Back to Journals » International Journal of Nanomedicine » Volume 17
Spermidine Crosslinked Gellan Gum-Based “Hydrogel Nanofibers” as Potential Tool for the Treatment of Nervous Tissue Injuries: A Formulation Study
Authors Vigani B, Valentino C, Sandri G ![]() , Caramella CM, Ferrari F
, Caramella CM, Ferrari F ![]() , Rossi S
, Rossi S ![]()
Received 1 April 2022
Accepted for publication 26 June 2022
Published 2 August 2022 Volume 2022:17 Pages 3421—3439
DOI https://doi.org/10.2147/IJN.S368960
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Barbara Vigani, Caterina Valentino, Giuseppina Sandri, Carla Marcella Caramella, Franca Ferrari, Silvia Rossi
Department of Drug Sciences, University of Pavia, Pavia, Italy
Correspondence: Silvia Rossi, Department of Drug Sciences, University of Pavia, Viale Taramelli 12, Pavia, 27100, Italy, Tel +39 0382987357, Fax +39 0382422975, Email [email protected]
Purpose: Aim of the work was to develop a potential neural scaffold, endowed with neuroprotective and neuroregenerative potential, to be applied at the site of nervous tissue injuries: nanofibers, consisting of gellan gum (GG), spermidine (SP) and gelatin (GL), were prepared via electrospinning. SP was selected for its neuroprotective activity and cationic nature that makes it an ideal GG cross-linking agent. GL was added to improve the scaffold bioactivity.
Methods: Mixtures, containing 1.5% w/w GG and increasing SP concentrations (0– 0.125% w/w), were prepared to investigate GG/SP interaction and, thus, to find the best mixture to be electrospun. Mixture rheological and mechanical properties were assessed. The addition of 0.1% w/w GL was also investigated. The most promising GG/SP/GL mixtures were added with poly(ethylene oxide) (PEO) and poloxamer (P407) and, then, electrospun. The resulting fibers were characterized in terms of size and mechanical properties and fiber morphology was observed after soaking in water for 24 hours. Nanofiber biocompatibility was assessed on Schwann cells.
Results: More and more structured GG/SP mixtures were obtained by increasing SP concentration, proving its cross-linking potential. After blending with PEO and P407, the mixture consisting of 1.5% w/w GG, 0.05% w/w SP and 0.1% w/w GL was electrospun. The resulting nanofibers appeared homogenous and characterized by a plastic behavior, suggesting a good mechanical resistance when applied at the injury site. Nanofibers were insoluble in aqueous media and able to form a thin gel layer after hydration. GG/SP/GL nanofibers showed a higher compatibility with Schwann cells than GG/SP ones.
Conclusion: SP and GL allowed the production of homogenous GG-based nanofibers, which preserved their structure after contact with aqueous media and showed a good compatibility with a neural cell line. After local application at the injury site, nanofibers should support and guide axonal outgrowth, releasing SP in a controlled manner.
Keywords: nervous tissue injuries, electrospinning, hydrogel nanofibers, gelatin, Schwann cells
Graphical Abstract:

Introduction
Over the last few decades, the electrospinning has been recognized as a valuable technique that enables the production of nanofibrous scaffolds for different biomedical purposes, among which the restoration of the broken neural circuitry at the site of nervous tissue injuries.1–3 Such lesions, caused in most cases by mechanical traumas, represent a challenging clinical issue worldwide and, nowadays, the treatments are often ineffective or provide only a partial functional recovery.4,5 Nervous defects and injuries may involve the central nervous system (CNS), specifically the spinal cord, and/or the somatic and autonomic components of the peripheral nervous one (PNS).6 The neurons of CNS and PNS differently respond to axotomy; the difference in CNS and PNS regenerative potential may be explained by the difference in neuroglia composition. In PNS, the presence of Schwann cells provides, at the site of injury, a pro-regenerative milieu by maintaining a constant nutrient supply to nervous tissue, myelinating nerve fibers, secreting neurotrophins and producing extracellular matrix (ECM) components. On the counter part, in CNS, axons do not regenerate in their native environment: astrocytes participate in the formation of a dense scar that circumscribes the site of damage and oligodendrocytes express inhibitory factors, which hinder axonal outgrowth.5,7,8
Despite the capacity of peripheral nerves to regenerate after damage, in the most severe cases, a combined therapeutic approach, ensuring both a neuroprotective and neuroregenerative effect, is required to restore the cavitary defects formed in nervous tissue. In the literature, a huge number of research articles have been published regarding the successful use of electrospun nanofibers as scaffolds for neural tissue applications.4,9–13
Once applied at the nervous tissue injury, nanofibers, consisting of biocompatible and biodegradable polymers, provide a bioinspired three-dimensional pro-regenerative environment, filling the lesion gap, supporting and guiding axonal outgrowth and, thus, promoting a proper functional recovery.14,15 The versatility of the electrospinning technique allows to design and obtain fibers with optimized architectural and mechanical features for the intended therapeutic use;16 in particular, it has been demonstrated that fiber size, texture, density and orientation could be affected by several parameters, related to the process (spinneret-collector distance, applied voltage and flow rate), to the collector geometry and, obviously, to the polymeric solution or blend to be electrospun.17–19
In a previous work of ours, a gellan gum (GG)-based composite system, loaded with a drug candidate with neuroprotective effect (RC-33), was developed for the local treatment of nervous tissue injuries. The design of the system, consisting of nanofibers embedded in a freeze-dried RC-33-loaded matrix, was functional to fill the lesion gap, control drug release and support axonal regrowth. Nanofibers, representing the inner structure of the composite system, were prepared by electrospinning a solution composed of GG, two different grades of poly(ethylene oxide) (PEO) and poloxamer; after cross-linking with calcium ions, the fibrous mat was soaked in a GG-RC-33 solution and, then, freeze-dried.20
Based on the experience gained with the electrospinning of GG-based solutions, the present work deals with the development of a fibrous scaffold to be implanted at the site of nervous tissue injuries. It consists of electrospun nanofibers based on GG, an attractive anionic polysaccharide in neural applications,21–27 and spermidine (SP), a biologic polyamine. SP was used as multifunctional agent for the preparation of nanofibers. In fact, SP cationic nature at physiological pH makes it an ideal cross-linking agent for anionic polysaccharides, such as GG. Moreover, SP is characterized by a neuroprotective activity: it preserves neurons from oxidative damage and modulates the over-expression of pro-inflammatory cytokines at the injury site.24,28
GG/SP mixtures, containing a fixed GG amount and increasing SP concentrations, were prepared and characterized to identify the GG/SP mixture most suitable for the preparation of the polymeric solutions to be electrospun. Due to the challenging GG electrospinnability, the GG/SP mixture selected for the prosecution of the work was blended with PEO, a spinning-enhancer synthetic polymer, and poloxamer, a surfactant agent. The addition of gelatin (GL) as bioactive molecule was also investigated to obtain electrospun nanofibers endowed with a greater compatibility with nervous cells and, thus, a higher capability to enhance cell attachment and colonization after in vivo application.29,30
To the best of our knowledge, this is the first time that nanofibers, containing GG cross-linked with SP, were prepared by the electrospinning technique. When nanofibers come into contact with aqueous fluids, the water-soluble synthetic fiber components (PEO and poloxamer) should dissolve, while GG/SP/GL, forming physical hydrogels, should preserve the original fibrous structure. Therefore, after application at the injury site, the resulting GG/SP/GL hydrogel nanofibers should support and guide axonal outgrowth and their degradation should be responsible for a SP controlled release.
Materials and Methods
Materials
Deacetylated gellan gum (GG, Gelrite®; Kelco Division of Merck & Co., Rahway, New Jersey, USA), spermidine trihydrochloride (SP; Sigma Aldrich, Milan, Italy), gelatin from porcine skin (GL, gel strength 90–110 g Bloom, Type A; Sigma Aldrich, Milan, Italy), poly(ethylene oxide) of high molecular weight (h-PEO, MW = 4000 kDa; Colorcon, Dartford, United Kingdom), poly(ethylene oxide) of low molecular weight (l-PEO, MW = 600 kDa; Sigma Aldrich, Milan, Italy) and Kolliphor P407 poloxamer (P407; Sigma Aldrich, Milan, Italy) were used for the preparation of the polymeric solutions to be electrospun.
