporting beta; DLAT, dihydrolipoamide S-acetyltransferase; FDX1, ferredoxin 1; LA-DLAT, lipoylated DLAT; LIAS, lipoyl synthase; SLC31A1, solute carrier family 31 member 1; TCA, tricarboxylic acid. |
Despite its importance, the precise molecular mechanisms underlying copper-induced toxicity and cell death remain elusive. Moreover, the known cellular machinery and enzymatic targets for copper do not fully explain cellular responses to copper toxicity. The discovery of cuproptosis emerged from efforts to understand how copper accumulation causes cellular toxicity.90 Tsvetkov et al, in 2019, described copper-dependent death while investigating the anticancer mechanism of elesclomol (ES), a copper ionophore.90 They demonstrated that ES sensitizes cancer cells to proteasome inhibitor-induced toxicity in a multiple myeloma mouse model. Mechanistically, ES-bound Cu2⁺ interacts with the mitochondrial enzyme ferredoxin 1 (FDX1), is reduced to Cu⁺, and consequently elevates ROS levels.90,95 Initial studies attributed ES lethality to lipid peroxidation.96 Subsequently, in 2022, the same group formally named this unique copper-dependent cell death pathway “cuproptosis”, characterized by the aggregation of lipoylated mitochondrial enzymes and loss of iron-sulfur (Fe-S) cluster proteins.4 Tsvetkov et al’s study revealed that copper-induced toxicity involves the specific disruption of mitochondrial metabolic enzymes, significantly advancing our understanding of how copper overload impairs mitochondrial function.4 Excess intracellular Cu2⁺ can be delivered to mitochondria by ionophores. FDX1 then reduces Cu2⁺ to Cu⁺. The increased Cu⁺ concentration directly binds to lipoylated dihydrolipoamide S-acetyltransferase (DLAT), causing aggregation of lipoylated proteins and destabilization of Fe-S cluster proteins. This leads to proteotoxic stress and ultimately, cuproptosis (Figure 4).4 Crucially, genetic or pharmacological inhibition of apoptosis, ferroptosis, and necroptosis failed to suppress ES-Cu complex-induced cell death across multiple cancer cell lines. Furthermore, other antioxidants, including N-acetylcysteine, α-tocopherol, ebselen, and JP4-039, could not rescue cells from ES-Cu-mediated growth inhibition, indicating that ROS (including mitochondrial ROS) are not required for cuproptosis. Interestingly, the hydrophilic antioxidant glutathione (GSH) inhibits ES-Cu-induced toxicity by chelating intracellular copper. These results strongly suggest that cuproptosis constitutes a novel cell death pathway distinct from previously identified mechanisms.4
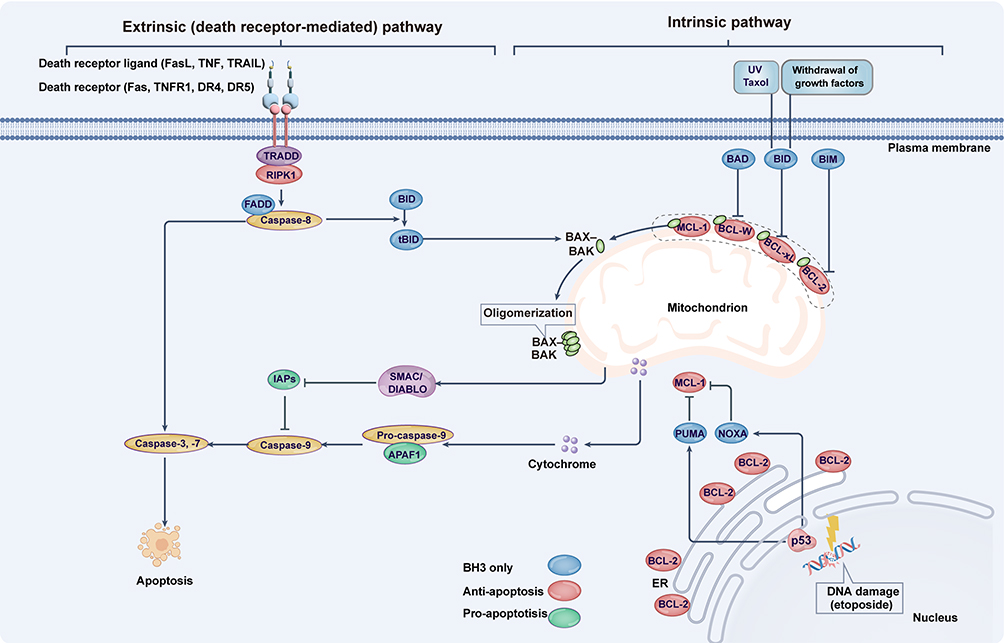 |
Figure 4 Core mechanisms of apoptosis. Extrinsic apoptosis (right) is triggered by the activation of plasma membrane death receptors—including TNFR1, Fas, and TRAIL receptors (DR4/DR5)—upon binding their cognate ligands (TNF, FasL, and TRAIL, respectively). In contrast, intrinsic apoptosis (left) can be initiated by growth factor withdrawal, mitochondrial or DNA damage, or chemotherapeutic agents like taxol. This pathway involves BH3-only BCL-2 family proteins (eg, p53-mediated transcriptional induction of NOXA/PUMA, post-translational modification of BAD /BIM, and caspase-8 cleavage of Bid), which neutralize pro-survival members (BCL-2, BCL-xL, MCL-1) and activate pro-death effectors BAX/BAK to oligomerize and disrupt mitochondria. Subsequent mitochondrial permeabilization releases cytochrome c and SMAC/DIABLO, driving APAF1-dependent caspase-9 activation. Caspase-9 then proteolytically processes executioner caspases-3 and −7 to execute apoptosis. These effector caspases further amplify the cascade through positive feedback activation of upstream caspases. Abbreviations: ER, endoplasmic reticulum; UV, ultraviolet. |
Tsvetkov et al also established a strong link between mitochondrial metabolism and copper toxicity sensitivity. Lung cancer cells reliant on galactose-mediated mitochondrial respiration were nearly 1000-fold more sensitive to ES-Cu-induced growth inhibition than cells relying on glucose-induced glycolysis. Accordingly, inhibitors of mitochondrial respiration—rotenone and antimycin A (targeting complexes I and III, respectively), UK5099 (inhibiting mitochondrial pyruvate uptake), or genetic inhibition of complex I—all blocked cuproptosis. While ES-Cu did not affect basal or adenosine 5′-triphosphate (ATP)-linked respiration, hypoxia (1% O2), which forces cells to rely on glycolysis rather than OXPHOS, reduced cancer cell sensitivity to cuproptosis. This metabolic dependency distinguishes cuproptosis from ferroptosis, which requires glucose uptake and pyruvate oxidation. Thus, cuproptosis and ferroptosis are associated with distinct alterations in energy metabolism and mitochondrial function. In summary, cuproptosis represents a novel form of regulated cell death that is copper-dependent, independent of oxidative stress, and initiated within the mitochondria.5
Apoptosis
The molecular machinery governing cell death was first characterized in Caenorhabditis elegans. Homologs of key C. elegans effector proteins—including caspases and BCL-2 family members—were subsequently identified in mammals. Mammalian apoptosis comprises two distinct pathways: extrinsic apoptosis, triggered by activation of plasma membrane-localized death receptors, and intrinsic apoptosis, which proceeds independently of death receptor signaling (Figure 4). These pathways diverge in their mechanisms for sensing and integrating apoptotic stimuli.97
Intrinsic apoptosis is activated by cellular alterations including DNA damage, growth factor withdrawal, and mitochondrial damage (Figure 4). This pathway encompasses specialized forms such as anoikis, triggered by detachment from the extracellular matrix via loss of integrin-mediated adhesion.98 The BCL-2 protein family serves as the principal upstream regulator of intrinsic apoptosis, with mammalian BCL-2—originally identified as an oncogene in follicular lymphomas with t(14;18) translocation—functioning as a survival promoter rather than a proliferation inducer.99 Structurally defined by 1–4 BCL-2 homology (BH) domains,100,101 this family comprises both anti-apoptotic (eg, BCL-2, BCL-xL, MCL-1, BCL-W) and pro-apoptotic members, functionally analogous to the C. elegans CED-9/EGL-1 system.101,102 Elevated anti-apoptotic BCL-2 protein levels suppress apoptosis, and genetic ablation of these proteins causes lethality or severe developmental defects in mice,103–105 underscoring apoptosis’ essential role in development and homeostasis. BH domain-mediated interactions between pro- and anti-apoptotic members regulate pathway activation. During cellular stress (DNA damage, oxidative stress, nutrient deprivation), transcriptional and post-translational activation of pro-apoptotic BCL-2 members—particularly BH3-only proteins (BIM, BID, BAD, NOXA, PUMA)—transduces death signals by neutralizing anti-apoptotic proteins while directly activating BAX/BAK. This triggers mitochondrial outer membrane permeabilization (Figure 4), releasing cytochrome c and SMAC/DIABLO to initiate caspase activation. Downstream, the APAF1 apoptosome (mammalian CED-4 homolog) activates caspase-9, which cleaves executioner caspases-3/7 to dismantle cellular structures.106–108 However, the traditional linear caspase cascade model requires refinement, as caspase-3/7 double-knockout delays mitochondrial damage and cell death,109 suggesting apoptosis operates as a self-amplifying circular cascade.
Conventional cancer radiation therapies and chemotherapies rely on activating the intrinsic apoptosis pathway through DNA damage caused by agents like ionizing irradiation, etoposide, or taxol. However, research over the past decade has revealed significant redundancy in mammalian cell death mechanisms. Notably, cells or mice with double knockout of Bax and Bak (Bax–/–Bak1–/–) exhibit profound resistance to apoptosis,7 yet a small percentage of these mutant mice can still be born and survive for months.110 This observation strongly suggests that other cell death mechanisms may be activated or selected for in Bax–/–Bak1–/– cells to eliminate unwanted cells when apoptosis fails during development. Furthermore, while DNA damage-induced cell death often proceeds through apoptosis by promoting mitochondrial damage and downstream caspase activation, it represents only part of the overall cell death response; severe DNA damage can also lead directly to cell death via mitotic catastrophe (discussed below). Consequently, when the programmed cell death mechanism is blocked, the disruption of cellular homeostatic pro-survival mechanisms likely contributes to cell elimination in mammals.
