Back to Journals » Cancer Management and Research » Volume 10
STAT1 inhibits STAT3 activation in esophageal squamous cell carcinoma
Authors Liu Z, Zhang Y, Chen Y, Lin Y, Lin Z, Wang H
Received 31 July 2018
Accepted for publication 4 November 2018
Published 29 November 2018 Volume 2018:10 Pages 6517—6523
DOI https://doi.org/10.2147/CMAR.S182105
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Harikrishna Nakshatri
Zhaoyong Liu,1,* Ying Zhang,2,* Yelong Chen,1 Youbin Lin,1 Zhen Lin,2 Hu Wang1
1Department of Orthopaedics, The First Affiliated Hospital of Shantou University Medical College, Shantou, Guangdong, China; 2Department of Pathology, Shantou University Medical College, Shantou, Guangdong, China
*These authors contributed equally to this work
Background: Signal transducer and activator of transcription (STAT) 1 is an important transcription factor and has been reported to be a tumor suppressor in many types of cancer. However, another STAT family member, STAT3, is considered to be an oncogene. The cross-talk between STAT1 and STAT3 in cancer has not been fully demonstrated.
Materials and methods: Esophageal squamous cell carcinoma (ESCC) was used as a model to examine STAT1-STAT3 cross-regulation in cancer. We detected STAT1-STAT3 binding by co-immunoprecipitation (co-IP) and measured the transcription activity by using a luciferase reporter gene. DNA binding was detected by a DNA probe. Expression of STAT1 and STAT3 in ESCC was detected by immunohistochemistry.
Results: We found that STAT1 attenuated STAT3 activity upon oncostatin M treatment by decreasing STAT3 transcription activity and DNA binding ability of STAT3. Furthermore STAT3 downregulation increased the phosphorylation and transcriptional activation of STAT1. Finally, STAT1 expression and STAT3 expression were negatively correlated in ESCC cases.
Conclusion: Altogether, this paper demonstrated STAT1 and STAT3 cross-regulation in ESCC and proposed that STAT3 downregulation and/or STAT1 accumulation may be a therapeutic approach to treat ESCC.
Keywords: STAT1, STAT3, esophageal squamous cell carcinoma, transcription activity
Introduction
Esophageal squamous cell carcinoma (ESCC) comprises 90% of all cases of esophageal cancer (EC) in China. Among all countries, China has the highest incidence of EC worldwide. However, the incidence of EC is regionally distributed throughout the nation. Specifically, the Chaoshan region has been a well-known high-risk area for EC. In this region, the annual average age-standardized incidence of EC is 74.5/100,000 people, which is ten times the global average of EC per capita.1 These findings suggest the possibility of potential genetic and/or environmental factors predisposing this population to EC.
Signal transducer and activator of transcription 1 (STAT1) mediates several cell functions in response to stimulation by cytokines, growth factors, and hormones, such as interferons (IFNs), interleukin (IL)-6, epidermal growth factor, vascular endothelial growth factor, and growth hormone.2 The essential role of STAT1 in IFN signaling has been demonstrated in multiple review papers.3,4 In our previous research, we found that STAT1 plays a tumor suppressor role in ESCC and that the constitutively activated STAT1 can induce apoptosis and inhibit cell growth in ESCC cells. ESCC patients with high STAT1 expression have a better survival than those with low STAT1 expression, with the underlying mechanisms involving inhibition of STAT3 activation and NF-kB pathway.5
STAT3, another STAT family member, performs both pro-apoptotic and pro-survival functions in normal cells and regulates genes that are involved in cell proliferation, cell death, and immunity.6 Previous studies have revealed that STAT3 can function as an oncogene, suggesting that STAT3 has an opposite role to that of STAT1.7 Cross-regulation of STAT1 and STAT3 has been demonstrated in several reports,4,8–10 including ours, which suggests that STAT1 activation can be regulated by STAT3. However, the mechanisms have not been clearly demonstrated. The main purpose of this study is to delineate the mechanisms by which STAT1 inhibits STAT3 activation and by which cross-talk occurs between STAT1 and STAT3 in ESCC.
