Back to Journals » Cancer Management and Research » Volume 13
Sevoflurane Limits Glioma Progression by Regulating Cell Proliferation, Apoptosis, Migration, and Invasion via miR-218-5p/DEK/β-Catenin Axis in Glioma
Authors Qi Y, Guo L, Liu Y, Zhao T, Liu X, Zhang Y
Received 24 June 2020
Accepted for publication 9 December 2020
Published 26 February 2021 Volume 2021:13 Pages 2057—2069
DOI https://doi.org/10.2147/CMAR.S265356
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yong Teng
Yingying Qi, Lina Guo, Yanchao Liu, Tonghang Zhao, Xianwen Liu, Yang Zhang
Department of Anesthesiology, Liaocheng People’s Hospital, Liaocheng, Shandong, People’s Republic of China
Correspondence: Yang Zhang
Department of Anesthesiology, Liaocheng People’s Hospital, No. 67, Dongchang West Road, Liaocheng, 252000, Shandong, People’s Republic of China
Tel +86-0635-8276414
Email [email protected]
Purpose: Sevoflurane (SEV) is a frequently used volatile anesthetic in cancer surgery. Sevoflurane treatment has been shown to suppress the migration and invasion of several human cancer cells. However, the effect of sevoflurane on glioma remains largely unclear.
Methods: Glioma cell lines (U251 and U343) were treated by various concentrations of sevoflurane. 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT), flow cytometry assay, and transwell assay were performed to detect the cell viability, apoptosis, migration and invasion. Western blot assay was employed to detect the protein levels of β-catenin, c-Myc, CyclinD1, β-catenin, N-cadherin, vimentin, and DEK. Moreover, quantitative real-time polymerase chain reaction (qRT-PCR) was used to examine the expression level of miR-218-5p. The target interaction between miR-218-5p and DEK was predicted through bioinformatics analysis and verified by dual-luciferase reporter assay system.
Results: We found that sevoflurane aberrantly inhibited the abilities on viability, migration, invasion, EMT and β-catenin signaling and promoted cell apoptosis in U251 and U343 cells in a dose-dependent manner. MiR-218-5p strikingly suppressed the abilities of proliferation, migration, invasion rather than apoptosis and activation of β-catenin signaling. Sevoflurane could facilitate the miR-218-5p expression, and its suppressing effects on glioma cells were reversed by pre-treatment with miR-218-5p inhibitors or pcDNA3.1/DEK in vitro and in vivo. Silencing of miR-218-5p reverted sh-DEK and sevoflurane-induced repression on proliferation, migration, invasion, and β-catenin signaling, and promotion on apoptosis in the glioma cells.
Conclusion: Our data showed that sevoflurane inhibited the proliferation, migration, invasion, and enhanced the apoptosis in glioma cells through regulating miR-218-5p/DEK/β-catenin axis.
Keywords: glioma, sevoflurane, miR-218-5p, DEK, β-catenin signaling pathway
Introduction
Glioma is one of the most common primary malignant brain tumors with poor prognosis in the world.1 Owing to the limitations of brain tissue function and structure and the formation of chemical resistance of tumor cells, most patients are easy to relapse, with less than 5 percentage of survival rates within 5 years.1 Currently, the treatment methods for glioma, including surgery, radiation therapy, and chemotherapy, while especially surgery, are still the primary means of treating glioma.2,3 Although glioma treatment has improved over the decades, there is still a long way to go for glioma diagnosis or treatment research. Sevoflurane (SEV) is an inhaled anesthetic gas commonly used in many surgical operations in cancer patients.4 Sevoflurane has been shown to inhibit cell growth, invasion, and migration, triggering morphological changes and apoptosis in several types of cancer cell lines.4–7 However, the function of sevoflurane in glioma needs to be further elucidated.
MicroRNAs (miRNAs) are small, non-coding RNAs that specifically inhibit translation and trigger mRNA degradation, thereby controlling genes involved in cellular processes such as inflammation, cell cycle regulation, differentiation, apoptosis, migration, and invasion.8,9 Previous studies have shown that many miRNAs are abnormally expressed under sevoflurane treatment. For example, miR-203 had the aberrant expression in sevoflurane-treated colorectal cancer cells.10 In addition, sevoflurane up-regulated miR-34a-5p expression in glioma.11 More importantly, mounting evidence has suggested that miRNAs are involved in influencing cell proliferation,12,13 apoptosis,14 migration, and invasion15 ability in glioma cells. Emerging evidence suggests that dysregulation of miRNAs may lead to cancer progression in humans.16 In human gliomas, abnormal expression of miRNAs or mutations in miRNA genes has been well characterized.17,18 miR-218 has been shown to be downregulated in many types of human malignancies.19–21 In addition, some studies demonstrated the inhibitory effect of miR-218 in gliomas because it inhibits cell growth and invasion and induces apoptosis.22,23 Yet, it is unknown the regulatory role of miR-218 in gliomas and the interaction between sevoflurane and miR-218.
DEK is preferentially expressed in active proliferating and malignant cells, and its function is closely related to many human diseases.24–27 The DEK of amplification and upregulated expression has been described in a variety of cancer types, including ovarian cancer,28 bladder cancer,29 and breast cancer.26 In addition, DEK promotes cell growth by inhibiting cell senescence and differentiation.30–33 Therefore, the overexpression of DEK is closely associated with tumor growth and pathological progression, with poor prognosis.34–36 However, the mechanism of action of DEK in gliomas is rarely reported.
Herein, we intended to investigate the role of sevoflurane on cell growth and metastasis of U251 and U343 cells, and explored the underlying mechanism in vitro and in vivo.
Methods
Cell Culture and Sevoflurane Administration
Human glioma cell lines U251 and U343 were obtained from Shanghai Innovation Biotechnology Co., Ltd. (Shanghai, China), with 1% penicillin/streptomycin (Beyotime Biotechnology Company, Shanghai, China), cultured as previously described.37 The cells were exposed to the different dosage of sevoflurane for 6 hours at a velocity of 6 L/min in a mixture of 95% air and 5% CO2 condition, respectively.6,38 The U251 and U343 cells were divided into 4 groups: control group (without sevoflurane), 1% sevoflurane group, 2% sevoflurane group, and 4% sevoflurane group. Glioma cells were exposed to 4% sevoflurane for 48 hours for subsequent experiments. Cells in log phase were seeded onto plates and incubated for 24 hours at 37°C. The cell culture plate was then placed at the inlet and connected to a sealed glass chamber of an anesthesia device (Cicero-EM 8060; Drager, Lübeck, Germany), and the other chamber was a gas monitor (PM8060; Drager) to monitor the concentration of sevoflurane. The anesthesia machine was connected to an anesthetic vaporizer (Sevorane; Abbott, Abbot Park, IL, USA) to supply sevoflurane. After treatment with sevoflurane, the cells were incubated for 24 hours at 37°C and then used for subsequent functional and molecular biology experiments.
3-(4,5-Dimethyl-2-Thiazolyl)-2,5-Diphenyl-2-H-Tetrazolium Bromide (MTT)
The proliferation capacity of glioma cells was evaluated through MTT assay. Briefly, transfected glioma cells (3×103/well) were seeded into 96-well plates (Corning Inc., Corning, NY, USA) and maintained in an incubator with 5% CO2 at 37°C for 24 h, 48 h, and 72 h. Then, MTT (20 μL) from Sigma was replenished to each well and kept for 4 h. Following this, the supernatant of each well was discarded, and DMSO (150 μL, Sigma) was added to dissolve formed formazan crystals. The Microplate Absorbance Reader (Thermo Fisher Scientific, Waltham, MA, USA) was executed for the assessment of the optical density value at 490 nm.
