Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 18
Serum Proteomic Profiling in Patients with Chronic Obstructive Pulmonary Disease
Authors Wu S, Huang K ![]() , Chang C, Chu X, Zhang K, Li B, Yang T
, Chang C, Chu X, Zhang K, Li B, Yang T ![]()
Received 7 April 2023
Accepted for publication 3 July 2023
Published 28 July 2023 Volume 2023:18 Pages 1623—1635
DOI https://doi.org/10.2147/COPD.S413924
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Sinan Wu,1– 4,* Ke Huang,1– 4,* Chenli Chang,1– 5 Xu Chu,1– 4,6 Kun Zhang,7 Baicun Li,1– 4 Ting Yang1– 4
1National Center for Respiratory Medicine, Beijing, People’s Republic of China; 2National Clinical Research Center for Respiratory Diseases, Beijing, People’s Republic of China; 3Institute of Respiratory Medicine, Chinese Academy of Medical Sciences, Beijing, People’s Republic of China; 4Department of Pulmonary and Critical Care Medicine, Center of Respiratory Medicine, China-Japan Friendship Hospital, Beijing, People’s Republic of China; 5China-Japan Friendship Hospital (Institute of Clinical Medical Sciences), Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, People’s Republic of China; 6Department of Pulmonary and Critical Care Medicine The First Affiliated Hospital of Henan University of Science and Technology, Luoyang, People’s Republic of China; 7Biotree-Shanghai, Focus Dream Park, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ting Yang; Baicun Li, Department of Pulmonary and Critical Care Medicine, Center of Respiratory Medicine, China-Japan Friendship Hospital, No. 2, East Yinghua Road, Chaoyang District, Beijing, 100029, People’s Republic of China, Tel +86 10-84206273, Email [email protected]; [email protected]
Purpose: Chronic obstructive pulmonary disease (COPD) is a heterogeneous disease with high morbidity and mortality rates. This study used proteomic profiling of serum to identify the differentially expressed proteins in COPD patients compared with healthy controls, to expand the knowledge of COPD pathogenesis and to ascertain potential new targets for diagnosis and treatment of COPD.
Methods: Serum samples were collected from 56 participants (COPD group n = 28; Healthy Control group n = 28). A data-independent acquisition quantitative proteomics approach was used to identify differentially expressed proteins (DEPs) between the two groups. Gene Ontology (GO) functional annotation, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway functional enrichment, and protein–protein interaction analyses of DEPs were conducted to identify their relevant biological processes, cellular components, and related pathways. We used a parallel reaction monitoring (PRM)-based targeted quantitative proteomics approach to validate those findings.
Results: Of 8484 peptides identified by searching the UniProtKB/Swiss-Prot knowledgebase, 867 proteins were quantifiable, of which 20 were upregulated and 35 were downregulated in the COPD group. GO functional annotation indicated that the subcellular localization of most DEPs was extracellular. The top three molecular functions of the DEPs were signaling receptor binding, antigen binding, and immunoglobulin receptor binding. The most relevant biological process was immune response. The transforming growth factor-β signaling pathway, Staphylococcus aureus infection, and hematopoietic cell lineage were the top three pathways identified in the KEGG pathway functional enrichment. Our PRM analyses confirmed the identification of 11 DEPs identified in our data-independent acquisition analyses, 8 DEPs were upregulated and 3 DEPs were downregulated.
Conclusion: This study using data-independent acquisition analyses with PRM confirmation of findings identified 11 DEPs in the serum of patients with COPD. These DEPs are potential diagnostic or prognostic biomarkers or may be future targets for the treatment of COPD.
Keywords: chronic obstructive pulmonary disease, proteomics, differentially expressed proteins, data-independent acquisition, parallel reaction monitoring
Introduction
Chronic obstructive pulmonary disease (COPD) is widespread and characterized by progressive airflow restriction and lung tissue destruction manifested as breathlessness, coughing, and sputum production. Airway inflammation, emphysema, and epithelial barrier dysfunction are all considered to be involved in the pathogenesis of COPD.1 Immune cells, such as neutrophils and macrophages, are also related to the airway inflammation and immune imbalance leading to the occurrence of COPD.1,2 The disease burden of COPD has increased in the last decades.3 Conclusions from the Global Burden of Disease 2019 study cohort showed that the global incidence of COPD increased by 85.89% from 1990 to 2019.4 In addition, 3,280,636 people died of COPD in 2019, compared with 2,520,219 in 1990, an increase of 30.17%.4 The pathogenesis and etiology of COPD have not been fully elucidated, and it is difficult to predict its development or progression. Therefore, mechanistic insights into the pathogenesis of COPD are urgently needed to enable discovery of reliable diagnostic and therapeutic targets for this disease.
