")
Back to Journals » Journal of Inflammation Research » Volume 14
Sodium Propionate Enhances Nrf2-Mediated Protective Defense Against Oxidative Stress and Inflammation in Lipopolysaccharide-Induced Neonatal Mice
Authors Chen D, Gao Z, Wang Y, Wan B, Liu G, Chen J, Wu Y, Zhou Q, Jiang S, Yu R , Pang Q
Received 28 January 2021
Accepted for publication 23 February 2021
Published 10 March 2021 Volume 2021:14 Pages 803—816
DOI https://doi.org/10.2147/JIR.S303105
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Monika Sharma
Dan Chen,1,* Zhi-qi Gao,1,* Ying-ying Wang,1 Bin-bin Wan,1 Gang Liu,1 Jun-liang Chen,1 Ya-xian Wu,1 Qin Zhou,2 Shan-yu Jiang,2 Ren-qiang Yu,2 Qing-feng Pang1
1Department of Physiopathology, Wuxi School of Medicine, Jiangnan University, Wuxi, 214122, Jiangsu Province, People’s Republic of China; 2Department of Neonatology, The Affiliated Wuxi Maternity and Child Health Care Hospital of Nanjing Medical University, Wuxi, 214002, Jiangsu Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ren-qiang Yu 48 Huaishu Lane, Liangxi District, Wuxi, Jiangsu Province, People’s Republic of China
Tel +86510-82709790
Fax +86-510-82725094
Email [email protected]
Qing-feng Pang 1800 Lihu Avenue, Binhu District, Wuxi, Jiangsu Province, People’s Republic of China
Tel +8651052430172
Fax +86-510-85329042
Email [email protected]
Background: Alveolar arrest and the impaired angiogenesis caused by chronic inflammation and oxidative stress are two main factors in bronchopulmonary dysplasia (BPD). Short-chain fatty acids (SCFAs), especially propionate, possess anti-oxidant and anti-inflammatory effects. The present study was designed to examine the roles of sodium propionate (SP) on lipopolysaccharide (LPS)-challenged BPD and its potential mechanisms.
Methods: WT, Nrf2-/- mice and pulmonary microvascular endothelial cells (HPMECs) were used in this study. LPS was performed to mimic BPD model both in vivo and vitro. Lung histopathology, inflammation and oxidative stress-related mRNA expressions in lungs involved in BPD pathogenesis were investigated. In addition, cell viability and angiogenesis were also tested.
Results: The increased nuclear factor erythroid 2-related factor (Nrf2) and decreased Kelch-like ECH-associated protein-1 (Keap-1) expressions were observed after SP treatment in the LPS-induced neonatal mouse model of BPD. In LPS-induced wild-type but not Nrf2-/- neonatal mice, SP reduced pulmonary inflammation and oxidative stress and exhibited obvious pathological alterations of the alveoli. Moreover, in LPS-evoked HPMECs, SP accelerated Nrf2 nuclear translocation presented and exhibited cytoprotective and pro-angiogenesis effects. In addition, SP diminished the LPS-induced inflammatory response by blocking the activation of nuclear factor-kappa B pathway. Moreover, pretreatment with ML385, an Nrf2 specific inhibitor, offsets the beneficial effects of SP on inflammation, oxidative stress and angiogenesis in LPS-evoked HPMECs.
Conclusion: SP protects against LPS-induced lung alveolar simplification and abnormal angiogenesis in neonatal mice and HPMECs in an Nrf2-dependent manner.
Keywords: sodium propionate, lipopolysaccharide, Nrf2, angiogenesis, bronchopulmonary dysplasia
Introduction
Bronchopulmonary dysplasia (BPD), the most frequent complication in premature infants, is a severe chronic lung disease. It is characterized by alveolar arrest and dysregulated angiogenesis, which then leads to persistent airway and pulmonary vascular disease, and eventually defects lung function.1 The pathogenesis of BPD is complex and involves a variety of causative factors. However, increasing evidence has revealed the increased proteobacterial abundance and endotoxin levels in the airways of infants with established BPD.2 Intra-amniotic endotoxin or neonatal intra-tracheal lipopolysaccharide (LPS) exposure remodeled the lung in rodents.3 Human mesenchymal stem cells promoted LPS-induced defective alveolarization and angiogenesis and alleviated BPD in rats.4 Moreover, angiogenesis actively promotes distal lung growth.5 In this context, it is of great importance to further explore a novel strategy to improve endotoxin-induced abnormal angiogenesis and prevent BPD.
Short-chain fatty acids (SCFAs), ie acetate, propionate and butyrate, are the major end products of bacterial fermentation in the large intestine. Nowadays, butyrate has been reported to induce angiogenesis and promote tissue remodeling in tendon-bones injury.6 Meanwhile, propionate has been described to possess potent anti-inflammatory and anti-oxidative stress effects.7 It has been reported that propionate normalized the increased serum levels of lipopolysaccharide (LPS) induced by high-fat diet and alleviated its induced endotoxaemia.8 In addition, propionate protected against LPS-induced mastitis in mice.9 In our previous study, we also found the anti-inflammatory effect of propionate against LPS-induced lung injury.10 However, whether propionate possesses a similar effect on angiogenesis is still unknown. The purpose of this study was designed to reveal the effect of propionate against LPS-induced BPD in neonatal mice and pulmonary microvascular endothelial cells and its underlying mechanisms.
Materials and Methods
Antibodies and Regents
LPS extracted from the membrane of Escherichia coli 0111:B4 was purchased from Sigma-Aldrich (St. Louis, MO, USA). Sodium propionate (SP) (HPLC≥99%) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Serum and lung superoxide dismutase (SOD) and hematoxylin and eosin were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). For immunohistochemical analysis, the antibody against von Willebrand factor (vWF) was obtained from Proteintech (Wuhan, China). For immunofluorescence staining, a primary antibody against Nrf2 was obtained from Proteintech (Wuhan, China). For Western blotting analysis, rabbit monoclonal antibody against Nrf2 was purchased from Cell Signaling Technology (Beverly, MA, USA). Rabbit polyclonal antibodies against vWF and Keap-1 were obtained from Proteintech (Wuhan, China). Mouse monoclonal antibody against GAPDH was obtained from Thermo Scientific (Waltham, MA, USA).
Experimental Animals
Timed pregnant C57BL/6J WT and Nrf2-/- mice on a C57BL/6 background were available from Jiangnan University (Wuxi, Jiangsu, China). Experimental procedures were approved by the Experimental Animal Care and Use Committee of Jiangnan University (JN.No20190930c0421020[234]) and the Guide for the Care and Use of Laboratory Animal published by the US National Institutes of Health (NIH publication, Eighth edition, 2011). The mice were housed on a 12-h light/dark cycle in a temperature-controlled room with standard chow and tap water ad libitum.
Animal Model and Study Design
A mouse model of LPS-challenged BPD was utilized as previously described.11 Briefly, at day 5 of life, mice randomized to per nursing dam were received an intraperitoneal injection of 1 mg/kg LPS (Sigma, St. Louis, MO), whereas the control mice received an equal volume injection of sterile saline solution. For experiments with the SP treatment, SP was injected intraperitoneally (1.2 mg/g) 2 hours after LPS injection. After SP was injected daily for 7 days, experiments were then carried out. Both male and female animals were used since pulmonary sequelae are similar in male and female mice in response to LPS.12
Hematoxylin and Eosin (H&E) Staining
The mice were anesthetized and the left lobes of lungs were collected, fixed in 10% buffered formalin and embedded in paraffin. Lung tissue sections (5 μm thick) were performed. To observe the morphology of lung injury, H&E staining was used following the manufacturer’s protocols (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). Quantification of the alveoli numbers and mean linear intercept (MLI) were calculated as previously reported.13
Immunohistochemistry
The mice were anesthetized and the left lobes of lungs were collected, fixed in 10% buffered formalin and embedded in paraffin. Lung tissue sections (5 μm thick) were performed. To determine vWF immunoreactivity, lung sections were immunostained with an antibody against vWF (Proteintech Group, Wuhan, China) at a dilution of 1:200 and then biotinylated secondary antibodies were used and followed by 3,3ʹ-diaminobenzidine (DAB) solution to detect the avidin-biotin complex signal.
