Back to Journals » Cancer Management and Research » Volume 14
Targeted Cancer Therapy Based on Acetylation and Deacetylation of Key Proteins Involved in Double-Strand Break Repair
Received 24 October 2021
Accepted for publication 13 January 2022
Published 22 January 2022 Volume 2022:14 Pages 259—271
DOI https://doi.org/10.2147/CMAR.S346052
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Seema Singh
Xiwen Wang, Jungang Zhao
Department of Thoracic Surgery, Shengjing Hospital of China Medical University, Shenyang, Liaoning, 110004, People’s Republic of China
Correspondence: Jungang Zhao, Department of Thoracic Surgery, Shengjing Hospital of China Medical University, Shenyang, Liaoning, 110004, People’s Republic of China, Tel/Fax +86 13889311066, Email [email protected]
Abstract: DNA double-strand breaks (DSBs) play an important role in promoting genomic instability and cell death. The precise repair of DSBs is essential for maintaining genome integrity during cancer progression, and inducing genomic instability or blocking DNA repair is an important mechanism through which chemo/radiotherapies exert killing effects on cancer cells. The two main pathways that facilitate the repair of DSBs in cancer cells are homologous recombination (HR) and non-homologous end-joining (NHEJ). Accumulating data suggest that the acetylation and deacetylation of DSB repair proteins regulate the initiation and progression of the cellular response to DNA DSBs, which may further affect the chemosensitivity or radiosensitivity of cancer cells. Here, we focus on the role of acetylation/deacetylation in the regulation of ataxia-telangiectasia mutated, Rad51, and 53BP1 in the HR pathway, as well as the relevant roles of PARP1 and Ku70 in NHEJ. Notably, several histone deacetylase (HDAC) inhibitors targeting HR or NHEJ have been demonstrated to enhance chemo/radiosensitivity in preclinical studies. This review highlights the essential role of acetylation/deacetylation in the regulation of DSB repair proteins, suggesting that HDAC inhibitors targeting the HR or NHEJ pathways that downregulate DNA DSB repair genes may be worthwhile cancer therapeutic agents.
Keywords: PTM, HR, NHEJ, chemotherapy, radiotherapy
Graphical Abstract:

Introduction
In eukaryotes and prokaryotes, lysine acetylation is a highly evolutionarily conserved protein post-translational modification (PTM), which is dynamically regulated by lysine acetyltransferases and deacetylases. The presence of acetyl groups on histones was first demonstrated by Allfrey and coworkers in 1964.1 Acetylation affects a wide array of biological functions by modifying histone and non-histone proteins. In particular, the acetylation of certain proteins plays an important role in DNA damage and repair in cancer.2 Several mechanisms regulate the DNA damage and repair processes in cancer, primarily by modulating chromatin structure and regulating the transcription and acetylation of DNA damage and repair-related proteins.3–5
DNA double-strand breaks (DSBs) are highly deleterious DNA lesions, which may lead to chromosomal translocations, genomic instability, and cell death in cancer.6,7 Conventional cancer therapies, such as radiation and chemotherapy, mainly exert their effects by inducing DSB-mediated cancer cell death. To counteract DSB damage and preserve genomic integrity, tumor cells have evolved to take advantage of highly conserved DSB repair pathways, including homologous recombination (HR) and non-homologous end-joining (NHEJ). HR and NHEJ repair are essential for maintaining genomic stability, and these pathways protect tumor cells against endogenous and exogenous DNA damaging factors, even though they may be deregulated in cancer cells.
In HR repair, the homologous chromosome or sister chromatid is used as a template to replace the missing information at the break. In contrast, NHEJ directly ligates the cognate broken ends without a homologous template, which is also called error-prone repair.
In the presence of the undamaged sister chromatid DNA template, error-free HR repair is regarded as accurate and efficient, and it generally occurs during the S and G2 phases of the cell cycle.8,9 HR repair begins with the recognition and binding of the double-strand DNA break by the MRE11–RAD50–NBS1 (MRN) complex.10 Following the activation of ataxia-telangiectasia mutated (ATM), the MRN complex recruits the carboxy-terminal binding protein-interacting protein (CtIP) endonuclease, as well as BRCA1/2, PALB2, and Rad51, initiating excision of the damaged DNA end. RAD52 also mediates the DNA-DNA interactions necessary for annealing of the complementary DNA strands.11–13 In the S and G2 phases, ataxia-telangiectasia-mutated and Rad3-related (ATR) activity is essential for CtIP recruitment,14 which mediates DSB end resection in concert with BRCA1 and the MRN complex.15,16
Unlike HR, the NHEJ pathways (canonical [c-NHEJ] and alternative [alt-NHEJ]) repair DSBs by directly ligating the two broken DNA ends. In cancer, this can result in error-prone repair17 and DNA sequence rearrangements.18,19 The NHEJ repair pathway is not dependent on the presence of replicated DNA, and it can therefore occur at any point in the cell cycle. In G1 phase, several negative regulators limit DSB end resection repair, including the Ku70/Ku80 heterodimer17 and the tumor suppressor p53-binding protein 1 (TP53BP1). Both of these proteins steadily bind the DSB broken ends, preventing resection20 and recruiting DNA-PK kinase and the endonuclease Artemis, which ultimately promotes repair via the NHEJ pathway in cancer.21,22 C-NHEJ is mainly executed by the Ku70/Ku80 heterodimer, DNA-PK, LIG4, XRCC4, and XLF, whereas CtIP, PARPs, LIG1/3, and XRCC1 are involved in the alt-NHEJ pathway. The Ku70/Ku80 heterodimer also activates DNA-PK, which subsequently promotes the activation of XRCC4 and Artemis.23,24 (Figure 1).
