Back to Journals » OncoTargets and Therapy » Volume 14
Silencing HIPPI Suppresses Tumor Progression in Non-Small-Cell Lung Cancer by Inhibiting DNA Replication
Authors Xie G, Li Y ![]() , Jiang Y, Ye X, Tang J, Chen J
, Jiang Y, Ye X, Tang J, Chen J ![]()
Received 10 February 2021
Accepted for publication 28 April 2021
Published 27 May 2021 Volume 2021:14 Pages 3467—3480
DOI https://doi.org/10.2147/OTT.S305388
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Takuya Aoki
Guanghui Xie,1,2,* Yongwen Li,1,* Yongjun Jiang,2,* Xian Ye,2 Jianfeng Tang,2 Jun Chen1
1Department of Lung Cancer Surgery, Tianjin Medical University General Hospital, Tianjin, People’s Republic of China; 2Department of Cardiothoracic Vascular Surgery, The Central Hospital of Yongzhou, Yongzhou, Hunan Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jun Chen
Department of Lung Cancer Surgery, Tianjin Medical University General Hospital, No. 154 Anshan Road, Heping District, Tianjin, 300052, People’s Republic of China
Email [email protected]
Introduction: Non-small cell lung cancer (NSCLC) is the most common form of lung cancer, accounting for approximately 80%– 85% of all cases of lung cancer. Huntingtin interacting protein-1 interacting protein (HIPPI) is a transcription regulator and plays an important role in apoptotic cell death. However, the role of HIPPI in NSCLC remains unclear.
Methods: Immunohistochemistry (IHC) and qRT-PCR were performed for expression analysis. The roles of HIPPI were studied using cell counting kit-8 (CCK-8), colony formation, flow cytometry, wound healing, Transwell invasion assays and mouse xenograft model. Gene microarray analysis and bioinformatics analysis were used to identify differentially expressed genes after HIPPI silencing.
Results: HIPPI is highly expressed in NSCLC tissues relative to adjacent normal tissues. Targeting HIPPI by RNA interference inhibits NSCLC cell proliferation in vitro and tumor growth in vivo. HIPPI silencing also attenuates cell migration and invasion and enhances cisplatin sensitivity in NSCLC cells. Mechanistic investigation suggests that HIPPI can positively regulate the expression of MCM2, MCM6 and MCM8, which are key regulators of DNA replication. Furthermore, consistent with HIPPI, MCM2, MCM6 and MCM8 are also upregulated in NSCLC tissues.
Conclusion: Our study highlights the importance of HIPPI for tumor biology in NSCLC and suggests that HIPPI may be a potential therapeutic target for NSCLC treatment.
Keywords: HIPPI, non-small cell lung cancer, MCM, DNA replication
Introduction
Lung cancer is one of the most commonly diagnosed cancers and the leading cause of cancer-related mortality worldwide. The 2018 Global Cancer Statistics has shown that there were approximately 1.8 million new lung cancer cases and 1.6 million lung cancer deaths (18.4% of all cancer deaths) in 2018.1 Lung cancer includes non–small cell lung cancer (NSCLC) and SCLC. NSCLC is the most common form of lung cancer, accounting for approximately 80%–85% of all cases of lung cancer. Although great progress has been made in therapeutic methods, the prognosis of patients with NSCLC remains far from satisfactory. The prognosis of patients depends largely on the stage in which the cancer is detected. Unfortunately, about 80% of patients with NSCLC were detected after cancer has already spread to regional lymph nodes or has metastasized, leading to a 5-year survival rate of 5% in patients with stage IV cancer. Therefore, it is necessary to explore novel markers and targets to provide more accurate and effective therapeutic strategies for patients.
HIPPI protein is a multifunctional protein that is involved in the regulation of apoptosis. HIPPI has been originally reported to be implicated in caspase proteins-mediated induction of neuronal apoptosis during the pathogenesis of Huntington’s disease.2 HIPPI interacts with HIP1 through death-effector domains to form HIPPI/HIP1 complex and translocates into the nucleus.3–5 In the nucleus, HIPPI acts as an important transcription regulator and regulates the expression of numerous downstream genes. Firstly, HIPPI interacts with the promoter sequence of caspase-1, −8 and −10 to induce the process of apoptosis.6,7 Rybp, Apoptin, and bifunctional apoptosis regulator (BAR) can enhance HIPPI-mediated apoptosis,8–10 whereas Homer1c prevents the pro-apoptotic effect of HIPPI.11 Further, HIPPI binds to the promoter of neuron-restrictive silencer factor (NRSF) and triggers its transcriptional activation and consequent repression of downstream genes of NRSF in the neuronal cell model of Huntington disease.12 There are few reports on the role of HIPPI in the tumor. Gdynia et al13 reported that HIPPI sensitized NCH89 glioblastoma cells to the pro-apoptotic actions of staurosporine and the death ligand TRAIL by enhancing caspase activation, cytochrome c release, and disruption of the mitochondrial membrane potential. Moreover, Datta et al14 showed that exogenous expression of HIPPI in HeLa cells altered the levels of several transcription factors including CBP, NRSF, C/EBP beta. Besides, HIPPI also interacted with P53 at the protein level, which was necessary for HIPPI mediated upregulation of the caspase-1 gene.
In the present study, we investigated the expression of HIPPI in NSCLC tissues and adjacent normal samples by IHC and analyzed the association between HIPPI and clinical parameters. We knocked down HIPPI in two NSCLC cell lines by short hairpin RNA (shRNA) and investigated its effect on cell proliferation, apoptosis, migration, invasion and cisplatin sensitivity. In addition, the effect of silencing HIPPI on tumor growth was examined in the xenograft tumor model. Lastly, preliminary explorations of the mechanism of HIPPI involving NSCLC were carried out by identifying co-expressed genes with global gene expression profiling.
Materials and Methods
Patients and Tissue Collection
Forty-two pairs of NSCLC tissues and matched normal tissues were obtained from Tianjin Medical University General Hospital between 2016 and 2018. The detailed clinicopathological data of patients are shown in Table 1. This study was approved by the Ethics Committee of Tianjin Medical University (Tianjin, China) and was performed in accordance with the Declaration of Helsinki. Written informed consent was obtained from the enrolled subjects.
