")
Back to Journals » Journal of Pain Research » Volume 13
Tapentadol Prolonged Release for Long-Term Treatment of Pain in Children
Authors Howard RF, Radic T , Sohns M, Eerdekens M , Waßmuth A
Received 21 July 2020
Accepted for publication 27 September 2020
Published 30 November 2020 Volume 2020:13 Pages 3157—3170
DOI https://doi.org/10.2147/JPR.S272751
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Michael Schatman
Richard F Howard,1 Tatjana Radic,2 Melanie Sohns,2 Mariëlle Eerdekens,2 Andrea Waßmuth2
1Department of Anaesthesia and Pain Medicine, Great Ormond Street Hospital and the GOS-UCL Institute of Child Health, London, UK; 2Grünenthal GmbH, Aachen, Germany
Correspondence: Richard F Howard
Department of Anaesthesia and Pain Medicine, Great Ormond Street Hospital and the GOS-UCL Institute of Child Health, London WC1N 3JH, UK
Tel +44 2078298865
Email [email protected]
Purpose: Investigation of the efficacy and safety of tapentadol prolonged release (PR) compared with morphine PR for long-term treatment of pain in children.
Patients and Methods: Children aged 6 to < 18 years requiring long-term treatment with opioids were studied in a 12-month, 2-part, multi-center trial: Part 1, 14-day open-label, randomized, active-controlled, parallel group non-inferiority trial comparing twice daily tapentadol PR with morphine PR; Part 2, open-label treatment with tapentadol PR for up to 12 months or no treatment “safety observation period”. Pain intensity was rated with visual analogue scale or Faces Pain Scale-Revised, and non-inferiority was assessed by comparison of “treatment responders” (those completing the 14-day treatment period and showing pre-defined changes in pain rating) in each group.
Results: Twenty-three of 48 centers enrolled 73 patients. In Part 1, 45 and 24 patients received tapentadol or morphine, respectively, of which 40 and 22 completed 14-day treatment. In Part 2, thirty-six and 58 patients entered the tapentadol PR or observation periods, respectively, with 20/36 completing at least 12 weeks of treatment; 10 of the 36 had received morphine in Part 1. Forty-four of the 58 patients in the safety observation period had received tapentadol. Tapentadol PR was non-inferior to morphine PR (lower limit of confidence interval above negative non-inferiority margin of − 0.2) in Part 1. Rates of adverse events were as expected with nausea (22.2%) and constipation (15.6%) in the tapentadol PR group, and with vomiting (33.3%), nausea and constipation (each 16.7%) in the morphine PR group. No new safety issues were identified; the safety profile of tapentadol over the 12 months treatment and observation periods was comparable to that established in subjects > 18 years old.
Conclusion: Tapentadol PR was well tolerated and equivalent to morphine PR for both efficacy and safety in children (6 to < 18 years old) requiring long-term treatment with opioids.
Keywords: tapentadol, analgesic, prolonged-release, pain, child, adolescent
Plain Language Summary
Tapentadol PR, a strong, long-acting, pain-relieving medicine with an opioid and a non-opioid component was compared to long-acting morphine in a group of 69 children who needed pain relief for at least 2 weeks. Tapentadol was found to be similar to morphine in its ability to reduce pain.
Some children continued to take tapentadol for up to 12 months with no change in its effectiveness. Both tapentadol and morphine caused side effects, but they were all as expected for these types of pain treatment medications. Within the limits of the trial, tapentadol was no less safe in children than it is currently known to be in adults.
Introduction
Tapentadol is an opioid analgesic with two synergistic mechanisms of action (μ-opioid receptor agonism [MOR] and noradrenaline reuptake inhibition [NRI])1 which provide a strong analgesic effect; the contribution of the NRI also allows for an overall reduced µ-load and thus a potentially lower incidence of opioid-related adverse effects compared to pure opioids.2 In addition, tapentadol has a favorable pharmacokinetic (PK) profile,3,4 and efficacy and good tolerability have been shown in the treatment of a range of acute and chronic pain conditions in adults.5,6 Trial data of up to 2 years of treatment in adults has demonstrated the maintenance of efficacy and good long-term safety of a prolonged release (PR) formulation of tapentadol.7
Tapentadol is the subject of a pediatric clinical development program for use in children and adolescents with both acute and chronic pain. Studies of single and multiple doses for the treatment of acute pain requiring opioid treatment in children 2 to <18 years of age have supported registration of tapentadol oral solution in children in the European Union.8–10 The trial presented here investigated the efficacy and safety of the PR formulation of tapentadol for long-term treatment of pain in children and adolescents (6 to <18 years).
Patients and Methods
Trial Design
A 2-part, open-label, multi-center, Phase II/III trial in children aged 6 to <18 years to assess the safety and efficacy of tapentadol PR compared with morphine PR for the treatment of pain of expected duration longer than 14 days. The trial consisted of a screening phase (≤14 days), followed sequentially by: Part 1; a 14-day open-label, randomized, active-controlled, parallel group trial comparing efficacy and safety of tapentadol PR with morphine PR, leading to: Part 2, an open-label period lasting for up to 12 months of either tapentadol treatment, the “tapentadol period”, or a no treatment “safety observation period”. Patients who were randomized to receive morphine PR in Part 1 could enter the tapentadol treatment period or the observation period of Part 2. In Part 1 patients completed weekly visits and an electronic diary recording twice daily assessment of pain intensity and their use of rescue analgesia.
In the tapentadol period of Part 2 patients completed monthly visits that included recording pain intensity and use of tapentadol PR. Safety laboratory assessments (hematology, clinical chemistry, and urinalysis) were every 3 months. In the observation period of Part 2 patients completed visits every 3 months, with the option of completing interim visits at 3, 6 and 9 months by telephone. Adverse events were recorded throughout the trial.
The trial was planned and initiated at 48 trial sites (hospitals and research institutes) in 9 European countries and Chile; the Supplementary Material lists the ethic committees of all sites which enrolled subjects. The trial was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice, and applicable local laws and regulations. Written informed consent was obtained from all parents/legal guardians, or participants if applicable. The trial is registered with ClinicalTrials.gov (identifier NCT02151682) and EudraCT (no. 2012–004360-22).
Participants
Male and female patients 6 to <18 years of age with a bodyweight of ≥17.5 kg presenting with a painful condition or procedure that was expected to require treatment with strong opioids for at least 14 days were eligible to enter the trial. Patients had to be able to swallow tablets of appropriate size and willing to comply with trial procedures. For patients previously taking opioids the last calculated morphine equivalent dose had to be <3.5 mg/kg bodyweight per day; there was no opioid wash-out phase before entering the trial.
The principal exclusion criteria were: history or current significant central nervous system (CNS) disorder, moderate to severe renal or hepatic impairment, abnormal pulmonary function or clinically relevant respiratory disease, planned surgery expected to require management in intensive care during the 14-day treatment period, or a pain indication that was considered unlikely to require or respond to opioids.