Flow Cytometry
Cells were collected by digesting with pancreatin and centrifuging. After that, cells were washed by iced phosphate-buffered saline (PBS) and resuspended with 1×binding buffer. Next, following the manufacturer’s instructions, Annexin V-fluorescein isothiocyanate propidium iodide (Annexin V-FITC/PI) kit (BD Pharmingen, San Diego, CA, USA) was conducted to steps subsequently. The flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) was applied to examine the apoptosis cells.
Transwell Assay
The rate of cell migration was detected by using the transwell chamber (Corning Inc.). At the same time, invasion assay was conducted with transwell chamber pre-coated with matrigel matrix (Corning Inc.). RPMI-1640 medium containing 10% FBS was added into the lower chamber, whereas the treated U251 and U343 cells in100 μL serum-free medium were injected into the upper one. And the whole steps were carried out according to the manufacturer’s instructions. The paraformaldehyde (PFA; Sigma) was used to attach cells located on the lower surface of the upper chamber. Cells were analyzed under a microscope after staining with crystal violet.
Western Blot
Total proteins from U251 and U343 cells were isolated using RIPA buffer (Solarbio, Beijing, China), followed by quantified with a NanoDrop 3000 (Thermo Fisher Scientific). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to separate proteins, and then proteins were transferred onto polyvinylidene fluoride (PVDF) membranes. Following blocked in skim milk for 2 h at 37°C, membranes were incubated with primary antibodies at 4°C overnight. After 2 h of incubation with secondary antibody [Goat Anti-Rabbit IgG H&L (HRP) (1:1000; ab205718, Abcam, Cambridge, UK)], an ECL detection kit (Beyotime) was employed to detect the chemiluminescence. The primary antibodies were as follows: anti-β-catenin (1:1000; ab16051, Abcam), anti-c-Myc (1:1000; ab32072, Abcam), anti-cyclinD1 (1:1000; ab226977, Abcam), anti-DEK (1:1000; ab245430, Abcam), anti-GAPDH (1:5000; ab37168, Abcam), anti-Vimentin (1:1000; ab92547, Abcam), anti-E-cadherin (1:1000; ab40772, Abcam), anti-N-cadherin (1:1000; ab76011, Abcam).
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
In this assay, TRIzol reagent (Life Technologies Corporation, Carlsbad, CA, USA) was used to extract the total RNAs from U251 and U343 cells. Primer-Script one-step RT-PCR kit (Takara, Shiga, Japan) or miRNA Reverse Transcription kit (GeneCopoeia, FulenGen, China) was employed to synthesize the first-strand complementary DNA of miR-218-5p and DEK. The levels of miR-218-5p and DEK were assessed via SYBR Premix Dimer Eraser Kit (Takara). The primer sequences used were presented as below:
miR-218-5p forward (5ʹ-TGCGGCGGCCCCACGCACCAG-3ʹ),
miR-218-5p reverse (5ʹ-CCAGTGCAGGGTCCGAGGT-3ʹ);
DEK forward (5ʹ-AAGAATGTGGGTCAGTTCAG-3ʹ),
DEK reverse (5ʹ-GTGCTGCTATCTGCTTTCTT-3ʹ);
U6 forward (5ʹ-GCTTCGGCAGCACATATACTAAAAT-3ʹ),
U6 reverse (5ʹ-CGCTTCACGAATTTGCGTGTCAT-3ʹ);
GAPDH forward (5ʹ-GACTCATGACCACAGTCCATGC-3ʹ),
GAPDH reverse (5ʹ-AGAGGCAGGGATGATGTTCTG-3ʹ).
According to the 2-ΔΔCt method, the expression levels of miR-218-5p and DEK were calculated, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or U6 snRNA was viewed as an internal control for DEK or miR-218-5p, respectively.
Transfection of U251 and U343 Cells
MiR-218-5p mimics, miR-218-5p inhibitors, short hairpin RNA (shRNA) targeting DEK (sh-DEK), pcDNA 3.1-DEK plasmid (pcDNA3.1/DEK), and controls (miR-NC mimics, miR-NC inhibitors, sh-control, pcDNA 3.1 plasmid) were all obtained from Thermo Fisher Scientific. According to the manufacturer’s instructions, Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) kit was used for transfection. The sequences were shown as follows: sh-DEK (sense: 5ʹ-GGAGAGATCAGGTGTAAATAG-3ʹ, anti-sense: 3ʹ-CTATTTACACCTGATCTCTCC-5ʹ).
Dual-Luciferase Reporter Assay
The TargetScan (http://www.targetscan.org) database was executed for the prediction of the binding sites between the miR-218-5p and DEK. Following this, the pGL3-control vector (Promega, Madison, WI, USA) with the wild type DEK sequence (DEK-WT) and mutant DEK sequence (DEK-MUT) (with predicted miR-218-5p binding sites or not) were constructed to verify the binding sites between miR-218-5p and DEK. Afterward, the miR-control or miR-218-5p was co-transfected into glioma cells with dual-luciferase reporter vectors for the execution of the dual-luciferase reporter assay, respectively. Finally, the luciferase activities of luciferase reporter vectors were tested through the dual-luciferase reporter assay kit (Promega).
Xenograft Mouse Model
Animal experiments were guided by the care and use of laboratory animals. The xenograft mouse model was established according to reference.39 U251 cells were transfected with miR-NC inhibitors or miR-218-5p inhibitors. Fifteen female nude mice (4-week-old) with no specific pathogen free (SPF) were randomly divided into three groups: Group1 was subcutaneously injected with 1×105 U251 cells; Group2 was injected with U251 cells transfected with miR-NC inhibitors; Group3 was injected subcutaneously with U251 cells transfected with miR-218-5p inhibitors. After feeding for 3 days, sevoflurane (50 mg/kg) was dissolved in 0.5 mL soybean oil and injected into Group2 and Group3 mice once a day and Group1 mice were injected with 0.5 mL soybean oil for 25 days. To analyze the tumor size, the tumor volumes were measured every 7 days and tumor weights were measured after the animals were sacrificed. All the animal studies were approved by the ethics committee of Liaocheng People’s Hospital.
Statistical Analysis
All data were expressed as mean ± standard deviation (SD) and analyzed by the SPSS 17.0 software. A P value less than 0.05 was regarded as statistically significant. Paired Student’s t-test or one-way ANOVA analysis of variance was used to compare the difference between groups.
Results
Sevoflurane Regulated the Abilities of Viability, Apoptosis, Migration, Invasion, and Suppressed the β-Catenin Signaling in Glioma Cells
To validate the role of sevoflurane on the progression of glioma cells, MTT assay was employed to detect the cell viability in U251 or U343 cells treated with various concentrations of sevoflurane. Compared with the control group, the U251 and U343 cells treated with sevoflurane showed decreased cell viability in a concentration-dependent way (Figure 1A and B). In terms of the rate of cell apoptosis, flow cytometry was performed when cells were administered with sevoflurane. Results showed that sevoflurane promoted cell apoptosis in a dose-dependent manner (Figure 1C). For migration and invasion assay, transwell assay was applied to measure the migration and invasion abilities of U251 or U343 cells treated with various concentrations of sevoflurane following 8 h of incubation. We found that treatment with sevoflurane weakened the cell migration and invasion abilities relative to the control group in a concentration-dependent manner (Figure 1D and E). It is well known that β-catenin signaling played essential roles in glioma progression by regulating cell proliferation, migration, invasion, and angiogenesis. Then, we explored the activation of β-catenin signaling by detecting the β-catenin pathways-associated proteins, and found that β-catenin, c-Myc, and CyclinD1 were downregulated in glioma cells under the increased dosage of sevoflurane (Figure 1F and G). Besides, Wnt/β-catenin signaling usually promotes the invasion and metastasis by regulating the process of epithelial-mesenchymal transition (EMT). We also detected the EMT markers (E-cadherin, N-cadherin, and Vimentin) in U251 or U343 cells treated with different concentrations of sevoflurane. As shown in Suppl. Figure S1A-B, E-cadherin protein levels were increased, while N-cadherin and Vimentin were decreased with the elevated concentrations of sevoflurane, suggesting that sevoflurane blocked β-catenin signaling pathway by inhibition of EMT.