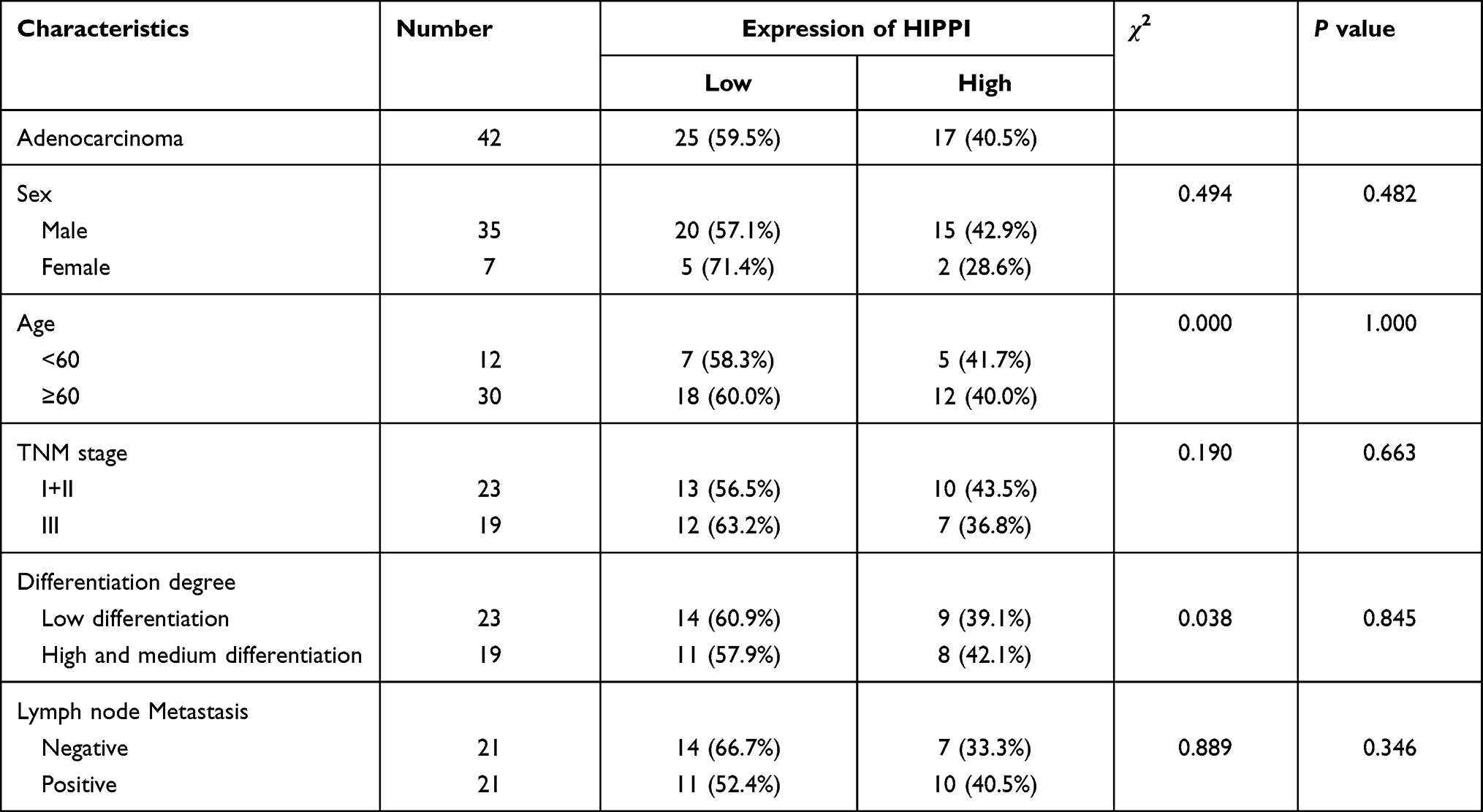 |
Table 1 Clinicopathologic Characteristics of Lung Adenocarcinoma Carcinomas |
IHC Staining
IHC staining for HIPPI was performed as described previously.15 Formaldehyde-fixed, paraffin-embedded tissue sections were used for the IHC experiment. Tissue sections were deparaffinized, and antigen retrieval was performed in 5 mM Tris-HCl for 10 min by microwave pretreatment. Endogenous peroxidase activity was quenched with 3% H2O2, and serum was used to block non-specific binding sites with the Endogenous Biotin-Blocking Kit (Thermo Fisher Scientific, USA). Then, the slides were incubated with an anti-HIPPI monoclonal primary antibody (1:50, Mouse, Thermo Fisher Scientific, USA) at 4°C for 22 h, washed and then incubated with a biotinylated anti-rabbit lgG secondary antibody for 30 min at room temperature. Slides were visualized with diaminobenzidine and followed by counterstaining with hematoxylin. The representative photographs were taken using an Olympus BX50 microscope (Japan). Slides were evaluated using light microscopy and a standard semi-quantitative immunoreactivity score as described previously. By recording the percentage of positive staining (1<25%, 2=25%–50%, 3=50%–75%, 4≥75%) and staining intensity (0=negative, 1=weak, 2=moderate, 3=strong) for each sample, immunoreactivity score (IRS) (0–12) was calculated by multiplying positive staining percentage with staining intensity. Low and high expressions were defined according to the median IRS.
Cell Culture
Human NSCLC cell lines H157 and A549 were obtained from American Type Culture Collection. Cells were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) at 37°C in a humidified incubator containing 5% CO2.
Cell Transfection
The short hairpin RNAs (shRNAs) against HIPPI (sh-HIPPI#1, sh-HIPPI#2, sh-HIPPI#3) and scrambled shRNA (sh-NC) were purchased from GenePharma Inc. (Shanghai, China) and transfected into cells with Lipofectamine 2000 reagent (Thermo Fisher Scientific, USA) following manufacturer’s instructions. These transfected cells were cultured in the RPMI-1640 medium supplemented with G418 antibiotic at 800 μg/mL for approximately 2 weeks to establish a stable shRNA-expressing cell clone.
Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
Total RNAs were extracted from cells and tissue samples using Trizol reagent (Invitrogen, Carlsbad, CA). Reverse transcription was conducted using the Applied Biosystem’s Power SYBR Green PCR Master Mix and the reactions were run on an ABI 7500 Fast Real-time PCR system. GAPDH was used as an internal control. The sequences of primers used in this study are shown in Table 2.