Randomization and Treatment
Eligible patients were randomly allocated (2:1) to either tapentadol PR or morphine PR using an interactive voice/web response system. Randomization followed computer-generated randomization lists balanced by blocks of treatments and stratified by age groups (6 to <12 years, 12 to <18 years) and cancer or non-cancer-related pain. Trial medication was administered twice daily with a dosing interval of approximately 12 h. Patients who completed the 14-day treatment with either tapentadol PR or morphine PR and who, in the opinion of the investigator, still required continued treatment with strong analgesics were offered entry to the open-label tapentadol period of Part 2.
Tapentadol and Morphine Dosage
Tapentadol PR was available as 25 mg and 100 mg tablets, morphine PR as 10 mg and 30 mg tablets. Starting doses were calculated according to the patients’ weight and titrated to clinically assessed therapeutic effect within a pre-defined range. Tapentadol PR dosage was based on a population PK model (data on file) which integrated data from adult tapentadol PR trials and from pediatric tapentadol oral solution trials. Simulations were performed to identify tapentadol PR doses that would produce steady state exposures in pediatric patients similar to those reported in adults. The approved adult therapeutic dose range (tapentadol PR 50 to 250 mg b.i.d.) was used for comparison.11 The simulations indicated that exposures to 1.25–1.5 mg/kg tapentadol PR bodyweight twice daily in children aged 6 to <18 years were in the range of exposures to tapentadol PR 50 mg and 100 mg b.i.d. in adults. Based on this, a starting dose of approx. 1.25–1.5 mg/kg every 12 h with a maximum dose of approx. 4.5 mg/kg every 12 h was chosen. Morphine PR dose selection was based on the above tapentadol PR dose simulation with the conversion factor (1:2.5). Patients with previous opioid treatment commenced at ≤70% of the equianalgesic dose of the previous treatment. Dose was increased at the investigator’s discretion, but not less than 2 days following a previous dose increase. Dose reduction was permitted at any time.
Patients taking tapentadol PR during Part 1 entered Part 2 taking the same dose as the last dose they received during Part 1. Patients taking morphine PR in Part 1 who chose to switch to the tapentadol period at Part 2, started Part 2 at ≤70% of the current morphine equivalent dose but using at least the minimum dose stipulated for their bodyweight.
Rescue Medication
In Part 1, morphine oral solution was available as rescue. The permitted dose per intake was 1/6 of the total daily dose of the scheduled trial medication intake. In Part 2, patients in the tapentadol period could take immediate release strong analgesics as rescue medication if needed.
Assessment of Efficacy
Efficacy was measured by self-reported change in pain intensity as determined using the visual analogue scale (VAS) and Faces Pain Scale-Revised (FPS-R)12. The VAS is a self-reporting 100 mm scale from 0=no pain to 100=worst imaginable pain, the FPS-R a self-reporting 6-point scale showing six faces with scores of 0, 2, 4, 6, 8, 10 (where 0=no pain and 10=very much pain). Baseline pain intensity, “pain right now”, was defined at the treatment allocation visit. Pain intensity was then rated twice daily during the treatment period immediately before the intake of trial medication using first VAS followed by the FPS-R. Patients used an electronic diary to record pain intensity and trial and any rescue medication dose in Part 1 of the trial. The primary efficacy endpoint was a binary variable “responder”; for definition see the section on statistics.
Assessment of Safety
Adverse events (AEs) were monitored throughout the trial. The assessment of both treatment-emergent adverse events (TEAE) and treatment-related TEAEs was a secondary endpoint of both randomized and extension periods; post-treatment AEs were recorded in the safety observation period. Safety measures included vital signs, physical examination, hematology and clinical chemistry; 12-lead-ECGs were performed at enrolment and end of randomized treatment.
Other Endpoints
Palatability and acceptability of the trial medication was assessed using a 5-point faces scale13 ranging from really good/really easy to really bad/really difficult, for both taste and ease of swallowing at the end of Part 1. Constipation was assessed at each weekly visit during Part 1, and at the end of the tapentadol and observation periods using the modified constipation assessment scale (mCAS,14 range 0–16, where 16 = worst possible constipation). Opioid withdrawal symptoms were assessed following the final dose of medication during both the Part 1 and the tapentadol period of Part 2 using the first 15 questions of the subjective opiate withdrawal assessment scale15 (range 0–60, where 60 = extreme withdrawal symptoms). Time to discontinuation of trial medication for both Part 1 (tapentadol and morphine) and tapentadol period was also recorded.
Statistical Analysis
The sample size estimate (SSE) was based on the rejection of the null hypothesis of inferiority of tapentadol PR to morphine PR with regard to the number of primary endpoint “treatment responders” who were defined by the following criteria:
- Completion of the 14-day treatment period
- Achievement of one of the following treatment goals during the last 3 days of treatment: either
- An average pain intensity of <50 on the VAS for patients 12 to <18 years, or <5 on the FPS-R for patients 6 to <12 years, or
- An average pain reduction from baseline of ≥20 on the VAS for patients 12 to <18 years, or ≥2 on the FPS-R for patients 6 to <12 years.
Based on previous trials and extrapolation to the current trial population, the proportion of responders in both treatment arms was estimated as 80%. Assuming a one-sided significance level of alpha=0.1, a power of at least 80% and a randomization ratio of 2:1 (tapentadol to morphine), a sample size of 69 subjects was required to show non-inferiority of tapentadol PR to morphine PR by a 20% non-inferiority margin. A one-sided alpha of 0.1was considered appropriate based on an approach using prior knowledge and the concept of extrapolation from a larger population to a small target population (ie, pediatric population) to reduce the burden of evidence in pediatrics by relaxing the Type I error, while controlling a certain posterior belief, ie, confidence after successful pediatric trials, in effectiveness of the drug in children.16
All analyses were performed with SAS version 9.4 (SAS Institute Inc., Cary, USA). All randomized patients who received at least one dose of trial medication were included in the primary efficacy analysis (full analysis set, FAS). The primary efficacy endpoint was summarized by treatment group. The standard maximum likelihood (ML) estimators for the proportion of subjects classified as responders in each treatment group were the estimated proportions adjusted for the baseline pain intensity, age group, and underlying pain condition. To obtain these estimators, a logistic regression model was fitted to the response using the baseline pain intensity, age group, treatment, and underlying pain condition as explanatory variables. The Farrington-Manning test was applied to the derived ML estimates, and a 2-sided 80% Farrington-Manning confidence interval (CI) of the difference in proportion between the two treatments was calculated.17 Non-inferiority of tapentadol compared with morphine was established if the lower limit of this 80% CI was above the negative non-inferiority margin –δ=−0.2. The primary efficacy endpoint “response” was derived based on missing pain assessments during the last 3 days of the treatment period imputed using multiple imputation (200 imputations). The logistic regression model and Farrington-Manning methodology were applied to each individual imputation, and the analysis results of the individual imputations were combined. To assess the robustness of these results, several sensitivity analyses were performed using: a Bayesian analysis, the per protocol set (PPS, ie, all FAS patients without major protocol deviations affecting the primary endpoint), the two pain scales independently for responder definition for the total population, no imputation of missing pain assessments during the last 3 days, a different completer definition (considering patients who discontinued treatment early because of no further need for it, as responder), baseline pain intensity not included as adjustment factor, pooled trial medication dose level added as adjustment factor, and exclusion of complex regional pain syndrome (CRPS) patients. All other endpoints were analyzed descriptively. The intake of rescue medication was calculated excluding one value resulting from a broken bottle (modified analysis).