For the experiments with Neuronal Schwann cells RT4-D6P2T (ATCC CRL-2768), the materials hereafter reported were used. Dulbecco’s Modified Eagle’s Medium (ATCC-30-2002) was purchased from LGC Standards S.r.L. (Milan, Italy), while inactivated Fetal Bovine Serum (FBS) from Biowest (Nuaillé, France). Phosphate Buffer Solution (PBS), antibiotic/antimycotic solution (100X; stabilized with 10,000 units penicillin, 10 mg streptomycin, and 25 g amphotericin B per mL) and trypsin–EDTA solution were purchased from VWR (Radnor, Pennsylvania, USA). Dimethyl sulfoxide (DMSO), MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) and Trypan Blue solution were purchased from Sigma-Aldrich (Milan, Italy).
Preparation of GG/SP Mixtures
Different GG/SP mixtures (GG/SP1-GG/SP6), containing 1.5% w/w GG and increasing concentrations of SP, were prepared in distilled water (Table 1). In details, 2% w/w GG solution, prepared at 60°C, was blended with different SP solutions according to 3:1 w/w ratio; each mixture was then maintained under vigorous magnetic stirring for 15 minutes at 60°C. A solution of pure GG, free of SP, was also prepared (Table 1).
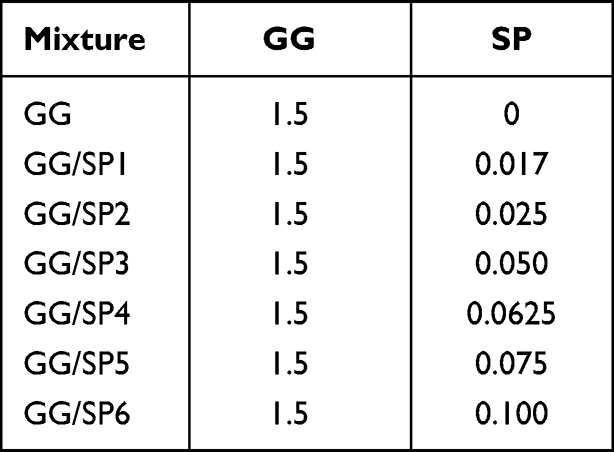 |
Table 1 Quali-Quantitative Composition of GG/SP Mixtures, Expressed as % w/w |
Characterization of GG/SP Mixtures
Rheological Analysis
Rheological analysis was carried out by means of a rotational rheometer (MCR 102, Anton Paar, Turin, Italy) equipped with a parallel-plate combination (PP50, ∅ = 50 mm) as measuring system. All the experiments were performed at 30°C and the gap was set at 0.5 mm; three replicates were considered for each sample.
Mixture viscosity was investigated in a wide shear rate interval (10–1000 s−1). In an attempt to evaluate, on a consistent basis, the increment of viscosity following the addition of increasing SP concentrations, the parameter ηGG/SP/ηGG was calculated for each GG/SP mixture at three different shear rates (50, 500 and 1000 s−1): at each value of shear rate considered, ηGG/SP and ηGG were the viscosity of GG/SP mixture and GG solution free of SP, respectively. Mixture viscoelasticity was investigated by dynamic oscillatory experiments, such as stress sweep test and oscillation test. In the stress sweep test, increasing stresses were applied at a constant frequency (1 Hz) and the elastic response of the sample, expressed as storage modulus G′, was measured as a function of stress; such a test allowed the identification of the linear viscoelastic region, characterized by constant G’ values on increasing stress. In the oscillation test, a shear stress, chosen in the linear viscoelastic region previously determined, was applied at increasing frequencies (1–20 Hz) and G’ (storage modulus) and G” (loss modulus) profiles were recorded. Loss tangent value (tgδ) was also calculated as G”/G’ for each GG/SP mixture at 10 Hz. Moreover, in an attempt to investigate the frequency dependence of G’ following the addition of increasing SP concentrations, the parameter ΔG’/G’1Hz was calculated for each GG/SP mixture, where ΔG’ was the difference between G’ measured at 20 Hz (G’20Hz) and G’ at 1 Hz (G’1Hz).
Texture Analysis
After rest for 24 hours at room temperature in a cylindrical mold, mixtures were subjected to a compression test by means of a TA.XT plus Texture Analyzer (Stable Micro Systems, Godalming, United Kingdom), equipped with a 1 kg load cell and a P/10 measuring system, consisting of a cylindrical probe with a diameter of 10 mm. Before testing, each sample was placed in a thermostatically controlled water bath at 30°C for 15 minutes. The probe was lowered with a test speed equal to 1.00 mm/s for a distance of 25 mm in order to determine an 80% sample deformation. Three replicates were carried out for each mixture; GG solution was considered as reference.
Compressive stress–strain curves were produced and the following parameters were considered: a) hardness that is the maximum compressive force (Fmax) per unit area required for sample destructuring; b) Young’s modulus (YM) that was calculated as the slope of the tangent at the first part of the compressive stress–strain curve (<15% of strain) in order to investigate the ability of each mixture to withstand changes in length when subjected to compression.
Preparation of the Polymeric Solutions for Electrospinning
GG/SP3 and GG/SP4 mixtures were added with 2.2% w/w poly(ethylene oxide) (PEO) at low molecular weight (l-PEO) and 0.5% w/w PEO at high molecular weight (h-PEO); poloxamer P407 was also added at the concentration of 2% w/w (Table 2). A polymeric solution free of SP (S), consisting of 1.5% w/w GG, 2.2% w/w l-PEO, 0.5% w/w h-PEO and 2% w/w P407, was prepared and used as reference. GG-containing polymeric solutions were prepared in distilled water and, then, maintained under magnetic stirring overnight at room temperature before electrospinning.
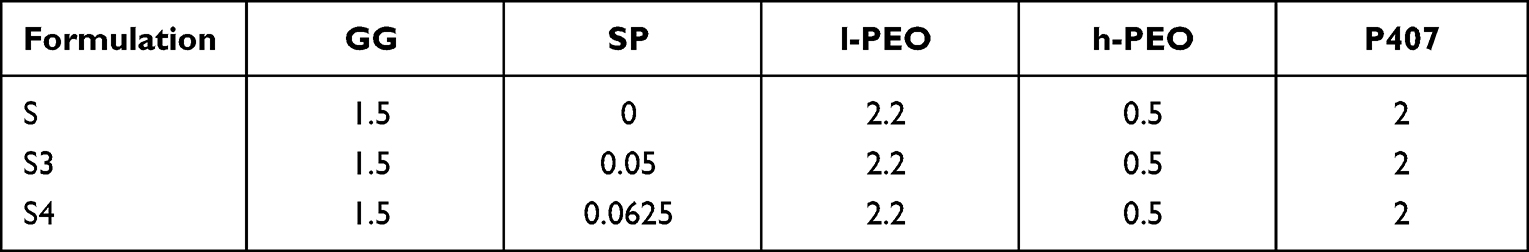 |
Table 2 Quali-Quantitative Composition of the Polymeric Solutions for Electrospinning, Expressed as % w/w |
The viscosity of all the polymeric solutions (S, S3 and S4) was assessed (see Supplementary Data).
GG/SP Nanofiber Preparation
All polymeric solutions were electrospun by using an electrospinning apparatus (STIKIT-40; Linari Engineering, Grosseto, Italy), equipped with a high-voltage power supply (Linari Engineering), a syringe pump (Razel Scientific, Saint Albans, VT, USA) and a collector plate (Linari Engineering); solutions were pumped at a rate of 0.8 mL/h through a 21 gauge needle with a length of 15 mm. Process parameters, such as spinneret–collector distance and applied voltage, were varied: the best electrospinning conditions were identified at 20 cm and 20–25 kV. The electrospinning process was performed at atmospheric pressure, maintaining temperature and relative humidity in a range of 30–35°C and 15–30%, respectively.
GG/SP Nanofiber Characterization
Morphological Properties
Fiber morphology was observed by means of a scanning electron microscope (Tescan Mira3 XMU, Brno, Czech Republic). Afterwards, fiber sizes were measured using the imaging analysis program ImageJ 2.0; thirty fibers were considered for each sample.
Moreover, GG/SP-containing fibers were soaked in distilled water for 24 hours and, then, dried at room temperature; a morphological investigation was carried out also on the resulting hydrated fibers.
Mechanical Properties: Resistance to Deformation
Fiber mechanical properties were assessed by means of a TA.XT plus Texture Analyzer, equipped with 5 kg load cell. Each sample was cut (1 × 3 cm) and then clamped on an A/TG tensile grips probe; an initial distance of 1 cm between the grips was set. The upper grip was raised at a constant speed of 10 mm/s for a distance of 50 mm. Before testing, fiber thickness was measured by means of a Sicutool 3955G-50 (Milan, Italy) apparatus. Tensile strength was calculated at different strain % (10%, 20%, 30%, 40% and 50%); three replicates were considered for each sample.