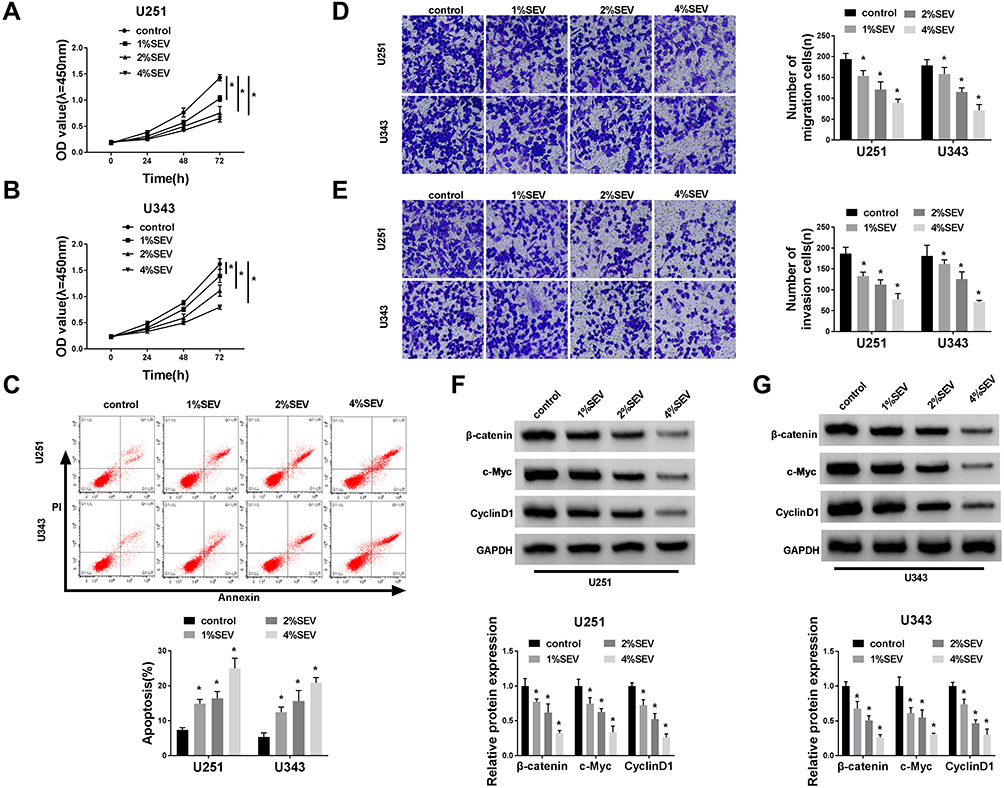 |
Figure 1 Sevoflurane regulated the abilities of viability, apoptosis, migration, invasion, and suppressed the β-catenin signaling in glioma cells. (A and B) MTT assay was used to manifest the cell viability of U251 or U343 cells at 24 h, 48 h, and 72 h after treatment of sevoflurane for various concentrations (1%, 2%, 4%). (C) Flow cytometry was carried out to measure the rate of apoptosis when cells administrated with sevoflurane. (D and E) Transwell assay was applied to evaluate migration and invasion abilities of U251 and U343 cells after treatment with sevoflurane (magnification: 100x, n=3). (F and G) Western blot assay was used to detect the expression of β-catenin signaling-related proteins, namely β-catenin, c-Myc, CyclinD1 in glioma cells. *P<0.05. |
Overexpression of miR-218-5p Repressed Cell Viability, Migration, Invasion, and β-Catenin Signaling, but Promoted the Apoptosis in Glioma Cells
Emerging evidence have demonstrated that miR-218-5p modulate glioma cells,13 however, the effects of sevoflurane on miR-218-5p in glioma cells are not yet known. To investigate sevoflurane function on miR-218-5p expression, we perform qRT-PCR assay to examine the expression of miR-218-5p in glioma cells treated with various concentrations of sevoflurane. And data suggested that the expression of miR-218-5p was increased versus the control group in a concentration-dependent manner (Figure 2A). And then, we up-regulated the expression of miR-218-5p by transfection with miR-218-5p mimics in U251 and U343 cells, qRT-PCR assay was carried out to verify transfection efficiency (Figure 2B). The cell viability was detected through MTT assay, and results indicated that overexpression of miR-218-5p induced obvious repression of cell viability in the two cell lines (Figure 2C and D). What is more, transfection of miR-218-5p mimics enlarged cell apoptosis, compared with miR-NC mimics transfection in U251 and U343 cells (Figure 2E). As shown in Figure 2F and G, the migration and invasion abilities of U251 and U343 cells were notably blocked by miR-218-5p mimics. Meanwhile, a significant decrease among β-catenin, c-Myc, CyclinD1 could be seen in cells transfected with miR-218-5p mimics (Figure 2H and I). miR-218-5p mimics also upregulated E-cadherin protein levels, but suppressed protein levels of N-cadherin, and Vimentin in contrast with the miR-NC group (Suppl. Fig. S1C-D).