All patients receiving at least one dose of trial medication were included in safety analyses. Underlying clinical diagnoses and concomitant diseases were encoded using the Medical Dictionary for Regulatory Activities (MedDRA version 17.1), adverse events were encoded using MedDRA version 20.1. Principal pain mechanism was estimated by the recruiting investigator as nociceptive, neuropathic or mixed.
Results
Patients: Trial Disposition and Baseline Characteristics
The trial was conducted between April 2015 (first subject in) and October 2018 (last subject out). Twenty-three of the 48 centers enrolled 73 subjects to the trial: three patients were not eligible or withdrew, and so 70 patients were randomized for treatment (Figure 1A). One patient did not receive trial medication and therefore 69 patients were included with 45 assigned to receive tapentadol PR, and 24 to receive morphine PR. Baseline characteristics, as well as pain type and intensity, underlying clinical diagnoses/reasons for pain were well matched between the treatment arms (Tables 1 and 2). Three children with a diagnosis of CRPS were recruited into the tapentadol arm of the trial before a protocol amendment made this an exclusion criterion on the basis that this condition was unlikely to respond to opioid treatment. These children were included in the main analysis but excluded in the relevant sensitivity analysis.
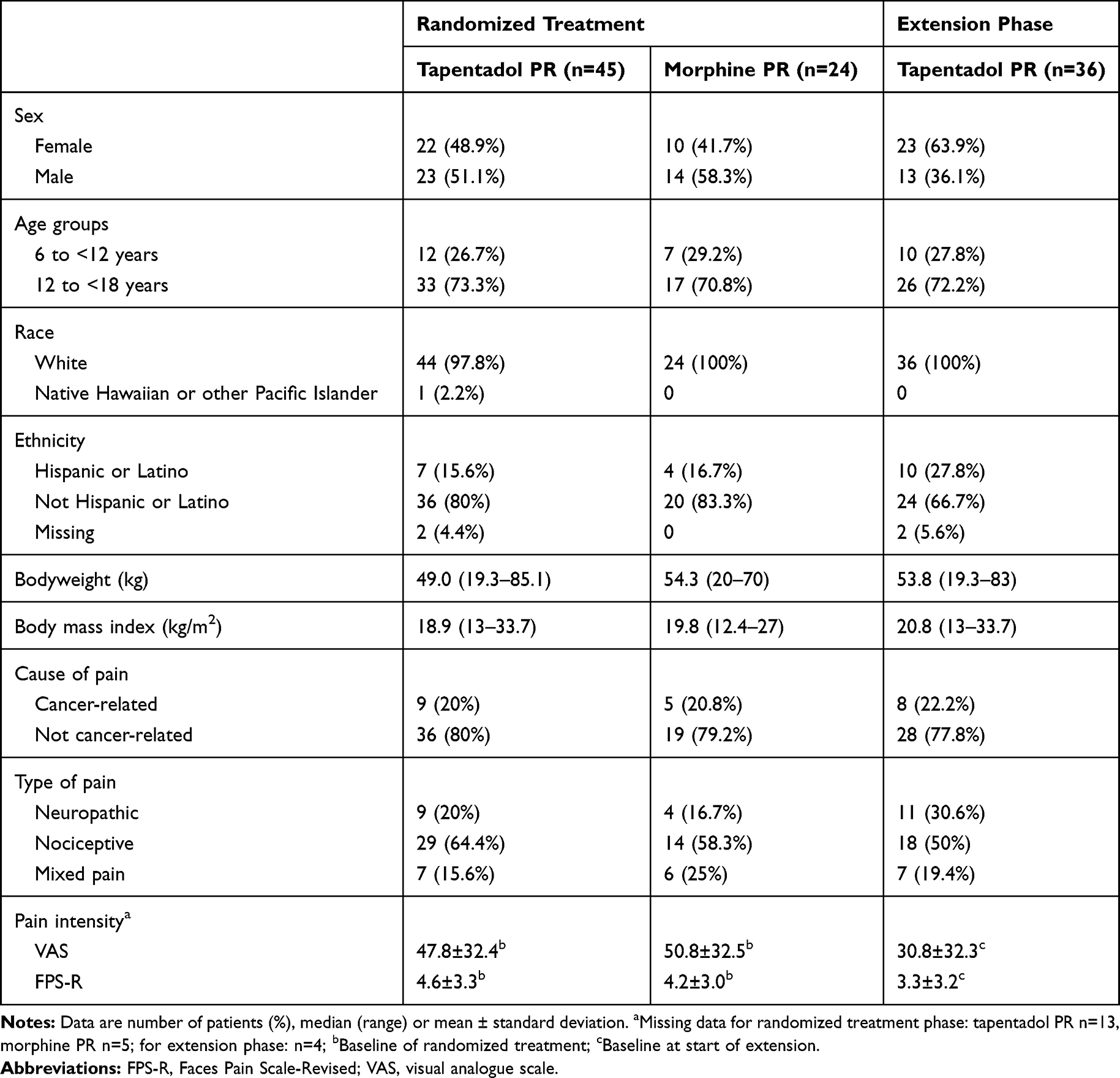 |
Table 1 Baseline Characteristics |
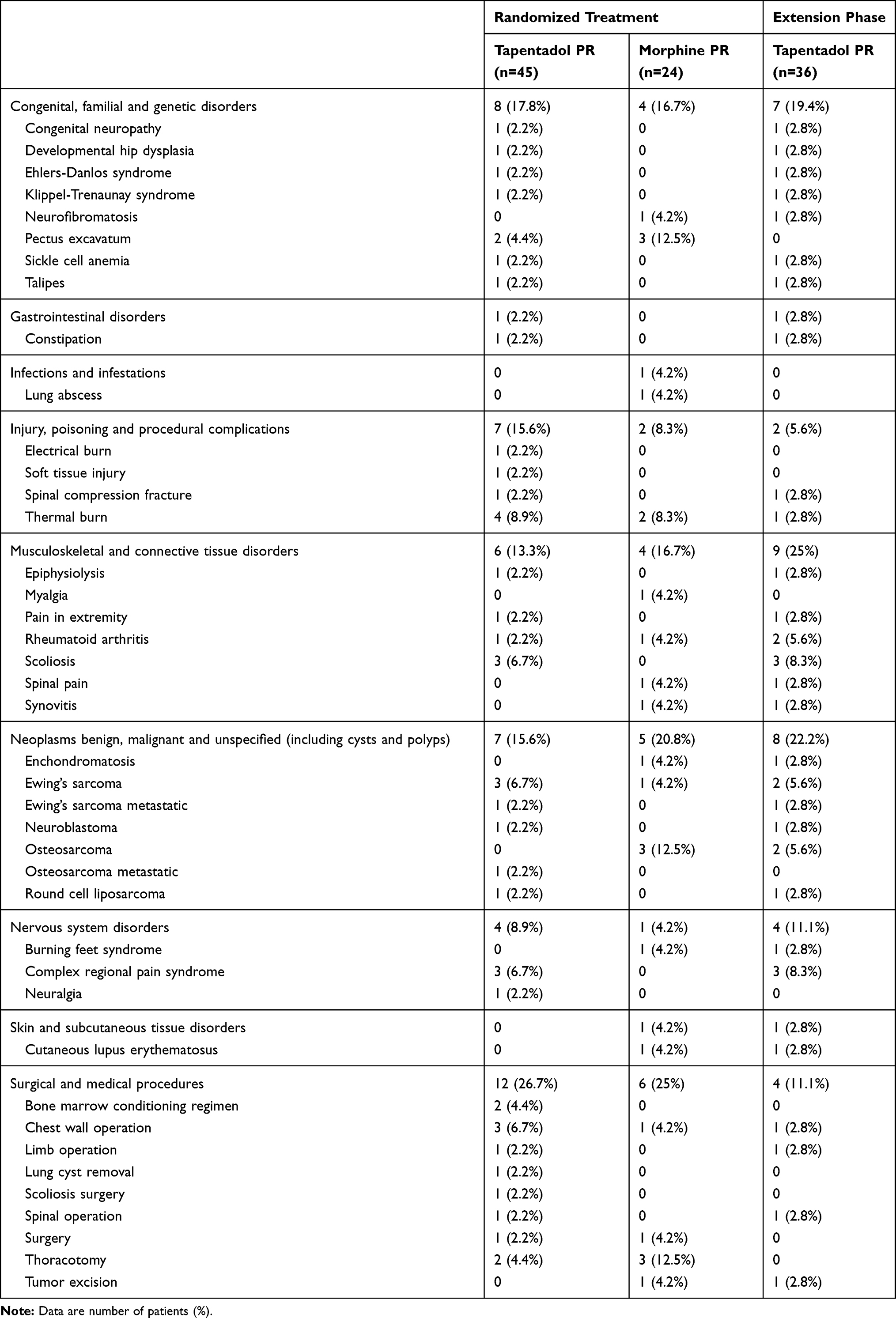 |
Table 2 Underlying Diagnosis/Reason for Pain |
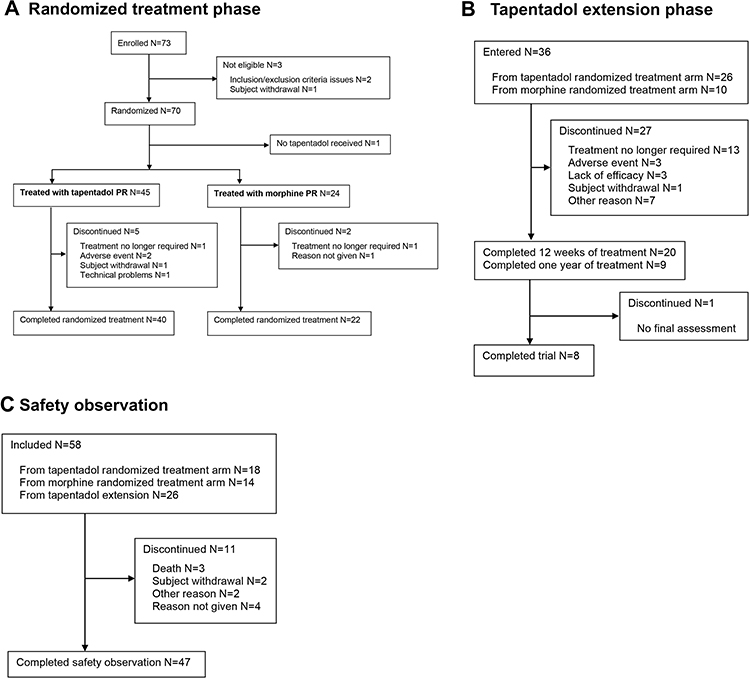 |
Figure 1 Patient flow chart. (A) Randomized treatment phase, (B) tapentadol extension phase, (C) safety observation. |
In Part 1, 40 patients completed randomized treatment with tapentadol PR (88.9%), and 22 patients (91.7%) with morphine PR, the reasons for discontinuation are given in Figure 1A; none of the patients discontinued due to lack of efficacy. Median time to discontinuation of trial medication was 4 days for tapentadol (n=5) and 11 days for morphine (n=2). Mean exposure to trial medication was comparable between the groups with 13.8 days (SD 3.2) for tapentadol PR and 14.5 days (SD 1.4) for morphine PR patients. Mean daily doses over the 14-day treatment period were 3.0 (SD 0.7) mg/kg tapentadol PR and 1.0 (SD 0.3) mg/kg morphine PR.
