Back to Journals » Infection and Drug Resistance » Volume 16
Structural Modelling of Platelet Activating Factor Acetyl Hydrolase in Leishmania donovani, Trypanosoma cruzi, and Trypanosoma brucei: Implications on Therapeutics for Leishmaniasis, Chagas Disease, and Sleeping Sickness
Authors Goswami A, Koley T, Rajan MV ![]() , Madhuri P, Upadhyay N
, Madhuri P, Upadhyay N ![]() , Das U, Kumar M, Ethayathulla AS, Hariprasad G
, Das U, Kumar M, Ethayathulla AS, Hariprasad G ![]()
Received 27 January 2023
Accepted for publication 28 March 2023
Published 11 April 2023 Volume 2023:16 Pages 2117—2128
DOI https://doi.org/10.2147/IDR.S403411
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Arunima Goswami, Tirthankar Koley, Madhan Vishal Rajan, Pathak Madhuri, Neelam Upadhyay, Uddipan Das, Manoj Kumar, Abdul Samath Ethayathulla, Gururao Hariprasad
Department of Biophysics, All India Institute of Medical Sciences, New Delhi, 110029, India
Correspondence: Gururao Hariprasad, Tel +91-11-26594240, Fax +91-11-26588663, Email [email protected]
Purpose: Leishmaniasis, Chagas disease, and sleeping sickness are caused by protozoa Leishmania donovani, Trypanosoma cruzi, and Trypanosoma brucei, respectively. Platelet activating factor acetyl hydrolase (PAF-AH) is an inflammatory protein implicated in pathogenesis of these three infections, thereby making them attractive drug targets.
Methods: PAF-AH sequences were retrieved from UniProt and aligned using Clustal Omega. Homologous models of parasitic proteins were built based on crystal structure of human PAF-AH and validated using PROCHECK server. Volumes of substrate-binding channel were calculated using the ProteinsPlus program. High throughput virtual screening using Glide program in Schrodinger was done with ZINC drug library against parasitic PAF-AH enzymes. Complexes with best hits were energy-minimized and subjected to 100 ns molecular dynamic simulation and analyzed.
Results: PAF-AH enzyme sequences from protozoa Leishmania donovani, Trypanosoma cruzi, Trypanosoma brucei, and human have a minimum of 34% sequence similarity with each other. Corresponding structures show a globular conformation consisting of twisted β-pleated sheets, flanked by α-helices on either side. Catalytic triad of serine-histidine-aspartate is conserved. Substrate-binding channel residues are conserved to an extent, with a lower channel volume in human as compared to target enzymes. Drug screening resulted in identification of three molecules that had better affinities than the substrate to the target enzymes. These molecules fulfill Lipinski’s rules for drug likeness and also bind with less affinity to the human counterpart, thereby establishing a high selective index.
Conclusion: Structures of PAF-AH from protozoan parasites and humans belong to the same family of enzymes and have a similar three-dimensional fold. However, they show subtle variations in residue composition, secondary structure composition, substrate-binding channel volume, and conformational stability. These differences result in certain specific molecules being potent inhibitors of the target enzymes while simultaneously having weaker binding to human homologue.
Keywords: platelet activating factor acetyl hydrolase, protozoa, drug target, potent inhibitor, leishmaniasis, Leishmania donovani, Chagas disease, Trypanosoma cruzi, sleeping sickness, Trypanosoma brucei, drug efficacy
Graphical Abstract:

Introduction
Protozoans are microscopic, unicellular, parasitic pathogens that cause disease in humans. Three such protozoan diseases are leishmaniasis, Chagas disease, and sleeping sickness that are caused by Leishmania donovani, Trypanosoma cruzi, and Trypanosoma brucei, respectively. These parasitic diseases have high morbidity and mortality rates globally, even after much progress in their prevention and treatment.1 These three diseases have been classified under Neglected Tropical Diseases by WHO with a mission to control, eliminate, and eradicate these diseases by 2030. A fatal form of leishmaniasis known as visceral leishmaniasis has an incidence of 50,000 to 90,000 annually, whereas cutaneous leishmaniasis was estimated to have an incidence of 600,000 to 1 million annually.2 Regarding Chagas disease, also known as American trypanosomiasis, it is estimated that 6 to 7 million people worldwide are infected with T. cruzi.3 The reported number of cases with sleeping sickness in 2020 was 663.4 The current mortality rates of leishmaniasis, Chagas disease, and sleeping sickness were 50,000, 2000, and 3500, respectively.5–7
Leishmania donovani affects the liver, spleen, and bone marrow, and clinical manifestations include hepato-splenomegaly, pyrexia, and weight loss.8,9 Trypanosoma cruzi infection, also called Chagas disease, mainly affects the heart and results in myocardial inflammation, dilated heart chambers, and dense fibroinflammatory plaques on the epicardium.10,11 Some of its symptoms include fever, headaches, swollen eyelids, and swelling at the site of bug bite.12 Trypanosoma brucei mainly affects the central nervous system resulting in increased levels of white cells and antibody IgM in CSF.13,14 Trypanosoma brucei results in a disease called sleeping sickness, whose symptoms are joint pain, itching, fatigue, and excessive sleepiness.15
Drugs used in the treatment of leishmaniasis include pentavalent antimonial, amphotericin B, miltefosine, paromomycin, and pentamidine. Though quite effective, these are known to have serious adverse effects like cardiotoxicity, nephrotoxicity, hepatotoxicity, teratogenicity, and dysglycemia.16 Two main drugs used in treatment of Chagas disease are benznidazole and nifurtimox and are known to cause skin rash, hepatitis, peripheral neuropathy, sleepiness, depression, and other psychiatric symptoms.17 While pentamidine and suramin are routinely used in the treatment of first-stage African trypanosomiasis, melarsoprol, eflornithine, and nifurtimox are used to treat second-stage disease. Suramin is known to cause nephrotoxicity, peripheral neuropathy, and agranulocytosis; eflornithine is associated with abdominal pain, hypertension, headache, myelosuppression, and seizure; and melarsoprol has been known to have life-threatening toxicity.18
The above data and observations make it important to look for newer targets and potent molecules for better therapeutic efficacy. Platelet-activating factor acetylhydrolase (PAF-AH) is a member of the phospholipase A2 (PLA2) family that hydrolyzes the acetyl group of PAF.19 This enzyme is crucial for the pathogenesis of infection by these protozoan parasites and executes multiple functions for the organism’s survival and virulence. Some of these are: (1) Leishmania PAF-AH binds and compromises the function of mammalian PAF that acts as an inflammatory signal for the leukocytes, thereby helping in the survival of viable parasites inside the host;20 (2) Trypanosoma cruzi PAF-AH lyses mammalian PAF that induces a pro-inflammatory signal to the macrophages for production of nitric oxide which has trypanocidal activity;20,21 (3) Trypanosoma brucei PAF-AH, by removing the sn-2 group, deacetylates PAF and phospholipids for the release of inflammatory arachidonic acid which is lethal to host cells.20,22 Our group has previously studied the PAF-AH enzyme in humans and its role in the mediation of physio-pathological functions.23,24 Human PAF-AH homolog participates in different biological processes like host defense, lipid digestion, and a vast number of housekeeping functions like homeostasis of cellular membranes and production of potent lipid mediators.25 It makes it imperative to develop potent molecules that can effectively inhibit these protozoal PAF-AH, while minimally affecting the human PAF-AH. In this study, we look to carry out the structural characterization of PAF-AH from parasitic protozoa for a rational approach towards drug designing.