With the development of biological technology, the methods for discovering new targets have gradually developed into large-scale high-throughput detection based on multi-omics, such as genomics, transcriptomics, proteomics and metabolomics. In essence, proteomics can study the characteristics of proteins on a large scale, including the expression level of proteins, post-translational modifications, protein–protein interactions, etc, so as to obtain a holistic and comprehensive understanding of the processes of disease occurrence and cell metabolism at the protein level. Previous studies conducting proteomic profiling of the serum of patients with COPD have detected some differentially expressed proteins (DEPs), including fibulin-3.5 However, there is a dearth of studies assessing DEPs in COPD that have used data-independent acquisition (DIA) quantitative proteomics, a relatively recently developed proteomics strategy based on global mass spectrometry (MS), combined with parallel reaction monitoring (PRM) for targeted quantitative verification in MS to confirm the DIA findings. PRM is currently the mainstream approach for targeted proteomics data acquisition. It is used to selectively detect specific peptides or target peptides (such as peptides undergoing post-translational modification). Thus, the relative or absolute quantification of the target protein/modified peptide segment can be achieved. Thus, to ascertain potential new targets for the diagnosis and treatment of COPD, we used DIA quantitative proteomics technology to distinguish DEPs in serum samples from COPD patients and healthy control participants. We then conducted Gene Ontology (GO) functional annotation, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway functional enrichment, and protein–protein interaction (PPI) analyses of DEPs to identify their associated biological processes, cellular components, and related molecular and signaling pathways. We then used PRM targeted quantitative proteomics technology to conduct a quantitative analysis of the DEPs (Figure 1).
 |
Figure 1 Schematic representation of the project workflow. |
Materials and Methods
Clinical Data and Serum Samples
Patients with COPD were recruited into the COPD group (n = 28) during follow-up visits to the China-Japan Friendship Hospital. Healthy individuals visiting the hospital for physical examinations were recruited into the Healthy Control group (n = 28). Peripheral blood was collected without coagulant. Samples were centrifuged at 3000 rpm for 15 min at 4 °C. The serum was subsequently separated and immediately frozen at −80 °C until use in analyses. Equal portions of serum samples from seven patients with COPD were combined into a single sample, and (separately) serum samples from seven healthy persons were combined into a single sample. Thus, we obtained four batches of the combined samples for the COPD group (identification numbers A1-A4) and four batches of the combined samples for the Healthy Control group (identification numbers B1-B4).
Inclusion criteria for patients with COPD were (1) aged ⩾20 years with a confirmed diagnosis of COPD in compliance with the GOLD guideline (forced expiratory volume in the first second after bronchodilator percentage of forced vital capacity [FEV1/FVC] < 70%), with or without chronic respiratory symptoms; (2) volunteered to participate in the study; and (3) had not participated in other clinical studies.
Exclusion criteria were (1) COPD accompanied by asthma, tuberculosis, malignant tumor, or occupational disease; (2) COPD accompanied by serious disease of the brain, heart, kidney, or liver; (3) COPD accompanied by mental disorder or cognitive disorder; and (4) had undergone chest, abdominal, or eye surgery in the last 3 months.
This study was approved by the ethics committees at the China-Japan Friendship Hospital (permit number 2022-KY-141). All participants provided written informed consent for inclusion in this study. The design and implementation process of the study complies with the Declaration of Helsinki.
Protein Extraction and Preparation for Proteomics
After thawing on ice, samples stored at −80 ° C were centrifuged at 12,000 × g for 10 min at 4 °C using a cryogenic high-speed centrifuge. After centrifugation, cell debris was removed, and the supernatant was transferred to a new centrifuge tube. Removal of highly abundance proteins was achieved using the Pierce™ Top 14 Abundant Protein Depletion Spin Columns Kit (Thermo Scientific) and the experimental procedure followed the instructions provided. Protein concentration was determined using bicinchoninic acid kits. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis was conducted to control sample quality.
The same amount of protein from each sample was used for enzymolysis. The volume was adjusted to be consistent with the lysate, and dithiothreitol was added to make the final concentration 5 mM, which was reduced at 56 °C for 30 min. Iodoacetamide was then added to make the final concentration 11 mM, and the samples were incubated in the dark at room temperature for 15 min. The alkylated samples were transferred to an ultrafiltration tube, centrifuged at 12,000 × g at room temperature for 20 min. The buffer was replaced three times with 8 M urea, and then three times with a replacement buffer. Trypsin was added at a ratio of 1:50 (trypsin:protein, m/m), and enzymolysis was performed overnight. The peptides were recovered by centrifugation at 12,000 × g at room temperature for 10 min. The peptides were recovered from the precipitate once more with ultra-pure water, and the peptide solution was combined.
Nano-Liquid Chromatography Tandem Mass Spectrometry (Nano-LC-MS/MS) Analyses
HPLC Fractionation: Eight groups of the trypsin-hydrolyzed peptides were mixed and graded into 12 fractions by high-pH reverse high-performance liquid chromatography. The iRT kit was added to all the fractions according to manufacturer’s instructions and dried by vacuum centrifuging.
Spectral Library Building—LC-MS/MS Analysis: The fraction samples were dissolved in liquid chromatography mobile phase A (Phase A is a water solution containing 0.1% formic acid and 2% acetonitrile) and separated using an EASY-nLC 1200 ultra-performance liquid system and injected into a nanospray ionization source for ionization. The data were collected using data-dependent acquisition mode with Thermo Q Exactive HF-X.
Data-independent Acquisition (DIA)—LC-MS/MS Analysis: The iRT kit was added to all the samples according to manufacturer’s instructions. The LC gradient was kept consistent with those in the spectral library building method. The separated peptides were analyzed in Q Exactive HF-X with a nano-electrospray ion source. The data acquisition was performed in DIA mode. The full MS scan resolution was set to 120,000 for a scan range of 385–1200 m/z. Each cycle contains one full scan followed by 70 DIA MS/MS scans with predefined precursor m/z range. The HCD fragmentation was performed at a normalized collision energy (NCE) of 27%. The fragments were detected in the Orbitrap at a resolution of 15,000. Fixed first mass was set as 200 m/z. Automatic gain control (AGC) target was set at 5E5.