Measurement of Superoxide Dismutase Activity
Lung tissues were collected and homogenized in saline. Lung and serum levels of superoxide dismutase (SOD) activity were examined according to the manufacturer’s instructions (SOD activity assay kit A001, Nanjing Jiancheng bioengineering Institute, China). Results were normalized by protein concentrations.
Cell Culture and Treatment
Human primary pulmonary microvascular endothelial cells (HPMECs) were obtained from ScienCell (Carlsbad, CA) and cultured in endothelial cell medium (ECM) supplemented with fetal bovine serum, antibiotics, and endothelial cell growth serum as recommended by the manufacturer (Scien Cell) in a humidified atmosphere at 37°C and 5% CO2. After 80% confluence, the cells were pretreated with LPS (1 μg/mL)14 or treated with SP (0.6 mM) 2 h after LPS stimulation.
Optical Microscopy
To obtain the Morphologic images, HPMECs were visualized and photographed using a phase-contrast microscope equipped with a digital camera (DP11, Olympus, Tokyo, Japan).
Cell Viability Examination
HPMECs cells were seeded on a 96-well plate at a density of 1×105cells/mL. After 24 h, the attached HPMECs cells were then cultured with a mixture of CCK-8 solution (Beyotime Institute of Biotechnology, Shanghai, China) and culture medium for 2 h at 37°C. The absorbance was measured at 450 nm with a microplate reader (ELX800, BioTek, Vermont, USA).
Tube Formation Assay
For tube formation assay, 50 μL growth factor reduced Matrigel (BD Bioscience, CA, USA) was planted on 96-well plates and incubated at 37 °C for 30 min. Approximately 5×104 cells/mL HPMECs were then seeded into a 96-well plate coated with Matrigel incubated at 37°C in 5% CO2 for 8 h. Tube formation was observed with a digital camera (DP11, Olympus, Tokyo, Japan).
Immunofluorescence Staining
HPMECs were seeded into the culture dish with coverslips at a density of 1×105 cells/mL. After 24 h, HPMECs were fixed with 4% paraformaldehyde for 15 min and permeabilized with 0.5% Triton X-100 for 20 min at room temperature. The dishes were then washed with phosphate-buffered saline (PBS) for three times, followed by incubation with 3% bovine serum album (BSA) for 30 min at room temperature. Primary antibody against Nrf2 (Proteintech; Wuhan, China) (1:100 dilution in PBS with 1% BSA) was then added overnight at 4°C. Cells were washed with cold PBS for three times and then incubated in Alexa 488-conjugated secondary antibody. The nuclei were stained with DAPI solution (1 mg/mL) for 5 min at room temperature. The slides were washed again and the images were collected on a zeiss LSM880 confocal microscope (Carl Zeiss, German).
Dihydroethidium Staining
Intracellular reactive oxygen species (ROS) in HPMECs were evaluated with dihydroethidium (DHE) staining as previously reported.15 Cells (3×105 cells/mL) in 96-well plates were incubated with DHE (10 μM) in PBS for 30 min in a dark and humidified container at 37°C and then washed twice with cold PBS. The fluorescence signals were obtained with a fluorescence spectrophotometer (Synergy H4, BioTek, Vermont, USA) and fluorescence microscopy under excitation at 518 nm and emission at 605 nm (DP70, Olympus Optical, Tokyo, Japan).
Real-Time Quantitative Polymerase Chain Reaction (qPCR)
Lung tissues or HPMECs were sonicated in Trizol reagent (Life Technologies, USA), and then homogenized according to the manufacturer’s protocols. Reverse transcriptase reactions were performed using the PrimeScript RT reagent kits. The reverse transcriptase-polymerase chain reaction (RT-PCR) was performed using quantitative PCR with SYBR Premix Ex Taq (Takara, Otsu, Shiga, Japan) and a LightCycler® 480 PCR detection system (Roche, Foster City, CA, USA). The expressions of mRNA were calculated using the comparative cycle threshold (Ct) method where the relative quantization of target transcript levels was determined by subtracting Ct values of target genes from Ct values of GAPDH. The sequences of primers for humans and mice are listed in Tables 1 and 2, respectively.
 |
Table 1 Primers for Real-Time Quantitative PCR Analysis in Humans |
 |
Table 2 Primers for Real-Time Quantitative PCR Analysis in Mice |
Western Blotting Analysis
Lung tissues or HPMECs were homogenized in a lysis buffer (radioimmunoprecipitation assay (RIPA): protease inhibitor = 100:1). A protein assay kit (BCA; Thermo Scientific, Waltham, MA, USA) was used to measure the total protein in samples. Equal total protein was separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to NC membranes in Tris-glycine-methanol buffer. The bands were visualized using enhanced chemiluminescence. The rabbit monoclonal antibody against Nrf2 (1:1000) was purchased from Cell Signaling Technology (Beverly, MA, USA). The rabbit monoclonal antibodies against Keap-1 (1:1000) and vWF (1:1000) were obtained from Proteintech (Wuhan, China). Glyceraldehyde 3-phosphate dehydrogenase (GADPH) was used as a loading control to normalize the data.
Statistical Analysis
All data were expressed as mean ± S.E. One-way and two-way analysis of variance (ANOVA) techniques were used for data analysis of more than two groups followed by Bonferroni’s post hoc analysis. A value of P ˂ 0.05 was considered statistically significant.
Results
SP Treatment Improves Alveolarization in LPS-Treated Antenatal Mice
Firstly, we tested the effect of LPS exposure on lung alveolar development. As shown in Figure 1A–C, LPS impaired alveolar growth indicated by the decrease of alveoli numbers and increase of MLI, which implied that alveoli were larger than those of saline-treated mice. Then, we examined the effect of SP treatment on LPS-induced alveolar simplification. As expected, neonatal mice receiving SP treatment showed a significantly increased alveoli numbers (approximately 45%) and significantly reduced MLI (approximately 12%) as compared with mice receiving LPS alone.
