Back to Archived Journals » Hypoxia » Volume 2
Signaling hypoxia by hypoxia-inducible factor protein hydroxylases: a historical overview and future perspectives
Authors Bishop T, Ratcliffe P ![]()
Received 17 September 2014
Accepted for publication 2 October 2014
Published 5 December 2014 Volume 2014:2 Pages 197—213
DOI https://doi.org/10.2147/HP.S47598
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Tammie Bishop, Peter J Ratcliffe
Nuffield Department of Medicine, University of Oxford, Oxford, UK
Abstract: By the early 1900s, the close matching of oxygen supply with demand was recognized to be a fundamental requirement for physiological function, and multiple adaptive responses to environment hypoxia had been described. Nevertheless, the widespread operation of mechanisms that directly sense and respond to levels of oxygen in animal cells was not appreciated for most of the twentieth century with investigators generally stressing the regulatory importance of metabolic products. Work over the last 25 years has overturned that paradigm. It has revealed the existence of a set of “oxygen-sensing” 2-oxoglutarate dependent dioxygenases that catalyze the hydroxylation of specific amino acid residues and thereby control the stability and activity of hypoxia-inducible factor. The hypoxia-inducible factor hydroxylase pathway regulates a massive transcriptional cascade that is operative in essentially all animal cells. It transduces a wide range of responses to hypoxia, extending well beyond the classical boundaries of hypoxia physiology. Here we review the discovery and elucidation of these pathways, and consider the opportunities and challenges that have been brought into focus by the findings, including new implications for the integrated physiology of hypoxia and therapeutic approaches to ischemic/hypoxic disease.
Keywords: hypoxia, oxygen-sensing, HIF, 2-oxoglutarate oxygenase, hydroxylase, cell signaling
Introduction
Provision of adequate levels of oxygen across different respiring tissues is a physiological challenge faced by all large multi-cellular organisms. Low or inadequate oxygen provision (hypoxia) is an important feature of most human disease. Much interest has therefore focused on the causes and consequences of “hypoxia” and on “oxygen-sensing” mechanisms that transduce adaptive responses in hypoxic cells.
Under physiological conditions, cells exist in highly diverse oxygen environments, and the term hypoxia, as referring to levels of oxygen that are low or sub-optimal in respect of cellular functions, has no exact physical correlate in terms of the partial pressure of oxygen (pO2) or O2 concentration. For instance, physiological oxygen tensions in humans range from 80–100 mmHg in the lung alveoli and arterial blood, down to 1–2 mmHg in tissues such as the renal papilla.1 Such regions are sometimes described as being physiologically hypoxic. Thus, “hypoxia”, may refer either to levels of oxygen that are simply low in a physical sense (physiological hypoxia) or to levels that are low in relation to those required for optimal physiological function (patho-physiological hypoxia). Similarly the term “oxygen-sensor” requires clarification. The function of many biological systems depends on oxygen, so that limitation of oxygen supply will have biological consequences. All such systems will clearly be “oxygen-sensitive”. To distinguish bona fide regulatory pathways, we define an “oxygen-sensing” system as one whose prime function is in biological control.
In this review we will outline major advances that have been made in the understanding of animal oxygen-sensing pathways, and consider new questions and opportunities raised by these insights.
Discovery of direct oxygen-sensing pathways in animal cells
The co-ordination of multiple physiological and developmental processes is required to ensure the delivery of appropriate quantities of oxygen to billions of respiring cells by the specialized oxygen delivery systems that characterize higher animals. In theory, control of such processes could be achieved by systems responding to oxygen levels per se, or to any one of many metabolic consequences of altered oxygen availability. Thus Krogh, in his landmark paper on the capillary density of muscle, noted that “the number of capillaries per square millimetre of striated muscle appears to be a function of metabolism”.2 A large body of subsequent work documented that capillary density was under dynamic control by interventions that altered the consumption or delivery of oxygen. However, almost invariably the investigators emphasized the importance of products of metabolism.3,4 Remarkably little attention was paid to the possibility that at least some of the responses might be directly regulated by levels of oxygen.
Nevertheless, direct regulation by oxygen was suggested by studies of regulation of red blood cell production by erythropoietin (Epo). In particular it had been observed that cobalt intoxication stimulates Epo production and erythropoiesis5 without obvious effects on metabolism, and that metabolic inhibitors could not mimic this effect.6 These findings strongly suggested that the regulatory process was distinct from the effects of hypoxia on metabolism and led to the idea of a specialized oxygen-sensing system, which was generally considered to be restricted to specialized cells mediating this strikingly dynamic response. Studies of the regulation of Epo by oxygen were initially held back by uncertainties about the molecular identity of the hormone and by the absence of a cellular system on which to base molecular analysis. In particular, the identity of the specific cells in kidney that produce Epo (now known to be a subset of the interstitial fibroblasts)7,8 was unclear and Epo-producing cultures of such cells could not be established.
In the late 1980s both of these obstacles were removed with the identification of the Epo gene,9 and the recognition that certain human hepatoma cells produce Epo in culture in an oxygen-regulated manner.10 Analysis of Epo gene regulation in these cells and in transgenic animals expressing a human Epo gene, led to the identification of Epo 3′ enhancer as the first hypoxia response element (HRE).11–13 Further studies of this enhancer radically opened the field in two distinct ways. Analysis of proteins binding to the HRE in hepatoma cells led to the identification of hypoxia-inducible factor-1 (HIF-1) by Semenza and Wang,14 whilst in our laboratory, transfection studies of the Epo 3′ enhancer in a wide range of non-Epo-producing cell types redirected the field by demonstrating (much contrary to the prevailing view) that this system of oxygen-sensing is widely operative in mammalian cells and extends well beyond the regulation of Epo.15 Subsequent work demonstrated that HIF-1 itself is indeed widely expressed, and defined genes encoding selected isoforms of glycolytic enzymes as the first non-Epo HIF target genes.16,17 Further work revealed the existence of a HIF-like DNA-binding complex in Drosophila melanogaster,18 which has neither Epo nor a blood vascular system, whilst studies in mammalian cells led to a rapidly expanding list of HIF target genes with diverse functions in biology, including glucose transporters,19 angiogenic growth factors,20,21 and iron transport proteins.22
Using oligonucleotide affinity chromatography based on the Epo HRE, the Semenza laboratory purified the HIF DNA-binding complex, revealing an α/β heterodimer of basic-helix-loop-helix-PAS (PER [period circadian protein]-AHR [aryl-hydrocarbon receptor]/ARNT [aryl-hydrocarbon-receptor nuclear translocator]-SIM [single-minded protein]) proteins.23 Whilst HIF-1β had previously been identified as the ARNT,24 which dimerizes with AHR to activate the xenobiotic response, HIF-1α was a newly identified protein conveying hypoxia-inducibility on the DNA-binding complex. Subsequently, bioinformatic analyses revealed paralogs of both HIF-1α and HIF-1β amongst an extensive family of basic-helix-loop-helix PAS proteins that had been identified by genomic analysis.25–27 Thus, mammalian genomes encode three HIF-α isoforms, each manifesting oxygen-sensitive activity: HIF-1α, HIF-2α (also termed EPAS-1)25 and HIF-3α (which generates multiple alternately spliced forms including molecules termed IPAS26 and NEPAS).28
Elucidation of the oxygen-sensing mechanism
Initial insights into the upstream pathways connecting HIF-α to the oxygen-sensing process were obtained by the definition of discrete domains that could confer oxygen sensitivity on heterologous transcription factors.29–31 Somewhat surprisingly, these analyses revealed the existence of multiple oxygen-sensitive domains. All of these retained the classical property of activation by cobaltous ions but appeared to operate in different ways: either by promoting changes in protein stability or changes in activity. It was generally believed that protein phosphorylation would be involved in signal transduction and hence impinge on these domains. Surprisingly, however, although it was clear that HIF-1α is heavily phosphorylated, mutation of every phospho-acceptor amino acid in one such isolated oxygen-regulated domain failed to ablate the oxygen-sensitivity, clearly indicating that some other mechanism of sensing/transduction was operating.29
Insights into this process ultimately came from the recognition that this domain (now known as the C-terminal oxygen degradation domain) was a binding site for the von Hippel-Lindau tumor suppressor protein (pVHL). Marked up-regulation of hypoxia-inducible genes had been noted in VHL-defective cells, but the mechanism was initially unclear.32,33 A key insight was therefore provided by the demonstration that pVHL interacted with and was necessary for the proteolytic regulation of both HIF-1α and HIF-2α,34 findings that were in keeping with the predicted function of pVHL as an ubiquitin E3 ligase. Subsequent work confirmed that pVHL is indeed an E3 ubiquitin ligase that directly targets two regions of a central oxygen-dependent degradation domain in HIF-α.35–38 These regions (the N-terminal oxygen degradation domain, and the C-terminal oxygen degradation domain) can act independently to mediate pVHL-dependent proteolytic destruction of HIF-α,38 but appear, in their physiological context, to act in a highly conserved co-operative manner to promote efficient oxygen-dependent destruction of HIF-α.39,40
Further analysis of the interaction between pVHL and HIF-α polypeptides revealed that the binding to pVHL required modification of the HIF polypeptide by an oxygen-dependent reaction. Biochemical analysis of this reaction and mutational/mass spectrometric studies of modified HIF-α peptides revealed the modification to be trans-4-hydroxylation of specific prolyl residues within each of the two pVHL-binding domains (Pro-402 and Pro-564 in human HIF-1α).38,41–43 The target prolyl hydroxylation sites did not resemble substrate sequences for the pro-collagen prolyl hydroxylases (then the only known human prolyl hydroxylases) and genetic studies excluded these enzymes as the source of the activity. However, the pro-collagen prolyl hydroxylases are members of the 2-oxoglutarate-dependent oxygenase (2-OG oxygenase) superfamily, and the properties of these enzymes fitted extremely well with the classical characteristics of Epo regulation and with the biochemical properties of the HIF-α modifying reaction. In particular, 2-OG oxygenases are dependent on molecular oxygen as co-substrate, and are markedly susceptible to inhibition by cobaltous ions. This suggested that one or more enzymes in this family might be responsible for generation of the oxygen-sensitive signal. Based on this hypothesis, a combination of structurally informed prediction and candidate testing defined three enzymes termed prolyl hydroxylase domain (PHD) 1, 2, and 3 (otherwise known as Egln 2, 1, and 3, respectively) that catalyze HIF prolyl hydroxylation.44,45 PHD activity was shown to be markedly sensitive to inhibition by hypoxia.44
Shortly after the elucidation of the HIF-PHD-pVHL pathway, a third site of hydroxylation (in this case asparaginyl hydroxylation) was defined by mass spectrometric analyses of the C-terminus of HIF-1α and HIF-2α and shown to regulate activity of the C-terminal activation domain,46 at least in part by preventing recruitment of p300/CREB (cAMP-response-element-binding protein) binding protein (CBP) co-activators. Bioinformatic prediction identified a molecule termed FIH (factor inhibiting HIF), which had been previously identified as inhibiting HIF by an unknown mechanism,47 as a second type of 2-OG oxygenase that catalyzes HIF asparaginyl hydroxylation.48,49 Thus, in mammalian cells a total of four 2-OG oxygenases generate oxygen-sensitive signals that regulate HIF by a dual process of prolyl and asparaginyl hydroxylation, promoting both destruction and inactivation of HIF-α in the presence of oxygen (Figure 1).
