Back to Journals » Infection and Drug Resistance » Volume 16
Seroprevalence and Molecular Characterization of Brucella abortus from the Himalayan Marmot in Qinghai, China
Authors Xue H, Li J, Ma L, Yang X, Ren L, Zhao Z, Wang J, Zhao Y, Zhao Z, Zhang X, Liu Z, Li Z
Received 18 September 2023
Accepted for publication 13 December 2023
Published 20 December 2023 Volume 2023:16 Pages 7721—7734
DOI https://doi.org/10.2147/IDR.S436950
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sandip Patil
Hongmei Xue,1 Jiquan Li,1 Li Ma,1 Xuxin Yang,1 Lingling Ren,1 Zhijun Zhao,1 Jianling Wang,1 Yuanbo Zhao,1 Zhongzhi Zhao,1 Xuefei Zhang,1 Zhiguo Liu,2 Zhenjun Li2
1Department of Brucellosis Prevention and Control, Qinghai Institute for Endemic Disease Prevention and Control, Xining, Qinghai, People’s Republic of China; 2National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, People’s Republic of China
Correspondence: Zhiguo Liu; Zhenjun Li, Email [email protected]; [email protected]
Objective: Brucellosis is a serious public health issue in Qinghai (QH), China. Surveying the seroprevalence and isolation of B. abortus strains from marmots is key to understanding the role of wildlife in the maintenance and spread of brucellosis.
Methods: In this study, a set of methods, including a serology survey, bacteriology, antibiotic susceptibility, molecular genotyping (MLST and MLVA), and genome sequencing, were employed to characterize the two B. abortus strains.
Results: The seroprevalence of brucellosis in marmots was 7.0% (80/1146) by serum tube agglutination test (SAT); one Brucella strain was recovered from these positive samples, and another Brucella strain from a human. Two strains were identified as B. abortus bv. 1 and were susceptible to all eight drugs examined. The distribution patterns of the accessory genes, virulence associated genes, and resistance genes of the two strains were consistent, and there was excellent collinearity between the two strains on chromosome I, but they had significant SVs in chromosome II, including inversions and translocations. MLST genotyping identified two B. abortus strains as ST2, and MLVA-16 analysis showed that the two strains clustered with strains from northern China. WGS-SNP phylogenetic analysis showed that the strains were genetically homogeneous with strains from the northern region, implying that strains from a common lineage were spread continuously in different regions and hosts.
Conclusion: Seroprevalence and molecular clues demonstrated frequent direct or indirect contact between sheep/goats, cattle, and marmots, implying that wildlife plays a vital role in the maintenance and spread of B. abortus in the Qinghai–Tibet Plateau.
Keywords: Brucella abortus, marmots, isolated, MLST, MLVA, WGS-SNP, QH
Introduction
Brucellosis is a zoonotic disease caused by Brucella spp., a gram-negative facultative intracellular coccobacillus.1 These microorganisms can infect livestock, wildlife, and humans, causing significant public concern and substantial agricultural economic loss. Currently, at least six novel Brucella species have been identified: Brucella pinnipedialis, Brucella ceti,2 Brucella papionis,3 Brucella microti,4 Brucella inopinata,5 and Brucella vulpis.6 However, Brucella melitensis, Brucella abortus, and Brucella suis remain the most important members of the genus because they are the strains largely responsible for human and animal diseases.7,8 Although brucellosis is generally considered an occupational disease in developed countries, in many undeveloped regions of the world, including the Middle East, Africa, Latin America, Central Asia, and several regions of the Mediterranean, it is endemic.9 In China, brucellosis has a high prevalence in humans and animals, and its geographic distribution is expanding annually, with a trend from northern to southern China.10,11 B. melitensis is responsible for most human brucellosis cases, whereas fewer cases are caused by B. abortus12 and B. suis.13
The Qinghai–Tibet Plateau region of China is a historic endemic region for human and animal brucellosis.14 Yak brucellosis is a serious condition in this region, with a pooled prevalence rate of 8.39%;15 along these lines, more than five B. abortus strains have been isolated from yaks in this area.16,17 Meanwhile, B. melitensis–related sequences were found in the guano of bats in India,18 and Brucella DNA sequences were detected in the spleens of two different species of bats from Georgia that were co-infected with Bartonella and Leptospira.19 In Africa, B. abortus, B. melitensis, B. inopinata, and B. suis were reported in wildlife; the pooled seroprevalence was 13.7% in buffalo, 7.1% in carnivores, and 2.1% in antelope.20 The elk in the Paradise Valley is a maintenance host for B. abortus and transmits the bacteria to cattle in the northern portion of the Greater Yellowstone Ecosystem.21 In Europe, more spillover events from wildlife to livestock, and possibly from wildlife to humans, are likely, particularly from the unrecognized wildlife reservoirs of Brucella spp.22 In France, an investigation showed that a B. melitensis strain spills from wildlife to domestic ruminants and the sustainability of the infection in Alpine ibex.23 Thus, wildlife reservoirs raise major concerns regarding brucellosis due to wild animals carrying or shedding Brucella strains, triggering a potential risk associated with transmission, persistence, and control in this population and domestic animals.24
Whole genome sequencing provides the most comprehensive collection of an individual’s genetic variation.25 A deeper comparative genetic analysis demonstrated important differences between the Ochrobactrum strains and different Brucella isolates.26 Genome sequencing and comparative genomics of strains from wildlife can characterize the role of wildlife in the maintenance and spread of brucellosis and the genetic diversity of B. abortus strains.27,28 Therefore, the purpose of this study was to characterize the seroprevalence and genetic profiles of two B. abortus strains isolated from marmots and humans in Qinghai, China.