GG/SP/GL Nanofiber Preparation and Characterization
GG/SP3 was added with 0.1% w/w GL (GG/SP/GL); a GG/GL mixture containing 1.5% w/w GG and 0.1% w/w GL was also prepared. GG/SP/GL was subjected to rheological and texture analyses as previously described in the subsections “Rheological analysis” and “Texture analysis”.
Thereafter, GG/SP/GL mixture, containing 1.5% w/w GG, 0.05% w/w SP and 0.1% w/w GL, was added with 2.2% w/w l-PEO, 0.5% w/w h-PEO and 2% w/w P407 and, then, electrospun as described in the section “GG/SP nanofiber preparation”. The resulting nanofibers (S3-G) were characterized for their morphological and mechanical properties (see section “GG/SP nanofiber characterization”).
Moreover, GG/SP and GG/SP/GL nanofibers were analyzed for their water retention capacity. Fibers were cut into pieces having a surface area of approximately 1 cm2, weighted and layered on a dialysis membrane in the apical chamber of a Franz cell (PermeGear, Bethlehem, Palestine). The receptor chamber, thermostated at 37°C, was filled with phosphate buffer saline (PBS, pH 7.4), mimicking biological fluids. After 24 hours, fiber weight was measured, and the Water Retention Capacity (WRC) calculated according to the following equation:

where Wf was the weight of the hydrated fibers (hydrogel nanofibers), while Wi was the weight of the dried one. For each sample, three replicates were performed.
In vitro Cell Culture Experiments: Schwann Cells
The cytotoxicity of the electrospun fibers (S3 and S3-G) was assessed on Schwann cells from 4th to 7th passage. Briefly, cells (50,000 cells/cm2) were seeded in the basolateral chamber of Transwell® Permeable Supports (Corning Incorporated, Corning, New York, USA) for 24 h. After UV-irradiation, fibers were cut into pieces having a surface area of approximately 1 cm2 and, then, placed in the apical chamber for other 24 h with the addition of 0.5 mL of complete culture medium (CM; DMEM ATCC-30-2002 with 10% FBS and 1% antibiotic/antimycotic solution). After 1, 3 and 7 days of treatment, cell morphology and fiber topological structure were observed by an inverted microscope (VWR International, Milan, Italy) and an MTT assay was performed.
Briefly, apical chambers were removed from each well and the medium in the basolateral chamber was discarded; the cells were rinsed with PBS. Subsequently, an MTT 2.5 μM solution prepared in DMEM without phenol red was added to each well and incubated for 3 h (37°C and 5% CO2). Finally, MTT solution was removed and DMSO was added to promote the complete dissolution of formazan crystals, obtained from MTT dye reduction by mitochondrial dehydrogenases of living cells. The solution absorbance was measured by means of a multi-mode microplate reader (FLUOstar Omega, Bio-Rad Laboratories Inc., Hercules, California, USA) at 570 nm and 690 nm wavelengths after 60s of mild shaking. Results were expressed as cell viability % by normalizing the absorbance measured after contact with each sample with that measured for CM, used as reference. Three replicates were performed for each sample.
Statistical Analyses
Whenever possible, experimental values of the various types of measurements were subjected to statistical analysis, carried out by means of the statistical package Statgraphics 5.0 (Statistical Graphics Corporation, Rockville, Maryland, USA). In particular, one-way ANOVA was carried out followed by a Multiple Range Test.
Results
Characterization of GG/SP Mixtures
Rheological Analysis
Preliminary rheological and mechanical investigations were performed on GG/SP mixtures, consisting of a fixed GG amount (1.5% w/w) and increasing SP concentrations (0–0.1% w/w), to identify the GG/SP mixture most suitable to be used for the preparation of polymeric solutions to be electrospun. Both rheological and mechanical analyses were performed at 30°C, that was the temperature at which the electrospinning process was conducted.
Depending on the concentration of the cationic polyamine added to the negatively charged polymer, the appearance, color and firmness of the obtained GG/SP mixtures was quite different: in particular, an excess of SP was responsible for syneresis.29
Figure 1A shows the flow curves of GG/SP mixtures in comparison with a solution of pristine GG at the same concentration present in the mixtures (1.5% w/w). It can be observed that the presence of SP results in a significant increase in the viscosity of GG/SP mixtures. In particular, for concentrations of SP ≥ 0.025% w/w (from GG/SP2 onwards), a more and more pronounced “spur” is observed in the flow curves as the concentration of SP increases, indicating a strong mixture structuring due to SP cross-linking action. Moreover, it must be underlined that the flow curve of GG/SP6 is characterized by a high variability, due to a poor sample homogeneity; in fact, for such sample a marked syneresis phenomenon is observed. In order to assess on a homogeneous basis the increase in the viscosity due to the presence of SP, the ηGG/SP/ηGG parameter was calculated for each mixture. In Figure 1B, ηGG/SP/ηGG values calculated at three different shear rates (low, medium, and high) for all GG/SP mixtures are reported. It can be observed that the presence of SP produces a marked increase in the ηGG/SP/ηGG parameter. Depending on SP concentration, the viscosity of GG/SP mixtures is 2–10 times greater than the viscosity of the GG solution free of SP.
 |
Figure 1 (A) Flow curves of GG/SP mixtures containing a fixed GG amount (1.5% w/w) and increasing SP concentrations (0.017–0.1% w/w); a solution of pristine GG, free of SP, was used as reference. Data are expressed as mean ± SE (n = 3). (B) ηGG/SP/ηGG values calculated at three shear rates (50, 500, and 1000 s−1) for all the GG/SP mixtures. Data are expressed as mean ± SE (n = 3). ANOVA one-way – Multiple Range Test (p-value ≤ 0.05): avsc-f; bvsc-f; cvse-f; dvsf; a’vsb’-f’; b’vsf’; c’vsf’; d’vsf’; e’vsf’; a”vsc”-f”; b”vsc”-f”. |
Since it is well known that GG is able to form self-supporting gels, it seems interesting to evaluate how the presence of SP could affect the viscoelastic behavior of GG/SP mixtures. As an example, in Figure 2, the storage elastic modulus (G’) and the loss viscous modulus (G”) profiles of GG solution and GG/SP3 mixture are reported as functions of frequency. Both GG and GG/SP3 are characterized by a typical hydrogel behavior in the whole range of frequencies: in particular, the G moduli vs frequency curves of both samples reveal a prevalence of the elastic component on the viscous one (G’>G”) that appears more evident in presence of SP.
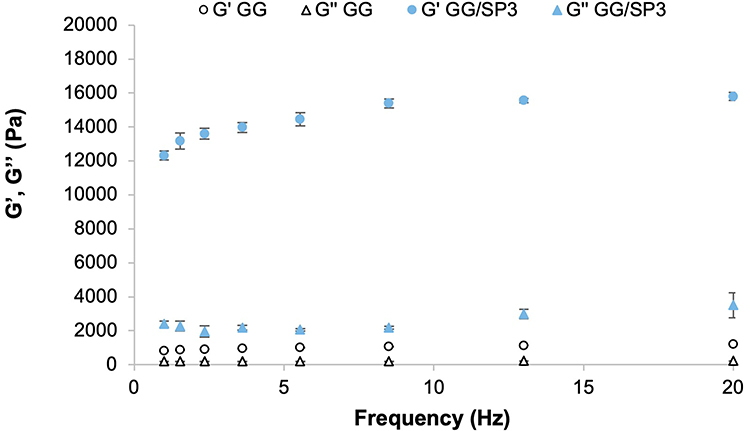 |
Figure 2 G’ and G” vs frequency profiles of 1.5% w/w GG solution in absence and in presence of 0.05% w/w SP. Data are expressed as mean ± SE (n = 3). |
A strict dependence of the mixture elasticity on the cationic polyamine concentration is also highlighted in Figures 3 and 4. The addition of increasing SP amount to the pristine GG solution is responsible for a progressive increase in G’ values in the whole frequency interval: at fixed GG amount, higher the SP concentration, greater is the number of cross-linking junctions, which can support the stress applied on the sample (Figure 3A and B). Moreover, all the samples (GG solution and GG/SP mixtures) possess loss tangent (tgδ) values lower than 1 at 10 Hz (Figure 3C). Since this parameter is calculated as the ratio G”/G’, values lower than 1 indicate a prevalence of the G’ over G”. In general, a decrease of tgδ is observed on increasing SP concentration, suggesting that the increase in SP concentration is responsible for a more pronounced prevalence of the elastic contribution of the sample to the viscous one.