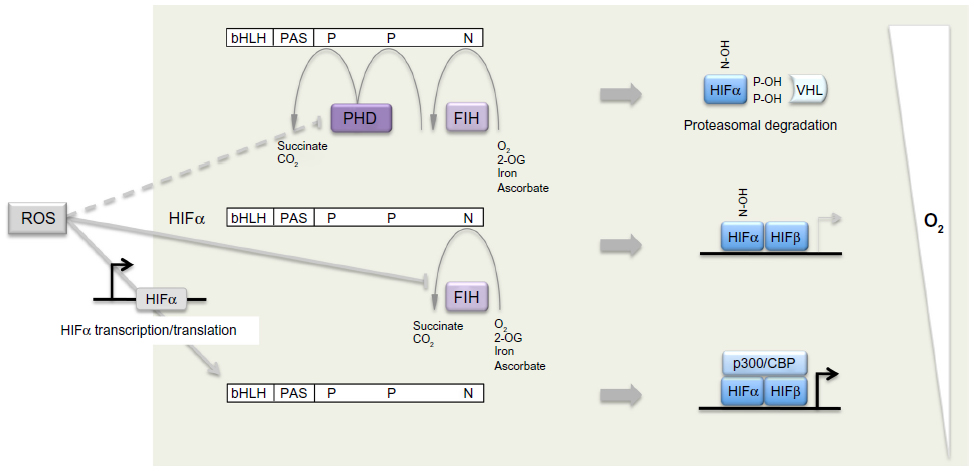 | Figure 1 Dual regulation of hypoxia-inducible factor (HIF)-α by oxygen-dependent prolyl and asparaginyl hydroxylation. |
These enzymes belong to a family of 60–70 human 2-OG oxygenases.50 They are characterized by a double-stranded β helix “jelly-roll” catalytic domain that co-ordinates the catalytic iron center through a 2-histidine-1-carboxylate “facial triad”.51 The binding of iron is labile, hence the enzymes are also Fe (II) dependent and inhibited by iron chelators. During catalysis, hydroxylation of HIF-α is coupled to the oxidative decarboxylation of 2-OG to succinate, in a reaction that splits molecular oxygen. Uncoupled cycles leave the enzyme in an oxidized, inactive, form and ascorbate (which is required non-stoichiometrically for full catalytic activity) acts at least in part to regenerate active enzyme. It is proposed that the inhibitory action of cobaltous ions and other transition metal ions arises from replacement of catalytic Fe (II) and/or effects on the availability of ascorbate.52 The extent to which the HIF hydroxylases are specialized among the 2-OG oxygenases for their “oxygen-sensing” function is not clear. Though high values for the Michaelis constant for oxygen (KmO2; >200 mmHg) have been described for PHD enzymes,53–55 the relevance is unclear since physiological pO2 levels are much lower and would support oxygen-limited kinetics even with a much lower KmO2. Nevertheless, unusual kinetic properties have been described for recombinant PHD2. First the PHDs, in particular PHD2, have an unusually high affinity for Fe (II),56 a property that is conserved in Trichoplax PHD,40 and which has also been reported for a different oxygen-sensing prolyl hydroxylase in Dictyostelium.57 Second, in the absence of HIF-α substrate, the PHD2.Fe.2-OG complex is unusually stable even in the presence of oxygen.56 Third, the reaction of the PHD2.Fe.2-OG.HIF-1α complex with oxygen is very slow in comparison with reaction rates of other 2-OG oxygenases.58 Such properties might be important in enabling the enzyme to avoid uncoupled turnover and operate accurately on very low levels of endogenous HIF-α substrates.
Evolution of the HIF hydroxylase system
Although oxygen-sensing systems are present in a wide range of prokaryotic and eukaryotic species, the HIF hydroxylase system appears to be restricted to animals, even though individual components of the system are present in many non-animal species.40 Thus, basic-helix-loop-helix proteins are found in all eukaryotes,59 whereas PAS domains are found in both prokaryotes and eukaryotes and commonly perform sensing functions, including oxygen-sensing.60 However basic-helix-loop-helix PAS proteins (the family of transcription factors to which HIF belongs) are restricted to animals. Very surprisingly, despite the role of PAS domains in other species, no sensing function has been defined for the PAS domains in HIF-α so far, although structural studies of one isoform, HIF-2α, have revealed a deep cavity in the PAS domain that has been postulated to interact with an, as yet undefined, endogenous ligand.61 Instead, as outlined above, oxygen-sensitive post-translational hydroxylation acts on sequences lying distal to the basic-helix-loop-helix PAS domains of HIF-α. Both types of HIF hydroxylase belong to groups of 2-OG dependent dioxygenases that manifest conservation across prokaryotes and eukaryotes and include members that perform oxygen-sensing functions in non-animal species.62–64 Thus, although the major components of the HIF hydroxylase pathway are represented widely across both animal and non-animal species, the present day HIF hydroxylase system appears to have emerged with the evolution of animals (Figure 2).
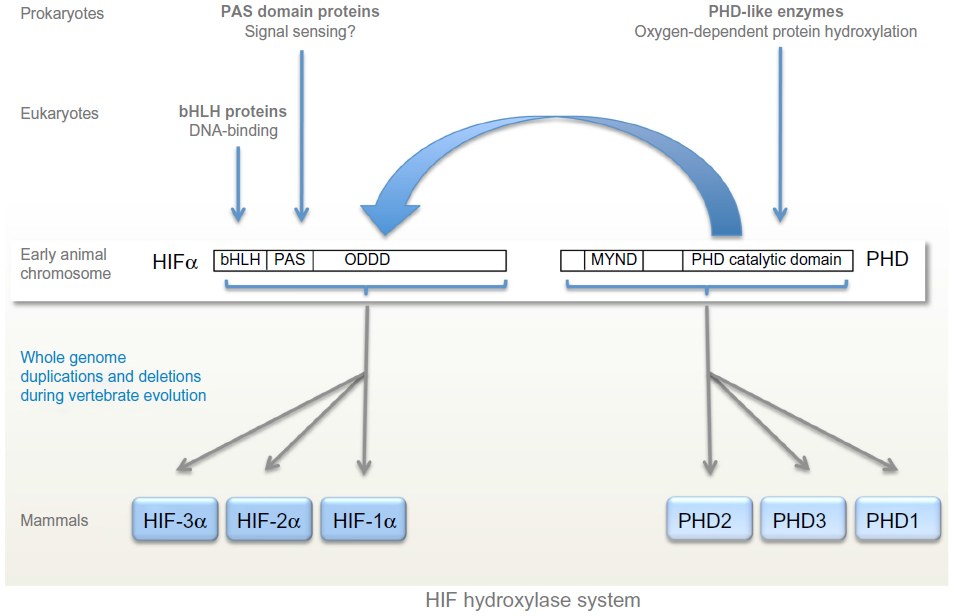 | Figure 2 Evolution of the hypoxia-inducible factor (HIF) hydroxylase system. |
At least one component of the basic HIF-PHD system has been identified in all animal species.40,65 The basic HIF hydroxylase system is represented by the HIF-1α/PHD2 couple; HIF-1α and PHD2 are the most widely expressed isoforms in higher animals and PHD2 makes the dominant contribution to the regulation of HIF-1α.66 The primitive PHD2 isoform is clearly marked by the presence of an N-terminal MYND finger motif that is conserved in all animal species.40 The multiple PHD and HIF isoforms seen in higher animals have arisen though gene duplication events at the base of vertebrate evolution. Mammalian genomes encode three PHD and three HIF isoforms, the product of two gene duplication events and one gene loss. The evolution of FIH is less clear; although all sequenced vertebrate genomes encode an FIH-like protein, the enzyme appears to have been lost from a number of invertebrate species including D. melanogaster and Caenorhabditis elegans.40,65
Although all the PHDs are active on both HIF-1α and HIF-2α, PHD1 and PHD3 are relatively more active on HIF-2α,67 suggesting that these less abundant, tissue-restricted, isoforms have evolved discrete functions. The regulation of HIF-3α is less well understood, but it contains at least one site of prolyl hydroxylation which promotes association with the pVHL E3 ligase.68 The HIF-3α gene forms multiple alternatively spliced and promoted variants, which are either transcriptionally inactive or weakly active, due to lack of transactivation domains. Some of these transcripts are themselves transcriptionally induced by HIF,69 and at least one (IPAS) can form non-productive heterodimers with HIF-1α to inhibit HIF transcription, potentially operating as a negative feedback control.26 HIF-1α and HIF-2α both mediate positive transcriptional responses to hypoxia, but despite recognizing identical DNA sequences, they manifest selectivity both in the distribution of binding sites across the genome and in the transcription activity of the DNA-bound isoform.70–72 Thus, pan-genomic analyses reveal in excess of 500 high stringency binding sites for each of HIF-1α and HIF-2α, about half of which are common to both isoforms.70,73,74
Insights into discrete physiological functions of HIF-1 and HIF-2 are intriguing in that although distinct functions have been described at every level of analysis (molecular, cellular, and organismal) a clear “headline” separation of over-arching physiological function has yet to emerge. Vertebrate evolution is characterized both by increasingly sophisticated oxygen delivery systems, and by the appearance of these additional HIF-PHD isoforms. In that context it is interesting that one of the clearest HIF-2α specific functions is in the regulation of Epo.75,76 Nevertheless, gene inactivation studies (eg, of cardiovascular development) clearly indicate that the primitive HIF-1α/PHD2 couple remains fundamentally involved in specialized vertebral oxygen delivery.77,78 Furthermore, although groups of transcriptional targets are discrete to one or other isoform (eg, glycolytic genes are discrete HIF-1α targets),79 other genes that apparently function in similar pathways (eg, glucose transporters) are apparently regulated by both isoforms.80 Whereas sometimes both isoforms contribute to activation of a response, in other settings the two isoforms act in opposition. For instance, macrophage differentiation to the M1 phenotype is promoted by HIF-1α whereas differentiation to the M2 phenotype is promoted by HIF-2α, with HIF-1α (inducible nitric oxide synthase) and HIF-2α (arginase) target genes having opposing functions in nitric oxide production and breakdown.81 Differences between HIF-1α and HIF-2α extend to their signal cross-talk and non-transcriptional functions. For instance, HIF-1α but not HIF-2α has been reported to antagonize Myc transcription by physical association with the HIF-1α PAS domain.82,83 Differential action of HIF-1α and HIF-2α is also observed in the regulation of protein translation. For instance, HIF-2α, but not HIF-1α, activates an alternative translational initiation process in hypoxic cells. This involves the recruitment of eIF4E2 to a set of messenger ribonucleic acids (mRNAs) via an interaction between a specific RNA sequence element, the RNA-binding protein RBM4, and HIF-2α.84
Overall, although HIF-2α generally displays slower and more prolonged induction in response to hypoxia than HIF-1α,85 mediates adaptive mechanisms such as hypoxia-specific translation,84 and has transcriptional targets that are more commonly associated with adaptive/reparative rather than cytostatic responses to hypoxia,74,86 their roles appear not to be clearly distinct. Rather, a complex mixture of overlapping and antagonistic functions has been described.
