Back to Journals » Journal of Inflammation Research » Volume 18
STK11 Alleviates Pulmonary Inflammation During Acute Lung Injury by Phosphorylating AMPK to Activate Autophagy in A549 Cell Model
Authors Xiao R, Qi Z, Chen T, Wei F
Received 9 July 2025
Accepted for publication 11 December 2025
Published 19 December 2025 Volume 2025:18 Pages 17789—17802
DOI https://doi.org/10.2147/JIR.S548864
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Cynthia Koziol-White
Rong Xiao,1 Zhimin Qi,1 Ting Chen,2 Fusheng Wei2
1Department of Anesthesiology and Operation, The Affiliated Stomatological Hospital, Jiangxi Medical College, Nanchang University, Jiangxi Provincial Key Laboratory of Oral Diseases, Jiangxi Provincial Clinical Research Center for Oral Diseases, Nanchang, 330000, People’s Republic of China; 2Department of Anesthesiology and Operation, the First Affiliated Hospital, Jiangxi Medical College, Nanchang University, Nanchang, 330000, People’s Republic of China
Correspondence: Fusheng Wei, Department of Anesthesiology and Operation, The First Affiliated Hospital of Jiangxi MedicaL College, Nanchang University, No. 1519, Dongyue Avenue, Xianghu New Town, Nanchang, 330000, People’s Republic of China, Email [email protected]
Purpose: To investigate if protein STK11 alleviates lipopolysaccharide-induced acute lung injury (ALI), hypothesizing it activates autophagy (harmful substance clearance) via AMPK’s “start signal”.
Methods: Researchers used a genomics-first approach: they integrated datasets GSE66890, GSE10474, and GSE32707, screened autophagy-related differentially expressed genes in ALI via bioinformatics, and confirmed STK11 as a key regulator through protein-protein interaction analysis. They treated human lung cells with 50 μg/mL lipopolysaccharide for 24 hours to establish ALI models, then overexpressed or silenced STK11 in the cells. They assessed cell function, apoptosis, inflammatory factors, autophagy activity, and AMPK activation.
Results: Researchers verified STK11 as a key regulator of autophagy in ALI. STK11 overexpression significantly improved cell function, reduced apoptosis (lower pro-apoptotic/higher anti-apoptotic proteins), decreased IL-6/IL-8/TNF-α (mRNA/protein levels), enhanced autophagy (elevated LC3B-II, reduced P62, more autophagosomes), and activated AMPK. STK11 silencing reversed these protective effects and inhibited AMPK.
Conclusion: STK11 mitigates lipopolysaccharide-induced lung cell damage by activating AMPK-mediated autophagy, emerging as a potential therapeutic target for ALI.
Keywords: STK11, AMPK, acute lung injury, inflammation, autophagy, LPS
Introduction
Acute lung injury (ALI) is a life-threatening inflammatory disease of the lungs, characterized by rapid onset of hypoxemia, alveolar-capillary barrier disruption, and extensive pulmonary inflammation, with a global mortality rate as high as 30–40%.1–5 Currently, the standard treatment for ALI remains primarily supportive care, including mechanical ventilation (to maintain oxygenation), fluid management (to balance tissue perfusion and avoid alveolar edema), and broad-spectrum antibiotics (for infection-related ALI).2,4 Although anti-inflammatory agents such as corticosteroids have been tested in clinical trials, their efficacy is inconsistent and may be associated with increased infection risk; notably, there are no targeted therapies approved to directly reverse ALI pathogenesis or modulate its core inflammatory pathways.3,5 This lack of specific interventions highlights an urgent need to identify novel molecular targets and regulatory mechanisms underlying ALI. The pathophysiological process of ALI involves multiple interconnected mechanisms, including excessive inflammatory response (driven by neutrophils and pro-inflammatory cytokines like IL-6, IL-8, and TNF-α), oxidative stress, endothelial/epithelial barrier dysfunction, apoptosis, and autophagy dysregulation.1–3 Among these, autophagy—a highly conserved lysosomal degradation pathway—plays a complex, context-dependent role in ALI progression.6–8 Moderate autophagy activation can alleviate cell damage by clearing damaged organelles and misfolded proteins, thereby reducing oxidative stress and inflammation;6,7 However, excessive or dysregulated autophagy may exacerbate epithelial cell death and lung injury.9 This dual role underscores the importance of elucidating the precise regulatory networks that control autophagy in ALI. STK11 (also known as LKB1), a serine/threonine kinase encoded by the gene on chromosome 19p13.3, is a well-recognized master regulator of cell energy homeostasis, metabolism, and growth.10 Beyond its established tumor-suppressive function in lung cancer (where STK11 mutations are frequent in non-small cell lung cancer),10,11 emerging evidence highlights STK11’s tissue-specific roles in lung epithelial cells: For instance, STK11 has been shown to maintain alveolar epithelial cell integrity by regulating metabolic pathways (eg, lipid metabolism and glucose utilization) under physiological conditions.12,13 In a mouse model of chronic obstructive pulmonary disease (COPD), STK11 deficiency in lung epithelial cells exacerbated cigarette smoke-induced inflammation and epithelial apoptosis, suggesting a protective role of STK11 in lung inflammatory disorders.14 Additionally, STK11 was recently reported to modulate epithelial barrier function by phosphorylating downstream targets (eg, MARK3) in bronchial epithelial cells.15 Despite these findings, no studies have investigated STK11’s role in ALI, nor its potential to regulate autophagy or inflammation in the context of LPS-induced lung epithelial injury—representing a critical knowledge gap. Notably, STK11’s role in autophagy regulation has been well documented in other disease models: It acts as an upstream activator of AMP-activated protein kinase (AMPK), a key sensor of cellular energy status, by directly phosphorylating AMPK at Thr172.16–19 In Alzheimer’s disease, STK11-mediated AMPK activation promoted autophagic clearance of neurotoxic amyloid-β aggregates;16 In breast cancer cells, STK11 induced cytoprotective autophagy via the AMPK-ULK1 axis.17 However, whether this STK11-AMPK-autophagy pathway operates in ALI—particularly in lung epithelial cells, the primary targets of LPS-induced injury—remains unknown. Our prior bioinformatics analysis (integrating GEO datasets GSE66890, GSE10474, and GSE32707) further supported STK11’s relevance to ALI: We identified STK11 as a core autophagy-related gene differentially expressed in ALI, with strong connectivity in protein-protein interaction (PPI) networks of ALI-associated genes. Building on this genomic insight, existing evidence of STK11’s protective roles in lung epithelium, and its established function in AMPK-mediated autophagy, we hypothesized that STK11 may alleviate LPS-induced ALI by activating AMPK-dependent protective autophagy, thereby reducing pulmonary inflammation. This study aimed to: (1) determine whether STK11 overexpression protects human lung adenocarcinoma A549 cells (a well-established model of alveolar epithelial cells) against LPS-induced injury; (2) investigate the anti-inflammatory effects of STK11 in LPS-stimulated A549 cells; (3) elucidate the role of autophagy activation in STK11-mediated protection; (4) confirm that STK11 exerts its protective effects through AMPK phosphorylation. The findings of this study will: provide the first experimental evidence for STK11’s involvement in ALI pathogenesis; clarify the molecular mechanism by which STK11 activates protective autophagy via AMPK; and establish STK11 as a potential therapeutic target for ALI. By systematically exploring the STK11-AMPK-autophagy axis in ALI, this work will offer new insights into the disease’s molecular basis and inform the development of targeted interventions for this fatal condition.
