Back to Journals » Journal of Inflammation Research » Volume 15
Structural Basis of the Pancreatitis-Associated Autoproteolytic Failsafe Mechanism in Human Anionic Trypsin
Authors Nagel F ![]() , Susemihl A
, Susemihl A ![]() , Geist N
, Geist N ![]() , Möhlis K, Palm GJ
, Möhlis K, Palm GJ ![]() , Lammers M, Delcea M
, Lammers M, Delcea M ![]()
Received 25 March 2022
Accepted for publication 24 May 2022
Published 24 June 2022 Volume 2022:15 Pages 3633—3642
DOI https://doi.org/10.2147/JIR.S367699
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Felix Nagel,1 Anne Susemihl,1,2 Norman Geist,1 Kevin Möhlis,1,3 Gottfried J Palm,4 Michael Lammers,4 Mihaela Delcea1
1Biophysical Chemistry, Institute of Biochemistry, University of Greifswald, Greifswald, Germany; 2Department of Hematology and Oncology, Internal Medicine C, University of Greifswald, Greifswald, Germany; 3Helmholtz Institute for Metabolic, Obesity and Vascular Research, Leipzig, Germany; 4Synthetic and Structural Biochemistry, Institute of Biochemistry, University of Greifswald, Greifswald, Germany
Correspondence: Mihaela Delcea, Biophysical Chemistry, Institute of Biochemistry, University of Greifswald, Greifswald, Germany, Tel +49 3834 420 4423, Fax +49 3834 420 4377, Email [email protected]
Objective: The pathophysiological mechanisms underlying chronic pancreatitis (CP) are still poorly understood. Human cationic (TRY1) and anionic (TRY2) trypsins are the two major trypsin isoforms and their activities are tightly regulated within pancreatic acinar cells. Typically, they exist in a molar ratio of 2:1 (cationic:anionic). This ratio is reversed during chronic alcohol abuse, pancreatic cancer, or pancreatitis due to selectively upregulated expression of TRY2, causing anionic trypsin to become the predominant isoform. The involvement of TRY2 in pancreatitis is considered limited due to the absence of disease-causing mutations and its increased prevalence for autoproteolysis. However, exacerbated pancreatitis in TRY2 overexpressing mice was recently demonstrated. Here, we aim to elucidate the molecular structure of human anionic trypsin and obtain insights into the autoproteolytic regulation of tryptic activity.
Methods: Trypsin isoforms were recombinantly expressed in E. coli, purified and refolded. Enzymatic activities of all trypsin isoforms were determined and crystals of TRY2 were grown using the vapor-diffusion method. The structure was solved by molecular replacement and refined to a resolution of 1.7 Å. Equilibration molecular dynamics simulations were used to generate the corresponding TRY1–TRY1 model.
Results: All trypsin isoforms display similar kinetic properties. The crystal structure of TRY2 reveals that the enzyme crystallized in the autoproteolytic state with Arg122 placed in the S1 binding pocket and the corresponding loop cleaved. The TRY2–TRY2 dimer confirms a previously hypothesized autoinhibitory state with an unexpectedly large binding interface.
Conclusion: We provide a structure of TRY2, which is the predominant trypsin isoform in chronic pancreatitis and pancreatic cancer. A proposed autoinhibition mode was confirmed and the structural basis of the autoproteolytic failsafe mechanism elucidated.
Keywords: pancreas, pancreatitis, serine protease, crystal structure, autoproteolysis, R122H
Introduction
Chronic pancreatitis (CP) is a progressive inflammatory disorder of the pancreas that leads to fibrosis and exocrine and endocrine insufficiencies.1–3 The underlying mechanisms for the development of the disease are diverse and are primarily caused by alcohol abuse or genetic risk factors but can also include smoking, autoimmunity and pancreatic injury.4 In order to prevent pancreatitis, the tryptic activity within zymogen granules of pancreatic acinar cells has to be tightly regulated. Premature autoactivation is prevented by expression as zymogens with the propeptide inhibiting trypsin. Enterokinase, which activates trypsinogen by cleavage of the propeptide, is spatially restricted to the duodenum. Simultaneously, prematurely activated trypsin is inhibited by the serine protease inhibitor Kazal-type 1 (SPINK1) or can be degraded and cleared by autoproteolysis.5,6 For the human cationic trypsinogen gene (PRSS1) a plethora of disease-causing mutations are known to play a role in hereditary pancreatitis.7 One of the most prominent mutations is R122H, which removes one highly exposed autocleavage site and thereby disables one of the failsafe mechanisms against pancreatitis.8–11 Simultaneously, the Arg122 containing loop was proposed to possess weak inhibitory activity, which is meaningful at high trypsinogen concentrations found inside zymogen granules of pancreatic acinar cells.12 Conversely, the gene for human anionic trypsin (PRSS2) is better known for its protective mutations.13 The G191R mutation, in particular, has been shown to be protective against pancreatitis by introducing an additional autocleavage site that makes the enzyme more sensitive to degradation. However, due to tighter control of TRY2 by chymotrypsin C (CTRC), the role of this isoform in hereditary chronic pancreatitis is limited.14,15 In healthy individuals the molar ratio of cationic trypsin (TRY1) and anionic trypsin (TRY2) is 2:1. During chronic alcohol abuse, pancreatic cancer and CP, this ratio inverts and TRY2 becomes the predominant isoform.16–19 Until recently, it was unclear whether increased TRY2 secretion predisposes to pancreatitis or alternatively has a protective role against it.16 However, transgenic expression of TRY2 in mice showed exacerbation of pancreatitis, clearly demonstrating the detrimental role of anionic trypsin.20 Because TRY2 is selectively up-regulated in many patients with pancreatitis, the authors urged that inhibitors designed for pancreatitis therapy be tested against all isoforms rather than focusing on TRY1.19,20 To understand the mechanisms by which TRY2 participates in pancreatitis, as well as to design novel inhibitors, a molecular model is indispensable. Probably due to the notoriously unstable nature of the PRSS2 isoform,16 no crystal structure is available to date. Here, we report successful and easy expression, purification and refolding of all three major trypsin isoforms using a conventional His6 tag. A crystal structure of active human anionic trypsin in a state representing the first step of autoproteolysis was solved. The TRY2 dimer represents an autoinhibited state of the enzyme, adding yet another mechanism by which tryptic activity is regulated inside pancreatic acinar cells.