The interface of molecular oxygen-sensing systems with integrated physiology
The massive diversity of HIF transcriptional targets revealed by cellular studies (Figure 3) immediately suggested a fundamental role in the integrated physiology of oxygen homeostasis. Phenotypes associated with targeted inactivation of the pathway in mice, and with human genetic polymorphism, have amply confirmed this.87,88 These studies not only demonstrated a major role in the classical cardiopulmonary, vascular, and metabolic responses to hypoxia, but also revealed many effects (such as on immune cell differentiation) that have not been extensively studied at the organismal level as part of the integrated physiology of hypoxia (Figure 4).
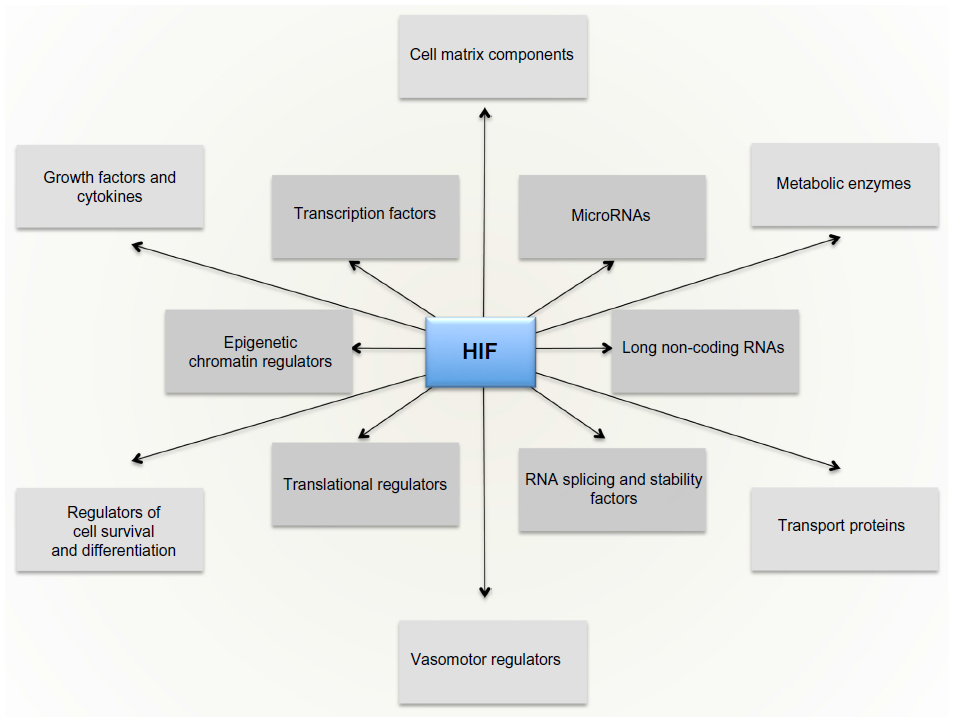 | Figure 3 The HIF transcriptional cascade. |
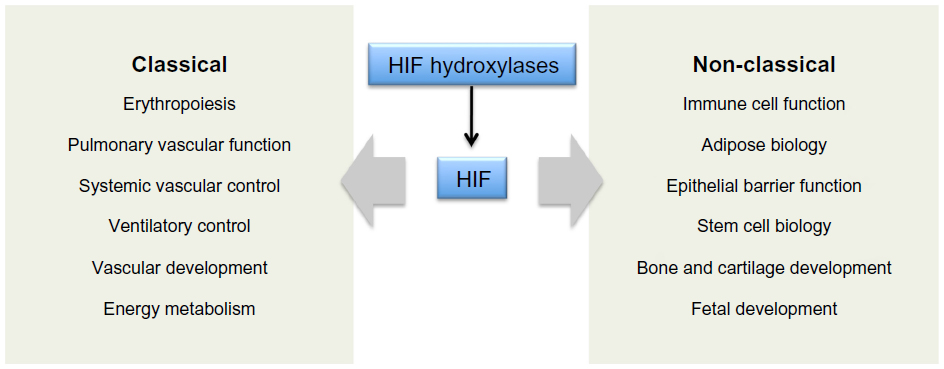 | Figure 4 Integrated physiological functions associated with HIF hydroxylase pathways. |
The findings suggest that hypoxia has a much wider role in the integrated physiology of the intact organism than was previously foreseen and raise a series of important new questions. How does the HIF hydroxylase system signal hypoxia over the wide range of oxygen tensions encountered by cells in the intact organism? Does the involvement of HIF signaling in these “new” physiological processes imply control by environmental hypoxia? How does fetal hypoxia impinge on development? Does activation of “oxygen-sensing” pathways in disease contribute to pathology, or do these pathways ameliorate disease by promoting adaptive responses? Can damaging and protective responses be distinguished and differentially activated therapeutically? These questions are largely unanswered, but we will summarize some of the major lines of research and consider the implications of findings to date.
The HIF pathway in developmental oxygen homeostasis
In most organisms rates of oxygen consumption are such that transfer of oxygen by diffusion is limiting, placing constraints on the mass and organization of tissues. Thus it might be predicted that inactivation of the HIF/PHD system would disrupt the development of even primitive animals and/or alter fundamental parameters such as cell size. Oddly, studies of invertebrate development show little evidence for this. The nematode worm C. elegans relies on simple diffusion of oxygen, whilst D. melanogaster employs a system of air passages known as the tracheal system to augment oxygen delivery. However, in neither organism is HIF induced to any major extent during normal development.89,90 Moreover, development of worms and flies (including fly tracheal development) occurs relatively normally in the presence of oxygen in the absence of a functional HIF gene.89–91 Nevertheless, the HIF/PHD system is clearly functional, as HIF-deficient developing flies and worms are extremely vulnerable to hypoxia.89,90 Moreover, PHD inactivation results in abnormal development (eg, excessive tracheal formation) that is corrected by additional inactivation of HIF.90 Thus, the HIF/PHD system is not essential for all aspects of developmental oxygen homeostasis. Rather it appears to operate on top of a developmental plan which is already compatible with oxygen homeostasis. This is illustrated by studies of the regulators of tracheal development in the fly (branchless and breathless, homologs respectively of the human fibroblast growth factor and its receptor). In contrast with the HIF/PHD system, disruption of the branchless/breathless pathway has more profound effects on fly tracheal development, in keeping with this pathway being controlled both by “hard-wired” developmental processes as well as being a target of the adaptive HIF/PHD system.92
This principle of HIF signaling operating on top of “hard-wired” developmental processes that are compatible with oxygen homeostasis is reflected in the early development of higher animals. Despite a physiologically hypoxic uterine environment, processes such as uterine implantation still take place in HIF deficient embryos. Furthermore, aspects of early embryonic development that are fundamental to oxygen homeostasis apparently occur independently of HIF signaling. For instance, neither HIF-1α nor HIF-2α is absolutely required for early vasculogenesis,77,93 which is normal even in HIF-1α deficient mouse embryos up to day 8.77 In marked contrast to flies and worms, however, later development of higher animals (as exemplified by studies of the mouse) absolutely requires a functional HIF/PHD system, and appears to be strongly affected by signals from developmental hypoxia. Thus, hypoxic cells expressing HIF are present during late placental development94 and inactivation of HIF-1α and HIF-1β are both associated with major defects in placental development.77,95 The role of HIF activation in specific developmental processes has been studied by targeted inactivation in relevant organs to avoid secondary hypoxia generated by placental disruption. For instance, profound hypoxia is observed in the developing heart prior to establishment of a coronary circulation. Both HIF-1α and HIF-2α are activated,96 patterns being spatially distinct with HIF-1α predominantly myocardial and HIF-2α predominantly endothelial. HIF-1α has been proposed to activate specific cardiac transcription factors and inactivation of HIF-1α in developing ventricular myocytes is associated with defective cardiac chamber development and hyperplasia of the ventricular myocardium.78 Similar studies of tissue-specific inactivation of HIF-1α in other organs have revealed a wide range of phenotypes supporting a central role in diverse developmental processes. These findings suggest the existence of time windows during which activation of HIF by physiological fetal hypoxia contributes to developmental programs. If true, studies of HIF in developing embryos might predict specific time windows during which systemic maternal hypoxia and/or placental ischemia might interact with these signals to generate particular developmental defects. Nevertheless the relationship between systemic hypoxia and regional hypoxia is difficult to predict in the intact organism.97 Surprisingly, despite abundant evidence that maternal hypoxia does indeed lead to developmental defects and adverse fetal outcome,98–100 there have been relatively few studies that attempt to define the precise nature of interaction on the HIF pathway and how this relates to specific developmental phenotypes.
Subversion of HIF pathways: more than oxygen homeostasis?
