Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
Suspected Transverse Myelitis with Normal MRI and CSF Findings in a Patient with Lupus: What to Do? A Case Series and Systematic Review
Authors Monahan RC, Beaart HJL, Fronczek R ![]() , Terwindt GM, Beaart-van de Voorde LJJ, de Bresser J, Kloppenburg M, van der Wee NJA, Huizinga TWJ
, Terwindt GM, Beaart-van de Voorde LJJ, de Bresser J, Kloppenburg M, van der Wee NJA, Huizinga TWJ ![]() , Steup-Beekman GM
, Steup-Beekman GM
Received 1 October 2020
Accepted for publication 27 November 2020
Published 22 December 2020 Volume 2020:16 Pages 3173—3186
DOI https://doi.org/10.2147/NDT.S267000
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Rory C Monahan,1 Hannelore JL Beaart,1 Rolf Fronczek,2,3 Gisela M Terwindt,2 Liesbeth JJ Beaart-van de Voorde,1 Jeroen de Bresser,4 Margreet Kloppenburg,1,5 Nic JA van der Wee,6 Tom WJ Huizinga,1 Gerda M Steup-Beekman1,7
1Department of Rheumatology, Leiden University Medical Center (LUMC), Leiden, the Netherlands; 2Department of Neurology, LUMC, Leiden, the Netherlands; 3Sleep-Wake Center SEIN, Heemstede, the Netherlands; 4Department of Radiology, LUMC, Leiden, the Netherlands; 5Department of Clinical Epidemiology, LUMC, Leiden, the Netherlands; 6Department of Psychiatry, LUMC, Leiden, the Netherlands; 7Department of Rheumatology, Haaglanden Medical Center, The Hague, the Netherlands
Correspondence: Rory C Monahan
Department of Rheumatology, Leiden University Medical Center (LUMC), Leiden 2333 ZA, the Netherlands
Tel +3171-5265762
Fax +3171-5266752
Email [email protected]
Purpose: To evaluate the use of immunosuppressive treatment, clinical outcome and diagnostic strategy in patients with systemic lupus erythematosus (SLE) presenting with clinical features of transverse myelitis (TM), but normal MRI of the spinal cord (sMRI) and normal cerebrospinal fluid (CSF) assessment, and to suggest a clinical guideline.
Patients and Methods: All patients with SLE and clinical features compatible with (sub)acute TM visiting the NPSLE clinic of the LUMC between 2007 and 2020 were included. Information on baseline characteristics, investigations, treatment and outcomes was collected from electronic medical records. In addition, a systematic review of individual participant data was performed up to April 2020 in PubMed, Embase and Web of Science, identifying all patients with TM, SLE and sMRI assessment. Data regarding sMRI, CSF analysis, treatment and outcome were extracted, and outcome was compared between patients with normal sMRI and CSF (sMRI-/CSF-) and patients with abnormalities.
Results: Twelve SLE patients with a clinical diagnosis of TM were identified: four sMRI-/CSF- and one sMRI- with CSF not available. All patients received immunosuppressive treatment, but outcome in sMRI-/CSF- patients was worse: no recovery (n=1) or partial recovery (n=3) compared to partial recovery (n=4) and (nearly) complete recovery (n=3) in MRI+ patients. The systematic literature review yielded 146 articles eligible for inclusion, 90% case reports. A total of 427 SLE patients with TM were identified, of which only four cases were sMRI-/CSF- (1%), showing no improvement (n=1), partial improvement (n=2) and complete recovery (n=1) after immunosuppressive treatment.
Conclusion: Outcome in SLE patients presenting with clinically suspected TM with normal sMRI and CSF is less favorable, despite treatment with immunosuppressive therapy. Taking a functional neurological disorder into consideration may be helpful in order to start other therapeutic strategies. We suggest prescribing immunosuppressive treatment for a restricted period of time to evaluate its effect in cases where a functional disorder initially is considered unlikely.
Keywords: lupus, neuropsychiatric, transverse myelitis, MRI, CSF
Introduction
Myelopathy is a rare but severe manifestation of neurological involvement in systemic lupus erythematosus (SLE), with an estimated prevalence of 1%.1 It is one of the nineteen 1999 ACR case definitions of neuropsychiatric syndromes in SLE.2 In the literature, myelopathy, transverse myelopathy, and acute transverse myelitis seem to be used interchangeably in SLE, but only (transverse) myelitis (TM) specifically refers to an inflammatory process. In this study, we focus solely on TM in patients with SLE.
A general set of diagnostic criteria for (sub)acute TM has been developed in 2002, including the following components: sensory, motor, or autonomic dysfunction attributable to the spinal cord should be present, a T2 hyperintense signal change on spinal magnetic resonance imaging (sMRI) should be visible and there should be no compressive cord lesion.3,4 These three elements are considered the most critical for the diagnosis. Other elements of the diagnostic criteria include the presence of bilateral signs and/or symptoms, a defined sensory level and inflammation defined by either specific cerebrospinal fluid (CSF) abnormalities (pleocytosis, elevated immunoglobulin G (IgG) index) or gadolinium enhancement on sMRI. Not all patients presenting with clinical signs of TM will fulfill all of the above criteria – especially the inflammatory features are not met by a significant percentage of individuals.5 The NPSLE case definition of myelopathy does not consider sMRI or CSF abnormalities mandatory,2 but the diagnosis of TM in patients with SLE without any evidence on sMRI or with CSF examination is challenging in clinical practice. A differential diagnosis in sMRI and CSF negative cases (sMRI-/CSF-) is a functional neurological disorder (FND). This distinction is important as treating patients with immunosuppressive medication can cause severe side-effects, and not initiating psychological or rehabilitation guidance in case of FND may delay clinical improvement.
In this article, we aim to gather available data on SLE patients presenting with clinical features of TM without MRI or CSF abnormalities (sMRI-/CSF-) compared to patients with MRI and/or CSF abnormalities in order to identify whether sMRI-/CSF- patients should receive immunosuppressive treatment. We focus on comparing treatment and outcome by sharing the experience from our tertial referral center for SLE patients with neuropsychiatric symptoms and performing a systematic literature review.
Patients and Methods
Part I: Case Series TM NPSLE Clinic
Study Population
The NPSLE clinic of the Leiden University Medical Center is a tertiary referral center for patients with (suspicion of) SLE and neuropsychiatric symptoms. All patients are evaluated in a multidisciplinary setting, as described in detail previously.6 All patients are evaluated in one day by a rheumatologist, neurologist, clinical neuropsychologist, psychiatrist, vascular internal medicine expert and an advanced nurse practitioner and undergo brain MRI and extensive laboratory investigation. CSF analysis and sMRI with gadolinium are performed on indication. sMRI met the following standards: at least 1.5T, visualization of the complete spine and spinal cord, a sagittal T1-weighted (before and after gadolinium) and T2-weighted sequence with a slice thickness of 2mm and a transverse T2-weighted sequence at the site of the suspected abnormality. After 2 weeks, a multidisciplinary team meeting takes place to reach a consensus-based diagnosis regarding the presence or absence of NPSLE. All patients who visited the NPSLE clinic between 2007 and May 2020 that had the diagnosis of SLE (based on clinical judgement), signed informed consent and were suspected of having TM attributed to SLE were included in this study. This study was conducted in accordance with the Declaration of Helsinki and approved by the Leiden-the Hague-Delft medical research ethical committee. Patients described in the case series provided informed consent for the details to be published.