Extrinsic apoptosis is initiated by the activation of plasma membrane-bound death receptors, characterized by an intracellular Death Domain (DD), such as Fas (CD95), TNFR1, TRAIL-R1 (DR4), and TRAIL-R2 (DR5), upon binding their cognate ligands.111,112 These pathways are clinically significant, as genetic defects in Fas or its ligand FasL cause autoimmune lymphoproliferative syndrome (ALPS) in humans—a rare autoimmune condition recapitulated in Fas- and FasL-deficient mouse models.113–116 During immune regulation, FasL expressed on activated T cells triggers apoptosis in target cells by engaging the Fas DD, thereby recruiting the adaptor protein FADD and activating caspase-8. Critically, ALPS-associated mutations in the Fas DD disrupt FADD recruitment and block caspase-8 activation, highlighting the indispensable role of caspase-8 in executing Fas-mediated extrinsic apoptosis.117
Lactylation Regulates Resistance to Regulated Cell Death
Lactylation Regulates Resistance to Ferroptosis
Lactylation functions as a master metabolic switch that universally enables ferroptosis resistance across malignancies. By hijacking histones, RNA modifiers, and antioxidant machinery, lactylation epigenetically activates cytoprotective genes, stabilizes key suppressors of lipid peroxidation, and rewires metabolic flux to synthesize radical-trapping molecules—collectively shielding cancer cells from iron-dependent death. Below, we dissect how lactylation of diverse substrates (H3K18, PRMT5, HDAC1, GCLM, SPRING, and others) orchestrates tissue-specific yet mechanistically convergent pathways to block ferroptosis execution, thereby sustaining tumor survival under therapeutic stress and microenvironmental challenges (Figure 5).
 |
Figure 5 Lactylation regulates resistance to ferroptosis. Histone lactylation epigenetically reinforces ferroptosis resistance by activating the METTL3/HIF1A /HMOX1 antioxidant axis to mitigate lipid peroxidation in Endometriosis. Lactylation sustains ferroptosis resistance by enabling PRMT5 to epigenetically suppress ALKBH5 and stabilize SLC7A11 expression in colorectal cancer (CRC). Lactylation of HDAC1-K412la stabilizes FSP1 mRNA via m6A modification to confer ferroptosis resistance in CRC. Lactylation of GCLM by ACAT2 enhances GSH synthesis to scavenge lipid peroxides, directly enabling ferroptosis resistance in KRASG12D-driven cancer. Histone lactylation (H3K18la) epigenetically upregulates NFS1 expression to reduce intracellular free iron and suppress lipid peroxidation, thereby conferring ferroptosis resistance in hepatocellular carcinoma. Histone lactylation (H3K18la) epigenetically upregulates NFS1 expression to reduce intracellular free iron and suppress lipid peroxidation, thereby conferring ferroptosis resistance in hepatocellular carcinoma. Histone lactylation epigenetically activates GCLC to suppress ferroptosis, enabling chemoresistance in colorectal cancer stem cells. Histone lactylation epigenetically activates GCLC to suppress ferroptosis, enabling chemoresistance in colorectal cancer stem cells. Thus, lactate-induced LSD1 lactylation epigenetically silences TFRC to inhibit ferroptosis, establishing a key mechanism of acquired resistance in melanoma. Lactylation confers ferroptosis resistance by activating the NSUN2-GCLC-GSH axis to suppress lethal lipid peroxidation. Lactate-induced NAA10-mediated lactylation of the RNA methyltransferase NSUN2 confers ferroptosis resistance by activating the GCLC-GSH axis to suppress lethal lipid peroxidation. Lactylation suppresses ferroptosis by enabling PRDX1-mediated activation of the NRF2 antioxidant pathway, promoting cancer cell survival under therapeutic stress. Lactylation of SPRING at K82, mediated by the pPCK1-pLDHA axis-induced lactate accumulation, activates the MVA pathway to synthesize radical-trapping antioxidants that suppress lipid peroxidation and confer ferroptosis resistance in AKT-hyperactivated intrahepatic cholangiocarcinoma (ICC). |
Histone Lactylation in Ferroptosis Resistance
Lactylation orchestrates ferroptosis resistance by hijacking m6A machinery through distinct mechanisms. Lactylation acts as a master switch that either activates m6A writers (METTL3) or inhibits m6A erasers (ALKBH5/FTO), driving hyper-methylation of pro-survival transcripts (HIF1A, SLC7A11, FSP1). This m6A-dependent mRNA stabilization amplifies antioxidant (HMOX1), cystine transporter (SLC7A11), and radical-trapping (FSP1) pathways, collectively desensitizing cells to lipid peroxidation. Elevated histone lactylation, specifically H3K18la, in endometriotic stromal cells (EESCs) drives ferroptosis resistance by activating the METTL3-mediated HIF1A/HMOX1 signaling axis. Mechanistically, lactate accumulation from enhanced glycolysis promotes H3K18la enrichment at the METTL3 promoter, increasing METTL3 expression and m6A RNA modification; METTL3 stabilizes HIF1A mRNA via m6A methylation, leading to HIF1A-dependent transcriptional upregulation of the antioxidant gene HMOX1, which suppresses lethal lipid peroxidation.118 Consequently, inhibiting histone lactylation (via 2-DG) restores ferroptosis sensitivity by disrupting this pathway.118 In summary, histone lactylation epigenetically reinforces ferroptosis resistance by activating the METTL3/HIF1A /HMOX1 antioxidant axis to mitigate lipid peroxidation.118
Huang et al revealed that insufficient microwave ablation (IMWA) induces sublethal heat stress (HS) in hepatocellular carcinoma (HCC), triggering increased glycolysis and lactate accumulation. This lactate surge drives histone H3 lysine 18 lactylation (H3K18la).12 Crucially, H3K18la enrichment at the promoter region epigenetically upregulates the expression of NFS1, a cysteine desulfurase essential for iron-sulfur cluster biosynthesis. Elevated NFS1 levels reduce intracellular free iron availability and subsequently suppress lipid peroxidation, the hallmark executioner of ferroptosis.12 Consequently, this H3K18la-NFS1 axis confers significant resistance to ferroptosis in HCC cells, enabling their survival and promoting metastasis post-IMWA.12 Knocking down NFS1 reverses this effect, increasing ferroptosis susceptibility and reducing metastasis. Furthermore, NFS1 deficiency synergizes with oxaliplatin (OXA) chemotherapy to enhance anti-tumor efficacy, partly by overcoming this ferroptosis resistance.12 Histone lactylation (H3K18la) epigenetically upregulates NFS1 expression to reduce intracellular free iron and suppress lipid peroxidation, thereby conferring ferroptosis resistance in hepatocellular carcinoma12 (Figure 5).
Histone lactylation, specifically at the H4K12 site (H4K12la), directly promotes chemoresistance in colorectal cancer stem cells (CCSCs) by inhibiting ferroptosis.119 Mechanistically, lactate-driven H4K12la, catalyzed by p300 and reversed by HDAC1, transcriptionally upregulates the expression of glutamate-cysteine ligase catalytic subunit (GCLC).119 Elevated GCLC enhances glutathione synthesis, thereby suppressing lipid peroxidation—the hallmark execution event of ferroptosis—and allowing CCSCs to survive chemotherapy-induced stress.119 Consequently, inhibition of the lactate-producing enzyme LDHA, the lactyltransferase p300, or GCLC itself (using BSO) restores ferroptosis sensitivity and overcomes chemoresistance.119 In summary, histone lactylation epigenetically activates GCLC to suppress ferroptosis, enabling chemoresistance in colorectal cancer stem cells119 (Figure 5).
Non-Histone Lactylation in Ferroptosis Resistance
Lactylation of the protein arginine methyltransferase PRMT5 at lysine 240 (PRMT5 K240lac) in colorectal cancer (CRC) cells promotes resistance to ferroptosis.120 Mechanistically, lactylated PRMT5 represses transcription of the m6A demethylase ALKBH5 through histone modifications (H4R3me2s and H3R8me2s), which stabilizes SLC7A11 mRNA (encoding the cystine/glutamate antiporter xCT) and enhances its expression in an m6A-dependent manner.120 Elevated SLC7A11 increases cystine uptake for glutathione synthesis, thereby suppressing lipid peroxidation and enabling CRC cells to evade ferroptosis.120 Crucially, mutating the lactylation site (PRMT5 K240R) abolishes this protective effect. Lactylation sustains ferroptosis resistance by enabling PRMT5 to epigenetically suppress ALKBH5 and stabilize SLC7A11 expression120 (Figure 5). Lactylation of HDAC1 at lysine 412 (HDAC1-K412la) drives ferroptosis resistance in colorectal cancer (CRC) by stabilizing the ferroptosis suppressor FSP1.121 Mechanistically, HDAC1-K412la enhances HDAC1-mediated suppression of the m6A erasers FTO and ALKBH5, leading to increased N6-methyladenosine (m6A) modification on FSP1 mRNA. This m6A enrichment recruits the reader protein IGF2BP1, which stabilizes FSP1 mRNA and boosts FSP1 protein expression, thereby inhibiting lipid peroxidation and ferroptosis.121 Critically, HDAC inhibitors (HDACi) like SAHA and TSA reduce HDAC1-K412la lactylation, destabilize FSP1 mRNA, and sensitize CRC to ferroptosis inducers.121 Lactylation of HDAC1-K412la stabilizes FSP1 mRNA via m6A modification to confer ferroptosis resistance in colorectal cancer121 (Figure 5). In conclusion, lactylation converges on m6A RNA modification to epigenetically and post-transcriptionally fortify antioxidant defenses, establishing a robust axis of ferroptosis resistance that spans histone, enzyme, and metabolic regulation. Critical gaps include how lactylation precisely recruits m6A regulators (eg, tissue-specific lactyltransferases targeting METTL3 vs PRMT5/HDAC1), whether lactylation-m6A crosstalk extends to other ferroptosis drivers (eg, GPX4, ACSL4), and if m6A readers (eg, YTHDFs) cooperate with lactylation to fine-tune mRNA stability. Future studies should: Develop dual-targeting inhibitors (eg, blocking lactyltransferases + m6A writers/readers); Map lactylation-m6A interactomes across cancer types to identify context-specific vulnerabilities; Explore lactate modulation (eg, LDHA inhibition) combined with m6A eraser activators to resensitize therapy-resistant tumors.
The oncogenic KRASG12D mutation drives lactate overproduction via MEK/ERK-mediated phosphorylation of LDHA, establishing a lactate-rich tumor microenvironment.9 Elevated lactate levels promote lactylation of the glutamate-cysteine ligase modifier subunit (GCLM) at lysine 34 (K34), catalyzed by the acetyltransferase ACAT29. GCLM lactylation enhances its interaction with the catalytic subunit GCLC, boosting glutamate-cysteine ligase (GCL) enzymatic activity. This increases glutathione (GSH) synthesis, which potently suppresses lipid peroxidation—a key executor of ferroptosis—thereby conferring resistance to ferroptosis inducers (eg, erastin, RSL3)9. Inhibition of ACAT2 or mutation of the GCLM K34 lactylation site ablates this protective effect, sensitizing KRASG12D-mutant cancers to ferroptosis. In summary, lactylation of GCLM by ACAT2 enhances GSH synthesis to scavenge lipid peroxides, directly enabling ferroptosis resistance in KRASG12D-driven cancers9 (Figure 5).
Li et al revealed that lactate accumulation in BRAF/MEK inhibitor (BRAFi/MEKi)-resistant melanoma drives lysine lactylation of the epigenetic regulator LSD1 at K503. Lactylated LSD1 forms a stable complex with the transcription factor FosL1, which recruits the LSD1/FosL1 complex to the promoter of the transferrin receptor gene TFRC.122 This recruitment facilitates LSD1-mediated demethylation of H3K4me2, leading to transcriptional repression of TFRC.122 Consequently, reduced TFRC expression diminishes cellular iron uptake, thereby suppressing ferroptosis—a critical vulnerability in therapy-resistant cells. Inhibition of LSD1 reverses TFRC repression, reactivates iron-dependent ferroptosis, and synergizes with anti-PD-1 immunotherapy to overcome resistance.122 Thus, lactate-induced LSD1 lactylation epigenetically silences TFRC to inhibit ferroptosis, establishing a key mechanism of acquired resistance in melanoma122 (Figure 5).