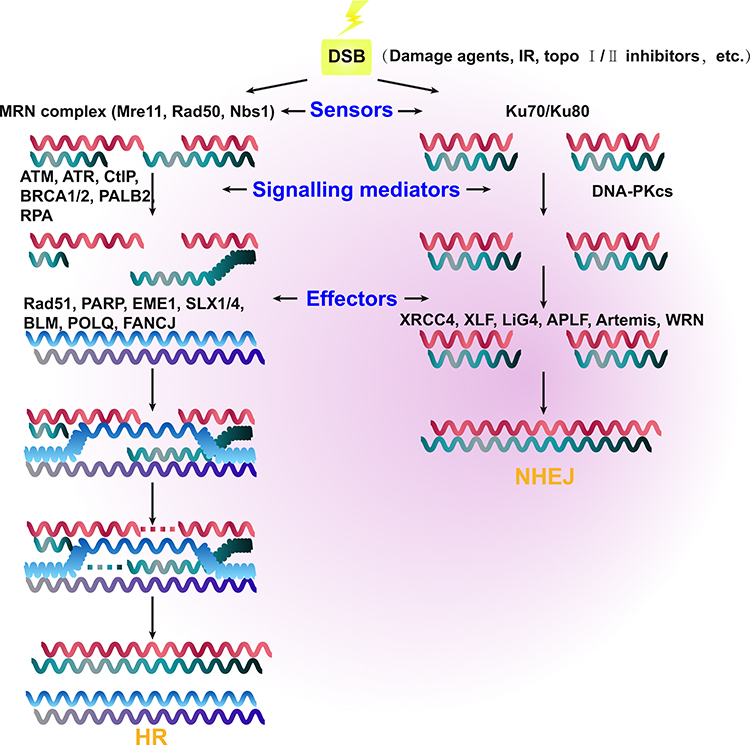 |
Figure 1 Schematic representation showing the mechanisms of the two main double-strand break (DSB) repair pathways in cancer, namely homologous recombination (HR) and non-homologous end-joining (NHEJ). The relevant sensors recognize the region of damaged DNA and initiate the HR and NHEJ repair pathways. The MRE11–RAD50–NBS1 (MRN) complex and Ku70/Ku80 are the major sensor proteins. DNA DSB signals are mediated by various proteins, including ATM, ATR, CtIP, BRCA1, BRCA2, PALB2, and RPA in HR, as well as DNA-PKcs in NHEJ. There are also various effector proteins, which include Rad51, PARP, EME1, SLX1/4, BLM, POLQ, and FANCJ in HR, as well as XRCC4, XLF, LiG4, APLF, Artemis, and WRN in NHEJ. |
Interestingly, it has been previously reported that WRN regulates pathway choice between classical and alternative non-homologous end joining;25 ATRX and RECQ5 also regulate distinct homologous recombination pathways;26 And BLM influences the DSB repair pathway choice by prevention of alt-NHEJ.27 Furthermore, the expression and localization of WRN and BLM helicases may be regulated by cellular essential enzyme SIRT1.28,29
Conventional cancer treatments such as radiotherapy and certain forms of chemotherapy30 exert killing effects on cancer cells by activating DSB pathways. Notably, cancer cells become even more dependent on DSB repair mechanisms to survive during these therapies.31 It has therefore been suggested that DSB repair-targeted therapy is particularly beneficial in combination with conventional radiotherapy and certain forms of chemotherapy.32 In recent years, several combination treatment strategies have been identified, including PARP inhibition (PARPi). Histone deacetylase (HDAC) inhibition (HDACi) is also successful in combination with chemo/radiotherapy.33 For instance, it was identified that pharmacological inhibition of CHK1 mediated homologous recombination repair (HRR), but not non-homologous end joining (NHEJ) repair, leading to hyper-radiosensitization of WRN-deficient cancers;34 It was also reported that a small molecule preferentially killed RECQL5-expressing breast cancers but not RECQL5-knockout.35
Acetylation and Deacetylation of Key Factors Mediating HR Repair
ATM
The kinase ATM plays an essential role in regulating DNA DSB repair. Under normal conditions, ATM exists as an inactive dimer, and it dissociates into active monomers upon autophosphorylation at serine (Ser) 1981. When cancer cells undergo DSBs, ATM signaling is enhanced, leading to activation of the HR and NHEJ repair pathways, the selection of which is determined by BRCA1 and 53BP1.36 ATR, another member of the phosphatidylinositol 3 kinase-like kinase family of Ser/Thr-protein kinases, also regulates various key proteins involved in DSB repair.
Blackford et al clearly demonstrated that Tip60-mediated acetylation of ATM (a critical regulator of self-activation and phosphorylation of various DSB repair factors23) was necessary for its kinase activity. The authors showed that ATM was acetylated and activated by Tip60 during HR repair and that the suppression of Tip60 interrupted the ATM-dependent phosphorylation of p53 and Chk2, which sensitized cancer cells to ionizing radiation. However, the recruitment of the ATM–Tip60 complex to DSB sites is independent of ATM kinase activity.37,38 The acetylation of ATM by Tip60 at Lys-3016 increases ATM kinase activity, and the MRN complex-dependent accumulation of UFL1 also promotes ATM activation by recruiting Tip60.39,40 According to recent findings, Tip60 is also activated and phosphorylated by various factors during DSB repair. The chromodomain of TIP60 has been shown to interact with H3K9me3, which activates the acetyltransferase activity of Tip60.41,42 Research has shown that the protein tyrosine kinase c-ABL also phosphorylates and regulates Tip60, which binds to H3K9me3 at DNA damage sites and activates ATM.43 Other regulators which have been identified as substrates of TIP60 (such as ATF2 and NOTCH1) also promote or inhibit ATM activation.44–46
Tang et al demonstrated that the Tip60-mediated acetylation of histone H4 prevented the interaction of the Tudor domain of 53BP1 with H4K20me2 during HR repair.47 Additionally, Tip60-catalyzed acetylation of H2AK15 also inhibited 53BP1 recruitment to H2AK15ub by preventing its ubiquitination.48 A previous study demonstrated that the Tip60 complex and its cofactor transformation/transcription-domain-associated protein (TRRAP) mediated the acetylation of histone H4 and the accumulation of the repair molecule Rad51 at DSB sites, which further facilitated HR repair.49 It is worth noting, however, that while the role of Tip60-mediated acetylation and activation of ATM in cancer cells has been well described, the dynamic regulation of ATM and ATR acetylation during HR repair is poorly understood.
MOF is a member of the highly conserved MYST family of histone acetyltransferases, which plays an important role in the DSB repair pathway, particularly in HR repair. Recent studies have shown that acetylation and ubiquitylation of chromatin modifiers and DNA repair proteins regulates their recruitment to sites of DNA damage.50 Gupta and colleagues showed that ionizing radiation (IR)-induced cancer cells exhibited MOF-mediated H4K16 hyperacetylation. Inhibiting this hyperacetylation led to decreased ATM autophosphorylation and kinase activity, suggesting that MOF is essential for the activation of ATM in this context.51 Conversely, ATM has also been shown to phosphorylate MOF at T392 during HR repair in cancer cells. In response, p-T392-MOF colocalized with γ-H2AX, ATM, and p53BP1, which further facilitated the recruitment of HR repair proteins such as BRCA1 and MDC1.52,53
Tang et al identified Sirtuin 7 (SIRT7) as a factor catalyzing the deacetylation of ATM at K3016, which led to the dephosphorylation and deactivation of ATM, thus impeding HR repair signals in human colon cancer cells.54 Furthermore, it has been demonstrated that SIRT7 expression correlates with the clinical response and prognosis of cancer chemotherapy.55 In addition, ATM interacts with HDAC1 through its LXCXE domain, while ATR has been shown to colocalize in a complex with HDAC2. ATM and ATR both phosphorylate BRCA1 in cells with DNA DSBs.56,57
Rad51 and 53BP1
During HR repair, Rad51 plays a central role in homologous strand exchange at damage sites, which is a key step in the DNA DSB repair process.58,59 It has been demonstrated that the inhibition of Rad51 may increase the chemosensitivity of cancer cells to cisplatin.60 A previous study showed that SIRT1 may interact with and deacetylate Rad51. In a lung cancer xenograft model, the inhibition of SIRT1 promoted the acetylation of Rad51, which further enhanced apoptosis in lung cancer cells induced by the WEE1 inhibitor MK-1775.61 In human colon cancer cells stimulated with IR, the HDAC inhibitor PCI-24781 impaired HR repair by blocking the formation of Rad51 subnuclear repair foci, which led to favorable therapeutic outcomes.62 In addition, in aggressive basal-like breast cancer cells, HDAC2 altered the expression of Rad51 by regulating the microRNA mir-182. The impaired HR efficiency promoted the progression of these breast cancer cells.63 In prostate cancer cells, an HDAC inhibitor was shown to downregulate E2F1-mediated expression of the DNA DSB repair genes Rad51, CHK1, and BRCA1, which lead to a downregulation of HR.64
It is worth taking note of TP53-binding protein 1 (53BP1), an NHEJ-promoting factor following DSBs. CBP-mediated acetylation of 53BP1 inhibits its recruitment to damaged chromatin, which results in the promotion of HR over NHEJ.65,66 (Figure 2).