Data Collection
Medical records were reviewed to collect data on demographics, clinical presentation, ACR 1997 SLE classification criteria, laboratory serum and CSF findings, brain and sMRI, treatment and clinical outcomes at the moment of initial presentation of neurological symptoms. All sMRIs were re-evaluated for signs of myelopathy or compression of the spinal cord by an experienced neuroradiologist (JdB). SLE activity was evaluated using the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI-2K, range: 0–105).7 As some patients presented with acute TM at a different hospital and were later referred to our NPSLE clinic additional information about the initial presentation was extracted from referral letters.
Analysis
Baseline characteristics of patients with a clinical presentation of TM without any abnormalities (sMRI-/CSF-) and with abnormalities (sMRI+ and/or CSF+) on additional investigation were compared. Patients with normal sMRI lacking CSF were not included in the comparison.
Part II: Systematic Review
Literature Search
Studies were identified in a literature search using PubMed, EMBASE and Web of Science, Cochrane and Emcare databases, restricted to the English, Dutch and German language. The search was developed and performed until 29–04-2020 in collaboration with an experienced librarian. The search strategies contained the keywords for ‘lupus’, “SLE” or ‘systemic lupus erythematosus’, combined with “myelopathy” or “transverse myelitis”. The full search strategy for PubMed is included in Supplementary materials. The systematic review was performed in line with the PRISMA guidelines for individual participant data (IPD).8
Study Selection
Study selection was independently carried out by two reviewers (RM and HB). Cases of disagreement were discussed until consensus was reached. First, all obtained titles were screened, and subsequently abstracts were reviewed after which full-text articles were screened for eligibility.
Articles were included if patients were ≥16 years or if the majority of the study population was adult, if clinical diagnosis of SLE or SLE according to the 1997 or 1982 revised classification criteria was present, if TM was addressed and if patients lacking abnormalities on lumbar puncture (LP) and/or sMRI were not excluded.9,10 In addition, patients with anterior spinal artery syndrome on sMRI assessment were excluded. Patients with SLE and neuromyelitis optica were included.
Data Extraction
Information was extracted from two types of articles by RM: case reports/case series and observational studies that presented information on patients with SLE and TM according to specific criteria for SLE and TM, over a specific amount of time in a hospital or region.
The following information was extracted from case reports/case series: age, sex, SLE duration and presence of sMRI abnormalities compatible with TM. sMRI compatible with TM was defined according to the diagnostic criteria.4 Treatment was only extracted in MRI-/CSF- cases. In addition, the presence and type of CSF abnormalities (pleocytosis, increased protein, low glucose, oligoclonal bands, increased IgG index) were assessed. If no reference values were provided for CSF analyses, pleocytosis was considered to be present in case of >5 white blood cells per μL or equivalent thereof, protein level increased if >45 mg/dl or equivalent thereof and low glucose if glucose <2.5 mmol/L or <45 mg/dL or a ratio serum glucose/CSF glucose of <0.5. If CSF was described as normal, pleocytosis, increased protein and low glucose were considered absent. Clinical outcome was divided into three time points: at hospital discharge or during hospital stay, short term: between hospital discharge and 6 months and long term: 6 months and longer. Clinical outcome status was defined as 1) complete recovery if no neurological symptoms remained, 2) near-complete recovery if symptoms remained without impairment of mobility/in daily life, 3) partial recovery if there were remaining symptoms other than 2 and 4) no recovery if neurological status was similar between presentation and at the end of follow-up time. If “remission” or “recovery” was stated, outcome was considered to be 1)/2). If “improved” without further detail was stated, outcome is considered 3). The final status was compared to the status at the first presentation for assessing the overall outcome.
For the observational studies, the same variables were extracted. In addition, information regarding the criteria used for the diagnosis of TM was obtained from these articles and divided in four categories: 1) TM based on clinical characteristics, 2) TM based on a combination of clinical characteristics and confirmation on additional assessment, 3) TM based on the 2002 criteria for TM4 and 4) TM based on the NPSLE 1999 ACR case definitions.2 Furthermore, the number of patients studied, age at diagnosis (range), number of patients without MRI abnormalities, CSF findings in patients without sMRI abnormalities and treatment and disease course in patients with no abnormalities. If individual data were not provided in the article, authors were contacted to obtain this information.
The following aspects were assessed for risk of bias: method of patient inclusion and clinical outcome definition. This is reported in Supplementary material.
Analysis
The primary outcome of this systematic review was the comparison of clinical outcome between sMRI-/CSF- patients and sMRI+ and/or CSF+ patients. If CSF was not available for sMRI- patients, these patients were excluded. If patients showed sMRI or CSF abnormalities later in the course of the disease, these were analyzed in the sMRI+ or CSF+ group. All summary data were calculated using Stata Statistical Software: Release 16 (2019).
Results
Part I
TM Patients NPSLE Clinic 2007–2020
Between 2007 and 2020, 560 patients attended the NSPLE clinic. Of these patients, 23 were suspected of having TM, of which eleven were excluded from this study because of clear alternative diagnoses (n = 6), no clinical diagnosis of SLE (n = 2), no clinical signs of TM (n = 2) and clinically isolated syndrome most likely due to multiple sclerosis (n = 1).
Clinical characteristics of all patients are provided in Table 1. sMRIs were made between 2 and 10 days after symptom onset in all patients and five patients had normal sMRI (sMRI-). Four sMRI- patients had CSF assessment (all normal). Nearly all patients were female (83%). Median age in sMRI- patients was 36 years (range: 23–47) and 55 years in sMRI+ patients (range: 36–67). Median SLE duration was 1 year in sMRI- (range: 0–6) and 4 years in sMRI+ patients (range: 0–17). Median SLEDAI score was 4 (range: 0–22) in sMRI- and 2 (range: 0–15) in sMRI+ patients.
 |
Table 1 Clinical Characteristics of SLE Patients Diagnosed and Treated for TM without (1–5) and with (6–12) sMRI Abnormalities in the NPSLE Clinic Between 2007 and 2020 |
Laboratory investigations are presented in Table 2. Increased ESR was only present in one sMRI- and one MRI+ patient. Antinuclear antibodies (ANA) were present in 2/5 sMRI- patients and 6/7 MRI+ patients. All patients that did not have ANA during this clinical presentation, had a positive ANA more than once in the past. Antiphospholipid antibodies (APLAs) were positive in 3/5 sMRI- patients and 2/7 MRI+ patients. Complement consumption was present in 1/5 sMRI- and 4/7 sMRI+ patients. Aquaporin-4 antibodies were tested in five patients, and positive in 1 sMRI+ patient.
 |
Table 2 Laboratory Investigation of SLE Patients Diagnosed with TM without (1–5) and with (6–12) sMRI Abnormalities at the LUMC NPSLE Clinic Between 2007 and 2020 |
Most patients were treated with methylprednisolone (MP) and/or prednisone at high dosage in combination with cyclophosphamide (5/5 sMRI-, 5/7 sMRI+), as shown in Table 3. Two patients with different treatment regimens received MP and IVIG in one case and rituximab and belimumab in the other case (because of a high cumulative dosage of cyclophosphamide due to previous TM). Outcome in sMRI-/CSF- patients was no recovery (n = 1) and partial recovery (n = 3) compared to partial recovery (n = 4) and (nearly) complete recovery (n = 3) in sMRI+ patients.