Niu et al showed that lactate-mediated lactylation of the RNA methyltransferase NSUN2 at lysine 508, catalyzed by the lactyltransferase NAA10, enhances NSUN2’s enzymatic activity.33 This activated NSUN2 specifically targets glutamate-cysteine ligase catalytic subunit (GCLC) mRNA, promoting m5C methylation at key cytosine sites (C13, C81, C972, C1135, C1136) which stabilizes GCLC mRNA and increases its expression.33 Elevated GCLC protein boosts glutathione (GSH) synthesis, reducing lipid peroxidation and enabling cancer cells to resist ferroptosis induced by agents like doxorubicin (Dox) or RSL3 in an acidic tumor microenvironment. Lactylation confers ferroptosis resistance by activating the NSUN2-GCLC-GSH axis to suppress lethal lipid peroxidation33 (Figure 5).
Lactylation of the antioxidant protein PRDX1 at lysine 67 (K67), driven by the transcription factor ZNF207, activates the NRF2 signaling pathway to suppress ferroptosis and confer resistance to the drug regorafenib in hepatocellular carcinoma (HCC).11 Specifically, ZNF207 upregulates lactate production and lactate dehydrogenase A (LDHA), increasing intracellular lactate levels that catalyze PRDX1 lactylation.11 Lactylated PRDX1 then facilitates NRF2 nuclear translocation and transcriptional activation of key antioxidant/ferroptosis-suppressing genes (SLC7A11, GPX4, HMOX1), thereby inhibiting lipid peroxidation and iron-dependent cell death.11 Disrupting ZNF207, PRDX1 lactylation, or NRF2 activity restores ferroptosis sensitivity and overcomes regorafenib resistance.11 In summary, lactylation suppresses ferroptosis by enabling PRDX1-mediated activation of the NRF2 antioxidant pathway, promoting cancer cell survival under therapeutic stress11 (Figure 5).
Zhu et al identified a critical axis (pPCK1-pLDHA-SPRINGlac) linking lactylation to ferroptosis resistance in AKT-hyperactivated intrahepatic cholangiocarcinoma (ICC).123 Phosphorylated PCK1 (pPCK1), driven by AKT hyperactivation, binds and phosphorylates LDHA at T248, enhancing glycolytic flux and lactate production. Elevated lactate activates KAT7, an acyltransferase that catalyzes lactylation of SPRING at K82 (SPRING lac).123 This lactylation event promotes retrograde transport of SCAP from the Golgi to the endoplasmic reticulum, facilitating SREBP2 translocation to the nucleus and subsequent upregulation of the mevalonate (MVA) pathway.123 The MVA pathway boosts synthesis of radical-trapping antioxidants (CoQ10H2 and menaquinone-4), which suppress lipid peroxidation and confer ferroptosis resistance. Disrupting this axis—via SPRING K82R mutation (lactylation-deficient) or simvastatin (MVA inhibitor)—sensitizes ICC cells to ferroptosis and overcomes chemo-immunotherapy resistance.123 In summary, lactylation of SPRING at K82, mediated by the pPCK1-pLDHA axis-induced lactate accumulation, activates the MVA pathway to synthesize radical-trapping antioxidants that suppress lipid peroxidation and confer ferroptosis resistance in AKT-hyperactivated ICC123 (Figure 5).
Lactylation drives ferroptosis resistance across cancers via diverse mechanisms, i.e. epigenetic regulation, enzyme modulation, metabolic adaptation, and iron homeostasis. Histone lactylation (H3K18la, H4K12la) upregulates antioxidant genes (HMOX1, GCLC, NFS1) to suppress lipid peroxidation. Lactylation of PRMT5, HDAC1, and NSUN2 stabilizes ferroptosis suppressors (SLC7A11, FSP1, GCLC) via RNA methylation or epigenetic silencing. Lactylation of metabolic enzymes (GCLM, PRDX1, SPRING) enhances glutathione synthesis, activates NRF2 signaling, or boosts mevalonate pathway-derived antioxidants (CoQ10H2), directly scavenging lipid peroxides. LSD1 lactylation represses TFRC to limit iron uptake, while NFS1 upregulation depletes free iron, jointly inhibiting ferroptosis execution. Key unresolved questions include the structural basis for lactylation’s target specificity (eg, why H3K18la dominates in some cancers vs *PRMT5-K240lac* in others), the role of tissue-specific “writer/eraser” enzymes (eg, ACAT2 for GCLM vs KAT7 for SPRING), and whether lactylation cross-talks with other PTMs (eg, phosphorylation, acetylation) to fine-tune ferroptosis sensitivity. Future research should develop isoform-specific lactyltransferase inhibitors (eg, targeting KAT7, ACAT2, NAA10) to disrupt lactylation-driven antioxidant pathways; Map lactylomes in therapy-resistant tumors to identify novel targets; Explore lactate depletion (via LDHA inhibition) combined with ferroptosis inducers (eg, GPX4 inhibitors) in in vivo models; Investigate lactylation’s role in regulating iron-sulfur cluster biogenesis (NFS1) and iron transport (TFRC) beyond antioxidant synthesis.
Non-Histone Lactylation Regulates Resistance to Cuproptosis
Lactylation, a lactate-derived post-translational modification, emerges as a master regulator of cuproptosis resistance across malignancies. By hijacking divergent signaling hubs—insulin-like growth factor receptor-1 (IGF-1R) in multiple myeloma and RNA processing factor NUDT21 in esophageal cancer—lactylation orchestrates molecular cascades that disable copper-induced cell death. These pathways converge on either stabilizing endogenous suppressors of cuproptosis (eg, CDKN2A) or depleting essential executors (eg, FDX1), ultimately enabling cancer cells to evade metabolic toxicity. Below, we dissect how lactylation subverts cuproptosis through non-histone protein modifications (Figure 6).
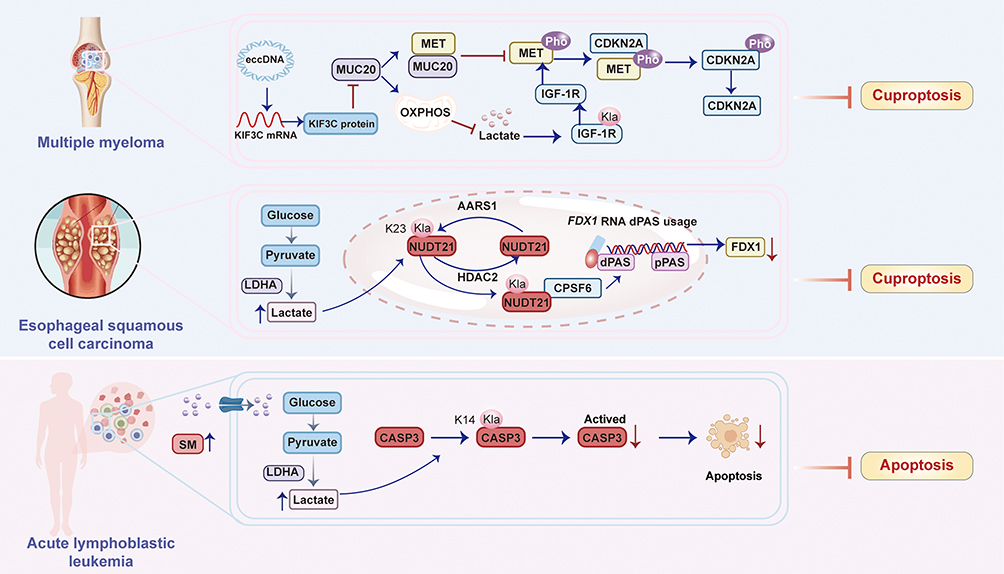 |
Figure 6 Lactylation regulates resistance to cuproptosis. Lactylation sustains cuproptosis resistance by stabilizing the IGF-1R/MET/CDKN2A signaling axis, enabling cancer cells to evade copper-induced death. Lactylation of Nudix hydrolase 21 (NUDT21) suppresses FDX1 expression through APA-mediated mRNA destabilization, thereby enabling cancer cells to evade cuproptosis in esophageal squamous cell carcinoma (ESCC). The sphingomyelin (SM)-driven lactate production mediates CASP3 K14 lactylation to prevent its proteolytic activation, conferring apoptosis resistance in leukemia. |
Wang et al revealed that lactylation of insulin-like growth factor receptor-1 (IGF-1R) promotes proteasome inhibitor (PI) resistance in multiple myeloma (MM) by suppressing cuproptosis.13 Specifically, downregulation of MUC20 in PI-resistant MM cells increases IGF-1R lactylation (likely via elevated lactate levels), which activates the MET proto-oncogene.13 Activated MET phosphorylates and stabilizes cyclin-dependent kinase inhibitor 2A (CDKN2A), a known suppressor of cuproptosis, ultimately blocking copper-dependent cell death. Conversely, restoring MUC20 expression reduces IGF-1R lactylation, disrupts MET/CDKN2A signaling, and reinstates cuproptosis sensitivity.13 Extrachromosomal circular DNA (eccDNA) amplifies KIF3C to downregulate MUC20, initiating this resistance cascade. In summary, lactylation sustains cuproptosis resistance by stabilizing the IGF-1R/MET/CDKN2A signaling axis, enabling cancer cells to evade copper-induced death13 (Figure 6).
Lin et al revealed that L-lactate-induced lactylation of Nudix hydrolase 21 (NUDT21), a core component of the 3′ end processing machinery at lysine 23 (K23), catalyzed by the “writer” enzyme AARS1 and reversed by the “eraser” HDAC2, reprograms alternative polyadenylation (APA) to promote resistance to cuproptosis in esophageal squamous cell carcinoma (ESCC).14 Specifically, NUDT21 lactylation enhances its interaction with CPSF6, facilitating CFIm complex formation and driving the selection of distal polyadenylation sites (dPAS) in the FDX1 transcript.14 This results in 3’ UTR lengthening of FDX1 mRNA, which destabilizes the transcript and reduces FDX1 protein expression. Since FDX1 is essential for cuproptosis (by reducing Cu2⁺ to Cu⁺ and inducing mitochondrial protein lipoylation), its downregulation directly confers resistance to copper-induced cell death.14 Clinically, elevated NUDT21 lactylation correlates with reduced FDX1 and worse ESCC prognosis, and combined targeting of lactate production (via LDHA inhibitor stiripentol) and copper delivery (via ionophore elesclomol) synergistically overcomes this resistance.14 In summary, lactylation of NUDT21 suppresses FDX1 expression through APA-mediated mRNA destabilization, thereby enabling cancer cells to evade cuproptosis14 (Figure 6).