 |
Figure 3 (A) Schematic representation of the ionic interactions between GG negatively charged chains and SP; the cross-linking junctions between GG chains and SP allows the formation of a three-dimensional network able to elastically respond to the applied stresses. (B) G’ modulus vs frequency profiles of GG/SP mixtures; GG solution free of SP was used as reference. Data are expressed as mean ± SE (n = 3). (C) Loss tangent (tgδ) values calculated at 10 Hz for all the GG/SP mixtures. Data are expressed as mean ± SE (n = 3). ANOVA one-way; Multiple Range Tests (p-value ≤ 0.05): avsc-g; bvsf-g; cvsg; dvsg; evsf-g; fvsg. |
 |
Figure 4 ΔG’/G’1Hz values calculated for all the GG/SP mixtures; GG solution was used as reference. Data are expressed as mean ± SE (n = 3). ANOVA one-way; Multiple Range Tests (p-value ≤ 0.05): avsd-g; bvsd-g; cvsd-g. |
Additionally, it is well known that a physical hydrogel, unlike a polymeric solution, is characterized by an elastic behavior that is independent from the frequency. Therefore, another rheological parameter, ΔG’/G’1Hz, was calculated in order to investigate the frequency dependence of G’ following the addition of increasing SP concentrations. Figure 4 shows that ΔG’/G’1Hz decreases on increasing SP concentration up to 0.05% w/w (GG/SP3), suggesting the formation of a gel-like structure due to the cross-linking effect of SP. For SP concentrations higher than 0.05% w/w, no significant differences are observed among ΔG’/G’ values.
Texture Analysis
Subsequently, the resistance to compression of each mixture was investigated through a compressive test by means of a texture analyzer; compressive stress–strain curves of GG solution and GG/SP mixtures are reported in Figure 5A. All the curves are characterized by two major portions: i) a linear region (where compressive stress is proportional to the applied strain), representing sample elastic deformation, for strain values lower than 20%, and ii) a curvilinear region (where compressive stress and strain are not characterized by any linear relationships), attributable to sample plastic deformation, for strain values higher than 20%. Contrary to elastic deformation, plastic deformation is irreversible since it occurs after gel failure that, in GG/SP5 and GG/SP6 mixtures, is also followed by water leaking through the gel-like structure.29 As shown in Figure 5A, the stress–strain curves of GG/SP5 and GG/SP6 mixtures are lower than GG/SP3 and GG/SP4 ones, suggesting their poor water holding capacity when exposed to external forces.
 |
Figure 5 (A) Compressive stress vs strain % curves of all the GG/SP mixtures are represented in comparison to GG solution. (B and C) Compressive parameters calculated for all the GG/SP mixtures: (B) hardness and (C) Young’s modulus; GG solution was used as reference. Below each graph, the SP concentration is reported as % w/w for each GG/SP mixture. Data are expressed as mean ± SE (n = 3). ANOVA one-way; Multiple Range Tests (p-value ≤ 0.05): (A) avsb-g, bvsc-g, cvsd-e, dvsf-g, evsf-g; (B) avsb-g, bvsc-g, cvsd-e, dvsf-g, evsf-g, fvsg. |
In an attempt to investigate the behavior of GG/SP mixtures when subjected to a uniaxial compressive force in more detail, two compressive parameters were determined: the hardness, that is the maximum compressive force required for gel failure, and the Young’s modulus, a parameter directly proportional to gel stiffness and calculated as the slope of the elastic region. As shown in Figure 5, GG/SP mixtures were characterized by two distinctive patterns of mechanical response below and above a critical SP concentration at which a maximum gel strength was recorded.30
Figure 5B and C suggest that the critical SP concentration was reached in GG/SP3 mixture (0.05% w/w); no significant differences can be appreciated between GG/SP3 and GG/SP4, in which SP concentration was equal to 0.0625% w/w. The addition of increasing SP concentrations beyond the critical value (>0.0625% w/w) was responsible for a decrease of both gel hardness (Figure 5B) and stiffness (Figure 5C).
The aforementioned results justify the use of GG/SP3 and GG/SP4 for the preparation of polymeric solutions to be electrospun. SP concentrations, present in GG/SP3 and GG/SP4 mixtures (0.05% and 0.0625% w/w, respectively) and recognized as critical, ensured that all the anionic groups on GG chains were occupied by cations, guaranteeing the maximum GG-SP interaction within the gel network.31,32 As evidenced by the rheological analysis above, SP concentrations <0.05% w/w exerted a mild cross-linking effect on GG chains (GG/SP1 and GG/SP2): not all the anionic sites of GG were occupied by SP and, thus, it could be assumed that such concentrations were not sufficient to guarantee the insolubility of GG-containing nanofibers after hydration in biologic fluids.
GG/SP Nanofiber Characterization
Morphological Properties
In an attempt to produce homogeneous nanofibers by electrospinning, GG/SP3 and GG/SP4 mixtures were added with a spinning adjuvant that was polyethylene oxide (PEO) at two different molecular weights, low (l-PEO) and high (h-PEO); moreover, a surfactant agent, poloxamer (P407), was added to the mixtures in order to reduce the surface tension of the polymeric solutions and, thus, to enhance the electrospinning process. The quali-quantitative composition of such solutions, named S3 and S4, was as follows: S3 consisted of 1.5% w/w GG, 0.05% w/w SP, 2.2% w/w l-PEO, 0.5% w/w h-PEO and 2% w/w P407, while S4 contained 1.5% w/w GG, 0.0625% w/w SP, 2.2% w/w l-PEO, 0.5% w/w h-PEO and 2% w/w P407. S solution derived from GG, namely a solution containing the same GG concentration but free of SP, was used as reference; it consisted of 1.5% w/w GG, 2.2% w/w l-PEO, 0.5% w/w h-PEO and 2% w/w P407. All the three polymeric solutions (S, S3 and S4) were electrospun maintaining 20 cm between spinneret and collector (plate geometry), applying a voltage equal to 20 kV and setting a flow rate of 0.793 mL/h. Such process parameters were identified as necessary to produce fibers from all the polymeric solutions considered in the present work.
Subsequently, the resulting fibers were characterized in terms of morphological properties: as reported in Figure 6A, S and S3 solutions produced homogenous nanofibers free of beads. Instead, the electrospinning process of S4 solution stopped continuously, probably due to the higher SP content that produced an increase in the viscosity of the polymeric solution; the resulting S4 fibers were not considered for the continuation of the work since they are not homogenous and characterized by a lot of defects.
 |
Figure 6 (A) SEM micrographs of S, S3 and S4 nanofibers at different magnifications. (B) S and S3 nanofiber dimensions (mean values ± SE; n = 30). ANOVA one-way; Multiple Range Tests (p-value ≤ 0.05): different symbols (* vs **) indicate significant differences; (C and D) macroscopic appearance and SEM micrograph of S3 nanofibers after soaking in distilled water for 24 hours. |
The presence of SP is responsible for the formation of nanofibers with a mean diameter of about 500 nm, significantly higher than that of the fibers obtained from S solution (Figure 6B). This result could be explained by a slight increase in the viscosity of S3 solution with respect to S (see Supplementary Data). Moreover, the presence of the cross-linker ensured that the nanofibers obtained from S3 result to be insoluble in distilled water and able to form a thin gel layer after hydration maintaining their structure. On the contrary, S fibers instantly dissolved when put in contact with aqueous fluids due to the absence of SP. Figure 6C and D show respectively the macroscopic appearance and the SEM micrograph of S3 nanofibers after 24 hours-hydration: it is evident that the soaking produces a statistically significant reduction in S3 fiber mean diameter (equal to 203.83 ± 9.06 nm; mean value ± SE, n = 30), attributable to the dissolution of the two grades of PEO and P407 in water. As already mentioned, after soaking, nanofibers preserved their structure.
Mechanical Properties: Resistance to Deformation
Finally, both the nanofibers obtained from S and S3 solutions were subjected to a tensile test. In Figure 7, it can be observed that S and S3 nanofibers show a completely different force-strain profile: SP-free fibers (S) are characterized by a sharp decrease in force on increasing deformation, while SP-containing fibers (S3) show tensile strength values significantly lower than those of S fibers and a progressive increase in force on strain increasing. This different behavior indicates that S fibers, free of SP, undergo a structure “breakdown” during the test, while S3 fibers, due to the presence of SP, deform plastically without breaking.
 |
Figure 7 Tensile strength values calculated at different strain % of S and S3 nanofibers (mean values ± SE; n = 3). |
GG/SP/GL Nanofiber Characterization
At first, the rheological and mechanical properties of GG/SP3 mixture added with gelatin (GL) at 0.1% w/w (GG/SP3/GL) were assessed to investigate how the presence of GL could affect the microstructure of the mixture. At acid and neutral pH, GL, characterized by an isoelectronic point in the pH range of 7–8, shows positive charges which can interact with the anionic groups of GG;33 considering that GG/SP3 mixture showed a pH equal approximately to 6, it may be expected that GG and GL form electrostatic complexes.