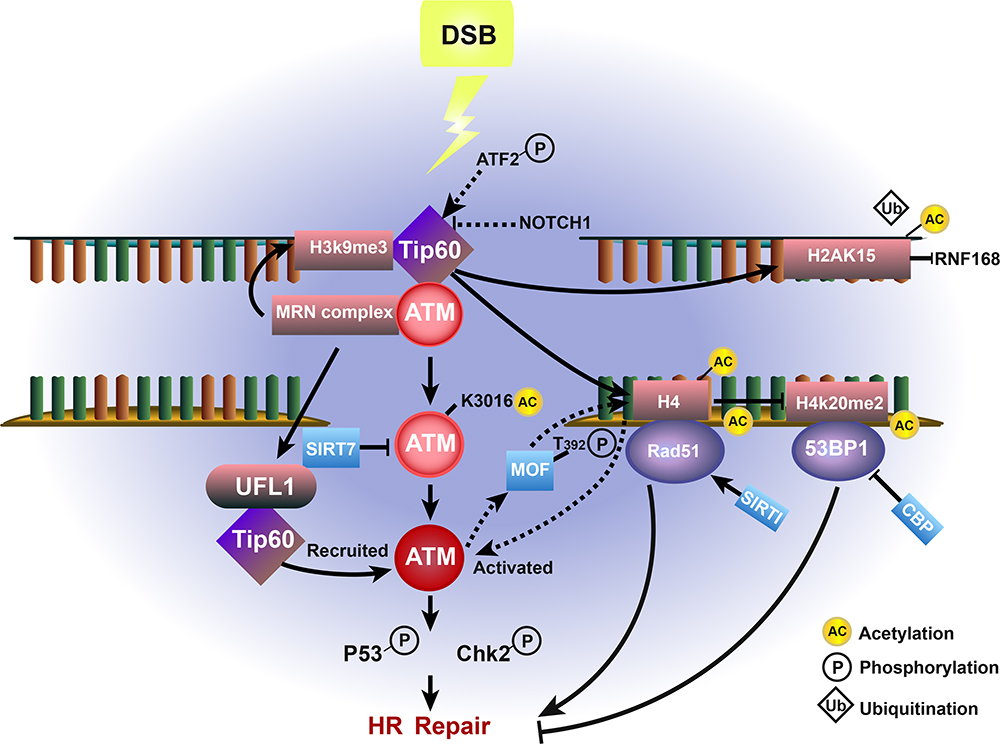 |
Figure 2 Effect of Tip60-ATM-associated acetylation and deacetylation on HR repair in cancer. Upon DSB damage, ATM is acetylated and activated by Tip60 at Lys-3016 and deacetylated by Sirtuin 7 (SIRT7), which promotes the ATM-dependent phosphorylation of p53 and Chk2, thereby enhancing HR repair. The MRN complex-dependent accumulation of UFL1 also promotes ATM activation by recruiting Tip60. H3K9me3 interacts with and activates Tip60 to further activate ATM. In addition, Tip60-mediated histone H4 acetylation prevents the interaction between H4K20me2 and 53BP1, while acetylation by CBP further inhibits HR repair. The Tip60-mediated interaction between H4-Ac and Rad51, which is deacetylated by SIRT1, promotes HR repair. The Tip60-catalyzed acetylation of H2AK15 also inhibit its ubiquitination. MOF-mediated H4 hyperacetylation is essential for the activation of ATM, which conversely phosphorylates MOF at T392. |
Acetylation and Deacetylation of Key Factors Involved in NHEJ Repair
PARP1
As a member of the PARP enzyme superfamily, the DNA damage sensor PARP1 catalyzes the synthesis of poly ADP-ribose polymers (PAR) by using NAD+ as a substrate. PARP1 is essential for DNA replication, chromatin remodeling, transcription, and the DNA damage response.67–69 PARP1 mainly participates in DNA base excision repair and DNA single-strand break repair, but it also plays an essential role in DNA DSB repair, participating in both the HR and NHEJ pathways. In HR, c-NHEJ, and alt-NHEJ, PARP1 facilitates the choice of repair pathway and the detection of DNA strand breaks by interacting with and regulating DNA repair factors.70–76
Various studies have demonstrated that several PTMs are involved in the regulation of PARP1, including PARylation, ubiquitination, and acetylation, and there is both competition and crosstalk between these modifications.77 More than 20 acetylation sites of PARP1 have been identified, including Lys498, Lys505, Lys508, Lys521, and Lys524, which regulate the transcriptional coactivator functions of PARP1. P300/CREB-binding protein-mediated acetylation of PARP-1 is essential for the activation of NF-κB-dependent genes, via enhanced interactions with p300 and the mediator complex.78 In particular, Lys498, Lys521, and Lys524 of PARP1 are simultaneously PARylated and acetylated at the same auto-modification domain. The dynamic PARylation of PARP1 regulates the recruitment and dissociation of critical DSB repair proteins at DSB sites and also provides the lysine residues as acceptor sites for ADP-ribosylation.79 In contrast, acetylation of these PARP1 lysine sites was shown to interrupt the binding of critical repair factors.81 Another functional study of PARP1 also demonstrated its acetylation at five lysine residues. PARP1 acetylation also promoted NF-κB-mediated chemoresistance of cancer cells to platinum compounds.80 In addition, the lysine residues K498, K521, and K524 were identified as acceptor sites for auto-PARylation, which might compete with the acetylation of these sites.79 Robert et al showed that the HDAC inhibitor trichostatin A (TSA) altered the acetylation level of PARP1, which impaired the NHEJ pathway. In HDACi-treated AML patients, more PAR was found to colocalize with DSBs. The authors demonstrated that the inhibition of HDAC increased PARP1 acetylation, which led to decreased NHEJ repair in leukemia cells, suggesting that PARP-1 acetylation inhibits DSB repair activity.81
In human breast cancer cells, PARP1 was shown to mediate the PARylation of MORC2 at DNA damage sites, which in turn promoted NAT10-mediated acetylation of PARP1 at Lys949. The acetylation of PARP1 impeded its own degradation, thus increasing the DSB repair ability of the cells.82,83 PARP inhibition (PARPi) has been used in the clinical treatment of ovarian, breast, prostate, and pancreatic cancers, especially in combination with other chemotherapy drugs in BRCA-deficient cancer cells.84–86 Together, the data suggest that acetylation of different PARP1 lysine sites can confer opposing functions of this protein in the DSB repair efficiency, suggesting that precise regulation of PARP1 acetylation may be a potentially novel therapeutic target.