Materials and Methods
B. abortus Strain Isolation and Phenotyping Identification
A total of 1446 serum samples of marmot were screened using the Rose Bengal plate test (RBPT) to select anti-Brucella serum-positive samples for further submission to bacteriological approaches. Brucella strains were isolated, and phenotypical characterization was performed in a biosafety level 3 laboratory (BSL-3), adhering to standard bacteriology and typing procedures.29,30 Briefly, liver, and spleen tissue samples collected from marmots that were serologically positive for brucellosis were minced, homogenized, and placed on selective agar medium plates for Brucella spp. analysis (Oxoid Ltd., Basingstoke, Hampshire, UK) in a rigorous sterile manner. Approximately 5–10 mL of fresh blood from 69 suspected patients infected with Brucella spp. was injected into a biphasic culture flask (BioMerieux, Marcy-l’Étoile, France) in a biosafety cabinet. These plates (or flasks) were then incubated at 37°C for at least 2 weeks with and without CO2. Suspicious Brucella spp. clones were sub-cultivated on a Brucella medium plate and further subjected to conventional bio-typing procedures.30 Gram and Kirschner staining were used to characterize the morphology of the strains. Biotyping reagents were purchased from the China Veterinary Drug Control Institute. Three vaccine strains, 104M (B. abortus), M5 (B. melitensis), and S2 (B. suis), were used as reference strains. All reagents were used in accordance with the manufacturer’s instructions and within the expiration date.
In vivo Antibiotics Susceptivity Analysis of Two Strains in This Study
In vivo, antibiotic susceptibility spectra of strains to nine first-line brucellosis treatment antibiotics were obtained based on the Clinical and Laboratory Standards Institute (CLSI) guidelines.31 Minimal Inhibitory Concentration (MIC) values of rifampin (RIF), ciprofloxacin (CIP), levofloxacin (LVX), tetracycline (TCY), streptomycin (STR), doxycycline (DOX), minocycline (MON), sparfloxacin (SPX), ceftriaxone sodium (CRO), and sulfamethoxazole (SMZ) to two B. abortus strains were determined using the E-test strip method according to a previous study,32 as well as testing procedure and results interpretation following the previous study,32 and B. abortus 2308 was used as a reference strain.
DNA Extraction and Molecular Identification of Two Strains
Subsequently, the genomic DNA of the two strains was extracted using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions, and the quality of the prepared DNA was detected using a fluorometer (Qubit ® 2.0; Thermo Fisher Scientific, Waltham, MA, USA). The strains were further subjected to BCSP-31 PCR33 and AMOS-PCR34 for genus and species determination, respectively. The MLST genotypes were deduced from the WGS data using PubMLST platform resources (https://pubmlst.org/organisms/brucella-spp).35 The MLVA-16 genotyping assay was conducted as previously described.36 MLVA genotyping characterized the value of 111 strains previously identified within China (Table S1), which were collected for genetic comparison using BioNumerics version 8.0 software (Applied Maths NV, Sint-Martens-Latem, Belgium) with the unweighted pair group method using the arithmetic average.
Library Construction, Sequencing, Assembly, and Annotation
The genome sequencing strategy of the strains was referenced as previously described.37 Briefly, sequencing libraries were constructed using the NEBNext Ultra DNA Library Prep Kit for Illumina (NEB, Ipswich, MA, USA) according to the manufacturer’s instructions. The whole genome of each strain was sequenced using the NovaSeq PE150 platform (Illumina, San Diego, CA, USA). Clean data were assembled using the SOAP Denovo 2.0 program,38 and the assembly results were integrated using CISA software.39 The GapCloser program40 was used to fill the gaps in the preliminary assembly results. Next, genome component prediction and gene function annotation were completed as previously reported,41,42 the GeneMarkS program,43 Tandem Repeats Finder,44 tRNAscan-SE version 2.0,45 RNAmmer version 1.2, IslandPath-DIOMB version 0.2,46 phiSpy version 2.3,47 CRISPRdigger version 1.0,48 and transposon PSI transposon traits program (https://transposonpsi.sourceforge.net/). In silico detection of AMR genes and virulence-associated determinants was performed using different databases, including the Virulence Factors of Pathogenic Bacteria Database (VFDB)49 (accessed on September 12, 2019), and antimicrobial resistance genes were assessed by searching the CARD50 and Resfinder version 4.0 databases.51
Comparison of Genome and SNP Phylogenetic Analyses
A total of 76 B. abortus strains (two strains from the present study and 74 from GenBank) (Table S2) were used for comparative genomic analysis. Genomic alignment between the 74 sample genomes and the B. abortus 2308 reference genome (NC_007618.1 and NC_007624.1) was performed using the MUMmer,52 and LASTZ53 tools and genomic synteny were analyzed based on the alignment results. The gene family assignment was analyzed using the Cluster R package, and pan-genes (including core, specific, and dispensable genes) were analyzed using the CD-HIT rapid clustering of similar proteins software54 (with a threshold of 50% pairwise identity and 0.7 length difference cutoff in amino acids). A Venn diagram and heatmap were then drawn to show the relationships among the samples. A phylogenetic tree was constructed using PhyML,55 and bootstraps were set to 1000 with orthologous genes.