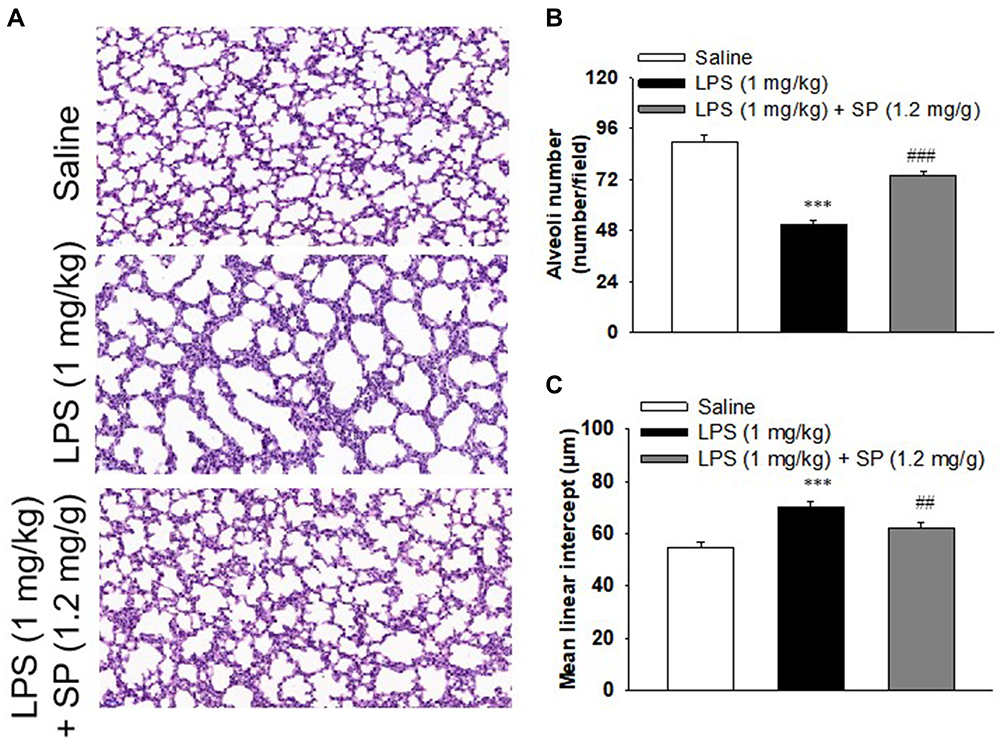 |
Figure 1 Sodium propionate (SP) treatment improves alveolarization in LPS-treated neonatal mice. When the newborn mice were at day of life 6, they received intraperitoneal injection of 1 mg/kg LPS, whereas the control mice received an equal volume injection of sterile saline solution. The LPS group were then randomly divided into two groups that received vehicle or sodium propionate (1.2 mg/g) for 7 d. (A) Representative H&E stained lung sections (magnification, ×200). (B) Quantification of alveoli numbers. (C) Quantification of mean linear intercept (MLI). ***P<0.001 vs saline; ##P<0.01 vs LPS; ###P<0.001 vs LPS. Values are mean±SE, n=6 per group. |
SP Mitigates LPS-Mediated Inflammation, Oxidative Stress and Promotes Angiogenesis During Lung Development
The organ indices of the lung and heart were observed. As shown in Figure 2A, lung weight (LW) to body weight (BW) ratio and heart weight (HW) to BW ratio were increased significantly in LPS group as compared to the Saline group. Luckily, SP treatment significantly reduced LW/BW and HW/BW ratio by approximately 20% and 12%, implying the potential effect of SP on easing lung congestion and cardiac hypertrophy, respectively. Moreover, we found that LPS-induced increase of pro-inflammatory cytokines (IL-1β, IL-6, TNF-α and IL-8) were attenuated by SP administration by approximately 32%, 54%, 35% and 62%, respectively (Figure 2B). In addition, we found that the reduced SOD activity induced by LPS stimuli was reversed with an increase of approximately 10% and 39% by SP treatment both in serum and lung tissues (Figure 2C and D), and the mRNA levels of antioxidant genes (SOD1, SOD2, Gclm and Txn) were also increased after SP administration by approximately 38%, 25%, 46% and 39%, respectively (Figure 2E). These results indicated the anti-inflammatory and anti-oxidant effects of SP against LPS-treated neonatal mice. What is more, as shown in Figure 2F–H, the protein expression of vWF, an indicator of vessel marker, was elevated approximately 62% after SP treatment, indicating the improvement of vascular development. It is generally known that Nrf2 plays a key role in the maintenance of anti-inflammation and anti-oxidant, and even involves in the regulation of developmental angiogenesis.16 Once activated, Nrf2 can release from Kelch-like ECH-associated protein-1 (Keap-1), then translocate into the nucleus to regulate downstream target gene transcription. We found that LPS-stimulated neonatal mice lung exhibited upregulated Keap-1 level, but downregulated Nrf2 protein level, while SP-treated lung displayed decreased Keap-1 (44%) and increased Nrf2 (85%) (Figure 2F and G), which suggested that SP could activate the Nrf2 pathway. Collectively, these above results illustrate a possibility that Keap-1/Nrf2 is involved in the protective effect of SP on vascular growth.
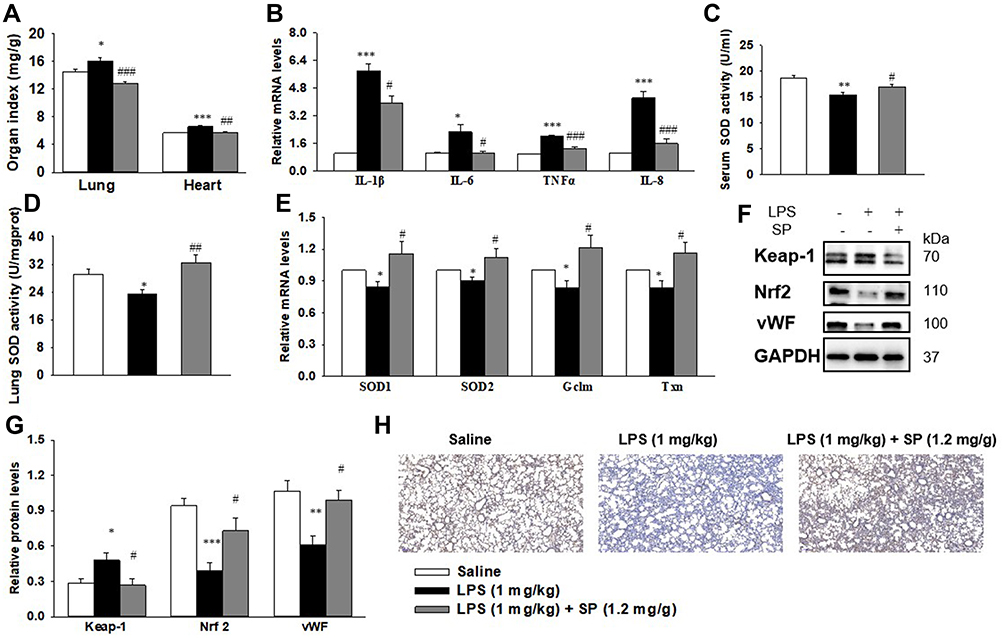 |
Figure 2 SP treatment dampens inflammation and oxidative stress, amplifies Nrf2 activation and vascularization in LPS-treated neonatal mice lung. When the newborn mice were at day of life 6, they received intraperitoneal injection of 1 mg/kg LPS, whereas the control mice received an equal volume injection of sterile saline solution. The LPS group were then randomly divided into two groups that received vehicle or sodium propionate (1.2 mg/g) for 7 d. (A) Effect of SP on lung and heart organ index. (B) Effect of SP on the mRNA expressions of inflammatory cytokines. (C and D) Effect of SP on serum and lung SOD activity, respectively. (E) Effect of SP on the mRNA expressions of SOD1, SOD2, Gclm and Txn. (F) Representative images of Western blots. (G) The protein levels of Keap-1, Nrf2 and vWF. (H) Immunohistochemistry for vWF in the lung section. *P<0.05 vs saline; **P<0.01 vs saline; ***P<0.001 vs saline; #P<0.05 vs LPS; ##P<0.01 vs LPS; ###P<0.001 vs LPS. Values are mean±SE, n=6 per group. |
The Beneficial Role of SP on LPS-Evoked Pulmonary Alveolar Remodeling is Nrf2-Dependent
Based on the aforementioned results, we further investigated whether Nrf2 was involved in the protective effect of SP on LPS-induced BPD mice. Herein, both wild-type (WT) and Nrf2 knockout (Nrf2-/-) mice were used to establish the mouse model of BPD for further studies. There was almost no Nrf2 protein level in lung tissues from Nrf2-/- mice (Figure 3A). The results of H&E staining (Figure 3B) and quantification of MLI (Figure 3C) showed that Nrf2 gene deletion exacerbated LPS-induced alveolar simplification, indicating the protective effect of Nrf2. However, SP failed to ameliorate LPS-induced alveolar simplification in Nrf2-/- mice. In addition, as shown in Figure 3E and F, SP could not suppress mRNA expressions of LPS-triggered pro-inflammatory cytokines (ie IL-6, TNF-α and IL-8) and not increase serum SOD activity and the mRNA expressions of antioxidant genes (SOD1, SOD2, Gclm and Txn) in Nrf2-/- mice. These data imply that SP partially prevents LPS-induced pulmonary alveolar remodeling dependent on the Nrf2 pathway. However, SP still reduced the LW/BW ratio and mRNA level of IL-1β in Nrf2-/- mice (Figure 3D and F).