Materials and Methods
Sequence Analysis
Protein sequences of PAF-AH from Leishmania donovani, Trypanosoma cruzi, Trypanosoma brucei, and human were obtained from UniProt protein sequence database.26 These sequences were aligned using the Clustal Omega web server at EMBL-EBI.27 This alignment was used as an input file to generate a guide tree with default parameters.
Model Building and Validation of Parasitic and Wild PAF-AH Enzymes
PAF-AH sequences of Leishmania donovani, Trypanosoma cruzi, and Trypanosoma brucei were submitted to SWISS-MODEL and GENO3D servers to generate homology models.28,29 3D X-ray crystal structure of human PAF-AH (PDB Id: 3D59) in RCSB was taken as the template. Models were validated using Ramachandran plot, VERIFY3D, and ERRAT Scores from the PROCHECK server.30–33 Human PAF-AH crystal structure (PDB Id: 3D59) was considered for comparative structural analysis.
Calculation of Substrate-Binding Channel Volume
The volume of the drug-binding pocket was calculated using ProteinsPlus protein-ligand interaction analysis server using DoGSiteScorer tool.34,35 The program calculates the difference in the Gaussian filter to predict the possible binding pockets based on the 3D structural features such as size, shape, and chemistry. To calculate the volume of the ligand binding site in Leishmania donovani, Trypanosoma cruzi, Trypanosoma brucei, and human PAF-AH coordinates of these structures, models were uploaded. Approximately 10 to 15 pocket sites were predicted for each model, and the total volume corresponding to the binding site was calculated.
Molecular Dynamics Simulation of Parasitic PAF-AH Enzymes
Molecular dynamics (MD) simulation was done by Desmond suite of programs using force field parameters of OPLS_2005.36 The best validated models were energy-minimized and then solvated using TIP3P water model in an orthorhombic box. The net electrostatic charges on the solvated model was neutralized by adding 6 Cl− ions for L. donovani, 4 Cl− ions for T. cruzi, and 6 Cl− ions for T. brucei. Solvated models were again energy-minimized with a threshold gradient of 1 kcal/mol/Å. MD simulation of each solvated model was performed for 100 ns under the NPT conditions with periodic boundary condition with the help of Nose-Hoover Thermostat and Martyna-Tobias-Klein barostat. The simulation was performed with step size of 2 fs in the presence of LINCS harmonic constraints, and the motion was integrated by RESPA integrator. A similar protocol was followed for crystal structure of hPAF-AH.37
High Throughput Virtual Screening (HTVS) and Molecular Dynamics Simulation of Protein-Ligand Complexes
Virtual screening of ZINC database against PAF-AH of Leishmania donovani, Trypanosoma cruzi, and Trypanosoma brucei was done using the Glide program.38 A grid was prepared for the three parasitic proteins by taking the catalytic residues as the centroid in the receptor grid generation program. Lipinski’s rule-compliant compounds from the ZINC biogenic library39 were subjected to molecular docking-based screening using virtual screening workflow in Schrodinger software package. In workflow, screening was initially performed in virtual high throughput docking mode, and top 10% of obtained hits were further subjected to standard precision (SP) docking mode. Finally top 10% of hits from SP docking was subjected to extra precision (XP) docking mode.40 Top-most hit from XP docking was selected for each of the PAF-AH enzymes from Leishmania donovani, Trypanosoma cruzi, and Trypanosoma brucei. The best docked poses were energy-minimized and then solvated using TIP3P water model in an orthorhombic box. Solvated complex was further energy-minimized with a threshold gradient of 1 kcal/mol/Å. These three complexes of the best hit molecule with their respective enzymes were then subjected to 100 ns molecular dynamics. Similarly, a grid was prepared in the human PAF-AH enzyme, and the same three hits were docked using Glide program with similar protocol.38 And these structures were also refined by similar MD simulation protocol. Binding affinity (KD) was estimated using the equation: ΔG=-nRT.lnKD, where ΔG stands for change in free energy of binding, R stands for Gas constant in calorie/mol/Kelvin, T stands for temperature in Kelvin, ln stands for natural log, and n stands for number of moles (calculated for 1 mol).
A pictorial depiction of the methodology followed in this study is provided in Figure 1.