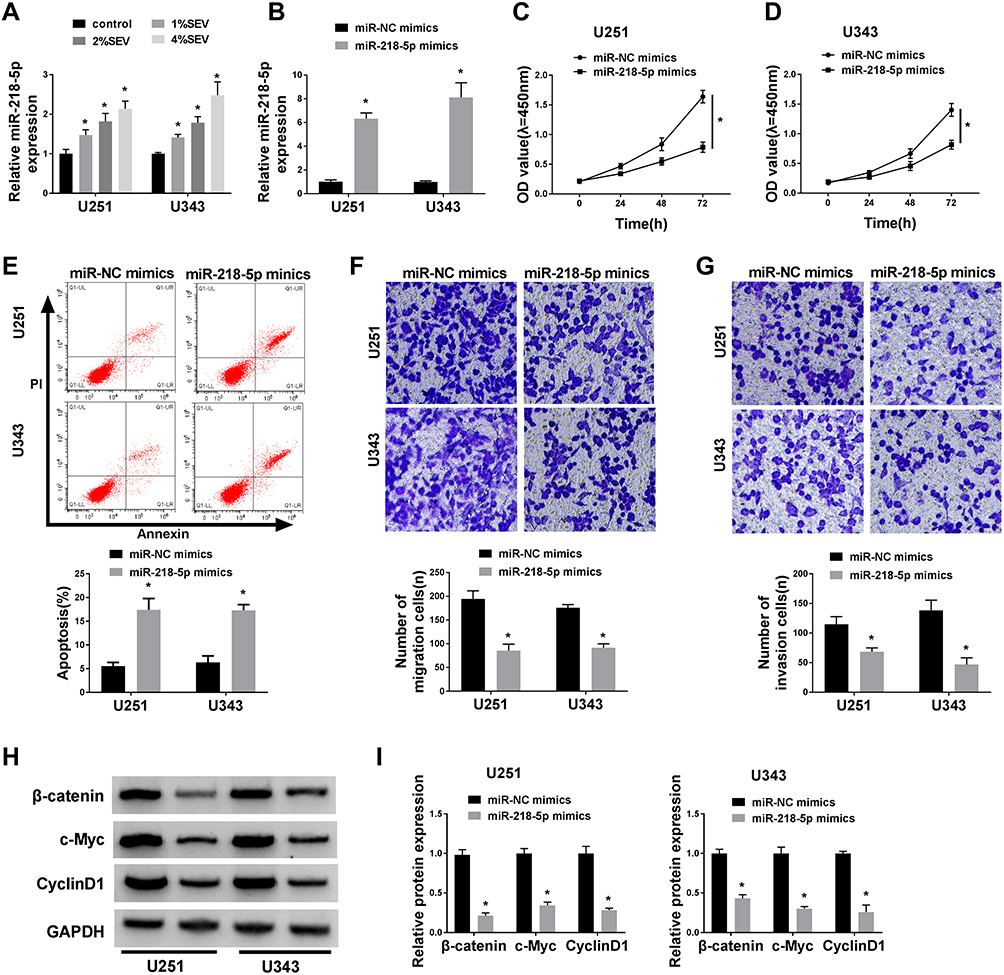 |
Figure 2 Overexpression of miR-218-5p repressed cell viability, migration, invasion, and β-catenin signaling, but promoted the apoptosis in glioma cells in vitro. (A) The mRNA expression level of miR-218-5p of U251 and U343 cells after treatment with sevoflurane was detected by qRT-PCR assay. U251 and U343 cells were transfected with miR-NC mimics or miR-218-5p mimics. (B) The miR-218-5p expression was tested via qRT-PCR assay at 48 h post-transfection in U251 and U343 cells. (C–E) The cell viability of U251 and U343 cells was measured by MTT assay at 24 h, 48 h and 72 h after transfection (C and D) and the rate of apoptosis was explored by flow cytometry after transfection for 48 h as well (E). (F and G) The number of migration and invasion cells was counted at 48 h following transfection through transwell chamber experiment (magnification: 100x, n=3). (H and I) The gray value (H) and analysis results (I) of protein immunoblots of β-catenin signal pathway-related proteins were shown.*P<0.05. |
Sevoflurane Regulated the Cell Viability, Apoptosis, Migration, Invasion, and Activation of β-Catenin Signaling by Targeting miR-218-5p
To explore the mechanism of sevoflurane in the progress of glioma cells, U251 and U343 cells were transfected with miR-NC inhibitors or miR-218-5p inhibitors before exposure to a concentration of 4% sevoflurane. Results showed that miR-218-5p inhibitors significantly reversed the promotion of miR-218-5p expression triggered by sevoflurane treatment (Figure 3A). MiR-218-5p inhibitors rescued the repressive effect on cell viability, migration, invasion, and promoted action on cell apoptosis of sevoflurane in both U251 and U343 cells (Figure 3B–F). Interestingly, silencing of miR-218-5p also inverted the low expression of β-catenin signaling-related proteins (Figure 3G and H) and the inhibition of EMT process induced by 4% sevoflurane (Suppl. Fig. S1E-F).
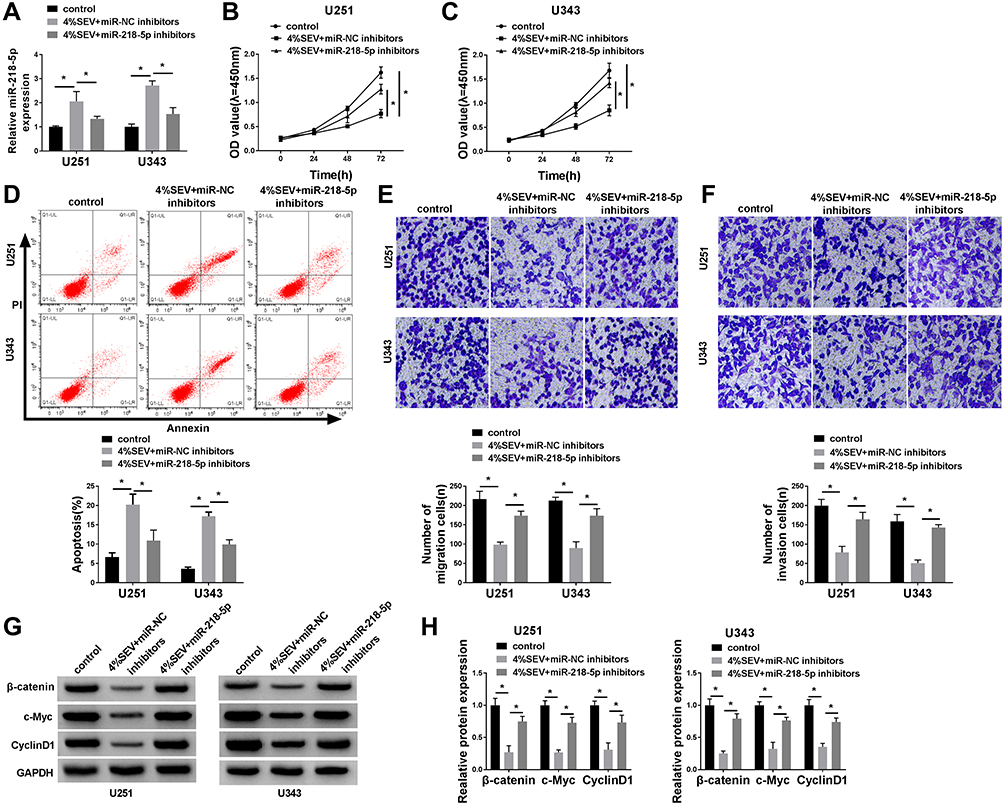 |
Figure 3 Sevoflurane regulated the cell viability, apoptosis, migration, invasion, and activation of β-catenin signaling by targeting miR-218-5p. (A) qRT-PCR assay showed the expression level of miR-218-5p in U251 and U343 cells after treatment with sevoflurane and miR-NC inhibitors or miR-218-5p inhibitors. (B and C) MTT assay showed cell viability of U251 and U343 cells after treatment. (D) Flow cytometry assay witnessed cell apoptosis of U251 and U343 cells after administration. (E and F) Transwell assay exhibited the number of migration and invasion cells after treatment (magnification: 100x, n=3). (G and H) Western blot analysis measured the activation of β-catenin signal pathways. *P<0.05. |
DEK Was a Direct Target of miR-218-5p and Negatively Regulated by miR-218-5p
To further investigate the underlying mechanism of miR-218-5p in glioma cells, Starbase and miR walk software were employed to predict the target mRNAs of miR-218-5p. As shown in Suppl. Fig. S2A, 130 mRNAs were predicted that existed the binding sites of miR-218-5p. By literature search and selection, 6 mRNAs (DEK; Nuclear Factor I A, NFIA; Homeodomain Interacting Protein Kinase 1, HIPK1; LIM Domain Only 3, LMO3; Chromobox 5, CBX5; and Cyclin D2, CCND2) were selected for further research. Then, dual-luciferase reporter assay was utilized to confirm the correlation relationship between miR-218-5p and 6 selected mRNAs. We found that the miR-218-5p reduced the luciferase activity of DEK-Wt and CCND2-Wt group in U251 cells, while it decreased the luciferase activity of DEK-Wt and HIPK1-Wt group in U343 cells (Suppl. Fig. S2B-C), thus DEK were selected for further research.
TargetScan was applied to identify the potential targets of miR-218-5p on the 3ʹ-UTR of DEK mRNA contains putative a binding site for miR-218-5p (Figure 4A). Following the dual-luciferase activity, the assay was conducted to verify the target relationship between miR-218-5p and DEK. Data demonstrated that the luciferase activity of DEK-WT was inhibited by miR-218-5p mimics (Figure 4B and C), elevated by miR-218-5p inhibitors in both U251 and U343 cells (Figure 4D and E). No distinct change of DEK-MUT luciferase activity was detected (Figure 4B–E). Based on the data from miRCancer Database, DEK was significantly increased in glioma tissues (N = 163) in contrast with the (N = 207) normal tissues (Suppl. Fig. S3). The protein level of DEK in U251 and U343 cells was measured by Western blot assay. Results showed that the DEK level was down-regulated by miR-218-5p mimics (Figure 4F and G), up-regulated by miR-218-5p inhibitors (Figure 4H and I).