Sequence Database Search
A spectral library was constructed by processing the generated data-dependent acquisition data using the MaxQuant search engine (v.1.6.6.0). The Pulsar search engine embedded in Spectronaut (v14.6) was searched using software default parameters. The database used was the human UniProtKB/Swiss-Prot (20395 sequences), and a decoy database was searched to calculate the false discovery rate caused by random matching.
DIA Data Analysis
All DIA data were analyzed in Skyline (v 20.1.0). The DDA search results were imported to Skyline to generate the spectral library, and the retention times were aligned to iRT reference values. Transition settings: precursor charges were set as 2, 3, 4, 5, ion charges were set as 1, 2. The ion match tolerance was set as 0.02 Da. Six most intense fragment ions from the spectral library were selected for each precursor. Decoy generation was based on shuffled sequences, and the FDR was estimated with the mProphet approach and set to 1%. Relative quantification of proteins was performed using the MSstats software package.
GO Functional Classification Annotation
GO is a bioinformatics analysis method that can organically link genes and various information about gene products (such as proteins) to provide statistical information. We used the GO annotation (GOA) program in the UniProt Knowledgebase (http://www.ebi.ac.uk/GOA/). GO annotation is divided into three categories—biological process, cellular component, and molecular function—to explain the biological effects of DEPs. Analyses of the Clusters of Orthologous Groups of proteins (prokaryotic homologous protein clusters/eukaryotic homologous protein clusters, COG/KOG) functional classification statistics for DEPs were conducted through database comparisons.
KEGG Pathway Functional Enrichment
The KEGG pathway enrichment analysis of DEPs was performed using two-tailed Fisher exact tests (p<0.05 was considered statistically significant) and the KEGG database. KEGG is an information network connecting known interactions between molecules, such as metabolic pathways, complexes, and biochemical reactions. KEGG pathways mainly include metabolism, genetic information processing, environmental information processing, cellular processes, human diseases, and drug development.
PPI Networks Analysis
The DEP database number or protein sequence screened from the comparison group was compared with the STRING (v.11.0) protein interaction network database. The interactions among DEPs were extracted with a confidence score >0.7, indicating high confidence. The R package networkD3 tool was then used to visualize the PPI networks of the DEPs. The circles in the visualized network represented DEPs, and the different colors represented different DEPs. The darker the color, the larger the multiple of the difference.
PRM Validation
PRM targeted quantitative proteomics methods were used to conduct quantitative analyses of the candidate COPD diagnostic marker proteome in serum samples. The process included protein extraction, protein enzymolysis, classification, extraction of characteristic peptide segments of target proteins, optimization of PRM MS detection conditions, PRM MS detection, and data analysis. The feasibility of a diagnostic marker proteome for COPD was confirmed, and the combination of protein markers having COPD diagnostic value was selected.
Statistical Analysis
The results are presented as the mean ± standard error of the mean (mean ± SEM), and a value of p < 0.05 was considered statistically significant. Differences among the groups were analyzed using Student’s t‐tests or one‐way analyses of variance. The primary software used for statistical analyses included GraphPad Prism (version 9.0.0) and SPSS (version 26.0).
Results
Clinical Characteristics of the Study Population
The clinical characteristics of all participants are given in Table 1. In total, 56 males were included in this study, and all participants were Han Chinese. There was no significant difference in age between the COPD (n = 28) and Healthy Control (n = 28) groups. In addition, no significant differences were found between the two groups for Forced expiratory volume in the first second (FEV1) and FEV1%pre, whereas there were significant differences between the groups for FVC and FEV1/FVC. Patients with COPD had a lower body mass index. These differences reflected the clinical features of patients with COPD, and the data indicated that patients in this study appeared to be in the early stage of the disease.
 |
Table 1 Clinical Diagnoses and Demographic Characteristics of Participants |
Protein Lysis, Quantification, and Enzymatic Digestion
The results of our sodium dodecyl sulfate–polyacrylamide gel electrophoresis analyses showed that the protein quality in the eight groups of samples was satisfactory for use in this study (Figure 2A). We identified 8484 peptides by searching the UniProtKB/Swiss-Prot knowledgebase. A comparison of these peptides with proteins yielded 906 identified proteins, of which 867 were quantifiable (Figure 2B). We generated a boxplot of the relative standard deviations (RSDs) of protein quantitative values among repeated samples and found that the overall RSD was small, indicating good quantitative repeatability (Figure 2C). Most peptide segments were 7–20 amino acids in length, which was consistent with proteolysis by trypsin and fragmentation of the higher-energy collision dissociation (HCD). The length distribution of the peptides identified by MS met the quality control requirements (Figure 2D). A correlation heatmap of the correlation coefficient matrix representing the degree of intra-sample repeatability and inter-sample correlation indicated that the experimental results were good (Figure 2E). Together, these findings suggested that the proteomic data were reliable and reproducible.