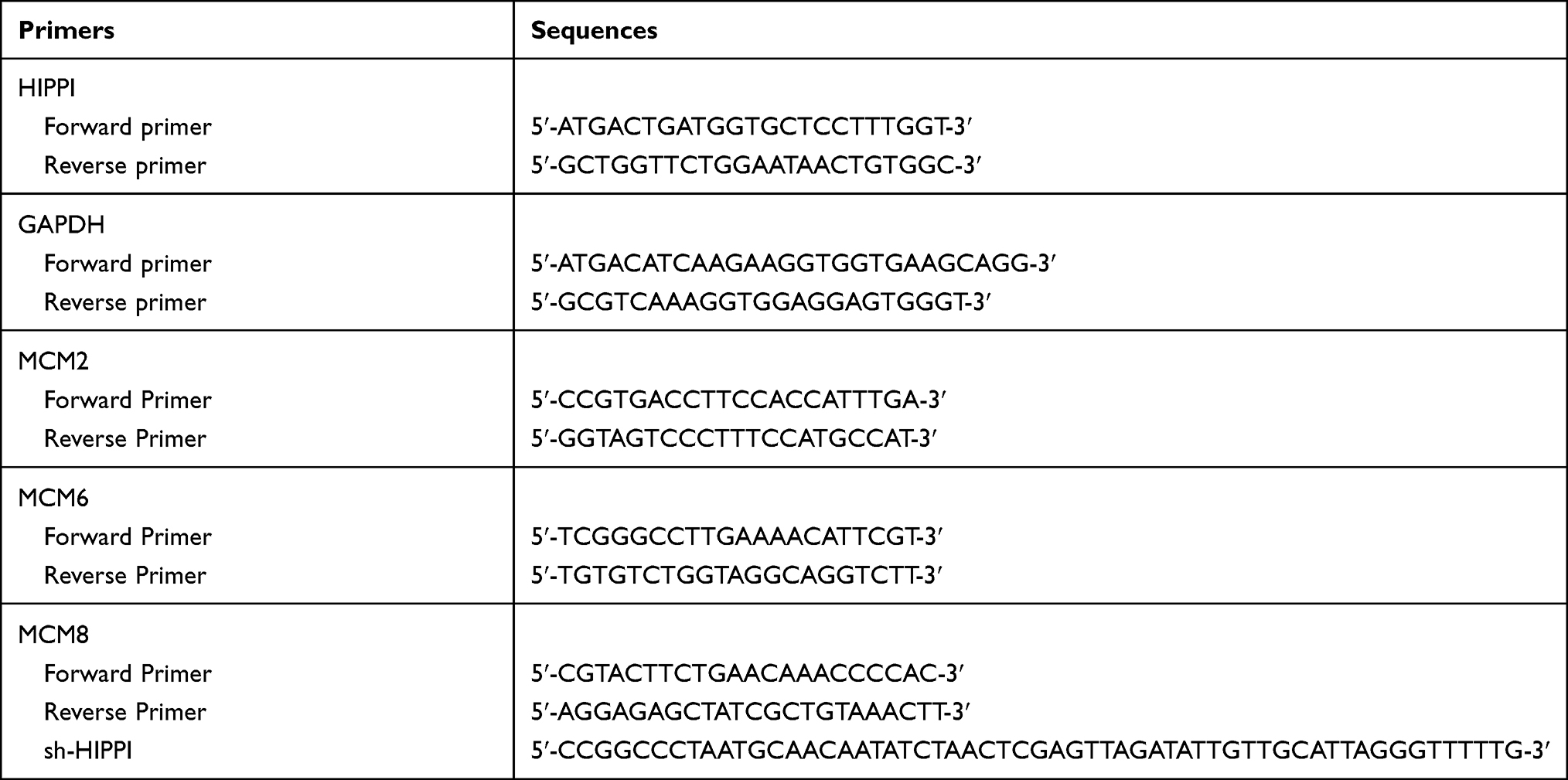 |
Table 2 The Sequences of Primers Used in This Study |
Cell Viability Assay
Cell viability was conducted by cell counting kit-8 kit (Genomeditech, Shanghai, China) according to the manufacturer’s instructions.
Colony Formation Assay
Cells were seed into 6-well plates at a density of 1×103 per well. The culture was continuously maintained for 14 days. Colonies were visualized and countered under a microscope.
Cell Cycle and Apoptosis Analysis
Cell cycle and apoptosis were detected using the Cell Cycle and Apoptosis Analysis Kit (Beijing 4A Biotech Co., Ltd). For cell cycle analysis, cells were harvested, washed with cold PBS, and fixed in ice-cold 95% ethanol at 4°C for 12 h. Fixed cells were then washed and incubated with propidium iodide (PI) for 30 min in the dark. Finally, samples were analyzed using a flow cytometer (Agilent Biosciences). For cell apoptosis analysis, cells were harvested, washed twice with PBS, and resuspended, followed by the addition of 5 µL Annexin V/FITC. After 5 min in the dark, cells were incubated with 10 µL PI (20 µg/mL) and 400 µL PBS. Finally, samples were analyzed using a flow cytometer (Agilent Biosciences).
Cell Migration Analysis
Cell migration was evaluated using wound healing test. 200 μL aseptic pipette was used to scratch the cells to get a cell-free area, PBS was used to rinse the cells, and a new culture medium was added for culture. At 0 h and 24 h after cell scratch, the cell migration ability was evaluated by microscope for scratches at three different positions.
Cell Invasion Assay
Matrigel chambers (BD Biosciences, CA, USA) were used for cell invasion assay according to the manufacturer’s instructions. Firstly, transfected cells at a density of 5000 cells/well in medium without serum were added into the hydrated matrigel chamber, and 500μL of RPMI 1640 medium containing 20% FBS was added into the lower chamber. The cells on the upper surface were cleaned after 48 h of culture at 37°C, whereas the invasive cells on the lower surface were fixed and stained with 0.1% crystal violet for 10 min after it was dried, and cell invasion was observed with a microscope.
Evaluation of Cisplatin Sensitivity
After transfection for 48 h, H157 and A549 cells were isolated to prepare the cell suspension and then seeded into 96-well plates at a density of 4×103 cells/well. After observation of cell adherence, the medium was replaced with mediums containing variable cisplatin concentrations (0, 2, 4, 6, 8 and 10μg/mL). After incubation for 24 h, 10μL CCK-8 solutions were added to each well. The cells were then cultured in an incubator for 4 h. The absorbance at 450 nm wavelength was measured using a microplate reader. Five duplicate wells were set in each group and the experiment was repeated three times. The half-maximal inhibitory concentration (IC50) was calculated according to the cell survival rate.
In vivo Tumorigenesis Study
All procedures were performed following the protocols approved by the Institutional Animal Care and Use Committee of the Tianjin Medical University. Four-week-old athymic nude male BALBL/c mice were obtained from Charles River Laboratories (Wilmington, MA). Before starting the experiments, mice were maintained for 5 days in the temperature and humidity-controlled condition. Mice were randomly allocated into three groups (sh-HIPPI, sh-NC and NC) of 8 animals. A total of 8×106 A549 transfected cells were subcutaneously injected into the right flanks. Tumor length and width were measured once a week. The average tumor volume in each group was calculated according to the equation of (length × width2)/2 (mm3). The animals were sacrificed 28 days post-graft and the tumors were removed and used for further analysis.
Gene Microarray
Gene microarray analysis was performed at RUIJIYIN Biotechnology co.LTD (Tianjin, China), using Affymetrix GeneChip Human Transcriptome 2.0 Array (Affymetrix, Santa Clara, CA, USA). Briefly, total RNAs were isolated from H157 and A549 cells and reverse transcribed to generate double-stranded cDNA that was amplified to produce cRNA. The cRNA generated was purified and subjected to 2nd -cycle single-stranded sense cDNA that was fragmented, labeled and hybridized to Affymetrix GeneChip® Human Transcriptome 2.0 array. The arrays were washed, stained in GeneChip Fluidics Station 450 and scanned using Affymetrix 3000 7G scanner.
The processed gene expression profiles were used to screen DEGs between sh-HIPPI transfected cell lines and control parental cell lines. DEGs screening was performed by using the limma package in R,16 with the criteria of P adjusted FDR < 0.01 and |log2 fold change| > 1.5. Gene Ontology (GO) annotation and KEGG enrichment for the proteins encoded by DEGs were performed using the Metascape database (http://metascape.org/gp/index.html#/main/step1).17
Statistical Analysis
The SPSS 22.0 software was used for analyses. All values were presented as the mean ± SEM from at least three independent experiments. Data were analyzed with two-sided unpaired Student’s t-test for samples with two groups and one-way analysis of variance (ANOVA) for samples with more than two groups. P < 0.05 was considered significant.