Lactylation drives cuproptosis resistance via distinct mechanisms across cancers: in multiple myeloma (MM), IGF-1R lactylation stabilizes MET/CDKN2A signaling to suppress cuproptosis, while in esophageal squamous cell carcinoma (ESCC), lactylation of NUDT21 reprograms alternative polyadenylation to destabilize FDX1 mRNA—a core cuproptosis executor—thus conferring copper tolerance. Both pathways converge on metabolic adaptation, where elevated lactate fuels lactylation-mediated evasion of copper-induced death. However, unresolved questions include the precise structural impact of lactylation on IGF-1R/NUDT21 function, tissue-specific drivers selecting divergent lactylation targets (eg, IGF-1R vs NUDT21), and whether lactylation broadly regulates other cuproptosis components (eg, DLAT, LIAS). Future studies should develop lactylation-specific inhibitors (eg, blocking “writer” enzymes like AARS1) to disrupt resistance pathways. Map lactylation interactomes in cuproptosis-sensitive cancers to identify novel targets. Explore lactate modulation (eg, LDHA inhibition) combined with copper ionophores in in vivo models to overcome therapy resistance. Future work must address context-dependent regulatory networks and translate mechanistic insights into targeted therapies and investigate cross-talk between lactylation and other post-translational modifications in fine-tuning cuproptosis sensitivity.
Non-Histone Lactylation Regulates Resistance to Apoptosis
This study reveals that elevated sphingomyelin (SM) in acute lymphoblastic leukemia (ALL) reprograms glucose metabolism by enhancing SLC2A1-mediated glucose uptake and glycolysis, leading to increased lactate production15 (Figure 6). The accumulated lactate drives lactylation of caspase-3 (CASP3) specifically at lysine 14 (K14). This K14 lactylation physically impedes the proteolytic activation of CASP3, thereby disabling its apoptotic function and conferring apoptosis resistance to ALL cells.15 Critically, depletion of SM reduces lactate levels, decreases CASP3 K14 lactylation, restores CASP3 activation, and sensitizes cells to apoptosis. In summary, SM-driven lactate production mediates CASP3 K14 lactylation to prevent its proteolytic activation, conferring apoptosis resistance in leukemia.15
Link Lactylation to Autophagy-Mediated Resistance to Regulated Cell Death
Certainly, several recent studies have established a direct molecular link between lactylation, the induction of autophagy, and subsequent resistance to regulated cell death. Lactylation is emerging as a direct regulator of autophagy that promotes resistance to regulated cell death. Recent evidence demonstrates that lactate, a glycolysis byproduct, serves as a precursor for lactylation, a post-translational modification that directly activates core components of the autophagy machinery. Specifically, lactylation of the key autophagy regulator Vps34 at lysine residues 356 and 781 enhances its lipid kinase activity by strengthening its interaction with regulatory subunits like Beclin-1.30,124,125 This Vps34 hyperlactylation significantly promotes autophagic flux and the endolysosomal degradation pathway.125 In the context of cellular stress, this lactate-driven autophagy can suppress cell death. For instance, in models of spinal cord injury, lactate-mediated lactylation of the Ran protein facilitates astrocyte polarization and survival by promoting STAT3 nuclear transport.126 Conversely, inhibiting this lactylation pathway reverses the protective autophagy and increases susceptibility to cell death. Furthermore, in stressed melanoma cells, the acidic microenvironment created by lactic acid was shown to suppress glucose deprivation-induced autophagic cell death.124 Therefore, lactylation functions as a critical metabolic switch that links glycolytic activity to pro-survival autophagy, conferring resistance to various forms of regulated cell death.
Targeting Lactylation Overcome Resistance to Regulated Cell Death
Against Ferroptosis Resistance
Inhibiting histone lactylation (via 2-DG) restores ferroptosis sensitivity by disrupting this pathway.118 Deoxy-D-glucose (2-DG) functions as a competitive glycolytic inhibitor by mimicking glucose structure yet lacking the 2-hydroxyl group essential for further glycolysis. This non-metabolizable glucose analog enters cells via glucose transporters (GLUTs) and undergoes phosphorylation by hexokinase to form 2-DG-6-phosphate. Unlike glucose-6-phosphate, this metabolite cannot be isomerized or further processed in glycolysis, resulting in intracellular accumulation that allosterically inhibits hexokinase activity and blocks glycolytic flux. In endometriotic stromal cells (EESCs), 2-DG significantly reduces lactate production by disrupting the Warburg effect—a hallmark metabolic shift in endometriosis where cells prioritize glycolysis over oxidative phosphorylation even under normoxic conditions.118 The consequent depletion of lactate, the substrate for histone lactylation, directly reduces global lysine lactylation (pan-Kla) and specifically diminishes histone H3 lysine 18 lactylation (H3K18la).118 This epigenetic modulation occurs via reduced enrichment of H3K18la at the promoter of METTL3, a key RNA methyltransferase, leading to decreased METTL3 expression and subsequent reduction of N6-methyladenosine (m6A) RNA modifications.118 Through this cascade, 2-DG disrupts the METTL3-regulated HIF1α/HMOX1 antioxidant signaling axis, thereby impairing cellular defenses against ferroptosis.118 Phenotypically, 2-DG treatment sensitizes EESCs to ferroptosis inducers (eg, erastin) by depleting glutathione (GSH), increasing lipid peroxidation (elevated MDA and Liperfluo staining), and accumulating intracellular Fe2⁺.118 In vivo, 2-DG synergizes with erastin to suppress endometriotic lesion growth in mice, demonstrating its therapeutic potential. This synergy arises from 2-DG’s dual role: metabolic disruption of lactate-dependent pathways and epigenetic silencing of pro-survival genes, ultimately overcoming ferroptosis resistance in endometriosis118 (Figure 7 and Table 2).
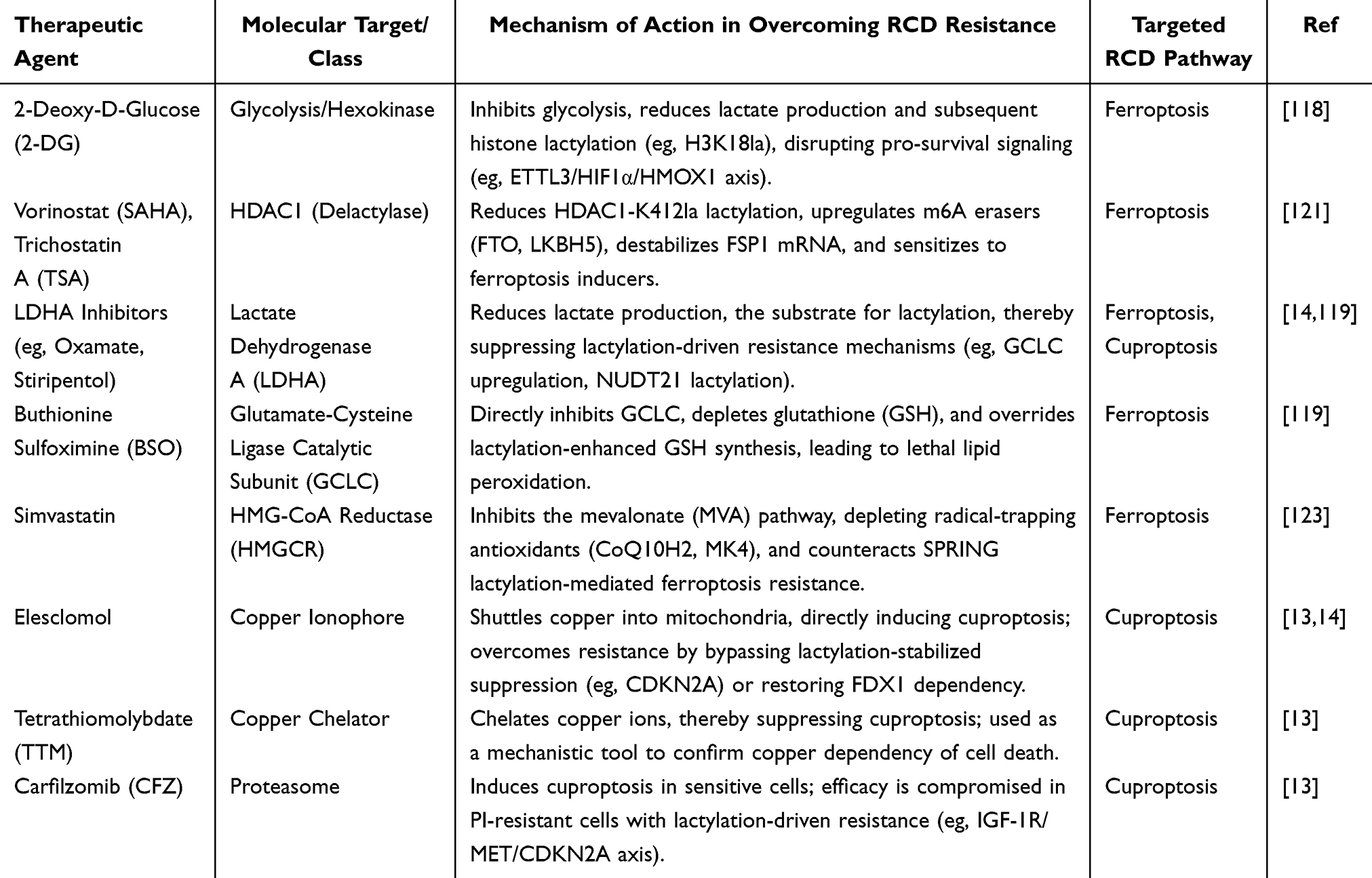 |
Table 2 Therapeutic Strategies Targeting Lactylation to Overcome Resistance to Regulated Cell Death (RCD) |
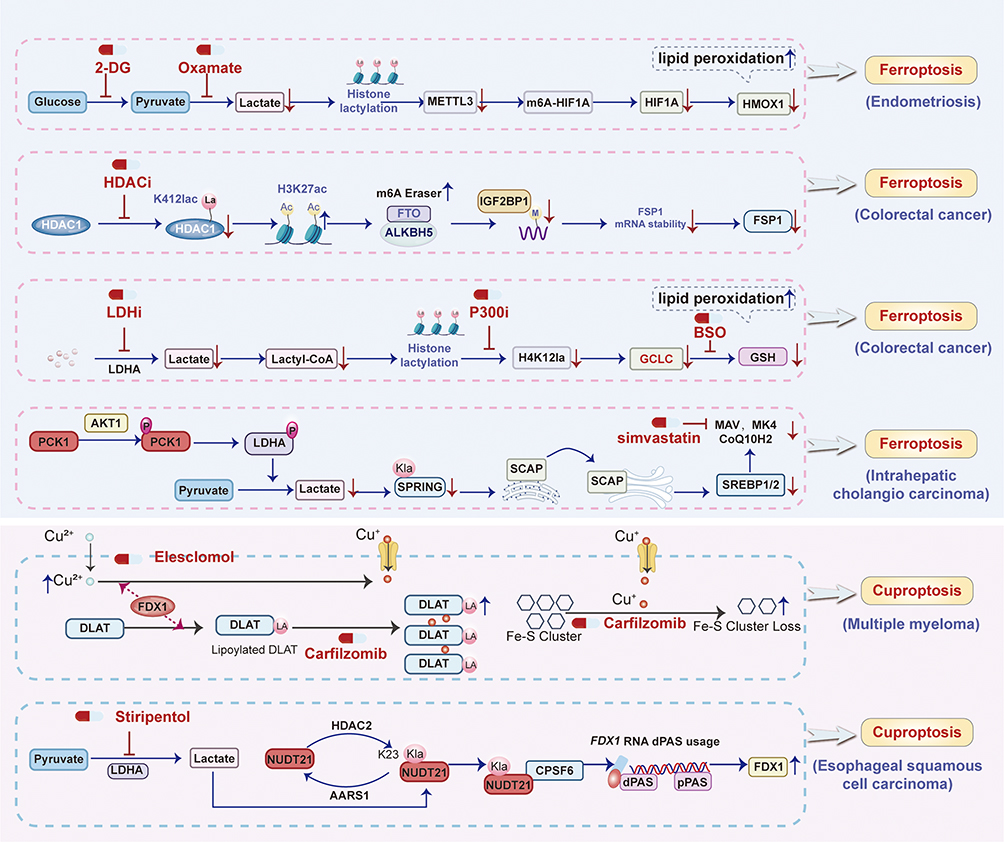 |
Figure 7 Pharmacologically targeting lactylation overcome resistance to ferroptosis and cuproptosis. |