 |
Figure 2 Process and quality control of DIA. Notes: Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) after removal of highly abundant proteins and data quality control (A). Lanes A1-A4 show serum proteins from COPD group. Lanes B1-B4 show serum proteins from Healthy Control group. Lane M shows the marker. Results of identified peptides, identified proteins and quantifiable proteins (B). The horizontal axis is the peptides or proteins, and the vertical axis is the amount. Boxplot for relative standard deviation (RSD) of protein quantification from all repeated samples (C). The horizontal axis is the sample group, and the vertical axis is the RSD. Diagram of peptide length distribution (D). The horizontal axis is the peptide length, and the vertical axis is the number of peptides. Heat map for correlation coefficients within protein quantification for every two samples (E). This value measures the degree of linear correlation between two groups: the closer the Pearson correlation coefficient is close to −1, the correlation is negative; the closer it is to 1, the correlation is positive; the closer it is to 0, the correlation is not correlated. |
DIA Analysis and Identification of DEPs
Our principal component analysis indicated a significant difference between the serum proteins detected in the COPD group and in the Healthy Control group (p< 0.05). Proteins derived from the COPD group and Healthy Control group were completely separated. The samples showed good clustering based on the two principal components (Figure 3A). Among the 867 quantifiable proteins, 20 were upregulated and 35 were downregulated (Figure 3B) in the COPD group compared with the Healthy Control group. We used a volcano map and a heatmap to show specific upregulated proteins (eg, LTA4H, which is a pro-inflammatory enzyme, and SFTPB, a protein involved in immune response and regulation) and specific downregulated proteins (eg, IGHG2, IGHG4, which are the heavy chains of IgG2 and IgG4, respectively) (Figure 3C, D and Supplemental Table 1).
 |
Figure 3 Results of differential expression proteins (DEPs) analysis. Notes: Principal-component analysis (PCA) of samples from COPD (A1-A4) and Healthy Control group (B1-B4) (A). The higher degree of aggregation between the duplicated samples, the better the quantitative repeatability. Histogram of DEPs from COPD and Healthy control group (B). Volcano plots of DEPs from COPD and Healthy control group (C). The horizontal axis is the relative quantitative protein value after Log2 transformation, and the vertical axis is the P value of difference significance test after Log10 transformation. Heat map of DEPs from COPD and Healthy Control group (D). Each column indicates a sample and each row indicates a protein. The red colors represent up-regulated proteins and the blue colors represent down-regulated proteins. The color intensity represents the ratio of relative protein content from COPD to Healthy control group. |
GO Annotation and KEGG Pathway Enrichment
We performed GO functional classification annotation for the DEPs in COPD group vs the Healthy Control group. For the cellular component category, 45 proteins were associated with the most frequent GO term extracellular region. The top three proteins were IGKV3D-11, HLA-A, and PSAP. For the molecular function category, the most frequent term, antigen binding, was associated with 14 proteins. The top three proteins were IGKV1D-33, IGKV1-17, and IGKV3-20. For the biological process category, antigen binding was the most frequent GO term, and it was associated with 22 proteins. The top three proteins were IGKV3D-11, HLA-A, and ST6GAL1 (Figure 4A). The top three subcellular localizations were the extracellular space (30 proteins, 54.55%), cytoplasm (12 proteins, 21.82%), and nucleus (5 proteins, 9.09%) (Figure 4B).
 |
Figure 4 Gene Ontology (GO) functional annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway functional enrichment of DEPs. Notes: The biological process, cellular component and molecular functional annotation of DEPs (A). The horizontal axis is the GO terms name, and the vertical axis is the number of proteins. Cellular component annotation of DEPs (B). Clusters of Orthologous Groups of proteins (prokaryotic homologous protein clusters/eukaryotic homologous protein clusters, COG/KOG) functional classification annotation of DEPs (C). The horizontal axis is the COG/KOG categories, and the vertical axis is the frequency of matched COG/KOG. KEGG pathway enrichment annotation of DEPs (D). The horizontal axis is the rich factor, and the vertical axis is the pathway. |
Our COG/KOG results indicated that the processes that the DEPs were most associated with included posttranslational modification, protein turnover, chaperones, signal transduction mechanisms, and defense mechanisms (Figure 4C). The transforming growth factor-β (TGF-β) signaling pathway, Staphylococcus aureus infection, and hematopoietic cell lineage were the top three pathways detected in the KEGG pathway functional enrichment (Figure 4D).
PPI Networks Analysis
PPI analyses of the DEPs were performed using the online database STRING. The PPI network of five upregulated DEPs and one downregulated DEP shown in Figure 5 indicates the relationships among the DEPs in this network. The PPIs of ITGAM and FCG were newly found in the database, whereas the PPIs of PSMA1 and PSMA3 had been identified by previous studies and were confirmed herein.
 |
Figure 5 Protein–protein interaction (PPI) networks analysis of DEPs. Note: Each node represents a protein and lines between the nodes represent the interaction between two proteins. |
PRM Validation of DEPs in COPD
To validate the results of our DIA LC-MS/MS strategy, we used PRM. The quality control chromatogram for PRM showed good retention time overlap and peak shape (Figure 6). As mentioned, of 11 DEPs identified through our DIA analysis, 8 were upregulated (HLA-A, S10A6, SAP, SFTPB, LKHA4, ECP, SIAT1, and PSMA3) and 3 were downregulated (IGHG2, IGHG4, and SG3A2) (Table 2). The results of the DEPS detected in our PRM analysis were consistent with those detected using our DIA LC-MS/MS strategy, although the DIA analysis indicated that HLA-A and PSMA3 were downregulated rather than upregulated.