Materials and Methods
Protein Expression and Purification
cDNA encoding for amino acids 16–247 of all trypsin isoforms was cloned in frame with an N-terminal hexahistidine tag (His6-tag) and an HRV3C cleavage site into a pET47b vector (Novagen, Darmstadt, Germany). N-terminal truncations were introduced to remove the signal peptide sequences for expression in E. coli. Inactivating S200A mutations were introduced in all trypsin isoforms by site directed mutagenesis using QuikChange XL (Agilent Technologies, Santa Clara, CA, USA).
Trypsin isoforms and their inactive S200A variants were overexpressed in BL21 (DE3) E. coli (New England Biolabs, Frankfurt am Main, Germany). Cultures were grown in terrific broth medium with 100 µg mL−1 kanamycin at 37 °C. Expression was induced using 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at an OD600 = 2 and carried out overnight at 25 °C. Cells were harvested by centrifugation (4000 g, 4 °C) and resuspended in 0.1 M Tris pH 8 and 5 mM EDTA (Tris-EDTA). Inclusion bodies were prepared essentially as described previously.6 In brief, cells were lyzed by sonication and inclusion bodies were washed three times with Tris-EDTA before solubilization in 6 M guanidine hydrochloride (Gdn-HCl), 0.1 M Tris pH 8, 2 mM EDTA, 30 mM dithiothreitol (DTT). After incubation at 37 °C for 30 min and centrifugation, the solubilized inclusion bodies were added to refolding solution (0.9 M Gdn-HCl, 0.1 M Tris pH 8, 2 mM EDTA, 1 mM L-cysteine, 1 mM L-cystine, 15 µg mL−1 benzamidine) using a syringe pump at 20 µL min−1 at 4 °C. After overnight incubation, the refolded trypsin was loaded onto a HisTrap excel column (Cytiva, Freiburg, Germany). The column was extensively washed using 20 mM Hepes pH 8, 150 mM NaCl, 20 mM imidazole, 15 µg mL−1 benzamidine and eluted using 20 mM Hepes pH 8, 150 mM NaCl, 500 mM imidazole. Purified trypsinogen isoforms were dialyzed into 2 mM HCl and 2 mM CaCl2 for storage before further use. During this step, residual benzamidine is also removed. All purification steps were carried out at 4 °C. Enterokinase activation was carried out at an enterokinase:trypsin ratio of 1:20,000 for WT and 1:50 for S200A variants in 20 mM Hepes pH 8, 50 mM NaCl and 2 mM CaCl2 and was completed after a few hours at room temperature. Homogeneity was confirmed by analytical size exclusion chromatography using an ÄKTA micro platform (GE Healthcare, Freiburg, Germany) with a Superdex 200 Increase 3.2/300 column (Cytiva, Freiburg, Germany). Calibration curves are reported elsewhere.21
Determination of Enzyme Activity
Michaelis-Menten constants (Km) and turnover numbers (kcat) were determined using L-BAPA (Nα-Benzoyl-L-arginine 4-nitroanilide hydrochloride, Sigma-Aldrich, Taufkirchen, Germany) as substrate. The enzyme concentration was 30 nM and the L-BAPA concentrations ranged from 0.02 mM to 9.5 mM. Assays were carried out in 20 mM Hepes pH 7.4, 150 mM NaCl, 2 mM CaCl2 and 0.05% Tween 20. Using the linear portion of the curves, initial conversion rates were determined at 37 °C. The amount of active enzyme was determined by active site titration using SPINK1. Absorbance of L-BAPA was monitored at 405 nm using a Cytation5 microplate reader (BioTek, VT, USA).
Crystallization and Data Collection
Enterokinase activated TRY2 was concentrated to 15 mg mL−1. Crystals were obtained using the hanging drop vapor diffusion method at 20 °C in a reservoir solution containing 22% polyethylene glycol (PEG) 4000, 0.1 M Hepes pH 7.5 and 0.1 M sodium acetate. Crystals with a size of around 200 µm were cryocooled in liquid nitrogen. The diffraction data were collected at the BESSY 14.2 beamline. Data collection parameters are reported in Table 1.