Results
Seroprevalence, Species/biovar Identified, and Drug Susceptibility of B. abortus Strains
The seroprevalence of brucellosis in marmots was 7.0% (80/1146) according to the SAT. XHM5 was obtained from these positive samples; XHM22 was isolated from one screened patient with brucellosis out of 69 from passive surveillance in outpatients who had a definite history of contact with animals and livestock and exhibited classical clinical manifestations, including fever, muscle, and joint pain, fatigue, and sweat. Colonies of suspicious strains appeared colorless, transparent, round, and slightly uplifted with smooth and homogeneous surfaces. Based on the staining results, gram-negative bacteria were observed (Figure 1A), and the strains were stained red using Kirschner staining (Figure 1B). According to the bio-typing tests, the two strains were identified as B. abortus bv.1 (Table 1). Special 223-bp and 498-bp bands were observed for BCSP-31 PCR and AMOS-PCR, respectively (Figure S1). The E-test results showed that the two strains were susceptible to all nine examined antibiotics, and no resistance phenotypes were detected (Table S3).
 |
Table 1 Bio-Tying Profile of the Two B. abortus Strains in This Study |
 |
Figure 1 Gram staining (A) and Koch staining (B) features of the strains in the present study. |
Genome Component and Comparison Genome Analysis of B. abortus Strains
The sequencing profiles of both strains are shown in Table S4. The genome size (bp), gene number (#), and GC% values of the two strains (XHM5 and XHM22) were 3,256,796 and 3,256,846 bp, 3258 and 3258, and 57.24 and 57.23, respectively. The gene components of the two strains were similar, including scattered repeat sequences, tandem repeats, ncRNAs, gene islands, and prophages (Table 2). The prophage number and total length were different between the strains; the number and length values were 2 and 73,903 bp, respectively, in XHM5 versus 