Critically, HDAC inhibitors (HDACi) like SAHA and TSA reduce HDAC1-K412la lactylation, destabilize FSP1 mRNA, and sensitize CRC to ferroptosis inducers. Lactylation of HDAC1-K412la stabilizes FSP1 mRNA via m6A modification to confer ferroptosis resistance in colorectal cancer121 (Figure 7 and Table 2). Vorinostat (SAHA) and Trichostatin A (TSA) both pan-histone deacetylase inhibitors (HDACis), exert their primary pharmacological action by selectively inhibiting HDAC1, a key epigenetic regulator.121 Their mechanism in sensitizing colorectal cancer (CRC) to ferroptosis involves a multi-step pathway initiated by the reduction of lactylation at lysine 412 (K412) on HDAC1 itself—a novel post-translational modification identified in this study. This diminished lactylation impairs HDAC1’s deacetylase function, leading to increased acetylation of histone H3 at lysine 27 (H3K27ac) specifically at the promoter regions of two critical RNA demethylases, FTO and ALKBH5.121 Consequently, the enhanced H3K27ac promotes transcriptional upregulation of FTO and ALKBH5. These enzymes, acting as m6A erasers, then reduce N6-methyladenosine (m6A) methylation on the mRNA of ferroptosis suppressor protein 1 (FSP1), a key defender against lipid peroxidation-driven cell death.121 The decreased m6A modification destabilizes FSP1 mRNA, accelerating its degradation and ultimately suppressing FSP1 protein expression. Since FSP1 maintains cellular resistance by regenerating the antioxidant ubiquinol (CoQH2) to neutralize lipid radicals, its downregulation cripples the CRC’s anti-ferroptosis defense system.121 This renders cancer cells profoundly vulnerable to ferroptosis inducers (eg, RSL3, erastin, IKE), causing synergistic cell death through accumulated lipid peroxidation. Importantly, SAHA and TSA demonstrate superior efficacy in reducing HDAC1[K412] lactylation compared to other HDAC1 inhibitors, correlating directly with their potency in ferroptosis sensitization. Pharmacologically, while both agents share this core mechanism, their clinical relevance is amplified by the finding that combining sublethal doses of these HDACis with ferroptosis inducers achieves potent antitumor effects in vivo (demonstrated in xenograft and AOM/DSS models) while potentially mitigating the dose-limiting toxicities (eg, myelosuppression, gastrointestinal effects) associated with higher doses of HDACis or conventional chemotherapy like 5-FU+oxaliplatin.121 Thus, their pharmacological role extends beyond traditional HDAC inhibition to encompass the regulation of lactylation-dependent epigenetic-to-RNA modification crosstalk, positioning them as strategic agents for combination therapy in ferroptosis-resistant CRC.121
Inhibition of the lactate-producing enzyme LDHA, the lactyltransferase p300, or GCLC itself (using BSO) restores ferroptosis sensitivity and overcomes chemoresistance119 (Figure 7). LDHA inhibitor (eg, oxamate) pharmacologically targets lactate dehydrogenase A (LDHA), a key enzyme in glycolysis that converts pyruvate to lactate. By inhibiting LDHA, LDHi reduces intracellular lactate production, which serves as the substrate for histone lactylation. This reduction in lactate availability decreases histone H4 lysine 12 lactylation (H4K12la) in colorectal cancer stem cells (CCSCs).119 Lower H4K12la levels diminish transcriptional activation of the ferroptosis-related gene GCLC (glutamate-cysteine ligase catalytic subunit), a critical enzyme in glutathione (GSH) synthesis.119 Consequently, impaired GSH biosynthesis weakens cellular antioxidant defenses, leading to accumulation of lipid peroxides and sensitization to ferroptosis.119 This mechanism enhances the efficacy of chemotherapeutic agents like oxaliplatin by overcoming CCSC chemoresistance, as validated both in vitro and in vivo through suppressed tumor growth and increased ferroptosis markers (eg, lipid ROS, MDA).119 Trichostatin A (TSA), an HDAC1 inhibitor, functions as a delactylase antagonist.119 By inhibiting HDAC1—identified as a histone delactylase—TSA increases global histone lactylation, including H4K12la, in CCSCs. Elevated H4K12la promotes GCLC transcription, enhancing GSH synthesis and inhibiting ferroptosis.119 Paradoxically, this confers chemoresistance; however, TSA’s pharmacological utility lies in its role as a tool to elucidate lactylation dynamics. When combined with lactate-depleting agents (eg, LDHi), TSA’s pro-lactylation effects are counteracted, indirectly supporting strategies to sensitize CCSCs to chemotherapy.119 Buthionine sulfoximine (BSO) directly inhibits GCLC, the rate-limiting enzyme in GSH synthesis. By depleting GSH, BSO disrupts the cellular redox balance, leading to uncontrolled lipid peroxidation and ferroptosis.119 Pharmacologically, BSO overrides the lactylation-driven GCLC upregulation in CCSCs, effectively reversing chemoresistance.119 In vitro, BSO synergizes with oxaliplatin to induce ferroptosis (evidenced by elevated lipid ROS/MDA); in vivo, it significantly suppresses tumor growth in xenograft models.119 This positions BSO as a therapeutic adjuvant to standard chemotherapy, particularly in GCLC-overexpressing, oxaliplatin-resistant CCSCs.119 Collectively, these agents target interconnected nodes of the lactylation-GCLC-ferroptosis axis: LDHi mitigates lactate-driven histone lactylation to suppress GCLC. TSA validates HDAC1’s role as a lactylation eraser but requires combinatorial use to avoid resistance. BSO directly inhibits GCLC to induce ferroptosis, offering a clinically translatable strategy to enhance chemosensitivity in colorectal cancer.119
Disrupting this axis—via SPRING K82R mutation (lactylation-deficient) or simvastatin (MVA inhibitor)—sensitizes ICC cells to ferroptosis and overcomes chemo-immunotherapy resistance.123 In summary, lactylation of SPRING at K82, mediated by the pPCK1-pLDHA axis-induced lactate accumulation, activates the MVA pathway to synthesize radical-trapping antioxidants that suppress lipid peroxidation and confer ferroptosis resistance.123
Simvastatin’s inhibition of HMGCR reverses this resistance by depleting CoQ10H2 and MK4, sensitizing ICC cells to ferroptosis.123 This effect is mechanistically validated by rescue experiments where exogenous MVA supplementation (MVA-Li) restores ferroptosis resistance, confirming the specificity of Simvastatin’s action on the MVA pathway.123 In preclinical models, Simvastatin synergizes with CIT by amplifying lipid peroxidation (increased MDA levels), enhancing CD8⁺ T-cell infiltration and activation (elevated TNFα⁺/IFNγ⁺ CD8⁺ T cells), and suppressing tumor growth in both subcutaneous and hydrodynamic ICC mouse models.123 Simvastatin’s ability to target this axis positions it as a rational adjunct therapy, particularly in AKT-hyperactivated ICC subtypes. This pharmacological strategy leverages statins’ established safety profile to overcome metabolic reprogramming-driven therapy resistance, offering a translatable approach to improve clinical outcomes.123
Against Cuproptosis Resistance
Carfilzomib (CFZ) functions as a second-generation proteasome inhibitor, core to MM treatment.13 It induces cell death in PI-sensitive MM cells primarily by triggering cuproptosis, a copper-dependent form of programmed cell death (Figure 7). Mechanistically, CFZ promotes the aggregation of lipoylated proteins (eg, DLAT) and loss of iron-sulfur cluster proteins, leading to proteotoxic stress.13 However, in PI-resistant MM cells, CFZ fails to elicit cuproptosis due to downregulated MUC20 and dysregulated downstream pathways (MET/CDKN2A). At concentrations of 10–20 nM, CFZ inhibits proteasome activity but only reduces viability in sensitive cells, highlighting its dependence on intact cuproptosis machinery.13
Tetrathiomolybdate (TTM) acts as a high-affinity copper chelator, disrupting cuproptosis by sequestering copper ions. Pharmacologically, TTM (at 50 nM) completely reverses CFZ-induced cell death in PI-sensitive MM cells, confirming copper’s essential role in this process. In rescue experiments, TTM neutralizes the pro-cuproptotic effects of MUC20 overexpression in resistant cells, demonstrating its utility in dissecting copper-dependent mechanisms. Its efficacy is dose-dependent, with 50 nM sufficient to abrogate cuproptosis without off-target cytotoxicity13 (Figure 7).