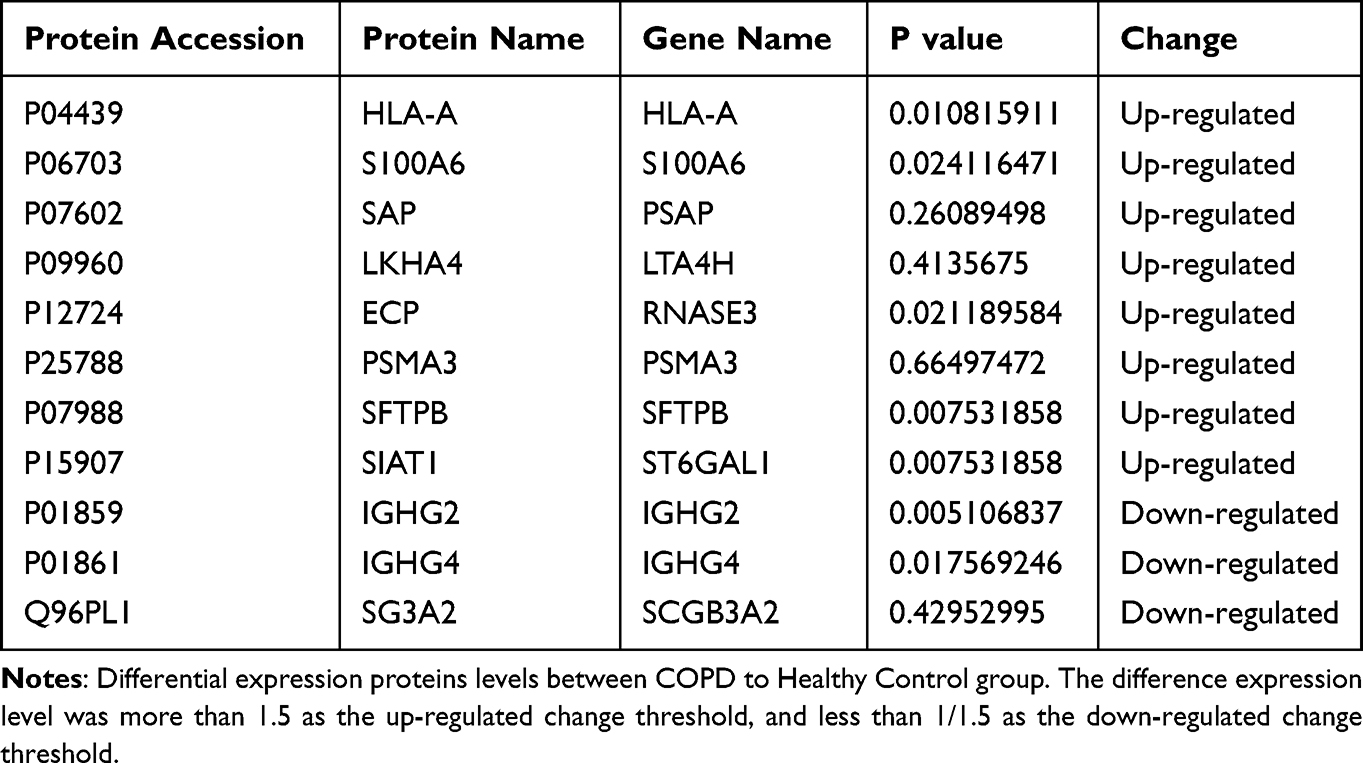 |
Table 2 Differential Expression Proteins in Serum Samples from COPD and Healthy Control Group |
 |
Figure 6 Chromatogram of quality control for parallel reaction monitoring (PRM) verification of DEPs. |
Discussion
This study is the first study, to our knowledge, to identify DEPs in the serum of patients with COPD compared with healthy individuals by using a DIA LC-MS/MS approach and to validate those findings using PRM. Of the initial 8484 peptides, 55 different proteins were identified using LC-MS/MC. Bioinformatics analyses, including GO, KEGG, and PPI, of these DEPs found that the most relevant biological processes were immune response and the most relevant molecular functions included signaling receptor binding, antigen binding, and immunoglobulin receptor binding. Using PRM, 11 proteins (3 downregulated and 8 upregulated) were identified that may be useful in understanding the pathogenesis and well as the diagnosis and treatment of COPD.
In our study, immunoglobulin heavy constant gamma 2 (IGHG2), IGHG4, and secretoglobin 3A2 (SGB3A2) were downregulated in the serum of patients with COPD compared with healthy individuals. IGHG2 and IGHG4 belong to the heavy chains of IgG2 and IgG4, respectively. As an important humoral immunity molecule, IgG plays major roles in the fight against bacterial and viral infections as well as in mediating antibody-dependent cell-mediated cytotoxicity and complement activation.6 IgG2 primarily provides a universal immune response against polysaccharide antigens, and IgG2 deficiency is associated with recurrent respiratory infections in children.6 Serum IgG subtypes were previously detected during hospitalization and after discharge of patients with COPD with acute exacerbation, and it was found that IgG and IgG4 levels were significantly decreased, which may be an important reason for the susceptibility to infection in these patients.7 In another study, a subgroup of persons with Down syndrome and IGHG4 deletion was susceptible to infection, suggesting that decreased IGHG4 protein expression is more likely to result in infection.8 In addition, the expression level of IGHG2 in sarcoma tumor cells is decreased and correlated with pyroptosis,9 a type of programmed cell death characterized by a robust inflammatory response.10 Zhu et al11 found that after cigarette smoke exposure of mice, alveolar macrophages showed an elevation of proteins indicative of pyroptosis. Mucus production and pro-inflammatory mediators, such as IL-6 and TNF-α, were also increased, whereas the phagocytic ability of the macrophages was decreased. These effects were alleviated by treatment with exosomes collected from adipose-derived stem cells. Consistent with the results of these studies, our study found that inflammation with an impaired innate immune response may be involved in the development of COPD. In addition, our enrichment and cluster analyses showed that the DEPs were closely related to inflammation signaling and bacterial defense mechanisms in COPD. IgG or IGHG functions are achieved via extracellular binding to antigens or microorganisms, which is consistent with our GO annotation results. Therefore, we suggest that the enhanced susceptibility to infection for persons with COPD may be attributable to decreases in levels of IGHG2 and IGHG4, which may be a causal factor in the onset of COPD.