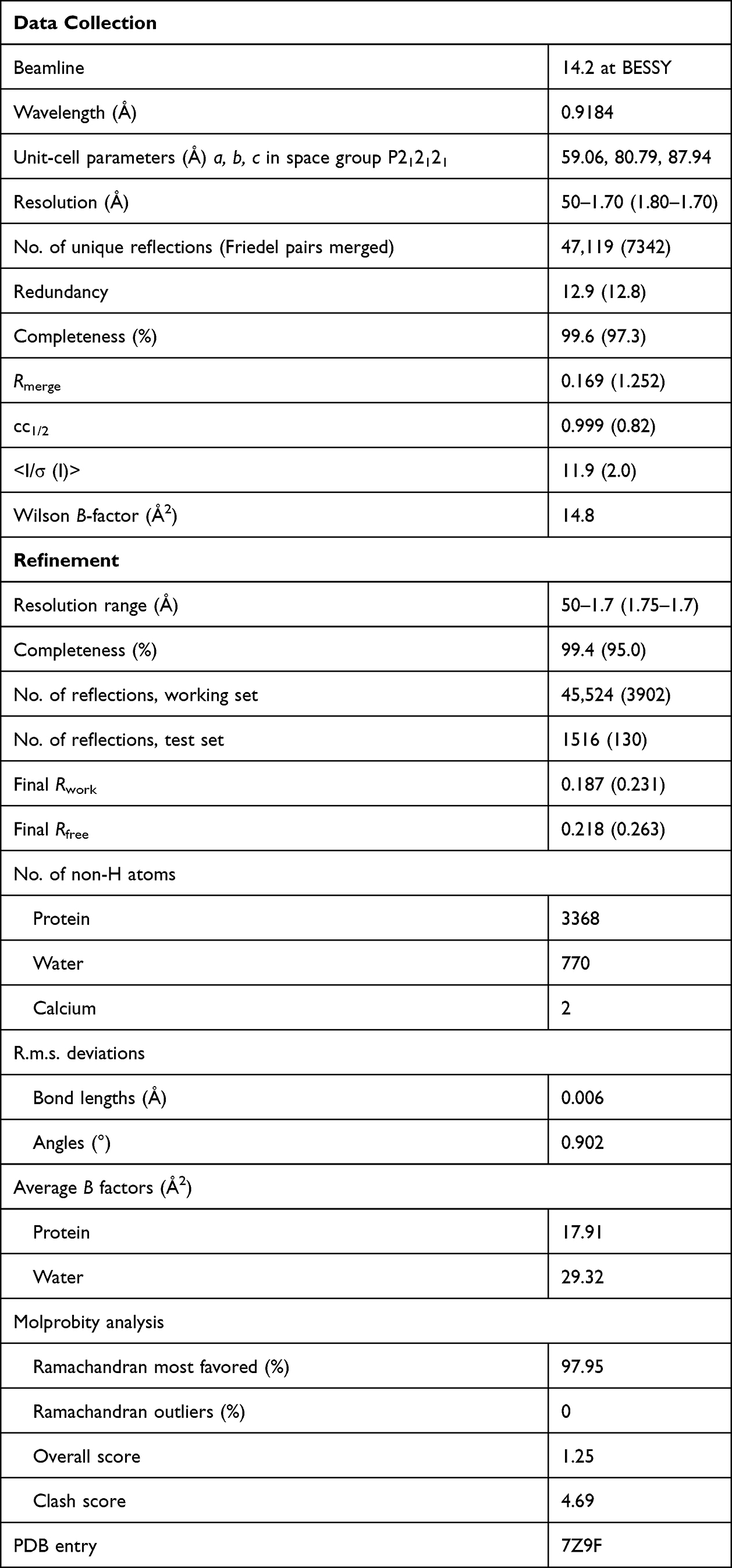 |
Table 1 Data Collection and Refinement Statistics of the TRY2 Homodimer |
Structure Determination
Data reduction was performed in XDS22 and molecular replacement was carried out in Phaser23 using human cationic trypsin (pdb: 1TRN) as search model. Phenix.refine24 was used for refinement, and the model was built in Coot.25 The crystal was isomorphous and contained two molecules per unit cell. NCS restraints were not applied. TLS refinement was used and refinement parameters are given in Table 1.
Molecular Dynamics Simulations
In order to generate a molecular model for the TRY1–TRY1 complex, the obtained crystal structure for TRY2 was mutated to the TRY1 sequence with Coot25 and subjected to equilibration molecular dynamics simulations with NAMD 2.14.26 The CHARMM36 forcefield was applied with CHARMM-GUI.27,28 VMD 1.9.3 was utilized to solvate the protein complex with the TIP3P water model in a cubic cell with side length 8 nm.29,30 NaCl concentrations were set to 0.15 M and the system was neutralized accordingly. The hydrogen mass repartitioning scheme (HMR) scaled the hydrogen masses in the protein to allow a longer timestep of 4 fs, while the SETTLE algorithm constrained the lengths of all bonds to hydrogen atoms. Pressures and temperatures were adjusted by a Langevin piston barostat at 100 fs period, 200 fs decay time and Langevin thermostat at 1 ps−1 damping time, respectively. Long-range electrostatic interactions were described by the smooth particle mesh Ewald method (sPME) and short-range interactions cutoffs were set to 1 nm with a switching function of 0.1 nm. The system was first subjected to standard energy minimization for 50k steps and then slowly heated from 100 K to 300 K in steps of 50 K, for 1 ns each. Afterward, the final production run proceeded at 310 K in an NPT ensemble for 2000 ns and samples were collected every 20 ps. RMSD values converged already after 100 ns, validating full equilibration of the model (Figure S1). RMSD and distance analyses were carried out with VMD.