Elesclomol operates as a copper ionophore that complexes with extracellular copper (Cu2⁺) and shuttles it into cells.13 At 50–150 nM, elesclomol potently restores cuproptosis in PI-resistant MM cells by overwhelming endogenous copper homeostasis. It synergizes with CFZ, significantly reducing proliferation and increasing programmed cell death (PCD) in resistant lines. In vivo, elesclomol (25 mg/kg) combined with CFZ suppresses tumor growth in xenograft models, validating its role in re-sensitizing resistant MM to PI therapy via copper delivery13 (Figure 7). This study revealed a dynamic balance wherein CFZ efficacy depends on functional cuproptosis, compromised in resistance. TTM and elesclomol serve as mechanistic tools: TTM suppresses cuproptosis by copper depletion, while elesclomol induces it by copper overload. Critically, elesclomol overcomes PI resistance by bypassing MUC20 deficiency and directly forcing copper-dependent cell death. This supports targeting copper metabolism as a strategy to overcome PI resistance, with elesclomol emerging as a promising adjuvant to proteasome inhibitors in refractory MM.13
Stiripentol, clinically approved for Dravet syndrome, functions as a potent inhibitor of lactate dehydrogenase A (LDHA), a key enzyme in the glycolytic pathway responsible for converting pyruvate to lactate. By inhibiting LDHA, stiripentol effectively reduces intracellular lactate production, thereby diminishing the substrate available for the lactylation modification catalyzed by AARS114 (Figure 7). This reduction in lactate levels consequently attenuates the lactylation of NUDT21 at lysine 23 (K23), a critical post-translational modification identified in the study.14 The suppression of NUDT21 lactylation disrupts its enhanced interaction with CPSF6 and the subsequent formation of the CFIm complex. Consequently, this prevents the NUDT21-mediated shift towards distal polyadenylation site (dPAS) usage in the FDX1 gene, leading to the restoration of FDX1 protein expression.14 FDX1 is essential for cuproptosis, as it reduces Cu2⁺ to the more toxic Cu⁺ and facilitates mitochondrial protein lipoylation, ultimately triggering copper-dependent cell death.14 Therefore, stiripentol’s primary pharmacological action in this context is metabolic reprogramming, sensitizing esophageal squamous cell carcinoma (ESCC) cells to copper-induced stress by reversing lactylation-driven resistance mechanisms.14
Elesclomol operates as a copper ionophore, forming a stable 1:1 complex with extracellular Cu2⁺ ions (eg, in the form of elesclomol-Cu2⁺). This complex facilitates the active transport and intracellular delivery of copper, specifically targeting mitochondria. Once inside the mitochondria, the released copper ions directly bind to lipoylated components of the tricarboxylic acid (TCA) cycle. This binding induces proteotoxic stress through the aggregation of lipoylated mitochondrial proteins and subsequent disruption of iron-sulfur cluster proteins, culminating in a unique form of regulated cell death termed cuproptosis. The efficacy of elesclomol is critically dependent on functional FDX1, which reduces the delivered Cu2⁺ to Cu⁺, the redox-active form responsible for initiating the lipoylated protein aggregation. Thus, elesclomol’s core pharmacological mechanism is the targeted induction of copper-dependent cytotoxicity via mitochondrial copper overload14 (Figure 7). The study demonstrated a powerful synergistic interaction between stiripentol and elesclomol in suppressing ESCC tumor growth in vivo.14 This synergy arises from their complementary mechanisms targeting the lactate-NUDT21-FDX1-cuproptosis axis. Stiripentol, by inhibiting lactate production and NUDT21 lactylation, restores FDX1 expression and removes the block to cuproptosis sensitivity.14 Elesclomol, concurrently, provides the cytotoxic copper payload. By overcoming lactylation-mediated resistance (via stiripentol) and effectively delivering the copper death signal (via elesclomol), the combination therapy achieves significantly greater anti-tumor efficacy than either agent alone.14 This synergistic effect highlights the therapeutic potential of co-targeting the metabolic adaptation (lactate/LDHA) and the cell death pathway (copper/cuproptosis) in cancers like ESCC characterized by elevated glycolysis, lactate, and NUDT21 expression. The combination was reported to be well-tolerated in the preclinical models described.14
Conclusions and Perspectives
This review consolidates the paradigm-shifting role of lysine lactylation (Kla) as a master metabolic regulator orchestrating resistance to regulated cell death (RCD) across diverse pathologies, particularly cancer. By dynamically modifying histones and non-histone proteins, lactylation establishes a robust shield against ferroptosis, cuproptosis, and apoptosis. The convergence of elevated lactate—a hallmark of pathological microenvironments—and specific lactyltransferases drives the reprogramming of epigenetic landscapes, RNA stability, metabolic fluxes, and metal homeostasis to sustain cell survival.
Lactylation epigenetically upregulates antioxidant genes (HMOX1, GCLC, NFS1), stabilizes suppressors (SLC7A11, FSP1) via RNA methylation, enhances glutathione synthesis (GCLM lactylation), activates NRF2 signaling (PRDX1 lactylation), and disrupts iron availability (LSD1-mediated TFRC repression; NFS1 upregulation). IGF-1R lactylation stabilizes the CDKN2A suppressor, while NUDT21 lactylation reprograms alternative polyadenylation to destabilize FDX1 mRNA, disabling mitochondrial copper toxicity. Caspase-3 lactylation directly impedes proteolytic activation, blocking apoptotic execution. These findings position lactylation as a central metabolic-epigenetic switch that rewires cellular stress responses to promote pathological resilience and therapeutic resistance. However, critical outstanding questions remain unresolved. What dictates the selective targeting of lactylation “writers” (eg, ACAT2 in KRAS-mutant cancer vs KAT7 in cholangiocarcinoma) and their substrate preferences in different disease contexts? How does lactylation integrate with other post-translational modifications (eg, acetylation, phosphorylation, ubiquitination) to fine-tune RCD sensitivity? Does lactylation compete with or reinforce these modifications? What is the precise structural impact of lactylation on enzyme function (eg, conformational changes in caspase-3 or IGF-1R) and how does lactylation alter reader protein engagement? Does lactylation regulate additional RCD components (eg, GPX4, DLAT, BCL-2 family members) or novel RCD modalities beyond the three discussed?
Future research directions should focus on translating these mechanistic insights into therapeutic strategies. Develop isoform-specific inhibitors of lactyltransferases (eg, KAT7, ACAT2, NAA10) or delactylases to disrupt lactylation-driven RCD resistance. Combinatorial approaches—pairing lactate depletion (via LDHA inhibitors) with RCD inducers (eg, ferroptosis inducers, copper ionophores)—hold promise for overcoming therapy-refractory diseases. Systematically map the “lactylome” across disease states and therapy-resistant niches to identify context-specific vulnerabilities. Integrate lactylation data with transcriptomic, metabolomic, and proteomic datasets to decipher regulatory networks. Investigate how lactate from stromal cells, immune infiltrates, or microbiota fuels lactylation in tumor cells, and whether modulating the microenvironment sensitizes cells to RCD. Establish genetically engineered models (eg, lactylation-site mutants) to rigorously test the causal role of lactylation in RCD resistance in vivo. Explore lactylation’s impact on antitumor immunity and its synergy with immunotherapy. Evaluate lactylation levels (eg, H3K18la, lactylated PRDX1) as prognostic markers or predictors of treatment response in clinical cohorts.
In conclusion, lactylation represents a fundamental link between cellular metabolism and cell fate determination. Targeting this axis offers a transformative strategy to dismantle the shield of RCD resistance, paving the way for novel therapies against cancer and other lactate-rich pathological conditions. Unlocking the full potential of lactylation modulation requires concerted efforts to address its contextual complexity and harness its therapeutic promise.
Highlights
- Lactylation serves as a master metabolic switch for regulated cell death (RCD) resistance.
- Diverse molecular mechanisms confer ferroptosis resistance.
- Lactylation subverts cuproptosis and apoptosis.
- Targeting lactylation regulators or combining lactate modulation with RCD inducers lactylation-mediated RCD resistance.
Data Sharing Statement
All data generated or analyzed during this study are included in this article.
Funding
This work was supported in part by the Beijing Natural Science Foundation (No. 7252174), Wu Jieping Medical Foundation (320.6750.2024-13-59), Natural Science Foundation of Inner Mongolia Autonomous Region (2025MS08029), Talent development plan for the future in Medical-Engineering Integration by BRA-CDCHE and ZTA (MBRC0012025029; MBRC0012025011), Science Foundation of ASCH (YN202305; YN202402; YN202423; YN202503), and the Science Foundation of AMHT (2024YK04; 2024YK05).
Disclosure
The authors declare no conflict of interest.
References
1. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi:10.1016/j.cell.2012.03.042
2. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22(7):381–396. doi:10.1038/s41568-022-00459-0
3. Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185:2401–2421. doi:10.1016/j.cell.2022.06.003
4. Tsvetkov P, Coy S, Petrova B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–1261. doi:10.1126/science.abf0529
5. Wang Y, Chen Y, Zhang J, et al. Cuproptosis: a novel therapeutic target for overcoming cancer drug resistance. Drug Resist Updat. 2024;72:101018. doi:10.1016/j.drup.2023.101018
6. Yin L, Liu P, Jin Y, Ning Z, Yang Y, Gao H. Ferroptosis-related small-molecule compounds in cancer therapy: strategies and applications. Eur J Med Chem. 2022;244:114861. doi:10.1016/j.ejmech.2022.114861
7. Lindsten T, Ross AJ, King A, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi:10.1016/S1097-2765(00)00136-2
8. Qiu Y, Shao X. Histone lactylation in diseases: regulation by Traditional Chinese medicine and therapeutic implications. Drug Des Devel Ther. 2025;19:6435–6459. doi:10.2147/DDDT.S524008
9. Chen Y, Yan Q, Ruan S, et al. GCLM lactylation mediated by ACAT2 promotes ferroptosis resistance in KRASG12D-mutant cancer. Cell Rep. 2025;44:115774. doi:10.1016/j.celrep.2025.115774
10. Liao Z, Chen B, Yang T, Zhang W, Mei Z. Lactylation modification in cardio-cerebral diseases: a state-of-the-art review. Ageing Res Rev. 2025;104:102631. doi:10.1016/j.arr.2024.102631