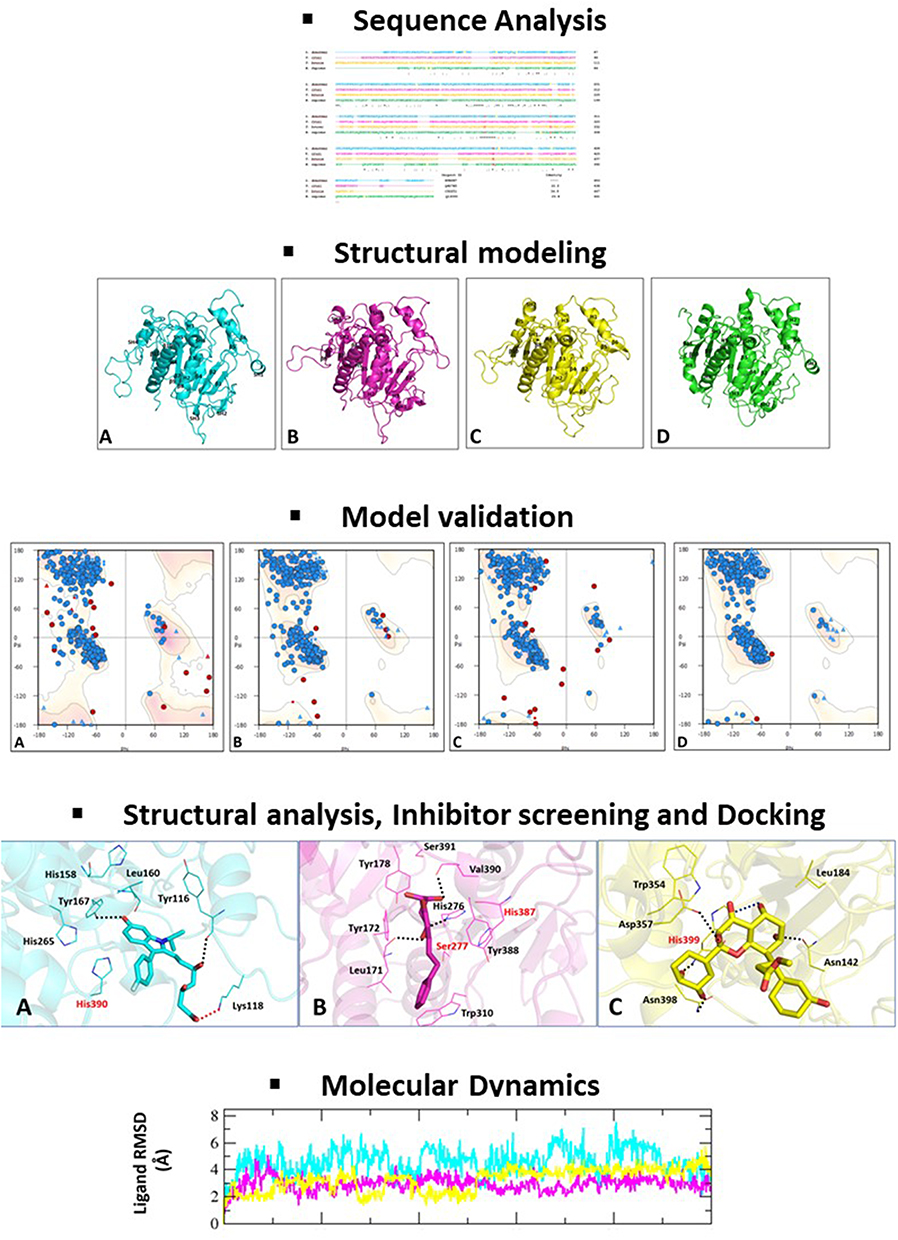 |
Figure 1 Diagrammatic overview of the methodology used in this study. |
Results and Discussion
Sequence Analysis
PAF-AH sequences from Trypanosoma cruzi, Trypanosoma brucei, and human have fair similarity with Leishmania donovani PAF-AH sequence. The least among these is the human analog as compared to the two other protozoan counterparts. Catalytic residues serine, aspartate, and histidine are conserved across all four sequences. PAF-AH sequences of Leishmania donovani, Trypanosoma cruzi, Trypanosoma brucei, and human have 11, 4, 10, and 7 cysteines, respectively. These cysteines are not at conserved positions in the enzymes, thereby suggesting that they are not involved in disulfide bond formation and in turn the stability of the proteins (Figure 2A). We have previously carried out detailed analysis of human PAF-AH enzyme structure.23,24 Residues such as leucine, histidine, phenylalanine, and tryptophan that are involved in substrate-binding are conserved. An aromatic residue, tryptophan, a known residue in the hydrophobic channel, is conserved in the protozoal sequences and is replaced by Phe322 in human. This could possibly be due to substrate-specific functions mediated by the parasitic and housekeeping human enzyme. Human PAF-AH has 32.4–38.1% similarity with the protozoal PAF-AH enzymes, making it a potential secondary drug target. Amongst the variations, some of the notable changes are the presence of charged residues in the human PAF-AH sequence. Some of them are: Arg82, Arg157, Glu178, Arg223, Arg264, and Arg288. Subsequent sections will look into the contribution of these residues in conferring stability to the overall fold of this enzyme. Guide tree provides a rough estimate of the relation between the four sequences from a evolutionary perspective (Figure 2B).
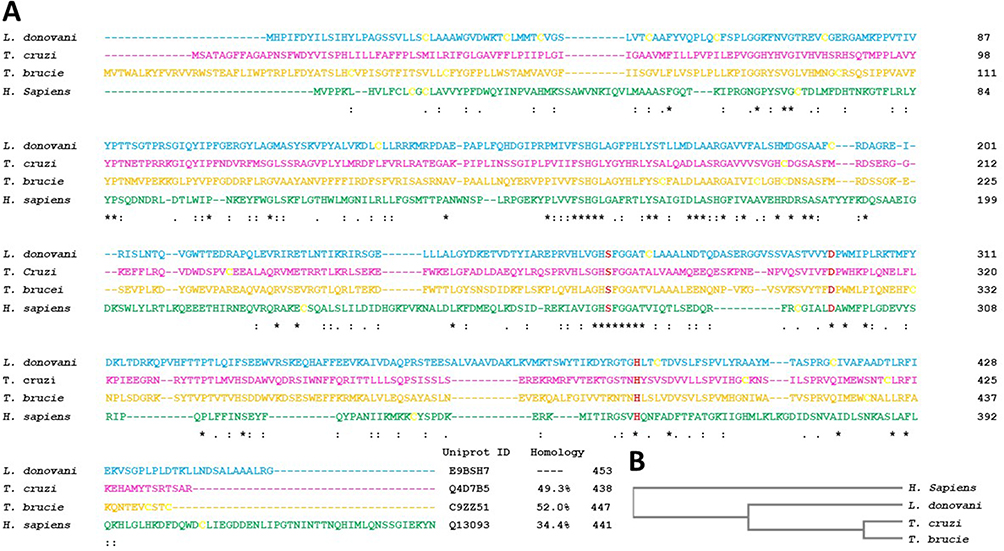 |
Figure 2 (A) Multiple sequence alignment of protozoan and human PAF-AH. The sequences in the order are Leishmania donovani PAF-AH (cyan), Trypanosoma cruzi PAF-AH (magenta), Trypanosoma brucei PAF-AH (yellow), and Homo sapiens PAF-AH (green). Conserved catalytic residues, serine, histidine, and aspartic acid are shown in bold red, and the cysteines are shown in bold yellow. The UniProt accession numbers and percentage identity are shown at the end of the sequences. The symbol “*” indicates identical residues, “:” indicates conserved substitutions, and “.” indicates semi-conserved substitutions. (B) Guide phylogeny tree. |
Model Building and Validation
A total of 24 homology models were generated for three parasitic PAF-AH proteins. Models of PAF-AH enzyme from Leishmania donovani, Trypanosoma cruzi, and Trypanosoma brucei with the best validation statistics were chosen for structural analysis (Supplementary Table 1). The chosen three models had a minimum of 98% of the residues in favored and allowed regions of the Ramachandran plot (Figure 3).