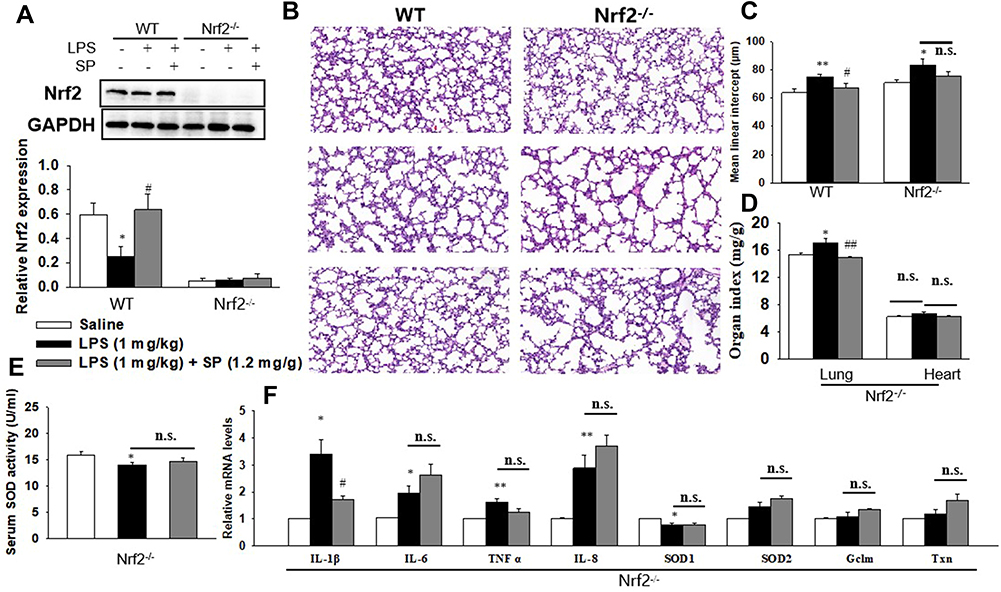 |
Figure 3 SP prevents LPS-evoked pulmonary alveolar remodeling depending on Nrf2. When the newborn mice (WT and Nrf2-/-) were at day of life 6, they received intraperitoneal injection of 1 mg/kg LPS, whereas the control mice received an equal volume injection of sterile saline solution. The LPS group were then randomly divided into two groups that received vehicle or sodium propionate (1.2 mg/g) for 7 d. (A) The protein expression of Nrf2. (B) Representative H&E stained lung sections (magnification, ×200). (C) Quantification of MLI. (D) Lung and heart organ index. (E) Serum SOD activity. (F) The mRNA levels of inflammatory cytokines and Nrf2 target genes. *P<0.05 vs saline; **P<0.01 vs saline; #P<0.05 vs LPS; ##P<0.01 vs LPS; n.s. vs no significance. Values are mean±SE, n=6 per group. |
SP Exposure Promotes Angiogenesis in HPMECs
Next, we wanted to elucidate the effect of SP on HPMECs in response to LPS stimulation. Here, SP (0.6 mM) was used according to our previous study.10 As shown in Figure 4A, SP treatment increased the cell viability by approximately 13% in HPMECs. Consistent with this, Figure 4B shows that under normal conditions, the edges of the HPMECs were smooth and spindle-shaped. After the LPS challenge, it displayed obvious vacuoles; the membrane integrity was impaired; cell boundaries were unclear and showing an irregular network structure, while SP treatment restored normal cell morphology. The tube formation ability of HPMECs was greatly impaired under LPS stimuli, whereas the addition of SP increased tube formation of HPMECs (Figure 4C). The vascular endothelial growth factor (VEGF) pathway governing vascular sprouting, proliferation and EC migration, is the primary pathway for the regulation of developmental angiogenesis.17 VEGFA, a dominant isoform of VEGF, is directly induced by HIF-1α and binds to VEGFR2, then mediates the extension of vascular networks and promotes angiogenesis.18 Thus, the mRNA expressions of the essential genes in VEGF pathway were assessed by qPCR. As shown in Figure 4D, the mRNA expressions of VEGFA, VEGFR2 and VEGFα inducer HIF-1α were decreased following exposure to LPS, but SP treatment significantly upregulated the VEGF pathway by approximately 84%, 124% and 54%, respectively. Therefore, the results implicate that SP mitigates LPS-evoked cell injury and the dysregulated angiogenesis in HPMECs, and the activated VEGF signaling is involved in the protective role of SP on vascular growth.
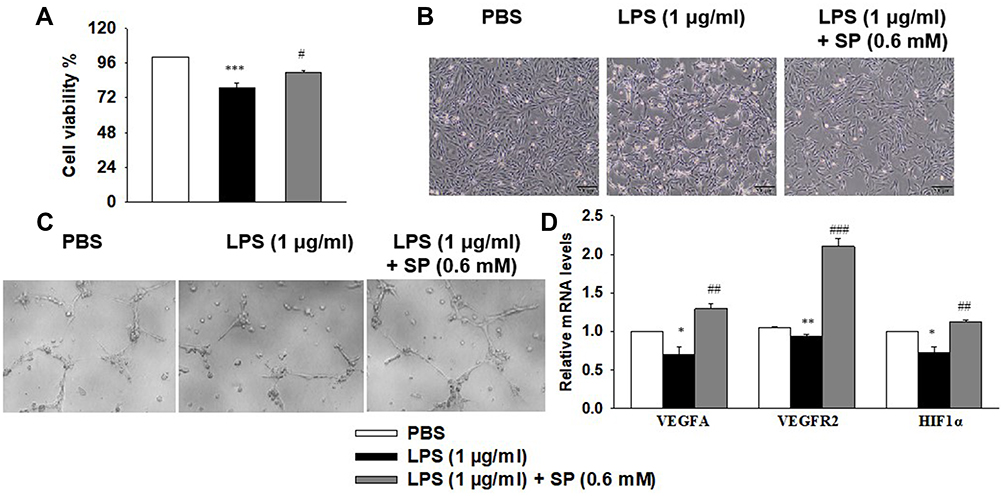 |
Figure 4 SP blocks LPS-evoked cell injury and the dysregulated angiogenesis in HPMECs. HPMECs were stimulated with LPS (1 μg/mL) for 2 h, followed by treated with SP (0.6 mM) for 24 h. (A) Cell viability was determined using the CCK-8 assay. (B) Representative cell morphology images. (C) Tube formation of HPMECs. (D) The mRNA levels of VEGFA, VEGFR1 and HIF-1α. *P<0.05 vs PBS; **P<0.01 vs PBS; ***P<0.001 vs PBS; #P<0.05 vs LPS; ##P<0.01 vs LPS; ###P<0.001 vs LPS. Values are mean±SE, n=3 per group. |
SP Administration Blocks LPS-Evoked Inflammation and NF-κB Activation in HPMECs
The anti-inflammatory effect of SP was then evaluated in HPMECs. As shown in Figure 5A, HPMECs that were stimulated with LPS showed a remarkable increase in pro-inflammatory factors, including IL-1β, IL-6, TNF-α and IL-8, while SP administration significantly inhibited the mRNA expression of these pro-inflammatory factors by approximately 39%, 59%, 67% and 61%, which strongly supported the anti-inflammatory effect of SP on LPS-induced HPMECs. Considering the production of these pro-inflammatory factors are closely regulated by transcription factor NF-κB, then the influence of SP on NF-κB signaling was investigated (Figure 5B). LPS treatment significantly elevated the phosphorylation level of NF-κB p65 compared to the PBS group (Figure 5C), which indicated that the NF-κB signaling pathway was activated by LPS in HPMECs. Importantly, SP administration significantly inhibited the phosphorylation of NF-κB p65 by approximately 27%. We then studied the subcellular translocation of NF-κB and found that NF-κB p65 levels in cytoplasmic fractions were greatly decreased and the levels in nuclear fractions were significantly enhanced in LPS-stimulated cells. From the cytosol to the nucleus, the translocation of NF-κB p65 was remarkably inhibited with an increase of approximately 28% in the cytosol and a decrease of approximately 35% in nuclear by SP administration (Figure 5D and E). In summary, these data show that SP inhibited LPS-induced NF-κB activation in HPMECs, which suggested that SP mediated the anti-inflammatory effect partly through inactivating the NF-κB pathway.
