Back to Journals » OncoTargets and Therapy » Volume 13
Sodium Butyrate Selectively Kills Cancer Cells and Inhibits Migration in Colorectal Cancer by Targeting Thioredoxin-1
Authors Wang W ![]() , Fang D, Zhang H, Xue J, Wangchuk D, Du J
, Fang D, Zhang H, Xue J, Wangchuk D, Du J ![]() , Jiang L
, Jiang L ![]()
Received 22 October 2019
Accepted for publication 7 May 2020
Published 27 May 2020 Volume 2020:13 Pages 4691—4704
DOI https://doi.org/10.2147/OTT.S235575
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianmin Xu
Wenqi Wang,1,2,* Daoquan Fang,3,* Hao Zhang,1 Jiao Xue,1 Drugyel Wangchuk,4 Jimei Du,1 Lei Jiang3
1Department of Microbiology and Immunology, School of Laboratory Medicine, Wenzhou Medical University, Wenzhou 325000, People’s Republic of China; 2Department of Laboratory Medicine, Shanghai University of Medicine & Health Sciences affiliated Zhoupu Hospital, Shanghai 201318, People’s Republic of China; 3Central Laboratory, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou 325000, People’s Republic of China; 4Department of General Surgery, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou 325000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lei Jiang
Central Laboratory, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou 325000, Zhejiang, People’s Republic of China
Tel +86-577-55579127
Fax +86-577-55578999
Email [email protected]
Jimei Du
Department of Microbiology and Immunology, School of Laboratory Medicine, Wenzhou Medical University, Wenzhou 325000, People’s Republic of China
Email [email protected]
Background: Sodium butyrate (NaB) is a short-chain fatty acid which is produced by bacterial fermentation of nondigestible dietary fiber and has been reported to exert anti-tumor effects in many tumors including colorectal cancer (CRC). However, the role of thioredoxin-1 (Trx-1) in NaB-induced anti-tumor effect has not been completely clarified.
Materials and Methods: Effects of NaB on the growth of CRC cell lines HT29 and SW480 were detected by the Cell Counting Kit-8 (CCK-8) and colony formation assays. The apoptotic cells were determined by flow cytometry, and cell migration was assessed by a Transwell assay. Western blot analysis was used to test the Trx-1 and epithelial-to-mesenchymal transition (EMT)-related proteins level. Reactive oxygen species (ROS) level was determined and N-acetylcysteine (NAC) recovery experiment was performed in CRC cells. In addition, mice xenograft model was established to test the effect of NaB on CRC growth in vivo. Further, the effects of NaB on CRC cells with overexpression or knockdown were tested by the CCK-8 and Transwell assays.
Results: NaB treatment significantly inhibited cell growth and decreased Trx-1 protein expression in CRC cells but not in normal colon epithelial cells. NaB also induced apoptosis, inhibited colony formation, migration and EMT in CRC cells. Besides, NaB increased ROS level in CRC cells and NAC reversed NaB-induced inhibition of cell proliferation. Moreover, downregulation of Trx-1 significantly enhanced NaB-induced inhibitory effects on cell growth and migration, whereas overexpression of Trx-1 attenuated NaB-induced inhibitory effects on growth and migration in CRC cells.
Conclusion: These findings indicate that the NaB-mediated anti-tumor effects on CRC cells are related to downregulation of Trx-1.
Keywords: anti-tumor effects, colorectal cancer, short-chain fatty acid, sodium butyrate, thioredoxin-1
Introduction
It is estimated that more than 1.8 million new cases of colorectal cancer (CRC) and 881,000 deaths occur in 2018, accounting for one-tenth of cancer cases and deaths.1 The overall five-year survival rate of CRC is gradually increasing with the advancement of surgical techniques and the emergence of new therapeutic drugs, but CRC is still one of the important causes of tumor-related death.2
The intestinal micro-ecological system is composed of the normal intestinal flora and the environment in which it lives.3 The tumor microenvironment induced by colorectal tumor lesions is significantly different from the normal intestinal environment.4 Short-chain fatty acids (SCFAs) are metabolites produced by intestinal microbial fermentation of undigested dietary fiber.5 CRC cells are more sensitive to SCFAs than normal intestinal epithelial cells, suggesting that SCFAs affect tumor cells through some certain pathways.6 As a member of SCFAs, Sodium Butyrate (NaB) induces histone acetylation as a deacetylase inhibitor.7 Many studies show that NaB acts as a negative regulator in various types of cancer.8–10 NaB has been found to inhibit cell proliferation and migration of colorectal cancer, promote CRC cell apoptosis and induce autophagy and cell death.10–14
The previously reported the correlation between NaB and Oxidative stress.15 Numerous studies have shown that NaB significantly induces reactive oxygen species (ROS)-mediated apoptosis in a variety of cancers.16–18 Trx-1, a small endogenous protein, widely participates in various redox reactions.19 This multifunctional protein acts to inhibit inflammation and cancer through its antioxidant function.20 Trx-1 is highly expressed in several human primary tumors including CRC.21–25 In our previous study, we demonstrated that Trx-1 induces epithelial–mesenchymal transition (EMT) and metastasis by activating Akt via interplay with S100P in CRC cells.25,26 However, little is known about the role of Trx-1 in NaB-induced anti-tumor effect in CRC.
In this study, we demonstrated the antitumor activity of NaB against CRC cells in vitro and in vivo. We also identified that NaB inhibits CRC cell proliferation and down-regulated Trx1 expression in CRC cells but not in normal colon epithelial cells. NaB down-regulated Trx1 expression and resulted in increased ROS production, and inhibits cell proliferation and migration in CRC cells.
Materials and Methods
Cell Culture
Human CRC cell lines HT-29 and SW480 were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Human colon epithelial cell line FHC was obtained from the American Type Culture Collection (Manassas, VA, USA). SW480 and FHC cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, MA, USA). HT-29 cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS. Cells were cultured in a humidified 37 °C incubator with 5% CO2.
Cell Viability Assays
The Cell-counting Kit-8 (CCK-8, Dojindo, Japan) assays were used to determine the effects of NaB treatment on cell viability. Briefly, the cells were seeded in a 96-well plate at densities of 5,000 cells/well and incubated with NaB for the indicated time. At the end of treatment, CCK-8 reagent (10 µL) was added to each well. After incubation for 2 h at 37 °C, the absorbance of each well was measured at a wavelength of 450 nm. The survival rate of NaB treatment was estimated in the base of the absence of NaB treated cells as the 100% of survival. Percent growth inhibition of cells induced by NaB was calculated as follows: Percentage of inhibition = 100 − (treated OD/untreated OD)×100.27 All experiments were performed in triplicate.
Cell Colony Formation Assays
CRC cells treated with NaB and control were seeded in a six-well plate at a density of 1,000 cells/well and cultured for 14 days to form colonies. Colonies were fixed with 4% paraformaldehyde and stained with 0.5% crystal violet. The assays were performed three times for each treatment.
Cell Apoptosis
Cells were treated with 0, 1.25, 2.5, or 5 mM NaB for 48 h and then apoptotic cell death were determined using an Annexin V Apoptosis Detection kit (Multi Sciences, China). Briefly, cells were resuspended in binding buffer at a density of 1×106 cells/mL and stained with Annexin V-FITC and PI. The cells were analyzed with the BD FACSCalibur flow cytometer (Franklin Lakes, NJ, USA). Data analysis was performed using FlowJo software (Treestar, OR, USA).
Transwell Assays
Migration assays of HT-29 and SW480 cells were performed in 8-µm 24-well Boyden chambers (Corning, Cambridge, MA, USA). CRC cells (1×105) suspended in 200 μL RPMI 1640 medium were added to the upper chamber. Medium with 20% FBS was added to the bottom chamber as a chemoattractant. SW480 and HT-29 cells were left to migrate for 24 h and 48 h, respectively. The migrated cells on the lower surface of the membrane were stained with 0.1% of crystal violet. Quantification was performed by counting the mean number of cells in five randomly selected microscopy fields per chamber.