5 and 203,525 bp, respectively, in XHM22 (Table 2). The SNP numbers for XHM5 and XHM22 were 1387, and 1408, respectively (Table 2). Compared with B. abortus 2308, multiple genome SVs were observed, including inversions, translocations, and indels, especially on chromosome II (Figure 2 and Table 2). Based on core pan-genome analysis, there were 1658 core genes among them (Figure S2), and the number of special genes was three in XHM5 and four in XHM5 (Figure S2). Meanwhile, the patterns and numbers of accessory genes and gene families of the two strains were consistent (Figure 3, marked in orange; Figure S3, marked in bold).
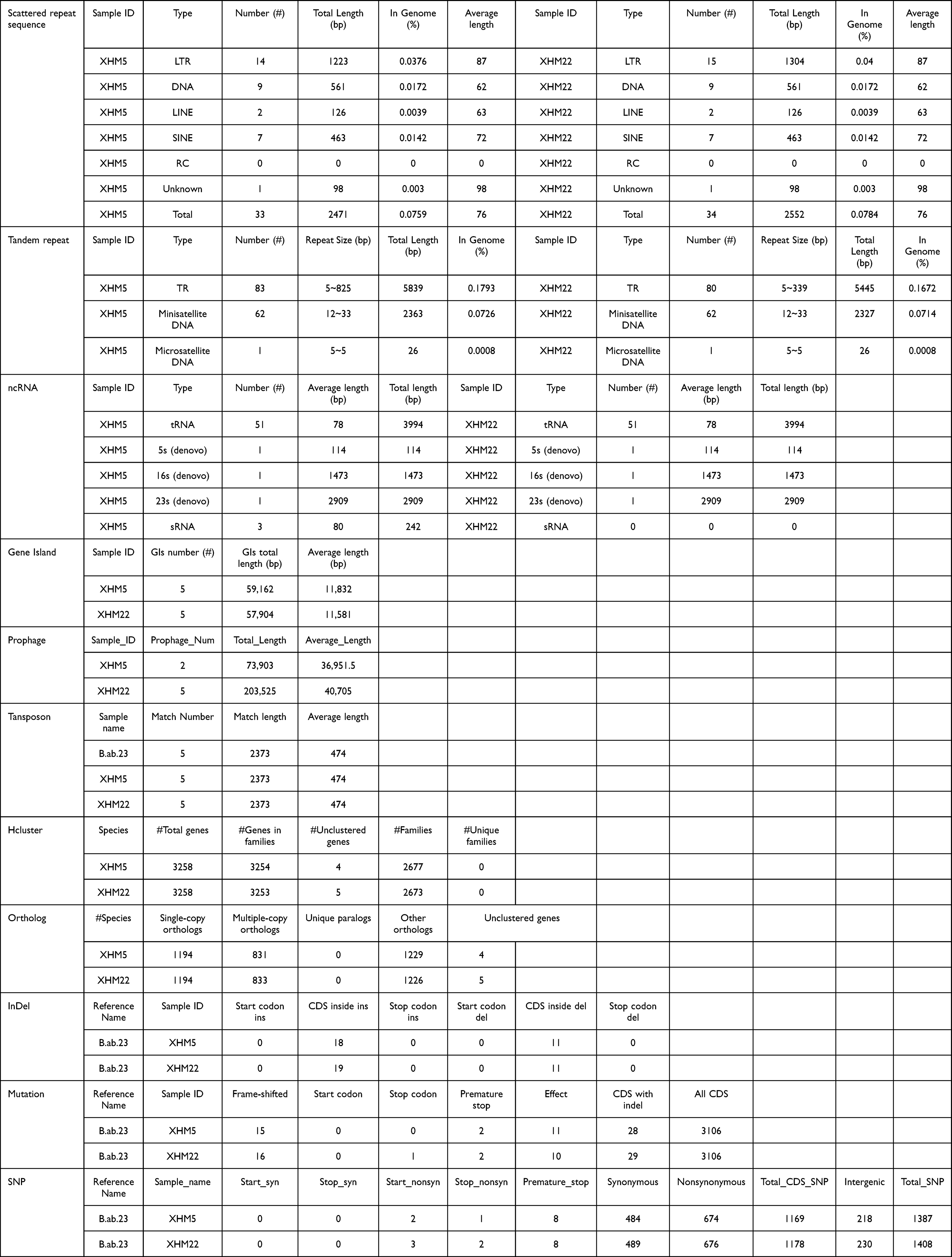 |
Table 2 Gene Components of Two Strain in This Study |
 |
Figure 2 Collinearity profiles of the genomes of the two strains in this study. |
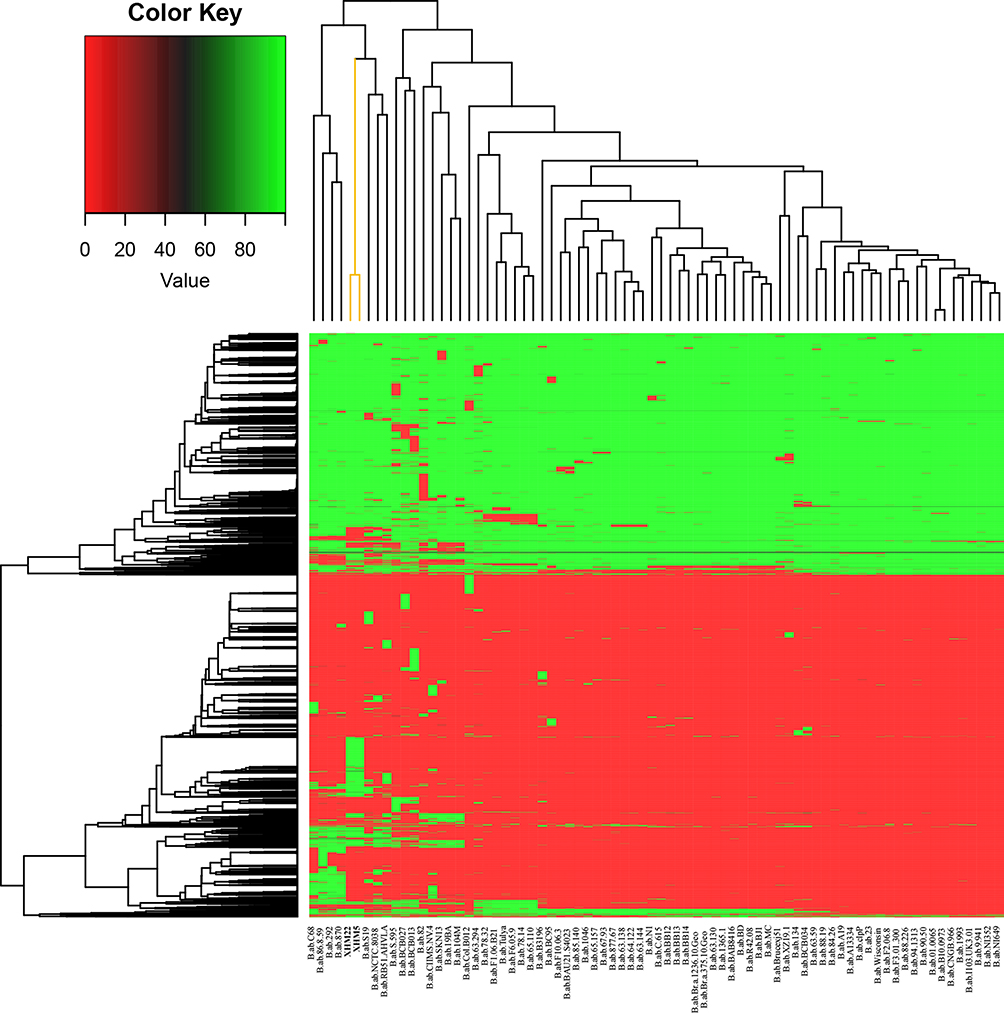 |
Figure 3 Cluster analysis of accessory genes in B. abortus from this study and GenBank. Note: Strains from this study are marked in yellow in the upper-right part of the image. |
Characteristics of Virulence-Associated Genes and Resistance Genes
The distribution patterns of virulence associated and resistance genes of the two strains were consistent. A total of 66 virulence-associated genes were observed, but bmaA and btaE were not observed (Figure S4). Moreover, the distribution profile of virulence-associated genes in the strains was identical to that of the majority of the compared strains from GenBank, but different from that of B.ab.clpP, B.ab.RB51, AHVLA, B.ab.NI352, B.ab.NI649, and B. ab. MC (Figure S4). In addition, Brucella_suis_mprF resistance genes were detected in both strains in the present study (Figure S5); the resistance gene profiles of the two studied strains were similar to those of the other strains, except for the B. ab. clpP strain. A resistance gene, APH (3’)-la, was found in the B. ab. clpP strain, but not in the other strains (Figure S5).