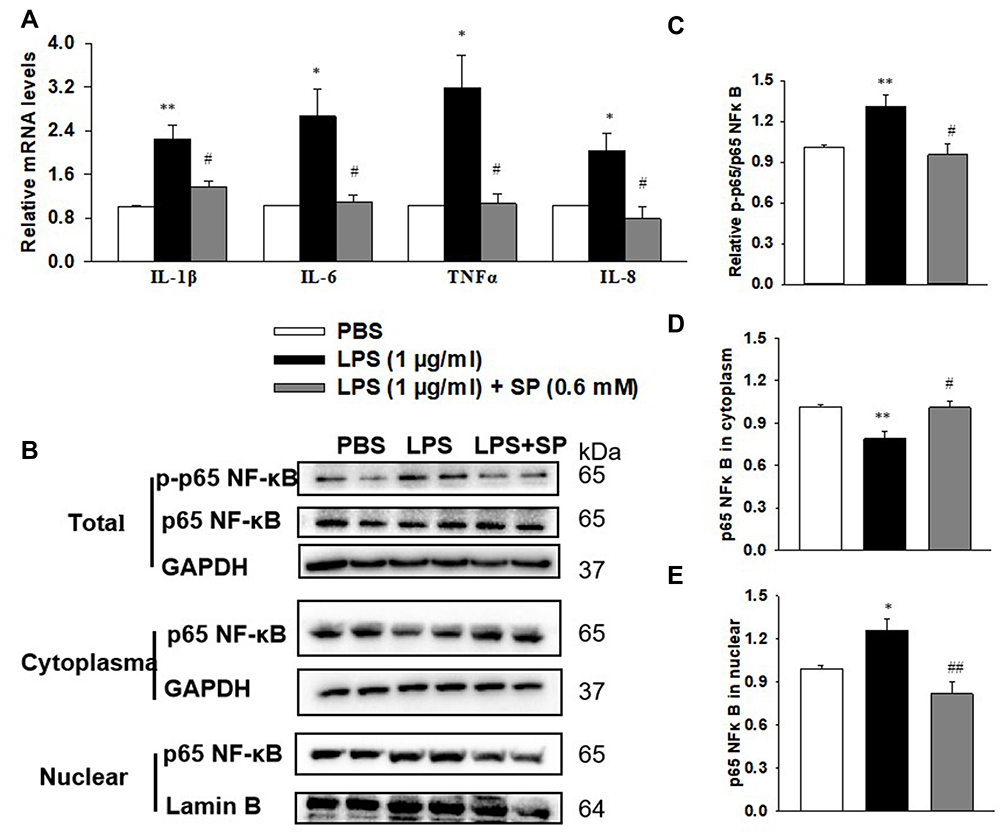 |
Figure 5 SP blocks LPS-stimulated inflammation and NF-κB activation in HPMECs. HPMECs were stimulated with LPS (1 μg/mL) for 2 h, followed by treated with SP (0.6 mM) for 24 h. (A) mRNA levels of inflammatory cytokines. (B) Representative images of Western blots. (C) Level of p65 NF-κB phosphorylation. (D) Content of p65 NF-κB in cytoplasm. (E) Content of p65 NF-κB in nuclear. *P<0.05 vs PBS; **P<0.01 vs PBS; #P<0.05 vs LPS; ##P<0.01 vs LPS. Values are mean ± SE, n=3 per group. |
SP Administration Inhibits LPS-Evoked Oxidative Stress in HPMECs
The antioxidant effect of SP was also examined in HPMECs. Intracellular ROS production was measured by DHE staining (Figure 6A and B). We found that SP significantly suppressed the generation of ROS under LPS-induced stressed conditions in HPMECs. Meanwhile, the mRNA expression of Nrf2 and its target antioxidant genes SOD1, Gclm and Txn in HPMECs were also increased by SP treatment when compared to treatment with LPS alone. Interestingly, SP had no effect on SOD2 mRNA level in HPMECs (Figure 6C). Consistent with the results of in vivo experiments, LPS-induced increased protein level of Keap-1 and decreased protein level of Nrf2 were also blocked by SP treatment in HPMECs (Figure 6D). Immunofluorescence staining showed a reduction of Nrf2 protein (green) in nuclear after stimulated by LPS, while SP treatment significantly accelerated the accumulation of Nrf2 in nuclear (Figure 6E). Taken together, these results suggest that the activated Nrf2 pathway contributes to the anti-oxidant effect of SP.
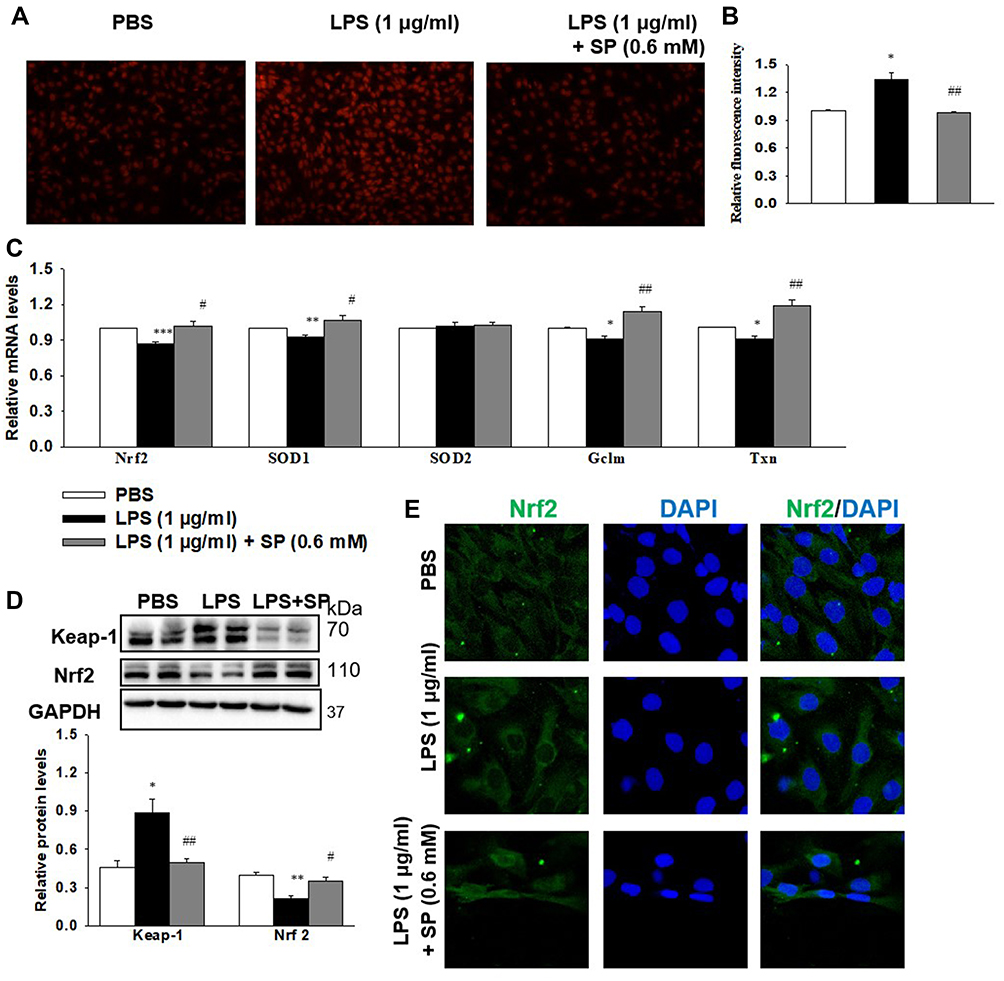 |
Figure 6 SP blocks LPS-induced oxidative stress in HPMECs. HPMECs were stimulated with LPS (1 μg/mL) for 2 h, followed by treated with SP (0.6 mM) for 24 h. (A) Intracellular ROS was determined using DHE staining. (B) Relative fluorescent intensity of DHE staining. (C) The mRNA levels of antioxidant genes. (D) Representative Western Blots and quantification of Keap-1 and Nrf2. (E) Representative images of immunofluorescence staining of Nrf2 (green). Nuclei were stained with DAPI (blue). *P<0.05 vs PBS; **P<0.01 vs PBS, ***P<0.001 vs PBS; #P<0.05 vs LPS; ##P<0.01 vs LPS. Values are mean±SE, n=3 per group. |
Inactivation of Nrf2 by ML385 Offsets the Beneficial Effect of SP Against Inflammation, Oxidative Stress and Dysregulated Angiogenesis in HPMECs
Considering the upregulation of Nrf2 activity after SP treatment in vascular development, we aimed to determine whether blocking Nrf2 activity can counteract SP’s beneficial effect on angiogenesis. ML385, a specific Nrf2 inhibitor, has been shown to significantly suppress Nrf2 activity and function. As shown in Figure 7A, the anti-inflammatory effect of SP was offset by pretreatment with ML385, since the mRNA levels of IL-1β, IL-6 and TNF-α remained no difference between the LPS and LPS+SP groups under ML385 stimuli. In addition, SP failed to increase the mRNA levels of Nrf2 target genes and angiogenesis-related genes (Figure 7B and C) when pretreatment with ML385. Moreover, there were still no changes in the production of ROS between the LPS and LPS+SP groups under ML385 stimuli (Figure 7D and E), indicating that the anti-oxidant and pro-angiogenesis effects of SP in LPS-stimulated HPMECs were all offset by pretreatment with ML385. Thus, the results reveal that SP inhibits inflammation, oxidative stress and promotes angiogenesis in an Nrf2-dependent manner.