Materials and methods
Patient samples
This study included 30 randomly selected patients with primary ESCC who underwent radical esophageal resection at the Shantou Cancer Hospital from 2010 to 2017. None of the patients received preoperative radiotherapy or chemotherapy. Among the patients, 24/30 (80%) were males and 6/30 (20%) were females. The median age was 57.9 years (range, 37–75 years). This study was approved by the ethical review committee of the Medical College of Shantou University. All participants involved in our study provided written informed consent.
Cell cultures and stable cell line generation
Human ESCC cell lines EC1 and EC109 were purchased from the Chinese Academy of Sciences Cell bank and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1× antibiotic mixture (Invitrogen, Carlsbad, CA). Generation of the STAT3C tetracycline (tet)-off stable cell clones was performed using the Tetracycline-Off system (Clontech, Palo Alto, CA, USA) according to the manufacturer’s instructions. Briefly, EC109 cells were first transfected with the pTet-Off vector that expresses the tetracycline-controlled transactivator, and stable cell clones were selected by exposure to 400 µg/mL Geneticin (Invitrogen). Then, the stable clones were transfected with pTRE2hyg-STAT3C vectors, and double stably transfected cell clones were selected using media containing 400 µg/mL hygromycin B (Invitrogen). These cells were cultured in DMEM supplemented with 10% FBS.
Western blot analysis
Western blot was performed as described previously.5 Antibodies against human β-actin (1:1000), STAT1 (1:1000), p-STAT1 (1:1000), STAT3 (1:1000), p-STAT3 (1:1000), and Flag (1:1000) were purchased from Cell Signaling Technology (Danvers, MA).
Immunohistochemistry (IHC)
IHC staining was performed using the Envision Labeled Peroxidase System (Dako, Carpinteria, CA) as described previously.11 IHC staining was examined by two pathologists who were blinded to the clinical outcome, and a high degree of concordance between two pathologists was indicated by an inter-rater agreement kappa value of 0.87. For STAT1 and STAT3 immunostaining, both the intensity and percentage of immunostained cells were evaluated. The percentages of positive-stained cells were assigned the following scores: 0 (<5% positive cells), 1 (6%–25% positive cells), 2 (26%–50% positive cells), 3 (51%–75% positive cells), or 4 (>75% positive cells). The staining intensity was scored on a scale of 0–3 as follows: 0, negative; 1, buff; 2, yellow; and 3, brown. The percentage of positive cells and the staining intensities were then multiplied to generate the immunoreactivity score for each case. Overall staining scores from 0 to 2, 3–6, and ≥7 were considered negative, weak, and strong, respectively. The weak and negative expressions were considered low, and the strong expression was considered high.
Chemicals, plasmid constructs, and transfection
Dimethyl sulfoxide and oncostatin M (OSM) were purchased from Sigma (St. Louis, MO). Constitutively active FLAG-tagged STAT1C cloned into the pcDNA3.1 vector was a gift from Dr Ouchi (University of New York). For each experiment, 1×106 ESCC cells were transiently transfected with 10 µg of STAT1C vector or empty vector in six-well plates, using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s suggested protocol.
Co-immunoprecipitation and immunoprecipitation
To detect the interaction between p-STAT3 and p-STAT1, whole-cell extracts were prepared by lysing the cells in immunoprecipitation buffer. Western blot analysis was then performed using standard techniques as previously described.5
Luciferase activity assay
STAT3 and STAT1 transcriptional activity analyses were performed. Transcription activity was measured by a luciferase reporter gene assay,12 using the Dual-Luciferase reporter assay system according to the manufacturer’s instructions (Promega Corporation, Madison, WI, USA).
Subcellular fractionation and DNA binding assays
Nuclear and cytoplasmic protein of ESCC cells was extracted using an NE-PER protein extraction kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions as described previously.12 Oligonucleotide pull-down assays were performed with an annealed nucleotide comprising the STAT3 or STAT1 consensus site with a biotin label.