Ku70
The Ku70 DNA end-joining protein binds damaged DNA during the NHEJ repair process, as well as during V(D)J recombination. Ku70 was identified as a suppressor of tumor cell apoptosis which is regulated by the interaction of its C-terminal linker domain with the proapoptotic factor Bax. Cohen et al has shown that Ku70 K542 is acetylated by CBP and PCAF in vivo. The hyperacetylation of Ku70 K539 or K542 abolished the Ku70 and Bax-mediated inhibition of apoptosis in cancer, suggesting that several acetyl-transferases might be potential tumor suppressors.87
Another study also suggested that SIRT1-mediated Ku70 deacetylation may promote the DSB repair capacity of cells.88 In prostate cancer cells, Ku70 K282, K338, K539, or K542 hyperacetylation was associated with DNA-binding affinity. The treatment of prostate cancer cells with HDAC inhibitors increased Ku70 acetylation and reduced cellular capability to repair DNA DSBs without disrupting Ku70/Ku80 heterodimer formation, thereby enhancing the chemotherapeutic effect.89 In neuroblastoma cells, IR-induced acetylation of nuclear Ku70 was determined by CBP and correlated with increased DNA repair activity. Cytoplasmic Ku70 was transferred to the nucleus following IR, indicating that Ku70 hyperacetylation may promote the effect of IR in neuroblastoma cells.90 In addition, it was reported that HDAC6 deacetylated Ku70 to regulate the Ku70–Bax interaction, which promoted cell death in neuroblastoma cells.91,92 In melanoma cell lines, the HDAC inhibitor sodium butyrate was shown to inhibit the expression of Ku70 and the DNA-PK catalytic subunit protein via deacetylation during NHEJ.93 In lung cancer A549 cells, epigallocatechin-3-gallate (EGCG) promoted cancer cell apoptosis by upregulating the expression of cleaved caspase-3 and Bax, as well as downregulating the expression of Bcl-xL. Mechanistically, it was established that EGCG induced K70 hyperacetylation in A549 cells and impaired the interaction of Bax with Ku70, thus promoting the apoptosis of the lung cancer cells.94 In hepatocellular carcinoma cells, the deacetylation of Ku70 K542 by SIRT6 inhibited the mitochondrial translocation of Bax and attenuated apoptosis, suggesting that the SIRT6-Ku70 pathway may serve as a promising therapeutic target for hepatocellular carcinoma in the future.95 In addition, NHEJ was also impaired by the differential acetylation of Ku70/Ku80 and PARP1 in response to TSA. These results suggest that IR-induced Ku70 acetylation promotes the apoptosis of cancer cells by reducing nuclear DNA DSB repair.81 P300/CBP was also implicated in the acetylation of H3 and H4, recruiting the Ku70/Ku80 complex to DSB sites during NHEJ repair.96 (Figure 3).
 |
Figure 3 Effect of PARP1 and Ku70-associated acetylation and deacetylation on NHEJ repair in cancer. Upon DSB damage, cisplatin-induced PARP1 acetylation activates NF-kB to promote chemo-resistance by enhancing NHEJ repair. Histone deacetylase inhibition (HDACi) and trichostatin A (TSA) may impair the NHEJ pathway by promoting PARP1 acetylation in leukemia cells. MORC2-mediated NAT10 acetylates PARP1 at K949 to promote its own stability and PARylation, which further enhances NHEJ repair in colon, breast, prostate, and pancreatic cancers. Furthermore, the interaction between Ku70 and Bax induces NHEJ repair, which may be inhibited by EGCG, CBP, and PCAF-mediated acetylation, and promoted by HDAC6-mediated deacetylation. SIRT1 and SIRT6 deacetylate Ku70 to promote NHEJ repair, and HDACi increases Ku70 acetylation to inhibit NHEJ repair. |
Taken together, the acetylation of Ku70 in DSB repair may be another potentially novel target for cancer therapy. The Ku70/Ku80 heterodimer also forms a DNA-PK complex with DNA-PKcs (Lys3241 and Lys3260) that serves as a docking site for other c-NHEJ proteins.97,98 Other c-NHEJ proteins that play an essential role in DNA end bridging and ligation include the LIG4XRCC4–XLF complex.99,100 However, the acetylation mediated functions involved in c-NHEJ still require further investigation.
Targeted Cancer Therapy
HDACs are a family of deacetylase enzymes that remove acetyl groups from histones and non-histone proteins. Pharmacological inhibition of HDAC activity using HDAC inhibitors causes significant changes to the chromatin acetylation landscape, resulting in transcriptome changes in cancers. It has been demonstrated that several HDAC small molecule inhibitors that modulate the acetylation of key proteins involved in the HR and NHEJ pathways hold promise for increasing chemo/radiosensitivity, which would further improve the survival rates of patients.
Combination of HDAC Inhibitors with Chemotherapy
Sulforaphane (SFN) is a natural HDAC inhibitor that has provided a new avenue for increasing the chemosensitivity of cancer cells by inhibiting HDAC activity101 and hindering DSB repair.102 In colon cancer cells, SFN may be effective in suppressing cancer cells as a chemotherapeutic agent alone or in combination with other HDAC inhibitors, and it is currently being tested in clinical trials.103 In a rat colon cancer model, novel tetrazole-containing SFN analogs have been shown to promote cell cycle arrest, apoptosis, and turnover of the DSB repair protein CtIP, which may lead to the deregulation of HR and NHEJ. These results provide a valuable avenue for synthetic lethality of HDAC inhibitors.104 Future investigations will need to clarify the value of these targets for combination chemotherapy protocols in clinical trials.105
Decreased HR repair capacity and increased cancer cell sensitization have been observed upon treatment with HDAC inhibitors, resulting in increased sensitivity to PARP inhibition,106,107 doxorubicin, and cisplatin.108,109 Decreased expression of Rad51 and attenuated HR activity was also observed in malignant melanoma cells when HDAC1 and HDAC2 were inhibited, which led to inefficient repair of DSBs and sensitization to temozolomide.110 Indeed, the synergistic combination of HDACi and classical chemotherapy in Phase I and Phase II clinical trials has demonstrated encouraging efficacy against cancer cells, while combination treatments with PARPi (such as the phase I trial NCT03742245) are worthy of future study.111–113
Combination of HDAC Inhibitors with Radiotherapy
Radiotherapy remains an important tool for treating a variety of cancer types. HDAC inhibitor-induced radiosensitization has been identified in various cancer cell lines, including breast, prostate, lung, colon, cervical, and head/neck.114 Several studies have demonstrated that HDAC inhibitors are essential for DSB repair in cancer cells. For example, the HDAC inhibitor vorinostat was shown to promote the expression of the DSB marker γ-H2AX in a transcription- and replication-dependent manner.115,116
Valproic acid has been shown to mediate the inhibition of class I HDACs and decrease the activity of key genes involved in HR repair, such as Rad51, CHK1, and BRCA1, leading to the enhanced sensitization of cancer cells to radiation and chemotherapy.64 Indeed, HDAC inhibitors are currently being tested in clinical trials in combination with radiotherapy, and there is evidence to suggest that the expression of ATM, p53, and BRCA1 may further enhance the antitumor efficacy of radiotherapy.117,118 In A549 lung cancer cells, the p53 inhibitor pifithrin-α was shown to inhibit the TSA-mediated synergistic effect of radiotherapy. In HeLa cervical cancer cells, leptomycin B mediated an increase in radiosensitization induced by TSA, by increasing p53 protein levels.119 The HR repair factor BRCA1 was shown to be downregulated by trichostatin A, which lead to increased radiosensitivity of the cancer cells.117
A previous study demonstrated that the class I and II HDAC inhibitor sodium butyrate acetylates histone H4 in melanoma cells, which lead to a decrease in KU70/KU80 and DNA-PKcs mRNA/protein levels, thereby enhancing sensitization to IR.120 The suppression of NHEJ resulted in the accumulation of DSBs due to HDACi-mediated H4K16 hyperacetylation and suppression of 53BP1 foci formation, which further increased radiosensitization.121
Recent studies examining human prostate cancer and lung adenocarcinoma cells have shown that the expression of critical components of the HR repair pathway is sensitive to HDAC inhibition. In particular, the HDAC inhibitor vorinostat was shown to induce DSBs by suppressing the transcription of RAD50 and MRE11, which increased radiosensitivity of the cells.122 In prostate cancer and glioma xenograft models, research has revealed that valproic acid and HDAC inhibitors promote increased radiosensitivity by inhibiting DNA DSB repair.123,124 Additionally, HDAC4, known as a biomarker of LBH589 activity, is reported to increase radiation-induced DNA DSBs in human non-small cell lung cancer (NSCLC).125 The HDAC inhibitor TSA may also increase radiosensitivity by downregulating DNA DSB repair proteins in NSCLC.126 In melanoma cells, gamma-H2AX phosphorylation is considered to be a predictive marker of the response to radiation, and HDAC inhibitors may sensitize cancer cells to radiation-induced DNA DSBs by inhibiting their capacity to repair DSBs.120 Vorinostat significantly improves the survival of patients with breast cancer who develop brain metastases, by enhancing radiosensitivity in vivo.127 In colorectal cancer models, vorinostat significantly reduced tumor volume in combination with radiotherapy, compared to the use of radiotherapy alone.128 A phase I trial that evaluated the effect of escalating doses of vorinostat with short-term palliative pelvic radiotherapy in patients with gastrointestinal carcinoma generated encouraging results.129 Consistent with these data, vorinostat also increases the radiosensitivity of osteosarcoma and rhabdomyosarcoma cells by downregulating Ku80 expression.130 (Table 1).