MLST and MLVA-16 Genotyping Profiles of B. abortus Strains
MLST (9) genotyping showed two strains belonging to ST2; ST is the same as strains isolated previously in QH (Ma10 and 64) (Figure 4). In MLVA-16, two new MLVA-8 and MLVA-11 genotypes were observed, and further comparison analysis of 111 strains (Table S1) from China found no completely matched MLVA-16 genotypes. However, the two strains were clustered in the same cluster and subdivided into two subclusters (SC I and II) (Figure 2). Furthermore, XHM5 and XHM22 were genetically related to strains from Inner Mongolia (GCA_001043295), Xinjiang (2013jiang#084, 86–90), and Hebei Province (2013jiang#085) (Figure 2).
 |
Figure 4 MVLA-16 dendrogram of 111 strains from this study. Note: The two strains from this study are marked in bold in the figure. |
Phylogenetic Analysis of Two B. abortus Strains from Qinghai on a Global Scale
Phylogenetic analysis showed that 76 strains were divided into eight clades (A–H) (Figure 5), and strains from China formed an independent clade and were placed in clade C; furthermore, two strains could be sorted into two sub-clades, namely C I (XHM5) and C II (XHM22). XHM5 (C I) was isolated from marmot and clustered with strains from Heilongjiang Province, while XHM22 (C II) was grouped with strains from Inner Mongolia, Hebei, Beijing, and Gansu Province (Figure 5). In addition, a close genetic relationship was observed between the strains from the present study and those from Russia.
 |
Figure 5 WGS-SNP phylogenetic analysis of B. abortus strains. Note: Strains from this study are marked in pink. |
Discussion
In the present study, a preliminary serology survey and molecular characterization of B. abortus strains from marmots and humans were conducted to provide novel insights into the epidemiology of brucellosis in wildlife. A high seroprevalence of brucellosis was detected in the marmots’ population, which demonstrates that marmots frequently come in direct or indirect contact with sheep/goats, cattle, or wildlife such as yaks. Marmots were contacted with abortion products, and their secretions of livestock (sheep/goats) and their consumption of aborted fetuses are potential reasons. In addition, animal husbandry is the main source of income for the local economy in Qinghai, and farmers and herders also frequently skin and eat marmots,56 creating opportunities for Brucella to jump hosts. B. abortus strains were isolated from sheep and yaks in northwest China,16 and B. abortus biovar 4 (XZ19-1) was first isolated from yaks in Tibet, China.57 B. abortus was also isolated from Himalayan marmots in the Xinjiang Uygur Autonomous Region and had similar MLVA-16 genotypes with strains from yaks and humans in Qinghai Province.58 These data verify that the ongoing distribution of B. abortus strains occurs in northern China. These areas have not only the same nomadic culture but also the same adjacency and shared grassland, thus providing a potential opportunity for the genetic homogeneity of strains. The most reported Brucella species in wildlife was B. abortus, with the highest prevalence rate recorded in the American bison, Bison bison (39.9%), followed by the Alpine ibex, Capra ibex (33%).59 The persistence of brucellosis in wildlife reservoirs poses a risk to the reintroduction of Brucella into domestic livestock and humans. The present study highlights that B. abortus infection events cannot be ignored, and strengthening wildlife surveillance and supervision is needed.
The distribution patterns of the AMR and virulence genes in the two B. abortus strains were consistent with the absence of classical AMR genes and virulence-associated factors. Similarly, a study showed that there was no clear difference in the distribution of AMR and virulence genes among both resistant and sensitive B. abortus and B. melitensis strains, even for those recovered from different hosts.60 The most identified pathogenicity-associated genes are involved in LPS production and type IV secretion systems, which play vital roles in cell entry, intracellular trafficking, and intracellular survival.61–63 These virulence-associated factors contribute to the pathogenesis of infections in humans and domestic animals, further triggering clinical manifestations. In addition, Brucella_suis_mprF resistance genes were detected in two strains, which potentially increased the virulence and resistance of methicillin-resistant Staphylococcus aureus (MRSA) and other pathogens to cationic host defense peptides and antibiotics.64 Similarly, a B. abortus strain isolated from marmots exhibited amikacin resistance and obtained amikacin resistance genes from Salmonella spp. through Tn3 family transposons.65 Although resistance genes are not always consistent with the strains’ displayed resistance, they exist and spread among strains, further posing a challenge to human brucellosis treatment. Further investigations should focus on phenotypic resistance mechanisms at the proteomic and transcriptomic levels. Prophages can contribute many biological properties to their bacterial hosts, such as virulence, biosynthesis, toxin secretion, genomic divergence, and evolution.66 There are differences in the prophage number and length between the two strains; that is, the number of prophages in strains from marmots is less than that in strains from humans, and they have significant genome SVs on chromosome II, which may reflect the different evolution patterns among strains. Further comparative analysis is necessary.