Determination of Cellular Reactive Oxygen Species (ROS)
To measure cellular ROS level, cells were stained with DCFH-DA (Nanjing Jiancheng Bioengineering Institute, China) which generates fluorescent signals when oxidized by ROS in the cells, and analyzed by flow cytometry (BD FACSCalibur). Data analysis was performed using FlowJo software. For expressing GFP cells, the ROS production was measured using the Cellular ROS Detection Assay Kit (Abcam) according to the manufacturer’s instructions.
Quantitative Reverse Transcription PCR (qRT-PCR)
Total RNA was isolated from cells using TRIzol reagent (Invitrogen). After detection of RNA concentration, 1 µg of total RNA was reverse-transcribed into cDNA with random primers, using the HiScript II Q Select RT SuperMix (Vazyme, China). The amplification reaction was performed using TB Green™ Premix Ex Taq™ (Takara, Japan) and an ABI 7500 Real-Time PCR System (Applied Biosystems, Warrington, U.K.). The human GAPDH gene was used as an internal control. The 2−ΔΔCT method was used to calculate the relative expression of target genes. All the samples were run in triplicate. The PCR primers sequences are as follows:25 Trx-1-F: 5ʹ-CAACCCTTTCTTTCATTCCCTCT-3ʹ, Trx-1-R: 5ʹ-CACCCA CCTTTTGTCCCTTCT-3ʹ; GAPDH-F: 5ʹ-CCAG CCGAGCCACATCGCTC-3ʹ, GAPDH-R: 5ʹ-ATGAGCCCCAGCCTTCTCCAT-3ʹ.
Western Blot Analysis
Protein was extracted from cells using RIPA buffer and separated by SDS–polyacrylamide gels and then transferred to PVDF membranes (Millipore, Billerica, MA, USA). Primary antibodies against Trx-1 (1:1000; #ab26320, Abcam, Cambridge, UK), E-Cadherin (1:1000; #3195, Cell Signaling Technology, Beverly, MA, USA), N-Cadherin (1:1000; #14215, Cell Signaling Technology), Vimentin (1:1000; #550513, BD Biosciences, NJ, USA), and GAPDH (1:1000; #ab011, Multi Sciences, China) were used. Peroxidase-conjugated secondary antibody (Cell Signaling Technology) was used, and the antigen–antibody reaction was visualized by enhance chemiluminescence assay (ECL, Thermo Fisher Scientific).
Lentiviral Vector Construction and Transduction
Lentiviral vector expressing human Trx-1 gene or shRNA targeting Trx-1 (shTrx-1: 5ʹ- GAC TGT CAG GAT GTT GCT TCA GAG TGT GA -3ʹ) was generated in our lab previously.25 Lentivirus production and transduction were performed according to the protocols described previously.25 Cells transduced with a lentiviral vector expressing GFP gene or shRNA targeting luciferase gene (shLuc) was as a control group in overexpression and knockdown experiments, respectively.
In vivo Tumorigenesis Assays
Six-week-old male athymic nude mice (nu/nu) were purchased from Shanghai SLAC Laboratory Animal Co. Ltd (Shanghai, China). All experiments were approved by the Animal Experimental Ethics Committee of Wenzhou Medical University (wydw2019-0507) and carried out according to the National Institutes of Health guidelines for the care and use of laboratory animals. SW480 cells (1×106) were subcutaneously into the right flanks of each mouse. The volume of the tumor (mm3) was calculated as follows: Volume = 0.5 × length × (width)2. When the tumor volume reached about 250 mm3, the mice were randomly divided into the control group (n = 5) and the experimental group (n = 5). The experimental group was injected with 0.2 mL NaB solution (600 mg/kg/d), and the control group was injected with saline of the same volume. Body weight and tumor volume were measured every 3 days. All mice were killed when the tumor volume reached 1000 mm3 in the control group.
Statistical Analysis