Inactivation of HIF-α isoforms (particularly HIF-1α) in specific tissues is also associated with a wide range of phenotypes specific to the adult mouse. These studies have implicated HIF in a range of functions that extend beyond the classical physiology of hypoxia (Figure 4). Interestingly, many of these functions, including immune/inflammatory cell biology,101–103 hematopoietic stem cell behavior,104 bone morphogenesis105 and epithelial barrier function,106 map to regions that are physiologically hypoxic.106–108 From studies of HIF inactivation alone it is difficult to distinguish whether HIF is playing a regulatory role or simply required as a permissive factor. However, mechanistic analyses have implicated HIF directly in the control of key molecules. For instance HIF-1α has been implicated in the control of the T-cell transcription factors, ROR-gt109 and FoxP3110 suggesting that micro-environmental hypoxia and HIF activation might indeed have a regulatory role in T-cell differentiation.
Although HIF has often been described as the master regulator of oxygen homeostasis, these and other studies indicate a substantially broader role in biology. Pan-genomic analysis of HIF transcriptional targets has also revealed many whose known functions are difficult to reconcile with a direct role in oxygen homeostasis.111 This is most clearly seen for metabolic target genes where the functions of the gene products are particularly well defined in relation to oxygen consumption. Thus, although some metabolic functions of HIF such as down-regulation of mitochondrial metabolism112,113 are easy to explain in terms of oxygen homeostasis, others are not. For instance HIF has been implicated in both glycogen synthesis114 and glycogen breakdown115 and in energy consuming steps in fatty acid synthesis,116,117 whereas the strongly isoform-specific regulation of glycolytic enzymes is more in keeping with a role in provision of biosynthetic intermediates than anaerobic provision of ATP.118,119
Taken together, these findings suggest that the physiology of oxygen homeostasis and that of the HIF pathway do not overlap precisely. First, the HIF hydroxylase system appears to be superimposed on a developmental plan that is already hard-wired to be broadly compatible with oxygen homeostasis. Second, the system appears to have been subsumed into functions that extend well beyond a direct role in oxygen homeostasis.
The “range finding” problem: integration of oxygen-dependent and non-oxygen-dependent signals
The lack of a fixed relationship between operating tissue pO2 and HIF signaling necessitates “range finding” mechanisms which modulate the relationship between the oxygen-dependent kinetics of the HIF hydroxylation reaction and the overall biological output of HIF activation (Figure 5; for review see Ratcliffe120). How this is achieved remains unclear and is one of the major unsolved problems in the field. Nevertheless, multiple signaling inputs operate at different levels in these pathways and potentially could contribute to the effects observed.
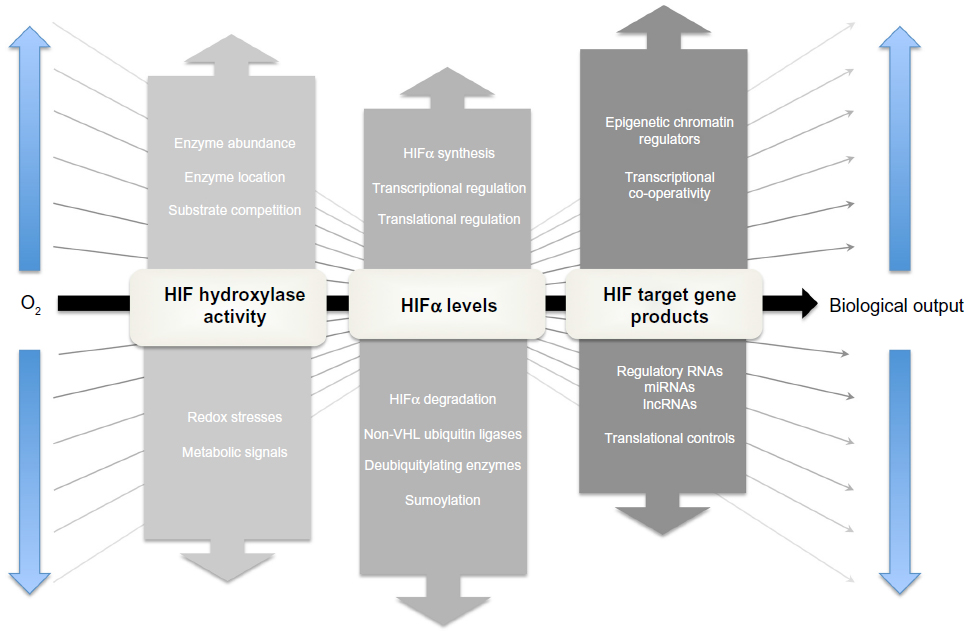 | Figure 5 The principle of “range-finding” processes in the HIF hydroxylase system. |
HIF hydroxylase activity
The co-substrate and co-factor requirements of the HIF hydroxylases (2-OG, iron, and ascorbate, as well as molecular oxygen) potentially provide inputs from metabolic and redox stresses that could modulate the signaling of hypoxia. Of particular interest is the interface with redox signaling. Several mechanisms potentially contribute to inactivation of the HIF hydroxylases by oxidant stress, including reduction in cytosolic Fe (II) levels, oxidation of the catalytic iron in PHD2, auto-hydroxylation of amino acid residues, and oxidation of protein thiols. In keeping with this, many investigators have described modulation of HIF hydroxylase pathways by redox stresses. In cultured cells, oxidant stresses reduce hydroxylation and activate HIF,121,122 whereas the reducing agents, in particular ascorbate, enhance hydroxylation and inactivate HIF.123 In this context, it has been proposed that increased reactive oxygen species (ROS) produced by hypoxic mitochondria, rather than reduced levels of molecular oxygen, are the main means of signaling hypoxia.124,125 However, this remains controversial. Whilst mitochondrial inhibitors block the production of ROS by hypoxic cells and reduce HIF activation, they also relieve hypoxia by reducing consumption of molecular oxygen, complicating mechanistic interpretation.126,127 It is also unclear how well studies in cell culture translate into effects in the intact organism. For instance, although supplementation with ascorbate strongly promotes HIF hydroxylation in cell lines, supplementation of ascorbate in scorbutic rats did not have major effects on the oxygen sensitivity of Epo production.128
In our view, the balance of evidence favors reduction in molecular oxygen itself as the principal cause of reduced HIF hydroxylation in hypoxic cells. Nevertheless, redox signals have the potential to inactivate the HIF hydroxylases and may modulate the response to hypoxia. Some insights into this have been gained through comparative studies of the PHDs and FIH.129,130 Whereas the PHDs are more sensitive to hypoxia than FIH,129 the reverse was observed for oxidant stress, with FIH being much more sensitive to inactivation by peroxide than the PHDs.130 These findings suggest that the two stimuli are distinct, but might interact. Thus partial inactivation of the enzyme by redox stress might alter the apparent kinetics in cells such that hydroxylation would be reduced at any given oxygen tension thus reducing the “gain” of the oxygen sensor in cells experiencing oxidant stress (Figure 1). In contrast, transcriptional induction of PHD2 and PHD3 in hypoxic cells increases the abundance of these enzymes, increasing the rate of hydroxylation at any given oxygen tension and hence increasing the “gain” in hypoxic cells.44,131
Steady-state levels of HIF-α
Levels of HIF-α sub-units are set by the balance between synthesis and degradation and changes in either process will contribute to the overall activity of the pathway. In the presence of oxygen the half-life of HIF-1α is extremely short, in the region of 1–2 minutes,132 whereas in hypoxic or VHL-defective cells where the PHD/pVHL degradation pathway is inactivated, HIF-1α half-life is greatly extended, in the region of 1–2 hours.34 In addition to this oxygen-regulated pathway, a wide range of PHD/pVHL-independent pathways have been described that modulate the degradation of HIF-α irrespective of oxygen. These include deubiquitylating enzymes that antagonize pVHL-mediated ubiquitylation, alternative ubiquitin ligases that target HIF-α domains independently of oxygen, and alterative modifications that alter HIF-α stability, such as sumoylation (for review see Kaelin and Ratcliffe, and Semenza).133,134 All of these processes have the potential to alter the relationship between oxygen and HIF-α levels, particularly in moderately hypoxic cells where oxygen-dependent degradation is already reduced.
Controls of HIF-α synthesis involve both transcriptional and post-transcriptional mechanisms. Take, for instance, the transcriptional activation of the HIF-1α gene by a nuclear factor kappa B (NF-kB) site in its promoter that mediates induction by cytokines, ROS, and other pro-inflammatory stimuli.135 HIF-2α mRNA levels are also regulated by oxidant stresses. For example, in VHL-defective renal cancer cells, the NADPH oxidase Nox4 has been reported to be essential for maintenance of HIF-2α mRNA levels. Translational controls are also of great importance in setting steady-state levels of HIF-α.47 In particular, in proliferating cells, HIF-α translation is augmented by mTOR dependent and mTOR independent pathways that are entrained by activation of the PI3K/PTEN/AKT and RAS/RAF/MAPK signaling cascades, leading to the activation of HIF even in the presence of oxygen (reviewed in Pouyssegur et al).136 In hypoxic cells, HIF-α sub-units escape translation suppression and are synthesized at rates that are either proportional to or exceed levels of the relevant mRNA. The mechanisms underlying this are unclear. Although internal ribosomal entry has been proposed137 this has not been confirmed by others.138 Interestingly, despite high rates of translation, HIF polypeptides have long GC rich 5´ untranslated regions (5´UTRs) suggesting the existence of transcript-specific controls of translation. Given the potential insights into range finding and therapeutic inhibition (see below) this appears to be an under-researched area.
Transcriptional co-operativity
The binding of HIF to HREs is highly selective across the genome. The human genome possesses approximately 1.1×106 HRE consensus sites of which somewhere in the region of 50,000 lie in unmethylated regions of DNA within open chromatin in a typical cell.74 Depending on the level of stringency applied to the Chromatin immunoprecipitation-sequencing (ChIP-seq) signals that have been used to define HIF-binding, only in the region of 500–2,500 of these sites (ie, <5%) actually bind HIF at a given time in hypoxic cells of a particular type.73,111 Across different cells the presence or absence of HIF-binding at a given locus correlates with the existence of a basal level of gene expression at that locus,73 site selectivity most probably reflecting co-operative interactions with other transcription factors that bind at the target locus. These effects underlie qualitative cell-type differences in HIF target gene selectivity. However, it also is likely that the distribution of HIF-binding across target sites is competitive (ie, there are more potential HIF-binding sites than HIF molecules). An important and so far unanswered question is whether there are quantitative differences between the level of HIF-α induction and the occupancy of specific sites, which might generate differences in levels of HIF induction required to promote specific physiological responses, or different sensitivity to oxygen, in different cell types.