 |
Table 1 DSB Repair Associated Experimental Evidence of the Combination Between HDACi and Chemo/Radiotherapy in Cancers |
Conclusions and Future Perspective
This article provides an overview of the recent progress that has been made in elucidating how acetylation/deacetylation of key HR and NHEJ repair proteins mediates the critical roles of these factors in cancer. We illustrated the mechanisms through which acetylation/deacetylation regulates the functions of key factors involved in DNA DSB repair, such as ATM, Rad51, and 53BP1 in HR, as well as PARP1 and Ku70 in NHEJ. ATM acetylation promotes DSB repair, whereas acetylation of Rad51 and Ku70 inhibits DSB repair in cancer cells. However, further research is needed to identify how acetylation might modify the functions of other DSB repair proteins. To date, several studies have revealed that HDAC inhibitors targeting HR and NHEJ repair enhance chemo/radiosensitivity in a variety of cancer types.
It is worth noting that different modification types, such as acetylation and ubiquitylation, can sometimes cooperate in the DNA DSB repair process. Notably, the interplay between acetylation and other modifications at the same or adjacent site in mediating the regulation of cancer DSB repair deserves further investigation. From a therapeutic perspective, HDAC inhibition potentiates the effect of chemotherapy and radiotherapy by inhibiting DSB repair in some cancer types. However, there is also a need to develop new HDACi drugs targeting DSB repair in other types of cancers. In parallel, more precise patient selection is also essential to ensure relevant treatment outcomes. This review illustrates the critical role of acetylation/deacetylation in the regulation of key DSB repair genes in cancer, highlighting the importance of discovering new anti-cancer target drugs to improve therapeutic effects.
Funding
This study was supported by 345 Talent Project of Shengjing Hospital (grant no. M0705).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci U S A. 1964;51(5):786–794. doi:10.1073/pnas.51.5.786
2. Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–840. doi:10.1126/science.1175371
3. Li Z, Zhu WG. Targeting histone deacetylases for cancer therapy: from molecular mechanisms to clinical implications. Int J Biol Sci. 2014;10(7):757–770. doi:10.7150/ijbs.9067
4. O’Hagan HM. Chromatin modifications during repair of environmental exposure-induced DNA damage: a potential mechanism for stable epigenetic alterations. Environ Mol Mutagen. 2014;55(3):278–291. doi:10.1002/em.21830
5. Roos WP, Krumm A. The multifaceted influence of histone deacetylases on DNA damage signalling and DNA repair. Nucleic Acids Res. 2016;44(21):10017–10030. doi:10.1093/nar/gkw922
6. Gad H, Koolmeister T, Jemth AS, et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature. 2014;508(7495):215–221. doi:10.1038/nature13181
7. Gaillard H, García-Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer. 2015;15(5):276–289. doi:10.1038/nrc3916
8. Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;11(3):196–207. doi:10.1038/nrm2851
9. Wright WD, Shah SS, Heyer WD. Homologous recombination and the repair of DNA double-strand breaks. J Biol Chem. 2018;293(27):10524–10535. doi:10.1074/jbc.TM118.000372
10. Ranjha L, Howard SM, Cejka P. Main steps in DNA double-strand break repair: an introduction to homologous recombination and related processes. Chromosoma. 2018;127(2):187–214. doi:10.1007/s00412-017-0658-1
11. Buisson R, Masson JY. PALB2 self-interaction controls homologous recombination. Nucleic Acids Res. 2012;40(20):10312–10323. doi:10.1093/nar/gks807
12. Liu J, Doty T, Gibson B, Heyer WD. Human BRCA2 protein promotes Rad51 filament formation on RPA-covered single-stranded DNA. Nat Struct Mol Biol. 2010;17(10):1260–1262. doi:10.1038/nsmb.1904
13. Honda M, Okuno Y, Yoo J, Ha T, Spies M. Tyrosine phosphorylation enhances RAD52-mediated annealing by modulating its DNA binding. EMBO J. 2011;30(16):3368–3382. doi:10.1038/emboj.2011.238
14. Reginato G, Cannavo E, Cejka P. Physiological protein blocks direct the Mre11-Rad50-Xrs2 and Sae2 nuclease complex to initiate DNA end resection. Genes Dev. 2017;31(23–24):2325–2330. doi:10.1101/gad.308254.117
15. Peterson SE, Li Y, Wu-Baer F, et al. Activation of DSB processing requires phosphorylation of CtIP by ATR. Mol Cell. 2013;49(4):657–667. doi:10.1016/j.molcel.2012.11.020
16. Ismail IH, Gagné JP, Genois MM, et al. The RNF138 E3 ligase displaces Ku to promote DNA end resection and regulate DNA repair pathway choice. Nat Cell Biol. 2015;17(11):1446–1457. doi:10.1038/ncb3259
17. Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol. 2017;18(8):495–506. doi:10.1038/nrm.2017.48
18. Ceccaldi R, Rondinelli B, D’Andrea AD. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26(1):52–64. doi:10.1016/j.tcb.2015.07.009
19. Radhakrishnan SK, Jette N, Lees-Miller SP. Non-homologous end joining: emerging themes and unanswered questions. DNA Repair. 2014;17:2–8. doi:10.1016/j.dnarep.2014.01.009
20. Marini F, Rawal CC, Liberi G, Pellicioli A. Regulation of DNA double strand breaks processing: focus on barriers. Front Mol Biosci. 2019;6:55. doi:10.3389/fmolb.2019.00055
21. Zhao X, Wei C, Li J, et al. Cell cycle-dependent control of homologous recombination. Acta Biochim Biophys Sin. 2017;49(8):655–668. doi:10.1093/abbs/gmx055
22. Gupta A, Hunt CR, Chakraborty S, et al. Role of 53BP1 in the regulation of DNA double-strand break repair pathway choice. Radiat Res. 2014;181(1):1–8. doi:10.1667/RR13572.1
23. Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell. 2017;66(6):801–817. doi:10.1016/j.molcel.2017.05.015
24. Pannunzio NR, Watanabe G, Lieber MR. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J Biol Chem. 2018;293(27):10512–10523. doi:10.1074/jbc.TM117.000374
25. Shamanna RA, Lu H, de Freitas JK, Tian J, Croteau DL, Bohr VA. WRN regulates pathway choice between classical and alternative non-homologous end joining. Nat Commun. 2016;7:13785. doi:10.1038/ncomms13785
26. Elbakry A, Juhász S, Chan KC, Löbrich M. ATRX and RECQ5 define distinct homologous recombination subpathways. Proc Natl Acad Sci U S A. 2021;118(3):e2010370118. doi:10.1073/pnas.2010370118
27. Grabarz A, Guirouilh-Barbat J, Barascu A, et al. A role for BLM in double-strand break repair pathway choice: prevention of CtIP/Mre11-mediated alternative nonhomologous end-joining. Cell Rep. 2013;5(1):21–28. doi:10.1016/j.celrep.2013.08.034