Statistical analysis was performed with SPSS19.0 software package (SPSS Company, Inc., Chicago, IL, USA). All data are presented as mean ± standard deviation. Comparisons of the continuous variables between 2 groups were performed with an independent samples t-test. All statistical tests were two sided, and P < 0.05 was considered to be statistically significant.
Result
NaB Inhibits Cell Growth and Protein Expression of Trx-1 in CRC Cells
To investigate the effects of NaB on cell growth of CRC cells and normal colon epithelial cells, CRC cell lines HT-29 and SW480, and a cell line came from human normal colorectal mucosa, FHC, were treated with NaB and CCK-8 assays were performed to assess the cell viability. As shown in Figure 1A, NaB decreased the viability of CRC HT-29 and SW480 cells in an apparent dose- and time-dependent manner. However, NaB had no significant cytotoxic effect on FHC cells at 24 h and 48 h (Figure 1A and B). The protein expression levels of Trx-1 were suppressed by NaB in HT-29 and SW480 cells but not in FHC cells (Figure 1C–E).
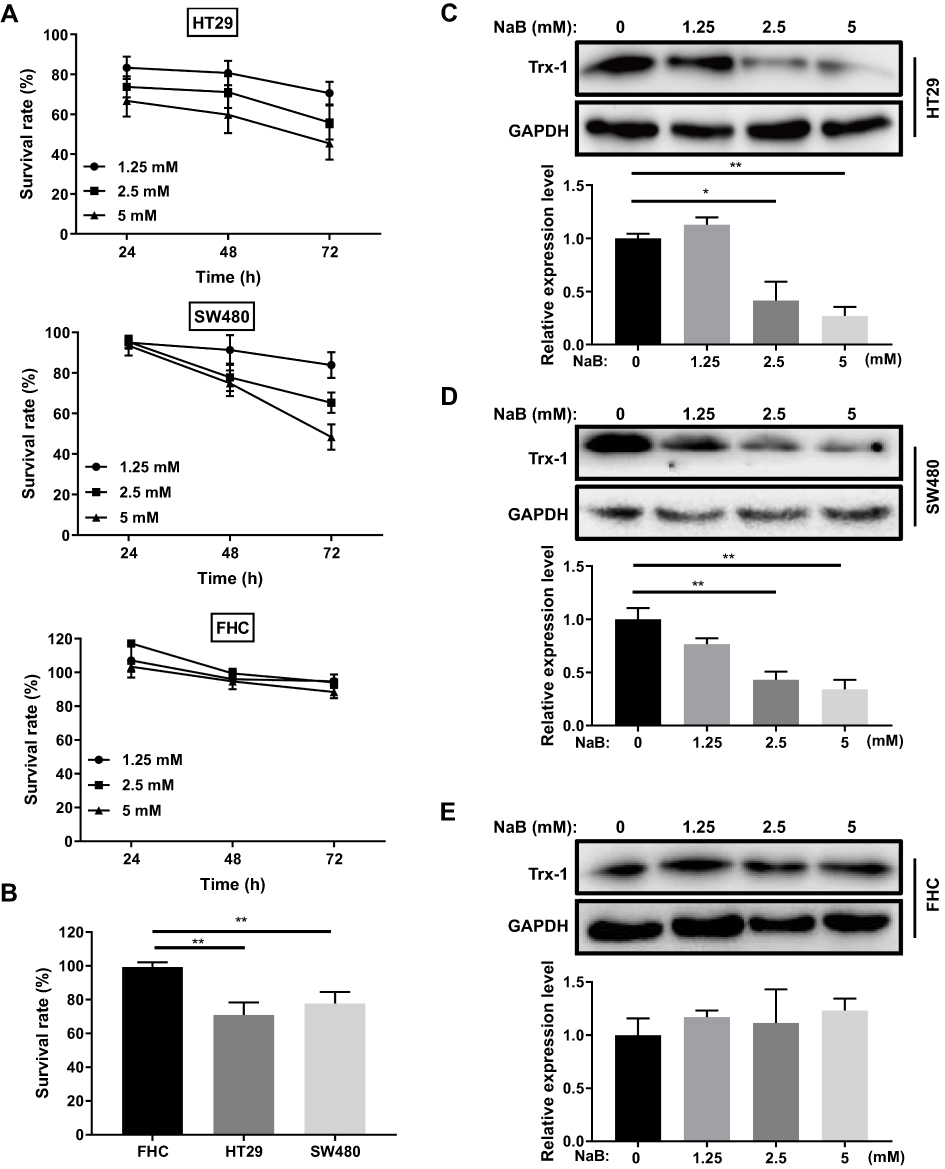 |
Figure 1 The effects of NaB on cell growth and Trx-1 expression in colorectal cancer cell lines and normal colon epithelial cell line. (A) Cell-counting Kit-8 assays were performed to determine the percentage of viable cells. Colorectal cancer cell lines (HT-29 and SW480) and normal colon epithelial cell line (FHC) were treated with different concentrations of NaB for 24 h, 48 h or 72 h. (B) NaB treatment induced growth inhibition in colorectal cancer cells but not in normal colon epithelial cells. Colorectal cancer cell lines (HT-29 and SW480) and normal colon epithelial cell line (FHC) were treated with NaB (2.5 mM) for 48 h. Cell viability was determined by Cell-counting Kit-8 assays. (C) The protein expression levels of Trx-1 were significantly inhibited by NaB treatment in HT-29 cells. (D) The protein expression levels of Trx-1 were significantly inhibited by NaB treatment in SW480 cells. (E) The protein expression levels of Trx-1 were not affected by NaB treatment in normal colon epithelial FHC cells. Cells were treated with the indicated concentrations of NaB for 48 h and then Trx-1 expression was detected by Western blotting. *P < 0.05; **P < 0.01. Abbreviations: GAPDH, glyceraldehyde 3-phosphate dehydrogenase; NaB, sodium butyrate; Trx-1, thioredoxin 1. |
NaB Induces Apoptosis and Inhibits Colony Formation, Cell Migration and EMT in CRC Cells
The level of cell apoptosis was detected by Annexin V-FITC and PI staining. We found that NaB treatment induced the apoptosis of HT-29 and SW480 cells in a dose-dependent manner (Figure 2A). When the cells were treated with 0, 2.5, 5 mM NaB for 48 h, the average apoptosis rate of HT-29 cells significantly increased from 5.17 ± 0.97% in control to 11.83 ± 1.28% (P < 0.01) and 19.57 ± 5.16% (P < 0.01), respectively; the average apoptosis rate of SW480 cells significantly increased from 7.98 ± 3.15% in control to 18.25 ± 4.27% (P < 0.05) and 27.74 ± 0.89% (P < 0.01), respectively (P-value compared with the control group). We also performed colony formation assays to evaluate the effects of NaB treatment on CRC cell growth. Compared with the control groups, HT29 and SW480 cells treated with NaB showed fewer and smaller colonies (Figure 2B). To determine the effects of NaB on cell migration, we performed the transwell assays in HT29 and SW480 cells. The assays showed that NaB treatment resulted in a significantly impaired migration ability of CRC cells (Figure 3A). The Western blot results presented in Figure 3B showed that the expression levels of mesenchymal markers (including N-cadherin and Vimentin) decreased while the expression level of the epithelial marker, E-cadherin, increased by NaB treatment, indicated that NaB treatment inhibited CRC cells EMT.