Both MLVA and WGS-SNP analyses highlighted that the two strains were genetically similar to strains from northern China, implying that the strains from a common lineage were spread continuously in different regions and hosts. Human activity has created a channel between livestock and wildlife for the spread and transmission of diseases. The control of human brucellosis mainly depends on the surveillance and control of the disease in domestic animals and livestock, and greater attention to farmers and butcher-related occupational populations is needed. Control measures are necessary to block the transmission of Brucella spp. among domestic and wild animals, and continuous and targeted molecular monitoring of wildlife is important to curb brucellosis in this region.
Conclusions
In the present study, survey data showed that there was frequent direct or indirect contact between sheep and goats, cattle, and marmots through livestock production activities, which represents a new challenge in controlling brucellosis in humans and livestock. Therefore, strengthening pathogen genome surveillance and collecting more strains from different hosts, as well as conducting more detailed epidemiological investigations and etiological analysis, will be helpful in determining the risk factors and infectious chains of brucellosis in Qinghai.
Abbreviations
WGS-SNP, Whole-genome sequencing single-nucleotide polymorphism; MLVA, Multilocus variable-number tandem repeat analysis.
Data Sharing Statement
The datasets used and/or analyzed in the current study are available in the manuscript and Supplementary Files. Moreover, genome sequence data have been deposited in the NCBI database (PRJNA930323 and PRJNA930316).
Ethics Approval and Consent to Participate
This study was conducted in accordance with the principles of the Declaration of Helsinki. The study protocol was approved by the Ethics Committee of the Institute for Endemic Disease Control and Prevention, Qinghai, China (No. 2022006). Informed consent was obtained from all patients, and Brucella spp. was isolated as a diagnostic approach for brucellosis. The animal sample collection plan was also checked and approved by the Ethics Committee of the Institute for Endemic Disease Control and Prevention, Qinghai (No. 2022006).
Consent for Publication
All authors have approved and agreed to the publication of this manuscript.
Funding
This study was supported by Key Research and Development, and Transformation Plan Project in Qinghai Province (2023-QY-202), Qinghai Province 2020 “Kunlun Elite High-end Innovative and Entrepreneurial Talents” Project, and Key Research and Development Project (No. 2021YFC2301001; Project name: Prevention and control of emerging infectious diseases, Institute No. 20072002). The funders had no role in the study design, data collection and analysis, decision to publish, or manuscript preparation.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
References
1. Corbel MJ. Brucellosis: an overview. Emerg Infect Dis. 1997;3(2):213–221. doi:10.3201/eid0302.970219
2. Foster G, Osterman BS, Godfroid J, Jacques I, Cloeckaert A. Brucella ceti sp. nov. and Brucella pinnipedialis sp. nov. for Brucella strains with cetaceans and seals as their preferred hosts. Int J Syst Evol Microbiol. 2007;57(Pt 11):2688–2693. doi:10.1099/ijs.0.65269-0
3. Whatmore AM, Davison N, Cloeckaert A, et al. Brucella papionis sp. nov., isolated from baboons (Papio spp.). Int J Syst Evol Microbiol. 2014;64(Pt 12):4120–4128. doi:10.1099/ijs.0.065482-0
4. Scholz HC, Hubalek Z, Sedlácek I, et al. Brucella microti sp. nov., isolated from the common vole Microtus arvalis. Int J Syst Evol Microbiol. 2008;58(Pt 2):375–382. doi:10.1099/ijs.0.65356-0
5. Scholz HC, Nöckler K, Göllner C, et al. Brucella inopinata sp. nov., isolated from a breast implant infection. Int J Syst Evol Microbiol. 2010;60(Pt 4):801–808. doi:10.1099/ijs.0.011148-0
6. Scholz HC, Revilla-Fernández S, Dahouk SA, et al. Brucella vulpis sp. nov., isolated from mandibular lymph nodes of red foxes (Vulpes vulpes). Int J Syst Evol Microbiol. 2016;66(5):2090–2098. doi:10.1099/ijsem.0.000998
7. Pisarenko SV, Kovalev DA, Volynkina AS, et al. Global evolution and phylogeography of Brucella melitensis strains. BMC Genomics. 2018;19(1):353. doi:10.1186/s12864-018-4762-2