28. Lee SY, Lee H, Kim ES, Park S, Lee J, Ahn B. WRN translocation from nucleolus to nucleoplasm is regulated by SIRT1 and required for DNA repair and the development of chemoresistance. Mutat Res. 2015;774:40–48. doi:10.1016/j.mrfmmm.2015.03.001
29. Vaitiekunaite R, Butkiewicz D, Krześniak M, et al. Expression and localization of Werner syndrome protein is modulated by SIRT1 and PML. Mech Ageing Dev. 2007;128(11–12):650–661. doi:10.1016/j.mad.2007.09.004
30. Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12(9):587–598. doi:10.1038/nrc3342
31. Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12(12):801–817. doi:10.1038/nrc3399
32. Toulany M. Targeting DNA double-strand break repair pathways to improve radiotherapy response. Genes. 2019;10(1). doi:10.3390/genes10010025
33. Minchom A, Aversa C, Lopez J. Dancing with the DNA damage response: next-generation anti-cancer therapeutic strategies. Ther Adv Med Oncol. 2018;10:1758835918786658. doi:10.1177/1758835918786658
34. Gupta P, Saha B, Chattopadhyay S, Patro BS. Pharmacological targeting of differential DNA repair, radio-sensitizes WRN-deficient cancer cells in vitro and in vivo. Biochem Pharmacol. 2021;186:114450. doi:10.1016/j.bcp.2021.114450
35. Chakraborty S, Dutta K, Gupta P, et al. Targeting RECQL5 functions, by a small molecule, selectively kills breast cancer in vitro and in vivo. J Med Chem. 2021;64(3):1524–1544. doi:10.1021/acs.jmedchem.0c01692
36. Jin MH, Oh DY. ATM in DNA repair in cancer. Pharmacol Ther. 2019;203:107391. doi:10.1016/j.pharmthera.2019.07.002
37. Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A. 2005;102(37):13182–13187. doi:10.1073/pnas.0504211102
38. Squatrito M, Gorrini C, Amati B. Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol. 2006;16(9):433–442. doi:10.1016/j.tcb.2006.07.007
39. Qin B, Yu J, Nowsheen S, et al. UFL1 promotes histone H4 ufmylation and ATM activation. Nat Commun. 2019;10(1):1242. doi:10.1038/s41467-019-09175-0
40. Sun Y, Xu Y, Roy K, Price BD. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol Cell Biol. 2007;27(24):8502–8509. doi:10.1128/MCB.01382-07
41. Sun Y, Jiang X, Xu Y, et al. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. 2009;11(11):1376–1382. doi:10.1038/ncb1982
42. Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, Xu Y, Price BD. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc Natl Acad Sci U S A. 2014;111(25):9169–9174. doi:10.1073/pnas.1403565111
43. Kaidi A, Jackson SP. KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature. 2013;498(7452):70–74. doi:10.1038/nature12201
44. Bhoumik A, Takahashi S, Breitweiser W, Shiloh Y, Jones N, Ronai Z. ATM-dependent phosphorylation of ATF2 is required for the DNA damage response. Mol Cell. 2005;18(5):577–587. doi:10.1016/j.molcel.2005.04.015
45. Bhoumik A, Singha N, O’Connell MJ, Ronai ZA. Regulation of TIP60 by ATF2 modulates ATM activation. J Biol Chem. 2008;283(25):17605–17614. doi:10.1074/jbc.M802030200
46. Adamowicz M, Vermezovic J, d’Adda Di Fagagna F. NOTCH1 inhibits activation of ATM by impairing the formation of an ATM-FOXO3a-KAT5/Tip60 complex. Cell Rep. 2016;16(8):2068–2076. doi:10.1016/j.celrep.2016.07.038
47. Tang J, Cho NW, Cui G, et al. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol. 2013;20(3):317–325. doi:10.1038/nsmb.2499
48. Jacquet K, Fradet-Turcotte A, Avvakumov N, et al. The TIP60 complex regulates bivalent chromatin recognition by 53BP1 through direct H4K20me binding and H2AK15 acetylation. Mol Cell. 2016;62(3):409–421. doi:10.1016/j.molcel.2016.03.031
49. Murr R, Loizou JI, Yang YG, et al. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol. 2006;8(1):91–99. doi:10.1038/ncb1343
50. van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009;19(5):207–217. doi:10.1016/j.tcb.2009.03.001
51. Gupta A, Sharma GG, Young CS, et al. Involvement of human MOF in ATM function. Mol Cell Biol. 2005;25(12):5292–5305. doi:10.1128/MCB.25.12.5292-5305.2005
52. Gupta A, Hunt CR, Hegde ML, et al. MOF phosphorylation by ATM regulates 53BP1-mediated double-strand break repair pathway choice. Cell Rep. 2014;8(1):177–189. doi:10.1016/j.celrep.2014.05.044
53. Li X, Corsa CA, Pan PW, et al. MOF and H4 K16 acetylation play important roles in DNA damage repair by modulating recruitment of DNA damage repair protein Mdc1. Mol Cell Biol. 2010;30(22):5335–5347. doi:10.1128/MCB.00350-10
54. Tang M, Li Z, Zhang C, et al. SIRT7-mediated ATM deacetylation is essential for its deactivation and DNA damage repair. Science Adv. 2019;5(3):eaav1118. doi:10.1126/sciadv.aav1118
55. Tang M, Lu X, Zhang C, et al. Downregulation of SIRT7 by 5-fluorouracil induces radiosensitivity in human colorectal cancer. Theranostics. 2017;7(5):1346–1359. doi:10.7150/thno.18804
56. Kim GD, Choi YH, Dimtchev A, Jeong SJ, Dritschilo A, Jung M. Sensing of ionizing radiation-induced DNA damage by ATM through interaction with histone deacetylase. J Biol Chem. 1999;274(44):31127–31130. doi:10.1074/jbc.274.44.31127
57. Schmidt DR, Schreiber SL. Molecular association between ATR and two components of the nucleosome remodeling and deacetylating complex, HDAC2 and CHD4. Biochemistry. 1999;38(44):14711–14717. doi:10.1021/bi991614n
58. Wyman C, Kanaar R. DNA double-strand break repair: all’s well that ends well. Annu Rev Genet. 2006;40:363–383. doi:10.1146/annurev.genet.40.110405.090451
59. Chailleux C, Tyteca S, Papin C, et al. Physical interaction between the histone acetyl transferase Tip60 and the DNA double-strand breaks sensor MRN complex. Biochem J. 2010;426(3):365–371. doi:10.1042/BJ20091329
60. Ito M, Yamamoto S, Nimura K, Hiraoka K, Tamai K, Kaneda Y. Rad51 siRNA delivered by HVJ envelope vector enhances the anti-cancer effect of cisplatin. J Gene Med. 2005;7(8):1044–1052. doi:10.1002/jgm.753
61. Chen G, Zhang B, Xu H, et al. Suppression of Sirt1 sensitizes lung cancer cells to WEE1 inhibitor MK-1775-induced DNA damage and apoptosis. Oncogene. 2017;36(50):6863–6872. doi:10.1038/onc.2017.297
62. Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJ. HDAC inhibitor PCI-24781 decreases Rad51 expression and inhibits homologous recombination. Proc Natl Acad Sci U S A. 2007;104(49):19482–19487. doi:10.1073/pnas.0707828104
63. Shan W, Jiang Y, Yu H, et al. HDAC2 overexpression correlates with aggressive clinicopathological features and DNA-damage response pathway of breast cancer. Am J Cancer Res. 2017;7(5):1213–1226.