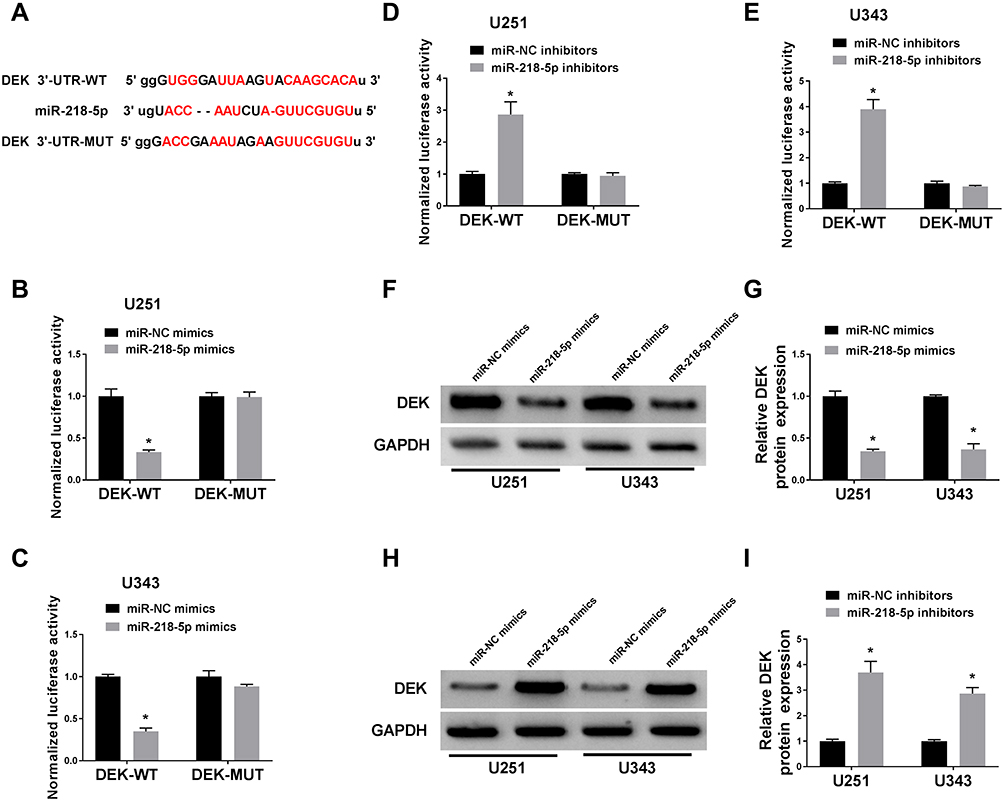 |
Figure 4 DEK was a direct target of miR-218-5p and negatively regulated by miR-218-5p. (A) The putative binding site and the mutant sites in of miR-218-5p on the 3ʹ-UTR of DEK mRNA were shown. (B–E) U251 and U343 cells were transfected with DEK-WT or DEK-MUT and miR-218-5p mimics or miR-NC mimics, miR-218-5p inhibitors or miR-NC inhibitors, followed by the measure of dual-luciferase activities at 48 h after transfection. (F–I) The protein level of DEK in the two cell lines was tested by Western blot after transfection. *P<0.05. |
MiR-218-5p Restored the Cell Viability, Apoptosis, Migration, Invasion Abilities and Revitalized β-Catenin Signaling by Targeting DEK
In order to figure out the inhibitory mechanism of miR-218-5p on glioma cell lines, we down-regulated the expression of miR-218-5p in U251 and U343 cells. Western blot assay exhibited that down-expression of miR-218-5p recuperated the repression of DEK expression by sh-DEK in the two cell lines (Figure 5A and B). Silencing of miR-218-5p also recovered the suppressive action of sh-DEK on cell viability, migration, and invasion, promoting apoptosis in U251 and U343 cells (Figure 5C–G). Unsurprisingly, down-regulated miR-218-5p also restored the inhibitory expression (β-catenin, c-Myc, CyclinD1) (Figure 5H and I) and the elevated E-cadherin protein levels, as well as the decreased N-cadherin, and Vimentin (Suppl. Fig. S1G-H) in the DEK silenced cells.
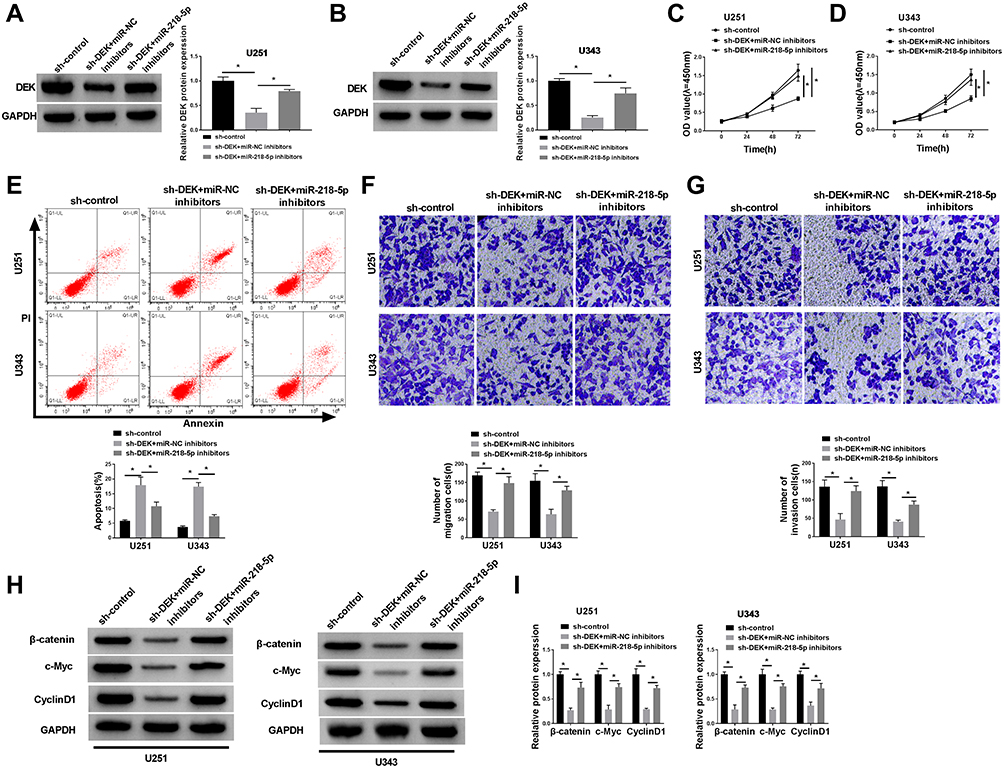 |
Figure 5 MiR-218-5p restored the cell viability, apoptosis, migration, invasion abilities and revitalized β-catenin signaling by targeting DEK. (A and B) Western blot assay revealed the protein level of DEK in U251 and U343 cells after co-transfection. (C and D) MTT assay showed cell viability of U251 and U343 cells after co-transfection. (E) Flow cytometry assessment performed cell apoptosis of U251 and U343 cells after treatment. (F and G) Transwell assay exhibited migration and invasion abilities of the two cell lines (magnification: 100x, n=3). (H and I) Western blot analysis uncovered the expression patterns upon β-catenin, c-Myc, CyclinD1 in glioma cells. *P<0.05. |
Upregulation of DEK Inverted Sevoflurane-Mediated Repression on Cell Viability, Migration, and Invasion and β-Catenin Signal Pathway, but a Promotion on Apoptosis in Glioma Cells
In order to further investigate the mechanism of sevoflurane in the progress of glioma cells, we transfected U251 and U343 cells with pcDNA3.1-DEK to up-regulate DEK after treatment with 4% sevoflurane. Results suggested that the overexpression of DEK strikingly relieved the repression of expression of DEK induced by sevoflurane in U251 and U343 cells (Figure 6A and B). We also demonstrated that the up-regulation of DEK also partially recovered the suppressive influence on cell viability, migration, and invasion, the promoted influence on apoptosis in both U251 and U343 cells (Figure 6C–G). In the meantime, the expression upon β-catenin, c-Myc, CyclinD1 recuperated by enhanced DEK could be observed in U251 and U343 cells (Figure 6H and I). Besides, the elevated DEK also reversed the promotion effect on E-cadherin and the inhibition effect on N-cadherin and Vimentin that induced by 4% sevoflurane (Suppl. Fig. S1I-J).