8. Garofolo G, Di Giannatale E, Platone I, et al. Origins and global context of Brucella abortus in Italy. BMC Microbiol. 2017;17(1):28. doi:10.1186/s12866-017-0939-0
9. Pappas G, Papadimitriou P, Akritidis N, Christou L, Tsianos EV. The new global map of human brucellosis. Lancet Infect Dis. 2006;6(2):91–99. doi:10.1016/s1473-3099(06)70382-6
10. Zhu X, Zhao Z, Ma S, et al. Brucella melitensis, a latent “travel bacterium”, continual spread and expansion from Northern to Southern China and its relationship to worldwide lineages. Emerg Microbes Infect. 2020;9(1):1618–1627. doi:10.1080/22221751.2020.1788995
11. Ran X, Chen X, Wang M, et al. Brucellosis seroprevalence in ovine and caprine flocks in China during 2000–2018: a systematic review and meta-analysis. BMC Vet Res. 2018;14(1):393. doi:10.1186/s12917-018-1715-6
12. Wang H, Xu WM, Zhu KJ, et al. Molecular investigation of infection sources and transmission chains of brucellosis in Zhejiang, China. Emerg Microbes Infect. 2020;9(1):889–899. doi:10.1080/22221751.2020.1754137
13. Liu ZG, Wang LJ, Piao DR, et al. Molecular investigation of the transmission pattern of Brucella suis 3 From Inner Mongolia, China. Front Vet Sci. 2018;5:271. doi:10.3389/fvets.2018.00271
14. Deqiu S, Donglou X, Jiming Y. Epidemiology and control of brucellosis in China. Vet Microbiol. 2002;90(1–4):165–182. doi:10.1016/s0378-1135(02)00252-3
15. Zhao B, Gong QL, Feng HF, et al. Brucellosis prevalence in yaks in China in 1980–2019: a systematic review and meta-analysis. Prev Vet Med. 2022;198:105532. doi:10.1016/j.prevetmed.2021.105532
16. Cao X, Li Z, Liu Z, et al. Molecular epidemiological characterization of Brucella isolates from sheep and yaks in northwest China. Transbound Emerg Dis. 2018;65(2):e425–e433. doi:10.1111/tbed.12777
17. Ma JY, Wang H, Zhang XF, et al. MLVA and MLST typing of Brucella from Qinghai, China. Infect Dis Poverty. 2016;5:26. doi:10.1186/s40249-016-0123-z
18. Banskar S, Bhute SS, Suryavanshi MV, Punekar S, Shouche YS. Microbiome analysis reveals the abundance of bacterial pathogens in Rousettus leschenaultii guano. Sci Rep. 2016;6(1):36948. doi:10.1038/srep36948
19. Bai Y, Urushadze L, Osikowicz L, et al. Molecular survey of bacterial zoonotic agents in bats from the Country of Georgia (Caucasus). PLoS One. 2017;12(1):e0171175. doi:10.1371/journal.pone.0171175
20. Simpson G, Thompson PN, Saegerman C, et al. Brucellosis in wildlife in Africa: a systematic review and meta-analysis. Sci Rep. 2021;11(1):5960. doi:10.1038/s41598-021-85441-w
21. O’Brien MP, Beja-Pereira A, Anderson N, et al. Brucellosis transmission between wildlife and livestock in the greater Yellowstone ecosystem: inferences from DNA genotyping. J Wildl Dis. 2017;53(2):339–343. doi:10.7589/2015-12-329
22. Yon L, Duff JP, Ågren EO, et al. Recent Changes In Infectious Diseases In European Wildlife. J Wildl Dis. 2019;55(1):3–43. doi:10.7589/2017-07-172
23. Mick V, Le Carrou G, Corde Y, Game Y, Jay M, Garin-Bastuji B. Brucella melitensis in France: persistence in wildlife and probable spillover from Alpine ibex to domestic animals. PLoS One. 2014;9(4):e94168. doi:10.1371/journal.pone.0094168
24. González-Espinoza G, Arce-Gorvel V, Mémet S, Gorvel JP. Brucella: reservoirs and niches in animals and humans. Pathogens. 2021;10(2):186. doi:10.3390/pathogens10020186
25. Ng PC, Kirkness EF. Whole genome sequencing. Gene Variat. 2010;628:215–226. doi:10.1007/978-1-60327-367-1_12
26. Holzer K, Hoelzle LE, Wareth G. Genetic comparison of Brucella spp. and Ochrobactrum spp. erroneously included into the genus Brucella confirms separate genera. Ger J Vet Res. 2023;3(1):31–37. doi:10.51585/gjvr.2023.1.0050
27. Suárez-Esquivel M, Chaves-Olarte E, Moreno E, Guzmán-Verri C. Brucella genomics: macro and micro evolution. Int J Mol Sci. 2020;21(20):7749. doi:10.3390/ijms21207749
28. Rajendhran J. Genomic insights into Brucella. Infect Genet Evol. 2021;87:104635. doi:10.1016/j.meegid.2020.104635
29. Yagupsky P, Morata P, Colmenero JD. Laboratory diagnosis of human brucellosis. Clin Microbiol Rev. 2019;33(1). doi:10.1128/CMR.00073-19
30. Alton GG, Jones LM, Angus RD, Verger JM. Techniques for the Brucellosis Laboratory. INRA Publications; 1988.
31. Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing.