Results
Human Anionic Trypsin Shows Specific Cleavage at the Arg122-Val123 Peptide Bond
Although the increased tendency for autoproteolysis of TRY2 is clearly established, the overall mechanism and sequence of digestion have remained elusive. To provide a structural framework for rationalizing the autoproteolytic failsafe mechanism, we determined the crystal structure of TRY2 in a state, representing the first step of autoproteolysis. After cleavage of the Arg122-Val123 peptide bond, Arg122 remains bound to TRY2 and is located in the S1 binding pocket (Schechter-Berger nomenclature).31 The structure was solved at 1.7 Å and two TRY2 molecules formed a homodimer in the asymmetric unit (Figure 1A, Table 1). Specific cleavage of the Arg122-Val123 bond was demonstrated by SDS-PAGE, while the catalytically inactive TRY2 S200A variant remained intact even after enterokinase activation (Figure 1B). Lower bands in the TRY2 S200A lane correspond to impurities introduced by the high amounts of enterokinase needed for activation, due to the absence of autoactivation in the inactive enzyme. Additional bands in the TRY2 WT lane are secondary products due to cleavage at the autolytic Lys193-Asp194 site. Homogeneity of anionic trypsinogen and trypsin was verified by analytical size exclusion chromatography (Figure 1C). Despite the cleavage of TRY2 into two chains, they remain attached by disulfide bonds and the enzyme maintains its activity. While intact TRY2 can be seen in the SDS-PAGE, no electron density for an intact Arg122-Val123 bond was visible in the crystal structure (Figure 2A). At 20 µM all trypsin isoforms elute as monomers with only a small peak visible at 1.55 mL corresponding to the size of a trypsin dimer. Based on the TRY2-TRY2 complex crystal structure, we built a model of the TRY1-TRY1 dimer (Figure S2). The equilibrated TRY1 model is highly similar to the TRY2 crystal structure. The distance between Arg122 and Val123 is ~8 Å in the crystal structure of TRY2 and on average ~11 Å for the TRY1 model. Over the course of 2000 ns the shortest distance observed was ~8 Å (Figure S3).
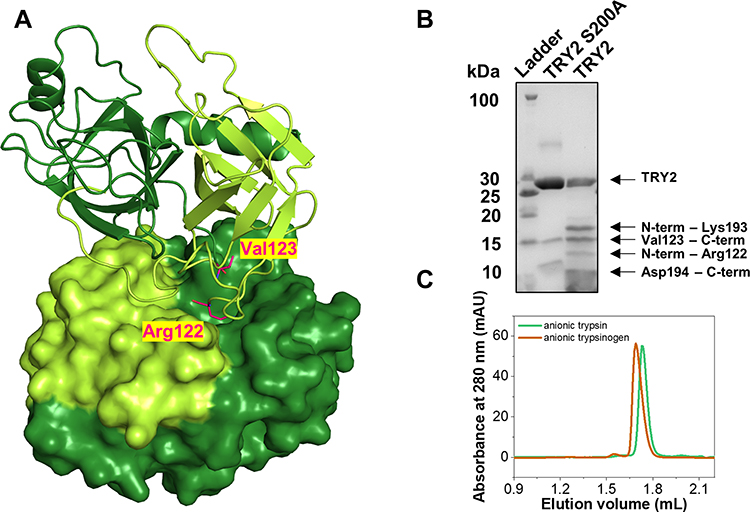 |
Figure 1 Structure of the TRY2 homodimer. (A) TRY2 N-terminus-Arg122 is colored yellow-green and Val123-C-terminus is colored in dark-green. The TRY2 – TRY2 complex shows one monomer in cartoon representation and one in surface representation. One monomer binds on top of TRY2 in a substrate-like manner and is rotated approximately 180° around the y-axis. Arg122 and Val123 are highlighted in pink. (B) SDS-PAGE of activated TRY2 WT and the catalytically inactive S200A variant. (C) Analytical size exclusion chromatograms of anionic trypsinogen and activated anionic trypsin. |
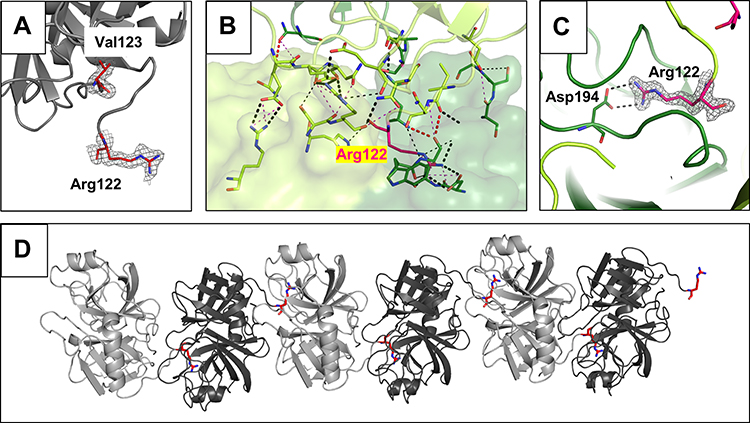 |
Figure 2 Arg122-Val123 cleavage site and specific interactions of the TRY2 dimer interface. (A) Close-up of the cleaved Arg122 loop. The 2Fo-Fc density map is shown 1.6 Å around Arg122 and Val123 and is contoured at 1.0 σ. No additional density for an intact loop is visible. (B) Interaction interface with interacting residues represented as sticks. The color scheme is the same as in Figure 1. (C) Arg122 specifically interacts with Asp194 in the S1 binding pocket. The 2Fo-Fc density is shown using the same parameters as in (A). (D) Crystallographic assembly of a TRY2 fiber with repetitive TRY2 dimers. Arg122 is highlighted in red. Two nicked TRY2 monomers (light and dark grey) form the asymmetric unit. |