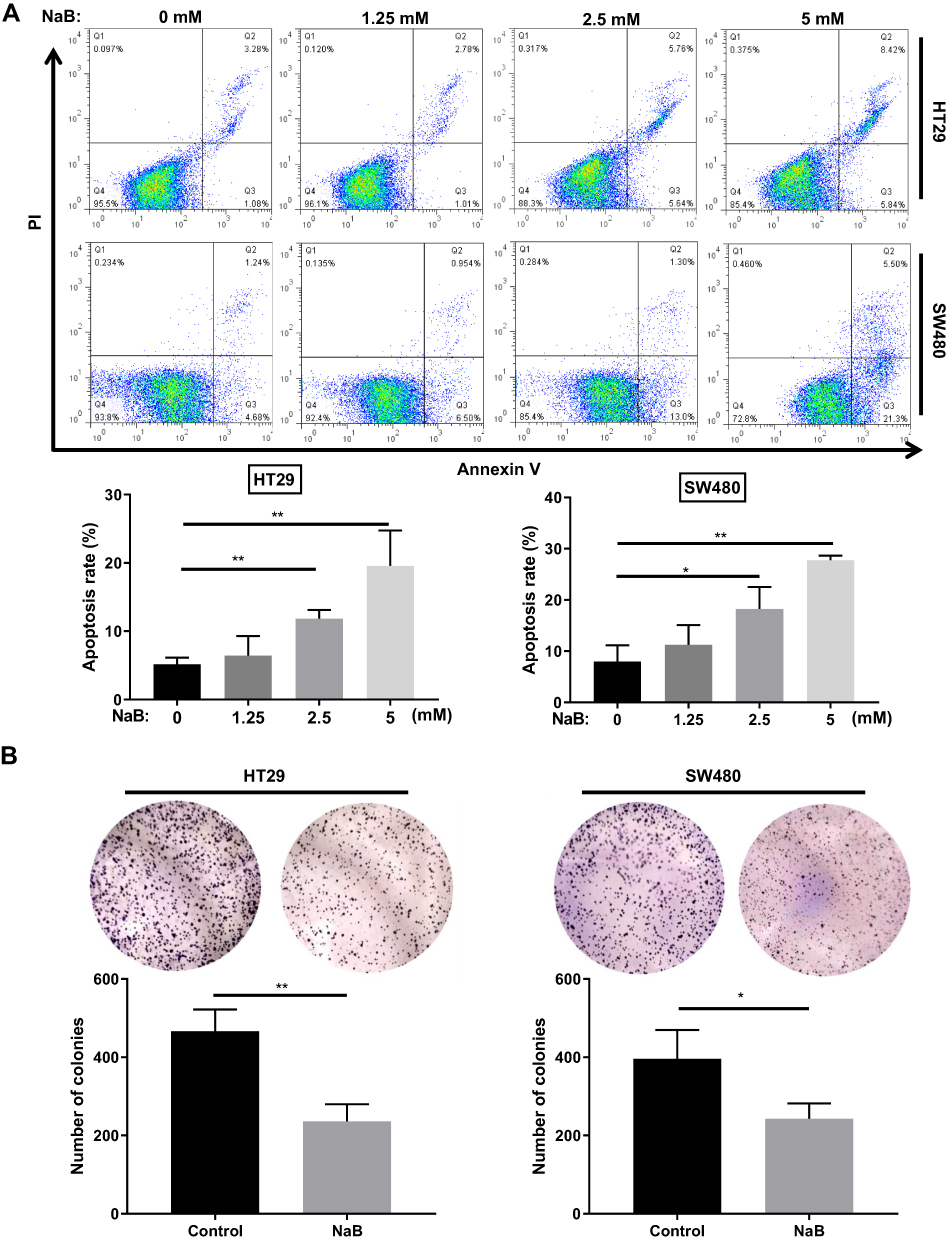 |
Figure 2 NaB induces apoptosis and inhibits colony formation in colorectal cancer cells. (A) NaB significantly increased the apoptosis in HT-29 and SW480 cells as demonstrated by Annexin V/PI staining. Cells were treated with 0, 1.25, 2.5, or 5 mM NaB for 48 h. (B) NaB significantly reduced the colony formation in HT-29 and SW480 cells. Cells were treated with 2.5 mM NaB for 48 h and then cell colony formation assay was performed. *P < 0.05; **P < 0.01. Abbreviations: NaB, sodium butyrate; PI, propidium iodide. |
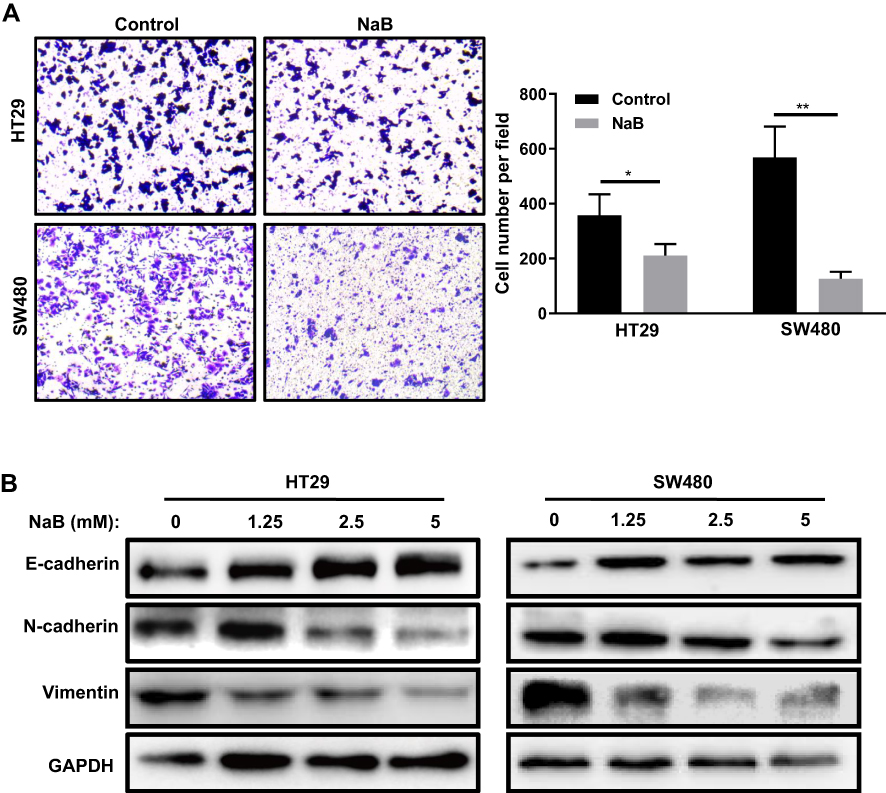 |
Figure 3 NaB inhibits cell migration and epithelial-to-mesenchymal transition in colorectal cancer cells. (A) NaB treatment significantly reduced cell migration in HT-29 and SW480 cells. Cells were treated with 2.5 mM NaB for 48 h and then the transwell cell migration assay was performed. (B) The expression levels of the epithelial-to-mesenchymal transition markers E-cadherin, N-cadherin and Vimentin were detected by Western blotting in HT-29 and SW480 cells treated with NaB (0, 1.25, 2.5, or 5 mM) for 48 h. GAPDH was used as an internal control. *P < 0.05; **P < 0.01. Abbreviations: GAPDH, glyceraldehyde 3-phosphate dehydrogenase; NaB, sodium butyrate. |
NaB Inhibits Tumor Growth and Protein Expression of Trx-1 in vivo
To examine the effects of NaB on tumor growth in vivo, nude mice were subcutaneously injected with SW480 cells and then were treated with NaB. NaB treatment significantly inhibited tumor growth in vivo (Figure 4A and B). The volume of tumors in NaB treatment group was significantly smaller than that in the control group at the end of treatment (Figure 4A, P < 0.05). We also examined the protein expression level of Trx-1 in tumor tissues and found that NaB treatment significantly decreased Trx-1 protein expressions (Figure 4C).
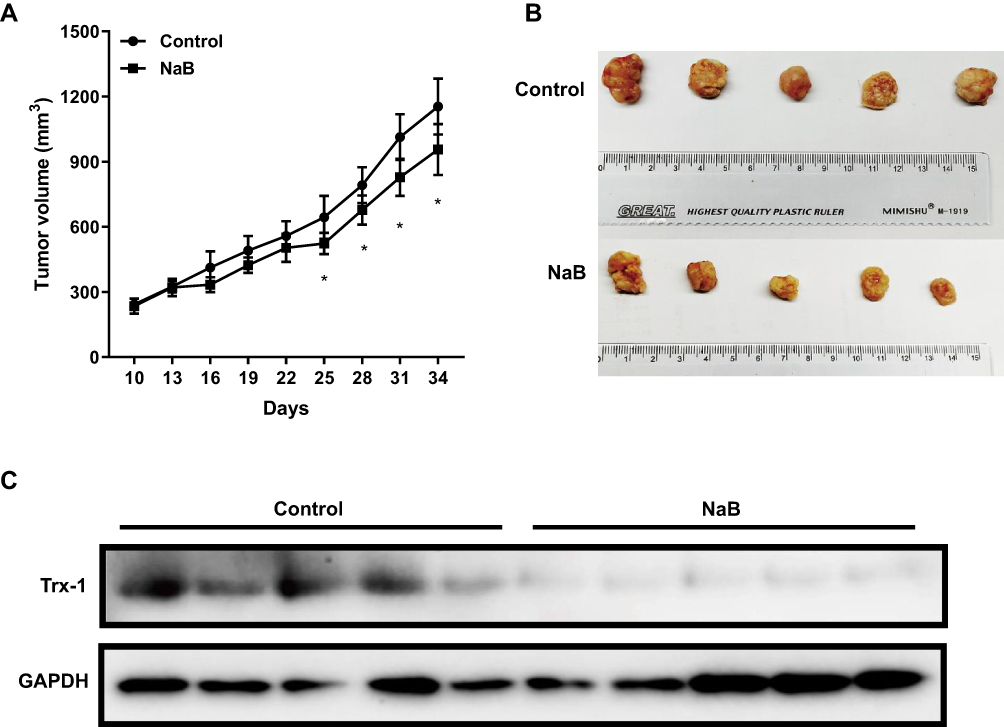 |
Figure 4 NaB inhibits tumor growth and Trx-1 protein expression in vivo. (A) SW480 cells (1×106) were injected subcutaneously in nude mice and xenograft tumor models were treated with saline or NaB (600 mg/kg/d) for 24 days. The tumor volume was measured every 3 days. (B) NaB treatment reduced the tumor size in SW480-inoculated nude mice. (C) Western blot analysis showed that treatment of NaB significantly decreased the expression of Trx-1 in tumor tissues taken from the nude mice. GAPDH was used as an internal control. *P < 0.05. Abbreviations: GAPDH, glyceraldehyde 3-phosphate dehydrogenase; NaB, sodium butyrate; Trx-1, thioredoxin 1. |
NaB Treatment Suppresses Trx-1 Transcription and Increased ROS Level in CRC Cells
NaB treatment significantly suppressed Trx-1 mRNA expression in HT-29 and SW480 cells (Figure 5A). Down-regulation of Trx-1 has been reported to increase ROS production,28 therefore we further investigated whether NaB could regulate ROS level in CRC cells. The intracellular ROS was measured by flow cytometry. As shown in Figure 5B, the ROS level was elevated after NaB treatment in HT-29 and SW480 cells. The rate of growth inhibition was 2.74 ± 1.46% in the NAC and NaB co-treatment group, and 15.73 ± 4.83% in the NaB treatment group in HT29 cells (P < 0.01); and 7.61 ± 1.52% in the NAC and NaB co-treatment group, and 14.76 ± 4.30% in the NaB treatment group in SW480 cells (P < 0.05). Supplement of NAC could partially block the NaB-induced growth inhibition of CRC cells (Figure 5C).