Therefore, in order to investigate the occurrence of a synergistic effect between SP and GL in improving the structure of GG solution, viscosity measurements were performed on GG/SP3/GL in comparison with GG/SP3. The ηMIX/ηGG parameter (where MIX indicates GG/SP3 and GG/SP3/GL mixtures) was calculated at three different shear rates (Figure 8). A synergistic effect of SP and GL can be observed in the whole shear rate interval: at 1000 s−1, that is the shear rate that best mimics the stress to which the polymeric solutions were subjected during the electrospinning process, the viscosity of GG/SP3/GL is 3 times greater than the viscosity of GG, while GG/SP3 is twice as viscous as GG. The increase in viscosity observed when GL was added to GG/SP3 suggested a strong structuring of GG/SP3/GL mixture due to the combined cross-linking action of SP and GL.
 |
Figure 8 ηMIX/ηGG values calculated at three shear rates (50, 500 and 1000 s−1) for GG/SP3, GG/GL and GG/SP3/GL mixtures. Data are expressed as mean ± SE (n = 3). ANOVA one-way – Multiple Range Test (p-value ≤ 0.05): avsb,c; bvsc; a’vsb’; b’vsc’; a”vsc”; b”vsc”. |
Table 3 reports the rheological (spur value, tgδ, ΔG’/G’1Hz) and mechanical (hardness and Young’s Modulus) parameters determined for GG/SP3 and GG/SP3/GL mixtures: it can be observed that the presence of both SP and GL produced a statistically significant increase in the spur value, suggesting a strengthening of the inner microstructure of the mixture. The viscoelastic parameters, tgδ and ΔG’/G’1Hz, were calculated for the mixtures: the addition of SP/GL seems to produce a statistically significant increase in the mixture elasticity, as shown by tgδ values. The addition of GL ensured the production of a gel-like structure with an elastic behavior higher than that of GG/SP3 mixture, probably due to the formation of an electrostatic complex between GG and GL. No significant differences are evidenced.
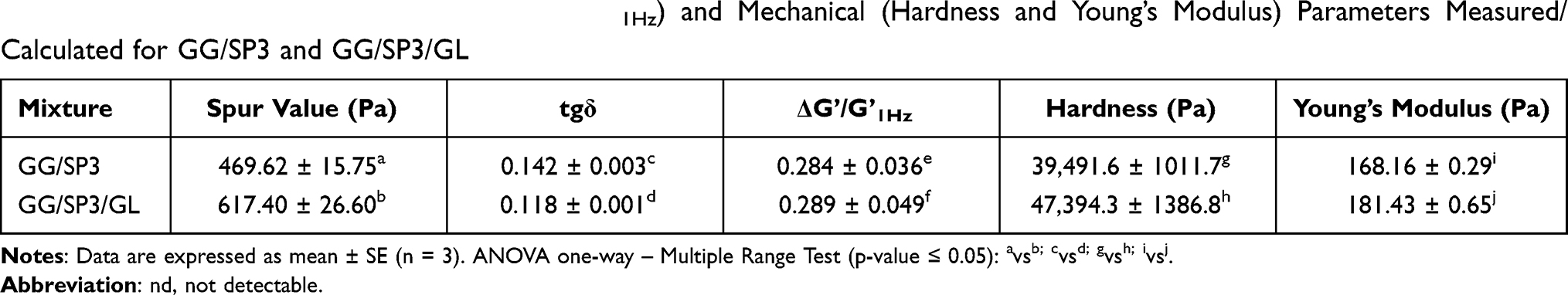 |
Table 3 Rheological (Spur Value, tgδ and ΔG’/G’1Hz) and Mechanical (Hardness and Young’s Modulus) Parameters Measured/Calculated for GG/SP3 and GG/SP3/GL |
All the mixtures were also subjected to a uniaxial compressive force by means of a texture analyzer; the compressive parameters, hardness and Young’s modulus were determined and reported in Table 3. Such parameters evidence that the addition of GL produced gels characterized by a reinforced inner microstructure and not susceptible to syneresis.
Therefore, GG/SP3/GL mixture was added with 2.2% w/w l-PEO, 0.5% w/w h-PEO and 2% w/w P407 in order to obtain a polymeric solution that could be electrospun. A slight modification in the process parameters appeared necessary to improve the electrospinning process; in particular, the applied voltage was increased from 20 to 25 kV. In Figure 9A, it can be appreciated the morphology of S3-G fibers, which appear homogenous and characterized by a mean diameter equal to 492.50 ± 25.23 (± SE; n = 30), not significantly different from S3 one.
 |
Figure 9 (A) SEM micrographs of S3-G nanofibers at different magnifications. (B) Tensile strength calculated for S3 and S3-G nanofibers; data are expressed as mean ± SE (n = 3). ANOVA one-way, Multiple Range Test (p < 0.05): different symbols (* vs **) indicate significant differences. |
Finally, the mechanical properties of S3-G were analyzed through a tensile test. The presence of GL increased the tensile strength of S3-G nanofibers with respect to S3 (Figure 9B), confirming the interaction of GG with both SP and GL. The formation of a polyelectrolyte complex between GG, SP and GL produced a reinforced nanofibrous structure that deformed plastically without breaking. After soaking in distilled water, the morphology of S3-G nanofibers (data not shown) was quite similar to that of S3.
The water retention capacity of both S3 and S3-G was calculated as PBS amount retained by nanofibers, normalized for nanofiber dry weight and expressed in percentage. Such calculation took into consideration the nanofiber mass loss due to the dissolution of PEO (low and high MW) and P407 after hydration. WRC% was equal to 522 + 22% and to 538 + 149% for S3 and S3-G, respectively.
In vitro Cell Culture Experiments: Schwann Cells
Finally, S3 and S3-G were tested on Schwann cells to evaluate their cytotoxicity during time; Figure 10 reports the percentage of living cells after treatment with S3 and S3-G nanofibers for different times (1, 3 and 7 days). These results demonstrate that the addition of GL improves the nanofiber compatibility with the cell substrate independently of the time considered: at each time, S3-G is characterized by a cell viability % comparable to complete culture medium (CM) and significantly higher than that of S3. The microscope observation of cell morphology after treatment with nanofibers (data not shown) confirmed MTT results. After treatment with S3-G, cells showed the typical neuronal morphology of Schwann cells that was similar to that of the untreated ones (CM). On the contrary, cells treated with S3 were characterized by a more rounded shape, suggesting a lower biocompatibility. The microscope observation revealed also that nanofibers preserved their topological structure after soaking in CM.
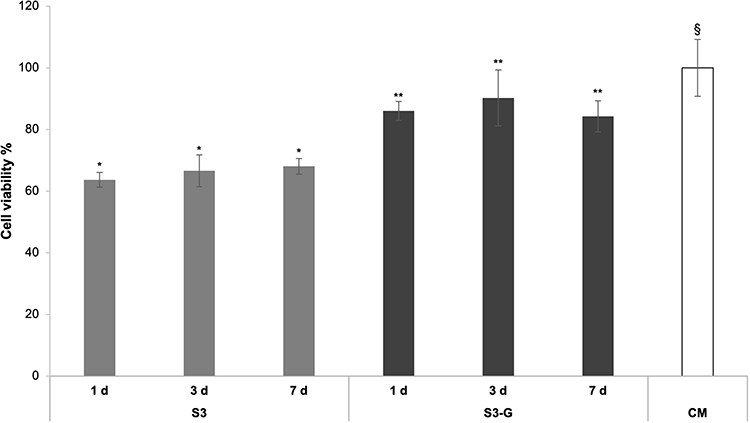 |
Figure 10 Viability % values calculated after cell indirect contact with S3 and S3-G for 1, 3 and 7 days; CM was used as reference. Data are expressed as mean ± SD (n = 3). ANOVA one-way, Multiple Range Test (p < 0.05): different symbols (* vs **, §) indicate significant differences. |
Discussion
In the present work, hydrogels in the form of nanofibers were proposed as potential tools for the treatment of nervous tissue injuries. Such nanosystems were obtained by the electrospinning technique through a single-step cross-linking approach: the cross-linker was dissolved into the polymeric solution to be electrospun without further treatment on the resulting fibrous membrane.