Materials and Methods
To Screen Out Differentially Expressed Autophagy-Related Genes in ALI
The acute lung injury related datasets GSE66890, GSE10474 and GSE32707 were downloaded from the GEO database. After merging the data, SVA package was used to remove batch effect, and limma package was used for differential expression analysis. Differentially expressed genes (DEGs) were screened according to screening conditions |log fold change (FC)| > 0.5 and P value < 0.05. Subsequently, the selected DEGs were intersected with the autophagy gene database (http://www.autophagy.lu/) to obtain autophagy-related differentially expressed genes. Then, ClusterProfiler software was used to perform KEGG pathway enrichment analysis and GO enrichment analysis of these genes. The PPI (protein interaction) network of intersection DEGs was constructed by STRING database (http://string-db.org/) and imported into Cytoscape 3.9.0 software for further visual analysis. Finally, the top 10 core genes with degree value were screened by the CytoHubba plug-in in Cytoscape software.
Main Reagents
In this study, recombinant Anti-LKB1 antibody (abcam, UK, ab199970), recombinant Anti-Bax antibody (abcam, UK, ab32503), recombinant Anti-Bcl-XL antibody (abcam, UK, ab32370), recombinant anti-Bax antibody (abcam, UK, ab32503), recombinant Anti-AMPK alpha 1(phospho S496) antibody (abcam, UK, ab92701), recombinant Anti-AMPK alpha 1 antibody (abcam, UK, ab32047), and Anti-LC3B antibody (abcam, UK, ab63817), Beta Actin Monoclonal antibody (proteintech, Wuhan, China,66009-1-lg), and recombinant Anti-SQSTM1 / p62 antibody (abcam, UK, ab207305).
Cell Culture
This study utilized publicly available anonymous data and commercially available cell lines, and no ethical approval was required. Human lung adenocarcinoma cell line A549 (ATCC CCL-185) was cultured in DMEM high glucose medium (Corning) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin (HyClone) in a 5% CO2 incubator at 37 ° C. The cells were subjected to LPS (50 μg/mL) stimulation for 24 h before analysis.
Plasmid Construction and Virus Packaging
Knockdown experiment: two specific shRNA sequences were designed for STK11 gene (sh-STK11#1: 5′-GCAGCTACATCCTCAAGATAA-3′; sh-STK11#2: 5′-GGATGCTTTGACTGCTATTAT-3′), which was linked to pLKO.1-puro vector (Addgene #8453). For negative control (sh-NC), a non-targeting sequence (5′-CAACAAGATGAAGAGCACCAA-3′) was used. For overexpression experiments, the full-length cDNA of human STK11 (NCBI RefSeq NM_000455.4) was cloned into the pCDH-CMV-MCS-EF1-Puro vector (System Biosciences), and the empty vector was used as a negative control (OE-NC). After sequencing verification, all plasmids were co-transfected into HEK293T cells with psPAX2/pMD2.G packaging plasmid by Lipofectamine 3000 (Invitrogen). After 48 hours, the viral supernatant was collected and filtered through a 0.45 μm filter.
Cell Transfection and Screening
Knockdown group: A549 cells were infected with lentivirus at MOI=5, and 72 hours later, 2 μg/mL puromycin (Sigma-Aldrich) was added for screening for 7 days. Overexpression group: infected with the same MOI, and the screening conditions were the same as those of knockdown group. The Untreated group was set as the baseline control.
Western Blot Analysis
1 Sample preparation The cells were collected, lysed with RIPA lysate containing protease/phosphatase inhibitors for 30 min on ice, centrifuged at 4 ° C (12,000 × g, 15 min), and the supernatant was removed. The protein concentration was quantified by BCA method, and after adjustment to consistency, 5× SDS loading buffer was added and denatured at 95 ° C for 5 min. 2. SDS-PAGE electrophoresis according to the molecular weight of the target protein, the separation gel (such as 10% gel) was prepared, and the concentration gel was 5%. 20–30 μg of protein was loaded into each well, the concentration gel was electrophorezed at 80 V for 30 min, and the separation gel was brought to the bottom of the bromophenol blue dot gel at 120 V. 3. The protein was transferred to PVDF membrane (containing 20% methanol membrane transfer buffer) by membrane wet transfer method, and the membrane was transferred for 1–2 h at constant current of 200–300 mA (adjusted according to molecular weight). 4. Block and antibody incubation membranes were blocked with 5% skim milk for 1 h, incubated with primary antibodies (such as anti-STK11 antibody, diluted 1:100–2000, overnight at 4 ° C), washed 3 times with TBST, incubated with HRP-labeled secondary antibodies (1:5000, 1 h at room temperature), and washed again with TBST. 5 After the assay was incubated with the imaging ECL reagent, the chemiluminescence imaging system such as ChemiDoc exposed and captured the bands. ImageJ software was used to analyze the gray-scale ratio of the target protein to the internal control (β-actin) and calculate the relative expression.
RNA Extraction and Qrt-Pcr
Total RNA was extracted from A549 cells using TRIzol reagent from BioTNT (China) according to the manufacturer’s instructions. One microgram of RNA was used for reverse transcription to synthesize cDNA, which was used as a quantitative real-time PCR template. The mRNA levels of IL-6, IL-8, and TNF-α were measured using the SYBRGreen Master Mix kit (Roche) according to the manufacturer’s instructions and standardized with β-actin as a loading control. Real-time PCR. Amplification is as follows: initial denaturation at 95°C for 30 seconds; 40 cycles of denaturation at 95°C3 seconds, annealing at 60°C for 30 seconds and extension at 72°C for 10 seconds; One cycle of 95°C for 15 seconds; 65°C for 1 minute; And a final cooling step of 40°C for 30 seconds. mRNA expression levels were calculated using the 2^ method. The primer sequences used for the amplification were: TNF-α, forward: TGACTTGTTCCTCAGCCTCTT, reverse: TGAGGTACAGGCCCTCTGAT. IL-6, forward: TGCGTCGTAGTTTCTCTTCTTCT, reverse: GGAATCTTCTCCTGGGGGTA; β-actin, forward: TGGCACCCAGCACAATGAAGATCA, Reverse: CTGCTTGCTGATCCACATCTGCT.
ELISA
Protein levels of IL-6, IL-8, and TNF-α in cell supernatants were determined using ELISA kits according to the manufacturer’s instructions.