Results
Expression and Clinical Significance of HIPPI in NSCLC
To investigate HIPPI expression pattern in NSCLC, IHC was performed on lung cancer tissues and matched to adjacent normal tissues. The clinical characteristics of 42 patients with NSCLC are summarized in Table 1. IHC analysis revealed that HIPPI localized in the cytoplasm and nucleus of lung cancer tissues (Figure 1A). There was a tendency towards higher HIPPI expression in the NSCLC than in the normal lung (Figure 1B). The positive rate of HIPPI in NSCLC tissues (40.5%, 17/42) was significantly higher than that in normal lung tissues (11.9%, 5/42) (P < 0.001, Figure 1C). Further, we analyzed the correlation between HIPPI expression and clinical characteristics. However, the statistical correlation was not observed between HIPPI expression and these clinical characteristics (Table 1).
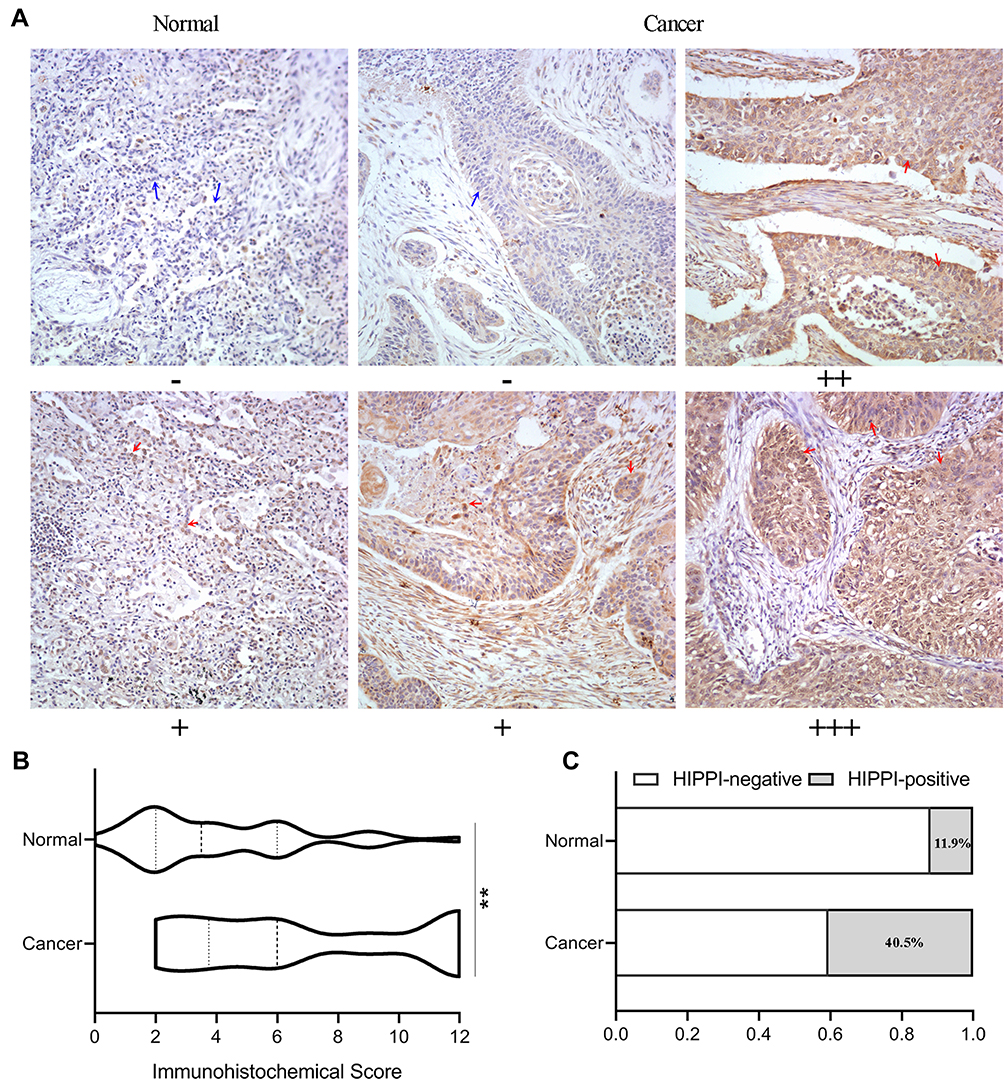 |
Figure 1 IHC analysis of HIPPI expression in lung tissues. (A) Representative images of IHC staining of HIPPI in NSCLC tissues and matched adjacent normal tissues. The red arrows indicate representative HIPPI-positive cells and the blue arrows indicate representative HIPPI-negative cells. (B) IHC score of normal and lung cancer samples. (C) The rate of HIPPI-high samples in normal and cancerous lung tissues. **p < 0.01. Abbreviation: IHC, Immunohistochemistry. |
HIPPI Knockdown Inhibits Cell Proliferation and Induces Cell Apoptosis and Cell Cycle Arrest
To assess the potential role of HIPPI in NSCLC progression, three HIPPI shRNAs (sh-HIPPI#1, sh-HIPPI#2, sh-HIPPI#3) were designed to specifically target different binding sites of HIPPI. In both H157 and A549 cell lines, sh-HIPPI#3 effectively inhibited the expression of HIPPI and was used for subsequent experiments (Figure 2A). We then investigated whether HIPPI affected cellular function by a series of cell experiments. The results of the CCK8 assay showed that HIPPI silencing significantly restrained H157 and A549 cell proliferation (Figure 2B). Meanwhile, colony formation assay revealed that HIPPI was positively associated with the proliferation of H157 and A549 cells (Figure 2C–D). Moreover, the results of cell apoptosis analysis demonstrated that sh-HIPPI-transfected cells showed a higher apoptotic rate than that of NC cells (Figure 2E and F). Also, cell cycle was analyzed by flow cytometry, as shown in Figure 2G and H, HIPPI silencing caused a significant accumulation in the G0/G1 phase compared to the negative control cells (p<0.001), while the proportions of these cells in G2/M phase was significantly decreased in sh-HIPPI-transfected cells compared to NC cells (p < 0.001).