The binding interface of the TRY2 dimer encompasses ~800 Å2. Arg122 interacts with the specificity determining residue Asp194 (Figure 2B and C). Due to the specific nature of the TRY2-TRY2 interaction, it is likely that the observed dimer not only shows autoproteolysis, but also demonstrates the autoinhibitory state of the enzyme. Inhibitory activity of the Arg122 loop has already been proposed and weak inhibition was demonstrated.12 Our structure shows that translation and 180° rotation about the translation axis is necessary to create the binding interface for a TRY2 dimer. Crystallographically, this results in an endless chain along the translation axis (Figure 2D). Due to the weakness of the interaction, only dimers, but no oligomers or even fibers were observed in solution. Unlike previous hypotheses, we found that the entire enzyme is involved in the autoinhibition interface, rather than just the Arg122 loop. In addition, the dimerization interface differs significantly from the already known α-chymotrypsin dimer, although the dimerization of α-chymotrypsin also involves an autocleavage site (Figure S4).32 TRY2 likely undergoes induced fit movements after cleavage of the Arg122-Val123 scissile bond. Similar to known Laskowski inhibitors, TRY2 potentially exists in an equilibrium between cleaved and re-ligated Arg122-Val123 as seen on SDS-PAGE (Figure 1B). However, because of the large distance between both residues after cleavage, the cleavage reaction is probably highly favored.
Trypsin Isoforms Differ in Surface Charge but Not in Kinetic Properties
We compared our TRY2 crystal structure with existing crystal structures of cationic trypsin and mesotrypsin (pdb: 7QE9 and 1H4W, respectively). Despite their high sequence identity (>90%), the three isoforms differ in their isoelectric point.33 Evolutionary, the existence of different trypsin isoforms is most likely rationalized by each isoform showing varying resistances to inhibitors, commonly encountered in foods. To identify local differences in surface charge, rather than the global charge of the entire enzyme, we used our newly obtained TRY2 structure to compare the overall charge distribution to the other trypsin isoforms (Figure 3). Mesotrypsin appears to be more positively charged near the S1 binding pocket compared to cationic trypsin, but shows a more negative charge on the backside of the enzyme. Anionic trypsin is the most negatively charged of the three isoforms with differences being most prominent on the backside of the enzyme as well.
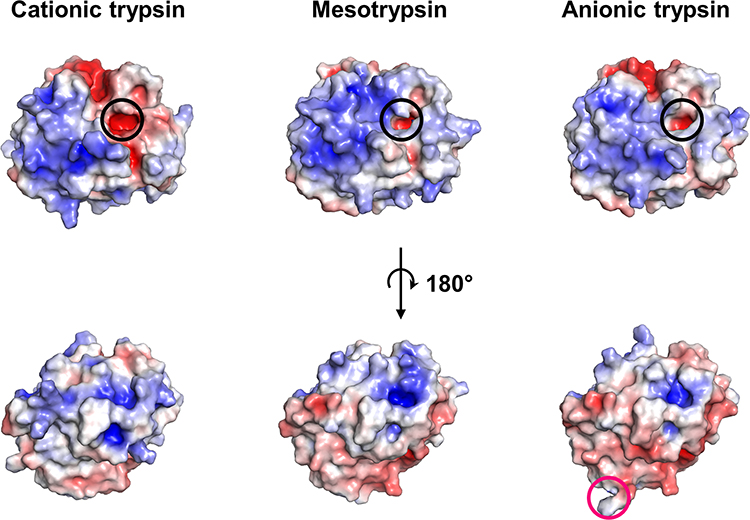 |
Figure 3 Surface charge comparison of the three major trypsin isoforms. Electrostatics were calculated at pH 7.4 and 150 mM NaCl concentration. The Pdb2pqr server and the adaptive poisson Boltzmann solver plugin in PyMOL were used.41 The S1 binding pocket is highlighted by a black circle, while Arg122 in TRY2 is highlighted by a pink circle. Color scales are from red (−5 kbT/ec) to blue (+5 kbT/ec). |
While there are differences in surface charge near the S1 binding pocket for all isoforms, the binding pocket itself remains negatively charged, which is reflected in all trypsin variants preferring either lysine or arginine as P1 residue. Despite their differences in charge, all isoforms are structurally very similar and show no variation in the overall scaffold (Figure 4A). In addition, their kinetic profiles are almost indistinguishable when small chromogenic substrates are used (Figure 4B and C). Using L-BAPA as model substrate, TRY1 and mesotrypsin (TRY3) display identical Michaelis-Menten constants (Km = 1.2 mM) and turnover numbers (kcat = 6 s−1). TRY2 shows slightly lower Km values (Km = 0.6 mM) and similar turnover numbers (kcat = 5.1 s−1). Although they are all kinetically similar when using a small chromogenic substrate, larger or proteinaceous substrates might be more affected by the electrostatic environment surrounding the S1 binding pocket of each isoform.