SCGB3A2 is a secreted protein that is highly expressed in airway epithelial cells. Previous studies have shown that SCGB3A2 significantly inhibits the upregulated inflammation in cells derived from the normal human bronchial epithelial (BEAS-2B) cell line after lipopolysaccharide stimulation. Moreover, SCGB3A2 inhibits the phosphorylation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in lipopolysaccharide-induced BEAS-2B cells, suggesting that SCGB3A2 reduces lipopolysaccharide-induced inflammation in bronchial epithelial cells by inhibiting the activation of ERK and JNK.12 The ERK signaling pathway is a pro-inflammatory pathway associated with many diseases, including COPD.13,14 In a mouse model of COPD, cigarette smoke and lipopolysaccharide induced a lung inflammatory response via ERK signaling.15 Our results show that SCGB3A2 expression levels were significantly reduced in the serum of patients with COPD, suggesting that the ability to inhibit inflammation may be reduced in part due to a decrease in SGB3A2 levels and thus result in the progression of airway inflammation in COPD.
Eight upregulated proteins were also identified in our study. Leukotriene A4 hydrolase (LTA4H), a pro-inflammatory enzyme, induces substrate LTA4 to produce the inflammatory mediator leukotriene B4 (LTB4), which promotes inflammation in disease. However, this enzyme contains both hydrolase and aminopeptidase activities, which play roles in pro-inflammatory and anti-inflammatory responses, respectively. In the pathogenesis of COPD, LTA4H acts as a double-edged sword. Studies have found that although LTA4 increases the production of LTB4 and has pro-inflammatory effects, it also promotes the degradation of proline in the tripeptide proline-glycine-proline (PGP), thus performing anti-inflammation activity to some extent.16 In another study,17 cigarette smoke promoted the development of emphysema by inhibiting the enzyme activity of LTA4H aminopeptidase in lung tissue and by accumulating proline-glycine-proline and neutrophils in the air cavity. Thus, LTA4H has attracted a lot of research interest in recent years for the development of novel drugs to treat chronic inflammatory diseases. Novel, highly potent and selective inhibitors of LTA4H are currently undergoing clinical trials. Thus, it is expected that LTA4H will play a role in the treatment of COPD.18
Human leukocyte antigen-A (HLA-A) plays important roles in immune response and regulation. Our study found that HLA-A expression levels were increased in patients with COPD, in line with the results of previous studies. For example, in the genomic analysis of patients with COPD, expression of the HLA-A gene was significantly increased.19 In another study, Chen et al explored copy number variations in susceptible regions of long noncoding RNAs associated with HLA-A and the development of COPD.20 They found that the variations increased the expression of HLA-A in COPD lung tissues compared with normal lung tissues. Another group showed that HLA-A was significantly increased in the airway of patients with COPD, and its expression was decreased after intervention with azithromycin, suggesting that HLA-A may be involved in the antibiotic effects of azithromycin in the treatment of COPD.21 Although there is currently a lack of research investigating the specific functions of HLA-A in COPD, it appears to warrant future research.
Eosinophil cationic protein (ECP) is a secreted protein with potent cytotoxic, antibacterial, and antiviral properties. ECPs are released by activated eosinophils and are considered a marker of eosinophilic inflammation. Previous studies have shown that ECP expression is significantly increased in patients with eosinophilic COPD, with the frequency of acute exacerbation and the risk of hospitalization being high in these patients.22 Eosinophilic inflammation may be evaluated to predict the treatment response to inhaled steroids, and ECP may be a biomarker. ECP has shown high sensitivity and specificity for inflammatory activity in mild COPD, with a strong association with sputum eosinophils counts.23 In a crossover study,24 the phosphodiesterase 4 inhibitor roflumilast significantly reduced the absolute number of eosinophils and levels of ECP in sputum from patients with COPD, suggesting that these anti-inflammatory effects may be involved in the treatment of COPD. Therefore, we believe that this protein may also be developed as a therapeutic target for specific types of COPD, but its role in non-eosinophilic COPD types still needs to be investigated.
The protein levels for proteasome 20S alpha 3 (PSMA3) were also found to be differentially elevated in our study. Proteasomes are distributed in high concentrations in eukaryotic cells and cleave peptides in non-lysosomal pathways in an ATP/ubiquitin-dependent process.25 Previous tumor studies have shown that increased PSMA3 levels lead to the proliferation and migration of pancreatic ductal adenocarcinoma cells.26 In the regulation of gene expression, a decrease in the RNAse activity of PSMA1 and PSMA3 is paralleled by changes in their phosphorylation and ubiquitylation status on hemin-induced differentiation of K562 erythroleukemia cells.27 Those findings indicated that the two proteins interacted with each other, which is consistent with the results of our study. Shi et al28 reported that fine particulate matter (PM2.5) induced an inflammatory response and lung toxicity in rats. To investigate the potential mechanism, they performed genome-wide DNA methylation and RNA transcript analysis using human bronchial epithelial cells (BEAS-2B). Their validation of the gene expression changes for several proteins, including PSMA3, indicated the functional participation of these proteins in biological processes critical to the pathogenesis of respiratory diseases. However, the role of PSMA3 in airway inflammation remains unclear. Further studies need to be performed to confirm whether PSMA1 and PSMA3 are good new candidate markers for the pathogenesis of COPD or as targets in the treatment of this disease.