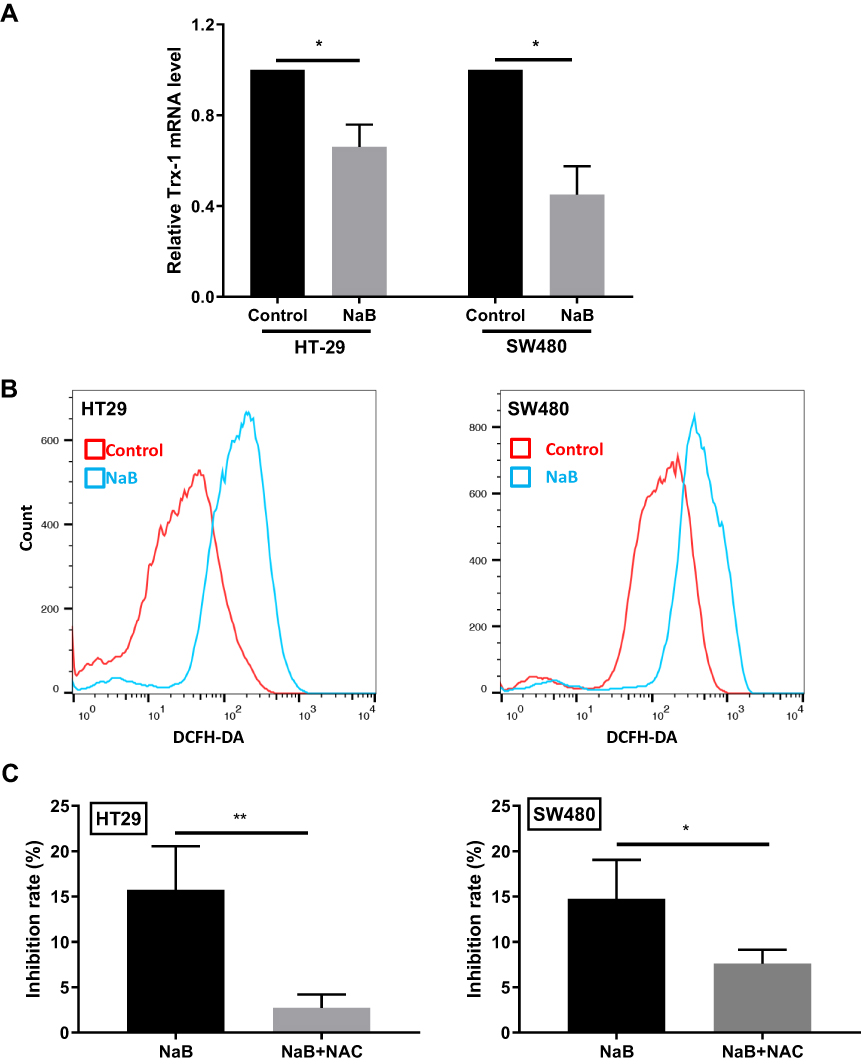 |
Figure 5 NaB treatment suppresses Trx-1 transcription and increased the ROS level in colorectal cancer cells. (A) qRT-PCR analysis showed that the mRNA levels of Trx-1 were inhibited by NaB treatment in HT-29 and SW480 cells. (B) An oxidation-sensitive fluorescent probe, DCFH-DA was used to measure intracellular ROS in HT-29 and SW480 cells treated with NaB (2.5 mM) for 2 days. (C) Supplement of antioxidant N-acetyl-L-cysteine (NAC) partially blocked the inhibition of cell proliferation caused by NaB treatment in HT-29 and SW480 cells. *P < 0.05; **P < 0.01. Abbreviations: DCFH-DA, 2ʹ,7ʹ-dichlorofluorescin diacetate; NAC, N-acetylcysteine; NaB, sodium butyrate; ROS, reactive oxygen species; Trx-1, thioredoxin 1. |
Knockdown of Trx-1 Promotes NaB-Induced Inhibition of Growth and Migration in CRC Cells
We further examined the effects of Trx-1 knockdown on NaB-induced inhibition of growth and migration in CRC cells. Trx-1 was knocked down by lentiviral vector carrying targeting Trx-1 shRNA in HT-29 and SW480 cells (Figure 6A). Knockdown of Trx-1 promoted the inhibitory effects of NaB on cell growth and migration in HT-29 and SW480 cells (Figure 6B and C).
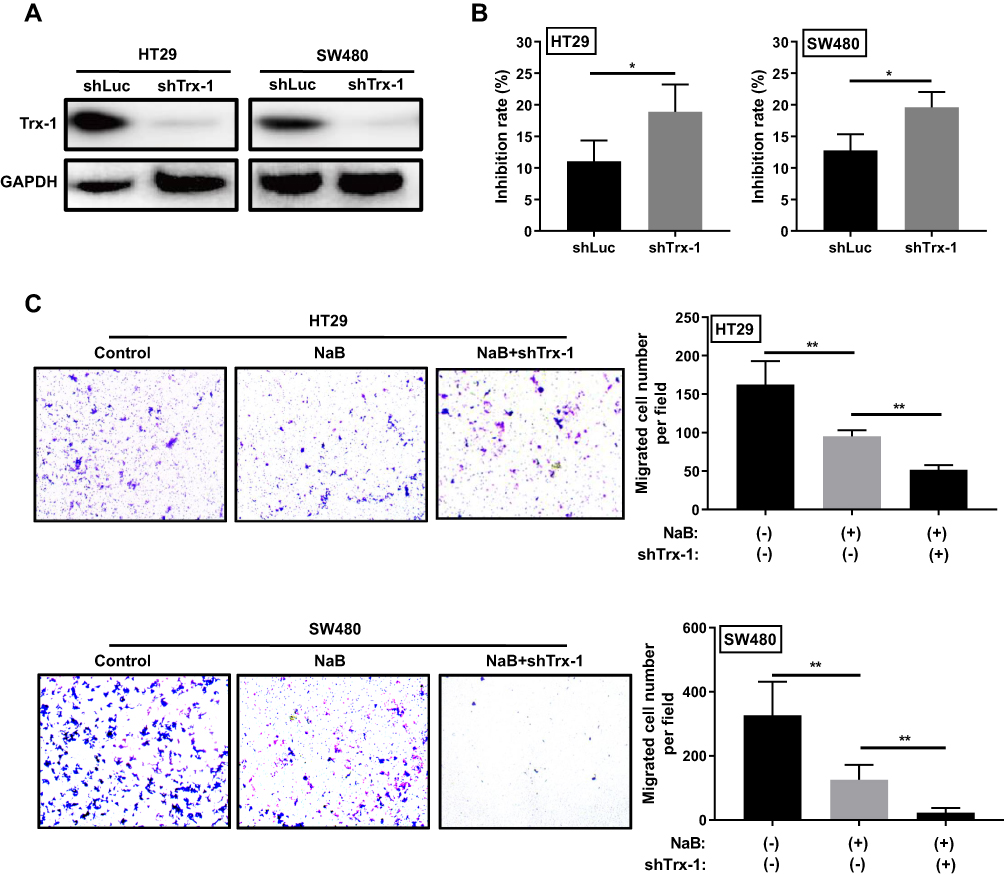 |
Figure 6 Knockdown of Trx-1 promotes NaB-induced inhibition of growth and migration in colorectal cancer cells. (A) Trx-1 knockdown by lentiviral vector-mediated shRNA targeting of Trx-1 (shTrx-1) in HT-29 and SW480 cells was verified by Western blot analysis. (B) Knockdown of Trx-1 increased cell growth inhibition induced by NaB (2.5 mM) in HT-29 and SW480 cells. (C) Knockdown of Trx-1 promoted NaB-induced cell migration inhibition in HT-29 and SW480 cells. *P < 0.05; **P < 0.01. Abbreviations: GAPDH, glyceraldehyde 3-phosphate dehydrogenase; shLuc, small hairpin RNA specific for luciferase gene; shTrx-1, small hairpin RNA specific for thioredoxin 1 gene; NaB, sodium butyrate; Trx-1, thioredoxin 1. |
Overexpression of Trx-1 Reversed NaB-Induced Inhibition of Growth and Migration in CRC Cells
To further investigate the role of Trx-1 in NaB-induced growth and migration inhibition in CRC cells, we examined the effect of overexpression of Trx-1 on cell growth following NaB treatment. The stably overexpressing Trx-1 in SW480 and HT-29 cells were verified by Western blotting (Figure 7A). Overexpression of Trx-1 in HT-29 and SW480 cells significantly decreased NaB-induced cell growth inhibition (Figure 7B). Similarly, overexpression of Trx-1 reversed the inhibitory effects of NaB on migration in HT29 and SW480 cells (Figure 7C). These results suggested that NaB treatment inhibited the growth and migration of CRC cells by inhibiting Trx-1 expression.