Quantitative RT-PCR
Using an RNeasy Mini Kit (Qiagen, Valencia, CA, USA), total cellular RNA was extracted from cells, and quantitative RT-PCR was performed. Primers for human IFN regulatory factor 1 (IRF1), human chemokine (C-X-C motif) ligand 10 (CXCL10), guanylate binding protein 2 (GBP2), intercellular adhesion molecule 10 (ICAM10), and transporter associated with antigen processing 1 (TAP1) were previously described.12
Colony formation assay
After STAT1C or empty vector transfection, doxycycline and control were added after 500 cells/well were plated in six-well plates and incubated for 10 days at 37°C. Cells were fixed with 4% buffered formalin for 15 minutes and then stained with 1% crystal violet (Sigma Aldrich) for 30 minutes. The plates were gently washed with PBS and dried before microscopic evaluation. Cell clusters with >30 cells were considered as a colony.
Immunofluorescence and confocal microscopy
Immunofluorescence was performed according to the manual. Cells were grown on cover slips coated with poly-l-lysine (Sigma Aldrich) in a six-well plate and fixed with 3% paraformaldehyde in PBS (pH 7.4). Cells were rinsed three times with PBS, permeabilized with Triton X-100, washed again with PBS, and incubated with 200 µL of anti-STAT1 and anti-STAT3 antibodies (1:50, Sigma Aldrich) overnight at room temperature in a humidified chamber. Cover slips were rinsed three times in PBS and incubated with secondary antibody conjugated with Alexa Fluor 488 or 568 (Invitrogen) at a 1:250 dilution for 1 hour at room temperature. After three rinses in PBS, coverslips were mounted on a slide using mounting media (Dako, Carpinteria, CA, USA). Cells were visualized with a Zeiss LSM 710 confocal microscope (Carl-Zeiss Company, Jena, Germany).
Statistical analysis
Data are expressed as the mean ± SD of at least three separate experiments. P<0.05 was considered as statistically significant. Differences among the treatment groups were assessed by ANOVA using statistical software (SPSS, IBM, Armonk, NY, USA).
Results
STAT1 downregulates STAT3 activation in ESCC cells
STAT1 was reported to decrease the expression of STAT3 after STAT1C transfection into ESCC cells, and siRNA knockdown of STAT1 substantially increases the expression level of STAT3 and p-STAT3. The STAT1C-induced change of STAT1 and STAT3 expression prompted us to further examine the interplay between STAT1 and STAT3 in ESCC cells. Enforced expression of STAT1C diminished STAT3 tyrosine 705 (Y705) phosphorylation upon OSM stimulation, as compared with cells transfected with empty vector (Figure 1A). Specifically, p-STAT3 (Y705) was maintained at an elevated level for at least 120 minutes after OSM exposure in empty vector-transfected cells, but significantly decreased and diminished to a faint level at 60 minutes in STAT1C-transfected cells (Figure 1A). Transfection efficiency of STAT1C was confirmed by the protein expression of p-STAT1 (Y701) and total STAT1.
 | Figure 1 STAT1 downregulates STAT3 activation in ESCC cells. Notes: (A) Protein expression was measured using Western blots in cells treated with OSM for various times. P-STAT3(Y705), STAT3, p-STAT1(Y701), and STAT1 were measured, and β-actin was used as the loading control. *P<0.05. (B) Binding of p-STAT1 to p-STAT3 was detected by co-immunoprecipitation. (C) Co-localization of STAT1 and STAT3 was detected by immunofluorescence (scale bar: 10 µm). (D) STAT3 luciferase reporter activity was measured 48 hours after transfection. **P<0.01. (E) STAT3 DNA-binding ability was determined using a pull-down assay with a STAT3 probe. Abbreviations: STAT, signal transducer and activator of transcription; ESCC, esophageal squamous cell carcinoma. |
Reciprocal co-immunoprecipitation experiments showed that p-STAT1 could form heterodimers with p-STAT3, and this interaction was enhanced by OSM stimulation (Figure 1B), suggesting that p-STAT1 and p-STAT3 could interact with each other. Specifically, upon OSM stimulation, p-STAT3 translocated to the nucleus and overlapped with the nuclear STAT1. Moreover, the co-localization of STAT3 and STAT1 was confirmed by confocal microscopy (Figure 1C). To further determine an interaction between STAT1 and STAT3, it was asssessed whether STAT1C affects transcription activation by STAT3. Transfection of STAT1C into cells expressing a STAT3 reporter gene dramatically decreased the transcriptional activity of STAT3 compared with the negative control (P<0.01). Comparable results were observed when cells were stimulated by OSM (Figure 1D).