CCK-8 Cell Viability Assay
A549 cells were treated according to the experimental protocol, seeded in 96-well plates at a density of 3×10 cells per well, and incubated overnight at 37°C in 5% CO2 for 24 hours. Three replicate Wells were used for each experimental group. After that, 10 μL of CCK-8 reagent was added to each well, and the mixture was incubated continuously for 2 hours at 37°C. Absorbance values at 450 nm were measured using an automated well plate colorimeter (Molecular Devices, USA).
Immunofluorescence Analysis
I. Principle of dual fluorescent autophagy tracing experiment: mRFP-GFP-LC3B is a fusion protein probe: GFP (green) and RFP (red) emit light at the same time to form yellow spots (red-green colocalization) in autophagosomes; In autolysosomes, GFP was degraded due to the acidic environment of lysosomes, and only RFP was retained, forming red spots (only red channel luminescence). By counting the number of the two spots, the dynamic changes of autophagy initiation (autophagosome) and degradation (autolysosome) can be analyzed. Plasmid transfection: mRFP-GFP-LC3B fusion protein expression plasmid was transfected into oe-NC, oe-STK11, sh-NC, sh-STK11 stable cell lines (A549 cells) to ensure that the cells expressed the probe. (2) Treatment to induce autophagy: After transfection, LPS was used to stimulate cells to induce autophagy. (3) Fixation and staining: cells were fixed with 4% paraformaldehyde (15–20 min at room temperature) to terminate the reaction; DAPI staining solution was added and incubated (5–10 min) to make the nucleus blue. Fluorescence imaging and analysis: three-channel images of GFP (green), RFP (red) and DAPI (blue) were collected by fluorescence microscope and combined to observe the structure of autophagy. The number of autophagosomes (yellow dots) and autolysosomes (red dots) were counted by image analysis software (such as ImageJ) to quantitatively compare the difference of autophagy activity in different groups (STK11 overexpression/knockdown). 2. Principle of LC3B immunofluorescence staining: LC3B is the core marker protein of autophagosome (LC3B-II is localized on the autophagic membrane), and the level of autophagy can be reflected by detecting the fluorescence intensity or spot aggregation of LC3B. Procedure: 1. Cell treatment: the same as above (transfected oe-NC/oe-STK11 or sh-NC/sh-STK11, LPS stimulation), and cells were seeded on coverslips or confocal chambers. (2) Fixation and rupture: fixation with 4% paraformaldehyde (room temperature, 15 min); The membrane was broken with buffer containing Triton X-100 (5–10 min) to increase the permeability of cell membrane and facilitate the entry of antibody. 3. Blocking and antibody incubation: Blocking: incubation with 5% BSA or serum (room temperature, 30 min) to block non-specific binding; Primary antibody incubation: anti-LC3B-specific antibody was added and incubated at 4°C overnight to make the antibody bind to intracellular LC3B. For secondary antibody incubation, fluorescence-labeled secondary antibody (such as Alexa Fluor 488, green) was added and incubated for 1 to 2 h at room temperature in the dark to allow the secondary antibody to bind to the primary antibody. 4. Nuclear staining and imaging: DAPI staining (the same steps as above); Images were collected by fluorescence microscopy, and the total fluorescence intensity of LC3B was quantified by software (such as ImageJ) to compare the autophagy activity of different groups (the higher the LC3B aggregation or expression, the more active autophagy).
Statistical Analysis
The sample size design for all cell experiments in this study was based on previous power analysis results: using G*Power 3.1 software, with the “difference in A549 cell viability between the LPS-treated group and the control group” as the primary outcome measure (preliminary experiments showed that the difference in cell viability between the two groups was approximately 30%), the significance level α=0.05, statistical power (1-β)=0.8, and effect size f=0.5 were set. The minimum sample size per group was calculated to be n=6. Researchers adopted a blinded design with “unknown group allocation” in the experiments. The number of independent biological replicates for all cell experiments was 3. All statistical analyses were performed using GraphPad Prism 8.0 (GraphPad Software, San Diego, California, USA), and all values are presented as mean ± standard error. Before conducting the two-tailed Student’s t-test or one-way ANOVA followed by Tukey’s test, the Shapiro–Wilk test was used to verify the normality of the data, and Levene’s test was used to verify the homogeneity of variances. The aforementioned parametric tests were only applied when the data met the prerequisites of normality and homogeneity of variances, and a difference was considered statistically significant at P< 0.05.
Results
The Core Gene STK11 Was Identified by Bioinformatics Analysis
Through bioinformatics analysis of gene expression data, batch effect-corrected data were generated (Figure 1A). A total of 350 intersection DEGS were obtained after screening, including 146 up-regulated genes and 204 down-regulated genes. Volcano map (Figure 1B) and hierarchical clustering heat map (Figure 1C) showed that genes in acute lung injury and control group had significant differences, indicating that the obtained genes had research significance. KEGG pathway enrichment analysis (Figure 1D) showed high expression in Autophagy and PI3K-Akt signaling pathway, and Small cell lung cancer, Longevity regulating pathway, Lipid and atherosclerosis (Figure 1D) atherosclerosis and its related Legionellosis also had relatively high expression values. GO enrichment analysis (Figure 1E) found that BP (biological process) was mainly a cellular response to external stimulus, Items such as programmed cell death-cell death had high counts, In the CC (cellular component), it is mainly related to nuclear related structures such as nucleoplasm and some protein complexes such as NF-kappaB complex. MF (molecular function) is mainly protein kinase activator activity (protein kinase activator activity), and the frequency of identical protein binding is high. Construct PPI networks (Figure 1F), According to Degree value, the top 10 key genes were 1 NFKB1 2CASP1 3STK11 4CDKN1A 5 NFE2L2 6NAMPT 7ATG7 8CCR2 9 PEA15 10ATG9A. It has been found that STK11, ATG7 and ATG9A play important roles in autophagy.