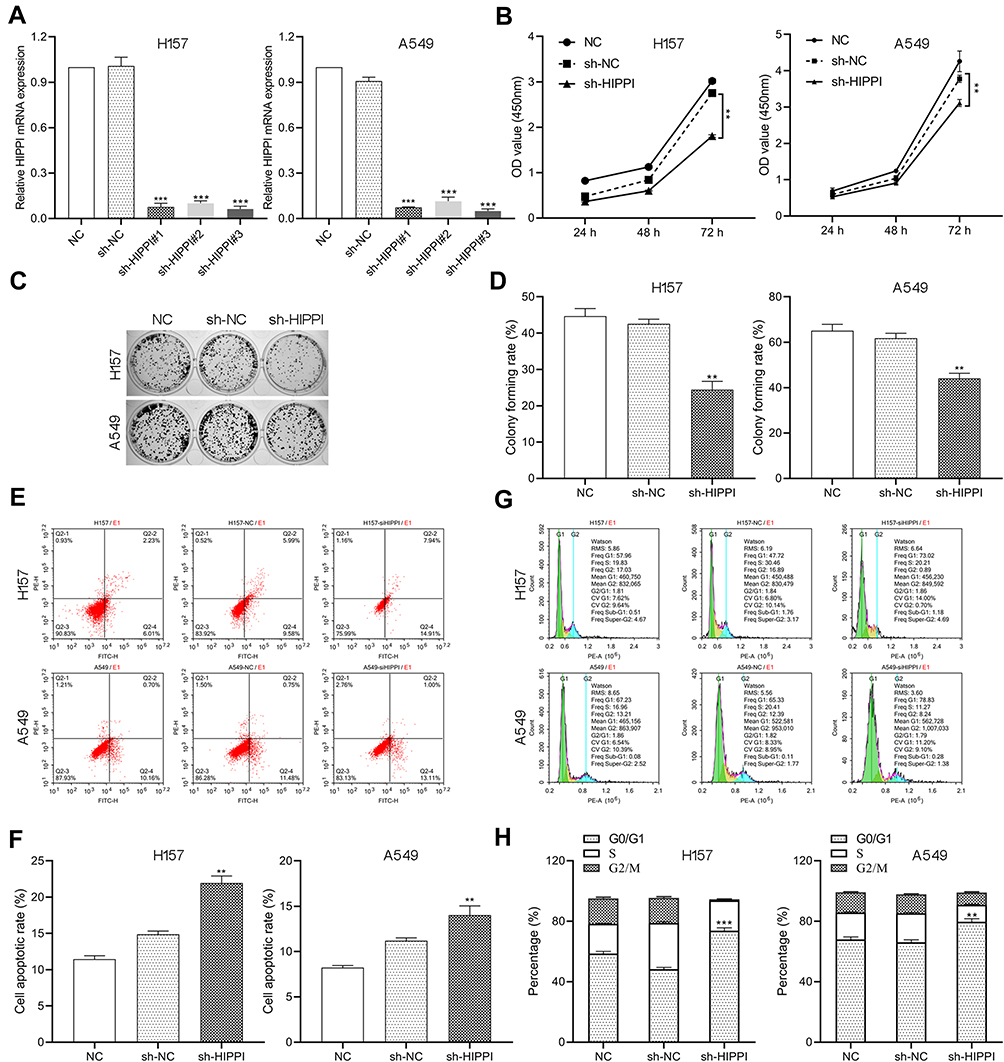 |
Figure 2 HIPPI knockdown inhibits cell proliferation, induces cell apoptosis and cell cycle arrest. H157 and A549 cells were transfected with sh-HIPPI or sh-NC and subjected to (A) qRT-PCR for the expression levels of HIPPI, (B) CCK8 assay for cell viability, (C and D) colony formation assay, (E and F) flow cytometry for cell apoptosis and (G and H) cell cycle analysis. **p < 0.01, ***p < 0.001 as compared with the NC groups. |
HIPPI Knockdown Suppresses Cell Invasion and Migration
We examined the effect of silencing HIPPI on cell migration and invasion by wound healing and transwell assays. As shown in Figure 3A and B, wound healing assay showed that the migratory potential of sh-HIPPI-transfected H157 and A549 cells were noticeably hindered compared with the NC groups. Besides, results from transwell assays indicated that the migratory and invasive capacities were greatly suppressed by knockdown of HIPPI in H157 and A549 cells, whereas NC transfected cells had similar cell migratory and invasive capacities to the negative control cells (Figure 3C and D). Together, these results demonstrate that knockdown of HIPPI inhibits migration and invasion in vitro.
 |
Figure 3 HIPPI knockdown suppresses cell invasion and migration. H157 and A549 cells were transfected with sh-HIPPI or sh-NC and subjected to (A and B) wound healing for cell migration, (C and D) transwell assay for cell migration and invasion. **p < 0.01, ***p < 0.001 as compared with the NC groups. |
Inhibition of HIPPI Enhances Cisplatin Sensitivity of Lung Cancer Cells
Cisplatin is the first-line chemotherapy drug for the treatment of NSCLC. However, cisplatin resistance has been a major problem in lung cancer, leading to difficulty in treatment and poor prognosis. Here, we asked whether HIPPI was involved in cisplatin resistance in NSCLC. Following transfection with sh-HIPPI or sh-NC, H157 and A549 cells were treated with different concentrations of cisplatin (0, 2, 4, 6, 8 and 10μg/mL). Subsequently, cell viability was examined by CCK8 assay. As shown in Figure 4A, the survival rate of cells in each group was significantly decreased with the increase in cisplatin concentration. When treated with 2, 4 and 6μg/mL of cisplatin, the relative survival rate in the sh-HIPPI-transfected cells was significantly decreased, as compared with that in the NC group. Furthermore, we calculated the cisplatin IC50 using GraphPad Prism 8.0. The results showed that the IC50 of H157 and A549 cells transfected with sh-HIPPI was 6.305 μg/mL and 5.422 μg/mL, respectively, while the IC50 of H157 and A549 cells in NC groups was 9.112 μg/mL and 12.11 μg/mL. The IC50 of H157 and A549 cells in sh-HIPPI was significantly lower than that of in NC groups (Figure 4B). These results indicate that knockdown of HIPPI enhances the sensitivity of NSCLC cells to cisplatin.