Previous results led us to ask whether decreased STAT3 transcriptional activity induced by STAT1C was caused by a reduction in STAT3 DNA binding. Pull-down experiments were performed using a probe containing a STAT3 consensus DNA-binding site. STAT1C markedly diminished the DNA binding of STAT3 when compared with the negative control (Figure 1E). These results support previous findings that activated STAT1 can downregulate STAT3 activity.
Knockdown of STAT3 increases STAT1 activation in ESCC cells
To further confirm the STAT1-STAT3 interaction in ESCC cells, a STAT3C tet-off stable cell was generated by using EC109 cells, which do not express STAT1. As shown in Figure 2A, p-STAT3 and STAT3 expression decreased in a dose-dependent manner according to the dose of doxycycline. These results demonstrated that at 24 hours after induction, STAT1C transfection and p-STAT1 expression gradually increase with doxycycline treatment. However, the control group did not express STAT1.
 | Figure 2 Knockdown of STAT3 increases STAT1 activation in ESCC cells. Notes: (A) Protein expression was measured using Western blots in EC109 tet-off cells treated with STAT1C transfection at different doses of doxycycline. Flag, p-STAT3(Y705), STAT3, p-STAT1(Y701), and STAT1 were measured, and β-actin was used as the loading control. (B) STAT1 luciferase reporter activity was measured 48 hours after adding doxycycline, and Western blots were performed in parallel. (**P<0.01, *P<0.05). (C) STAT1 DNA-binding activity was determined using a pull-down assay with a STAT1 probe. (D) The mRNA expression levels of STAT1 downstream genes, IRF1, TAP1, GBP2, ICAM1, and CXCL10, were measured by qRT-PCR. (E) Correlation of STAT1/STAT3 with colony formation. STAT1C or empty vector was transfected, and then cells were treated with doxycycline or control. Then, the cells were cultured for 10 days. Similar results were observed in three independent experiments (*P<0.05). Abbreviations: EV, empty vector; doxy, doxycycline; tet, tetracycline; STAT, signal transducer and activator of transcription; ESCC, esophageal squamous cell carcinoma. |
Furthermore, to detect STAT1 activation in STAT3 tet-off ESCC cells, the DNA binding ability and transcription activation of STAT1 after adding doxycycline were assessed. Using a STAT1 luciferase reporter, the effect of STAT3 on the transcriptional activity of STAT1 in ESCC cell lines was assessed. As shown in Figure 2B, STAT3 knockdown increased the transcriptional activity of STAT1 (P<0.05) as compared to that of empty vector. Again, to test whether the increased transcriptional activity of STAT1 mediated by STAT3 transfection was caused by an increase in STAT1-DNA binding, a pull-down experiment using a biotinylated probe containing the STAT1 DNA-binding consensus sequence was performed. Decreased STAT3 gradually enhanced the DNA binding of STAT1 when compared with the negative control (Figure 2C).
To confirm the effect of STAT3 on transcription of the STAT1 gene, quantitative RT-PCR was performed. EC109 tet-off cells were treated with doxycycline and transfected with STAT1C, and mRNA expression levels of several known STAT1 downstream targets (including IRF1, TAP1, CXCL10, GBP2, and ICAM10) were analyzed. The results are illustrated in Figure 2D. Compared to the control, STAT3 downregulation at 24 hours significantly increased the mRNA expression of all five target genes examined. The STAT1/STAT3 correlation was examined in a colony formation assay (Figure 2E). STAT1C or doxycycline treatment diminished the clonogenic ability of ESCC cells (P<0.05). These results suggest that STAT3 downregulation could increase STAT1 activation.
Correlation of STAT1 and STAT3 expression in ESCC
Results from in vitro studies led us to hypothesize that expression of STAT3 may inversely correlate with that of STAT1 in ESCC tumors. By performing IHC analysis on a series of paraffin-embedded tumor samples, the expression of STAT3 and STAT1 in 30 case-matched benign esophageal tissues and tumors was assessed (Figure 3A). STAT1 and STAT3 expression was found in both nuclei and cytoplasm, and the stain intensity was categorized into high and low (including negative). STAT1-positive immunoreactivity was found in 26 (86.7%) cases of benign esophageal epithelial tissues and 20 (66.7%) ESCC tumors. In 20 positive STAT1-expressed ESCC samples, 10 tumors expressed weak STAT1 levels, whereas 10 tumors expressed strong levels of STAT1. Thus, 10 cases were considered as STAT1-high and 20 cases were STAT1-low. In five benign esophageal epithelial tissues, no STAT3 was detected, but all tumors had positive STAT3 staining, with 23 tumors being high and seven being low (Figure 3A).