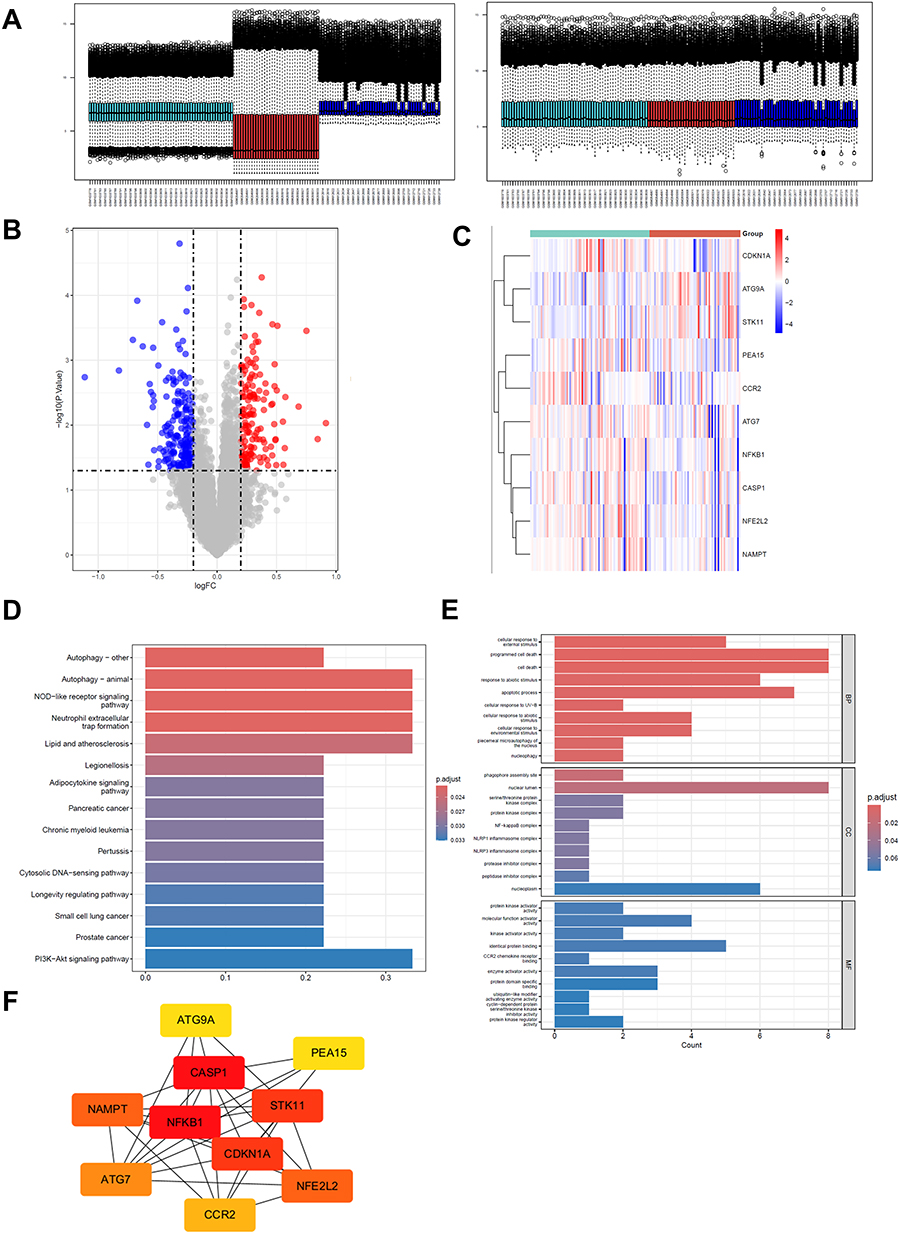 |
Figure 1 Bioinformatics analysis screened differentially expressed autophagy-related key genes in ALI. (A) The variation plot illustrating the data distribution after batch effect removal. (B) volcano plot of gene chip, blue and red dots represent differentially expressed genes (red dots represent up-regulated genes, blue dots represent down-regulated genes) (C) Hierarchical clustering heat map of gene chip (red is highly expressed genes, blue is low expressed genes, Group refers to Sepsis and sepsis-ALI groups) (D) KEGG enrichment analysis bar graph, the red color indicates the higher significance of the pathway. (E) GO functional enrichment analysis bar chart, divided into three parts: biological process (BP), cellular component (CC) and molecular function (MF), and the red color indicates the higher significance. (F) protein interaction network diagram, the red color indicates the higher connectivity. |
Overexpression of STK11 Protected the Viability of Lung Epithelial Cells Under LPS Treatment
To explore the effect of STK11 on cell viability, lung epithelial cells (A549 cells) were treated with LPS. The inhibitory effect of LPS on cell viability was concentration-dependent, in the sense that the inhibition was stronger at higher concentrations, and also time-dependent, in the sense that the inhibition was more pronounced at longer treatment times (Figure 2A). A549 cells were transfected with sh-STK11#1, sh-STK11#2, and OE-STK11, respectively, and the transfection effect of STK11 gene in A549 cells was significant (Figure 2B). Western blot results showed that STK11 levels in sh-STK11#1 and sh-STK11#2 cells were significantly decreased compared with the sh-NC group, and STK11 levels in OE-STK11 cells were significantly increased compared with the OE-NC group (Figure 2C). We found that the viability of A549 cells in the sh-STK11#1 group was significantly reduced, but OE-STK11 could increase the viability of A549 cells (Figure 2D). These results indicated that overexpression of STK11 inhibited LPS-induced decline in lung epithelial cell viability and had a protective effect.
 |
Figure 2 Overexpression of STK11 protected the viability of lung epithelial cells treated with LPS. (A) LPS had a concentration - and time-dependent effect on cell viability. (B) The relative mRNA expression level of STK11 gene in A549 cells. (C) A549 cells were transfected with sh-STK11#1, sh-STK11#2, OE-STK11 plasmids or control plasmids for 48 hours, and STK11 levels were measured by Western blot. (D) A549 cells were transfected with sh-STK11#1, OE-STK11 plasmids or control plasmids for 48 hours and subsequently treated with LPS (50 μg/mL) for 12 hours. The survival ability of A549 cells was detected by CCK-8 assay. The experiment was repeated three times (three independent experiments). Data shown are mean ± standard error. Different letters (a, b, c, d, e) indicate statistical differences between groups (P < 0.05). **P < 0.01. |
STK11 Inhibited LPS-Induced Apoptosis of Lung Epithelial Cells
Further exploring the effect of STK11 on LPS-induced apoptosis of lung epithelial cells, the results of flow cytometry analysis showed that in the OE group, the apoptosis rate of OE-STK11 was slightly higher than that of OE-NC group, but the difference was not significant, while SH-STK11 #1 increased the apoptosis rate in the sh group (Figure 3A). Western blot results showed that the expression of BAX was decreased while the expression of BCL-XL was increased in the OE-STK11 group, and the apoptosis was reduced in the OE-STK11 group, while the expression of BAX was increased while the expression of BCL-XL was decreased in the sh-STK11# group, and the apoptosis was increased in the sh-STK11 group (Figure 3B). These findings indicated that STK11 inhibited apoptosis in lung epithelial cells in response to LPS.
 |
Figure 3 STK11 inhibited apoptosis of lung epithelial cells in response to LPS. (A) Flow cytometry analysis of cell apoptosis. (B) Western blot analysis was used to detect the expression of STK11, BAX, BCL-XL and other proteins in A549 cells treated with LPS. The experiment was repeated three times (three independent experiments). Data shown are mean ±SEM. **P < 0.01. |
STK11 Inhibited LPS-Induced Inflammatory Response in Lung Epithelial Cells
To investigate the effect of STK11 on the production of inflammatory cytokines in LPS-treated lung epithelial cells, A549 cells were transfected with sh-STK11#1, OE-STK11 plasmids and negative control plasmids. After 48 hours of transfection, the cells were incubated with 50 μg/mL LPS. Overexpression of STK11 inhibited the mRNA expression of IL-6, IL-8, and TNF-α, but knockdown of STK11 promoted the mRNA expression of inflammatory factors (Figure 4A and B). These data indicated that STK11 inhibited LPS-induced inflammatory responses in lung epithelial cells.