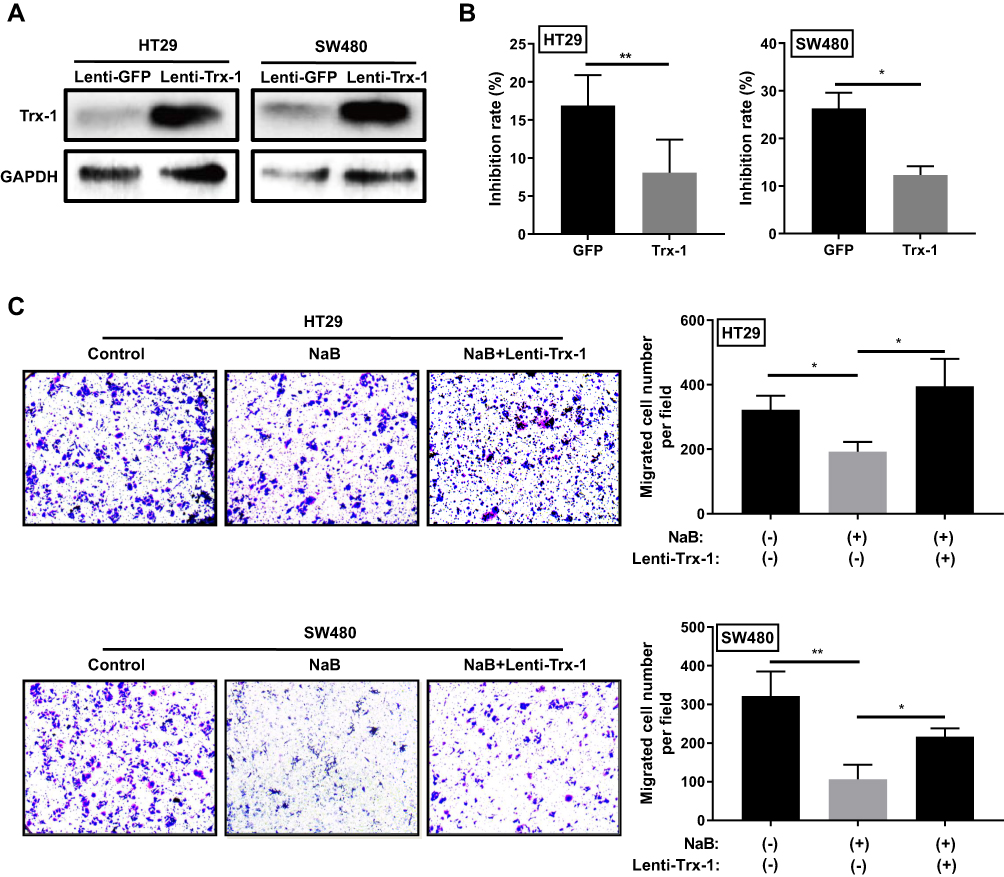 |
Figure 7 Overexpression of Trx-1 reversed NaB-induced inhibition of growth and migration in colorectal cancer cells. (A) Lentiviral vector-mediated overexpression of Trx-1 in HT-29 and SW480 cells was verified by Western blot analysis. (B) Overexpression of Trx-1 decreased cell growth inhibition induced by NaB (2.5mM) in HT-29 and SW480 cells. (C) Overexpression of Trx-1 partially restored NaB-induced cell migration inhibition in HT-29 and SW480 cells. *P < 0.05; **P < 0.01. Abbreviations: GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GFP, green fluorescent protein; lenti, lentivirus; NaB, sodium butyrate; Trx-1, thioredoxin 1. |
Effect of Trx-1 on Intracellular Generation of ROS
The ROS production in Trx-1 knockdown and Trx-1 overexpressed SW480 cells was measured by the Cellular ROS Detection Assay Kit. We observed that knockdown of Trx-1 significantly increased intracellular generation of ROS (Figure 8A), and overexpression of Trx-1 decreased intracellular generation of ROS in SW480 cells (Figure 8B).
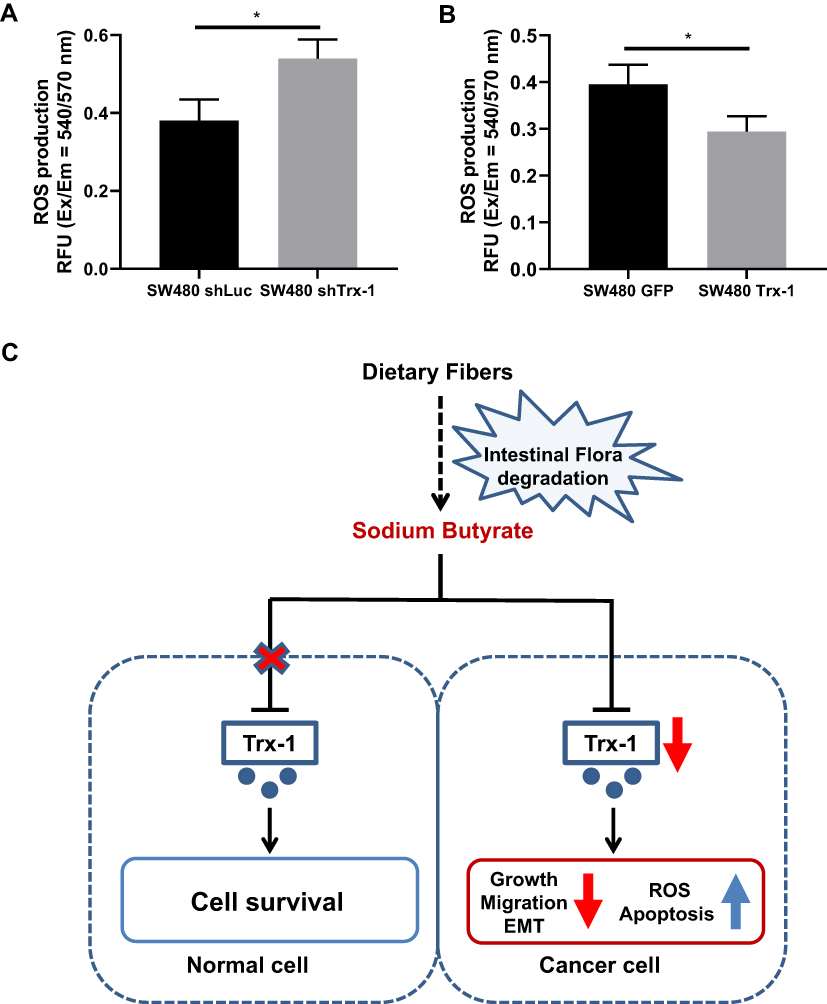 |
Figure 8 Mechanism of antitumor effects of sodium butyrate. (A, B) The ROS production in Trx-1 knockdown and Trx-1 overexpressed SW480 cells was measured. (C) Sodium butyrate significantly inhibits cell growth and decrease Trx-1 protein expression in colorectal cancer cells but not in normal colon epithelial cells. Sodium butyrate increased ROS level, induces apoptosis, and inhibits growth, migration, and EMT in colorectal cancer cells by regulating Trx-1 transcription. *P < 0.05. Abbreviations: EMT, epithelial–mesenchymal transition; Ex, excitation; Em, emission; GFP, green fluorescent protein; RFU, relative fluorescence unit; ROS, reactive oxygen species; shLuc, small hairpin RNA specific for luciferase gene; shTrx-1, small hairpin RNA specific for thioredoxin 1 gene; Trx-1, thioredoxin 1. |
Discussion
Diet has an important role in the development of CRC and could influence the composition and function of gut microbiota.29 Alteration of intestinal flora plays an extremely vital role in the progression of CRC.30,31 Intestinal microbes and their metabolites regulate the cancer-promoting effects of chronic inflammation.32 SCFA are generally produced by the fermentation of anaerobic bacteria in the colon.33 Butyrate, which has a high proportion in SCFA, has been reported to play an important role in CRC.34 NaB inhibits the enzyme activities of histone deacetylases (HDACs) leading to hyperacetylation of histones, and this function is related to the anti-cancer effects of NaB.7 Also, NaB could induce autophagy through LKB1/AMPK signaling.14 Although there are multiple mechanisms involved in the antitumor activity of NaB, the role of Trx-1 in the antitumor effects of NaB against CRC remains unclear.