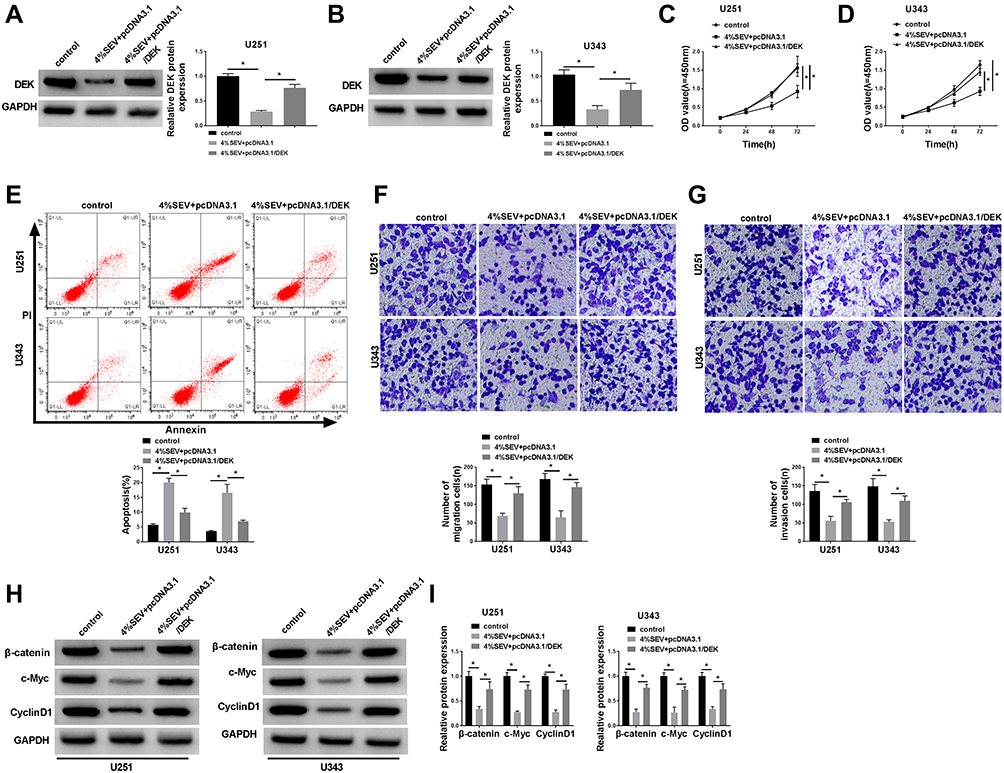 |
Figure 6 Upregulation of DEK inverted sevoflurane-mediated repression on cell viability, migration, and invasion and β-catenin signal pathway, but promotion on apoptosis in glioma cells. U251 and U343 cells were transfected with pcDNA3.1 vector or pcDNA3.1-DEK after treatment with 4% sevoflurane. (A and B) The protein level of DEK in U251 and U343 cells, (C and D) cell viability of U251 and U343 cells and apoptosis (E), migration (F), and invasion (G) of cell numbers in U251 and U343 cells, supplemented with the expression of β-catenin signal pathway-associated proteins (H and I) were shown.*P<0.05. |
Sevoflurane Reduced the Growth of Tumor by Regulating miR-218-5p/DEK Axis
To analyze the function of miR-218-5p in glioma in vivo, U251 cells with stable transfection of in-miR-NC, in-miR-218-5p or control were utilized for xenograft model. As shown in Figure 7A and B, tumor volume and weight were significantly reduced under sevoflurane treatment, and miR-218-5p inhibitors have reversed the effects on tumor size. In addition, miR-218-5p was markedly increased under the sevoflurane treatment and decreased in SEV+miR-218-5p inhibitor group (Figure 7C). Furthermore, the protein expression levels of DEK were reduced under sevoflurane treatment, while DEK protein expression levels were up-regulated in SEV+miR-218-5p group in vivo (Figure 7D).
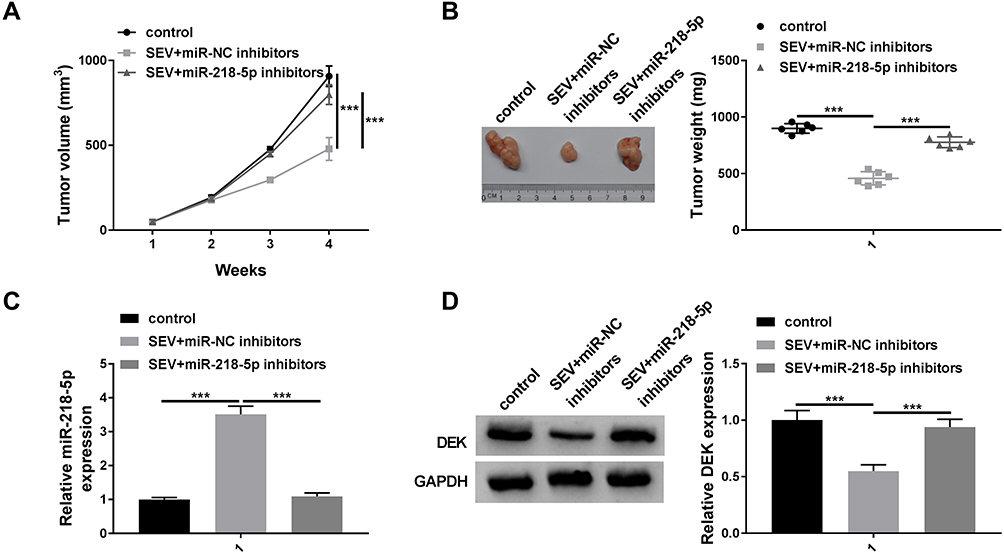 |
Figure 7 Knockdown of miR-218-5p reversed sevoflurane-treated effects on tumor growth in vivo. U251 cells transfected with in-miR-NC or in-miR-218-5p were injected into the nude mice for 3 days, mice were injected of 50 mg/kg sevoflurane or 0.5 mL soybean oil for 25 days. After the mice were sacrificed, the tumor was measured. Tumor volume (A) and Tumor weight (B) were detected. (C) The expression level of miR-218-5p was detected by qRT-PCR. (D) The protein expression level of DEK was measured by Western blot assay. ***P<0.001. |
Discussion
The novel role of sevoflurane was found as a tumor-suppressor in recent years. It worked as a malignancy-inhibitor, and participated in colorectal cancer,40 breast cancer,10 and osteosarcoma.10 Liu et al showed that sevoflurane could restrain the proliferation ability in breast cancer cells via promoting the expression of miRNA-203 in vitro.40 Also, in Fan et al research, sevoflurane inhibited the migration and invasion via deactivation of ERK/MMP-9 signaling by up-regulating miR-203 in colorectal cancer cells.10 Besides, Gao et al reported that in osteosarcoma cells, sevoflurane showed the suppressed effects on cell growth and metastasis.41 In addition, sevoflurane suppressed glioma cell migration and invasion via regulating miR-637.7
In our study, we first tested the changed phenotype in tumor cells (U251 and U343) treated with different sevoflurane concentrations. Through MTT assay, transwell assay, flow cytometry assay, individually, sevoflurane was found to be able to inhibit the proliferation, migration, and invasion, and promoted apoptosis in glioma cells, which is consistent with that reported in previous articles.10,40,41 It is well known that Wnt/β-catenin signaling usually promotes the invasion and metastasis as well as cell migration and proliferation during the development of cancer through epithelial-mesenchymal transition (EMT).42,43 We have subsequently examined the expression levels of β-catenin signaling pathway-associated molecular marker proteins and EMT markers at the exposure of sevoflurane and found the β-catenin signaling pathway was significantly activated by sevoflurane.