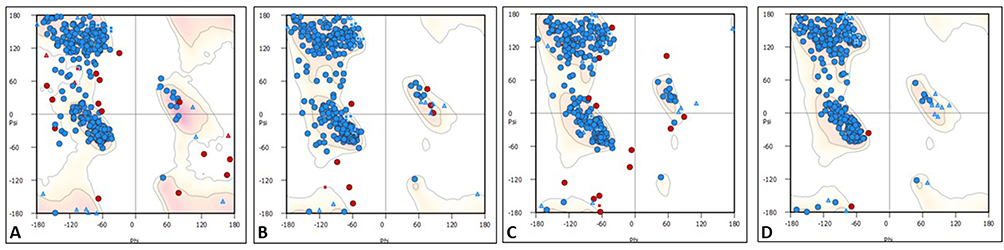 |
Figure 3 Ramachandran plots for PAF-AH proteins. (A) Leishmania donovani PAF-AH, (B) Trypanosoma cruzi PAF-AH, (C) Trypanosoma brucei PAF-AH, and (D) Human PAF-AH. |
Biophysical Parameters
Analysis of molecular dynamics trajectory of the four structures indicates that the systems reached equilibrium quickly during MD simulation (Supplementary Figure 1a–c). RMSD plot shows that once protein structure converged, fluctuation observed is within the thermal vibration range (Supplementary Figure 1d). It also shows that structure of Trypanosoma cruzi PAF-AH converged around 50 ns, while the other three structures converged within 10 ns of simulation. This plot also shows that conformation of Leishmania donovani and Trypanosoma cruzi changed more than Trypanosoma brucei and human PAF-AH compared to their respective conformation before simulation. However, these variations are only about 2 Å which is within the limit of thermal vibration at 300 K. The radius of gyration plot indicates that human PAF-AH is more compact compared to the three parasitic PAF-AH (Supplementary Figure 1e). Plots of total energy of the native protein models are energetically stable throughout the MD simulations (Supplementary Figure 1f). The total energy, potential energy, and volume of four proteins in the molecular dynamic phase are shown (Table 1).
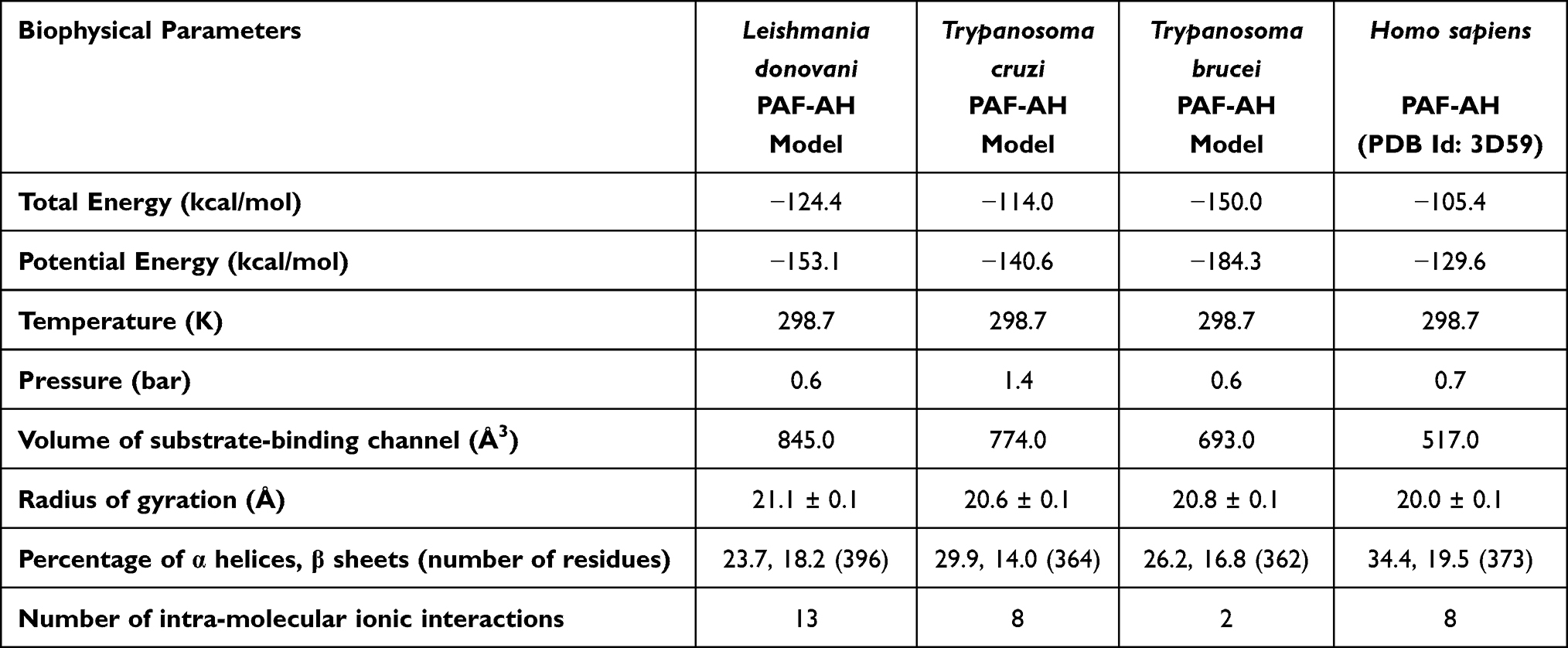 |
Table 1 Biophysical Parameters of Platelet Activating Factor-Acetyl Hydrolase Structures |
Structural Analysis
Modeled structures of Leishmania donovani, Trypanosoma cruzi, Trypanosoma brucei, and the crystal structure of human PAF-AH all have globular fold consisting of α-helices, short helices, and β-sheets (Figure 4). The core of the enzyme consists of twisted β-pleated sheet flanked by helices, connected by loops on either side. Cysteine residues are mostly seen on the surface of the molecules and are not involved in disulfide bond formation. It is mostly the polar character of these cysteine residues that contribute to the stability. Each of the enzymes has a substrate-binding channel, wherein the helices and loops form the walls and the catalytic site is nestled deep in the pocket. Charged residues on the enzymes are seen to be making several intra-molecular salt bridges. These interactions contribute to the stability of the structures.