Taken together, these findings provide multiple means by which the biological output of the basic oxygen-sensitive signal provided by the HIF hydroxylases could, in theory, be modulated to meet signaling requirements in the intact organism. Key questions remain, however, as to which of these processes actually function physiologically in this way, and whether such an understanding would illuminate therapeutic approaches to the manipulation of specific components as opposed to general activity of the pathway.
Therapeutic potential of manipulating the HIF hydroxylase system
In keeping with the central involvement of hypoxia in most human diseases, elucidation of the HIF hydroxylase oxygen-sensing system has opened a major new therapeutic arena. Detailed accounts of this rapidly expanding field have been given in other recent reviews.139–141 Here we will highlight some of the opportunities and challenges. The rationale for therapeutic manipulation of HIF encompasses two basic perspectives: either that augmenting HIF signaling will improve the outcome of ischemic/hypoxic diseases or that inhibiting HIF activity will improve the outcome of diseases such as cancer that utilize the system to support their development. However, it is likely that this perspective is overly simple. First, in different contexts, activation of HIF can have both pro- and anti-tumorigenic effects, suggesting that the outcome of general blockade of HIF in cancer will depend on the balance of these effects (for reviews see Ratcliffe, Semenza, and Shen and Kaelin).120,142,143 At least in some circumstances, inactivating specific components of the pathway (for instance, specific inactivation of HIF-2α in clear cell renal cancer)143,144 might be the most effective strategy. Second, the complexity of the role of HIF in patho-physiological processes such as immune cell differentiation102,139 combined with the potentially opposing effects of HIF-1 and HIF-2 (for instance on macrophage differentiation)81 make the effects of general activation difficult to predict in other contexts. Many routes to activation or inactivation of HIF pathways have been proposed (reviewed in Semenza, and Fraisl et al).140,141 However, the most clinically advanced approaches aim to reduce HIF activity by inhibiting protein synthesis, or activate HIF by inhibition of the PHDs.
Targeting HIF protein synthesis
The extreme dependence of HIF activation on protein synthesis suggests that agents that modulate protein synthesis would be of value in restricting HIF activation. Indeed, it is possible that existing drugs that target protein synthesis such as mTOR inhibitors and agents that act on upstream oncogenic kinases such as PI3K/Akt and MAP kinase pathways act at least in part through inhibition of HIF.136,140,145 Interestingly, HIF-1α and HIF-2α are differentially dependent on mTOR complexes. Thus, HIF-1α is dependent on both mTORC1 and mTORC2, whilst HIF-2α is dependent on mTORC2.146 Differential targeting of HIF-2α versus HIF-1α might therefore be possible through kinase inhibitors with differential effects on these pathways and might be a valuable approach in HIF-2 dependent VHL-defective renal cancer.143,147
Another interesting lead has been provided by studies of digoxin, which appears to act specifically on translation of HIF-1α.148 Though the doses used in the experimental studies are high in relation to those that inhibit the classic target (Na/K ATPase) and could be dose-limiting, a better understanding of the effects of digoxin of HIF protein synthesis might enable the development of new molecules with greater specificity.
Targeting the HIF prolyl hydroxylases
Inhibition of the HIF hydroxylases by 2-OG analogs and related compounds efficiently induces HIF in normoxic cells and has opened an extensive and very exciting novel therapeutic area (for reviews see Fraisl et al, Selvaraju et al, and Rabinowitz).141,149,150 Given the history of the field and the extreme sensitivity of Epo to regulation by hypoxia, it is fitting that the first target has been the therapy of anemia.
Recombinant Epo is an effective and relatively safe treatment for the anemia of renal disease and other conditions characterized by relative Epo deficiency.151 Nevertheless, some individuals do not respond and concerns have been raised about enhanced rates of cardiovascular events in patients receiving high Epo doses.152 Although mechanism(s) are not fully understood, it is likely that very high plasma Epo levels associated with high dose recombinant Epo have adverse effects on vascular function. HIF prolyl hydroxylase inhibitors augment endogenous Epo levels and have other effects such as promotion of iron absorption that enable efficient stimulation of erythropoiesis at much lower Epo levels.151 Very interestingly, some kidney disease patients appear to be particularly sensitive to prolyl hydroxylase inhibitors and respond more strongly even than control subjects with normal kidneys.153 Therefore, this approach has the potential to avoid Epo-related toxicity and such agents are now in late-stage clinical trials. Many experimental studies suggest that PHD inhibitors have the potential to improve the outcome of ischemic diseases, raising the possibility that rather than simply avoiding cardiovascular toxicity, these agents may in fact improve vascular outcomes in patients being treated for anemia.
A related question is whether PHD inhibitors might be used in the primary treatment of vascular diseases. Although a large number of experimental studies using either genetic or pharmacological intervention of the PHDs have demonstrated benefits in different experimental settings,154–159 long-term, complete inactivation of PHD2 and PHD3, or overexpression of a stabilized HIF-1α protein, has been associated with cardiomyopathy, possibly due to down regulation of mitochondrial function upon activation of HIF.160–162 In the clinical setting, the generation of excessive erythropoiesis could also be problematic. These considerations mandate careful analysis of the temporal and dosing windows that might be deployed to obtain the optimal therapeutic outcome (ie, anti-ischemia versus erythropoietic effects and vice versa). Similar considerations apply to the development of clinical protocols for the use of PHD inhibitors in inflammatory and immune-mediated pathologies.
Importantly, quite subtle differences in drug exposure and drug action could have a major impact on medical outcome. For instance, in anemia it appears likely that the concentration of the drug in the liver and kidneys163 (the Epo producing organs) plays a major role in obtaining a differential effect on anemia versus other effects of HIF activation. Molecules that are less restricted in their organ distribution might offer enhanced anti-ischemic effects in relation to erythropoiesis. It is also likely that agents with modest differences in selectivity amongst the PHDs (or against other 2-OG oxygenases that input to the pathway) would offer therapeutic differentiation. For instance, since HIF-1α and HIF-2α have opposing actions on some outputs81,164 and PHDs 1 and 3 have relatively more action on HIF-2α than HIF-1α,67 small differences in selectivity amongst the PHDs could be clinically significant. In our view, these considerations strongly support the clinical testing of chemically diverse PHD inhibitors.
Finally, given potential difficulties arising from general activation of HIF, it will be important to test non-systemic therapies in the clinic. Experimental studies provide strong support for non-systemic agents in dermatological conditions such as ischemic ulceration,165,166 and gastro-intestinal diseases such as colitis.167–169
Wider role of protein hydroxylation in biology
Though protein hydroxylation had an established role in the structural modification of collagen, its role in signaling, as revealed by discovery of the HIF hydroxylases, was unprecedented. Thus, the identification of the HIF hydroxylases, in particular the recognition that FIH (known to be a member of an extensive group of jumonji-domain containing, Jumonji C [JmjC] proteins)47 was a 2-OG oxygenase that catalyzes protein hydroxylation immediately raised the possibility that hydroxylation of intracellular proteins was more widespread than had been previously considered.48
Subsequently, many JmjC proteins were shown to catalyze demethylation of histone proteins, in a reaction in which hydroxylation of the methyl group leads to demethylation following the release of formaldehyde.170,171 Dynamic regulation of histone methylation has attracted widespread interest and is modulated by hypoxia,172,173 raising questions as to whether the changes induced by hypoxia are indeed mediated by the JmjC 2-OG oxygenases, which enzyme(s) are involved, and what are the physiological consequences. For instance, an interesting possibility is that oxygen-dependent changes in histone methylations modulate the transcriptional output of the HIF pathway. Other studies have indicated that 2-oxygenase catalyzed protein hydroxylation is widespread. First, non-HIF substrates of the HIF hydroxylases have been identified and, second, new types of protein hydroxylase have been defined that are closely related to both the PHD and FIH enzymes.
One important caveat to this work is that secure detection of biologically relevant protein hydroxylation is difficult. Immunological detection of specific hydroxylated epitopes (for instance in HIF-1α) by hydroxy-residue specific antibodies is possible,39,129 but the production of high-fidelity generic hydroxy-residue specific reagents for prospective discovery of novel sites of protein hydroxylation is challenging. Mass spectrometry methods are prone to artifactual oxidations, including on proline, so that rigorous confirmation of enzymatic dependence of proposed sites of hydroxylation is essential. Thus, although a great deal of indirect cell biology evidence has been provided for the existence of non-HIF substrates of the HIF hydroxylases and is in some cases supported by mass spectrometry (MS) evidence of hydroxylation (reviewed in Myllyharju),174 the extent of non-HIF hydroxylation catalyzed by the HIF hydroxylases, particularly the PHDs, remains unclear. However for FIH, studies of the hydroxylation of ankyrin repeat domain (ARD)-containing proteins do provide clear evidence of widespread protein hydroxylation.175–177 The human genome encodes in the region of 300 ARD-containing proteins with highly diverse roles in biology. FIH-catalyzed hydroxylation of asparagine residues within the ankyrin consensus has been demonstrated both in vivo and in vitro and the target hydroxylation has been visualized by nuclear magnetic resonance (NMR) and X-ray methods as well as MS.178,179 Interestingly, within the context of ARD proteins, FIH can catalyze the hydroxylation of a range of amino acid residues including aspartate and histidine in addition to asparagine.180,181
Aside from protein hydroxylation by the HIF hydroxylases, recent work has revealed the existence of new groups of protein hydroxylase that are closely related to the HIF hydroxylases but target sites on the ribosome.62,63,182,183 These ribosomal hydroxylases include a novel prolyl hydroxylase (OGFOD1) that catalyzes hydroxylation of a highly conserved residue on RPS23 that lies at the coding center63,182 and the FIH-like enzymes Mina53 and No66 that catalyze histidinyl hydroxylations on RPL27a and RPL8 respectively.62 Whether these hydroxylations mediate signaling functions akin to the HIF hydroxylases remains to be resolved. However, they are remarkably conserved. For instance, Mina53 and No66 are orthologs of the bacterial hydroxylase ycfD, which catalyzes arginyl hydroxylation at an analogous position in the prokaryotic ribosome.184
Thus, it is now clear that hydroxylation of intracellular proteins extends beyond HIF signaling and is widespread in biology. This raises new questions as to its physiological roles, in particular in signaling oxygen levels, or metabolic/redox stresses. The precision with which signaling is required to sustain physiological oxygen homeostasis raises another fundamental, but largely unanswered, question. That is whether the oxygen-dependent kinetics of HIF hydroxylation in cells is modulated either by substrate competition or by the association of HIF hydroxylases with proteins that bind at the catalytic site but are poor substrates. Future research into the biochemistry, biology, and therapeutic manipulation of these hypoxia signaling pathways can be expected to be rewarding.