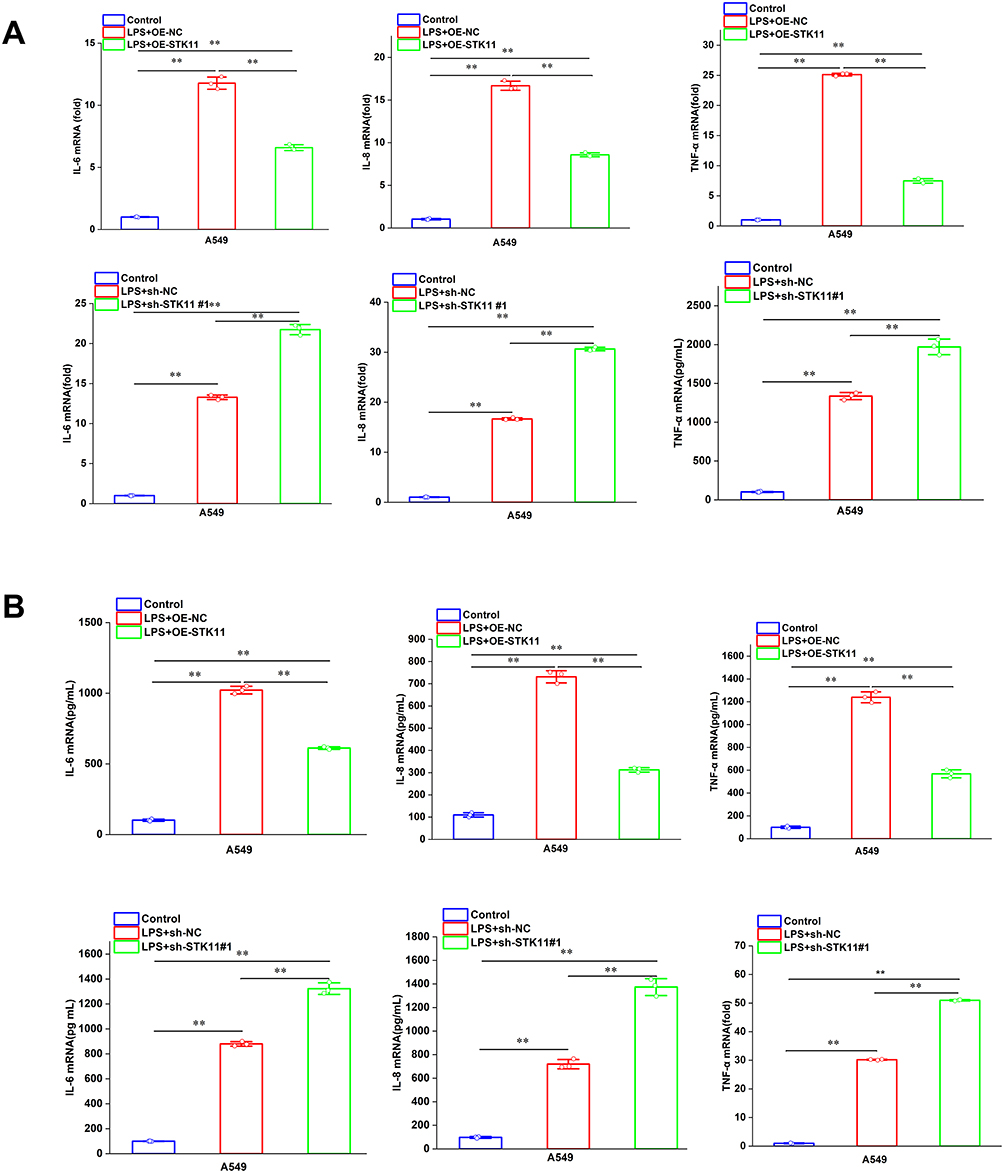 |
Figure 4 STK11 attenuates the LPS-induced inflammatory response in pulmonary epithelial cells. A549 cells were transfected with sh-STK11#1, OE-STK11 plasmid, or negative control plasmid for 48 hours, and subsequently exposed to LPS (50 μg/mL) for 12 hours. (A) The relative mRNA expression levels of IL-6, IL-8, and TNF-α in the cells were determined through quantitative reverse transcription polymerase chain reaction (qRT-PCR). (B) The protein secretion levels of IL-6, IL-8, and TNF-α in the cell culture supernatant were quantitatively assessed using ELISA.The experiment was repeated three times (three independent experiments). Data shown are mean ±SEM.**P < 0.01. |
STK11 Overexpression Alleviated LPS-Induced Lung Epithelial Cell Injury by Activating Autophagy
To clarify the effect of STK11 on autophagy-related proteins in LPS-induced lung epithelial cells, the protein levels of STK11, LC3B and P62 were detected by Western blotting. Overexpression of STK11 enhanced LC3B expression and decreased P62 levels, whereas knockdown of STK11 had the opposite effect (Figure 5A). This suggests that STK11 may affect cell function by promoting autophagy. In addition, the efficiency of autophagic flux could be assessed by observing the relative intensity of red fluorescence (RFP) and green fluorescence (GFP) as shown by cell fluorescence microscopy images and quantitative analysis (Figure 5B). The number of autophagosomes and autolysosomes in OE-STK11 group was significantly higher than that in OE-NC group, indicating that overexpression of STK11 promoted the autophagy process. However, the number of autophagosomes and autolytic lysosomes was significantly reduced in the sh-STK11#1 group compared with the sh-NC group, indicating that the knockdown of STK11 inhibited the autophagy process (Figure 5C). The green fluorescence intensity in OE-STK11 group was significantly higher than that in OE-NC group, indicating that overexpression of STK11 enhanced autophagy. However, the green fluorescence intensity in the sh-STK11 group was lower than that in the sh-NC group, but the difference was relatively small, indicating that the knockdown of STK11 attenuated cell autophagy (Figure 5D). The above results indicated that overexpression of STK11 alleviated LPS-induced lung epithelial cell injury by activating autophagy.