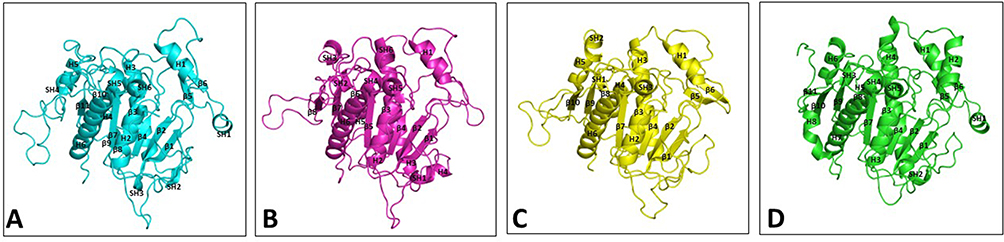 |
Figure 4 Ribbon diagram showing overall structure of PAF-AH enzymes. (A) Leishmania donovani PAF-AH model (cyan), (B) Trypanosoma cruzi PAF-AH model (magenta), (C) Trypanosoma brucei PAF-AH model (yellow), and (D) Homo sapiens PAF-AH crystal structure (PDB Id 3D59) (green). Secondary structures in all the four proteins are labeled and numbered from the N-terminus to the C-terminus. Abbreviations: H, helices; SH, short helices; β, beta-strands. |
Catalytic Site and Substrate-Binding Channel
The catalytic triad consisting of serine, histidine, and aspartate is within interacting distance in all the four PAF-AH proteins (Figure 5A–D). These residues, distantly located on the primary structure, come in close proximity in the three-dimensional structure. This is an important requisite for the functionality of the enzyme and thereby validates the accuracy of model structures. Here, serine acts as a catalytic nucleophile created by the resonating electron formation on the nitrogen atom in histidine; furthermore, that histidine nitrogen resonating electron is induced by the effect of the deprotonated carboxylic group resonance of aspartic acid.41 After a 100 ns molecular dynamics simulation of Leishmania donovani PAF-AH, Trypanosoma cruzi PAF-AH, Trypanosoma brucei PAF-AH, and human PAF-AH, the serine residue of the catalytic triad forms a hydrogen bond with histidine, and the histidine makes a hydrogen bond with the aspartic acid residue in all four structures. This shows that the overall structure of the enzyme across the four species is similar, confirming that the proteins have a functional fold. The substrate-binding channel is hydrophobic in nature and occupies the lipid head at the catalytic site. It has 19 amino acid residues along the walls. Four of these residues, leucine, phenylalanine, histidine, and tryptophan are conserved across all four enzymes. The 322nd residue in the human protein is a phenylalanine, which is replaced by tryptophan in Leishmania donovani, Tryptophan cruzi, and Tryptophan brucei at 335, 342, and 354 positions, respectively (Figure 6). A narrow volume was observed in the substrate-binding channel of human as compared to protozoal PAF-AH. The possible reasons for the reduced volume which appears to hinder the entry and occupancy of the molecules into the channel are: (1) presence of a bulky phenylalanine at position 357 as compared to valine at the corresponding position; (2) conformation of the loop formed by Ala184-Ser185-Ala186 and stabilized by polar interaction with a neighboring loop (Ser185 Oγ…O Gly154); (3) short helical conformation of residues from Gln211 to Glu213, as compared to loop conformations in target PAF-AH structures (Figure 7).
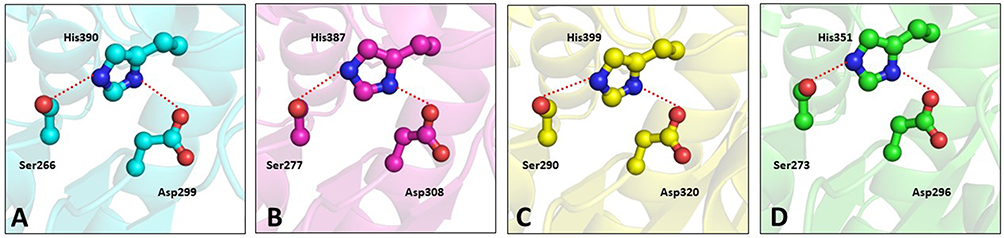 |
Figure 5 Ball and stick diagram showing catalytic site residues in the PAF-AH enzymes. (A) Leishmania donovani PAF-AH (cyan), (B) Trypanosoma cruzi PAF-AH (magenta), (C) Trypanosoma brucei PAF-AH (yellow), and (D) Homo sapiens PAF-AH (green). |
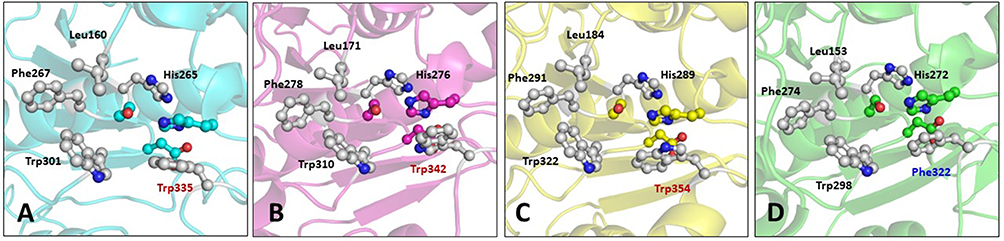 |
Figure 6 Ball and stick diagram showing residues in the substrate-binding channel in the PAF-AH enzymes. (A) Leishmania donovani PAF-AH (cyan), (B) Trypanosoma cruzi PAF-AH (magenta), (C) Trypanosoma brucei PAF-AH (yellow), and (D) Homo sapiens PAF-AH (green). |
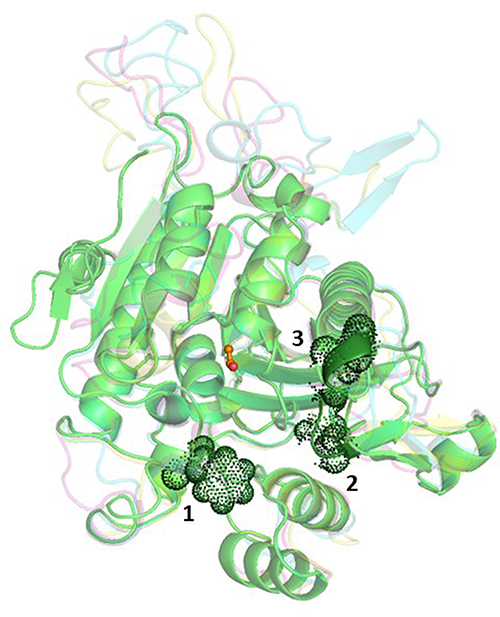 |
Figure 7 Ribbon diagram showing super-imposed structures of PAF-AH enzymes. Fold conformations of Leishmania donovani PAF-AH, Trypanosoma cruzi PAF-AH, Trypanosoma brucei PAF-AH, and human PAF-AH are shown in cyan, pink, yellow, and green, respectively. Catalytic serine in the channel is marked in orange. Factors responsible for a smaller channel volume in human PAF-AH are shown as dot rendering. |