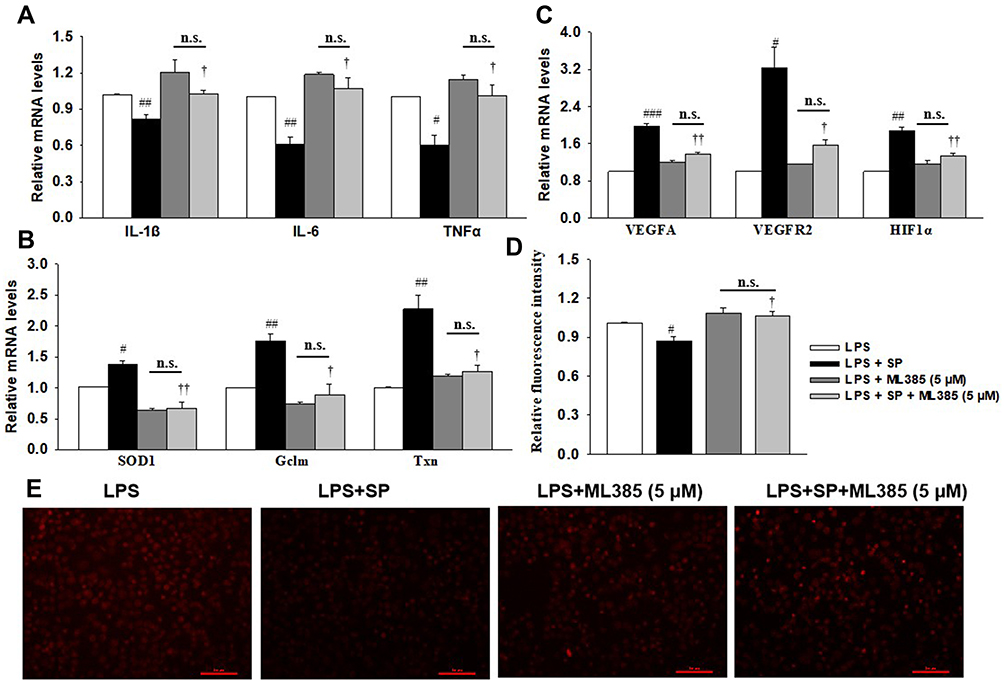 |
Figure 7 SP blocks LPS-triggered HPMECs injury depending on Nrf2 activation. HPMECs were stimulated with ML358 (5 μM) for 1 h, followed by stimulated with LPS (1 μg/mL) for 2 h and treated with SP (0.6 mM) for additional 24 h. (A) mRNA levels of inflammatory cytokines. (B) mRNA levels of antioxidant genes. (C) The mRNA levels of angiogenesis-related genes. (D) Relative fluorescent intensity of DHE staining. (E) Representative DHE staining images. #P<0.05 vs LPS; ##P<0.01 vs LPS; ###P<0.001 vs LPS; †P<0.05 vs LPS + SP; ††P<0.01 vs LPS + SP; n.s. vs no significance. Values are mean ± SE, n=3 per group. |
Discussion
In clinical setting, risk factors associated with BPD, such as hyperoxia and sepsis, trigger the production of ROS in the premature lung, which lead to alveolar simplification and impaired angiogenesis.1 Gram-negative bacterial sepsis is a common complication of preterm birth and is associated with an acute increase in ROS, lung inflammation, and the development of BPD.19 The major findings of the present study are that SP inhibits oxidative stress and NF-κB-mediated inflammation in an Nrf2-dependent manner and protects against LPS-induced lung alveolar simplification and vascular dysplasia in neonatal mice and HPMECs.
So far, several experimental mouse models have been established to mimic BPD-like disease, including hyperoxia, hypoxia, invasive ventilation, inflammation and genetic manipulation.20 However, it is sometimes complicated to develop new therapies for BPD by the lack of a standardized animal model. Clinically, hyperoxia is one of the many contributors to BPD. It has been reported that the continuous exposure of newborn mouse pups to 85% O2 from P1 to P14 led to the most pronounced impact on the gas exchange structure (cumulatively reflected by a change in a total number of alveoli, alveolar density and gas exchange surface area) as well as disturbances to alveolar septal wall thickness.21 However, models using prolonged hyperoxia may not be optimal, considering the possibility that animals exposed to noxious stimuli in the alveolar stage may have already shown signs of recovery.20 In addition to hyperoxia, neonatal sepsis is a major postnatal risk factor for developing BPD.22 Persistent perinatal lung inflammation and episodes of postnatal sepsis have consistently been identified as independent risk factors for the development of BPD.23 LPS is the major biologically active component and primary recognition structure of gram-negative bacteria and has been widely used to model sepsis in experimental animals.24 Moreover, in those studies aimed at exploring the effects of postnatal LPS exposure, LPS was mostly administered during the alveolar phase of lung development.25 Here, in our study, LPS was intraperitoneally injected during the alveolar phase of lung development (5-day-old). We found that LPS induced alveolar simplification (with fewer alveoli numbers and increased MLI) and pulmonary blood vessel density (with decreased vWF-stained lung blood vessels), indicating that LPS exposure during the alveolar phase of lung development leads to BPD in preterm infants.
SCFAs are produced mainly through saccharolytic fermentation of carbohydrates, which are not digested and absorbed in the small intestine. The major products are acetate, propionate and butyrate. It has been reported that propionate plays a key role in regulating inflammation in the airway and lung under normal and pharmacological settings. For example, propionate treatment reduced house dust mite-induced allergic inflammation in the lungs. Mice treated with propionate are protected against the development of allergic airway inflammation.26 Sodium propionate can suppress M1 Mψ and increase M2 Mψ polarization.27 Propionate levels can distinctly modulate lung immune responses in vitro and in vivo and that gut microbiome increased production of propionate is associated with reduced lung inflammation.28 The transcription factor NF-κB, a master regulator of the inflammatory response, has an important role in the pathogenesis of inflammation and angiogenesis.29 NF-κB activation is triggered by IκB phosphorylation and subsequent degradation, which causes NF-κB translocation to the nucleus and subsequent transcription of several target genes. The activation of NF-κB is closely linked to the pathogenesis of LPS-induced BPD.30 In the present study, LPS increased p65 phosphorylation and p65 NF-κB nuclear translocation, which were prevented by SP administration in HPMECs. Similarly, we found that newborn mice and HPMECs exposed to LPS showed increased mRNA expressions of pro-inflammatory factors IL-1β, TNF-α, IL-6 and IL-8, which were the important biomarkers for the prediction of adverse pulmonary outcomes in preterm infants31 and were known to be associated with the activation of NF-κB. However, SP administration reduced these mRNA levels of pro-inflammatory factors. These results show that SP inhibited NF-κB nuclear translocation and subsequently decreased the release of pro-inflammatory cytokines.
Preterm infants are particularly exposed to oxidative stress due to the transition from intrauterine hypoxia to extrauterine hyperoxia and have an increased risk of BPD. Anti-oxidative therapy strategies represent the possibility of preventing BPD.32 We found that SP administration attenuated LPS-induced alveolar simplification. Meanwhile, the activities of SOD both in serum and lung tissues and the mRNA levels of antioxidant genes (SOD1, SOD2, Gclm and Txn) were all elevated after SP administration in vivo. In addition, SP also reduced the generation of ROS and the mRNA levels of antioxidant genes in HPMECs. Interestingly, there were no changes in SOD2 mRNA expression among these groups. We guessed that SOD2 is probably specifically non-inducible in HPMECs under inflammatory conditions. These results imply that the beneficial effect of SP on alveolar development may partially attribute to its depressor effect on oxidative stress and provide a basis for SP on preventing BPD.