The so-called “hydrogel nanofibers” combine the advantages of both hydrogels and nanofibers.34 Hydrogels are porous, soft and elastic matrices able to incorporate high amounts of water, providing a three-dimensional micro-architecture that resembles the extracellular matrix of several tissue types,35,36 including nervous one.37 Due to their porosity, hydrogels represent ideal substrates for cell proliferation and colonization;38 in particular, there is evidence that hydrogels offer physical guiding cues to axonal growth, creating a pro-regenerative environment for the repair and the restoration of nervous tissue defects.39 On the counter part, nanofibers have been demonstrated as effective therapeutic platforms in neural tissue regeneration due to their unique morphological and mechanical properties, such as large surface area-to-volume ratio, small pore size, interconnected structure and flexibility. In the last two decades, the electrospinning technique has gained increasing interest as simple, economic and versatile method to obtain nanofibers from polymeric solutions.15,17,40,41
To date, only a few works concerning the development of electrospun hydrogel nanofibers are reported in the literature.34 Moreover, they refer to nanofibers subjected to a post-spinning cross-linking. In the 2018, alginate/gelatin hydrogel nanofibers were prepared by wet electrospinning: the polymeric solution was electrospun into a Ca2+-containing bath instead of a solid collector, producing physical hydrogels in nanofibrous form due to the rapid alginate gelation.42 Core-shell electrospinning was also used for the preparation of hydrogel nanofibers. Two-layer fibers, consisting in an alginate core and a polyvinyl pyrrolidone (PVP) shell, were electrospun and, subsequently, soaked in a CaCl2 ethanol/water solution: PVP dissolved, and alginate-based hydrogel nanofibers were obtained.43 The same procedure was exploited by Naganuma et al, who produced core-shell nanofibers characterized by a core of cellulose acetate and a shell of polyvinyl alcohol (PVA) that, when immersed in an aqueous bath, formed an hydrogel layer.44 In 2020, Kim and Kim developed glucose-responsive transparent hydrogel nanofibrous patches by a two-step approach: electrospun fibers were subsequent heated at 110°C to obtain chemical hydrogels in nanofibrous form.45
In the present work, gellan gum (GG) was selected as the main structural component of hydrogel nanofibers; GG is a negatively charged polysaccharide that can interact with different cationic compounds producing physical hydrogels. In the pharmaceutical field, the latter are generally preferred to chemical gels since the lack in covalent bonds facilitates system biodegradation and elimination from the organism.
In 2013, Lopez-Cebral et al demonstrated, for the first time that endogen polyamines (for example, putrescine, spermine and spermidine) were capable to cross-link GG in a more effective manner than the inorganic cations generally used for the production of polyanion-containing gels, such as Ca2+ and K+.37 Besides being highly valuable molecules, such polyamines result as potent GG cross-linkers, preserving a good gelling capability at concentrations lower that those necessary for inorganic cations.
Among the polyamines, spermidine (SP) appears the most interesting one to produce physical hydrogels intended for nervous tissue repair/regeneration. SP is characterized by neuroprotective activity: it preserves neurons from oxidative damage and modulates the over-expression of pro-inflammatory cytokines at the injury site.24 Therefore, GG/SP mixtures, containing a fixed GG amount (1.5% w/w) and increasing SP concentrations (0–0.1% w/w), were prepared and characterized to identify the GG/SP mixture most suitable for the preparation of polymeric solutions to be electrospun. In fact, it is widely recognized that, during the electrospinning process, the rheological properties of the starting polymeric solutions play a crucial role in the morphology and homogeneity of the resulting nanofibers.18
In the presence of SP, GG-based hydrogels are formed spontaneously by ionotropic gelation that represents the simplest and no time-consuming method for the preparation of physical hydrogels due to the electrostatic interaction between the negatively charged polymer and the cationic polyamine. The rheological analysis (viscosity and viscoelasticity measurements) performed on the GG/SP mixtures confirmed that SP acts as a potent cross-linking agent of GG: the addition of increasing SP concentrations to a solution of pristine GG produced more and more structured and elastic mixtures, consisting of three-dimensional branched polymeric networks with entrapped water.46
In addition to being characterized in terms of viscosity and viscoelasticity, GG/SP mixtures were investigated for their textural properties, as well as for their deformation ability, through a compressive test. It is well known in the literature that GG is able to form both elastic and brittle gels depending on the concentration of the cationic compound used as cross-linker. Such a concentration may affect the microstructure of the network of GG chains forming the physical gel and, thus, the mechanical behavior of the gel when subjected to uniaxial compressive forces. In the present work, texture analysis revealed that GG/SP mixtures were characterized by two distinctive patterns of mechanical response below and above a critical SP concentration at which a maximum gel strength was recorded.30
Mao et al30 demonstrated that the composition of GG/Ca2+ gels affected their mechanical properties. In particular, the authors have supposed a relationship between the microstructure, the textural properties and the water holding capacity (related to the syneresis phenomenon) of GG/Ca2+ gels, which were all strictly influenced by the concentration of the inorganic cation. According to the literature, the results obtained in the present work proved that the textural properties of GG/SP mixtures are correlated to the gel capability to retain water that, in turn, depends on SP concentration. GG/SP mixtures containing SP concentrations higher than the critical value (from GG/SP5 onwards) appeared brittle and exhibited syneresis during compression. When such mixtures were subjected to external forces, the water entrapped within the polymeric network was squeezed out of the gel: the loss of water resulted in a shrinking of the gel with a subsequent change in its textural properties. On the contrary, GG/SP mixtures containing SP concentrations equal or lower than the critical value were soft, easily deformable during compression and not susceptible to syneresis.
The compressive parameters, shown in the present work, were characterized by higher values than those reported in the literature for hydrogels consisting of GG and SP. Such parameters strongly depend on GG concentration that, in the present work, was fixed at 1.5% w/w (vs GG concentrations ≤0.5% w/w reported in the literature for GG/SP gels). Such a GG concentration was selected since it was proved to be optimal to produce homogenous electrospun nanofibers,20 as well as hydrogel nanofibers after soaking into biologic fluids. As previously described, Lopez-Cebral et al prepared physical hydrogels containing GG and SP; albumin and chondroitin sulfate were also added in the formulation as interesting endogen molecules. Texture analysis evidenced a slight increase in the mechanical parameters (hardness, cohesiveness and Young’s modulus) on increasing SP concentration up to 0.017% in presence of 0.14% w/w GG.; no significant differences were recorded after the addition of a double amount of SP (from 0.017% to 0.034% w/w). Instead, a significant decrease in gel strength was highlighted by halving the GG content (from 0.14% to 0.07% w/w).37 A year later, the same research group proposed such physical hydrogels as drug delivery systems for the controlled release of cloxacillin, a low molecular weight antibiotic. A neuro-fuzzy logic tool, used to investigate the effect of the hydrogel composition on the textural properties, revealed that the hardest hydrogels were prepared by blending high GG amount and low SP concentration and, vice versa, the softest ones resulted from low GG concentration and high SP content.47
Therefore, as confirmed by a review of the literature, GG represented the main structuring agent of GG/SP mixtures and, depending on GG content, the cross-linker concentration could affect the mechanical properties of the resulting gel.24 According to our results, the use of low SP concentrations, close to a critical value, were sufficient to cross-link 1.5% w/w GG and to obtain physical hydrogels easily deformable and characterized by a GG chain entanglement that should be sufficient to sustain the electrospinning process. GG/SP3 and GG/SP4 were selected for the continuation of the experiments.
In a previous work of ours, homogeneous electrospun GG nanofibers were prepared by adding two grades of poly(ethylene oxide) (PEO) (low MW, l-MW, and high MW, h-PEO) and poloxamer (P407) to a GG aqueous solution;20 the polymer concentrations employed were as follows: 1.5% w/w GG, 2.2% w/w l-PEO, 0.15% w/w h-PEO and 2% w/w P407. Based on our experience in the electrospinning, the use of spinning synthetic non-ionic polymers, such as PEO, is necessary to overcome the limitations related to the intrinsic properties of polysaccharides, which could hinder the process. In recent years, we have demonstrated that the addition of a blend, mainly consisting of l-PEO and containing a small amount of h-PEO, to an aqueous solution of anionic polysaccharides (alginate and GG) was functional to obtain electrospun nanofibers with optimal mechanical properties.19 Moreover, the addition of a surfactant (P407) was proposed to modulate the surface tension of the polymeric solution and, thus, to enhance its electrospinnability.20 Therefore, GG/SP3 and GG/SP4, selected as the mixtures with the best rheological and textural properties, were added to a blend of two grades of PEO and P407 and, then, electrospun. S3 solution, obtained starting from GG/SP3 mixture, allowed the production of homogenous nanofibers. On the contrary, the electrospinning of the polymeric solution, from GG/SP4 mixture, stopped continuously, probably due to the higher SP content responsible for an excessive increase in viscosity; the resulting S4 fibers were not homogenous, characterized by a lot of defects and, thus, not suitable for the continuation of the work.