In Part 2, thirty-six patients entered the tapentadol treatment period, 26 from the tapentadol PR and 10 from the morphine PR treatment arms of Part 1 (Figure 1B). Baseline characteristics and underlying diagnosis/reasons for pain are described in Tables 1 and 2, respectively. Twenty of the patients entering the tapentadol extension period (55.6%) completed 3 months of tapentadol PR treatment, nine patients (25%) completed a year of treatment. “Treatment no longer required” was the main reason for treatment discontinuation (n=13). Patients received tapentadol PR on average for 175.8 days (SD 139.4) with an overall mean daily dose of 3.5 mg/kg (SD 1.7).
The observation period in Part 2 was completed by 47 of the 58 patients who entered (Figure 1C).
Efficacy
In Part 1, mean pain intensity was comparable in both treatment arms at baseline (Table 1) and decreased at a similar rate during the randomized period irrespective of pain scale used. Mean change from baseline at day 14 was −19.0 (SD 40.9) for tapentadol PR and −24.6 (SD 42.8) for morphine PR on the VAS and −2.0 (SD 4.1) and −2.1 (SD 3.7), respectively, on the FPS-R (“as observed” data; tapentadol n=23, morphine n=15). Baseline scores were higher for the younger age group, with higher mean changes in pain intensity at the last six assessments before final administration of trial medication compared to the adolescent group (Figure 2).
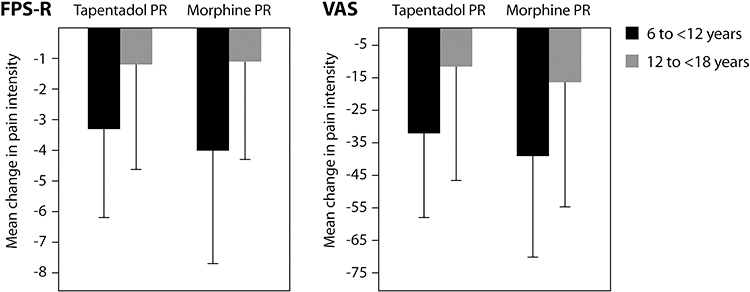 |
Figure 2 Mean change in pain intensity (SD) from baseline to the last six assessments before final administration of trial medication (randomized treatment phase). |
A total of 71% of tapentadol PR and 79.2% of morphine PR patients met the criteria for the primary endpoint “treatment responders”: ML estimates (80% CI) were 0.76 (0.64, 0.85) and 0.83 (0.69, 0.91), respectively. The difference between the groups was −0.06 (−0.19, 0.06; p=0.079); the lower CI bound was thus above the negative non-inferiority margin of −0.2 demonstrating non-inferiority of tapentadol PR treatment to morphine PR treatment. When the 3 CRPS patients were excluded, a difference of −0.02 (−0.15, 0.1; p=0.0332) was observed. The primary endpoint result was supported by the majority of sensitivity analyses (Figure 3).