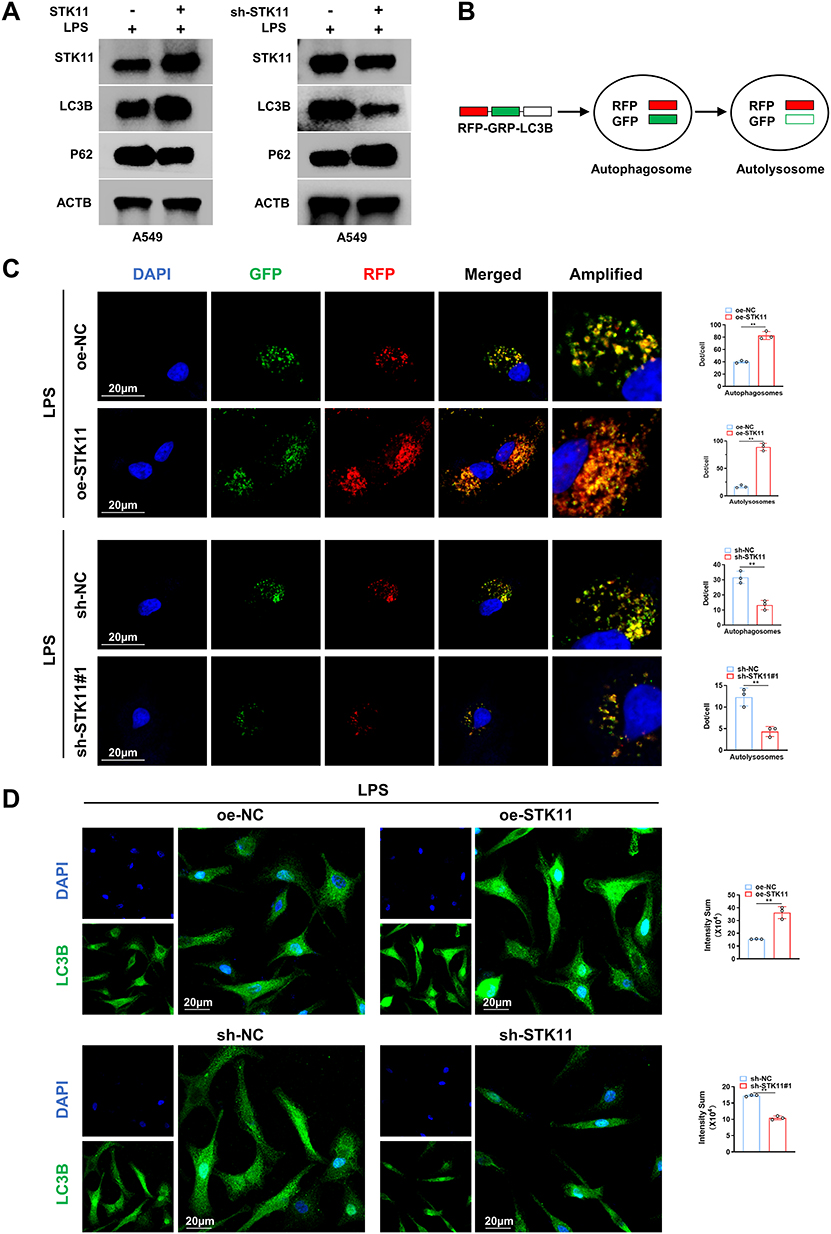 |
Figure 5 Overexpression of STK11 attenuated LPS-induced lung epithelial cell injury by activating autophagy. (A) Protein levels of STK11, LC3B, and P62 were determined by Western blot analysis. (B) Dynamic changes of marker proteins of the autophagic process, and the efficiency of autophagic flow was evaluated by observing the relative intensity of red and green fluorescence. (C) Representative images and quantitative analysis of fluorescence. In LPS-induced A549 cells, DAPI (blue) shows nuclei, GFP (green) usually marks autophagosomes (autophagosomes), and RFP (red) usually marks autolysosomes. Merged is the combined image of DAPI, GFP, and RFP. Amplified is the magnified image. Scale bar =20 μm. (D) Representative images and quantitative analysis of fluorescence. In LPS-induced A549 cells, DAPI (blue) shows the nucleus, and LC3B (green) is an autophagy marker protein. Scale bar =20 μm. The experiment was repeated three times (three independent experiments). Data shown are mean ±SEM. **P < 0.01. |
STK11 Activates Autophagy by Phosphorylating AMPK to Alleviate Acute Lung Injury
To further investigate the effect of STK11 on AMPK phosphorylation, in LPS-induced A549 cells, Western blot analysis showed that the expression level of STK11 in OE-STK11 group was significantly higher than that in OE-NC group, indicating that STK11 was successfully overexpressed. However, the expression level of STK11 in sh-#1 group was significantly lower than that in sh-NC group, indicating that STK11 was successfully knocked down. There was no significant difference in the total protein expression level of AMPK among the groups. The expression level of P-AMPK was higher in OE-STK11 group than in OE-NC group, indicating that STK11 overexpression promoted AMPK phosphorylation, while the expression level of P-AMPK was lower in sh-#1 group than in SH-NC group, indicating that STK11 knockdown inhibited AMPK phosphorylation (Figure 6). This study showed that STK11 promoted AMPK phosphorylation to activate autophagy to alleviate acute lung injury.
 |
Figure 6 STK11 activates autophagy by phosphorylating AMPK to alleviate acute lung injury. Western Blot analysis was used to detect the expression levels of STK11, AMPK and its phosphorylated form (P-AMPK). |
Discussion
Previous studies have shown that STK11 mutations promote cell proliferation and inhibit M1 macrophage differentiation, apoptosis, and the AMPK signaling pathway.20 Previous studies have also reported that CSE enhances the degradation of STK11 protein in airway epithelial cells through the FBXL19-mediated ubiquitin-proteasome pathway, leading to increased cell death.21 Studies have shown that STK11 deficiency impairs antioxidant defenses, leading to elevated levels of reactive oxygen species (ROS) and Snail stabilization in KL cells.22 Therefore, the inhibitory effect of STK11 on LPS-induced cell inflammation should be considered as a target for mitigating inflammatory damage. Based on this, we hypothesized that STK11 may be a novel target for ALI. We found for the first time that STK11 overexpression can inhibit LPS-induced survival rate of lung epithelial cells. Experiments further revealed that STK11 inhibited apoptosis of lung epithelial cells under LPS stimulation. In addition, we also demonstrated that STK11 inhibited the inflammatory response of LPS-induced lung epithelial cells. Previous studies have shown that STK11 silencing attenuates the cytoprotective autophagy of breast cancer cells after HNK treatment, and breast cancer cells undergo autophagy in an STK11-dependent manner.23,24 In recent years, STK11/LKB1 has played an important role in ADIPOQ/adiponectin-mediated cytotoxic autophagy and tumor growth suppression.25 Therefore, STK11 plays a dominant role in the regulation of autophagy. STK11 protein function recovery triggers immunogenic cell death through reactivated apoptosis and autophagy-induced DAMPs release, STK11 can activate the apoptosis process, STK11 nanocomplexes can effectively induce autophagy, STK11 can increase the release of pro-inflammatory factors, including TNF-α, IL-6, and INF-γ, and reduce the release of anti-inflammatory factors, including TGF-β, which provides a theoretical basis for anti-inflammation in vivo.10 Contrary to reports in cancer models where STK11 may promote inflammation,10 our data demonstrate its anti-inflammatory role in ALI. This discrepancy may stem from cell type-specific effects (epithelial vs immune cells) or disease context (acute injury vs chronic malignancy. AMPK-mediated STK11 phosphorylation activates autophagy, while TORC1-mediated STK11 phosphorylation inhibits autophagy. AMPK-activated STK11 phosphorylates type III phosphatidylinositol 3-kinase (PtdIns3K) complex factors, which in turn generate phosphatidylinositol-3-phosphate (PtdIns3P) in the endoplasmic reticulum (ER), a prerequisite for autophagosome formation.26 However, in sepsis-induced acute lung injury (ALI), there is no evidence that STK11 is related to autophagy.