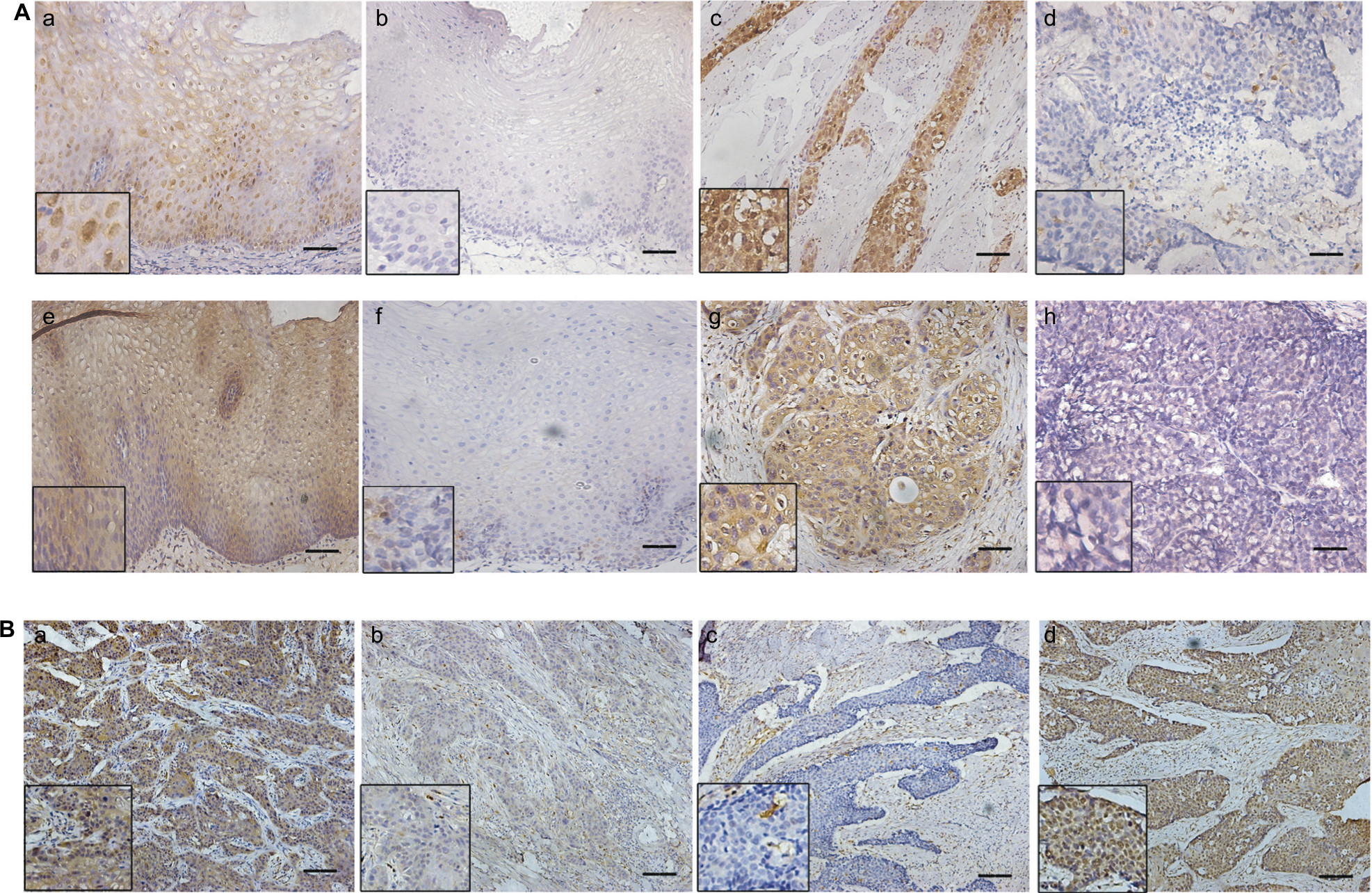 | Figure 3 Expression of STAT1 and STAT3 in 30 ESCC samples. Notes: (A) (a) STAT1-high expression in esophageal epithelial. (b) STAT1-low expression in esophageal epithelial. (c) STAT1-high expression in ESCC. (d) STAT1-low expression in ESCC. (e) STAT3-high expression in esophageal epithelial. (f) STAT3-low expression in esophageal epithelial. (g) STAT3-high expression in ESCC. (h) STAT3-low expression in ESCC. (B) In the same ESCC sample (a,b), STAT1 expression is high (a) and STAT3 expression is low (b). In another ESCC sample, STAT1 expression is low (c) and STAT3 expression is high (d). Scale bar: 100 μm. Abbreviations: STAT, signal transducer and activator of transcription; ESCC, esophageal squamous cell carcinoma. |
In most cases (25/30), expression of STAT1 was higher in normal tissues than in tumor tissues, and STAT3 expression was higher in tumor tissues than in case-matched normal epithelial in 18/30 tissue. Moreover, STAT1 expression inversely correlated with STAT3 in ESCC tissues (Figure 3B, Table 1).
 | Table 1 Correlation between STAT1 and STAT3 in 30 ESCC cases Abbreviations: STAT, signal transducer and activator of transcription; ESCC, esophageal squamous cell carcinoma. |
Discussion
It has been previously shown that STAT1 is a tumor suppressor in ESCC and that constitutively activated STAT1(STAT1C) can induce apoptosis and inhibit cell invasion. Moreover, most (75.4%) ESCC samples examined showed downregulation of STAT1 as compared to case-matched, benign epithelial tissues adjacent to the tumors. Patients with low STAT1-expressing tumors always had poor prognosis.5 The mechanism by which STAT1 inhibited cell growth involves inhibition of STAT3 phosphorylation and its expression. Based on a previous study5 which showed that STAT1 can interact with STAT3 and decrease its expression in ESCC cells, we tested the mechanisms by which STAT1 inhibits STAT3 activation.