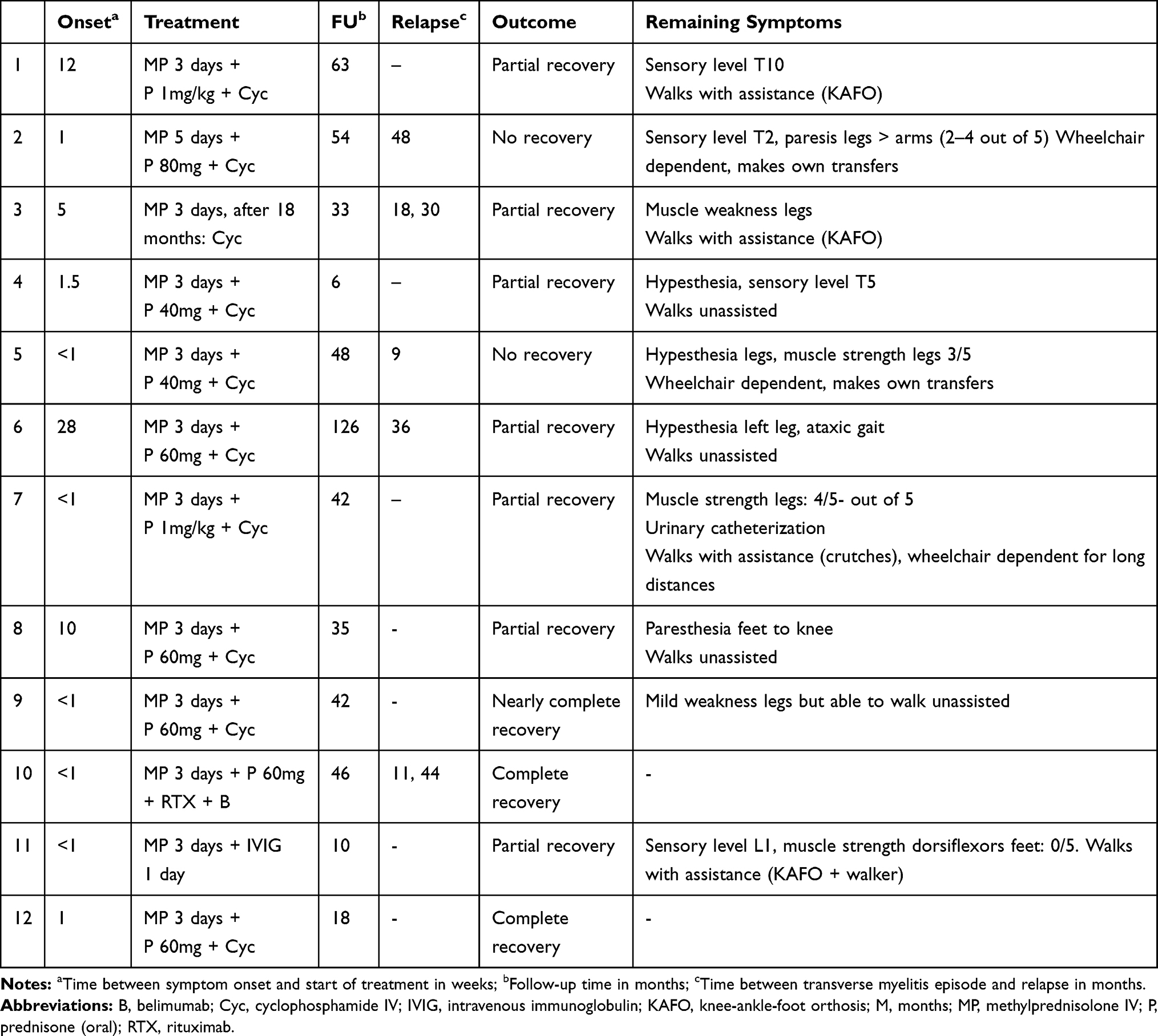 |
Table 3 Treatment and Outcome of SLE Patients Diagnosed with TM Without (1–5) and Without (6–12) sMRI Abnormalities at the LUMC NPSLE Clinic Between 2007 and 2020 |
Case Series sMRI-/CSF- Patients
A 42-year-old man was admitted to the Department of Neurology because of decreased sensation and strength in his lower limbs, which started 10 days previously. His medical history showed hernia decompression surgery (L4-L5) and an episode of paraparesis of the legs that spontaneously disappeared 5 years previously. The diagnosis of SLE was established several months before presentation based on antinuclear antibodies (ANA+), lupus anticoagulant (LAC+), oral ulcers, photosensitivity, arthralgia and fatigue. Neurological examination at admission showed a sensory level at T10, bilateral paresis of the lower limbs (3/5) and hypotonia of the legs with normal tendon reflexes, including plantar reflexes. Neurological exam showed discrepancies of muscle strength of the lower limbs over time. Several days after admission, urinary retention requiring catheterization occurred. Elaborate investigation, including sMRI, CSF assessment, motor evoked potential (MEP) and somatosensory evoked potential (SSEP) did not reveal any abnormalities. SLEDAI at presentation was 0. A FND was suspected and psychological counseling and rehabilitation were initiated. After 2 months, the patient was referred to our NPSLE clinic. At that time, no improvement of his neurological status had occurred and all investigations remained normal. Because it was considered that TM due to SLE could not be fully ruled out, treatment with MP was initiated, after which a subjective decrease of numbness of the legs occurred, but no objective changes were found. Treatment with cyclophosphamide was started and after 6 months, the patient reported additional improvement of the strength of the upper legs. Cyclophosphamide was continued for another year and at 18 months, no further neurological improvement had occurred. At his last follow-up six years later he was able to walk with assistance (knee-ankle-foot-orthosis, KAFO), but anesthesia of the legs remained.