To the best of our knowledge, it is the first time that homogenous electrospun nanofibers (S3), containing GG cross-linked with SP, were obtained. After S3 nanofiber soaking in an aqueous medium, the water-soluble synthetic fiber components (l-PEO, h-PEO and P407) dissolved and hydrogel GG/SP nanofibers were formed, as confirmed by SEM micrographs. In fact, the presence of the cross-linker ensured that the GG/SP nanofibers (S3) resulted to be insoluble in aqueous fluids and formed a thin gel layer (hydrogel nanofibrous membrane) after hydration; on the contrary, SP-free fibers (S) instantly dissolved when put in contact with aqueous media. Moreover, the tensile test revealed that S3 nanofibers, contrary to S fibers, did not undergo a structure “breakdown” during the test, but got a plastic deformation without breaking. These results suggest that membranes consisting of S3 nanofibers can be easily handled during the surgical procedure or rolled up to produce cylindrical systems to apply at the site of injury, filling the gap generated by the nervous tissue defect. Furthermore, after application, the mechanical properties of S3 nanofibers should ensure that they could withstand external stresses, while preserving their structure.
Since it is well known the relatively bioinert nature of GG, it was assumed that the addition of small amounts of gelatin (GL) into S3 polymeric solution could allow to obtain electrospun nanofibers endowed with a greater compatibility with nervous cells and, thus, a higher capability to enhance cell attachment and colonization after in vivo application.48,49 In fact, GL is a bioactive molecule derived from collagen, that is the main component of the extracellular matrix of various tissues, and it is widely used to produce three-dimensional scaffolds for regenerative purposes. Preliminary rheological and mechanical analyses were performed on GG/SP3 mixture added with 0.1% w/w GL (GG/SP3/GL) in order to assess how the GG-based hydrogel properties could be affected by the addition of small amounts of GL. A synergistic effect of SP and GL was highlighted in increasing hydrogel textural properties. Such a result suggested a strong structuring of GG/SP3/GL mixture due to the cross-linking action of both SP and GL; the use of a small polycationic compound (SP) and a positive charged polypeptide (GL) ensured a sufficient number of cross-linking junctions and a fair amount of chain entanglements to generate a continuous jet during the electrospinning process. Such a hypothesis was confirmed by the electrospinning of a polymeric solution consisting of GG/SP3/GL mixture, 2.2% l-PEO, 0.5% w/w h-PEO and 2% w/w P407: homogenous nanofibers, turning into hydrogel nanofibers after hydration, were obtained. Regarding fiber mechanical properties, the formation of a polyelectrolyte complex between GG and GL reinforced the nanofibrous structure while preserving its plastic behavior. Finally, in vitro studies performed on Schwann cells, used as neural cell model, evidenced that the presence of GL enhanced the nanofiber compatibility with the cell substrate.
Conclusion
Hydrogels in the form of nanofibers, consisting of GG and SP, were prepared for the first time by the electrospinning technique and proposed as a potential tool for the treatment of nervous tissue injuries.
Different GG/SP mixtures, containing a fixed GG amount and increasing SP concentrations, were prepared by ionotropic gelation. SP, a cationic polyamine, acts as a potent cross-linking agent of GG, that is an anionic polysaccharide: the addition of low SP concentrations was sufficient to cross-link GG and to obtain physical hydrogels easily deformable and characterized by a GG chain entanglement that should be sufficient to sustain the electrospinning process.
The GG/SP mixture with the best rheological and textural properties was added to a blend of two grades of PEO, as spinnable enhancers, and P407, as surfactant, and, then, electrospun. The resulting nanofibers were homogeneous and characterized by optimal mechanical properties: they can be easily handled during the surgical procedure or rolled up to produce cylindrical systems to apply at the site of injury, filling the gap generated by the nervous tissue defect. After nanofiber soaking in aqueous fluids, the water-soluble synthetic fiber components dissolved and GG/SP hydrogel nanofibers were formed.
In order to improve the bioactivity of hydrogel nanofibers, GL was added. A synergistic effect of SP and GL in reinforcing the nanofibrous structure was highlighted, without affecting its plastic behavior. Finally, in vitro studies performed on a neural cell model evidenced that the presence of GL enhanced the hydrogel nanofiber compatibility with the cell substrate.
Acknowledgments
University of Pavia (Fondo Ricerca Giovani, FRG) is kindly acknowledged. Moreover, the authors would like to thank Dr. Ilenia Tredici and Dr. Alessandro Girella (CISRiC Arvedi, University of Pavia) for their assistance in SEM analyses.
Author Contributions
All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, have agreed on the journal to which the article has been submitted, and agree to be accountable for all aspects of the work. SR and BV conceived and designed the experiments and wrote the paper. BV and CV performed the experiments and acquired experimental data. SR, BV, GS, CMC and FF analyzed and interpreted the experimental data.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ye K, Kuang H, You Z, et al. Electrospun nanofibers for tissue engineering with drug loading and release. Pharmaceutics. 2019;11:182. doi:10.3390/pharmaceutics11040182
2. Xie X, Chen Y, Wang X, et al. Electrospinning nanofiber scaffolds for soft and hard tissue regeneration. J Mater Sci Technol. 2020;59:243–261. doi:10.1016/j.jmst.2020.04.037
3. Rahmati M, Mills DK, Urbanska AM, et al. Electrospinning for tissue engineering applications. Prog Mater Sci. 2021;117:100721.
4. Kabu S, Gao Y, Kwon BK, et al. Drug delivery, cell-based therapies, and tissue engineering approaches for spinal cord injuries. J Control Release. 2015;219:141–154. doi:10.1016/j.jconrel.2015.08.060
5. Vijayavenkataraman S. Nerve guide conduits for peripheral nerve injury repair: a review on design, materials and fabrication methods. Acta Biomater. 2020;106:54–69. doi:10.1016/j.actbio.2020.02.003
6. Tian L, Prabhakaran MP, Ramakrishna S. Strategies for regeneration of components of nervous system: scaffolds, cells and biomolecules. Regen Biomater. 2015;2:31–45. doi:10.1093/rb/rbu017
7. Zhang N, Wen X. Neural tissue engineering and regenerative medicine. In: Meyer U, Meyer T, Handschel J, editors. Fundamentals of Tissue Engineering and Regenerative Medicine. Heidelberg, Germany: Springer; 2009:271–288.