In this study, our results showed that NaB treatment inhibited proliferation, migration, and EMT in CRC cells. We also found that NaB treatment suppressed Trx-1 protein expression in CRC cells but not the normal colon epithelial cells. This may be the reason why NaB selectively inhibits CRC cell growth with few effects on normal colon epithelial cells. Consistent with our finding, it has been reported that SAHA, an HDAC inhibitor (HDACi), induces the expression of TBP-2, which is a Trx-1 binding protein and reduces Trx-1 activity, causing downregulation of Trx-1 in cancer cells.35,36 However, Endo H et al observed Trx-1 induction by NaB in human hepatoma HepG2 cells.37 Our study further show that NaB could downregulate Trx-1 expression, which results in increased ROS production that in turn inhibits cell proliferation and migration of CRC cells. Although NaB was also shown to significantly inhibit tumor growth in vivo, the effects of NaB treatment on tumor growth appeared to be weak. A potential explanation for this apparent may be the low plasma level of NaB due to delivery and short half-life of NaB in vivo.38,39 We think that a much higher total dosage may be needed for treatment in vivo.
Trx-1 is involved in various redox reactions, and its reversible oxidation of the active center catalyzes the dithiol-disulfide exchange reaction.40 As an auxiliary factor of intracellular antioxidant and reductant, Trx-1 is widely involved in several functions of cells including protein disulfifide reduction, oxidative stress regulation and regulation of transcription factor.41–43 Our previous study has shown that Trx-1 has an important role in CRC and the elevation of Trx-1 promoted cancer invasion and metastasis by promoting the EMT in CRC.25 EMT is an intricate process during which cells lose epithelial characteristics, gain mesenchymal properties and increased motility, and is associated with cancer invasion and metastasis in CRC.44,45 Trx-1 was highly expressed in CRC tissues and high Trx-1 expression was associated with tumor lymph node metastasis and poor overall survival.25 Our results suggest that downregulation of Trx-1 expression may be an important mechanism, at least in part, by NaB caused inhibition of growth, migration, and EMT of CRC cells.
HDACi has been reported to cause an accumulation of ROS in transformed cells.46 We also found that NaB treatment increased ROS level in CRC cells. NAC, a scavenger of ROS prevented NaB-induced inhibition of cell proliferation in CRC cells. Previous studies proved that ROS level was inhibited by Trx-1 with the antioxidant function.47 Downregulation of Trx-1 enhanced cancer cell death by increasing ROS level.28,48 Consistent with previous findings, we observed that knockdown of Trx-1 significantly increased intracellular ROS level, and overexpression of Trx-1 reduced intracellular ROS level in CRC cells. Therefore, our results suggested that NaB-mediated growth inhibition may be related to Trx-1 downregulation caused high levels of ROS. Moreover, downregulation of Trx-1 significantly enhanced NaB-induced inhibitory effects on CRC cell growth and migration, whereas overexpression of Trx-1 attenuated NaB-induced inhibitory effects on growth and migration in CRC cells. These findings indicate that NaB inhibits cell proliferation and migration of CRC cells through downregulation of Trx-1 expression (Figure 8C).
In conclusion, our study reveals that NaB inhibits cell proliferation, migration, and EMT, and induces Trx-1 downregulation in CRC cells. Moreover, NaB increases ROS level and NAC could reverse NaB-induced growth inhibition of CRC cells. Trx-1 plays an important role in NaB-induced inhibition of growth and migration in CRC cells. Our data suggest that NaB could be used as an inhibitor of Trx-1, inhibits growth and metastasis of CRC.
Abbreviations
CCK-8, Cell-counting Kit-8; CRC, colorectal cancer; EMT, epithelial–mesenchymal transition; HDACi, HDAC inhibitor; HDACs, histone deacetylases; NaB, sodium butyrate; NAC, N-acetylcysteine; ROS, reactive oxygen species; SCFA, short-chain fatty acid.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Nelson RS, Thorson AG. Colorectal cancer screening. Curr Oncol Rep. 2009;11(6):482–489. doi:10.1007/s11912-009-0065-8
3. Ma Q, Li Y, Li P, et al. Research progress in the relationship between type 2 diabetes mellitus and intestinal flora. Biomed Pharmacother. 2019;117:109138. doi:10.1016/j.biopha.2019.109138
4. Azcarate-Peril MA, Sikes M, Bruno-Barcena JM. The intestinal microbiota, gastrointestinal environment and colorectal cancer: a putative role for probiotics in prevention of colorectal cancer? Am J Physiol Gastrointest Liver Physiol. 2011;301(3):G401–424. doi:10.1152/ajpgi.00110.2011
5. Sanna S, van Zuydam NR, Mahajan A, et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat Genet. 2019;51(4):600–605. doi:10.1038/s41588-019-0350-x
6. Canani RB, Costanzo MD, Leone L, Pedata M, Meli R, Calignano A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J Gastroenterol. 2011;17(12):1519–1528. doi:10.3748/wjg.v17.i12.1519
7. Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell. 2012;48(4):612–626. doi:10.1016/j.molcel.2012.08.033
8. Shuwen H, Miao D, Quan Q, et al. Protective effect of the “food-microorganism-SCFAs” axis on colorectal cancer: from basic research to practical application. J Cancer Res Clin Oncol. 2019;145(9):2169–2197. doi:10.1007/s00432-019-02997-x
9. Nakagawa H, Yui Y, Sasagawa S, Itoh K. Evidence for intrathecal sodium butyrate as a novel option for leptomeningeal metastasis. J Neurooncol. 2018;139(1):43–50. doi:10.1007/s11060-018-2852-2
10. Verma SP, Agarwal A, Das P. Sodium butyrate induces cell death by autophagy and reactivates a tumor suppressor gene DIRAS1 in renal cell carcinoma cell line UOK146. In Vitro Cell Dev Biol Anim. 2018;54(4):295–303. doi:10.1007/s11626-018-0239-5
11. Coradini D, Pellizzaro C, Marimpietri D, Abolafio G, Daidone MG. Sodium butyrate modulates cell cycle-related proteins in HT29 human colonic adenocarcinoma cells. Cell Prolif. 2000;33(3):139–146. doi:10.1046/j.1365-2184.2000.00173.x
12. Xu Z, Tao J, Chen P, et al. Sodium butyrate inhibits colorectal cancer cell migration by downregulating Bmi-1 through enhanced miR-200c expression. Mol Nutr Food Res. 2018;62(6):e1700844. doi:10.1002/mnfr.201700844
13. Pattayil L, Balakrishnan-Saraswathi HT. In vitro evaluation of apoptotic induction of butyric acid derivatives in colorectal carcinoma cells. Anticancer Res. 2019;39(7):3795–3801. doi:10.21873/anticanres.13528
14. Luo S, Li Z, Mao L, Chen S, Sun S. Sodium butyrate induces autophagy in colorectal cancer cells through LKB1/AMPK signaling. J Physiol Biochem. 2019;75(1):53–63. doi:10.1007/s13105-018-0651-z
15. Fauser JK, Matthews GM, Cummins AG, Howarth GS. Induction of apoptosis by the medium-chain length fatty acid lauric acid in colon cancer cells due to induction of oxidative stress. Chemotherapy. 2013;59(3):214–224. doi:10.1159/000356067
16. Chang MC, Tsai YL, Chen YW, et al. Butyrate induces reactive oxygen species production and affects cell cycle progression in human gingival fibroblasts. J Periodontal Res. 2013;48(1):66–73. doi:10.1111/j.1600-0765.2012.01504.x
17. Salimi V, Shahsavari Z, Safizadeh B, Hosseini A, Khademian N, Tavakoli-Yaraki M. Sodium butyrate promotes apoptosis in breast cancer cells through reactive oxygen species (ROS) formation and mitochondrial impairment. Lipids Health Dis. 2017;16(1):208. doi:10.1186/s12944-017-0593-4
18. Pant K, Yadav AK, Gupta P, Islam R, Saraya A, Venugopal SK. Butyrate induces ROS-mediated apoptosis by modulating miR-22/SIRT-1 pathway in hepatic cancer cells. Redox Biol. 2017;12:340–349. doi:10.1016/j.redox.2017.03.006