STAT1 is the first member discovered in the family of STAT proteins, which was identified as the key mediator for type I and type II IFNs.13 STAT3 is another important key factor in the STATs family and is considered a tumor promoter. STAT3 plays an opposite role in that it promotes cell invasion and proliferation in tumor cells.7 STAT1 and STAT3 fulfill distinct roles in cancer cell proliferation, apoptosis, inflammation, and tumorigenesis. Specifically, STAT1 induces anti-proliferative and pro-apoptotic genes that directly hamper tumor growth, while STAT3 promotes cell survival and proliferation.3,6 Cross-regulation of STAT1 and STAT3 has been demonstrated in many papers thus, the expression and/or activity ratio of STAT1 and STAT3 constitute important mechanisms in controlling responses to cytokines and growth factors.9 For instance, STAT3 directly blocks the DNA binding of STAT1 in human gastric cancer cells,14 and IL-6 prolongs STAT1 activation and induces expression of multiple IFN-γ-dependent genes in STAT3-deficient MEF cells.15 Investigators have also suggested that reciprocal regulation between STAT1 and STAT3 might exist in tumor cells and that the ratio of STAT1 and STAT3 is a determinant of cancer progression.8,9 It is similar to results found for colorectal cancer, whereby colorectal cancer cells with high STAT1 and low STAT3 levels grow slower in xenografts than other types of cells, and the ratio of STAT1 and STAT3 reflects the clinical outcome of colorectal cancer patients.16
The mechanisms by which STAT1 and STAT3 oppose each other have not been clearly demonstrated till now. Previous research has suggested that STAT1 and STAT3 may compete for the same receptor docking sites, target gene promoters, and other cofactors.17 Another possible explanation involves heterodimers of STAT1 and STAT3, which are transcriptionally inactive or reduced. In this paper, STAT1 inhibits STAT3 DNA binding ability and the expression of downstream genes. Based on the previous results, activated STAT1 can promote the formation of STAT1-STAT3 heterodimers and inhibit STAT3 phosphorylation and expression. This extensively demonstrated the mechanisms underlying the STAT1-STAT3 cross-talk.