Previous studies have shown that miR-218-5p was decreased in pancreatic carcinoma, and its overexpression repressed the biological functions of tumor growth, migration, and invasion.44 In Liu et al research, miR-218-5p targets SNHG16 limited the cell proliferation, migration, and invasion in pancreatic cancer cells.45 Here, we were surprised to find that sevoflurane upregulated miR-218-5p level, and the over expressed miR-218-5p showed significant inhibition on biological function (consistent with the above reports44,45) and activation of the β-catenin signaling. To demonstrate that the up-regulation of miR-218-5p is due to exposure of sevoflurane, the expression of miR-218-5p was silenced by a loss-functional experiment, and the data presented that miR-218-5p inhibitors restore the above tumor-suppressive effects of sevoflurane treatment.
Aim to find the target gene of miR-218-5p, we predicted the downstream target gene of miR-218-5p and verified by dual-luciferase reporter gene experiment. The gene DEK was identified as a potential target of miR-218-5p. Followed by, we explored the regulatory relationship between them with each other. Results suggested that DEK level was decreased with the increase of miR-218-5p, and increased with the decrease of miR-218-5p, which was verified through the loss- and gain-of function assay. Also, miR-218-5p inhibitors reversed the biological function on proliferation, apoptosis, migration, and invasion and limitation of β-catenin signaling pathways and EMT in sh-DEK treated cells.
In breast cancer, the oncogene DEK was regarded as a stimulator of β-catenin signaling pathways, contributing to a series of biofunctional effects on invasion and mammosphere formation.46 Privette Vinnedge, in her assignments, confirmed that the expression of DEK stimulated the production and secretion of Wnt ligands to sustain an autocrine/paracrine canonical β-catenin signaling loop.47 In order to examine whether sevoflurane inhibited the development of gliomas through the miR-218-5p/DEK/β-catenin axis, we examined the tumor-suppressive action of sevoflurane on DEK overexpressed glioma cells. And data suggested that overexpressed DEK perfectly restored the malignant degree on stated biological functions in tumor as well. In addition, sevoflurane suppressed the glioma progression via miR-218-5p/DEK axis, which has also been verified in vivo.
There are some limitations to this experiment. For example, this experiment did not involve clinical samples. Also, the targeting relationship between miR-218-5p and DEK still needs to be supplemented with the results of RNA pull-down or RNA immunoprecipitation.
Conclusion
In summary, we identified that sevoflurane could inhibit the glioma progression in vitro and in vivo. Furthermore, our study firstly highlighted the role of miR-218-5p/DEK/β-catenin axis in glioma and revealed that sevoflurane could suppress glioma progression by regulating miR-218-5p/DEK/β-catenin axis.
Acknowledgments
The authors would like to thank the participants in this study.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Sun Q, Xu R, Xu H, et al. Extracranial metastases of high-grade glioma: the clinical characteristics and mechanism. World J Surg Oncol. 2017;15(1):181. doi:10.1186/s12957-017-1249-6
2. Kumar AJ, Leeds NE, Fuller GN, et al. Malignant gliomas: MR imaging spectrum of radiation therapy- and chemotherapy-induced necrosis of the brain after treatment. Radiology. 2000;217(2):377–384. doi:10.1148/radiology.217.2.r00nv36377
3. Weller M, Wick W, Aldape K, et al. Glioma. Nat Rev Dis Primers. 2015;1(1):15017. doi:10.1038/nrdp.2015.17
4. Kvolik S, Dobrosevic B, Marczi S, et al. Different apoptosis ratios and gene expressions in two human cell lines after sevoflurane anaesthesia. Acta Anaesthesiol Scand. 2009;53(9):1192–1199. doi:10.1111/j.1399-6576.2009.02036.x
5. Kvolik S, Glavas-Obrovac L, Bares V, et al. Effects of inhalation anesthetics halothane, sevoflurane, and isoflurane on human cell lines. Life Sci. 2005;77(19):2369–2383. doi:10.1016/j.lfs.2004.12.052
6. Liang H, Gu M, Yang C, et al. Sevoflurane inhibits invasion and migration of lung cancer cells by inactivating the p38 MAPK signaling pathway. J Anesth. 2012;26(3):381–392. doi:10.1007/s00540-011-1317-y
7. Yi W, Li D, Guo Y, et al. Sevoflurane inhibits the migration and invasion of glioma cells by upregulating microRNA-637. Int J Mol Med. 2016;38(6):1857–1863. doi:10.3892/ijmm.2016.2797
8. Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11(4):441–450. doi:10.1016/j.devcel.2006.09.009
9. Di Leva G, Garofalo M, Croce CM. MicroRNAs in cancer. Annu Rev Pathol. 2014;9(1):287–314. doi:10.1146/annurev-pathol-012513-104715
10. Fan L, Wu Y, Wang J, He J, Han X. Sevoflurane inhibits the migration and invasion of colorectal cancer cells through regulating ERK/MMP-9 pathway by up-regulating miR-203. Eur J Pharmacol. 2019;850:43–52. doi:10.1016/j.ejphar.2019.01.025
11. Wu R, Zhao B, Ren X, et al. MiR-27a-3p targeting GSK3β promotes triple-negative breast cancer proliferation and migration through Wnt/β-catenin pathway. Cancer Manag Res. 2020;12:6241–6249. doi:10.2147/CMAR.S255419
12. Ionescu C. The first printed Romanian medical bulletin. Viata Med Rev Inf Prof Stiint Cadrelor Medii Sanit. 1989;37(12):281–284.