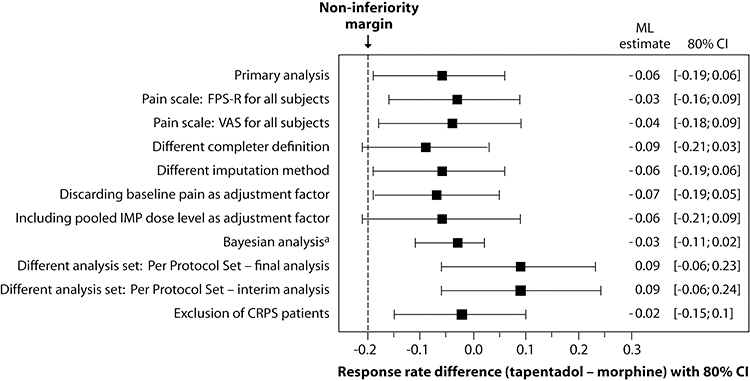 |
Figure 3 Forest plot of primary endpoint analysis and sensitivity analyses of the difference in treatment response between tapentadol PR and morphine PR (14-day randomized treatment period). |
Forty percent of tapentadol PR patients and 25% of morphine PR patients received rescue medication. Time to first intake in the tapentadol group was a mean 74.6 h (SD 94.5) and a median 29.3 h (Q1 16.4, Q3 135.1). For the morphine group, a mean time of 39.7 h (SD 63.8) with a median of 15 h (Q1 11.1, Q3 18.1) was documented. The modified average daily dose of rescue medication was 0.11 mg/kg (SD 0.2) for tapentadol patients, 0.08 mg/kg (SD 0.2) for morphine patients.
For patients entering the tapentadol treatment period of Part 2 the mean pain intensity scores at the end of Part 1 (baseline) were 30.8 (SD 32.3) on the VAS and 3.3 (SD 3.2) on the FPS-R. Further slight reductions were observed but overall, pain intensity remained stable over the 12-month treatment period for both VAS and FPS-R (Figure 4).
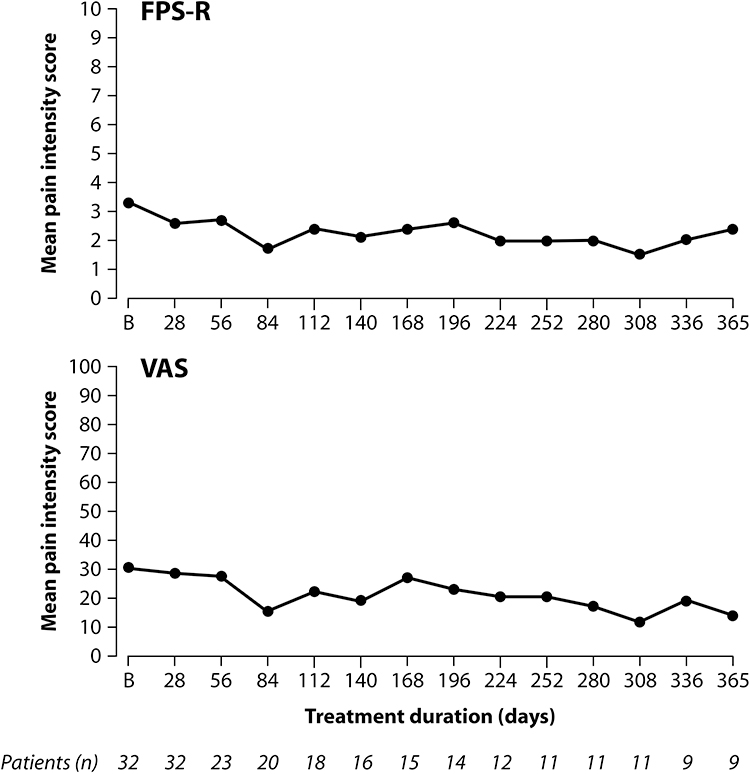 |
Figure 4 Mean pain intensity under long-term tapentadol PR treatment (“as observed” data). |
Safety
In Part 1, TEAEs were documented for 57.8% of tapentadol PR patients, and 50% of morphine PR patients (Table 3), most of them mild or moderate in intensity (tapentadol PR 94.6%, morphine PR 96.2%). Three patients taking tapentadol PR had a single serious TEAE of cystitis, malignant neoplasm progression, and acute kidney injury; one patient on morphine PR experienced four serious TEAEs (diarrhea, vomiting, mucosal inflammation, clostridium difficile infection). None of these events was considered related to the trial medication by the investigator. Premature discontinuation of tapentadol PR due to TEAEs was documented in 3 patients: 2 were judged to be possibly or certainly related to trial medication including intense vomiting in a patient following surgery for pectus excavatum, and diarrhea in a patient with metastatic disease who was receiving chemotherapy.
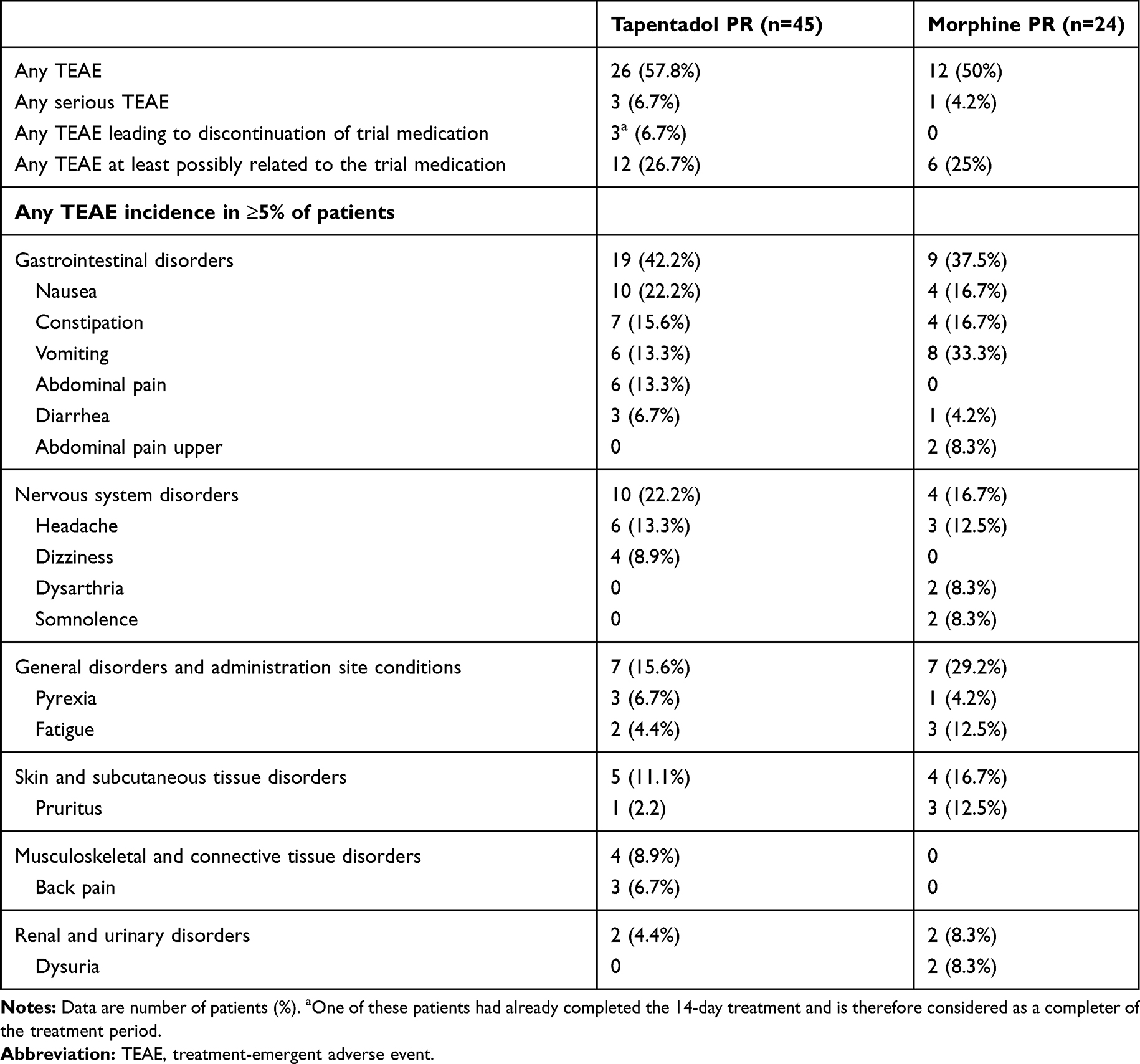 |
Table 3 Treatment-Emergent Adverse Event Profile During the Randomized Treatment Period (Safety Set) |
Small fluctuations in clinical chemistry and hematology laboratory parameters and in vital signs over the treatment period, as well as the occurrence of minor abnormalities in ECG recordings, were not considered to be clinically relevant. There was no indication of clinically relevant withdrawal symptoms.