A 36-year-old woman presented at the Department of Neurology because of bilateral progressive loss of sensation, starting at the fingers and toes. She had SLE based on ANA+, arthritis, photosensitivity, oral ulcers, pleuritis, Raynaud and secondary antiphospholipid syndrome. In addition, she experienced multiple transient ischemic attacks in the previous year and suffered from an anxiety disorder. At neurological examination she initially showed decreased sensibility of the feet to knees and hands up to elbows, which increased to sensory level T6 during admission. Tendon reflexes were brisk bilaterally and plantar reflexes were normal. Muscle strength was preserved. Some discrepancies in the neurological exam were present over the course of the hospital admission, with a varying level of sensory deficit. LP and SSEP did not reveal any abnormalities and sMRI revealed a mild bulging of the disc at level C3/C4 and a discrete protrusion of the disc at level T6-T7 with slight impression on the spinal cord, but without myelopathy or signs of myelitis. SLEDAI at presentation was 8, based on the presence of oral ulcers, alopecia and mild arthritis. Because it was considered that TM due to SLE could not be fully ruled out, treatment with MP was initiated, followed by high-dose oral steroids. After 1 week she was assessed at the NPSLE clinic, at which time the patient mentioned subjective improvement of neurological symptoms. Neurological examination showed a sensory level at T4 and cyclophosphamide was started. After 6 months, subjective improvement of the arms was reported, but a sensory level at T4 remained and cyclophosphamide was continued. After 18 months, no further changes were observed and the patient remained dependent on the wheelchair for mobilization. After 4 years, a relapse occurred with a complete tetraparesis, once again without any compatible abnormalities on sMRI or in CSF. The patient was treated with rituximab and after 6 months recovered to the state before the tetraparesis, with hypesthesia of the lower legs and arms and wheelchair dependency.
A 26-year-old woman presented at the emergency room because of paresthesia of the feet and legs, reduced strength of the legs and difficulty in urinating. The diagnosis SLE was established two years earlier based on ANA+, positive anti-double-stranded DNA (anti-dsDNA+), photosensitivity, malar rash and polyarthritis. In addition, she was recently analyzed for the presence of epileptic seizures and suffered from an anxiety disorder. Neurological examination showed reduced muscle strength bilaterally (3/5) of the lower limbs and normal tendon reflexes, including the plantar reflex. In addition, a sensory level at T9 and urinary retention were present. Additional investigation, including sMRI and CSF assessment, did not show any abnormalities. SLEDAI was 4 based on mild arthritis of the hands. No treatment was initiated initially, because of diagnostic uncertainty. Several weeks later, paresis of the legs increased and a Babinski sign was present bilaterally. In the course of the disease, there was uncertainty regarding the presence of a Babinski sign: it was described by some (but not all) neurologists, both unilaterally and bilaterally. Because of persistent uncertainty regarding the diagnosis, the patient was treated with MP which led to some improvement of muscle strength. Rehabilitation was started as well. After 10 months, a relapse occurred in which the left leg was predominantly affected and patient was treated with MP again. Twelve months after symptom onset, she was re-evaluated at the NPSLE clinic. Neurological examination showed a T10 sensory level, brisk reflexes but no evident Babinski sign. Additional investigation (CSF, sMRI) remained normal and auto-immune serology including ANA was negative at this time. Patient was referred for a third opinion because of remaining uncertainty regarding the diagnosis. At the referral center, treatment with cyclophosphamide was initiated, which led to clinical improvement initially, but after 6 months neurological symptoms recurred. Corticosteroid treatment was continued and years after symptom onset, improvement of walking ability started to occur. Patient currently is able to walk a maximum distance of 1 km with KAFO and still needs catheterization.
A 23-year-old woman was diagnosed with SLE based on ANA+, anti-dsDNA+, anti-Smith antibodies (anti-Sm+), antiphospholipid antibodies, complement consumption, malar rash, oral ulcers, hair loss, arthralgia, fatigue and lupus nephritis class II. A week after the diagnosis, she visited the outpatient clinic of the Department of Neurology because of reduced strength and paresthesia of her legs since 1 week. Neurological examination showed slightly reduced strength of the leg muscles (4/5) and a sensory level at T6. Elaborate investigation, including EMG, visual evoked potential (VEP), SSEP, MEP and CSF assessment did not reveal any abnormalities. sMRI revealed slight impression of the left nerve root S1 due to disc protrusion and showed a left paracentral disc protrusion at level T5/T6 and T7/T8 and a central disc protrusion at level C5/C6, but these findings were not compatible with the clinical symptoms. Normal tendon reflexes (including plantar reflexes) were present. sMRI was repeated after a week and remained normal. As there were some discrepancies at the neurological examination, the differential diagnosis was TM or an FND. During hospital admission, the patient developed mononuclear diplopia and bilateral papilledema. SLEDAI at that time was 22 based on visual disturbance, hematuria, rash, complement consumption and anti-dsDNA positivity. Treatment with MP and cyclophosphamide was initiated for NPSLE as it was considered that TM could not be excluded completely. After 4 months, sMRI remained normal and muscle strength had recovered, but hypesthesia at sensory level T5 persisted. Uncertainty regarding the presence of TM remained and therefore cyclophosphamide was stopped after 4 months. This was the clinical status during the last available follow-up visit, several months ago.
Part II: Systematic Review
The systematic literature review yielded a total of 1391 citations. After removal of duplicates, title and abstract evaluation identified 229 papers as suitable for full-text review. After full-text review, 146 articles were included (see Supplementary Figure 1), of which 131 case reports and 15 observational studies.
Case Reports
The 131 case reports described a total of 168 patients with SLE and TM, as shown in Supplementary Table 1. Of these cases, 151 were female (90%) and median age was 32 years (range: 16–92). Of these patients, 119 patients met ACR criteria for SLE (71%), 23 patients only had the clinical diagnosis of SLE (14%) and for 25 patients details on the diagnosis of SLE were not provided (15%). Median SLE duration was available for 149 patients and was 3 years (range: 0–35).
sMRI-abnormalities were described in 154/168 patients (92%). Information on CSF was available for 107 patients, of which 91 had abnormalities (85%). Outcome information was available for 151 patients (90%). Death was reported in 9 patients (6%), no improvement in 31 patients (21%), partial improvement in 58 patients (38%), partial or complete improvement in 1 (1%) and (near) complete recovery in 52 patients (34%). Time at which the outcome was reported varied: during hospital stay or at hospital discharge (n = 18), short term (n = 29), long term (n = 86) and unknown (n = 18).