The redox-sensitive transcription factor Nrf2 is a major regulator of antioxidant response element-(ARE-) driven cytoprotective genes. The activation of Nrf2 signaling plays an essential role in preventing cells and tissues from injury induced by oxidative stress. The possible beneficial role of Nrf2 in BPD of preterm infants has been elucidated.33 Beyond that, curcumin, an Nrf2 activator, was reported to attenuate hyperoxic lung injury in a newborn rat model of BPD.34 Sulforaphane, a well-recognized Nrf2 inducer, could inhibit hyperoxia-induced lung inflammation in neonatal mice.35 Aurothioglucose attenuated hyperoxia-induced lung developmental deficits in newborn mice via Nrf2-dependent mechanisms.36 Here, we found that deficiency of Nrf2 and exposure to LPS stimulation in newborn mice increase the severity of alveolar development inhibition, implying the beneficial role of Nrf2 against LPS toxicity. Moreover, SP treatment significantly accelerated the accumulation of Nrf2 in nuclear, activated Nrf2 activity and promoted the expressions of Nrf2 and its target genes (SOD1, Gclm and Txn) in HPMECs. What is more, whether pretreatment with Nrf2 inhibitor ML385 in HPMECs or knockout of Nrf2 in mice, inhibiting Nrf2 can equally offset the anti-inflammatory and anti-oxidant effects of SP. These results suggest that the protective role of SP against sepsis-induced abnormal development of alveoli is Nrf2-dependent. Surprisingly, SP still reduced the LW/BW ratio and mRNA level of IL-1β in Nrf2-/- mice. As we knew, IL-1β was one of the key factors in pulmonary congestive effusion, the decreased IL-1β expression and LW/BW ratio indicating the alleviated pulmonary congestion in Nrf2-/- mice after SP administration. These results imply that the protective role of SP against LPS-induced lung injury was partly dependent on Nrf2.
In addition to alveolar simplification, abnormal angiogenesis is another major feature of BPD. It has been suggested that blood vessels in the lung actively promote normal alveolar development and contribute to the maintenance of alveolar structures throughout postnatal life.37 Abnormal angiogenesis was influenced by vascular endothelial growth factors (VEGF) and their receptors, which is essential for normal blood vessel development, and its dysregulation has a major role in BPD pathogenesis.38 VEGFA, a predominant isoform of VEGF, is directly induced by HIF-1α and binds to VEGFR2. The activation of VEGFA/VEGFR2 mediates VEGF-dependent angiogenesis and increases vessel permeability.18 Impaired VEGF signaling has been implicated in the pathogenesis of BPD and lung capillary density and the expression of VEGF and its receptor are significantly decreased both in infants dying with BPD and BPD animal models. Moreover, increased VEGF inhibits hyperoxia-induced alveolar disruption. It has been reported that erythropoietin in combination with mesenchymal stem/stromal cells significantly attenuated lung injury in a neonatal mouse model of BPD by promoting angiogenesis.39 Omega-3 polyunsaturated fatty acids (PUFA ω-3) can reverse the reduced levels of VEGFA and VEGFR2 and then significantly improve alveolarization, vascular remodeling and vascular density in a hyperoxia-induced rat model.40 Aerosolized deferoxamine administration in a mouse model of BPD activated downstream VEGF-induced angiogenesis and promoted the pulmonary vascularization and alveolarization.41 Here, in our study, we found that SP administration significantly reversed the decreased VEGF signaling pathway and promoted angiogenesis in HPMECs and in vivo under LPS stimuli. Actually, Nrf2 played an important role in the regulation of developmental angiogenesis and activation of Nrf2 blocked VEGF induction of VEGFR2-PI3K/Akt in ECs.16 In consistent with this, we found that inhibition of Nrf2 by ML385 abrogated the improvement of VEGF signaling due to SP exposure in HPMECs, indicating the involvement of the Nrf2-dependent VEGF signaling pathway in improving angiogenesis in the BPD model. These results imply that the beneficial role of SP against LPS-induced alveolar simplification and angiogenesis may be, at least partially, attributed to the upregulated Nrf2-mediated anti-inflammatory and anti-oxidant effects.
Conclusion
SP can improve alveolar simplification and abnormal angiogenesis by activating Nrf2-mediated anti-inflammatory and anti-oxidant pathway in sepsis-treated neonatal mice and HPMECs.
Abbreviations
BPD, bronchopulmonary dysplasia; GAPDH, glyceraldehyde phosphate dehydrogenase; Gclm, glutamate-cysteine ligase modifier subunit; HPMECs, human primary pulmonary microvascular endothelial cells; HIF-1α, hypoxia-inducible factor-1α; Keap-1, kelch-like ECH-associated protein-1; LDH, lactate dehydrogenase; LPS, lipopolysaccharide; MPO, myeloperoxidase; MLI, mean linear intercept; Nrf2, nuclear factor erythroid 2-related factor; NF-κB, nuclear factor-kappa B; SCFAs, short-chain fatty acids; SOD, superoxide dismutase; SP, sodium propionate; Txn, thioredoxin; TNF-α, tumor necrosis factor-α; ROS, reactive oxygen species; vWF, von Willebrand factor; VEGF, vascular endothelial growth factor.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81901522; 81702799). China Postdoctoral Science Foundation (Grant No. 2019M661730; 2020M671347). Natural Science Foundation of Jiangsu Province (BK20200602). Jiangsu Commission of Health and Family Planning (Z2020042). Wuxi health and family planning commission (Z201810); Wuxi Young and Middle-aged Medical Talents Project (BJ202075); Wuxi Key Medical Discipline (ZDXK003); Fundamental Research Funds for the Central Universities (JUSRP11955); Public Health Research center at Jiangnan University (JUPH201805).
Disclosure
The authors have no conflicts of interest to report.
References
1. Thebaud B, Goss KN, Laughon M, et al. Bronchopulmonary dysplasia. Nat Rev Dis Primers. 2019;5(1):78.
2. Lal CV, Travers C, Aghai ZH, et al. The airway microbiome at birth. Sci Rep. 2016;6(1):31023. doi:10.1038/srep31023
3. Benjamin JT, Smith RJ, Halloran BA, Day TJ, Kelly DR, Prince LS. FGF-10 is decreased in bronchopulmonary dysplasia and suppressed by toll-like receptor activation. Am J Physiol Lung Cell Mol Physiol. 2007;292(2):L550–558. doi:10.1152/ajplung.00329.2006
4. Chou HC, Li YT, Chen CM. Human mesenchymal stem cells attenuate experimental bronchopulmonary dysplasia induced by perinatal inflammation and hyperoxia. Am J Transl Res. 2016;8(2):342–353.