In the tapentadol period of Part 2, 30 of 36 patients (83.3%) experienced 226 TEAEs (Table 4), which were mostly mild or moderate in intensity (91.1%). Thirteen patients (36.1%) experienced 23 serious TEAEs, none of them related to tapentadol PR treatment: 3 events of white blood cell count decreased, 2 events of cutaneous lupus erythematosus, and one event each of appendicitis, application site infection, breakthrough pain, dissociation, fall, herpes zoster, infection, limb operation, lymphopenia, malaise, malignant neoplasm progression, movement disorder, neuralgia, pain, pyrexia, sickle cell anemia with crisis, somnolence, and superficial thrombophlebitis. Premature discontinuation of tapentadol PR due to TEAEs was documented in 4 patients (11.1%). Reasons for withdrawal were dissociation, nightmares, fatigue and bradypnea (of moderate intensity that was considered by the investigator to be “certainly related” to tapentadol PR treatment) in one patient each. Again, there was no indication of clinically relevant withdrawal symptoms after the end of treatment –see below.
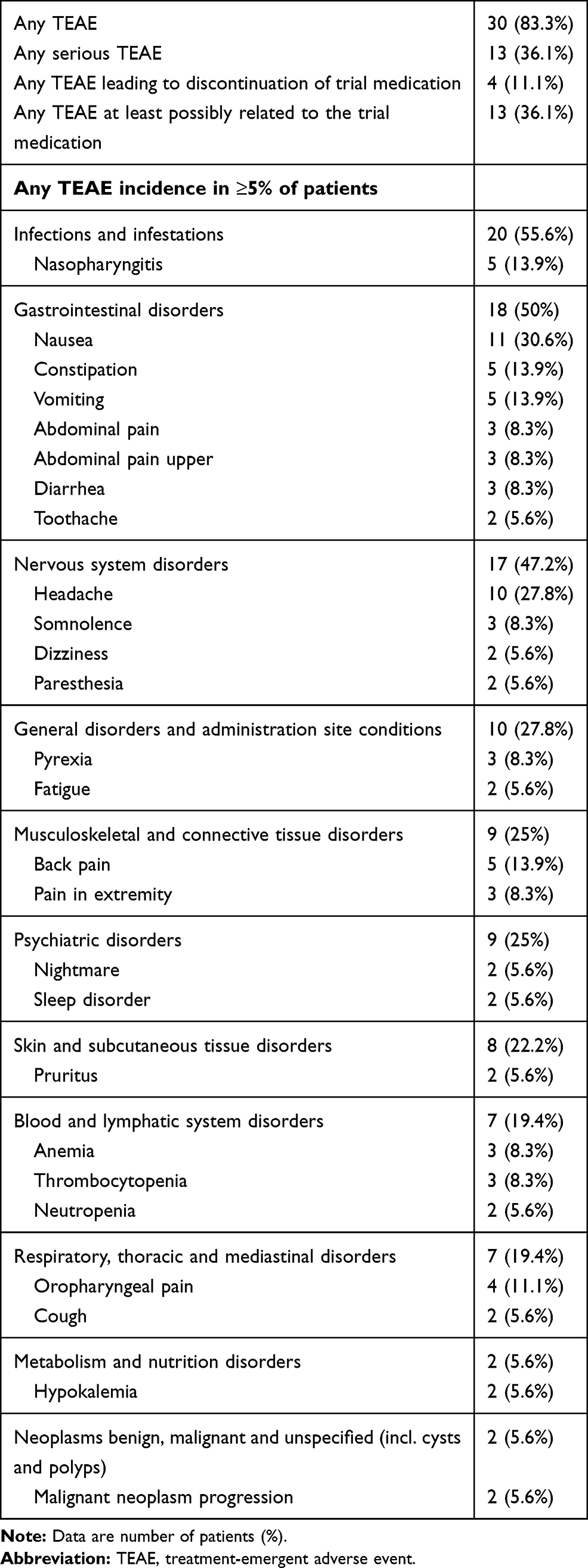 |
Table 4 Treatment-Emergent Adverse Event Profile During the Tapentadol PR Extension Phase (Safety Set, n=36) |
Adverse events occurring after the final dose of trial medication in the observation period were documented for 27 of the 58 patients (46.6%): Infections and infestations (27.6% of patients), nervous system disorders (17.2%), general disorders and administration site conditions (13.8%), and musculoskeletal and connective tissue disorders (13.8%) were most frequently reported; none were considered long-term consequences of the trial medication. Three patients died due to underlying diseases (progression of Ewing’s sarcoma, sarcoma, and osteosarcoma). Physical examination findings, clinical chemistry and hematology laboratory values, and vital signs were either unchanged or were as expected due to underlying clinical condition.
Other Trial Outcomes
Palatability and acceptability of the trial medication: Swallowing was rated as “easy/really easy” by 79.6% of patients in the tapentadol PR group and 87.5% in the morphine PR group. Taste was rated “good/really good” by 45.5% and 62.5%, respectively.
Constipation scores were stable and low throughout Part 1 at 1.5 (SD 1.4)-1.8 (SD 2.0)/16 for the tapentadol PR group and 2.7 (SD 3.0)-2.7 (SD 2.4)/16 for morphine PR at the start and end of treatment. Constipation scores fell by 0.7 points during the tapentadol treatment period of Part 2 from 2.3 (SD 2.4) at baseline to 1.8 (SD 2.7) at the end of treatment.