Of the 14 patients with normal sMRI, CSF was available for 11 patients.11–22 9/11 showed abnormalities in CSF: pleocytosis (4/10), increased protein (7/11), low glucose (3/9), oligoclonal bands (2/6) and an increased IgG index (3/3). Only two patients were MRI-/CSF-.17,22 One MRI-/CSF- patient was treated with MP 1000mg IV for 3 days followed by prednisone 1mg/kg for 1 month. This patient showed recovery from hypotonia grade 1–2/5 right and 4/5 left to 4/5 bilaterally at hospital discharge (partial improvement).17 No information on MRI at follow-up is available in this report. In the other MRI-/CSF- patient, sMRI showed abnormalities at follow-up: spinal atrophy was visible after 1 year. The patient received treatment with hydrocortisone 100mg daily for 5 days followed by prednisolone 30mg/day for 1 month and showed complete and persistent recovery 5 days after treatment initiation.22
Observational Studies
The characteristics of the observational studies, including patient data, are summarized in Table 4. In these studies, a total of 282 patients with TM were assessed, with ages ranging from 9 to 77 years. For 268 patients, information on sex was available and the majority of patients were female (93%). Data on SLE according to the ACR definition were available for 267 patients, of which 263 fulfilled these criteria. sMRI data were present for 259 patients (92%). Of these patients, 238 showed abnormalities compatible with TM (92%). Data on CSF were available for 111 patients, of which 81 showed abnormalities (73%). Outcome data were available for 247 patients (88%). Death was reported in 4 patients (2%), no improvement in 61 patients (25%), no or partial improvement in 20 patients (8%), partial improvement in 41 patients (17%), partial or complete improvement in 76 patients (31%) and (nearly) complete improvement in 45 patients (18%).
 |
Table 4 Overview of Observational Studies Included from Systematic Review Regarding Characteristics of SLE Patients with Transverse Myelitis |
Of the 23 patients with normal sMRI, CSF analysis was present for 17 patients, of which 14 patients showed abnormalities (82%). The following CSF abnormalities were present: pleocytosis in 4/13, increased protein in 10/17 and low glucose in 5/12. Information of two of the three MRI-/CSF- patients was not available in the article and later provided by the authors.23,24 Of the three patients, one patient was treated with MP IV for 3 days and cyclophosphamide IV 1x followed by treatment with azathioprine and showed partial improvement at 15 months.23 Another patient was treated with MP IV followed by cyclophosphamide and recovered completely after 5 days.24 The last patient was treated with MP and cyclophosphamide IV and showed no clinical improvement after 10 years of follow-up.1
Summary Outcome
Combined clinical outcomes in sMRI+ or CSF+ patients from the case reports/case series, observational studies and our own cohort (n = 401) showed 13 deaths (3%), 91 patients with no improvement (23%), 21 with no or partial recovery (5%), 101 with partial improvement (26%), 76 with partial to complete recovery (19%) and 99 with (nearly) complete recovery (25%). Clinical outcome in all sMRI-/CSF- patients (n = 8) was no recovery in 3 (37.5%), partial recovery in 4 (50%) and complete recovery in 1 patient (12.5%).
Discussion
In this study, we aimed to collect diagnostic and outcome data to guide the clinical approach to patients with SLE presenting with clinical features compatible with (sub) acute TM, but with normal sMRI and CSF assessment by studying cases from our tertiary referral center and cases reported in the literature. Our data suggest that clinical signs of TM in the absence of sMRI and CSF abnormalities in SLE patients have a less favorable outcome, despite similar immunosuppressive treatment as patients with abnormalities. This leads to the question whether immunosuppressive therapy is appropriate for sMRI-/CSF- patients. However, as so few cases of sMRI-/CSF- TM in SLE patients have been described in literature, a clear clinical approach cannot be identified based on the literature review.
The most important consideration in the clinical approach to patients with features of TM, but normal sMRI and CSF, is whether the assumption that TM is caused by an immune SLE-related origin is correct. This uncertainty is emphasized by the difference in outcome between sMRI-/CSF- and MRI+ patients in our cohort. Inflammation without any sign on sMRI or CSF seems unlikely, although a recent study also describes eleven patients with a clinical presentation mimicking MS, without any abnormalities on conventional brain MRI or sMRI.25 The immunological pathophysiology is assumed in these patients because of the increased frequency of plasmablasts in peripheral blood and good response to treatment with IV methylprednisolone and plasmapheresis. In addition, these patients were shown to have abnormalities in structural integrity of the white matter not visible on conventional MRI, but characterized by decreased fractional anisotropy (FA) values on diffusion tensor (DTI) MRI analysis of the brain. Perhaps TM- patients have abnormalities that cannot be detected with conventional MRI modalities, in accordance with a substantial percentage of CNS-NPSLE patients with normal conventional MRI, that have abnormalities detectable with quantitative MRI techniques.26–28 However, the limited response to immunosuppressive therapy in sMRI-/CSF- TM makes this less likely.
A remaining differential diagnosis is a FND: when retrospectively reviewing these cases in detail, several uncertainties about the clinical presentation and neurological evaluation were noted. Firstly, only 1/5 sMRI- patients showed a Babinski sign, as opposed to 5/7 in the sMRI+ group. Secondly, discrepancies were noticed such as variation in muscle strength during different examinations or extreme complaints such as complete lack of gnostic sensibility from toes up to the jawline. Therefore, in retrospect, an important differential diagnosis remains a FND in these patients. FNDs are defined as the presence of one or more symptoms that affect voluntary motor or sensory function or cause transient loss of consciousness, but are not found to be due to a general medical condition. These symptoms are among the most common causes of neurological disability and require specific diagnostic features. Unfortunately, specific tests for FND such as described by Stone et al.29 were not documented in our cases. Little is known about the prevalence of FND in patients with SLE and the mechanism of FND is also unknown. Only one case-report on this topic is available, describing the presence of an FND at the time of diagnosis of SLE in a young patient.30 Although this was suspected immediately, immunosuppressive treatment was initiated due to the progressive nature of her neurological symptoms, without leading to any clinical improvement. Months later, psychotherapy was started, which led to a complete clinical recovery. This case report underlines the necessity of an adequate diagnosis: not only can immunosuppressive therapy cause unnecessary harm through side-effects but not treating for FND may also negatively affect recovery.
There are, however, also uncertainties regarding the possible diagnosis of FND in the sMRI-/CSF- patients in our cohort. Firstly, most patients presented with specific neurological symptoms that contributed to the decision to start immunosuppressive treatment for TM. Three out of four sMRI-/CSF- patients presented with urinary retention requiring catheterization for at least 6 months (case 1, 2 and 3). In theory, urinary retention can also be part of a FND, although the prevalence of FND as cause of urinary retention is unknown. Unfortunately, urodynamic studies cannot help clinicians distinguish FND from TM as both can present with similar abnormalities.31 In general, urinary retention is considered a supportive symptom for myelopathy.32,33 Secondly, the absence of abdominal reflexes were described in one patient (case 1). Although this may be indicative of TM, abdominal reflexes are absent in 20% of normal individuals, in particular in those with lax abdominal tone, obesity, women who have borne children and elderly. Unilateral absence of abdominal reflex, on the other hand, is considered abnormal.34,35 Lastly, apart from neurological features, there were other reasons to start treatment for TM in these patients, such as SLE disease activity. However, it is important to note that TM in sMRI+/CSF+ cases does not necessarily present with a high disease activity: approximately one-third of sMRI+/CSF+ cases present at low disease activity (SLEDAI ≤4).36
Delaying or omitting immunosuppressive treatment in uncertain cases might have the drawback of leading to untreated (irreversible) damage. Several small studies have suggested a better outcome of TM in sMRI+ cases when treatment was initiated early,37–40 but the amount of cases studied is limited and primarily involves patients with MRI abnormalities. The largest study uses a post-hoc analysis to suggest a better outcome when treatment is initiated <2 weeks, but only 3 of the 14 patients studied were actually treated in that time frame (of which two had a good outcome).40 In a different study, poor outcomes are described despite treatment initiated early.41 Therefore, clear evidence is lacking and may indicate that it is advisable to take full considerations of pros and cons before starting treatment in suspected sMRI-/CSF- TM cases.