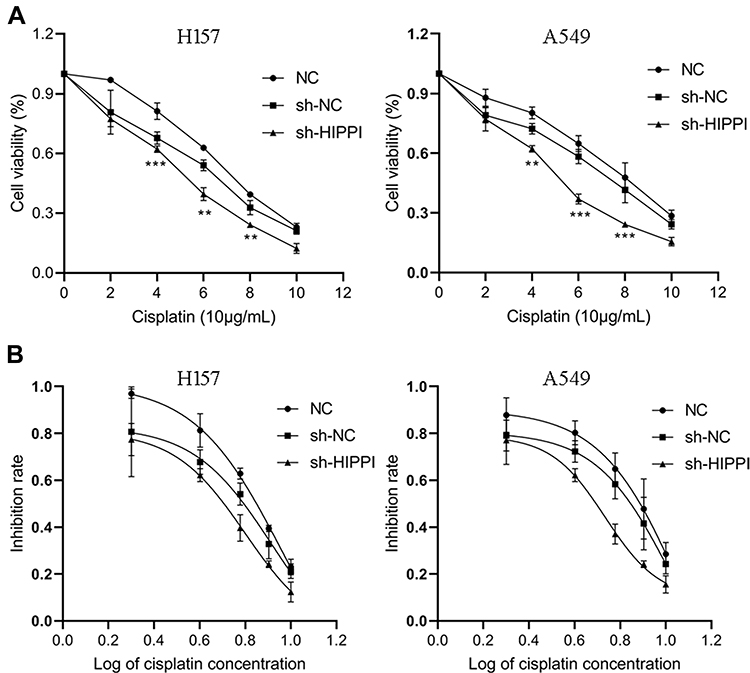 |
Figure 4 Inhibition of HIPPI enhances cisplatin sensitivity of NSCLC cells. (A) Cell viability was examined by CCK8 assay. (B) The cisplatin IC50 in each group was calculated using GraphPad Prism 8.0. **p < 0.01, ***p < 0.001 as compared with the NC groups. |
HIPPI Knockdown Suppresses Tumor Growth in vivo
To further evaluate the effect of HIPPI on tumor growth in vivo, an in vivo A549 cell line-derived xenograft tumor model was conducted in nude mice. The subcutaneous tumor volume reached 138.98±29.52 mm3 at day 28 post-graft in the NC group, while the tumors in the sh-HIPPI group reached 35.11±10.84 mm3 at day 28. HIPPI knockdown significantly inhibited the tumor volume on days 14, 21 and 28 (Figure 5A and B). These results were correlated with the tumor weight at day 28, which was significantly decreased in the sh-HIPPI group compared to the NC group (P < 0.001, Figure 5C).
 |
Figure 5 HIPPI knockdown suppresses tumor growth in vivo. Xenograft assay of A549 with indicated treatments was performed on nude mice. (A) The nude mice carrying tumors from respective groups were shown. (B) Tumors growth curves. (C) Tumors and tumor weight were shown. ***p < 0.001 as compared with the NC groups. |
Global Gene Expression Profiling Identified Potential Downstream Target of HIPPI
To identify potential targets of the HIPPI and to decipher the molecular mechanism underlying the observed phenomena upon silencing of the HIPPI gene, the Affymetrix GeneChip® Human Transcriptome 2.0 Array was employed to identify global gene expression changes in H157 and A549 cells, following transfection with sh-HIPPI or sh-NC. Using |log2 fold change| of > 1.5 and an adjusted P-value < 0.05 as thresholds, a total of 1845 genes were found to be up-regulated and 1776 genes were down-regulated in the sh-HIPPI-transfected H157 cells as compared to NC cells, whereas 1638 genes were found to be up-regulated and 1975 genes were down-regulated in the sh-HIPPI-transfected A549 cells when compared to NC cells (Figure 6A). By combining these data, a total of 841 differentially expressed genes (DEGs), including 328 up-regulated genes and 513 down-regulated genes in both sh-HIPPI-transfected H157 and A549 cells, were identified (Figure 6B, Supplementary Table S1).
 |
Figure 6 Global gene expression profiling identified potential downstream target of HIPPI. (A) Differentially expressed genes in HIPPI-silencing cells compared to negative control. Red represents upregulated genes, blue represents down-regulated genes, and gray represents no significantly differentially expressed genes. (B) Hierarchical clustering of overlapped genes in HIPPI silenced H157 and A549 cells. (C) GO enriched terms of downregulated DEGs in HIPPI-silencing cells (Metascape), colored by p-values. (D) KEGG pathways enriched terms of downregulated DEGs in HIPPI-silencing cells, colored by p-values. |
The functions of downregulated DEGs were predicted by analyzing GO and KEGG. The cellular components and molecular functions enrichment analysis of downregulated DEGs showed that HIPPI related genes mainly distributed in microtubule organizing center, chromosomal region, centrosome and exerted transcriptional regulation activities and participated in biological processes, such as DNA replication, cell cycle phase transition, mitotic cell cycle phase transition, double-strand break repair and DNA conformation change (Figure 6C). Further, KEGG enrichment revealed that these DEGs were significantly enriched in DNA replication, homologous recombination (HR), insulin signaling pathway, p53 signaling pathway and cell cycle pathways (Figure 6D). Significant changes were observed in the DNA replication, HR and cell cycle pathways, which were closely related to tumorigenesis, after HIPPI silencing. Notably, minichromosome maintenance proteins (MCM) family members, including MCM2, MCM6 and MCM8, were identified that intersected in DNA replication, HR and cell cycle pathways, indicating the importance of the MCM family in the DNA replication, HR and cell cycle pathways with HIPPI-mediated oncogenic activity in NSCLC.
The MCM Family Members are Highly Expressed and Positively Correlated with HIPPI in NSCLC
The global gene expression profiling analysis suggested that MCM2, MCM6 and MCM8 might be involved in the process of the DNA replication, HR and cell cycle pathways in NSCLC cell lines. To verify the relationship between these genes and HIPPI, we tested the expression of MCM2, MCM6 and MCM8 in cells after transfection with sh-HIPPI by qRT-PCR. Results showed that the levels of MCM2, MCM6 and MCM8 were significantly down-regulated in sh-HIPPI-transfected H157 and A549 (Figure 7A). Next, we tested NSCLC samples from 15 patients and matched the adjacent tissues and found that the expression of MCM2, MCM6 and MCM8 was aberrantly elevated at the mRNA levels in NSCLC tissues (Figure 7B). To further investigate the correlation of MCM levels with the survival of NSCLC patients, we searched the GEPIA database that contained 347 normal lung tissue samples and 483 LUAD tissue samples. First, the results indicated that MCM2 was expressed at higher levels in NSCLC tissues than in normal lung tissues, and MCM6 and MCM8 showed increased patterns without a statistical difference (Figure 7C). Additionally, we found that high levels of MCM2, MCM6 and MCM8 were all associated with shorter overall survival of LUAD patients (Figure 7D).