The mean opiate withdrawal score (0–60) was 6.8 (SD 7.4) for tapentadol PR and 4.0 (SD 3.8) for morphine PR after last trial medication intake in those patients who stopped trial medication during or at the end of Part 1, decreasing by 5 and 2.7 points respectively by day 7 after trial treatment discontinuation. For patients who received their final tapentadol PR dose in Part 2, the mean score after treatment discontinuation was 6.0 (SD 5.9) decreasing by 1.7 points at day 7.
Discussion
The results of the current trial show, for the first time, the efficacy and safety of tapentadol PR in the treatment of pain lasting at least 2 weeks and requiring strong opioid treatment in children and adolescents. Tapentadol PR demonstrated non-inferiority to the active comparator morphine PR over the 14-day randomized phase, and pain intensity scores remained low and stable during the subsequent 12-month period of tapentadol treatment. The adverse event profile was in line with the safety profile known from adult tapentadol PR trials;6 and no new safety issues or tapentadol-related side effects were identified in this 6 to <18 years pediatric population. Tapentadol PR was generally well tolerated with no clinically relevant withdrawal symptoms on termination of treatment; no adverse events that occurred after the final dose were considered to be long-term consequences of treatment.
Tapentadol is an opioid analgesic (MOR-NRI) combining both µ-opioid receptor agonist and noradrenaline reuptake inhibitory activity in one molecule with these two mechanisms appearing to act synergistically.1 Although the analgesic effect of tapentadol is similar to that of classical µ-opioid agonists such as morphine and oxycodone, there is relatively lower µ-agonist activity. The NRI activity of tapentadol leads to activation of adrenergic inhibitory mechanisms in the brain and spinal cord, and recent experimental data also implies that tapentadol may show enhanced adrenergic inhibitory activity in neuropathic pain, contributing to a stronger theoretical basis for its potential effectiveness in nociceptive, neuropathic and mixed mechanism pain.18 Consequently, in the adult, the use of tapentadol is being described and researched for a broad variety of indications including cancer pain and chronic musculoskeletal pain.19–22 Notwithstanding the recent focus on the suitability of long-term opioid therapy for some types of chronic pain, opioids remain amongst the most effective analgesics available, and are clearly indicated where the benefits and risks are in favor of their use in both adults and children, including chronic pain indications.23,24 In the light of this data, and in the spirit of initiatives designed to ensure that children are neither excluded from the benefits of new or existing treatments, nor are exposed to compounds that have not been adequately assessed, the drug development plan for tapentadol included a desire to assess its efficacy and safety for chronic pain in children.25,26
However, pediatric trials, especially in pain research, are known to have many challenges that include trial design constraints due to ethical and logistical considerations, and enrolment of a sufficient number of trial subjects with a suitably broad age range and compatible diagnoses within a reasonable time frame.26–28 These difficulties are evidenced by the extremely low number of publications available, especially for pediatric chronic pain.26,29 In recognition of these and other obstacles a number of ameliorating strategies relevant to the current study have been proposed and were initially implemented here as far as possible.27,30,31 Nevertheless, recruitment of the targeted pediatric population requiring treatment of sufficient duration with a strong PR opioid analgesic was thought to be potentially very challenging, despite the relatively low recruitment target (SSE = 69) due to careful trial design. Chronic pain is highly prevalent in children but such a non-specific indication would be likely to include many patients for whom long-term opioid therapy would not be appropriate.32 This problem was addressed by an agreed change in the pain indication for inclusion in the trial from “severe chronic pain” to “severe long-term pain requiring PR opioid treatment for at least 14 days”, this allowed the additional inclusion of a broader variety of indications (eg, cancer, postsurgical pain, burns) whilst still maintaining trial objectives.
In the event, only 23 (48%) of all invited 48 trial sites were able to enroll patients within the recruitment period of 3.5 years, clearly illustrating the difficulties faced by such trials despite the use of problem mitigating strategies. Nevertheless, a broad range of diagnoses and relevant underlying pain mechanisms (nociceptive, neuropathic, and mixed) are included here.
Of further interest, of the 36 patients who entered the longer-term tapentadol treatment phase 16 (44.4%) did not complete the first 3 months, mainly because opioid treatment was no longer required. Only 9 patients, all ≥10 years of age, received tapentadol PR treatment for a year, in line with previous suggestions that eligible children with severe chronic pain do not require opioid analgesics for longer time periods, and that the need for around-the-clock opioid treatment for severe pain for more than 4 weeks is rare.27,33,34
When evaluating these findings the small sample size and the open-label trial design need to be considered and that the use of rescue medication could be a confounding factor for efficacy. However, overall, the use of rescue medication in the study was very low in both groups and the study was not designed or powered to evaluate such differences. In this regard, the efficacy outcomes in Part 2 also need to be carefully interpreted as only a few patients completed this part of the trial and results may have been influenced by the natural course of their underlying disease process, enabling patients to discontinue from Part 2 as treatment was no longer required.
Conclusions
The results of this randomized, parallel group, non-inferiority trial show that tapentadol PR was equivalent to morphine PR in terms of both efficacy and safety in children aged 6 to <18 years requiring long-term opioid analgesia for at least 14 days. No decline in efficacy, new safety issues, long-term opioid adverse effects or adverse reactions were identified during the trial period of duration up to one year.
Abbreviations
AE, adverse event; CI, confidence interval; CNS, central nervous system; CRPS, complex regional pain syndrome; ECG, electrocardiogram; FAS, full analysis set; FPS-R, Faces Pain Scale-Revised; mCAS, modified constipation assessment scale; ML, maximum likelihood; MOR, μ-opioid receptor agonism; NRI, noradrenaline reuptake inhibition; PK, pharmacokinetic; PPS, per protocol set; PR, prolonged release; SSE, sample size estimate; TEAE, treatment-emergent adverse event; VAS, visual analogue scale.
Data Sharing Statement
Any request for data should be submitted via the following website: https://www.grunenthal.com/en/r-d-vision-mission/clinical-trials/data-sharing-clinical-trials.
Acknowledgments
The authors thank all patients, parents/legal guardians, investigators, and the trial site teams involved in this investigation. The trial was funded by Grünenthal GmbH. Editorial assistance was provided by Elke Grosselindemann and Birgit Brett and was paid for by Grünenthal GmbH.
Disclosure
RH provided consultancy advice to Grünenthal GmbH during trial development and was co-ordinating investigator during the trial; in addition he reports personal fees from Grünenthal GmbH during the conduct of the study, and personal fees from Wockhardt UK and consultancy for Regeneron UK Ltd outside the submitted work. AW was an employee of Grünenthal GmbH, at the time the trial was conducted. TR, MS, and ME are current employees of Grünenthal GmbH. The authors report no other potential conflicts of interest for this work.
References
1. Tzschentke TM, Christoph T, Kögel BY. The mu-opioid receptor agonist/noradrenaline reuptake inhibition (MOR-NRI) concept in analgesia: the case of tapentadol. CNS Drugs. 2014;28(4):319–329.