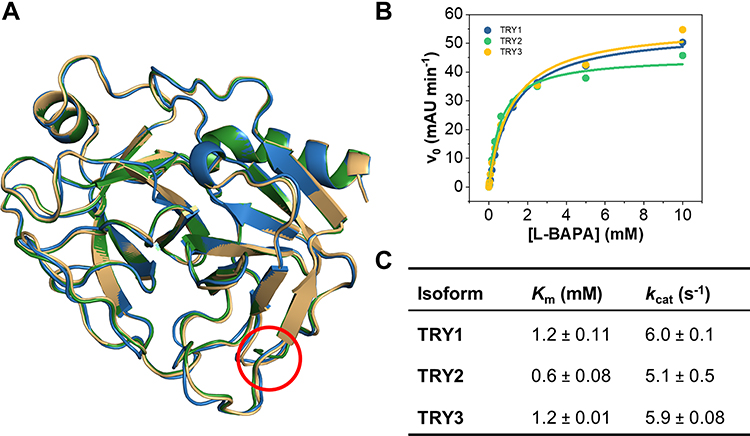 |
Figure 4 Structural and kinetic comparison of the three major trypsin isoforms. (A) Superposition of TRY1-3. TRY1, 2 and 3 are colored blue, green and yellow, respectively. The location of the Arg122 residue is highlighted by a red circle. (B) Representative Michaelis-Menten kinetics of all three isoforms using L-BAPA as substrate. (C) Kinetic parameters for each isoform. Reported values represent mean ± s.d. of three independent experiments. |
Interestingly, Arg193 in mesotrypsin, which usually prevents inhibitor binding by steric hindrance, is oriented in a way that would not interfere with the formation of a trypsin dimer in the autoinhibited state.34 Under physiological conditions, Tyr154 is sulfated, which causes a small decrease in affinity for the trypsin inhibitor SPINK1.35 In the context of trypsin dimerization however, sulfation of this residue might even be beneficial by stabilizing the cleaved Arg122 loop and potentially the newly generated N-terminus (Figure S5). Stabilization of the loop after cleavage might aid in resynthesis of the peptide bond. Our data thereby support a model in which cleavage of the Arg122-Val123 bond is accompanied by self-inhibition of all trypsin isoforms at high concentrations while not affecting the overall activity of the enzyme at low concentrations.
Discussion
We demonstrate that human anionic trypsin selectively cleaves itself at the Arg122-Val123 bond. This cleavage and resulting interactions are of physiological relevance as cleavage of this bond is considered the first step of the autoproteolytic failsafe mechanism. In order to understand the molecular basis of this interaction, we solved the structure of a TRY2–TRY2 dimer at 1.7 Å and used it as a template to build a model of the corresponding TRY1–TRY1 dimer. We observe induced fit movements of Arg122 after cleavage, which is then deeply burrowed within the S1 binding pocket. SDS-PAGE confirms the formation of two fragments of appropriate size and suggests the existence of an equilibrium between cleavage and re-ligation of the Arg122 loop. Even after cleavage, analytical size exclusion chromatography and enzymatic assays show a functional enzyme.
Our finding that TRY2 cleaves the Arg122-Val123 loop is consistent with biochemical assays involving different concentrations of NaCl and CaCl2, different pH values, and mutagenesis studies involving R122H or R122A mutations.13,14,16 The importance of this specific loop is also reflected in R122H being the most prominent pancreatitis-causing mutation in the cationic trypsin gene (PRSS1). R122H or R122C suppress the autolytic site within TRY1, thereby disrupting autoproteolysis and clearance of prematurely activated trypsin.8–11 Hence, a molecular model of a TRY1–TRY1 dimer was built on the basis of the TRY2–TRY2 dimer. The observed structures and interactions are essentially the same in both complexes and the findings of the anionic trypsin dimer can be translated to the cationic trypsin dimer.
Previous studies report that the Arg122 loop is not only the main autolytic site, but also functions as a weak inhibitory loop.12 Given the conformation observed in our crystal structure, it is apparent that binding of Arg122 blocks entry into the S1 binding pocket and thereby inhibits further cleavage. Our SDS-PAGE also shows intact TRY2 even after very long incubation times, which further supports previous hypotheses, where the Arg122-Val123 can be re-ligated by trypsin via a similar mechanism observed in many canonical serine protease inhibitors following the Laskowski mechanism.36 In addition to providing proof for this proposed mechanism, we also show that not only the Arg122 loop is involved in trypsin autoinhibition, but also the entire backside of the enzyme. Although the entire enzyme participates in inhibition, there are few specific interactions, which probably explain the high Ki values, estimated at around 80 µM.12 At these Ki values, meaningful autoinhibition can only occur within zymogen granules of pancreatic acinar cells, where trypsin concentrations can exceed 1 mM and enzymes exist in a semi-crystalline state.18,37
While the Arg122 loop functions similarly to known Laskowski inhibitors, there are several distinguishing features. Most importantly, in Laskowski inhibitors like SPINK1, the binding loop is very rigid, whereas the Arg122 loop is not stabilized by disulfide bonds or other intramolecular interactions. This causes the Arg122 and Val123 residues to be separated by more than 8 Å after cleavage, which is far from a conformation that would allow efficient re-ligation. Furthermore, during our 2 µs MD simulation of the TRY1 dimer, Arg122 and Val123 never got closer than 8 Å. We therefore assume that, while re-ligation of this bond is possible, the equilibrium is tilted towards cleavage, which is in accordance with the reported weak inhibitory potential of the Arg122 loop.