We also found that several biological processes were associated with COPD. The TGF-β signaling pathway, which regulates a wide variety of cellular processes, such as pro-inflammation, inhibition of effects on immune cells, and induction of fibrosis29,30 was enriched, suggesting that this signaling pathway plays a vital role in COPD, fibrosis, and cancer. Previous study results have suggested that the levels of TGF-β signaling via SMAD are increased in macrophages collected from a mouse model of COPD.31 TGF-β pathway activation is also involved in the progression of COPD by regulating fibroblasts in lung tissue and small airway remodeling.32,33 Our findings were consistent with the previous study results.
In the present study, proteomic methods were used to find and verify DEPs in the serum of patients with COPD. However, this study had some limitations. Because we combined several samples within each participant group and analyzed the combined samples, we were unable to conduct association analyses between the DEPs and each participant’s phenotypes. All data analyzed in this study were obtained from serum samples; further experimental evidence of mechanisms underlying COPD development or occurrence associated with the observed protein level changes is needed using cell lines and animal models. In this study, the subjects were all male and mainly patients with mild COPD. Multicenter studies with larger sample sizes, all genders, and all stages are needed.
Conclusions
A DIA quantitative proteomics study with PRM confirmation of results was conducted to compare proteomic changes in the serum of patients with COPD. We identified DEPs associated with the occurrence and development of COPD. Our bioinformatics analysis results indicated that the DEPs were associated with the inflammatory process, infection, and immune response in COPD. The DEPs identified in this study may serve as potential diagnostic and prognostic markers for COPD phenotypes in the future and provide a basis for generating optimized treatment strategies.
Acknowledgments
The authors would like to thank all the patients who participated in this research. We are grateful for the DIA proteomic experiment MoloSight Technology Co., Ltd (Suzhou, China).
Funding
This study was funded by grants from the National Natural Science Foundation of China (81970043, 82270038), National Key Research and Development Program of China (2022YFF0710803, 2022YFF0710800), CAMS Innovation Fund for Medical Sciences (CIFMS) (No. 2021-I2M-1–049). The funder had no role in the design of the study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Christenson SA, Smith BM, Bafadhel M, Putcha N. Chronic obstructive pulmonary disease. Lancet. 2022;399(10342):2227–2242. doi:10.1016/S0140-6736(22)00470-6
2. Staples KJ. Breaching the defenses? Mucosal-associated invariant T cells, smoking, and chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2023;68(1):9–10. doi:10.1165/rcmb.2022-0393ED
3. Stolz D, Mkorombindo T, Schumann DM, et al. Towards the elimination of chronic obstructive pulmonary disease: a Lancet Commission. Lancet. 2022;400(10356):921–972. doi:10.1016/S0140-6736(22)01273-9
4. Li HY, Gao TY, Fang W, et al. Global, regional and national burden of chronic obstructive pulmonary disease over a 30-year period: estimates from the 1990 to 2019 Global Burden of Disease Study. Respirology. 2023;28(1):29–36. doi:10.1111/resp.14349
5. Koba T, Takeda Y, Narumi R, et al. Proteomics of serum extracellular vesicles identifies a novel COPD biomarker, fibulin-3 from elastic fibres. ERJ Open Res. 2021;7(1):00658–2020. doi:10.1183/23120541.00658-2020
6. Wahn V, von Bernuth H. IgG subclass deficiencies in children: facts and fiction. Pediatr Allergy Immunol. 2017;28(6):521–524. doi:10.1111/pai.12757
7. Ba Ta T, Tran Viet T, Xuan Nguyen K, et al. Changes in serum immunoglobulin G subclasses during the treatment of patients with chronic obstructive pulmonary disease with infectious exacerbations. Adv Respir Med. 2022;90(6):500–510. doi:10.3390/arm90060056
8. Jeraiby MA. Molecular basis of immunoglobulin heavy constant G4 gene (IGHG4)-related low serum IgG4 subclasses in Down syndrome. Saudi Med J. 2021;42(9):975–980.
9. Wen H, Guo D, Zhao Z, et al. Novel pyroptosis-associated genes signature for predicting the prognosis of sarcoma and validation. Biosci Rep. 2022;42(12):57.