Screening for Potential Inhibitory Molecules
The three protozoal PAF-AH enzyme drug targets were screened based on virtual screening, and the top 10% hits resulted in 143 molecules which fulfilled the Lipinski 5 criteria. The best hit among these was: ZINC95617672 [7-[3-(4-fluorophenyl)-6-hydroxy-1-propan-2-ylindol-2-yl]-3,5-dihydroxyhept-6-enoic acid] for Leishmania donovani, ZINC33830752 [5,6,7,8-tetrahydroxy-2-phenethyl-5,6,7,8-tetrahydrochromen-4-one] for Trypanosoma cruzi, and ZINC96115307 [methyl (3R)-3-[2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxy-4-oxochromen-8-yl]-3hydroxyphenyl)] for Trypanosoma brucei, and these molecules will henceforth be referred to as D1, C1, and B1, respectively, in the paper. Lipinski 5 parameters for these three molecules are presented in Table 2. The fact that three different molecules are the best inhibitors for the three target enzymes from the same family indicates that subtle differences in the residue composition of the channels and their volumes play a role in inhibitor selection and binding. Analysis of MD trajectory of the docked complexes shows the simulation system is stable (Supplementary Figure 2a–c). Protein RMSD plots of the docked complexes indicate that the backbone of proteins converged at around 30 ns (Supplementary Figure 1d). Similarly, RMSD of the ligands in the docked complexes were also stable during simulations after convergence (Supplementary Figure 1e). Also, the PAF-AH complexes of Trypanosoma cruzi and Trypanosoma brucei are found to be more compact than the Leishmania donovani complex (Supplementary Figure 1f).
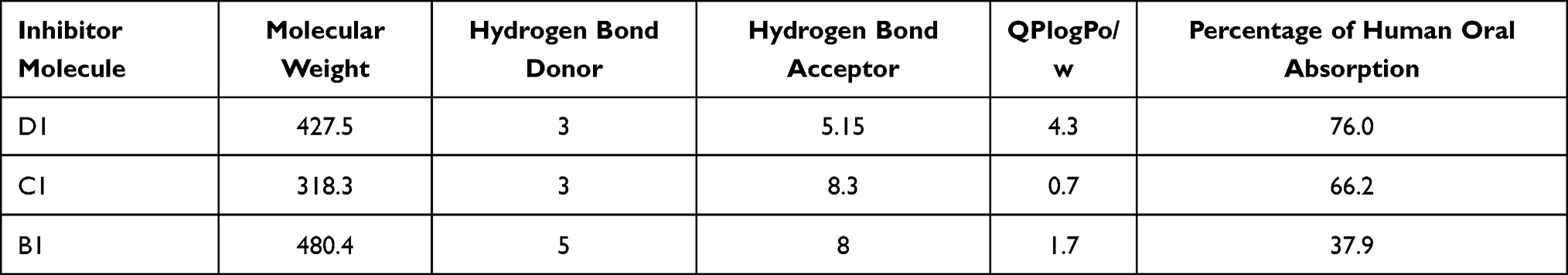 |
Table 2 Lipinski Criteria for the Screened Molecules Against the PAF-AH Target |
Structural Analysis of Protozoal PAF-AH Enzymes with Inhibitory Molecules
In order to develop inhibitory molecules against protozoal PAF-AH enzymes, it was imperative to understand the binding parameters of the substrate to these enzymes. Platelet activating factor, a substrate, was also docked to the three parasitic enzymes. While the polar head group was in the vicinity of the catalytic site, the hydrophobic tail occupied the substrate-binding channel, making interactions with residues that form the walls of the channel. The binding affinities of the substrate to these enzymes were in a range of 0.03 μM–0.3 μM (Table 3). These biophysical data were used to define the binding pocket, occupancy of the channel, and ideal potencies of screened inhibitor molecules.
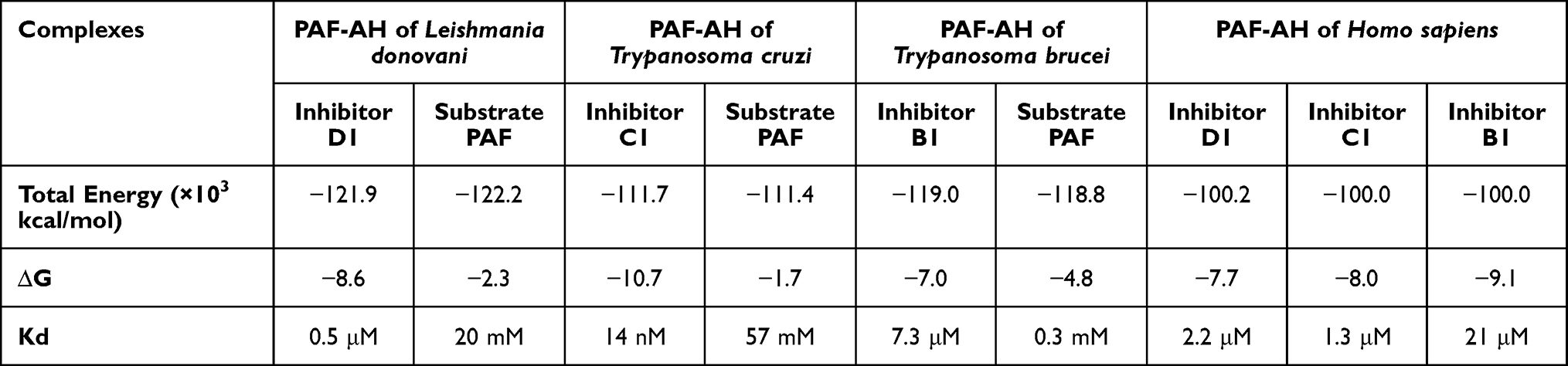 |
Table 3 Binding Parameters of Inhibitors and Substrate to PAF-AH Enzymes |
In the Leishmania donovani PAF-AH-D1 complex, the fluorophenyl ring makes a van der Waal contact with Cδ of catalytic His390. The molecule also makes van der Waals interactions with Leu160 and His265 which are present in the substrate-binding channel. Charge on the carboxyl oxygen atom of enoic acid forms a salt bridge with the nitrogen atom of Lys118 (D1 OH…N Lys118 = 2.7 Å). The hydroxyl group at the third position of the molecule makes a hydrogen bond with the main chain oxygen of Tyr116 (D1 O1…O Tyr116). Also, the hydroxyl group on the 1-propan-2-ylindol-2-yl group makes a hydrogen-bonded interaction with the hydroxyl group of Try167. The planar hydrogen bond is highly stable due to the resonation of d-pi electrons present there in the benzene group. Though D1 is not deep in the hydrophobic channel, the delineated interactions account for the potent binding (Figure 8A).