11. Yang T, Zhang S, Nie K, et al. ZNF207-driven PRDX1 lactylation and NRF2 activation in regorafenib resistance and ferroptosis evasion. Drug Resist Updat. 2025;82:101274. doi:10.1016/j.drup.2025.101274
12. Huang J, Xie H, Li J, et al. Histone lactylation drives liver cancer metastasis by facilitating NSF1-mediated ferroptosis resistance after microwave ablation. Redox Biol. 2025;81:103553. doi:10.1016/j.redox.2025.103553
13. Wang X, Shi Y, Shi H, et al. MUC20 regulated by extrachromosomal circular DNA attenuates proteasome inhibitor resistance of multiple myeloma by modulating cuproptosis. J Exp Clin Cancer Res. 2024;43:68. doi:10.1186/s13046-024-02972-6
14. Lin J, Yin Y, Cao J, et al. NUDT21 lactylation reprograms alternative polyadenylation to promote cuproptosis resistance. Cell Discov. 2025;11:52. doi:10.1038/s41421-025-00804-1
15. Lin Z, Long F, Liu J, et al. Metabolic reprogramming promotes apoptosis resistance in acute lymphoblastic leukemia through CASP3 lactylation. Mol Cancer. 2025;24:204. doi:10.1186/s12943-025-02392-w
16. Wang X, Wang N, Wang W, et al. Histone lactylation boosts reparative gene activation post-myocardial infarction. Circ Res. 2022;131:893–908. doi:10.1161/CIRCRESAHA.122.320488
17. Wang Y, Chen J, Wang Y, Zhao J, Zhang J, Wang H. Pharmacologically targeting protein lactylation to overcome cancer drug resistance. Eur J Med Chem. 2025;296:117905. doi:10.1016/j.ejmech.2025.117905
18. Gao X, Pang C, Fan Z, Wang Y, Duan Y, Zhan H. Regulation of newly identified lysine lactylation in cancer. Cancer Lett. 2024;587:216680. doi:10.1016/j.canlet.2024.216680
19. He Y, Song T, Ning J, et al. Lactylation in cancer: mechanisms in tumour biology and therapeutic potentials. Clin Transl Med. 2024;14:e70070. doi:10.1002/ctm2.70070
20. Li H, Sun L, Gao P, Hu H. Lactylation in cancer: current understanding and challenges. Cancer Cell. 2024;42:1803–1807. doi:10.1016/j.ccell.2024.09.006
21. Lv M, Huang Y, Chen Y, Ding K. Lactylation modification in cancer: mechanisms, functions, and therapeutic strategies. Exp Hematol Oncol. 2025;14:
22. Shi H, Zou Y, Jin S, Wu J, Liu B. Lactate-induced lactylation: from basic research to clinical perspectives. Front Pharmacol. 2025;16:1586973. doi:10.3389/fphar.2025.1586973
23. Iozzo M, Pardella E, Giannoni E, Chiarugi P. The role of protein lactylation: a kaleidoscopic post-translational modification in cancer. Mol Cell. 2025;85:S1097–2765(25)00142–00142X[pii]. doi:10.1016/j.molcel.2025.02.011
24. Zhang N, Zhang Y, Xu J, et al. α-myosin heavy chain lactylation maintains sarcomeric structure and function and alleviates the development of heart failure. Cell Res. 2023;33(9):679–698. doi:10.1038/s41422-023-00844-w
25. Zhang D, Tang Z, Huang H, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574(7779):575–580. doi:10.1038/s41586-019-1678-1
26. Zhu R, Ye X, Lu X, et al. ACSS2 acts as a lactyl-CoA synthetase and couples KAT2A to function as a lactyltransferase for histone lactylation and tumor immune evasion. Cell Metab. 2025;37:361–376.e7. doi:10.1016/j.cmet.2024.10.015
27. Chen Y, Wu J, Zhai L, et al. Metabolic regulation of homologous recombination repair by MRE11 lactylation. Cell. 2024;187:294–311.e21. doi:10.1016/j.cell.2023.11.022
28. Yang K, Fan M, Wang X, et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. 2022;29(1):133–146. doi:10.1038/s41418-021-00841-9
29. Li H, Li Y, Chen H, et al. NBS1 lactylation is required for efficient DNA repair and chemotherapy resistance. Nature. 2024;631:663–669. doi:10.1038/s41586-024-07620-9
30. Sun W, Jia M, Feng Y, et al. Lactate is a bridge linking glycolysis and autophagy through lactylation. Autophagy. 2023;19(12):3240–3241. doi:10.1080/15548627.2023.2246356
31. Xie B, Zhang M, Li J, et al. KAT8-catalyzed lactylation promotes eEF1A2-mediated protein synthesis and colorectal carcinogenesis. Proc Natl Acad Sci U S A. 2024;121:e2314128121. doi:10.1073/pnas.2314128121
32. Yuan J, Yang M, Wu Z, et al. The lactate-primed KAT8‒PCK2 axis exacerbates hepatic ferroptosis during ischemia/reperfusion injury by reprogramming OXSM-dependent mitochondrial fatty acid synthesis. Adv Sci. 2025;12(11):e2414141. doi:10.1002/advs.202414141
33. Niu K, Chen Z, Li M, et al. NSUN2 lactylation drives cancer cell resistance to ferroptosis through enhancing GCLC-dependent glutathione synthesis. Redox Biol. 2025;79:103479. doi:10.1016/j.redox.2024.103479
34. Ju J, Zhang H, Lin M, et al. The alanyl-tRNA synthetase AARS1 moonlights as a lactyltransferase to promote YAP signaling in gastric cancer. J Clin Invest. 2024;134:e174587. doi:10.1172/JCI174587
35. Li H, Liu C, Li R, et al. AARS1 and AARS2 sense L-lactate to regulate cGAS as global lysine lactyltransferases. Nature. 2024;634:1229–1237. doi:10.1038/s41586-024-07992-y
36. Zong Z, Xie F, Wang S, et al. Alanyl-tRNA synthetase, AARS1, is a lactate sensor and lactyltransferase that lactylates p53 and contributes to tumorigenesis. Cell. 2024;187:2375–2392.e33. doi:10.1016/j.cell.2024.04.002
37. Mao Y, Zhang J, Zhou Q, et al. Hypoxia induces mitochondrial protein lactylation to limit oxidative phosphorylation. Cell Res. 2024;34:13–30. doi:10.1038/s41422-023-00864-6
38. Liu R, Ren X, Park YE, et al. Nuclear GTPSCS functions as a lactyl-CoA synthetase to promote histone lactylation and gliomagenesis. Cell Metab. 2025;37:377–394.e9. doi:10.1016/j.cmet.2024.11.005
39. Du R, Gao Y, Yan C, et al. Sirtuin 1/sirtuin 3 are robust lysine delactylases and sirtuin 1-mediated delactylation regulates glycolysis. Iscience. 2024;27:110911. doi:10.1016/j.isci.2024.110911
40. Sun Y, Xu Y, Zhang Y, et al. Genetic encoding of ε-N-L-lactyllysine for detecting delactylase activity in living cells. Chemical Communications. 2022;58:8544–8547. doi:10.1039/D2CC02643K
41. Jennings EQ, Ray JD, Zerio CJ, et al. Sirtuin 2 regulates protein lactoyllys modifications. Chembiochem. 2021;22:2102–2106. doi:10.1002/cbic.202000883
42. Zu H, Li C, Dai C, et al. SIRT2 functions as a histone delactylase and inhibits the proliferation and migration of neuroblastoma cells. Cell Discov. 2022;8:54. doi:10.1038/s41421-022-00398-y
43. Fan Z, Liu Z, Zhang N, et al. Identification of SIRT3 as an eraser of H4K16la. Iscience. 2023;26:107757. doi:10.1016/j.isci.2023.107757
44. Jin J, Bai L, Wang D, et al. SIRT3-dependent delactylation of cyclin E2 prevents hepatocellular carcinoma growth. EMBO Rep. 2023;24:e56052. doi:10.15252/embr.202256052
45. Chen C, Zhang Y, Zang Y, et al. SIRT3 functions as an eraser of histone H3K9 lactylation to modulate transcription for inhibiting the progression of esophageal cancer. Mol Cell Proteomics. 2025;24(5):100973. doi:10.1016/j.mcpro.2025.100973
46. Moreno-Yruela C, Zhang D, Wei W, et al. Class I histone deacetylases (HDAC1-3) are histone lysine delactylases. Sci Adv. 2022;8:eabi6696. doi:10.1126/sciadv.abi6696
47. Li F, Si W, Xia L, et al. Positive feedback regulation between glycolysis and histone lactylation drives oncogenesis in pancreatic ductal adenocarcinoma. Mol Cancer. 2024;23:90. doi:10.1186/s12943-024-02008-9
48. Zessin M, Meleshin M, Praetorius L, Sippl W, Bařinka C, Schutkowski M. Uncovering robust delactoylase and depyruvoylase activities of HDAC isoforms. ACS Chem Biol. 2022;17:1364–1375. doi:10.1021/acschembio.1c00863
49. Hu X, Huang X, Yang Y, et al. Dux activates metabolism-lactylation-MET network during early iPSC reprogramming with Brg1 as the histone lactylation reader. Nucleic Acids Res. 2024;52:5529–5548. doi:10.1093/nar/gkae183
50. Zhai G, Niu Z, Jiang Z, et al. DPF2 reads histone lactylation to drive transcription and tumorigenesis. Proc Natl Acad Sci U S A. 2024;121:e2421496121. doi:10.1073/pnas.2421496121
51. Passarella S, de Bari L, Valenti D, et al. Mitochondria and l -lactate metabolism. FEBS Lett. 2008;582(25–26):3569–3576. doi:10.1016/j.febslet.2008.09.042
52. Chen XJ, Wang X, Kaufman BA, et al. Aconitase couples metabolic regulation to mitochondrial DNA maintenance. Science. 2005;307(5710):714–717. doi:10.1126/science.1106391
53. Schell JC, Olson KA, Jiang L, et al. A role for the mitochondrial pyruvate carrier as a repressor of the Warburg effect and colon cancer cell growth. Mol Cell. 2014;56(3):400–413. doi:10.1016/j.molcel.2014.09.026
54. Wang Y, Wu S, Li Q, Sun H, Wang H. Pharmacological inhibition of ferroptosis as a therapeutic target for neurodegenerative diseases and strokes. Adv Sci. 2023;10:e2300325.doi:doi: 10.1002/advs.202300325
55. Wang Y, Wu S, Wang CX, et al. Interplay of cGAS-STING and ferroptosis: crosstalk, molecular mechanisms, and therapeutic prospects. Arch Toxicol. 2025;2025:1–23.doi:doi: 10.1007/s00204-025-04150-9
56. Wang Y, Wu X, Ren Z, et al. Overcoming cancer chemotherapy resistance by the induction of ferroptosis. Drug Resist Updat. 2023;66:100916. doi:10.1016/j.drup.2022.100916
57. Tang D, Kroemer G. Ferroptosis. Curr Biol. 2020;30:R1292–1292R1297. doi:10.1016/j.cub.2020.09.068
58. Chen X, Kang R, Kroemer G, Tang D. Ferroptosis in infection, inflammation, and immunity. J Exp Med. 2021;218:e20210518. doi:10.1084/jem.20210518
59. Hadian K, Stockwell BR. SnapShot: ferroptosis. Cell. 2020;181(5):1188–1188.e1. doi:10.1016/j.cell.2020.04.039
60. Wang R, Chen X, Li X, Wang K. Molecular therapy of cardiac ischemia-reperfusion injury based on mitochondria and ferroptosis. J Mol Med. 2023;101(9):1059–1071. doi:10.1007/s00109-023-02346-z
61. Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2021;17:2054–2081. doi:10.1080/15548627.2020.1810918
62. Dixon SJ, Winter GE, Musavi LS, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10:1604–1609. doi:10.1021/acschembio.5b00245
63. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–98. doi:10.1038/nchembio.2239
64. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. 2019;35:830–849. doi:10.1016/j.ccell.2019.04.002
65. Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90. doi:10.1038/nchembio.2238
66. Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell. 2022;82(12):2215–2227. doi:10.1016/j.molcel.2022.03.022
67. Shah R, Shchepinov MS, Pratt DA. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent Sci. 2018;4(3):387–396. doi:10.1021/acscentsci.7b00589
68. Zou Y, Li H, Graham ET, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16:302–309. doi:10.1038/s41589-020-0472-6
69. Jacquemyn J, Ralhan I, Ioannou MS. Driving factors of neuronal ferroptosis. Trends Cell Biol. 2024;S0962-8924(24):00022–00029[pii].
70. Bao WD, Pang P, Zhou XT, et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021;28(5):1548–1562. doi:10.1038/s41418-020-00685-9
71. Chen PH, Wu J, Ding CC, et al. Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death Differ. 2020;27(3):1008–1022. doi:10.1038/s41418-019-0393-7
72. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021–1032. doi:10.1038/cr.2016.95
73. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin regulate ferroptosis. Mol Cell. 2015;59(2):298–308.