Considering the lack of convincing support for early treatment, especially in sMRI- cases, we suggest the following steps in patients with the diagnosis or suspicion of SLE presenting with neurologic signs consistent with TM (Figure 1). Firstly, an extensive patient history and physical examination should be conducted, followed by an sMRI with gadolinium of good quality (as described in the Method section) and a LP to look for signs of inflammation. If either sMRI or CSF shows signs of inflammation and no alternative diagnosis is more likely (such as infection), treatment for TM should be initiated. If both are normal, other diagnoses should be considered. At this point, FND should also be considered. Furthermore, there is no evidence that immunosuppressive treatment needs to be initiated immediately.42,43 After 2–7 days, neurological examination, sMRI and CSF could be repeated if initial investigations were in the first week of symptom presentation. If all remain normal, additional tests can be considered such as EMG, MEP, SSEP, VEP. If strong suspicion of TM remains, immunosuppressive treatment might be started, but needs to be re-evaluated after 3 months. If sMRI and CSF analysis remain normal and treatment has not led to any objective improvement, consider to stop immunosuppressive treatment, as our cases and cases of literature demonstrated that continuing treatment in sMRI-/CSF- patients generally does not result in complete recovery. Even before that point, attention towards intensive rehabilitation including physical therapy, patient education, psychosocial support and if indicated psychotherapy seems of great importance. Thus, to evaluate all patients multidisciplinary and start rehabilitation early in the course of the disease, regardless of the underlying cause of the symptoms, is advisable.
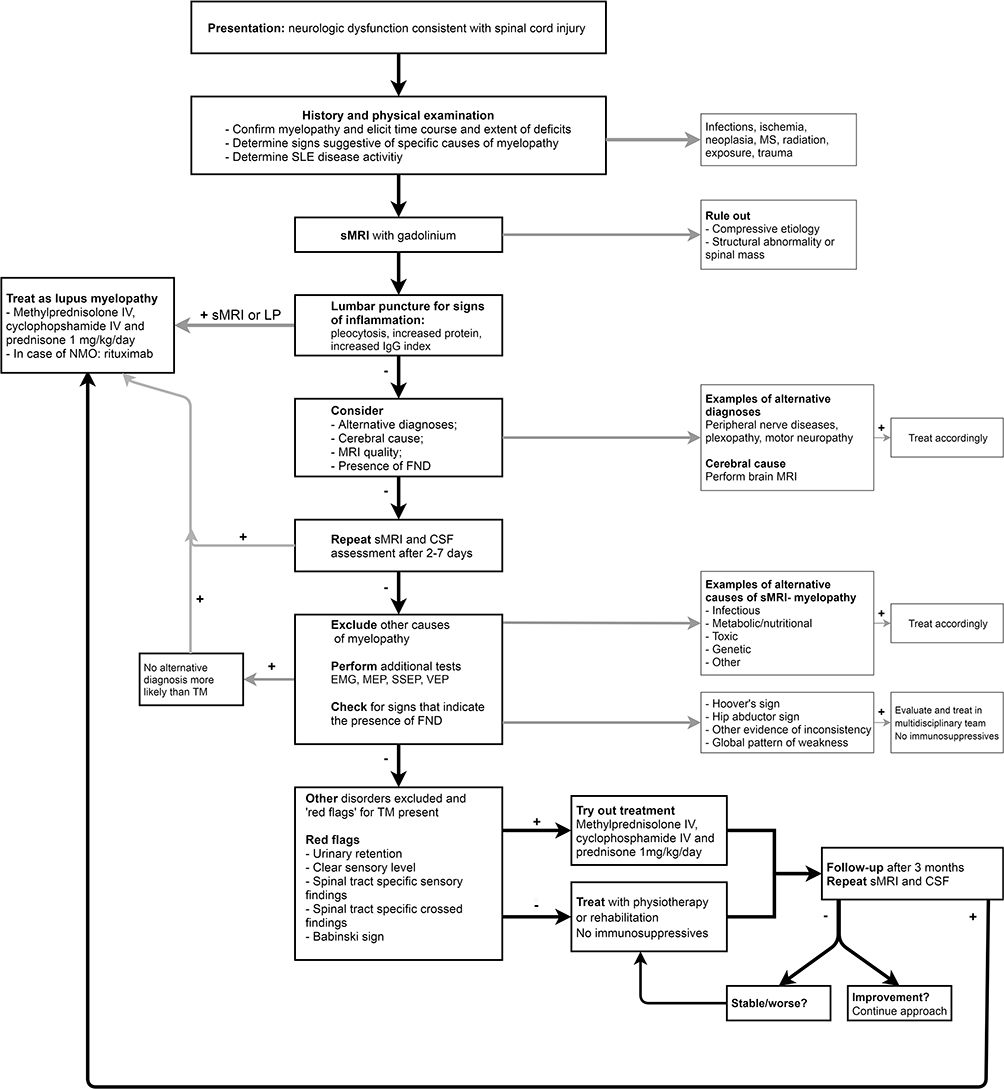 |
Figure 1 Flow chart for patients diagnosed with or suspicion of SLE that present with neurologic dysfunction consistent with spinal cord injury. Abbreviations: ALS, amyotrophic lateral sclerosis; EMG, electromyography; FND, functional neurological disorder; MEP, magnetic evoked potential; MS, multiple sclerosis; NMO, neuromyelitis optica; SEP, somatosensory evoked potential; sMRI, MRI of the spinal cord; TM, transverse myelitis; VEP, visual evoked potential. Notes: +, present or abnormal; -, absent or normal. |
Future research should focus on bringing this approach in practice and reporting more about sMRI-/CSF- patients, in order increase knowledge and improve clinical approach to these patients. In addition, the value of additional sMRI investigation (such as DTI) should be studied in these patients.
This is the first study that focuses on patients with SLE and a clinical presentation of TM, but without any abnormalities on additional investigation. Our literature study has confirmed that there is a knowledge gap regarding this topic, which might have led to incorrect treatment of these patients. This specific clinical presentation leads to uncertainty amongst physicians and we attempt to provide an overview and approach on how to handle this situation in clinical practice based on our clinical experience. As all our patients were assessed in a multidisciplinary setting including a psychiatrist, their clinical presentation and disease course could be studied in detail, providing more insight into this rare manifestation of SLE. There are, however, some limitations to our study. Due to the retrospective nature of our study not all patients underwent all additional investigations as suggested in our flowchart; therefore, absolute certainty about the diagnosis remains impossible. For example, specific tests for FND, repeated sMRI and elaborate psychiatric assessment focused on the presence of FND have not been performed in all patients. However, this is exactly the challenge we are addressing in this study and has enabled us to create this flowchart after years of experience. The most important limitation remains the difficult comparison between sMRI-/CSF- and other patients due to the probable publication bias. We hope that in the future more information regarding these patients will be shared.