13. Li Z, Qian R, Zhang J, Shi X. MiR-218-5p targets LHFPL3 to regulate proliferation, migration, and epithelial-mesenchymal transitions of human glioma cells. Biosci Rep. 2019;39(3):BSR20180879. doi:10.1042/BSR20180879
14. Cullen BR. MicroRNAs as mediators of viral evasion of the immune system. Nat Immunol. 2013;14(3):205–210. doi:10.1038/ni.2537
15. White NM, Fatoohi E, Metias M, et al. Metastamirs: a stepping stone towards improved cancer management. Nat Rev Clin Oncol. 2011;8(2):75–84. doi:10.1038/nrclinonc.2010.173
16. Zhang B, Pan X, Cobb GP, et al. MicroRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302(1):1–12. doi:10.1016/j.ydbio.2006.08.028
17. Xiao B, Tan L, He B, et al. MiRNA-329 targeting E2F1 inhibits cell proliferation in glioma cells. J Transl Med. 2013;11(1):172. doi:10.1186/1479-5876-11-172
18. Hui W, Yuntao L, Lun L, et al. MicroRNA-195 inhibits the proliferation of human glioma cells by directly targeting cyclin D1 and cyclin E1. PLoS One. 2013;8(1):e54932. doi:10.1371/journal.pone.0054932
19. Zhang C, Ge S, Hu C, et al. MiRNA-218, a new regulator of HMGB1, suppresses cell migration and invasion in non-small cell lung cancer. Acta Biochim Biophys Sin (Shanghai). 2013;45(12):1055–1061. doi:10.1093/abbs/gmt109
20. Jin J, Cai L, Liu ZM, et al. miRNA-218 inhibits osteosarcoma cell migration and invasion by down-regulating of TIAM1, MMP2 and MMP9. Asian Pac J Cancer Prev. 2013;14(6):3681–3684. doi:10.7314/apjcp.2013.14.6.3681
21. Prudnikova TY, Mostovich LA, Kashuba VI, et al. miRNA-218 contributes to the regulation of D-glucuronyl C5-epimerase expression in normal and tumor breast tissues. Epigenetics. 2012;7(10):1109–1114. doi:10.4161/epi.22103
22. Xia H, Yan Y, Hu M, et al. MiR-218 sensitizes glioma cells to apoptosis and inhibits tumorigenicity by regulating ECOP-mediated suppression of NF-kappaB activity. Neuro Oncol. 2013;15(4):413–422. doi:10.1093/neuonc/nos296
23. Zhang JM, Sun CY, Yu SZ, et al. Relationship between miR-218 and CDK6 expression and their biological impact on glioma cell proliferation and apoptosis. Zhonghua Bing Li Xue Za Zhi. 2011;40(7):454–459.
24. Riveiro-Falkenbach E, Ruano Y, Garcia-Martin RM, et al. DEK oncogene is overexpressed during melanoma progression. Pigment Cell Melanoma Res. 2017;30(2):194–202. doi:10.1111/pcmr.12563
25. Nakashima T, Tomita H, Hirata A, et al. Promotion of cell proliferation by the proto-oncogene DEK enhances oral squamous cell carcinogenesis through field cancerization. Cancer Med. 2017;6(10):2424–2439. doi:10.1002/cam4.1157
26. Liu S, Wang X, Sun F, et al. DEK overexpression is correlated with the clinical features of breast cancer. Pathol Int. 2012;62(3):176–181. doi:10.1111/j.1440-1827.2011.02775.x
27. Kondoh N, Wakatsuki T, Ryo A, et al. Identification and characterization of genes associated with human hepatocellular carcinogenesis. Cancer Res. 1999;59(19):4990–4996.
28. Hacker KE, Bolland DE, Tan L, et al. The DEK oncoprotein functions in ovarian cancer growth and survival. Neoplasia. 2018;20(12):1209–1218. doi:10.1016/j.neo.2018.10.005
29. Xu X, Zou L, Yao Q, et al. Silencing DEK downregulates cervical cancer tumorigenesis and metastasis via the DEK/p-Ser9-GSK-3beta/p-Tyr216-GSK-3beta/beta-catenin axis. Oncol Rep. 2017;38(2):1035–1042. doi:10.3892/or.2017.5721
30. Privette Vinnedge LM, Kappes F, Nassar N, et al. Stacking the DEK: from chromatin topology to cancer stem cells. Cell Cycle. 2013;12(1):51–66. doi:10.4161/cc.23121
31. Riveiro-Falkenbach E, Soengas MS. Control of tumorigenesis and chemoresistance by the DEK oncogene. Clin Cancer Res. 2010;16(11):2932–2938. doi:10.1158/1078-0432.CCR-09-2330
32. Pease NA, Wise-Draper T, Privette Vinnedge L. Dissecting the potential interplay of DEK functions in inflammation and cancer. J Oncol. 2015;2015:106517. doi:10.1155/2015/106517
33. Grottke C, Mantwill K, Dietel M, et al. Identification of differentially expressed genes in human melanoma cells with acquired resistance to various antineoplastic drugs. Int J Cancer. 2000;88(4):535–546. doi:10.1002/1097-0215(20001115)88:4<535::aid-ijc4>3.0.co;2-v
34. Wu Q, Li Z, Lin H, et al. DEK overexpression in uterine cervical cancers. Pathol Int. 2008;58(6):378–382. doi:10.1111/j.1440-1827.2008.02239.x
35. Shibata T, Kokubu A, Miyamoto M, et al. DEK oncoprotein regulates transcriptional modifiers and sustains tumor initiation activity in high-grade neuroendocrine carcinoma of the lung. Oncogene. 2010;29(33):4671–4681. doi:10.1038/onc.2010.217
36. Piao J, Shang Y, Liu S, et al. High expression of DEK predicts poor prognosis of gastric adenocarcinoma. Diagn Pathol. 2014;9(1):67. doi:10.1186/1746-1596-9-67
37. Liu Q, Liu H, Cheng H, et al. Downregulation of long noncoding RNA TUG1 inhibits proliferation and induces apoptosis through the TUG1/miR-142/ZEB2 axis in bladder cancer cells. Onco Targets Ther. 2017;10:2461–2471. doi:10.2147/OTT.S124595
38. Wang L, Wang T, Gu JQ, et al. Volatile anesthetic sevoflurane suppresses lung cancer cells and miRNA interference in lung cancer cells. Onco Targets Ther. 2018;11:5689–5693. doi:10.2147/OTT.S171672
39. Zhang D, Zhou XH, Zhang J, et al. Propofol promotes cell apoptosis via inhibiting HOTAIR mediated mTOR pathway in cervical cancer. Biochem Biophys Res Commun. 2015;468(4):561–567. doi:10.1016/j.bbrc.2015.10.12940
40. Liu J, Yang L, Guo X, et al. Sevoflurane suppresses proliferation by upregulating microRNA-203 in breast cancer cells. Mol Med Rep. 2018;18(1):455–460. doi:10.3892/mmr.2018.8949
41. Gao K, Su Z, Liu H, et al. Anti-proliferation and anti-metastatic effects of sevoflurane on human osteosarcoma U2OS and Saos-2 cells. Exp Mol Pathol. 2019;108:121–130. doi:10.1016/j.yexmp.2019.04.005
42. Heuberger J, Birchmeier W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb Perspect Biol. 2010;2(2):a002915. doi:10.1101/cshperspect.a002915
43. Li Y, Welm B, Podsypanina K, et al. Evidence that transgenes encoding components of the Wnt signaling pathway preferentially induce mammary cancers from progenitor cells. Proc Natl Acad Sci U S A. 2003;100(26):15853–15858. doi:10.1073/pnas.2136825100
44. Wang A, Dai H, Gong Y, et al. ANLN-induced EZH2 upregulation promotes pancreatic cancer progression by mediating miR-218-5p/LASP1 signaling axis. J Exp Clin Cancer Res. 2019;38(1):347. doi:10.1186/s13046-019-1340-7
45. Liu S, Zhang W, Liu K, et al. LncRNA SNHG16 promotes tumor growth of pancreatic cancer by targeting miR-218-5p. Biomed Pharmacother. 2019;114:108862. doi:10.1016/j.biopha.2019.108862
46. Privette Vinnedge LM, Mcclaine R, Wagh PK, et al. The human DEK oncogene stimulates beta-catenin signaling, invasion and mammosphere formation in breast cancer. Oncogene. 2011;30(24):2741–2752. doi:10.1038/onc.2011.2
47. Privette Vinnedge LM, Benight NM, Wagh PK, et al. The DEK oncogene promotes cellular proliferation through paracrine Wnt signaling in Ron receptor-positive breast cancers. Oncogene. 2015;34(18):2325–2336. doi:10.1038/onc.2014.173
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.