74. Geng N, Shi BJ, Li SL, et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci. 2018;22(12):3826–3836. doi:10.26355/eurrev_201806_15267
75. Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–1428. doi:10.1080/15548627.2016.1187366
76. Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. 2019;15:1137–1147. doi:10.1038/s41589-019-0408-1
77. Dai E, Chen X, Linkermann A, et al. A guideline on the molecular ecosystem regulating ferroptosis. Nat Cell Biol. 2024;26:1447–1457. doi:10.1038/s41556-024-01360-8
78. Guo D, Cai S, Deng L, et al. Ferroptosis in pulmonary disease and lung cancer: molecular mechanisms, crosstalk regulation, and therapeutic strategies. MedComm. 2025;6(3):e70116. doi:10.1002/mco2.70116
79. He Y, Lin Y, Song J, et al. From mechanisms to medicine: ferroptosis as a therapeutic target in liver disorders. Cell Commun Signaling. 2025;23(1):125. doi:10.1186/s12964-025-02121-2
80. Huo L, Liu C, Yuan Y, Liu X, Cao Q. Pharmacological inhibition of ferroptosis as a therapeutic target for sepsis-associated organ damage. Eur J Med Chem. 2023;257:115438. doi:10.1016/j.ejmech.2023.115438
81. Lei P, Walker T, Ayton S. Neuroferroptosis in health and diseases. Nat Rev Neurosci. 2025;26:497–511. doi:10.1038/s41583-025-00930-5
82. Li L, Zhang H, Tang J. Pathogenesis mechanism of ferroptosis and pharmacotherapy in kidney diseases: a review. Nephron. 2025;2025:1–22.
83. Maremonti F, Tonnus W, Gavali S, et al. Ferroptosis-based advanced therapies as treatment approaches for metabolic and cardiovascular diseases. Cell Death Differ. 2024;31(9):1104–1112. doi:10.1038/s41418-024-01350-1
84. Stockwell BR, Jiang X. The chemistry and biology of ferroptosis. Cell Chem Biol. 2020;27(4):365–375. doi:10.1016/j.chembiol.2020.03.013
85. Wang Y, Wang W, Zhang Y, Fleishman JS, Wang H. Targeting ferroptosis offers therapy choice in sepsis-associated acute lung injury. Eur J Med Chem. 2024;283:117152. doi:10.1016/j.ejmech.2024.117152
86. Lin Q, Hou S, Dai Y, Jiang N, Lin Y. Monascin exhibits neuroprotective effects in rotenone model of Parkinson’s disease via antioxidation and anti-neuroinflammation. Neuroreport. 2020;31:637–643. doi:10.1097/WNR.0000000000001467
87. Beratis NG, Yee M, LaBadie GU, Hirschhorn K. Effect of copper on Menkes’ and normal cultured skin fibroblasts. Dev Pharmacol Ther. 1980;1(5):305–317. doi:10.1159/000455548
88. Bégin ME, Ells G, Horrobin DF. Polyunsaturated fatty acid-induced cytotoxicity against tumor cells and its relationship to lipid peroxidation. J Natl Cancer Inst. 1988;80(3):188–194. doi:10.1093/jnci/80.3.188
89. Li X, Wang Q, Xu C, et al. Ferroptosis inducers kill mesenchymal stem cells affected by neuroblastoma. Cancers. 2023;15:1301. doi:10.3390/cancers15041301
90. Tsvetkov P, Detappe A, Cai K, et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat Chem Biol. 2019;15:681–689. doi:10.1038/s41589-019-0291-9
91. Tsang T, Davis CI, Brady DC. Copper biology. Curr Biol. 2021;31(9):R421–R427. doi:10.1016/j.cub.2021.03.054
92. Chen L, Min J, Wang F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct Target Ther. 2022;7(1):378. doi:10.1038/s41392-022-01229-y
93. Xiong C, Ling H, Hao Q, Zhou X. Cuproptosis: p53-regulated metabolic cell death. Cell Death Differ. 2023;30(4):876–884. doi:10.1038/s41418-023-01125-0
94. Ge EJ, Bush AI, Casini A, et al. Connecting copper and cancer: from transition metal signalling to metalloplasia. Nat Rev Cancer. 2022;22:102–113. doi:10.1038/s41568-021-00417-2
95. Nagai M, Vo NH, Shin Ogawa L, et al. The oncology drug elesclomol selectively transports copper to the mitochondria to induce oxidative stress in cancer cells. Free Radic Biol Med. 2012;52(10):2142–2150. doi:10.1016/j.freeradbiomed.2012.03.017
96. Gao W, Huang Z, Duan J, Nice EC, Lin J, Huang C. Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol Oncol. 2021;15:3527–3544. doi:10.1002/1878-0261.13079
97. Yuan J, Ofengeim D. A guide to cell death pathways. Nat Rev Mol Cell Biol. 2023;25:379–395. doi:10.1038/s41580-023-00689-6
98. Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–626. doi:10.1083/jcb.124.4.619
99. Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi:10.1038/335440a0
100. Green DR. The mitochondrial pathway of apoptosis part II: the BCL-2 protein family. Cold Spring Harb Perspect Biol. 2022;14:a041046. doi:10.1101/cshperspect.a041046
101. Kelekar A, Thompson CB. Bcl-2-family proteins: the role of the BH3 domain in apoptosis. Trends Cell Biol. 1998;8:324–330. doi:10.1016/S0962-8924(98)01321-X
102. Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–656. doi:10.1038/nrc883
103. Motoyama N, Wang F, Roth KA, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–1510. doi:10.1126/science.7878471
104. Rinkenberger JL, Horning S, Klocke B, et al. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 2000;14(1):23–27. doi:10.1101/gad.14.1.23
105. Veis DJ, Sorenson CM, Shutter JR, et al. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell. 1993;75(2):229–240. doi:10.1016/0092-8674(93)80065-M
106. Julien O, Wells JA. Caspases and their substrates. Cell Death Differ. 2017;24:1380–1389. doi:10.1038/cdd.2017.44
107. Li Y, Zhou M, Hu Q, et al. Mechanistic insights into caspase-9 activation by the structure of the apoptosome holoenzyme. Proc Natl Acad Sci U S A. 2017;114:1542–1547. doi:10.1073/pnas.1620626114
108. Yuan S, Yu X, Asara JM, et al. The holo-apoptosome: activation of procaspase-9 and interactions with caspase-3. Structure. 2011;19(8):1084–1096. doi:10.1016/j.str.2011.07.001
109. Lakhani SA, Masud A, Kuida K, et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–851. doi:10.1126/science.1115035
110. Whelan RS, Konstantinidis K, Wei AC, et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci U S A. 2012;109:6566–6571. doi:10.1073/pnas.1201608109
111. Krammer PH. CD95’s deadly mission in the immune system. Nature. 2000;407:789–795. doi:10.1038/35037728
112. Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi:10.1016/S0092-8674(00)81874-7
113. Fisher GH, Rosenberg FJ, Straus SE, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–946. doi:10.1016/0092-8674(95)90013-6
114. Rieux-Laucat F, Le Deist F, Hivroz C, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–1349. doi:10.1126/science.7539157
115. Takahashi T, Tanaka M, Brannan CI, et al. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell. 1994;76:969–976. doi:10.1016/0092-8674(94)90375-1
116. Watanabe-Fukunaga R, Brannan CI, Copeland NG, et al. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356(6367):314–317. doi:10.1038/356314a0
117. Martin DA, Zheng L, Siegel RM, et al. Defective CD95/APO-1/Fas signal complex formation in the human autoimmune lymphoproliferative syndrome, type Ia. Proc Natl Acad Sci U S A. 1999;96:4552–4557. doi:10.1073/pnas.96.8.4552
118. Liang Z, Liu J, Gou Y, et al. Elevated histone lactylation mediates ferroptosis resistance in endometriosis through the METTL3-regulated HIF1A/HMOX1 signaling pathway. Adv Sci. 2025;12(31):e08220. doi:10.1002/advs.202408220
119. Deng J, Li Y, Yin L, et al. Histone lactylation enhances GCLC expression and thus promotes chemoresistance of colorectal cancer stem cells through inhibiting ferroptosis. Cell Death Dis. 2025;16(1):193. doi:10.1038/s41419-025-07498-z
120. Qu S, Feng B, Xing M, et al. PRMT5 K240lac confers ferroptosis resistance via ALKBH5/SLC7A11 axis in colorectal cancer. Oncogene. 2025;44(32):2814–2830. doi:10.1038/s41388-025-03457-2
121. Yang Z, Su W, Zhang Q, et al. Lactylation of HDAC1 confers resistance to ferroptosis in colorectal cancer. Adv Sci. 2025;12:e2408845. doi:10.1002/advs.202408845
122. Li A, Gong Z, Long Y, et al. Lactylation of LSD1 is an acquired epigenetic vulnerability of BRAFi/MEKi-resistant melanoma. Dev Cell. 2025;60:S1534–5807(25)00121–2[pii]. doi:10.1016/j.devcel.2025.02.016
123. Zhu J, Xiong Y, Zhang Y, et al. Simvastatin overcomes the pPCK1-pLDHA-SPRINGlac axis-mediated ferroptosis and chemo-immunotherapy resistance in AKT-hyperactivated intrahepatic cholangiocarcinoma. Cancer Commun. 2025;45(8):1038–1071. doi:10.1002/cac2.70036
124. Matsuo T, Sadzuka Y. Extracellular acidification by lactic acid suppresses glucose deprivation-induced cell death and autophagy in B16 melanoma cells. Biochem Biophys Res Commun. 2018;496(4):1357–1361. doi:10.1016/j.bbrc.2018.02.022
125. Jia M, Yue X, Sun W, et al. ULK1-mediated metabolic reprogramming regulates Vps34 lipid kinase activity by its lactylation. Sci Adv. 2023;9(22):eadg4993. doi:10.1126/sciadv.adg4993
126. Wang Z, Li M, Guo S, et al. Lactate-mediated Ran lactylation at lysine 123 promotes astrocytes polarization after oxygen-glucose deprivation/reoxygenation. Int Immunopharmacol. 2025;168(Pt 2):115861. doi:10.1016/j.intimp.2025.115861
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Programmed Cell Death in Liver Fibrosis
Gao R, Tang H, Mao J
Journal of Inflammation Research 2023, 16:3897-3910
Published Date: 1 September 2023
Programmed Cell Death in Rheumatoid Arthritis
Tong L, Qiu J, Xu Y, Lian S, Xu Y, Wu X
Journal of Inflammation Research 2025, 18:2377-2393
Published Date: 18 February 2025
Inhibitory Effects of Paclitaxel-Loaded Iron Oxide Nanoparticles on Non-Small Cell Lung Cancer by Enhancing Autophagy-Dependent Ferroptosis and Apoptosis Pathways
Deng R, Liang G, Chen W, Nie Q, Wen J
Cancer Management and Research 2025, 17:541-555
Published Date: 11 March 2025
New Targets for Immune Inflammatory Response in Rheumatoid Arthritis: Focus on the Potential Significance of N6-Methyladenosine, Ferroptosis and Cuproptosis
Wang S, Wan L, Zhang M, Yan D, Li F
Journal of Inflammation Research 2025, 18:8085-8106
Published Date: 19 June 2025
Machine Learning-Based Identification and Experimental Validation of Hub Ferroptosis-Related Cuproptosis Genes in Lupus Nephritis
Zhang S, Hu W, Zhang Y, Huang C, He Z, Xu J, Lin S, Yang B, Chen X
Journal of Inflammation Research 2025, 18:11335-11353
Published Date: 18 August 2025