The clinical presentation of (sub) acute TM with negative sMRI and CSF findings in SLE patients is barely reported, but is a challenge in clinical practice. The most important question is whether the clinical signs of TM in this patient group are caused by an immune SLE-related origin. Our data indicate that immunosuppressive therapy in this patient group is of limited value. It is possible that these patients suffer from other disorders, including a FND, and elaborate investigations should be done in order to avoid misdiagnosis and -management. Therefore, we suggest giving immunosuppressive treatment for a maximum of 3 months in case of remaining uncertainty, to stop if no clinical improvement occurs and start rehabilitation early in the course of the disease. Multidisciplinary evaluation is extremely important in these patients and patients should be followed-up closely.
Acknowledgments
We thank J.W. Schoones for assisting with the literature search. Furthermore, we thank all members of the NPSLE multidisciplinary team, authors that provided additional information for this project and especially thank all the patients for their participation.
Disclosure
Gisela Terwindt reports grants or consultancy support from Novartis, Lilly, Teva, Allergan, and independent support from Netherlands Organization for Health Research and Development (NWO, and ZonMW), NIH, European Community, Dutch Heart Foundation, and Dutch Brain Foundation (no funding was received for this work). Margreet Kloppenburg reports fee for consultancy and local investigator of industry-driven trial; all paid to institution from AbbVie, fees for consultancy paid to institution from Pfizer, Levicept, GlaxoSmithKline, Merck-Serono, Kiniksa, Flexion, Galapagos, and CHDR, personal fees and royalties (UptoDate) from Wolters Kluwer, and personal fees and royalties (Rheumatology and Clinical Immunology) from Springer-Verlag, outside the submitted work. The authors report no other potential conflicts of interest in this work.
References
1. Kovacs B, Lafferty TL, Brent LH, DeHoratius RJ. Transverse myelopathy in systemic lupus erythematosus: an analysis of 14 cases and review of the literature. Ann Rheum Dis. 2000;59(2):120–124. doi:10.1136/ard.59.2.120
2. Liang MH, Corzillius M, Bae SC, et al. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999;42(4):599–608.
3. Group TMCW. Proposed diagnostic criteria and nosology of acute transverse myelitis. Neurology. 2002;59(4):499–505. doi:10.1212/WNL.59.4.499
4. Krishnan C, Greenberg B. Transverse myelitis. In: UpToDate. Waltham, MA: UptoDate;
5. de Seze J, Lanctin C, Lebrun C, et al. Idiopathic acute transverse myelitis: application of the recent diagnostic criteria. Neurology. 2005;65(12):1950–1953. doi:10.1212/01.wnl.0000188896.48308.26
6. Zirkzee EJ, Steup-Beekman GM, van der Mast RC, et al. Prospective study of clinical phenotypes in neuropsychiatric systemic lupus erythematosus; multidisciplinary approach to diagnosis and therapy. J Rheumatol. 2012;39(11):2118–2126. doi:10.3899/jrheum.120545
7. Gladman D, Ibañez D, Urowitz M. Systemic lupus erythematosus disease activity index 2000. J Rheumatol. 2002;29:288–291.
8. Stewart LA, Clarke M, Rovers M, et al. PRISMA-IPD development group. preferred reporting items for systematic review and meta-analyses of individual participant data: the PRISMA-IPD statement. JAMA. 2015;313(16):1657–1665. doi:10.1001/jama.2015.3656
9. Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25(11):1271–1277. doi:10.1002/art.1780251101
10. Hochberg M. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus (letter). Arthritis Rheum. 1997;40(9):1725. doi:10.1002/art.1780400928
11. Provenzale J, Bouldin TW. Lupus-related myelopathy: report of three cases and review of the literature. J Neurol Neurosurg Psychiatry. 1992;55(9):830–835.
12. Marabani M, Zoma A, Hadley D, Sturrock RD. Transverse myelitis occurring during pregnancy in a patient with systemic lupus erythematosus. Ann Rheum Dis. 1989;48(2):160–162. doi:10.1136/ard.48.2.160
13. Valesini G, Priori R, Francia A, et al. Central nervous system involvement in systemic lupus erythematosus: a new therapeutic approach with intrathecal dexamethasone and methotrexate. Springer Semin Immunopathol. 1994;16(2–3):313–321. doi:10.1007/BF00197524
14. Smyth AE, Bruce IN, McMillan SA, Bell AL. Transverse myelitis: a complication of systemic lupus erythematosus that is associated with the antiphospholipid syndrome. Ulster Med J. 1996;65(1):91–94.
15. Osawa H, Yamabe H, Kaizuka M, et al. Systemic lupus erythematosus associated with transverse myelitis and parkinsonian symptoms. Lupus. 1997;6(7):613–615. doi:10.1177/096120339700600710
16. Bonnet F, Mercie P, Morlat P, et al. Devic’s neuromyelitis optica during pregnancy in a patient with systemic lupus erythematosus. Lupus. 1999;8(3):244–247. doi:10.1191/096120399678847696
17. Salmi HH, Shalaby MA, Ageely HM, Moustafa F. A case of neuropsychiatric lupus with myelopathy successfully treated with corticosteroids. West Afr J Med. 2004;23(2):173–175.
18. Mok CC, Mak A, To CH. Mycophenolate mofetil for lupus related myelopathy. Ann Rheum Dis. 2006;65(7):971–973. doi:10.1136/ard.2005.046615
19. Rodrigues CEM, de Carvalho JF. Clinical, radiologic, and therapeutic analysis of 14 patients with transverse myelitis associated with antiphospholipid syndrome: report of 4 cases and review of the literature. Semin Arthritis Rheum. 2011;40(4):349–357. doi:10.1016/j.semarthrit.2010.05.004
20. Hamming L, van der Meulen R, Vergouwen A, Siegert C. Myelopathy in systemic lupus erythematosus: a case report and a review of the literature. Neth J Med. 2015;73(6):290–292.
21. Chen HC, Lai JH, Juan CJ, Kuo SY, Chen CH, Chang DM. Longitudinal myelitis as an initial manifestation of systemic lupus erythematosus. Am J Med Sci. 2004;327(2):105–108. doi:10.1097/00000441-200402000-00011
22. Salmaggi A, Lamperti E, Eoli M, et al. Spinal cord involvement and systemic lupus erythematosus: clinical and magnetic resonance findings in 5 patients. Clin Exp Rheumatol. 1994;12(4):389–394.