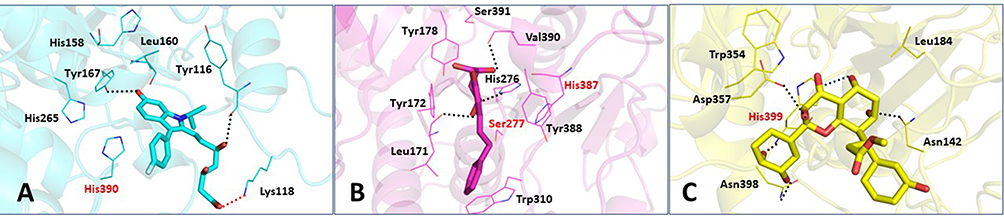 |
Figure 8 Binding interactions of parasitic PAF-AH proteins with their respective inhibitor molecules. Binding interactions of: (A) Leishmania donovani PAF-AH with D1; (B) Trypanosoma cruzi PAF-AH with C1; (C) Trypanosoma brucei PAF-AH with B1. Catalytic residues are shown in red. Hydrogen bonds are shown as black dotted lines, and ionic bonds are shown as red dotted lines. |
In the Trypanosoma cruzi PAF-AH-C1 complex, the phenyl ring of the phenethyl group makes a van der Waals interaction with the hydroxyl group of the catalytic residue Ser277. In addition, this phenyl ring of the phenethyl group makes a hydrophobic interaction with Trp310, a substrate-binding channel residue. Hydroxyl groups of C1 also make hydrogen bonds with His276 and Leu171 that line the channel. One of the hydroxyl groups on the phenyl ring of the phenethyl group makes a hydrogen-bonded interaction with valine (C1 OH…N Val390). The binding of C1 to this enzyme is further stabilized by an array of hydrophobic interactions with aromatic residues Tyr388, Try172, and Tyr178. These interactions establish that C1 could be a potential inhibitor of the Trypanosoma cruzi PAF-AH enzyme with a good binding affinity (Figure 8B).
In the Trypanosoma brucei PAF-AH and B1 complex, it is seen that molecule B1 forms two hydrogen bonds: one with the nitrogen atom of the side chain of the catalytic residue His399 and another with the nitrogen atom of its side chain. A van der Waals interaction is observed between the chromen-4-one group and Leu184. Another van der Waals bond is formed between the phenyl ring at the 2nd position of the chromen-4-one ring and Trp354. Both Leu184 and Trp354 are residues of the hydrophobic channel. Other hydrogen bonds which stabilize this complex are between (1) the hydroxyl group of the chromen-4-one group and Asn142; (2) the 3rd position hydroxyl group of the phenyl ring at the 2nd position of the chromen-4-one group and Asn398; and (3) the 3rd position hydroxyl group of the chromen-4-one group and Asp357 (Figure 8C).
Therapeutic Potential of the Identified Inhibitory Molecules
The therapeutic value of a drug is governed by maximum efficacy and minimum side effects against the disease. This would require optimum potency of the molecules against the primary drug target and minimal binding to the host protein. The proposed inhibitor molecules of protozoan PAF-AH enzyme targets show the following features: (1) have better binding affinity to the enzymes than substrate molecules; (2) engage the catalytic histidine residue; (3) make interactions with residues in the substrate-binding channel; and (4) are orally bioavailable as they fulfill all the Lipinski rules.
Since human PAF-AH has 42% or more sequence homology and the main chain RMSD of up to 3 Å compared to parasitic PAF-AH, the screened inhibitory molecules were docked to the human PAF-AH to estimate the extent of binding. It is seen that the molecules bind with a lower affinity to the human PAF-AH enzyme as compared to the parasitic enzymes (Table 3). Analysis of the complexes implies that the decreased volume of the human PAF-AH substrate-binding channel is responsible for the lower affinity. Human PAF-AH will therefore continue to perform its intended housekeeping functions; which include reducing oxidative stress and generating bioactive fatty acids in mast cells which play an important role in inflammatory reactions and immune response.21 This result, therefore, augurs well for the development of these molecules as potential drug candidates against leishmaniasis, Chagas disease, and sleeping sickness.
Additional Information
Coordinates for models of platelet activating factor acetyl hydrolase of Leishmania donovani, Trypanosoma cruzi, and Trypanosoma brucei and their complexes with inhibitors D1, C1, and B1 are available at: https://www.aiims.edu/index.php?option=com_content&view=article&id=721&lang=en.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Custodio H. Protozoan Parasites. Pediatr Rev. 2016;37(2):59–69;quiz 70–71. doi:10.1542/pir.2015-0006
2. World Health Organization. Leishmaniasis; 2023. Available from: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis.
3. World Health Organization. Chagas disease (also known as American trypanosomiasis); 2022. Available from: https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis).
4. World Health Organization. Trypanosomiasis, human African (sleeping sickness); 2022. Available from: https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness).