64. Kachhap SK, Rosmus N, Collis SJ, et al. Downregulation of homologous recombination DNA repair genes by HDAC inhibition in prostate cancer is mediated through the E2F1 transcription factor. PLoS One. 2010;5(6):e11208. doi:10.1371/journal.pone.0011208
65. Sivanand S, Rhoades S, Jiang Q, et al. Nuclear acetyl-CoA production by ACLY promotes homologous recombination. Mol Cell. 2017;67(2):252–265.e256. doi:10.1016/j.molcel.2017.06.008
66. Guo X, Bai Y, Zhao M, et al. Acetylation of 53BP1 dictates the DNA double strand break repair pathway. Nucleic Acids Res. 2018;46(2):689–703. doi:10.1093/nar/gkx1208
67. Li M, Yu X. The role of poly(ADP-ribosyl)ation in DNA damage response and cancer chemotherapy. Oncogene. 2015;34(26):3349–3356. doi:10.1038/onc.2014.295
68. Kim MY, Mauro S, Gévry N, Lis JT, Kraus WL. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell. 2004;119(6):803–814. doi:10.1016/j.cell.2004.11.002
69. Aredia F, Scovassi AI. Poly(ADP-ribose): a signaling molecule in different paradigms of cell death. Biochem Pharmacol. 2014;92(1):157–163. doi:10.1016/j.bcp.2014.06.021
70. Leppard JB, Dong Z, Mackey ZB, Tomkinson AE. Physical and functional interaction between DNA ligase IIIalpha and poly(ADP-Ribose) polymerase 1 in DNA single-strand break repair. Mol Cell Biol. 2003;23(16):5919–5927. doi:10.1128/MCB.23.16.5919-5927.2003
71. Prasad R, Lavrik OI, Kim SJ, et al. DNA polymerase beta -mediated long patch base excision repair. Poly(ADP-Ribose)polymerase-1 Stimulates Strand Displacement DNA Synthesis. J Biol Chem. 2001;276(35):32411–32414.
72. Liu Y, Kadyrov FA, Modrich P. PARP-1 enhances the mismatch-dependence of 5’-directed excision in human mismatch repair in vitro. DNA Repair. 2011;10(11):1145–1153. doi:10.1016/j.dnarep.2011.08.012
73. Pines A, Vrouwe MG, Marteijn JA, et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J Cell Biol. 2012;199(2):235–249. doi:10.1083/jcb.201112132
74. Hochegger H, Dejsuphong D, Fukushima T, et al. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J. 2006;25(6):1305–1314. doi:10.1038/sj.emboj.7601015
75. Luijsterburg MS, de Krijger I, Wiegant WW, et al. PARP1 links CHD2-mediated chromatin expansion and H3.3 deposition to DNA repair by non-homologous end-joining. Mol Cell. 2016;61(4):547–562. doi:10.1016/j.molcel.2016.01.019
76. Ray Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol. 2017;18(10):610–621. doi:10.1038/nrm.2017.53
77. Wu Q, Cheng Z, Zhu J, et al. Suberoylanilide hydroxamic acid treatment reveals crosstalks among proteome, ubiquitylome and acetylome in non-small cell lung cancer A549 cell line. Sci Rep. 2015;5:9520. doi:10.1038/srep09520
78. Hassa PO, Haenni SS, Buerki C, et al. Acetylation of poly(ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-kappaB-dependent transcription. J Biol Chem. 2005;280(49):40450–40464. doi:10.1074/jbc.M507553200
79. Altmeyer M, Messner S, Hassa PO, Fey M, Hottiger MO. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. 2009;37(11):3723–3738. doi:10.1093/nar/gkp229
80. Lagunas VM, Meléndez-Zajgla J. Nuclear factor-kappa B as a resistance factor to platinum-based antineoplasic drugs. Met Based Drugs. 2008;2008:576104. doi:10.1155/2008/576104
81. Robert C, Nagaria PK, Pawar N, et al. Histone deacetylase inhibitors decrease NHEJ both by acetylation of repair factors and trapping of PARP1 at DNA double-strand breaks in chromatin. Leuk Res. 2016;45:14–23. doi:10.1016/j.leukres.2016.03.007
82. Li DQ, Nair SS, Ohshiro K, et al. MORC2 signaling integrates phosphorylation-dependent, ATPase-coupled chromatin remodeling during the DNA damage response. Cell Rep. 2012;2(6):1657–1669. doi:10.1016/j.celrep.2012.11.018
83. Zhang L, Li DQ. MORC2 regulates DNA damage response through a PARP1-dependent pathway. Nucleic Acids Res. 2019;47(16):8502–8520. doi:10.1093/nar/gkz545
84. Maintenance olaparib new standard in pancreatic cancer? Cancer Discov. 2019;9(8):Of6. doi:10.1158/2159-8290.CD-NB2019-065
85. Mateo J, Lord CJ, Serra V, et al. A decade of clinical development of PARP inhibitors in perspective. Ann Oncol. 2019;30(9):1437–1447. doi:10.1093/annonc/mdz192
86. Lin KY, Kraus WL. PARP inhibitors for cancer therapy. Cell. 2017;169(2):183. doi:10.1016/j.cell.2017.03.034
87. Cohen HY, Lavu S, Bitterman KJ, et al. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol Cell. 2004;13(5):627–638. doi:10.1016/S1097-2765(04)00094-2
88. Jeong J, Juhn K, Lee H, et al. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp Mol Med. 2007;39(1):8–13. doi:10.1038/emm.2007.2
89. Chen CS, Wang YC, Yang HC, et al. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Res. 2007;67(11):5318–5327. doi:10.1158/0008-5472.CAN-06-3996
90. Subramanian C, Hada M, Opipari AW
91. Subramanian C, Jarzembowski JA, Opipari AW
92. Subramanian C, Opipari AW
93. Thurn KT, Thomas S, Moore A, Munster PN. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol. 2011;7(2):263–283. doi:10.2217/fon.11.2
94. Li M, Li JJ, Gu QH, et al. EGCG induces lung cancer A549 cell apoptosis by regulating Ku70 acetylation. Oncol Rep. 2016;35(4):2339–2347. doi:10.3892/or.2016.4587
95. Tao NN, Ren JH, Tang H, et al. Deacetylation of Ku70 by SIRT6 attenuates Bax-mediated apoptosis in hepatocellular carcinoma. Biochem Biophys Res Commun. 2017;485(4):713–719. doi:10.1016/j.bbrc.2017.02.111
96. Ogiwara H, Ui A, Otsuka A, et al. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene. 2011;30(18):2135–2146. doi:10.1038/onc.2010.592
97. Davis AJ, Chen BP, Chen DJ. DNA-PK: a dynamic enzyme in a versatile DSB repair pathway. DNA Repair. 2014;17:21–29. doi:10.1016/j.dnarep.2014.02.020
98. Davis AJ, Lee KJ, Chen DJ. The N-terminal region of the DNA-dependent protein kinase catalytic subunit is required for its DNA double-stranded break-mediated activation. J Biol Chem. 2013;288(10):7037–7046. doi:10.1074/jbc.M112.434498
99. Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124(2):301–313. doi:10.1016/j.cell.2005.12.031
100. Sulkowski PL, Scanlon SE, Oeck S, Glazer PM. PTEN regulates nonhomologous end joining by epigenetic induction of NHEJ1/XLF. Mol Cancer Res. 2018;16(8):1241–1254. doi:10.1158/1541-7786.MCR-17-0581
101. Myzak MC, Karplus PA, Chung FL, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64(16):5767–5774. doi:10.1158/0008-5472.CAN-04-1326