19. Holmgren A, Bjornstedt M. Thioredoxin and thioredoxin reductase. Methods Enzymol. 1995;252:199–208.
20. Ohashi S, Nishio A, Nakamura H, et al. Protective roles of redox-active protein thioredoxin-1 for severe acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2006;290(4):G772–781. doi:10.1152/ajpgi.00425.2005
21. Raffel J, Bhattacharyya AK, Gallegos A, et al. Increased expression of thioredoxin-1 in human colorectal cancer is associated with decreased patient survival. J Lab Clin Med. 2003;142(1):46–51. doi:10.1016/S0022-2143(03)00068-4
22. Chang LC, Fan CW, Tseng WK, et al. The ratio of thioredoxin/Keap1 protein level is a predictor of distant metastasis in colorectal cancer. Biomark Med. 2017;11(12):1103–1111. doi:10.2217/bmm-2017-0107
23. Sun Y, Rigas B. The thioredoxin system mediates redox-induced cell death in human colon cancer cells: implications for the mechanism of action of anticancer agents. Cancer Res. 2008;68(20):8269–8277. doi:10.1158/0008-5472.CAN-08-2010
24. Shang W, Xie Z, Lu F, et al. Increased thioredoxin-1 expression promotes cancer progression and predicts poor prognosis in patients with gastric cancer. Oxid Med Cell Longev. 2019;2019:9291683. doi:10.1155/2019/9291683
25. Lin F, Zhang P, Zuo Z, et al. Thioredoxin-1 promotes colorectal cancer invasion and metastasis through crosstalk with S100P. Cancer Lett. 2017;401:1–10. doi:10.1016/j.canlet.2017.04.036
26. Zuo Z, Zhang P, Lin F, et al. Interplay between Trx-1 and S100P promotes colorectal cancer cell epithelial-mesenchymal transition by up-regulating S100A4 through AKT activation. J Cell Mol Med. 2018;22(4):2430–2441. doi:10.1111/jcmm.13541
27. Shahneh FZ, Valiyari S, Azadmehr A, et al. Inhibition of growth and induction of apoptosis in fibrosarcoma cell lines by echinophora platyloba DC: in vitro analysis. Adv Pharmacol Sci. 2013;2013:512931. doi:10.1155/2013/512931
28. Samaranayake GJ, Troccoli CI, Huynh M, et al. Thioredoxin-1 protects against androgen receptor-induced redox vulnerability in castration-resistant prostate cancer. Nat Commun. 2017;8(1):1204. doi:10.1038/s41467-017-01269-x
29. Yang J, Yu J. The association of diet, gut microbiota and colorectal cancer: what we eat may imply what we get. Protein Cell. 2018;9(5):474–487. doi:10.1007/s13238-018-0543-6
30. Hold GL, Allen-Vercoe E. Gut microbial biofilm composition and organisation holds the key to CRC. Nat Rev Gastroenterol Hepatol. 2019;16(6):329–330. doi:10.1038/s41575-019-0148-4
31. Zhang H, Chang Y, Zheng Q, Zhang R, Hu C, Jia W. Altered intestinal microbiota associated with colorectal cancer. Front Med. 2019;13(4):461–470. doi:10.1007/s11684-019-0695-7
32. Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The role of short-chain fatty acids in health and disease. Adv Immunol. 2014;121:91–119.
33. Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther. 2008;27(2):104–119. doi:10.1111/j.1365-2036.2007.03562.x
34. Scheppach W, Weiler F. The butyrate story: old wine in new bottles? Curr Opin Clin Nutr Metab Care. 2004;7(5):563–567. doi:10.1097/00075197-200409000-00009
35. Ungerstedt JS, Sowa Y, Xu WS, et al. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102(3):673–678. doi:10.1073/pnas.0408732102
36. Butler LM, Zhou X, Xu WS, et al. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc Natl Acad Sci U S A. 2002;99(18):11700–11705. doi:10.1073/pnas.182372299
37. Endo H, Niioka M, Kobayashi N, Tanaka M, Watanabe T. Butyrate-producing probiotics reduce nonalcoholic fatty liver disease progression in rats: new insight into the probiotics for the gut-liver axis. PLoS One. 2013;8(5):e63388. doi:10.1371/journal.pone.0063388
38. Miller AA, Kurschel E, Osieka R, Schmidt CG. Clinical pharmacology of sodium butyrate in patients with acute leukemia. Eur J Cancer Clin Oncol. 1987;23(9):1283–1287. doi:10.1016/0277-5379(87)90109-X
39. Frese KK, Neesse A, Cook N, et al. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012;2(3):260–269. doi:10.1158/2159-8290.CD-11-0242
40. Yodoi J, Matsuo Y, Tian H, Masutani H, Inamoto T. Anti-inflammatory thioredoxin family proteins for medicare, healthcare and aging care. Nutrients. 2017;9(10):1081. doi:10.3390/nu9101081
41. Powis G, Montfort WR. Properties and biological activities of thioredoxins. Annu Rev Pharmacol Toxicol. 2001;41(1):261–295. doi:10.1146/annurev.pharmtox.41.1.261
42. Mukherjee A, Martin SG. The thioredoxin system: a key target in tumour and endothelial cells. Br J Radiol. 2008;81(No special_issue_1):S57–68. doi:10.1259/bjr/34180435
43. Espinosa B, Arner ESJ. Thioredoxin-related protein of 14 kDa as a modulator of redox signalling pathways. Br J Pharmacol. 2019;176(4):544–553. doi:10.1111/bph.14479
44. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127(4):679–695. doi:10.1016/j.cell.2006.11.001
45. Vu T, Datta PK. Regulation of EMT in colorectal cancer: a culprit in metastasis. Cancers. 2017;9(12):171. doi:10.3390/cancers9120171
46. Marks PA. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin Investig Drugs. 2010;19(9):1049–1066. doi:10.1517/13543784.2010.510514
47. Huh KH, Cho Y, Kim BS, et al. The role of thioredoxin 1 in the mycophenolic acid-induced apoptosis of insulin-producing cells. Cell Death Dis. 2013;4(7):e721. doi:10.1038/cddis.2013.247
48. Sun X, Wang W, Chen J, et al. The natural diterpenoid isoforretin a inhibits thioredoxin-1 and triggers potent ros-mediated antitumor effects. Cancer Res. 2017;77(4):926–936. doi:10.1158/0008-5472.CAN-16-0987
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.