5. Bi K, Chen Y, Zhao S, Kuang Y, John Wu CH. Current visceral leishmaniasis research: a research review to inspire future study. Biomed Res Int. 2018;2018:1–13. doi:10.1155/2018/9872095
6. Echeverria LE, Morillo CA. American trypanosomiasis (Chagas disease). Infect Dis Clin North Am. 2019;33(1):119–134. doi:10.1016/j.idc.2018.10.015
7. Ozioko K, Okoye C, Obiezue R, et al. Accelerating towards human African trypanosomiasis elimination: issues and opportunities. J Vector Borne Dis. 2020;57(2):105. doi:10.4103/0972-9062.310860
8. van Griensven J, Diro E. Visceral leishmaniasis. Infect Dis Clin North Am. 2012;26(2):309–322. doi:10.1016/j.idc.2012.03.005
9. Papadopoulou C, Kostoula A, Dimitriou D, Panagiou A, Bobojianni C, Antoniades G. Human and canine leishmaniasis in asymptomatic and symptomatic population in Northwestern Greece. J Infect. 2005;50(1):53–60. doi:10.1016/j.jinf.2004.05.004
10. Marin-Neto JA, Cunha-Neto E, Maciel BC, Simões M. Pathogenesis of chronic Chagas heart disease. Circulation. 2007;115(9):1109–1123. doi:10.1161/CIRCULATIONAHA.106.624296
11. Bonney KM, Luthringer DJ, Kim SA, Garg NJ, Engman DM. Pathology and pathogenesis of Chagas heart disease. Annu Rev Pathol. 2019;14:421–447. doi:10.1146/annurev-pathol-020117-043711
12. Rassi A, Rassi A, Marin-Neto JA. Chagas disease. Lancet. 2010;375(9723):1388–1402. doi:10.1016/S0140-6736(10)60061-X
13. Carvalho T, Trindade S, Pimenta S, Santos AB, Rijo-Ferreira F, Figueiredo LM. Trypanosoma brucei triggers a marked immune response in male reproductive organs. PLoS Negl Trop Dis. 2018;12(8):e0006690. doi:10.1371/journal.pntd.0006690
14. Greenwood BM, Whittle HC. The pathogenesis of sleeping sickness. Trans R Soc Trop Med Hyg. 1980;74(6):716–725. doi:10.1016/0035-9203(80
15. Kennedy PGE, Rodgers J. Clinical and neuropathogenetic aspects of human African trypanosomiasis. Front Immunol. 2019;10:39. doi:10.3389/fimmu.2019.00039
16. Pradhan S, Schwartz RA, Patil A, Grabbe S, Goldust M. Treatment options for leishmaniasis. Clin Exp Dermatol. 2022;47(3):516–521. doi:10.1111/ced.14919
17. Lascano F, García Bournissen F, Altcheh J. Review of pharmacological options for the treatment of Chagas disease. Br J Clin Pharmacol. 2022;88(2):383–402. doi:10.1111/bcp.14700
18. Büscher P, Cecchi G, Jamonneau V, Priotto G. Human African trypanosomiasis. Lancet. 2017;390(10110):2397–2409. doi:10.1016/S0140-6736(17
19. Pawlowic MC, Zhang K. Leishmania parasites possess a platelet-activating factor acetylhydrolase important for virulence. Mol Biochem Parasitol. 2012;186(1):11–20. doi:10.1016/j.molbiopara.2012.08.005
20. Shapiro J, Lui H. Vaniqa--eflornithine 13.9% cream. Skin Therapy Lett. 2001;6(7):1–3, 5.
21. Kono N, Arai H. Platelet-activating factor acetylhydrolases: an overview and update. Biochimica Et Biophysica Acta. 2019;1864(6):922–931.
22. Ridgley EL, Ruben L. Phospholipase from Trypanosoma brucei releases arachidonic acid by sequential sn-1, sn-2 deacylation of phospholipids. Mol Biochem Parasitol. 2001;114(1):29–40. doi:10.1016/s0166-6851(01
23. Hariprasad G, Kaur P, Srinivasan A, Singh TP, Kumar M. Structural analysis of secretory phospholipase A2 from Clonorchis sinensis: therapeutic implications for hepatic fibrosis. J Mol Model. 2012;18(7):3139–3145. doi:10.1007/s00894-011-1333-8
24. Khan MI, Hariprasad G. Structural modeling of wild and mutant forms of human plasma platelet activating factor-acetyl hydrolase enzyme. J Inflamm Res. 2020;13:1125–1139. doi:10.2147/JIR.S274940
25. Machado FS, Mukherjee S, Weiss LM, Tanowitz HB, Ashton AW. Bioactive lipids in Trypanosoma cruzi infection. Adv Parasitol. 2011;76:1–31. doi:10.1016/B978-0-12-385895-5.00001-3
26. UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506–D515. doi:10.1093/nar/gky1049
27. Sievers F, Higgins DG. Clustal Omega, Accurate Alignment of Very Large Numbers of Sequences. Springer; 2014:105–116. doi:10.1007/978-1-62703-646-7_6
28. Waterhouse A, Bertoni M, Bienert S, et al. Swiss-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi:10.1093/nar/gky427
29. Combet C, Jambon M, Deleage G, Geourjon C. Geno3D: automatic comparative molecular modelling of protein. Bioinformatics. 2002;18(1):213–214. doi:10.1093/bioinformatics/18.1.213
30. Bowie JU, Lüthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253(5016):164–170. doi:10.1126/science.1853201
31. Lüthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356(6364):83–85. doi:10.1038/356083a0
32. Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2(9):1511–1519. doi:10.1002/pro.5560020916
33. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26(2):283–291. doi:10.1107/S0021889892009944
34. Schöning-Stierand K, Diedrich K, Fährrolfes R, et al. ProteinsPlus: interactive analysis of protein–ligand binding interfaces. Nucleic Acids Res. 2020;48(W1):W48–W53. doi:10.1093/nar/gkaa235
35. Volkamer A, Griewel A, Grombacher T, Rarey M. Analyzing the topology of active sites: on the prediction of pockets and subpockets. J Chem Inf Model. 2010;50(11):2041–2052. doi:10.1021/ci100241y
36. Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27(3):221–234. doi:10.1007/s10822-013-9644-8
37. Bowers KJ, Sacerdoti FD, Salmon JK, et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In:
38. Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. doi:10.1021/jm0306430
39. Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: a free tool to discover chemistry for biology. J Chem Inf Model. 2012;52(7):1757–1768. doi:10.1021/ci3001277
40. Shaikh F, Siu SWI. Identification of novel natural compound inhibitors for human complement component 5a receptor by homology modeling and virtual screening. Med Chem Res. 2016;25(8):1564–1573. doi:10.1007/s00044-016-1591-1
41. Carter P, Wells JA. Dissecting the catalytic triad of a serine protease. Nature. 1988;332(6164):564–568. doi:10.1038/332564a0
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
State-of-the-Art in the Drug Discovery Pathway for Chagas Disease: A Framework for Drug Development and Target Validation
Gabaldón-Figueira JC, Martinez-Peinado N, Escabia E, Ros-Lucas A, Chatelain E, Scandale I, Gascon J, Pinazo MJ, Alonso-Padilla J
Research and Reports in Tropical Medicine 2023, 14:1-19
Published Date: 14 June 2023