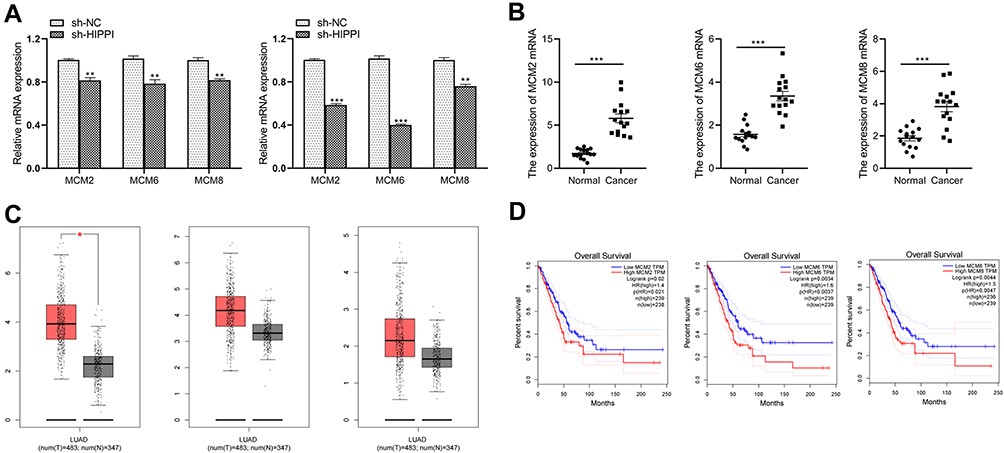 |
Figure 7 MCM2, MCM6 and MCM8 are highly expressed and positively correlated with HIPPI in NSCLC (A) The qRT-PCR assays were performed to detect the expression of MCM2, MCM6 and MCM8 on the control and HIPPI-depleted H157 and A549 cells. (B) MCM2, MCM6 and MCM8 mRNA expression in human NSCLC and control normal tissues (n=15). (C) MCM2, MCM6 and MCM8 expression in LUAD tissues from TCGA. Images were obtained from the GEPIA online database (http://gepia.cancer-pku.cn). (D) The prognostic value of the expression levels of MCM2, MCM6 and MCM8 in LUAD patients. LUAD, lung adenocarcinoma; *p < 0.05, **p < 0.01, ***p < 0.001 as compared with corresponding control groups. Abbreviations: T, lung adenocarcinoma tissues; N, normal lung tissue. |
Discussion
NSCLC is a heterogeneous disease of multiple distinct subtypes with different genetic, pathological, and clinical manifestations.18 Over the past two decades, the disease biology and mechanisms of tumor progression have been studied and understood in greater depth and significant advancements have been made in the treatment of NSCLC.19,20 Despite these molecular advances, advanced-stage NSCLC remains largely incurable due to therapeutic resistance. However, due to disease complexity, drug resistance, and delay in diagnosis, the overall cure and survival rates for NSCLC remain low.21 In this study, we found that HIPPI was elevated in NSCLC, and knockdown of HIPPI inhibited NSCLC progression in vitro and in vivo. These data indicate that HIPPI may be a potential therapeutic target in NSCLC.
The HIPPI protein is a transcription regulator and has the ability to bind DNA sequence motif 5ʹ-AAAGACATG-3ʹ present in the promoter of genes, such as CASP1, CASP8 and CASP10. BLOC1S2 and HIPPI sensitize NCH89 glioblastoma cells to the pro-apoptotic actions of staurosporine and the death ligand TRAIL by enhancing caspase activation, cytochrome c release, and disruption of the mitochondrial membrane potential.13 Cheng et al reported that HIPPI and Apoptin were perfectly colocalized in the cytoplasm of normal human HEL cells, whereas in cancerous HeLa cells, most Apoptin and HIPPI were located separately in the nucleus and cytoplasm,10 indicating that HIPPI may function differently in distinct cellular contexts. In the present study, we found that the expression of HIPPI was elevated in NSCLC tissues compared with corresponding normal tissues. Cell experiments showed that down-regulated HIPPI inhibited cell proliferation, induced cell apoptosis and cell cycle arrest at G0/G1 phase, suppressed migration and invasion. Further, the xenograft tumor model experiment confirmed that HIPPI knockdown inhibited tumor growth in vivo. These results indicate HIPPI promotes cell proliferation, migration and invasion by regulating cell cycle in NSCLC.
Enabling replicative immortality and uncontrolled cell cycle are hallmarks of cancer cells. The MCM proteins act as the DNA replicative helicase to unwind duplex DNA and initiate DNA replication and play vital roles in DNA replication and cell cycle progression.22 Recent studies have suggested that dysregulated MCMs lead to tumor initiation, progression, and chemoresistance via modulating cell cycle and DNA replication stress.23,24 Overexpressed MCMs are detected in various cancer tissues and are potential biomarkers for the diagnosis and prognosis of many kinds of cancers. Wu et al25 found that high levels of MCM2 were observed in LUSC and associated with adverse tumor features. Dehan et al26 confirmed the upregulation of MCM2 and MCM6 in NSCLC. Besides, MCM2 and MCM6 were also found to be upregulated in other cancers, such as HCC,27 pancreatic cancer,28 gastric cancer29 and triple-negative breast cancer.30 MCM8 expression was undoubtedly up-regulated in gastric cancer and high expression of MCM8 was associated with shorter overall survival (OS) and progression-free survival in patients with gastric cancer.31 Consistent with these findings, our study also found that MCM2, MCM6 and MCM8 were up-regulated in NSCLC tissues, and overexpression of MCM2, MCM6 and MCM8 correlated with shorter OS of NSCLC patients. Further cellular experiments revealed that knockdown of HIPPI decreased the expression of MCM2, MCM6 and MCM8 at transcriptional level. Hence, given the crucial function of the MCM family in tumor development and progression, we suggest that HIPPI activates DNA replication and cell cycle pathways by up-regulating the expression of MCM2, MCM6 and MCM8, thereby exerting an oncogenic role in NSCLC.
There may be some possible limitations in this study. First, due to the short follow-up time of the cases in this study, it was not sufficient for prognostic analysis. Although the prognostic analysis of the expression levels of HIPPI in LUAD patients from TCGA showed no statistically significant difference (Figure S1), long-term follow-up of patients is needed for prognostic analysis. Second, we conducted a loss-of-function experiment on HIPPI. Although these results confirmed that HIPPI plays an important role in lung cancer, the supplement of gain-of-function experiments may be more convincing, and we will continue to do it in future experiments. In addition, HIPPI may have a broad transcriptional regulation effect. This study only focused on three genes related to DNA replication. Therefore, more experiments are needed to further explore the regulatory relationships between HIPPI and other genes.
Conclusion
In conclusion, our study revealed the oncogenic role of HIPPI via regulating DNA replication in NSCLC. Overexpressed HIPPI is involved in the development and chemotherapy sensitivity, suggesting that HIPPI may be a potential therapeutic target in NSCLC.
Abbreviations
HIPPI, huntingtin Interacting Protein-1 Interacting Protein; NSCLC, non-small cell lung cancer; IHC, immunohistochemistry; qRT-PCR, quantitative real-time polymerase chain reaction; CCK-8, cell counting kit-8 assay; BAR, bifunctional apoptosis regulator; NRSF, neuron-restrictive silencer factor; IRS, immunoreactivity score; shRNA, short hairpin RNA; sh-HIPPI, short hairpin RNA targeting HIPPI; NC, negative control; RIPA, radioimmunoprecipitation assay; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; FITC, fluorescein isothiocyanate; PVDF, polyvinylidene difluoride; PBS, phosphate-buffered saline; PI, propidium iodide; IC50, inhibitory concentration; ANOVA, analysis of variance; MCM, minichromosome maintenance proteins.
Ethics Approval and Consent to Participate
This study was approved by the Medical Ethics Committee of Tianjin Medical University (Tianjin, China), and was performed in accordance with the Declaration of Helsinki. Written informed consent was obtained from the enrolled subjects. All animal experiments were followed the National Institutes of Health guide for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978).