Acknowledgment
Work in the authors’ laboratory was supported by the Wellcome Trust, and the Ludwig Institute for Cancer Research.
Disclosure
PJR is a scientific co-founder and holds equity in ReOx Ltd, a university spin-out company that seeks to develop therapeutic inhibitors of the HIF hydroxylases. TB has no conflicts of interest to disclose.
References
Leichtweiss HP, Lübbers DW, Weiss C, Baumgartl H, Reschke, W. The oxygen supply of the rat kidney: measurements of intrarenal pO2. Pflugers Archi. 1969;309(4):328–349. | |
Krogh A. The number and distribution of capillaries in muscles with calculations of the oxygen pressure head necessary for supplying the tissue. J Physiol. 1919;52(6):409–415. | |
Clark E, Clark EL. Microscopic observations on the growth of blood capillaries in the living mammal. American Journal of Anatomy. 1939;64(2):251–301. | |
Ashton N. Retinal vascularization in health and disease: Proctor Award Lecture of the Association for Research in Ophthalmology. Am J Ophthalmol. 1957;44(4 Pt 2):7–17. | |
Barron AG, Barron ESG. Mechanism of cobalt polycythemia. Effect of ascorbic acid. Proceedings of the Society of Experimental Biology and Medicine. 1936;35:407–409. | |
Necas E, Thorling EB. Unresponsiveness of erythropoietin – producing cells to cyanide. American Journal of Physiology. 1972;222(5):1187–1190. | |
Maxwell PH, Osmond MK, Pugh CW, et al. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney International. 1993;44:1149–1162. | |
Bachmann S, Le Hir M, Eckardt KU. Co-localization of erythropoietin messenger RNA and ecto-5′-nucleotidase immunoreactivity in peritubular cells of rat renal cortex indicates that fibroblasts produce erythropoietin. J Histochem Cytochem. 1993;41(3):335–341. | |
Lin FK, Suggs S, Lin CH, et al. Cloning and expression of the human erythropoietin gene. Proc Natl Acad Sci U S A. 1985;82(22):7580–7584. | |
Goldberg MA, Glass GA, Cunningham JM, Bunn HF. The regulated expression of erythropoietin by two human hepatoma cell lines. Proc Natl Acad Sci U S A. 1987;84(22):7972–7976. | |
Beck I, Ramirez S, Weinmann R, Caro J. Enhancer element at the 3′-flanking region controls transcriptional response to hypoxia in the human erythropoietin gene. J Biol Chem. 1991;266(24):15563–15566. | |
Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc Natl Acad Sci U S A. 1991;88(13):5680–5684. | |
Pugh CW, Tan CC, Jones RW, Ratcliffe PJ. Functional analysis of an oxygen-regulated transcriptional enhancer lying 3′ to the mouse erythropoietin gene. Proc Natl Acad Sci U S A. 1991;88(23):10553–10557. | |
Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12(12):5447–5454. | |
Maxwell PH, Pugh CW, Ratcliffe PJ. Inducible operation of the erythropoietin 3′ enhancer in multiple cell lines: evidence for a widespread oxygen sensing mechanism. Proc Natl Acad Sci U S A. 1993;90(6):2423–2427. | |
Firth JD, Ebert BL, Pugh CW, Ratcliffe PJ. Oxygen-regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase A genes: similarities with the erythropoeitin 3′ enhancer. Proc Natl Acad Sci U S A. 1994;91(14):6496–6500. | |
Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. 1994;269(38):23757–23763. | |
Nagao M, Ebert BL, Ratcliffe PJ, Pugh CW. Drosophila melanogaster SL2 cells contain a hypoxically inducible DNA binding complex which recognises mammalian HIF-1 binding sites. FEBS Lett. 1996;387(2–3):161–166. | |
Ebert BL, Firth JD, Ratcliffe PJ. Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter-1 via distinct Cis-acting sequences. J Biol Chem. 1995;270(49):29083–29089. | |
Levy AP, Levy NS, Wegner S, Goldberg MA. Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J Biol Chem. 1995;270(22):13333–13340. | |
Forsythe JA, Jiang BH, Iyer NV, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16(9):4604–4613. | |
Rolfs A, Kvietikova I, Gassmann M, Wenger RH. Oxygen-regulated transferrin expression is mediated by hypoxia-inducible factor-1. J Biol Chem. 1997;272(32):20055–20062. | |
Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92(12):5510–5514. | |
Hoffman EC, Reyes H, Chu FF, et al. Cloning of a factor required for activity of the Ah (Dioxin) receptor. Science. 1991;252(5008):954–958. | |
Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes and Development. 1997;11:72–82. | |
Makino Y, Cao R, Svensson K, et al. Inhibitory PAS domain protein is a negative regulator of hypoxia – inducible gene expression. Nature. 2001;414:550–554. | |
Hogenesch JB, Chan WK, Jackiw VH, et al. Characterization of a subset of the basic-helix-loop-helix-PAS superfamily that interacts with components of the dioxin signaling pathway. Journal of Biological Chemistry. 1997;272:8581–8593. | |
Yamashita T, Ohneda O, Nagano M, et al. Abnormal heart development and lung remodeling in mice lacking the hypoxia – inducible factor-related basic helix-loop-helix PAS protein NEPAS. Molecular and Cell Biology. 2008;28(4):1285–1297. | |
Pugh CW, O’Rourke JF, Nagao M, Gleadle JM, Ratcliffe PJ. Activation of hypoxia inducible factor – 1; definition of regulatory domains within the α subunit. Journal of Biological Chemistry. 1997;272:11205–11214. | |
Jiang B-H, Zheng JZ, Leung SW, Roe R, Semenza GL. Transactivation and inhibitory domains of hypoxia-inducible factor 1α. Modulation of transcriptional activity by oxygen tension. Journal of Biological Chemistry. 1997;272:19253–19260. | |
Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1α is mediated by an oxygen-dependent domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1998;95(14):7987–7992. | |
Gnarra JR, Zhou S, Merrill MJ, et al. Post-transcriptional regulation of vascular endothelial growth factor mRNA by the product of the VHL tumor suppressor gene. Proc Natl Acad Sci U S A. 1996;93(20):10589–10594. | |
Iliopoulos O, Levy AP, Jiang C, Kaelin WG Jr, Goldberg MA. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci U S A. 1996;93(20):10595–10599. | |
Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. | |
Cockman ME, Masson N, Mole DR, et al. Hypoxia inducible factor-α binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. Journal of Biological Chemistry. 2000;275:25733–25741. | |
Ohh M, Park CW, Ivan M, et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the β-domain of the von Hippel-Lindau protein. Nature Cell Biology. 2000;2:423–427. | |
Tanimoto K, Makino Y, Pereira T, Poellinger L. Mechanism of regulation of the hypoxia-inducible factor-1α by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000;19(16):4298–4309. | |
Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. EMBO J. 2001;20(18):5197–5206. | |
Chan DA, Sutphin PD, Yen SE, Giaccia AJ. Coordinate regulation of the oxygen-dependent degradation domains of hypoxia-inducible factor 1 α. Mol Cell Biol. 2005;25(15):6415–6426. | |
Loenarz C, Coleman ML, Boleininger A, et al. The hypoxia-inducible transcription factor pathway regulates oxygen sensing in the simplest animal, Trichoplax adhaerens. EMBO Rep. 2011;12(1):63–70. | |
Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–472. | |
Ivan M, Kondo K, Yang H, et al. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292(5516):464–468. | |
Yu F, White SB, Zhao Q, Lee FS. HIF-1α binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci U S A. 2001;98(17):9630–9635. | |
Epstein AC, Gleadle JM, McNeill LA, et al. C elegans EGL-9 and mammalian homologues define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107(1):43–54. | |
Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294(5545):1337–1340. | |
Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain: a hypoxic switch. Science. 2002;295(5556):858–861. | |
Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15(20):2675–2686. | |
Hewitson KS, McNeill LA, Riordan MV, et al. Hypoxia inducible factor (HIF) asparagine hydroxylase is identical to Factor Inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem. 2002;277(29):26351–26355. | |
Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16(12):1466–1471. | |
Loenarz C, Schofield CJ. Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2 – oxoglutarate oxygenases. Trends Biochem Sci. 2011;36(1):7–18. | |
McDonough MA, Loenarz C, Chowdhury R, Clifton IJ, Schofield CJ. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr Opin Struct Biol. 2010;20(6):659– 672. | |
Salnikow K, Donald SP, Bruick RK, Zhitkovich A, Phang JM, Kasprzak KS. Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J Biol Chem. 2004;279(39):40337–40344. | |
Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor HIF. J Biol Chem. 2003;278(33):30772–30780. | |
Koivunen P, Hirsila M, Kivirikko KI, Myllyharju J. The length of peptide substrates has a marked effect on hydroxylation by the hypoxia-inducible factor prolyl 4 hydroxylases. J Biol Chem. 2006;281(39):28712–28720. | |
Ehrismann D, Flashman E, Genn DN, et al. Studies on the activity of the hypoxia – inducible factor hydroxylases using an oxygen consumption assay. Biochem J. 2007;401(1):227–234. | |
McNeill LA, Flashman E, Buck MR, et al. Hypoxia-inducible factor prolyl hydroxylase 2 has a high affinity for ferrous iron and 2-oxoglutarate. Mol Biosyst. 2005;1(4):321–324. | |
van der Wel H, Ercan A, West CM. The Skp1 prolyl hydroxylase of dictyostelium is related to the HIFa-class of animal prolyl 4-hydroxylases. J Biol Chem. 2005;280(15):14645–14655. | |
Flashman E, Hoffart LM, Hamed RB, Bollinger JM Jr, Krebs C, Schofield CJ. Evidence for the slow reaction of hypoxia – inducible factor prolyl hydroxylase 2 with oxygen. FEBS J. 2010;277(19):4089–4099. | |
Atchley WR, Fitch WM. A natural classification of the basic helix-loop-helix class of transcription factors. Proc Natl Acad Sci U S A. 1997;94(10):5172–5176. | |