2. Raffa RB, Elling C, Tzschentke TM. Does ‘strong analgesic’ equal ‘strong opioid’? Tapentadol and the concept of ‘µ-load’. Adv Ther. 2018;35(10):1471–1484. doi:10.1007/s12325-018-0778-x
3. Göhler K, Brett M, Smit JW, Rengelshausen J, Terlinden R. Comparative pharmacokinetics and bioavailability of tapentadol following oral administration of immediate- and prolonged-release formulations. Int J Clin Pharmacol Ther. 2013;51(4):338–348. doi:10.5414/CP201722
4. Smit JW, Oh C, Rengelshausen J, et al. Effects of acetaminophen, naproxen, and acetylsalicylic acid on tapentadol pharmacokinetics: results of two randomized, open-label, crossover, drug-drug interaction studies. Pharmacotherapy. 2010;30(1):25–34. doi:10.1592/phco.30.1.25
5. Xiao JP, Li AL, Feng BM, Ye Y, Wang GJ. Efficacy and safety of tapentadol immediate release assessment in treatment of moderate to severe pain: a systematic review and meta-analysis. Pain Med. 2017;18(1):14–24. doi:10.1093/pm/pnw154
6. Baron R, Eberhart L, Kern KU, et al. Tapentadol prolonged release for chronic pain: a review of clinical trials and 5 years of routine clinical practice data. Pain Pract. 2017;17(5):678–700. doi:10.1111/papr.12515
7. Buynak R, Rappaport SA, Rod K, et al. Long-term safety and efficacy of tapentadol extended release following up to 2 years of treatment in patients with moderate to severe, chronic pain: results of an open-label extension trial. Clin Ther. 2015;37(11):2420–2438.
8. Finkel JC, Goldberg J, Rosenburg R, et al. First evaluation of tapentadol oral solution for the treatment of moderate to severe acute pain in children aged 6 to <18. J Pain Res. 2019;12:1925–1936.
9. Muse D, Tarau E, Lefeber C, et al. Pharmacokinetics, safety, and efficacy of tapentadol oral solution for treating moderate to severe pain in pediatric patients. J Pain Res. 2019;12:1777–1790.
10. Beuter C, Volkers G, Radic T, Goldberg J, van den Anker J. Efficacy and safety of multiple doses of tapentadol oral solution in the treatment of moderate to severe acute pain in children aged 2 to <18 years - a randomized, double-blind, placebo-controlled trial. J Pain Res. 2019;12:3099–3112.
11. Electronic Medicines Compendium. Palexia SR Prolonged Release Tablets. Summary of Product Characteristics. Datapharm PLC; 2020. Available at https://www.medicines.org.uk/emc/product/5158/smpc.
12. Hicks CL, von Baeyer CL, Spafford PA, van Korlaar I, Goodenough B. The Faces Pain Scale-Revised: toward a common metric in pediatric pain measurement. Pain. 2001;93(2):173–183. doi:10.1016/S0304-3959(01)00314-1
13. Guinard JX. Sensory and consumer testing with children. Trends Food Sci Technol. 2001;11(8):273–283. doi:10.1016/S0924-2244(01)00015-2
14. Woolery M, Carroll E, Fenn E, et al. A constipation assessment scale for use in pediatric oncology. J Pediatr Oncol Nurs. 2006;23:65–74.
15. Handelsman L, Cochrane KJ, Aronson M, Ness R, Rubinstein KJ, Kanof PD. Two new rating scales for opiate withdrawal. Am J Drug Alcohol Abuse. 1987;13(3):293–308. doi:10.3109/00952998709001515
16. Hlavin G, Koenig F, Male C, Posch M, Bauer P. Evidence, eminence and extrapolation. Stat Med. 2016;35(13):2117–2132. doi:10.1002/sim.6865
17. Farrington CP, Manning G. Test statistics and sample size formulae for comparative binomial trials with null hypothesis of non-zero risk difference or non-unity relative risk. Stat Med. 1990;9(12):1447–1454. doi:10.1002/sim.4780091208
18. Romualdi P, Grilli M, Canonico PL, Collino M, Dickenson AH. Pharmacological rationale for tapentadol therapy: a review of new evidence. J Pain Res. 2019;12:1513–1520. doi:10.2147/JPR.S190160
19. Billeci D, Coluzzi F. Tapentadol extended release for the management of chronic neck pain. J Pain Res. 2017;10:495–505. doi:10.2147/JPR.S129056
20. Kress HG, Coluzzi F. Tapentadol in the management of cancer pain: current evidence and future perspectives. J Pain Res. 2019;12:1553–1560. doi:10.2147/JPR.S191543
21. Coluzzi F, Polati E, Freo U, Grilli M. Tapentadol: an effective option for the treatment of back pain. J Pain Res. 2019;12:1521–1528. doi:10.2147/JPR.S190176
22. Coluzzi F, Pergolizzi JV
23. Ballantyne JC. Opioids for the treatment of chronic pain: mistakes made, lessons learned, and future directions. Anesth Analg. 2017;125(5):1769–1778. doi:10.1213/ANE.0000000000002500
24. Krane EJ, Weisman SJ, Walco GA. The national opioid epidemic and the risk of outpatient opioids in children. Pediatrics. 2018;142(2):e20181623. doi:10.1542/peds.2018-1623
25. European Medicines Agency. Assessment of the paediatric needs - Pain. 2005. Available from: https://www.ema.europa.eu/en/documents/other/assessment-paediatric-needs-pain_en.pdf.
26. Davies EH, Ollivier CM, Saint Raymond A. Paediatric investigation plans for pain: painfully slow! Eur J Clin Pharmacol. 2010;66(11):1091–1097. doi:10.1007/s00228-010-0886-2
27. Berde CB, Walco GA, Krane EJ, et al. Pediatric analgesic clinical trial designs, measures, and extrapolation: report of an FDA scientific workshop. Pediatrics. 2012;129(2):354–364.
28. Pica N, Bourgeois F. Discontinuation and nonpublication of randomized clinical trials conducted in children. Pediatrics. 2016;138(3):e20160223. doi:10.1542/peds.2016-0223
29. Eccleston C, Fisher E, Cooper TE, et al. Pharmacological interventions for chronic pain in children: an overview of systematic reviews. Pain. 2019;160(8):1698–1707. doi:10.1097/j.pain.0000000000001609
30. US Food and Drug Administration. CDER Conversation: pediatric pain management options. 2015. Available from: https://www.fda.gov/drugs/news-events-human-drugs/cder-conversation-pediatric-pain-management-options.
31. Eerdekens M, Beuter C, Lefeber C, van den Anker J. The challenge of developing pain medications for children: therapeutic needs and future perspectives. J Pain Res. 2019;12:1649–1664.
32. King S, Chambers CT, Huguet A, et al. The epidemiology of chronic pain in children and adolescents revisited: a systematic review. Pain. 2011;152(12):2729–2738. doi:10.1016/j.pain.2011.07.016
33. Finkel J, Finley A, Greco C, Weisman S, Zeltzer L. Transdermal fentanyl in the management of children with chronic severe pain: results from an international study. Cancer. 2005;104(12):2847–2857. doi:10.1002/cncr.21497
34. US Food and Drug Administration. Pediatric pain and the approach to studying opioid analgesics in pediatric populations.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.