23. Chiganer EH, Lessa CF, Di Pace JL, et al. Transverse myelitis in systemic lupus erythematosus: clinical features and prognostic factors in a large cohort of Latin American patients. J Clin Rheumatol. 2020. doi:10.1097/RHU.0000000000001322
24. Zhang S, Wang Z, Zhao J, et al. Clinical features of transverse myelitis associated with systemic lupus erythematosus. Lupus. 2020;29(4):389–397. doi:10.1177/0961203320905668
25. Takewaki D, Lin Y, Sato W, et al. Normal brain imaging accompanies neuroimmunologically justified, autoimmune encephalomyelitis. Neurol Neuroimmunol Neuroinflamm. 2018;5(3):e456. doi:10.1212/NXI.0000000000000456
26. Bosma GP, Middelkoop HA, Rood MJ, Bollen EL, Huizinga TW, van Buchem MA. Association of global brain damage and clinical functioning in neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. 2002;46(10):2665–2672. doi:10.1002/art.10574
27. Emmer BJ, Steens SC, Steup-Beekman GM, et al. Detection of change in CNS involvement in neuropsychiatric SLE: a magnetization transfer study. J Magn Reson Imaging. 2006;24(4):812–816. doi:10.1002/jmri.20706
28. Steens SC, Admiraal-Behloul F, Bosma GP, et al. Selective gray matter damage in neuropsychiatric lupus. Arthritis Rheum. 2004;50(9):2877–2881. doi:10.1002/art.20654
29. Stone J. The bare essentials: functional symptoms in neurology. Pract Neurol. 2009;9(3):179–189.
30. Nakamura M, Tanaka S, Inoue T, et al. Systemic lupus erythematosus and Sjögren’s syndrome complicated by conversion disorder: a case report. Kurume Med J. 2018;64:97–101. doi:10.2739/kurumemedj.MS644005
31. Sakakibara R, Uchiyama T, Awa Y, et al. Psychogenic urinary dysfunction: a uro-neurological assessment. Neurourol Urodyn. 2007;26(4):518–524. doi:10.1002/nau.20321
32. Schmalstieg WF, Weinshenker B. Approach to acute or subacute myelopathy. Neurology. 2010;75:S2–S8. doi:10.1212/WNL.0b013e3181fb3638
33. Smith MD, Seth JH, Fowler CJ, Miller RF, Panicker JN. Urinary retention for the neurologist. Pract Neurol. 2013;13(5):288–291. doi:10.1136/practneurol-2012-000478
34. Campbell WW. DeJong’s the Neurologic Examination.
35. Alpert JN. Reflex aberrations. In: The Neurologic Diagnosis: A Practical Bedside Approach. Springer; 2012:90.
36. Li XY, Xiao P, Xiao HB, et al. Myelitis in systemic lupus erythematosus frequently manifests as longitudinal and sometimes occurs at low disease activity. Lupus. 2014;23(11):1178–1186. doi:10.1177/0961203314541690
37. Harisdangkul V, Doorenbos D, Subramony SH. Lupus transverse myelopathy: better outcome with early recognition and aggressive high-dose intravenous corticosteroid pulse treatment. J Neurol. 1995;242(5):326–331. doi:10.1007/BF00878876
38. Propper DJ, Bucknall RC. Acute transverse myelopathy complicating systemic lupus erythematosus. Ann Rheum Dis. 1989;48(6):512–515. doi:10.1136/ard.48.6.512
39. Sherer Y, Hassin S, Shoenfeld Y, et al. Transverse myelitis in patients with antiphospholipid antibodies–the importance of early diagnosis and treatment. Clin Rheumatol. 2002;21(3):207–210. doi:10.1007/s10067-002-8287-2
40. Lu X, Gu Y, Wang Y, Chen S, Ye S. Prognostic factors of lupus myelopathy. Lupus. 2008;17(4):323–328. doi:10.1177/0961203307088005
41. Costallat BL, Ferreira DM, Costallat LT, Appenzeller S. Myelopathy in systemic lupus erythematosus: clinical, laboratory, radiological and progression findings in a cohort of 1,193 patients. Rev Bras Reumatol Engl Ed. 2016;56(3):240–251. doi:10.1016/j.rbr.2015.12.006
42. Wong SH, Boggild M, Enevoldson TP, Fletcher NA. Myelopathy but normal MRI: where next? Pract Neurol. 2008;8(2):90–102. doi:10.1136/jnnp.2008.144121
43. Jacob A, Weinshenker BG. An approach to the diagnosis of acute transverse myelitis. Semin Neurol. 2008;28(1):105–120. doi:10.1055/s-2007-1019132
44. Chan KF, Boey ML. Transverse myelopathy in SLE: clinical features and functional outcomes. Lupus. 1996;5(4):294–299. doi:10.1177/096120339600500409
45. Mok CC, Lau CS, Chan EY, Wong RW. Acute transverse myelopathy in systemic lupus erythematosus: clinical presentation, treatment, and outcome. J Rheumatol. 1998;25(3):467–473.
46. D’Cruz DP, Mellor-Pita S, Joven B, et al. Transverse myelitis as the first manifestation of systemic lupus erythematosus or lupus-like disease: good functional outcome and relevance of antiphospholipid antibodies. J Rheumatol. 2004;31(2):280–285.
47. Birnbaum J, Petri M, Thompson R, Izbudak I, Kerr D. Distinct subtypes of myelitis in systemic lupus erythematosus. Arthritis Rheum. 2009;60(11):3378–3387. doi:10.1002/art.24937
48. Schulz SW, Shenin M, Mehta A, Kebede A, Fluerant M, Derk CT. Initial presentation of acute transverse myelitis in systemic lupus erythematosus: demographics, diagnosis, management and comparison to idiopathic cases. Rheumatol Int. 2012;32(9):2623–2627. doi:10.1007/s00296-011-2053-1
49. Saison J, Costedoat-Chalumeau N, Maucort-Boulch D, et al. Systemic lupus erythematosus-associated acute transverse myelitis: manifestations, treatments, outcomes, and prognostic factors in 20 patients. Lupus. 2015;24(1):74–81. doi:10.1177/0961203314547795
50. Hryb JP, Chiganer E, Contentti EC, Di Pace JL, Lessa C, Perassolo MB. Myelitis in systemic lupus erythematosus: clinical features, immunological profile and magnetic resonance imaging of five cases. Spinal Cord Ser Cases. 2016;2:16005. doi:10.1038/scsandc.2016.5
51. Quintanilla-Gonzalez L, Atisha-Fregoso Y, Llorente L, Fragoso-Loyo H. Myelitis in systemic lupus erythematosus: clinical characteristics and effect in accrual damage. A single-center experience. Lupus. 2017;26(3):248–254. doi:10.1177/0961203316659152
52. Ahn SM, Hong S, Lim DH, et al. Clinical features and prognoses of acute transverse myelitis in patients with systemic lupus erythematosus. Korean J Intern Med. 2019;34(2):442–451. doi:10.3904/kjim.2016.383
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.