5. Woik N, Kroll J. Regulation of lung development and regeneration by the vascular system. Cell Mol Life Sci. 2015;72(14):2709–2718. doi:10.1007/s00018-015-1907-1
6. Liu D, Andrade SP, Castro PR, Treacy J, Ashworth J, Slevin M. Low concentration of sodium butyrate from ultrabraid+nabu suture, promotes angiogenesis and tissue remodelling in tendon-bones injury. Sci Rep. 2016;6(1):34649. doi:10.1038/srep34649
7. Filippone A, Lanza M, Campolo M, et al. The anti-inflammatory and antioxidant effects of sodium propionate. Int J Mol Sci. 2020;21(8):8. doi:10.3390/ijms21083026
8. Song B, Zhong YZ, Zheng CB, Li FN, Duan YH, Deng JP. Propionate alleviates high-fat diet-induced lipid dysmetabolism by modulating gut microbiota in mice. J Appl Microbiol. 2019;127(5):1546–1555. doi:10.1111/jam.14389
9. Wang J, Wei Z, Zhang X, Wang Y, Yang Z, Fu Y. Propionate protects against lipopolysaccharide-induced mastitis in mice by restoring blood-milk barrier disruption and suppressing inflammatory response. Front Immunol. 2017;8:1108. doi:10.3389/fimmu.2017.01108
10. Chen D, Qiu YB, Gao ZQ, et al. Sodium propionate attenuates the lipopolysaccharide-induced epithelial-mesenchymal transition via the PI3K/Akt/mTOR signaling pathway. J Agric Food Chem. 2020;68(24):6554–6563. doi:10.1021/acs.jafc.0c01302
11. Menden HL, Xia S, Mabry SM, Navarro A, Nyp MF, Sampath V. Nicotinamide adenine dinucleotide phosphate oxidase 2 regulates LPS-induced inflammation and alveolar remodeling in the developing lung. Am J Respir Cell Mol Biol. 2016;55(6):767–778. doi:10.1165/rcmb.2016-0006OC
12. Nguyen L, Castro O, De Dios R, Sandoval J, McKenna S, Wright CJ. Sex-differences in LPS-induced neonatal lung injury. Sci Rep. 2019;9(1):8514. doi:10.1038/s41598-019-44955-0
13. Liu C, Chen Z, Li W, Huang L, Zhang Y. Vitamin D enhances alveolar development in antenatal lipopolysaccharide-treated rats through the suppression of interferon-gamma production. Front Immunol. 2017;8:1923. doi:10.3389/fimmu.2017.01923
14. Ding R, Zhao D, Li X, Liu B, Ma X. Rho-kinase inhibitor treatment prevents pulmonary inflammation and coagulation in lipopolysaccharide-induced lung injury. Thromb Res. 2017;150:59–64. doi:10.1016/j.thromres.2016.12.020
15. Chen D, Zang YH, Qiu Y, et al. BCL6 attenuates proliferation and oxidative stress of vascular smooth muscle cells in hypertension. Oxid Med Cell Longev. 2019;2019:5018410.
16. Wei Y, Gong J, Thimmulappa RK, Kosmider B, Biswal S, Duh EJ. Nrf2 acts cell-autonomously in endothelium to regulate tip cell formation and vascular branching. Proc Natl Acad Sci U S A. 2013;110(41):E3910–3918. doi:10.1073/pnas.1309276110
17. Wu JB, Tang YL, Liang XH. Targeting VEGF pathway to normalize the vasculature: an emerging insight in cancer therapy. Onco Targets Ther. 2018;11:6901–6909. doi:10.2147/OTT.S172042
18. Pitulescu ME, Schmidt I, Giaimo BD, et al. Dll4 and notch signalling couples sprouting angiogenesis and artery formation. Nat Cell Biol. 2017;19(8):915–927. doi:10.1038/ncb3555
19. Guo RF, Ward PA. Role of oxidants in lung injury during sepsis. Antioxid Redox Signal. 2007;9(11):1991–2002. doi:10.1089/ars.2007.1785
20. Berger J, Bhandari V. Animal models of bronchopulmonary dysplasia. The term mouse models. Am J Physiol Lung Cell Mol Physiol. 2014;307(12):L936–947. doi:10.1152/ajplung.00159.2014
21. Nardiello C, Mizikova I, Silva DM, et al. Standardisation of oxygen exposure in the development of mouse models for bronchopulmonary dysplasia. Dis Model Mech. 2017;10(2):185–196. doi:10.1242/dmm.027086
22. Shrestha AK, Bettini ML, Menon RT, et al. Consequences of early postnatal lipopolysaccharide exposure on developing lungs in mice. Am J Physiol Lung Cell Mol Physiol. 2019;316(1):L229–L244. doi:10.1152/ajplung.00560.2017
23. Van Marter LJ, Dammann O, Allred EN, et al. Chorioamnionitis, mechanical ventilation, and postnatal sepsis as modulators of chronic lung disease in preterm infants. J Pediatr. 2002;140(2):171–176. doi:10.1067/mpd.2002.121381
24. Fink MP. Animal models of sepsis. Virulence. 2014;5(1):143–153. doi:10.4161/viru.26083
25. Menden H, Welak S, Cossette S, Ramchandran R, Sampath V. Lipopolysaccharide (LPS)-mediated angiopoietin-2-dependent autocrine angiogenesis is regulated by NADPH oxidase 2 (Nox2) in human pulmonary microvascular endothelial cells. J Biol Chem. 2015;290(9):5449–5461. doi:10.1074/jbc.M114.600692
26. Trompette A, Gollwitzer ES, Yadava K, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. 2014;20(2):159–166. doi:10.1038/nm.3444
27. Wang Z, Zhang X, Zhu L, et al. Inulin alleviates inflammation of alcoholic liver disease via SCFAs-inducing suppression of M1 and facilitation of M2 macrophages in mice. Int Immunopharmacol. 2020;78:106062. doi:10.1016/j.intimp.2019.106062
28. Tian X, Hellman J, Horswill AR, Crosby HA, Francis KP, Prakash A. Elevated gut microbiome-derived propionate levels are associated with reduced sterile lung inflammation and bacterial immunity in mice. Front Microbiol. 2019;10:159. doi:10.3389/fmicb.2019.00159
29. Iosef C, Alastalo TP, Hou Y, et al. Inhibiting NF-kappaB in the developing lung disrupts angiogenesis and alveolarization. Am J Physiol Lung Cell Mol Physiol. 2012;302(10):L1023–1036. doi:10.1152/ajplung.00230.2011
30. Dodson RB, Powers KN, Gien J, et al. Intrauterine growth restriction decreases NF-kappaB signaling in fetal pulmonary artery endothelial cells of fetal sheep. Am J Physiol Lung Cell Mol Physiol. 2018;315(3):L348–L359. doi:10.1152/ajplung.00052.2018
31. Bose CL, Dammann CE, Laughon MM. Bronchopulmonary dysplasia and inflammatory biomarkers in the premature neonate. Arch Dis Child Fetal Neonatal Ed. 2008;93(6):F455–461. doi:10.1136/adc.2007.121327
32. Poggi C, Dani C. Antioxidant strategies and respiratory disease of the preterm newborn: an update. Oxid Med Cell Longev. 2014;2014:721043. doi:10.1155/2014/721043
33. Cho HY, van Houten B, Wang X, et al. Targeted deletion of nrf2 impairs lung development and oxidant injury in neonatal mice. Antioxid Redox Signal. 2012;17(8):1066–1082. doi:10.1089/ars.2011.4288
34. Sakurai R, Villarreal P, Husain S, et al. Curcumin protects the developing lung against long-term hyperoxic injury. Am J Physiol Lung Cell Mol Physiol. 2013;305(4):L301–311. doi:10.1152/ajplung.00082.2013
35. McGrath-Morrow SA, Lauer T, Collaco JM, et al. Transcriptional responses of neonatal mouse lung to hyperoxia by Nrf2 status. Cytokine. 2014;65(1):4–9. doi:10.1016/j.cyto.2013.09.021
36. Li Q, Wall SB, Ren C, et al. Thioredoxin reductase inhibition attenuates neonatal hyperoxic lung injury and enhances nuclear factor e2-related factor 2 activation. Am J Respir Cell Mol Biol. 2016;55(3):419–428. doi:10.1165/rcmb.2015-0228OC
37. Thebaud B, Ladha F, Michelakis ED, et al. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: evidence that angiogenesis participates in alveolarization. Circulation. 2005;112(16):2477–2486. doi:10.1161/CIRCULATIONAHA.105.541524
38. Levesque BM, Kalish LA, Winston AB, et al. Low urine vascular endothelial growth factor levels are associated with mechanical ventilation, bronchopulmonary dysplasia and retinopathy of prematurity. Neonatology. 2013;104(1):56–64. doi:10.1159/000351040
39. Sun C, Zhang S, Wang J, et al. EPO enhances the protective effects of MSCs in experimental hyperoxia-induced neonatal mice by promoting angiogenesis. Aging (Albany NY). 2019;11(8):2477–2487. doi:10.18632/aging.101937
40. Zhong Y, Catheline D, Houeijeh A, et al. Maternal omega-3 PUFA supplementation prevents hyperoxia-induced pulmonary hypertension in the offspring. Am J Physiol Lung Cell Mol Physiol. 2018;315(1):L116–L132. doi:10.1152/ajplung.00527.2017
41. Chen Y, Gao S, Yan Y, Qian J, Chen H. Aerosolized deferoxamine administration in mouse model of bronchopulmonary dysplasia improve pulmonary development. Am J Transl Res. 2018;10(1):325–332.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.