Expression of STAT1 and STAT3 in the same cohort of ESCC patient samples was detected to support our in vitro results. We showed that STAT1 expression was significantly higher in normal tissues than that in case-matched cancer tissues, which is consistent with our previous results.5 The expression of STAT3 was higher in ESCC tissues than in case-matched normal tissues in most cases. The results further showed that STAT3 and STAT1 expression in ESCC tissues was negatively correlated with each other, supporting the hypothesis that STAT1 could inhibit STAT3 expression and activation in ESCC.
Taken together, this paper reported that STAT1 can inhibit STAT3 expression and activation. Furthermore, STAT3 downregulation results in elevating the expression and activation of STAT1, a tumor suppressor in ESCC, which may provide as a new therapeutic approach to treat ESCC by targeting both STAT1 and STAT3.
Acknowledgment
This work was supported by Natural Science Foundation of Guangdong (2018A030310219).
Disclosure
The authors report no conflicts of interest in this work.
References
Su M, Liu M, Tian DP, et al. Temporal trends of esophageal cancer during 1995-2004 in Nanao Island, an extremely high-risk area in China. Eur J Epidemiol. 2007;22(1):43–48. | ||
Kim HS, Lee MS. STAT1 as a key modulator of cell death. Cell Signal. 2007;19(3):454–465. | ||
Meissl K, Macho-Maschler S, Müller M, Strobl B. The good and the bad faces of STAT1 in solid tumours. Cytokine. 2017;89:12–20. | ||
Gujam FJ, Mcmillan DC, Edwards J. The relationship between total and phosphorylated STAT1 and STAT3 tumour cell expression, components of tumour microenvironment and survival in patients with invasive ductal breast cancer. Oncotarget. 2016;7(47):77607–77621. | ||
Zhang Y, Molavi O, Su M, Lai R. The clinical and biological significance of STAT1 in esophageal squamous cell carcinoma. BMC Cancer. 2014;14:791. | ||
Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736–746. | ||
Chai EZ, Shanmugam MK, Arfuso F, et al. Targeting transcription factor STAT3 for cancer prevention and therapy. Pharmacol Ther. 2016;162:86–97. | ||
Dimberg LY, Dimberg A, Ivarsson K, et al. Stat1 activation attenuates IL-6 induced Stat3 activity but does not alter apoptosis sensitivity in multiple myeloma. BMC Cancer. 2012;12:318. | ||
Friedrich K, Dolznig H, Han X, Moriggl R. Steering of carcinoma progression by the YIN/YANG interaction of STAT1/STAT3. Biosci Trends. 2017;11(1):1–8. | ||
Han W, Carpenter RL, Cao X, Lo HW. STAT1 gene expression is enhanced by nuclear EGFR and HER2 via cooperation with STAT3. Mol Carcinog. 2013;52(12):959–969. | ||
Zhang Y, Wang H, Bi C, Xiao Y, Liu Z. Expression of CDX2 in gastric cardia adenocarcinoma and its correlation with H. pylori and cell proliferation. Oncotarget. 2016;7(34):54973–54982. | ||
Zhang Y, Chen Y, Yun H, Liu Z, Su M, Lai R. STAT1β enhances STAT1 function by protecting STAT1α from degradation in esophageal squamous cell carcinoma. Cell Death Dis. 2017;8(10):e3077. | ||
Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23(2):96–101. | ||
Ehret GB, Reichenbach P, Schindler U, et al. DNA binding specificity of different STAT proteins. Comparison of in vitro specificity with natural target sites. J Biol Chem. 2001;276(9):6675–6688. | ||
Costa-Pereira AP, Tininini S, Strobl B, et al. Mutational switch of an IL-6 response to an interferon-gamma-like response. Proc Natl Acad Sci U S A. 2002;99(12):8043–8047. | ||
Nivarthi H, Gordziel C, Themanns M, et al. The ratio of STAT1 to STAT3 expression is a determinant of colorectal cancer growth. Oncotarget. 2016;7(32):51096–51106. | ||
Zhang Y, Liu Z. STAT1 in cancer: friend or foe? Discov Med. 2017;24(130):19–29. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.