In this study, we found that STK11 activates autophagy, STK11 overexpression enhanced LC3B expression and reduced P62 levels, while STK11 knockdown produced the opposite effect, STK11 overexpression alleviated LPS-induced lung epithelial cell injury by activating autophagy, the number of autophagosomes and autolysosomes was significantly increased, and STK11 knockdown produced the opposite effect. The results of this study also found that STK11 alleviated LPS-induced lung epithelial cell injury by activating AMPK-mediated autophagy. STK11 overexpression enhanced the expression of LC3B and reduced the level of P62, and the autophagy process was activated. STK11 knockdown produced the opposite effect and inhibited the autophagy process. Our study has some limitations: 1. Limitations of bioinformation analysis. Although bioinformation analysis screened out core genes such as STK11, these analyses are mainly based on existing databases and algorithms, and have certain predictability and uncertainty. Therefore, these analysis results need to be further verified in experimental studies. 2. Limitations of in vitro experiments. This study was mainly conducted in in vitro cell lines. Although it can provide preliminary mechanistic evidence, in vitro experiments cannot completely simulate the complex in vivo environment. Therefore, the role of STK11 in acute lung injury needs to be further verified in in vivo animal models. 3. In terms of the depth of mechanism research, although this study revealed the mechanism by which STK11 alleviates acute lung injury by activating AMPK-dependent autophagy, the specific role of this mechanism in different etiologies and different cell types still needs to be further studied. In addition, the interaction between STK11 and other signaling pathways (such as NF-κB, PI3K-Akt, etc.) and its comprehensive regulatory mechanism in acute lung injury also need to be further explored.
This study has the limitation of lacking in vivo experimental data. All current experiments are based on the in vitro model of the human lung adenocarcinoma cell line A549, which only simulates the single injury scenario of alveolar epithelial cells during the pathological process of acute lung injury (ALI). However, the pathophysiological environment of in vivo ALI is complex, involving the interaction of multiple cell types such as alveolar epithelial cells, pulmonary vascular endothelial cells, and immune cells (eg, neutrophils, macrophages), as well as the synergistic effects of multiple factors including systemic inflammatory response, oxidative stress, and pulmonary hemodynamic changes. None of these can be reproduced by a single in vitro cell model. Next, further exploration can be conducted on how these in vitro research results can be translated in animal models or clinical settings.
Conclusions
This study revealed that STK11 alleviates pulmonary inflammation in acute lung injury by activating AMPK-dependent autophagy, providing a new potential target for the treatment of ALI. These findings not only provide new insights into understanding the molecular basis of ALI, but also lay the foundation for developing new therapeutic strategies. To promote the translation of this mechanism into clinical applications in the future, in-depth research should be carried out in the following directions:1. In vivo verification: Using LPS-induced acute lung injury (ALI) mouse models, achieve lung-specific STK11 overexpression via adenoviruses. Detect lung histopathology, cytokines in bronchoalveolar lavage fluid (BALF), lung wet/dry weight ratio, and mouse survival rate to verify the in vivo protective effect of STK11.2. STK11 small-molecule modulators: Screen compounds that interfere with FBXL19-mediated STK11 degradation; explore known upstream activators such as phenformin and metformin analogs, and evaluate their potential in enhancing pulmonary STK11-AMPK signaling.3. Patient-derived biomarkers: Detect STK11 expression and P-AMPK levels in lung biopsies or blood samples from patients with ALI/acute respiratory distress syndrome (ARDS), and correlate these indicators with clinical parameters; stratify patients based on STK11 status to identify populations that are most likely to benefit from STK11-targeted therapy.4. Safety considerations: Regarding the pleiotropy of STK11, evaluate the toxicity of its activators on non-pulmonary tissues, with particular attention to potential metabolic side effects in metabolically active organs.
Ethics Approval
This study utilized publicly available data obtained legally, and the information used was anonymized. It did not cause harm to the human body, did not involve sensitive personal information or commercial interests, and thus could be exempted from ethical approval. Measures for the Administration of Ethical Review of Biomedical Research Involving Humans, Article 32, National Health Commission of the People’s Republic of China, 2023, https://www.nhc.gov.cn/. “Article 32 For human-related life science and medical research involving the use of human information data or biological samples that falls under the following circumstances—causing no harm to the human body, and not involving sensitive personal information or commercial interests—ethical review may be exempted. This aims to reduce unnecessary burdens on researchers and promote the development of human-related life science and medical research. (1) Conducting research using legally obtained public data, or data generated through observation without interfering with public behavior; (2) Conducting research using anonymized information data”.
Acknowledgments
We would like to express my sincere gratitude to Professor Wei for his meticulous guidance, to my colleagues in the research group for their selfless assistance, and also to my family for their warm support.
Funding
This study was supported by Jiangxi Provincial Natural Science Foundation (20232BAB206080).
Disclosure
The authors declare that they have no competing interests.
References
1. Shen Y, He Y, Pan Y, Liu L, Liu Y, Jia J. Role and mechanisms of autophagy, ferroptosis, and pyroptosis in sepsis-induced acute lung injury. Front Pharmacol. 2024;15:1415145. doi:10.3389/fphar.2024.1415145
2. Jiang T, Liu E, Li Z, et al. SIRT1-Rab7 axis attenuates NLRP3 and STING activation through late endosomal-dependent mitophagy during sepsis-induced acute lung injury. Int J Surg. 2024;110(5):2649–2668. doi:10.1097/JS9.0000000000001215
3. Zhang J, Guo Y, Mak M, Tao Z. Translational medicine for acute lung injury. J Transl Med. 2024;22(1):25. doi:10.1186/s12967-023-04828-7
4. Yehya N, Smith L, Thomas NJ, et al. Definition, incidence, and epidemiology of pediatric acute respiratory distress syndrome: from the second pediatric acute lung injury consensus conference. Pediatr Crit Care Med. 2023;24(Suppl):S87–S98. doi:10.1097/PCC.0000000000003161
5. Gu W, Zeng Q, Wang X, Jasem H, Ma L. Acute lung injury and the NLRP3 inflammasome. J Inflamm Res. 2024;17:3801–3813. PMID: 38887753; PMCID: PMC11182363. doi:10.2147/JIR.S464838
6. Zhao J, Liang Q, Fu C, Cong D, Wang L, Xu X. Autophagy in sepsis-induced acute lung injury: friend or foe? Cell Signal. 2023;111:110867. doi:10.1016/j.carolcarrollellsig.2023.110867
7. Liu Q, Wu J, Zhang X, et al. Circulating mitochondrial DNA-triggered autophagy dysfunction via STING underlies sepsis-related acute lung injury. Cell Death Dis. 2021;12(7):673. doi:10.1038/s41419-021-03961-9
8. Tian L, Jin J, Lu Q, et al. Bidirectional modulation of extracellular vesicle-autophagy axis in acute lung injury: molecular mechanisms and therapeutic implications. Biomed Pharmacother.2024;180:117566.