Due to the tight control of TRY2 by chymotrypsin C, no disease-causing mutations are known within the PRSS2 gene.14 Nevertheless, TRY2 contributes to the pancreatitis risk, which is demonstrated by the G191R mutation. By introducing an additional autolytic site, the mutation, which is enriched in the healthy control, causes a small protective effect.13 In addition, the expression of TRY2 in transgenic mice revealed exacerbated pancreatitis, clearly demonstrating the involvement of TRY2 in pancreatitis.20 The authors stressed that inhibitors directed at treating pancreatitis should be designed and tested against all trypsin isoforms.
To this day, no pharmacological therapy for pancreatitis exists. Treatment is mostly supportive, including administration of fluids, bowel rest, antibiotics and pain control.38,39 Due to the complexity and elusiveness of the disease, development of therapeutics is challenging and results obtained from animal models are often not transferable to clinical studies.40 Our findings add to the understanding of this highly complex disease and provide a structural framework from which novel therapeutics could be developed.
In addition to autoproteolysis, we show that tryptic activity is also regulated by self-inhibition following Arg122 cleavage, adding another mechanism, by which the pancreas elegantly protects itself from prematurely activated trypsin.
Conclusion
We solved the crystal structure of human anionic trypsin in a state representing the first step of autoproteolysis. The structural basis underlying the pathogenicity of the R122H variant in cationic trypsin was elucidated and a previously hypothesized autoinhibitory mechanism confirmed. Our data support a model in which autoproteolysis of the Arg122 loop is accompanied by self-inhibition, which contributes to the tight regulation of tryptic activity in pancreatic acinar cells.
Acknowledgments
We acknowledge access to beamline BL 14.1 and 14.2 of the BESSY II storage ring (Berlin, Germany) via the Joint Berlin MX-Laboratory sponsored by the Helmholtz Zentrum Berlin für Materialien und Energie, the Freie Universität Berlin, the Humboldt-Universität zu Berlin, the Max-Delbrück Centrum, and the Leibniz-Institut für Molekulare Pharmakologie.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Beyer G, Habtezion A, Werner J, Lerch MM, Mayerle J. Chronic pancreatitis. Lancet. 2020;396(10249):499–512. doi:10.1016/S0140-6736(20)31318-0
2. Angelopoulos N, Dervenis C, Goula A, et al. Endocrine pancreatic insufficiency in chronic pancreatitis. Pancreatology. 2005;5(2–3):122–131. doi:10.1159/000085264
3. Capurso G, Traini M, Piciucchi M, Signoretti M, Arcidiacono PG. Exocrine pancreatic insufficiency: prevalence, diagnosis, and management. Clin Exp Gastroenterol. 2019;12:129–139. doi:10.2147/CEG.S168266
4. Weiss FU, Laemmerhirt F, Lerch MM. Etiology and risk factors of acute and chronic pancreatitis. Visc Med. 2019;35(2):73–81. doi:10.1159/000499138
5. Hirota M, Ohmuraya M, Baba H. The role of trypsin, trypsin inhibitor, and trypsin receptor in the onset and aggravation of pancreatitis. J Gastroenterol. 2006;41(9):832–836. doi:10.1007/s00535-006-1874-2
6. Nagel F, Palm GJ, Geist N, et al. Structural and biophysical insights into SPINK1 bound to human cationic trypsin. Int J Mol Sci. 2022;23(7):3468. doi:10.3390/ijms23073468
7. Hegyi E, Sahin-Tóth M. Genetic risk in chronic pancreatitis: the trypsin-dependent pathway. Dig Dis Sci. 2017;62(7):1692–1701. doi:10.1007/s10620-017-4601-3
8. Whitcomb DC, Gorry MC, Preston RA, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet. 1996;14(2):141–145. doi:10.1038/ng1096-141
9. Teich N, Bauer N, Mössner J, Keim V. Mutational screening of patients with nonalcoholic chronic pancreatitis: identification of further trypsinogen variants. Am J Gastroenterol. 2002;97(2):341–346. doi:10.1111/j.1572-0241.2002.05467.x
10. Gorry MC, Gabbaizedeh D, Furey W, et al. Mutations in the cationic trypsinogen gene are associated with recurrent acute and chronic pancreatitis. Gastroenterology. 1997;113(4):1063–1068. doi:10.1053/gast.1997.v113.pm9322498
11. Witt H, Luck W, Becker M. A signal peptide cleavage site mutation in the cationic trypsinogen gene is strongly associated with chronic pancreatitis. Gastroenterology. 1999;117(1):7–10. doi:10.1016/s0016-5085(99)70543-3
12. Kukor Z, Tóth M, Pál G, Sahin-Tóth M. Human cationic trypsinogen: Arg (117) is the reactive site of an inhibitory surface loop that controls spontaneous zymogen activation. J Biol Chem. 2002;277(8):6111–6117. doi:10.1074/jbc.M110959200
13. Witt H, Sahin-Tóth M, Landt O, et al. A degradation-sensitive anionic trypsinogen (PRSS2) variant protects against chronic pancreatitis. Nat Genet. 2006;38(6):668–673. doi:10.1038/ng1797