8. Mahumane GD, Kumar P, du Toit LC, et al. 3D scaffolds for brain tissue regeneration: architectural challenges. Biomater Sci. 2018;6:2812–2837. doi:10.1039/C8BM00422F
9. Quan Q, Chang B, Meng HY, et al. Use of electrospinning to construct biomaterials for peripheral nerve regeneration. Rev Neurosci. 2016;27:761–768. doi:10.1515/revneuro-2016-0032
10. Chen C, Tang J, Gu Y, et al. Bioinspired hydrogel electrospun fibers for spinal cord regeneration. Adv Funct Mater. 2019;29:1806899. doi:10.1002/adfm.201806899
11. Puhl DL, Funnell JL, Nelson DW, et al. Electrospun fiber scaffolds for engineering glial cell behavior to promote neural regeneration. Bioengineering. 2020;8:4. doi:10.3390/bioengineering8010004
12. Vigani B, Rossi S, Sandri G, et al. Dual-functioning scaffolds for the treatment of spinal cord injury: alginate nanofibers loaded with the sigma 1 receptor (S1R) agonist RC-33 in chitosan films. Mar Drugs. 2020;18:21. doi:10.3390/md18010021
13. Ghane N, Khalili S, Khorasani SN, et al. Regeneration of the peripheral nerve via multifunctional electrospun scaffolds. J Biomed Mater Res A. 2021;109:437–452. doi:10.1002/jbm.a.37092
14. Faccendini A, Vigani B, Rossi S, et al. Nanofiber scaffolds as drug delivery systems to bridge spinal cord injury. Pharmaceuticals. 2017;10:63. doi:10.3390/ph10030063
15. Vigani B, Rossi S, Sandri G, et al. Design and criteria of electrospun fibrous scaffolds for the treatment of spinal cord injury. Neural Regen Res. 2017;12:1786–1790. doi:10.4103/1673-5374.219029
16. Kennedy KM, Bhaw-Luximon A, Jhurry D. Cell-matrix mechanical interaction in electrospun polymeric scaffolds for tissue engineering: implications for scaffold design and performance. Acta Biomater. 2017;50:41–55. doi:10.1016/j.actbio.2016.12.034
17. Rogina A. Electrospinning process: versatile preparation method for biodegradable and natural polymers and biocomposite systems applied in tissue engineering and drug delivery. Appl Surf Sci. 2014;296:221–230. doi:10.1016/j.apsusc.2014.01.098
18. Vigani B, Rossi S, Sandri G, et al. Coated electrospun alginate-containing fibers as novel delivery systems for regenerative purposes. Int J Nanomed. 2018;13:6531–6550. doi:10.2147/IJN.S175069
19. Vigani B, Rossi S, Milanesi G, et al. Electrospun alginate fibers: mixing of two different poly(ethylene oxide) grades to improve fiber functional properties. Nanomaterials. 2018;8:971. doi:10.3390/nano8120971
20. Vigani B, Valentino C, Cavalloro V, et al. Gellan-based composite system as a potential tool for the treatment of nervous tissue injuries: cross-linked electrospun nanofibers embedded in a RC-33-loaded freeze-dried matrix. Pharmaceutics. 2021;13:164. doi:10.3390/pharmaceutics13020164
21. Silva NA, Cooke MJ, Tam RY, et al. The effects of peptide modified gellan gum and olfactory ensheathing glia cells on neural stem/progenitor cell fate. Biomaterials. 2012;33:6345–6354. doi:10.1016/j.biomaterials.2012.05.050
22. Lozano R, Stevens L, Thompson BC, et al. 3D printing of layered brain-like structures using peptide modified gellan gum substrates. Biomaterials. 2015;67:264–273. doi:10.1016/j.biomaterials.2015.07.022
23. Gomes ED, Mendes SS, Leite-Almeida H, et al. Combination of a peptide-modified gellan gum hydrogel with cell therapy in a lumbar spinal cord injury animal model. Biomaterials. 2016;105:38–51. doi:10.1016/j.biomaterials.2016.07.019
24. Koivisto JT, Joki T, Parraga JE, et al. Bioamine-crosslinked gellan gum hydrogel for neural tissue engineering. Biomed Mater. 2017;12:025014. doi:10.1088/1748-605X/aa62b0
25. Carvalho CR, Wrobel S, Meyer C, et al. Gellan gum-based luminal fillers for peripheral nerve regeneration: an in vivo study in the rat sciatic nerve repair model. Biomater Sci. 2018;6:1059–1075. doi:10.1039/C7BM01101F
26. Kornev VA, Grebenik EA, Solovieva AB, et al. Hydrogel-assisted neuroregeneration approaches towards brain injury therapy: a state-of-the-art review. Comput Struct Biotechnol J. 2018;16:488–502. doi:10.1016/j.csbj.2018.10.011
27. Gomes ED, Ghosh B, Lima R, et al. Combination of a gellan gum-based hydrogel with cell therapy for the treatment of cervical spinal cord injury. Front Bioeng Biotechnol. 2020;8:984. doi:10.3389/fbioe.2020.00984
28. Madeo F, Eisenberg T, Pietrocola F, et al. Spermidine in health and disease. Science. 2018;359:eaan2788. doi:10.1126/science.aan2788
29. Banerjee S, Bhattacharya S. Compressive textural attributes, opacity and syneresis of gels prepared from gellan, agar and their mixtures. J Food Eng. 2011;102:287–292. doi:10.1016/j.jfoodeng.2010.08.025
30. Mao R, Tang J, Swanson BG. Water holding capacity and microstructure of gellan gels. Carbohyr Polym. 2001;46:365–371. doi:10.1016/S0144-8617(00)00337-4
31. Tang J, Lelievre J, Tung MA, Zeng Y. Polymer and ion concentration effects on gellan gum strength and strain. J Food Sci. 1994;59:216–220. doi:10.1111/j.1365-2621.1994.tb06934.x
32. Tang J, Tung MA, Zeng Y. Compression strength and deformation of gellan gels formed with mono- and divalent cations. Carbohyr Polym. 1996;29:11–16. doi:10.1016/0144-8617(95)00124-7
33. Zheng Y, Liang Y, Zhang D, et al. Gelatin-based hydrogels blended with gellan as an injectable wound dressing. ACS Omega. 2018;3:4766–4775. doi:10.1021/acsomega.8b00308
34. Ghosh T, Das T, Purwar R. Review of electrospun hydrogel nanofiber system: synthesis, properties and applications. Polym Eng Sci. 2021;61:1887–1911. doi:10.1002/pen.25709
35. Zhou L, Zhao J, Chen Y, et al. MoS2-ALG-Fe/GOx hydrogel with Fenton catalytic activity for combined cancer photothermal, starvation, and chemodynamic therapy. Colloids Surf B Biointerfaces. 2020;195:111243. doi:10.1016/j.colsurfb.2020.111243
36. Chen Z, Wu H, Wang H, et al. An injectable anti-microbial and adhesive hydrogel for the effective noncompressible visceral hemostasis and wound repair. Mater Sci Eng C Mater Biol Appl. 2021;129:112422. doi:10.1016/j.msec.2021.112422
37. López-Cebral R, Paolicelli P, Romero-Caamaño V, et al. Spermidine-cross-linked hydrogels as novel potential platforms for pharmaceutical applications. J Pharm Sci. 2013;102:2632–2643. doi:10.1002/jps.23631
38. López-Cebral R, Civantos A, Ramos V, et al. Gellan gum based physical hydrogels incorporating highly valuable endogen molecules and associating BMP-2 as bone formation platforms. Carbohydr Polym. 2017;167:345–355. doi:10.1016/j.carbpol.2017.03.049
39. Madhusudanan P, Raju G, Shankarappa S. Hydrogel systems and their role in neural tissue engineering. J R Soc Interface. 2020;17:20190505. doi:10.1098/rsif.2019.0505
40. Xue J, Wu T, Dai Y, Xia Y. Electrospinning and electrospun nanofibers: methods, materials, and applications. Chem Rev. 2019;119:5298–5415. doi:10.1021/acs.chemrev.8b00593
41. Xu X, Wang S, Wu H, et al. A multimodal antimicrobial platform based on MXene for treatment of wound infection. Colloids Surf B Biointerfaces. 2021;207:111979. doi:10.1016/j.colsurfb.2021.111979
42. Majidi SS, Slemming-Adamsen P, Hanif M, et al. Wet electrospun alginate/gelatin hydrogel nanofibers for 3D cell culture. Int J Biol Macromol. 2018;118:1648–1654. doi:10.1016/j.ijbiomac.2018.07.005
43. Fujita S, Wakuda Y, Matsumura M, et al. Geometrically customizable alginate hydrogel nanofibers for cell culture platforms. J Mater Chem B. 2019;7:6556–6563. doi:10.1039/C9TB01353A
44. Naganuma C, Moriyama K, Suye S, et al. One-step surface immobilization of protein A on hydrogel nanofibers by core-shell electrospinning for capturing antibodies. Int J Mol Sci. 2021;22:9857. doi:10.3390/ijms22189857
45. Kim GJ, Kim KO. Novel glucose-responsive of the transparent nanofiber hydrogel patches as a wearable biosensor via electrospinning. Sci Rep. 2020;10:18858. doi:10.1038/s41598-020-75906-9
46. Petcharat T, Benjakul S, Hemar Y. Improvement of gel properties of fish gelatin using gellan. Int J Food Eng. 2017;13:20160410.
47. López-Cebral R, Romero-Caamaño V, Seijo B, et al. Spermidine cross-linked hydrogels as a controlled release biomimetic approach for cloxacillin. Mol Pharm. 2014;11:2358–2371. doi:10.1021/mp500067z
48. Lee KY, Shim J, Bae IJ, Cha J, Park CS, Lee HG. Characterization of gellan/gelatin mixed solutions and gels. LWT. 2003;36:795–802796. doi:10.1016/S0023-6438(03)00116-6
49. Koivisto JT, Gering C, Karvinen J, et al. Mechanically biomimetic gelatin-gellan gum hydrogels for 3D culture of beating human cardiomyocytes. ACS Appl Mater Interfaces. 2019;11:20589–20602. doi:10.1021/acsami.8b22343
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.