9. Qiao X, Yin J, Zheng Z, Li L, Feng X. Endothelial cell dynamics in sepsis-induced acute lung injury and acute respiratory distress syndrome: pathogenesis and therapeutic implications. Cell Commun Signal. 2024;22(1):241. doi:10.1186/s12964-024-01620-y
10. Pons-Tostivint E, Lugat A, Fontenau JF, Denis MG, Bennouna J. STK11/LKB1 modulation of the immune response in lung cancer: from biology to therapeutic impact. Cells. 2021;10(11):3129. doi:10.3390/cells10113129
11. Long LL, Ma SC, Guo ZQ, et al. PARP inhibition induces synthetic lethality and adaptive immunity in LKB1-mutant lung cancer. Cancer Res. 2023;83(4):568–581. doi:10.1158/0008-5472.CAN-22-1740
12. Besse B, Pons-Tostivint E, Park K, et al. Biomarker-directed targeted therapy plus durvalumab in advanced non-small-cell lung cancer: a Phase 2 umbrella trial. Nat Med. 2024;30(3):716–729. doi:10.1038/s41591-024-02808-y
13. Proteau S, Krossa I, Husser C, et al. LKB1-SIK2 loss drives uveal melanoma proliferation and hypersensitivity to SLC8A1 and ROS inhibition [published correction appears in EMBO Mol Med. 2024;16(9):2262–2267]. EMBO Mol Med. 2023;15(12):e17719. doi:10.15252/emmm.202317719
14. Qian Y, Galan-Cobo A, Guijarro I, et al. MCT4-dependent lactate secretion suppresses antitumor immunity in LKB1-deficient lung adenocarcinoma. Cancer Cell. 2023;41:1363–80.
15. Shen X, He L, Cai W. Role of lipopolysaccharides in the inflammation and pyroptosis of alveolar epithelial cells in acute lung injury and acute respiratory distress syndrome. J Inflamm Res. 2024;17:5855–5869. PMID: 39228678; PMCID: PMC11370780. doi:10.2147/JIR.S479051
16. Wani A, Al Rihani SB, Sharma A, et al. Crocetin promotes clearance of amyloidβ by inducing autophagy via the STK11/LKB1-mediated AMPK pathway. Autophagy. 2021;17(11):3813–3832. doi:10.1080/15548627.2021.1872187
17. Fujisawa F, Kunimasa K, Kano-Fujiwara R, et al. STK11 loss drives rapid progression in a breast cancer patient resulting in pulmonary tumor thrombotic microangiopathy. Breast Cancer. 2021;28(3):765–771. doi:10.1007/s12282-020-01200-1
18. Feng Y, Chen Y, Wu X, et al. Interplay of energy metabolism and autophagy. Autophagy. 2024;20(1):4–14. doi:10.1080/15548627.2023.2247300
19. Choi EJ, Oh HT, Lee SH, et al. Metabolic stress induces a double-positive feedback loop between AMPK and SQSTM1/p62 conferring dual activation of AMPK and NFE2L2/NRF2 to synergize antioxidant defense. Autophagy. 2024;20(11):2490–2510. doi:10.1080/15548627.2024.2374692
20. Zhang Q, Feng J, Liu K, Yang X, Huang Y, Tang B. STK11 mutation impacts CD1E expression to regulate the differentiation of macrophages in lung adenocarcinoma. Immun Inflamm Dis. 2023;11(7):e958. doi:10.1002/iid3.958
21. Li X, Lakshmi SP, Uemasu K, et al. FBXL19 targeted STK11 degradation enhances cigarette smoke-induced airway epithelial cell cytotoxicity. COPD. 2024;21(1):2342797. doi:10.1080/15412555.2024.2342797
22. Han JH, Kim YK, Kim H, et al. Snail acetylation by autophagy-derived acetyl-coenzyme A promotes invasion and metastasis of KRAS-LKB1 co-mutated lung cancer cells. Cancer Commun. 2022;42(8):716–749. doi:10.1002/cac2.12332
23. Muniraj N, Siddharth S, Shriver M, et al. Induction of STK11-dependent cytoprotective autophagy in breast cancer cells upon honokiol treatment. Cell Death Discov. 2020;6:81. doi:10.1038/s41420-020-00315-w
24. Cui Y, Li Y, Meng S, Song Y, Xie K. Molecular hydrogen attenuates sepsis-induced cardiomyopathy in mice by promoting autophagy. BMC Anesthesiol. 2024;24(1):72. doi:10.1186/s12871-024-02462-4
25. Chung SJ, Nagaraju GP, Nagalingam A, et al. ADIPOQ/adiponectin induces cytotoxic autophagy in breast cancer cells through STK11/LKB1-mediated activation of the AMPK-ULK1 axis. Autophagy. 2017;13(8):1386–1403. doi:10.1080/15548627.2017.1332565
26. Bakula D, Mueller AJ, Proikas-Cezanne T. WIPI β-propellers function as scaffolds for STK11/LKB1-AMPK and AMPK-related kinase signaling in autophagy. Autophagy. 2018;14(6):1082–1083. doi:10.1080/15548627.2017.1382784
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Inhibition of miRNA-1-Mediated Inflammation and Autophagy by Astragaloside IV Improves Lipopolysaccharide-Induced Cardiac Dysfunction in Rats
Wang Q, Chen W, Yang X, Song Y, Sun X, Tao G, Wang H, Zhao N, Huang Y, Chai E, Tang F
Journal of Inflammation Research 2022, 15:2617-2629
Published Date: 23 April 2022
IFN-γ and LPS Induce Synergistic Expression of CCL2 in Monocytic Cells via H3K27 Acetylation
Akhter N, Kochumon S, Hasan A, Wilson A, Nizam R, Al Madhoun A, Al-Rashed F, Arefanian H, Alzaid F, Sindhu S, Al-Mulla F, Ahmad R
Journal of Inflammation Research 2022, 15:4291-4302
Published Date: 27 July 2022
The Role and Mechanism of Metformin in Inflammatory Diseases
Lin H, Ao H, Guo G, Liu M
Journal of Inflammation Research 2023, 16:5545-5564
Published Date: 23 November 2023
Capsaicin Attenuates LPS-Induced Acute Lung Injury by Inhibiting Inflammation and Autophagy Through Regulation of the TRPV1/AKT Pathway
Hu Q, Liu H, Wang R, Yao L, Chen S, Wang Y, Lv C
Journal of Inflammation Research 2024, 17:153-170
Published Date: 9 January 2024
Quercetin: A Flavonoid with Potential for Treating Acute Lung Injury

Huang M, Liu X, Ren Y, Huang Q, Shi Y, Yuan P, Chen M
Drug Design, Development and Therapy 2024, 18:5709-5728
Published Date: 6 December 2024