Taylor BL, Zhulin IB. PAS domains: internal sensors of oxygen, redox potential, and light. Microbiol Mol Biol Rev. 1999;63(2):479–506. | |
Scheuermann TH, Tomchick DR, Machius M, Guo Y, Bruick RK, Gardner KH. Artificial ligand binding within the HIF2α PAS-B domain of the HIF2 transcription factor. Proc Natl Acad Sci U S A. 2009;106(2):450–455. | |
Ge W, Wolf A, Feng T, et al. Oxygenase-catalyzed ribosome hydroxylation occurs in prokaryotes and humans. Nat Chem Biol. 2012;8(12):960–962. | |
Singleton RS, Liu-Yi P, Formenti F, et al. OGFOD1 catalyzes prolyl hydroxylation of RPS23 and is involved in translation control and stress granule formation. Proc Natl Acad Sci U S A. 2014;111(11):4031–4036. | |
Hughes BT, Espenshade PJ. Oxygen-regulated degradation of fission yeast SREBP by Ofd1, a prolyl hydroxylase family member. EMBO J. 2008;27(10):1491–1501. | |
Hampton-Smith RJ, Peet DJ. From polyps to people: a highly familiar response to hypoxia. Ann N Y Acad Sci. 2009;1177:19–29. | |
Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alha in normoxia. EMBO J. 2003;22(16):4082–4090. | |
Appelhoff RJ, Tian YM, Raval RR, et al. Differential function of the prolyl hydroxylases PHD1, PHD2 and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279(37):38458–38465. | |
Maynard MA, Qi H, Chung J, et al. Multiple splice variants of the human HIF-3 α locus are targets of the von Hippel-Lindau E3 ubiquitin ligase complex. J Biol Chem. 2003;278(13):11032–11040. | |
Makino Y, Uenishi R, Okamoto K, et al. Transcriptional up-regulation of inhibitory PAS domain protein gene expression by hypoxia-inducible factor 1 (HIF-1): a negative feedback regulatory circuit in HIF-1-mediated signaling in hypoxic cells. J Biol Chem. 2007;282(19):14073–14082. | |
Mole DR, Blancher C, Copley RR, et al. Genome-wide association of hypoxia-inducible factor (HIF)-1α and HIF-2α DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. 2009;284(25):16767–16775. | |
Lau KW, Tian YM, Raval RR, Ratcliffe PJ, Pugh CW. Target gene selectivity of hypoxia-inducible factor-α in renal cancer cells is conveyed by post-DNA-binding mechanisms. Br J Cancer. 2007;96(8):1284–1292. | |
Hu CJ, Sataur A, Wang L, Chen H, Simon MC. The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1α and HIF-2α. Mol Biol Cell. 2007;18(11):4528–4542. | |
Xia X, Lemieux ME, Li W, et al. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc Natl Acad Sci U S A. 2009;106(11):4260–4265. | |
Schodel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. 2011;117(23):e207–e217. | |
Warnecke C, Zaborowska Z, Kurreck J, et al. Differentiating the functional role of hypoxia-inducible factor (HIF)-1α and HIF-2α (EPAS-1) by the use of RNA interference: erythropoietin is a HIF-2α target gene in HepB and Kelly cells. FASEB J. 2004;18(12):1462–1464. | |
Franke K, Gassmann M, Wielockx B. Erythrocytosis: the HIF pathway in control. Blood. 2013;122(7):1122–1128. | |
Iyer NV, Kotch LE, Agani F, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 α. Genes Dev. 1998;12(2):149–162. | |
Krishnan J, Ahuja P, Bodenmann S, et al. Essential role of developmentally activated hypoxia – inducible factor 1α for cardiac morphogenesis and function. Circ Res. 2008;103(10):1139–1146. | |
Hu C-J, Wang L-Y, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1 α (HIF-1 α) and HIF-2 α in hypoxic gene regulation. Molecular and Cellular Biology. 2003;23(24):9361–9374. | |
Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J, Gleadle JM. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate dependent dioxygenase inhibition; the role of HIF-1α, HIF-2α and other pathways. J Biol Chem. 2006;281(22):15215–15226. | |
Takeda N, O’Dea EL, Doedens A, et al. Differential activation and antagonistic function of HIF-α isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24(5):491–501. | |
Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1α induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004;23(9):1949–1956. | |
Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2α promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11(4):335–347. | |
Uniacke J, Holterman CE, Lachance G, et al. An oxygen-regulated switch in the protein synthesis machinery. Nature. 2012;486(7401):126–129. | |
Holmquist-Mengelbier L, Fredlund E, Lofstedt T, et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell. 2006;10(5):413–423. | |
Covello KL, Kehler J, Yu H, et al. HIF-2α regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20(5):557–570. | |
Smith TG, Robbins PA, Ratcliffe PJ. The human side of hypoxia-inducible factor. Br J Haematol. 2008;141(3):325–334. | |
Prabhakar NR, Semenza GL. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev. 2012;92(3):967–1003. | |
Jiang H, Guo R, Powell-Coffman JA. The Caenorhabditis elegans hif-1 gene encodes a bHLH-PAS protein that is required for adaptation to hypoxia. Proc Natl Acad Sci U S A. 2001;98(14):7916–7921. | |
Centanin L, Ratcliffe PJ, Wappner P. Reversion of lethality and growth defects in Fatiga oxygen-sensor mutant flies by loss of Hypoxia-Inducible Factor-α/Sima. EMBO Rep. 2005;6(11):1070–1075. | |
Bishop T, Lau KW, Epstein AC, et al. Genetic analysis of pathways regulated by the von Hippel-Lindau tumor suppressor in Caenorhabditis elegans. PLoS Biol. 2004;2(10):1549–1560. | |
Ghabrial A, Luschnig S, Metzstein MM, Krasnow MA. Branching morphogenesis of the Drosophila tracheal system. Annu Rev Cell Dev Biol. 2003;19:623–647. | |
Tian H, Hammer RE, Matsumoto AM, Russell DW, McKnight SL. The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes and Development. 1998;12:3320–3324. | |
Lee YM, Jeong CH, Koo SY, et al. Determination of hypoxic region by hypoxia marker in developing mouse embryos in vivo: a possible signal for vessel development. Dev Dyn. 2001;220(2):175–186. | |
Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon MC. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386(6623):403–407. | |
Dunwoodie SL. The role of hypoxia in development of the Mammalian embryo. Dev Cell. 2009;17(6):755–773. | |
Schaffer L, Vogel J, Breymann C, Gassmann M, Marti HH. Preserved placental oxygenation and development during severe systemic hypoxia. Am J Physiol Regul Integr Comp Physiol. 2006;290(3):R844–R851. | |
Ingalls TH, Curley FJ, Prindle RA. Experimental production of congenital anomalies; timing and degree of anoxia as factors causing fetal deaths and congenital anomalies in the mouse. N Engl J Med. 1952;247(20):758–768. | |
Moore LG, Shriver M, Bemis L, et al. Maternal adaptation to high-altitude pregnancy: an experiment of nature – a review. Placenta. 2004;25 Suppl A:S60–S71. | |
Patterson AJ, Zhang L. Hypoxia and fetal heart development. Curr Mol Med. 2010;10(7):653–666. | |
Cramer T, Yamanishi Y, Clausen BE, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 2003;112(5):645–657. | |
Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. 2009;9(9):609–617. | |
Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364(7):656–665. | |
Takubo K, Suda T. Roles of the hypoxia response system in hematopoietic and leukemic stem cells. Int J Hematol. 2012;95(5):478–483. | |
Schipani E, Ryan HE, Didrickson S, et al. Hypoxia in cartilage: HIF-1α is essential for chondrocyte growth arrest and survival. Genes Dev. 2001;15(21):2865–2876. | |
Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114(8):1098–1106. | |
Parmar K, Mauch P, Vergilio JA, Sackstein R, Down JD. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci U S A. 2007;104(13):5431–5436. | |
Caldwell CC, Kojima H, Lukashev D, et al. Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J Immunol. 2001;167(11):6140–6149. | |
Dang EV, Barbi J, Yang HY, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146(5):772–784. | |
Clambey ET, McNamee EN, Westrich JA, et al. Hypoxia-inducible factor-1 α-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci U S A. 2012;109(41):E2784–E2793. | |
Schodel J, Mole DR, Ratcliffe PJ. Pan-genomic binding of hypoxia-inducible transcription factors. Biol Chem. 2013;394(4):507–517. | |
Zhang H, Gao P, Fukuda R, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11(5):407–420. | |
Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129(1):111–122. | |
Pescador N, Villar D, Cifuentes D, et al. Hypoxia promotes glycogen accumulation through hypoxia inducible factor (HIF)-mediated induction of glycogen synthase 1. PLoS One. 2010;5(3):e9644. | |
Favaro E, Bensaad K, Chong MG. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab. 2012;16(6):751–764. | |
Furuta E, Pai SK, Zhan R, et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008;68(4):1003–1011. | |
Krishnamoorthy S, Honn KV. Eicosanoids and other lipid mediators and the tumor hypoxic microenvironment. Cancer Metastasis Rev. 2011;30(3–4):613–618. | |
Masson N, Ratcliffe PJ. Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer Metab. 2014;2(1):3. | |
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. | |
Ratcliffe PJ. Oxygen sensing and hypoxia signalling pathways in animals: the implications of physiology for cancer. J Physiol. 2013;591(Pt 8):2027–2042. | |
Gerald D, Berra E, Frapart YM, et al. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118(6):781–794. | |
Pan Y, Mansfield KD, Bertozzi CC, et al. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol. 2007;27(3):912–925. | |
Knowles HJ, Raval RR, Harris AL, Ratcliffe PJ. Effect of ascorbate on the activity of hypoxia inducible factor (HIF) in cancer cells. Cancer Res. 2003;63(8):1764–1768. | |
Brunelle JK, Bell EL, Quesada NM, et al. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1(6):409–414. | |
Bell EL, Klimova TA, Eisenbart J, et al. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol. 2007;177(6):1029–1036. | |
Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1α. Science. 2003;302(5652):1975–1978. | |
Doege K, Heine S, Jensen I, Jelkmann W, Metzen E. Inhibition of mitochondrial respiration elevates oxygen concentration, but leaves regulation of hypoxia-inducible factor (HIF) intact. Blood. 2005;106(7):22311–2317. | |
Nytko KJ, Maeda N, Schläfli P, et al. Vitamin C is dispensable for oxygen sensing in vivo. Blood. 2011;117(20):5485–5493. | |
Tian YM, Yeoh KK, Lee MK, et al. Differential sensitivity of HIF hydroxylation sites to hypoxia and hydroxylase inhibitors. J Biol Chem. 2011;286(15):13041–13051. | |
Masson N, Singleton RS, Sekirnik R, et al. The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity. EMBO Rep. 2012;13(3):251–257. | |
Stiehl DP, Wirthner R, Köditz J, Spielmann P, Camenisch G, Wenger RH. Increased Proyl 4-Hydroxylase Domain Proteins Compensate for Decreased Oxygen Levels. J Biol Chem. 2006;281(33):23482–23491. | |
Jewell UR, Kvietikova I, Scheid A, Bauer C, Wenger RH, Gassmann M, et al. Induction of HIF-1α in response to hypoxia is instantaneous. FASEB J. 2001;15(7):1312–1314. | |
Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol Cell. 2008;30(4):393–402. | |
Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda). 2009;24:97–106. | |
Bonello S, Zähringer C, BelAiba RS, et al. Reactive oxygen species activate the HIF-1α promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol. 2007;27(4):755–761. | |
Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441(7092):437–443 (2006). | |
Lang KJ, Kappel A, Goodall GJ. Hypoxia-inducible factor-1α mRNA contains an internal ribosome entry site that allows efficient translation during normoxia and hypoxia. Mol Biol Cell. 2002;13(5):1792–1801. | |
Young RM, Wang SJ, Gordan JD, Ji X, Liebhaber SA, Simon MC. Hypoxia-mediated selective mRNA translation by an internal ribosome entry site-independent mechanism. J Biol Chem. 2008;283(24):16309–16319. | |
Scholz CC, Taylor CT. Targeting the HIF pathway in inflammation and immunity. Curr Opin Pharmacol. 2013;13(4):646–653. | |
Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408. | |
Fraisl P, Aragones J, Carmeliet P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat Rev Drug Discov. 2009;8(2):139–152. | |
Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–732. | |
Shen C, Kaelin WG Jr. The VHL/HIF axis in clear cell renal carcinoma. Semin Cancer Biol. 2013;23(1):18–25. | |
Scheuermann TH, Li Q, Ma HW, Key J, et al. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat Chem Biol. 2013;9(4):271–276. | |
Kaelin WG Jr. Treatment of kidney cancer: insights provided by the VHL tumor-suppressor protein. Cancer. 2009;115(Suppl 10):2262–2272. | |
Toschi A, Lee E, Gadir N, Ohh M, Foster DA. Differential dependence of hypoxia-inducible factors 1 α and 2 α on mTORC1 and mTORC2. J Biol Chem. 2008;283(50):34495–34499. | |
Raval RR, Lau KW, Tran MG, et al. Contrasting Properties of Hypoxia-Inducible Factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-Associated Renal Cell Carcinoma. Mol Cell Biol. 2005;25(13):5675–5686. | |
Zhang H, Qian DZ, Tan YS, et al. Digoxin and other cardiac glycosides inhibit HIF – 1α synthesis and block tumor growth. Proc Natl Acad Sci U S A. 2008;105(50):19579–19586. | |
Selvaraju V, Parinandi NL, Adluri RS, et al. Molecular mechanisms of action and therapeutic uses of pharmacological inhibitors of HIF-prolyl 4-hydroxylases for treatment of ischemic diseases. Antioxid Redox Signal. 2014;20(16):2631–2665. | |
Rabinowitz MH. Inhibition of hypoxia-inducible factor prolyl hydroxylase domain oxygen sensors: tricking the body into mounting orchestrated survival and repair responses. J Med Chem. 2013;56(23):9369–9402. | |
Muchnik E, Kaplan J. HIF prolyl hydroxylase inhibitors for anemia. Expert Opin Investig Drugs. 2011;20(5):645–656. | |
McCullough PA, Barnhart HX, Inrig JK, et al. Cardiovascular toxicity of epoetin-alfa in patients with chronic kidney disease. Am J Nephrol. 2013;37(6):549–558. | |
Bernhardt WM, Wiesener MS, Scigalla P, et al. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J Am Soc Nephrol. 2010;21(12):2151–2156. | |
Katschinski DM. In vivo functions of the prolyl-4-hydroxylase domain oxygen sensors: direct route to the treatment of anaemia and the protection of ischaemic tissues. Acta Physiol (Oxf). 2009;195(4):407–414. | |
Karuppagounder SS, Ratan RR. Hypoxia-inducible factor prolyl hydroxylase inhibition: robust new target or another big bust for stroke therapeutics? J Cereb Blood Flow Metab. 2012;32(7):1347–1361. | |
Nangaku M, Izuhara Y, Takizawa S, et al. A novel class of prolyl hydroxylase inhibitors induces angiogenesis and exerts organ protection against ischemia. Arterioscler Thromb Vasc Biol. 2007;27(12):2548–2554. | |
Wang Z, Schley G, Türkoglu G, et al. The protective effect of prolyl-hydroxylase inhibition against renal ischaemia requires application prior to ischaemia but is superior to EPO treatment. Nephrol Dial Transplant. 2012;27(3):929–936. | |
Chen RL, Nagel S, Papadakis M, et al. Roles of individual prolyl-4-hydroxylase isoforms in the first 24 hours following transient focal cerebral ischaemia: insights from genetically modified mice. J Physiol. 2012;590(Pt 16):4079–4091. | |
Bao W, Qin P, Needle S, et al. Chronic inhibition of hypoxia-inducible factor prolyl 4-hydroxylase improves ventricular performance, remodeling, and vascularity after myocardial infarction in the rat. J Cardiovasc Pharmacol. 2010;56(2):147–155. | |
Minamishima YA, Moslehi J, Bardeesy N, Cullen D, Bronson RT, Kaelin WG Jr. Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood. 2008;111(6):3236–3244. | |
Minamishima YA, Moslehi J, Padera RF, Bronson RT, Liao R, Kaelin WG Jr. A feedback loop involving the Phd3 prolyl hydroxylase tunes the mammalian hypoxic response in vivo. Mol Cell Biol. 2009; 29(21):5729–5741. | |
Bekeredjian R, Walton CB, MacCannell KA, et al. Conditional HIF-1α expression produces a reversible cardiomyopathy. PLoS One. 2010;5(7):e11693. | |
Safran M, Kim WY, O’Connell F, et al. Mouse model for noninvasive imaging of HIF prolyl hydroxylase activity: Assessment of an oral agent that stimulates erythropoietin production. Proc Natl Acad Sci U S A. 2006;103(1):105–110. | |
Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12(1):9–22. | |
Kalucka J, Ettinger A, Franke K, et al. Loss of epithelial hypoxia-inducible factor prolyl hydroxylase 2 accelerates skin wound healing in mice. Mol Cell Biol. 2013;33(17):3426–3438. | |
Zimmermann AS, Morrison SD, Hu MS, et al. Epidermal or dermal specific knockout of PHD-2 enhances wound healing and minimizes ischemic injury. PLoS One. 2014;9:e93373. | |
Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology. 2008;134(1):145–155. | |
Taniguchi CM, Miao YR, Diep AN, et al. PHD inhibition mitigates and protects against radiation-induced gastrointestinal toxicity via HIF2. Sci Transl Med. 2014;6(236):236ra264. | |
Tambuwala MM, Cummins EP, Lenihan CR, et al. Loss of prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology. 2010;139(6):2093–2101. | |
Trewick SC, McLaughlin PJ, Allshire RC. Methylation: lost in hydroxylation? EMBO Rep. 2005;6(4):315–320. | |
Whetstine JR, Nottke A, Lan F, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125(3):467–481. | |
Johnson AB, Denko N, Barton MC. Hypoxia induces a novel signature of chromatin modifications and global repression of transcription. Mutat Res. 2008;640(1–2):174–179. | |
Chervona Y, Costa M. The control of histone methylation and gene expression by oxidative stress, hypoxia, and metals. Free Radic Biol Med. 2012;53(5):1041–1047. | |
Myllyharju J. Prolyl 4-hydroxylases, master regulators of the hypoxia response. Acta Physiol (Oxf). 2013;208(2):148–165. | |
Cockman ME, Lancaster DE, Stolze IP, et al. Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc Natl Acad Sci U S A. 2006;103(40):14767–14772. | |
Zheng X, Linke S, Dias JM, et al. Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc Natl Acad Sci U S A. 2008;105(9):3368–3373. | |
Wilkins SE, Karttunen S, Hampton-Smith RJ, Murchland I, Chapman-Smith A, Peet DJ. Factor inhibiting HIF (FIH) recognises distinct molecular features within hypoxia inducible factor (HIF)-α versus ankyrin repeat substrates. J Biol Chem. 2012;287(12):8769–8781. | |
Hardy AP, Prokes I, Kelly L, Campbell ID, Schofield CJ. Asparaginyl β-hydroxylation of proteins containing ankyrin repeat domains influences their stability and function. J Mol Biol. 2009;392(4):994–1006. | |
Coleman ML, McDonough MA, Hewitson KS, et al. Asparaginyl hydroxylation of the Notch ankyrin repeat domain by factor inhibiting hypoxia-inducible factor. J Biol Chem. 2007;282(33):24027–24038. | |
Yang M, Ge W, Chowdhury R, Claridge TD, et al. Asparagine and aspartate hydroxylation of the cytoskeletal ankyrin family is catalyzed by factor-inhibiting hypoxia-inducible factor. J Biol Chem. 2011;286(9):7648–7660. | |
Yang M, Chowdhury R, Ge W, et al. Factor-inhibiting hypoxia-inducible factor (FIH) catalyses the post-translational hydroxylation of histidinyl residues within ankyrin repeat domains. FEBS J. 2011;278(7):1086–1097. | |
Loenarz C, Sekirnik R, Thalhammer A, et al. Hydroxylation of the eukaryotic ribosomal decoding center affects translational accuracy. Proc Natl Acad Sci U S A. 2014;111(11):4019–4024. | |
Katz MJ, Acevedo JM, Loenarz C, et al. Sudestada1, a Drosophila ribosomal prolyl-hydroxylase required for protein translation, cell homeostasis and organ growth. Proc Natl Acad Sci U S A. 2014;111(11):4025–4030. | |
Chowdhury R, Sekirnik R, Brissett NC, et al. Ribosomal oxygenases are structurally conserved from prokaryotes to humans. Nature. 2014;510(7505):422–426. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.