Acknowledgment
This work was supported by grants from the National Natural Science Foundation of China (81773207), the Natural Science Foundation of Tianjin (19YFZCSY00040), and the Special support program for High Tech Leader & Team of Tianjin (TJTZJH-GCCCXCYTD-2-6). Funding sources had no role in study design, data collection, and analysis; in the decision to publish; or in the preparation of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
2. Wanker EE. Hip1 and hippi participate in a novel cell death-signaling pathway. Dev Cell. 2002;2:126–128. doi:10.1016/S1534-5807(02)00121-1
3. Bhattacharyya NP, Banerjee M, Majumder P. Huntington’s disease: roles of huntingtin-interacting protein 1 (HIP-1) and its molecular partner HIPPI in the regulation of apoptosis and transcription. FEBS J. 2008;275:4271–4279. doi:10.1111/j.1742-4658.2008.06563.x
4. Majumder P, Chattopadhyay B, Mazumder A, Das P, Bhattacharyya NP. Induction of apoptosis in cells expressing exogenous hippi, a molecular partner of huntingtin-interacting protein Hip1. Neurobiol Dis. 2006;22:242–256. doi:10.1016/j.nbd.2005.11.003
5. Majumder P, Choudhury A, Banerjee M, Lahiri A, Bhattacharyya NP. Interactions of HIPPI, a molecular partner of huntingtin interacting protein HIP1, with the specific motif present at the putative promoter sequence of the caspase-1, caspase-8 and caspase-10 genes. FEBS J. 2007;274:3886–3899. doi:10.1111/j.1742-4658.2007.05922.x
6. Majumder P, Chattopadhyay B, Sukanya S, et al. Interaction of HIPPI with putative promoter sequence of caspase-1 in vitro and in vivo. Biochem Biophys Res Commun. 2007;353:80–85. doi:10.1016/j.bbrc.2006.11.138
7. Niu Q, Ybe JA. Crystal structure at 2.8 A of Huntingtin-interacting protein 1 (HIP1) coiled-coil domain reveals a charged surface suitable for HIP1 protein interactor (HIPPI). J Mol Biol. 2008;375:1197–1205. doi:10.1016/j.jmb.2007.11.036
8. Stanton SE, Blanck JK, Locker J, Schreiber-Agus N. Rybp interacts with hippi and enhances hippi-mediated apoptosis. Apoptosis. 2007;12:2197–2206. doi:10.1007/s10495-007-0131-3
9. Backendorf C, Visser AE, de Boer AG, et al. Apoptin: therapeutic potential of an early sensor of carcinogenic transformation. Annu Rev Pharmacol Toxicol. 2008;48:143–169. doi:10.1146/annurev.pharmtox.48.121806.154910
10. Cheng CM, Huang SP, Chang YF, Chung WY, Yuo CY. The viral death protein apoptin interacts with hippi, the protein interactor of huntingtin-interacting protein 1. Biochem Biophys Res Commun. 2003;305:359–364. doi:10.1016/S0006-291X(03)00764-2
11. Sakamoto K, Yoshida S, Ikegami K, et al. Homer1c interacts with hippi and protects striatal neurons from apoptosis. Biochem Biophys Res Commun. 2007;352:1–5. doi:10.1016/j.bbrc.2006.10.167
12. Datta M, Bhattacharyya NP. Regulation of RE1 protein silencing transcription factor (REST) expression by HIP1 protein interactor (HIPPI). J Biol Chem. 2011;286(39):33759–33769. doi:10.1074/jbc.M111.265173
13. Gdynia G, Lehmann-Koch J, Sieber S, et al. BLOC1S2 interacts with the HIPPI protein and sensitizes NCH89 glioblastoma cells to apoptosis. Apoptosis. 2008;13:437–447. doi:10.1007/s10495-007-0176-3
14. Datta M, Choudhury A, Lahiri A, Bhattacharyya NP. Genome wide gene expression regulation by HIP1 protein interactor, HIPPI: prediction and validation. BMC Genomics. 2011;12:463. doi:10.1186/1471-2164-12-463
15. Regzedmaa O, Li Y, Li Y, et al. Prevalence of DLL3, CTLA-4 and MSTN expression in patients with small cell lung cancer. Onco Targets Ther. 2019;12:10043–10055. doi:10.2147/OTT.S216362
16. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi:10.1093/nar/gkv007
17. Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10:1523. doi:10.1038/s41467-019-09234-6
18. Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the evolution of non-small-cell lung cancer. N Engl J Med. 2017;376:2109–2121. doi:10.1056/NEJMoa1616288
19. Herbst R, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–454. doi:10.1038/nature25183
20. Huang W, Yan Y, Liu Y, et al. Exosomes with low miR-34c-3p expression promote invasion and migration of non-small cell lung cancer by upregulating integrin α2β1. Signal Transduct Target Ther. 2020;5:39. doi:10.1038/s41392-020-0133-y
21. Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. 2017;17:637–658.
22. Li Z, Xu X. Post-translational modifications of the mini-chromosome maintenance proteins in DNA replication. Genes. 2019;10:331.
23. Jang N, Baek J, Ko Y, Song P, Gu M. High MCM6 expression as a potential prognostic marker in clear-cell renal cell carcinoma. In Vivo. 2021;35:299–306. doi:10.21873/invivo.12259
24. Yu S, Wang G, Shi Y, Xu H, Zheng Y, Chen Y. MCMs in cancer: prognostic potential and mechanisms. Anal Cell Pathol. 2020;2020:3750294. doi:10.1155/2020/3750294
25. Wu W, Wang X, Shan C, Li Y, Li F. Minichromosome maintenance protein 2 correlates with the malignant status and regulates proliferation and cell cycle in lung squamous cell carcinoma. Onco Targets Ther. 2018;11:5025–5034. doi:10.2147/OTT.S169002
26. Dehan E, Ben-Dor A, Liao W, et al. Chromosomal aberrations and gene expression profiles in non-small cell lung cancer. Lung Cancer. 2007;56:175–184. doi:10.1016/j.lungcan.2006.12.010
27. Cao T, Yi S, Wang L, et al. Identification of the DNA replication regulator MCM complex expression and prognostic significance in hepatic carcinoma. Biomed Res Int. 2020;2020:3574261. doi:10.1155/2020/3574261
28. Deng Y, Ma H, Hao J, Xie Q, Zhao R. MCM2 and are potential biomarkers for the diagnosis and prognosis of pancreatic cancer. Biomed Res Int. 2020;2020:8604340. doi:10.1155/2020/8604340
29. Liu M, Li J, Tian D, Huang B, Rosqvist S, Su M. MCM2 expression levels predict diagnosis and prognosis in gastric cardiac cancer. Histol Histopathol. 2013;28(4):481–492. doi:10.14670/HH-28.481
30. Issac M, Yousef E, Tahir M, Gaboury L. MCM2, MCM4, and MCM6 in breast cancer: clinical utility in diagnosis and prognosis. Neoplasia. 2019;21(10):1015–1035. doi:10.1016/j.neo.2019.07.011
31. Huang B, Lin M, Lu L, et al. Identification of mini-chromosome maintenance 8 as a potential prognostic marker and its effects on proliferation and apoptosis in gastric cancer. J Cell Mol Med. 2020;24(24):14415–14425. doi:10.1111/jcmm.16062
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.