10. Feng Y, Li M, Yangzhong X, et al. Pyroptosis in inflammation-related respiratory disease. J Physiol Biochem. 2022;78(4):721–737. doi:10.1007/s13105-022-00909-1
11. Zhu Z, Lian X, Su X, Wu W, Zeng Y, Chen X. Exosomes derived from adipose-derived stem cells alleviate cigarette smoke-induced lung inflammation and injury by inhibiting alveolar macrophages pyroptosis. Respir Res. 2022;23(1):5. doi:10.1186/s12931-022-01926-w
12. Wang X, Tanino Y, Sato S, et al. Secretoglobin 3A2 attenuates lipopolysaccharide-induced inflammation through inhibition of ERK and JNK pathways in bronchial epithelial cells. Inflammation. 2015;38(2):828–834. doi:10.1007/s10753-014-9992-0
13. Su Y, Han W, Kovacs-Kasa A, Verin AD, Kovacs L. HDAC6 activates ERK in airway and pulmonary vascular remodeling of chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2021;65(6):603–614. doi:10.1165/rcmb.2020-0520OC
14. Xu X, Li J, Zhang Y, Zhang L. Arachidonic acid 15-lipoxygenase: effects of its expression, metabolites, and genetic and epigenetic variations on airway inflammation. Allergy Asthma Immunol Res. 2021;13(5):684–696. doi:10.4168/aair.2021.13.5.684
15. Shin NR, Kim SH, Ko JW, et al. HemoHIM, a herbal preparation, alleviates airway inflammation caused by cigarette smoke and lipopolysaccharide. Lab Anim Res. 2017;33(1):40–47. doi:10.5625/lar.2017.33.1.40
16. Wells JM, O’Reilly PJ, Szul T, et al. An aberrant leukotriene A4 hydrolase-proline-glycine-proline pathway in the pathogenesis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;190(1):51–61. doi:10.1164/rccm.201401-0145OC
17. Paige M, Wang K, Burdick M, et al. Role of leukotriene A4 hydrolase aminopeptidase in the pathogenesis of emphysema. J Immunol. 2014;192(11):5059–5068. doi:10.4049/jimmunol.1400452
18. Röhn TA, Numao S, Otto H, Loesche C, Thoma G. Drug discovery strategies for novel leukotriene A4 hydrolase inhibitors. Expert Opin Drug Discov. 2021;16(12):1483–1495. doi:10.1080/17460441.2021.1948998
19. Wei L, Xu D, Qian Y, et al. Comprehensive analysis of gene-expression profile in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2015;10:1103–1109. doi:10.2147/COPD.S68570
20. Chen X, Lu X, Chen J, et al. Association of nsv823469 copy number loss with decreased risk of chronic obstructive pulmonary disease and pulmonary function in Chinese. Sci Rep. 2017;7:40060. doi:10.1038/srep40060
21. Płusa T. Azithromycin in the treatment of patients with exacerbation of chronic obstructive pulmonary disease. Pol Merkur Lekarski. 2020;48(283):65–68.
22. Li T, Gao L, Ma HX, et al. Clinical value of IL-13 and ECP in the serum and sputum of eosinophilic AECOPD patients. Exp Biol Med (Maywood). 2020;245(14):1290–1298. doi:10.1177/1535370220931765
23. Fens N, de Nijs SB, Peters S, et al. Exhaled air molecular profiling in relation to inflammatory subtype and activity in COPD. Eur Respir J. 2011;38(6):1301–1309. doi:10.1183/09031936.00032911
24. Grootendorst DC, Gauw SA, Verhoosel RM, et al. Reduction in sputum neutrophil and eosinophil numbers by the PDE4 inhibitor roflumilast in patients with COPD. Thorax. 2007;62(12):1081–1087. doi:10.1136/thx.2006.075937
25. Atta H, Alzahaby N, Hamdy NM, Emam SH, Sonousi A, Ziko L. New trends in synthetic drugs and natural products targeting 20S proteasomes in cancers. Bioorg Chem. 2023;133:106427. doi:10.1016/j.bioorg.2023.106427
26. Bi J, Liang W, Wang Y, Tian W, Cao S, Liu P. Long noncoding RNA PSMA3 antisense RNA 1 promotes cell proliferation, migration, and invasion in pancreatic ductal adenocarcinoma via targeting microRNA-154-5p to positively modulate karyopherin subunit alpha 4. Pancreas. 2022;51(8):1037–1046. doi:10.1097/MPA.0000000000002136
27. Mittenberg AG, Moiseeva TN, Kuzyk VO, Barlev NA. Regulation of endoribonuclease activity of alpha-type proteasome subunits in proerythroleukemia K562 upon hemin-induced differentiation. Protein J. 2016;35(1):17–23. doi:10.1007/s10930-015-9642-x
28. Shi Y, Zhao T, Yang X, et al. PM(2.5)-induced alteration of DNA methylation and RNA-transcription are associated with inflammatory response and lung injury. Sci Total Environ. 2019;650(Pt 1):908–921. doi:10.1016/j.scitotenv.2018.09.085
29. Morikawa M, Derynck R, Miyazono K. TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol. 2016;8(5):a021873. doi:10.1101/cshperspect.a021873
30. Peng D, Fu M, Wang M, Wei Y, Wei X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol Cancer. 2022;21(1):104. doi:10.1186/s12943-022-01569-x
31. He S, Xie L, Lu J, Sun S. Characteristics and potential role of M2 macrophages in COPD. Int J Chron Obstruct Pulmon Dis. 2017;12:3029–3039. doi:10.2147/COPD.S147144
32. Ong J, Faiz A, Timens W, et al. Marked TGF-β-regulated miRNA expression changes in both COPD and control lung fibroblasts. Sci Rep. 2019;9(1):18214. doi:10.1038/s41598-019-54728-4
33. Liu G, Philp AM, Corte T, et al. Therapeutic targets in lung tissue remodelling and fibrosis. Pharmacol Ther. 2021;225:107839. doi:10.1016/j.pharmthera.2021.107839
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Plasma Proteomics Study Between the Frequent Exacerbation and Infrequent Exacerbation Phenotypes of Chronic Obstructive Pulmonary Disease
Yang C, Yang L, Yang L, Li S, Ye L, Ye J, Chen C, Zeng Y, Zhu M, Lin X, Peng Q, Wang Y, Jin M
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:1713-1728
Published Date: 9 August 2023