102. Rajendran P, Delage B, Dashwood WM, et al. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Mol Cancer. 2011;10:68. doi:10.1186/1476-4598-10-68
103. Rajendran P, Kidane AI, Yu TW, et al. HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary isothiocyanates. Epigenetics. 2013;8(6):612–623. doi:10.4161/epi.24710
104. Chernikova SB, Game JC, Brown JM. Inhibiting homologous recombination for cancer therapy. Cancer Biol Ther. 2012;13(2):61–68. doi:10.4161/cbt.13.2.18872
105. Okonkwo A, Mitra J, Johnson GS, et al. Heterocyclic analogs of sulforaphane trigger DNA damage and impede DNA repair in colon cancer cells: interplay of HATs and HDACs. Mol Nutr Food Res. 2018;62(18):e1800228. doi:10.1002/mnfr.201800228
106. Chao OS, Goodman OB
107. Min A, Im SA, Kim DK, et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances anti-tumor effects of the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in triple-negative breast cancer cells. Breast Cancer Res. 2015;17:33. doi:10.1186/s13058-015-0534-y
108. Lopez G, Liu J, Ren W, et al. Combining PCI-24781, a novel histone deacetylase inhibitor, with chemotherapy for the treatment of soft tissue sarcoma. Clin Cancer Res. 2009;15(10):3472–3483. doi:10.1158/1078-0432.CCR-08-2714
109. Maiso P, Colado E, Ocio EM, et al. The synergy of panobinostat plus doxorubicin in acute myeloid leukemia suggests a role for HDAC inhibitors in the control of DNA repair. Leukemia. 2009;23(12):2265–2274. doi:10.1038/leu.2009.182
110. Krumm A, Barckhausen C, Kücük P, et al. Enhanced histone deacetylase activity in malignant melanoma provokes Rad51 and FANCD2-triggered drug resistance. Cancer Res. 2016;76(10):3067–3077. doi:10.1158/0008-5472.CAN-15-2680
111. Münster P, Marchion D, Bicaku E, et al. Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: a clinical and translational study. J Clin Oncol. 2007;25(15):1979–1985. doi:10.1200/JCO.2006.08.6165
112. Ramalingam SS, Kummar S, Sarantopoulos J, et al. Phase I study of vorinostat in patients with advanced solid tumors and hepatic dysfunction: a National Cancer Institute Organ Dysfunction Working Group study. J Clin Oncol. 2010;28(29):4507–4512. doi:10.1200/JCO.2010.30.2307
113. Ramalingam SS, Parise RA, Ramanathan RK, et al. Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clin Cancer Res. 2007;13(12):3605–3610. doi:10.1158/1078-0432.CCR-07-0162
114. Nome RV, Bratland A, Harman G, Fodstad O, Andersson Y, Ree AH. Cell cycle checkpoint signaling involved in histone deacetylase inhibition and radiation-induced cell death. Mol Cancer Ther. 2005;4(8):1231–1238. doi:10.1158/1535-7163.MCT-04-0304
115. Lee JH, Choy ML, Ngo L, Foster SS, Marks PA. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci U S A. 2010;107(33):14639–14644. doi:10.1073/pnas.1008522107
116. Conti C, Leo E, Eichler GS, et al. Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer Res. 2010;70(11):4470–4480. doi:10.1158/0008-5472.CAN-09-3028
117. Zhang Y, Carr T, Dimtchev A, Zaer N, Dritschilo A, Jung M. Attenuated DNA damage repair by trichostatin A through BRCA1 suppression. Radiat Res. 2007;168(1):115–124. doi:10.1667/RR0811.1
118. Kim IA, Kim IH, Kim HJ, Chie EK, Kim JS. HDAC inhibitor-mediated radiosensitization in human carcinoma cells: a general phenomenon? J Radiat Res. 2010;51(3):257–263. doi:10.1269/jrr.09115
119. Kim IA, Shin JH, Kim IH, et al. Histone deacetylase inhibitor-mediated radiosensitization of human cancer cells: class differences and the potential influence of p53. Clin Cancer Res. 2006;12(3 Pt 1):940–949. doi:10.1158/1078-0432.CCR-05-1230
120. Munshi A, Kurland JF, Nishikawa T, et al. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin Cancer Res. 2005;11(13):4912–4922. doi:10.1158/1078-0432.CCR-04-2088
121. Hsiao KY, Mizzen CA. Histone H4 deacetylation facilitates 53BP1 DNA damage signaling and double-strand break repair. J Mol Cell Biol. 2013;5(3):157–165. doi:10.1093/jmcb/mjs066
122. Groselj B, Sharma NL, Hamdy FC, Kerr M, Kiltie AE. Histone deacetylase inhibitors as radiosensitisers: effects on DNA damage signalling and repair. Br J Cancer. 2013;108(4):748–754. doi:10.1038/bjc.2013.21
123. Camphausen K, Cerna D, Scott T, et al. Enhancement of in vitro and in vivo tumor cell radiosensitivity by valproic acid. Int J Cancer. 2005;114(3):380–386. doi:10.1002/ijc.20774
124. Camphausen K, Scott T, Sproull M, Tofilon PJ. Enhancement of xenograft tumor radiosensitivity by the histone deacetylase inhibitor MS-275 and correlation with histone hyperacetylation. Clin Cancer Res. 2004;10(18 Pt 1):6066–6071. doi:10.1158/1078-0432.CCR-04-0537
125. Geng L, Cuneo KC, Fu A, Tu T, Atadja PW, Hallahan DE. Histone deacetylase (HDAC) inhibitor LBH589 increases duration of gamma-H2AX foci and confines HDAC4 to the cytoplasm in irradiated non-small cell lung cancer. Cancer Res. 2006;66(23):11298–11304. doi:10.1158/0008-5472.CAN-06-0049
126. Zhang F, Zhang T, Teng ZH, Zhang R, Wang JB, Mei QB. Sensitization to gamma-irradiation-induced cell cycle arrest and apoptosis by the histone deacetylase inhibitor trichostatin A in non-small cell lung cancer (NSCLC) cells. Cancer Biol Ther. 2009;8(9):823–831. doi:10.4161/cbt.8.9.8143
127. Baschnagel A, Russo A, Burgan WE, et al. Vorinostat enhances the radiosensitivity of a breast cancer brain metastatic cell line grown in vitro and as intracranial xenografts. Mol Cancer Ther. 2009;8(6):1589–1595. doi:10.1158/1535-7163.MCT-09-0038
128. Folkvord S, Ree AH, Furre T, Halvorsen T, Flatmark K. Radiosensitization by SAHA in experimental colorectal carcinoma models-in vivo effects and relevance of histone acetylation status. Int J Radiat Oncol Biol Phys. 2009;74(2):546–552. doi:10.1016/j.ijrobp.2009.01.068
129. Ree AH, Dueland S, Folkvord S, et al. Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: the Pelvic Radiation and Vorinostat (PRAVO) Phase 1 study. Lancet Oncol. 2010;11(5):459–464. doi:10.1016/S1470-2045(10)70058-9
130. Blattmann C, Oertel S, Ehemann V, et al. Enhancement of radiation response in osteosarcoma and rhabdomyosarcoma cell lines by histone deacetylase inhibition. Int J Radiat Oncol Biol Phys. 2010;78(1):237–245. doi:10.1016/j.ijrobp.2010.03.010
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.