14. Jancsó Z, Sahin-Tóth M. Tighter control by chymotrypsin C (CTRC) explains lack of association between human anionic trypsinogen and hereditary Pancreatitis*♦. J Biol Chem. 2016;291(25):12897–12905. doi:10.1074/jbc.M116.725374
15. Weiss FU, Skube ME, Lerch MM. Chronic pancreatitis: an update on genetic risk factors. Curr Opin Gastroenterol. 2018;34(5):322–329. doi:10.1097/MOG.0000000000000461
16. Kukor Z, Tóth M, Sahin-Tóth M. Human anionic trypsinogen: properties of autocatalytic activation and degradation and implications in pancreatic diseases. Eur J Biochem. 2003;270(9):2047–2058. doi:10.1046/j.1432-1033.2003.03581.x
17. Guy O, Lombardo D, Bartelt DC, Amic J, Figarella C. Two human trypsinogens. purification, molecular properties, and N-terminal sequences. Biochemistry. 1978;17(9):1669–1675. doi:10.1021/bi00602a014
18. Rinderknecht H, Renner IG, Carmack C. Trypsinogen variants in pancreatic juice of healthy volunteers, chronic alcoholics, and patients with pancreatitis and cancer of the pancreas. Gut. 1979;20(10):886–891. doi:10.1136/gut.20.10.886
19. Rinderknecht H, Stace NH, Renner IG. Effects of chronic alcohol abuse on exocrine pancreatic secretion in man. Dig Dis Sci. 1985;30(1):65–71. doi:10.1007/BF01318373
20. Wan J, Haddock A, Edenfield B, Ji B, Bi Y. Transgenic expression of human PRSS2 exacerbates pancreatitis in mice. Gut. 2020;69(11):2051–2052. doi:10.1136/gutjnl-2019-320399
21. Susemihl A, Nagel F, Grabarczyk P, Schmidt CA, Delcea M. Easy expression and purification of fluorescent N-terminal BCL11B CCHC zinc finger domain. Molecules. 2021;26:24. doi:10.3390/molecules26247576
22. Kabsch W. XDS. Acta Crystallogr Sect D. 2010;66(2):125–132. doi:10.1107/S0907444909047337
23. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40(4):658–674. doi:10.1107/S0021889807021206
24. Liebschner D, Afonine PV, Baker ML, et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr Sect D. 2019;75(10):861–877. doi:10.1107/S2059798319011471
25. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr Sect D Biol Crystallogr. 2010;66(4):486–501. doi:10.1107/S0907444910007493
26. Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802. doi:10.1002/jcc.20289
27. Huang J, MacKerell AD Jr. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem. 2013;34(25):2135–2145. doi:10.1002/jcc.23354
28. Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem. 2008;29(11):1859–1865. doi:10.1002/jcc.20945
29. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–38. doi:10.1016/0263-7855(96)00018-5
30. Hopkins CW, Le Grand S, Walker RC, Roitberg AE. Long-time-step molecular dynamics through hydrogen mass repartitioning. J Chem Theory Comput. 2015;11(4):1864–1874. doi:10.1021/ct5010406
31. Schechter I, Berger A. On the size of the active site in proteases. I Papain. Biochem Biophys Res Commun. 1967;27(2):157–162. doi:10.1016/S0006-291X(67)80055-X
32. Blevins RA, Tulinsky A. The refinement and the structure of the dimer of alpha-chymotrypsin at 1.67-A resolution. J Biol Chem. 1985;260(7):4264–4275. doi:10.2210/pdb5cha/pdb
33. Scheele G, Bartelt D, Bieger W. Characterization of human exocrine pancreatic proteins by two-dimensional isoelectric focusing/sodium dodecyl sulfate gel electrophoresis. Gastroenterology. 1981;80(3):461–473. doi:10.1016/0016-5085(81)90007-X
34. Katona G, Berglund GI, Hajdu J, Gráf L, Szilágyi L. Crystal structure reveals basis for the inhibitor resistance of human brain trypsin11. J Mol Biol. 2002;315(5):1209–1218. doi:10.1006/jmbi.2001.5305
35. Szabó A, Toldi V, Gazda LD, Demcsák A, Tőzsér J, Sahin-Tóth M. Defective binding of SPINK1 variants is an uncommon mechanism for impaired trypsin inhibition in chronic pancreatitis. J Biol Chem. 2021;296:100343. doi:10.1016/j.jbc.2021.100343
36. Zakharova E, Horvath MP, Goldenberg DP. Structure of a serine protease poised to resynthesize a peptide bond. Proc Natl Acad Sci USA. 2009;106(27):11034–11039. doi:10.1073/pnas.0902463106
37. Goncz KK, Behrsing R, Rothman SS. The protein content and morphogenesis of zymogen granules. Cell Tissue Res. 1995;280(3):519–530. doi:10.1007/BF00318356
38. Kambhampati S, Park W, Habtezion A. Pharmacologic therapy for acute pancreatitis. World J Gastroenterol. 2014;20(45):16868–16880. doi:10.3748/wjg.v20.i45.16868
39. Leppäniemi A, Tolonen M, Tarasconi A, et al. 2019 WSES guidelines for the management of severe acute pancreatitis. World J Emerg Surg. 2019;14(1):27. doi:10.1186/s13017-019-0247-0
40. Pezzilli R. Pharmacotherapy for acute pancreatitis. Expert Opin Pharmacother. 2009;10(18):2999–3014. doi:10.1517/14656560903382630
41. Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci. 2001;98(18):10037–10041. doi:10.1073/pnas.181342398
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.