32. Liu ZG, Di DD, Wang M, et al. In vitro antimicrobial susceptibility testing of human Brucella melitensis isolates from Ulanqab of Inner Mongolia, China. BMC Infect Dis. 2018;18(1):43. doi:10.1186/s12879-018-2947-6
33. Matar GM, Khneisser IA, Abdelnoor AM. Rapid laboratory confirmation of human brucellosis by PCR analysis of a target sequence on the 31-kilodalton Brucella antigen DNA. J Clin Microbiol. 1996;34(2):477–478. doi:10.1128/jcm.34.2.477-478.1996
34. Yuan HT, Wang CL, Liu LN, et al. Epidemiologically characteristics of human brucellosis and antimicrobial susceptibility pattern of Brucella melitensis in hinggan league of the inner mongolia autonomous Region, China. Infect Dis Poverty. 2020;9(1):79. doi:10.1186/s40249-020-00697-0
35. Jolley KA, Bray JE, Maiden MCJ. Open-access bacterial population genomics: bIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018;3:124. doi:10.12688/wellcomeopenres.14826.1
36. Liu ZG, Di DD, Wang M, et al. MLVA genotyping characteristics of human Brucella melitensis isolated from Ulanqab of Inner Mongolia, China. Front Microbiol. 2017;8:6. doi:10.3389/fmicb.2017.00006
37. Xue H, Zhao Z, Wang J, et al. Native circulating Brucella melitensis lineages causing a brucellosis epidemic in Qinghai, China. Front Microbiol. 2023;14:1233686. doi:10.3389/fmicb.2023.1233686
38. Li R, Yu C, Li Y, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966–1967. doi:10.1093/bioinformatics/btp336
39. Lin SH, Liao YC, Watson M. CISA: contig integrator for sequence assembly of bacterial genomes. PLoS One. 2013;8(3):e60843. doi:10.1371/journal.pone.0060843
40. Xu M, Guo L, Gu S, et al. TGS-GapCloser: a fast and accurate gap closer for large genomes with low coverage of error-prone long reads. Gigascience. 2020;9(9):9. doi:10.1093/gigascience/giaa094
41. Liu ZG, Cao XA, Wang M, et al. Whole-Genome Sequencing of a Brucella melitensis Strain (BMWS93) isolated from a bank clerk and exhibiting complete resistance to rifampin. Microbiol Resour Announc. 2019;8(33). doi:10.1128/MRA.01645-18
42. Brambila-Tapia AJ, Armenta-Medina D, Rivera-Gomez N, Perez-Rueda E, Cloeckaert A. Main functions and taxonomic distribution of virulence genes in Brucella melitensis 16 M. PLoS One. 2014;9(6):e100349. doi:10.1371/journal.pone.0100349
43. Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29(12):2607–2618. doi:10.1093/nar/29.12.2607
44. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–580. doi:10.1093/nar/27.2.573
45. Chan PP, Lin BY, Mak AJ, Lowe TM. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021;49(16):9077–9096. doi:10.1093/nar/gkab688
46. Hsiao W, Wan I, Jones SJ, Brinkman FS. IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics. 2003;19(3):418–420. doi:10.1093/bioinformatics/btg004
47. Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39(suppl):W347–52. doi:10.1093/nar/gkr485
48. Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–7. doi:10.1093/nar/gkm360
49. Chen L, Xiong Z, Sun L, Yang J, Jin Q. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 2012;40:D641–5. doi:10.1093/nar/gkr989
50. Alcock BP, Raphenya AR, Lau TTY, et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020;48(D1):D517–D525. doi:10.1093/nar/gkz935
51. Bortolaia V, Kaas RS, Ruppe E, et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J Antimicrob Chemother. 2020;75(12):3491–3500. doi:10.1093/jac/dkaa345
52. Marçais G, Delcher AL, Phillippy AM, Coston R, Salzberg SL, Zimin A. MUMmer4: a fast and versatile genome alignment system. PLoS Comput Biol. 2018;14(1):e1005944. doi:10.1371/journal.pcbi.1005944
53. Harris RS. Improved Pairwise Alignment of Genomic DNA. The Pennsylvania State University; 2007.
54. Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–1659. doi:10.1093/bioinformatics/btl158
55. Criscuolo A. morePhyML: improving the phylogenetic tree space exploration with PhyML 3. Mol Phylogenet Evol. 2011;61(3):944–948. doi:10.1016/j.ympev.2011.08.029
56. Xi J, Duan R, He Z, et al. First case report of human plague caused by excavation, skinning, and eating of a hibernating marmot (Marmota himalayana). Front Public Health. 2022;10:910872. doi:10.3389/fpubh.2022.910872
57. Sun M, Liu M, Zhang X, et al. First identification of a Brucella abortus biovar 4 strain from yak in Tibet, China. Vet Microbiol. 2020;247:108751. doi:10.1016/j.vetmic.2020.108751
58. Yan B, Zhu Q, Xu J, et al. Brucella in Himalayan Marmots (Marmota himalayana). J Wildl Dis. 2020;56(3):730–732. doi:10.7589/2019-09-237
59. Dadar M, Shahali Y, Fakhri Y, Godfroid J. The global epidemiology of Brucella infections in terrestrial wildlife: a meta-analysis. Transbound Emerg Dis. 2021;68(2):715–729. doi:10.1111/tbed.13735
60. Dadar M, Alamian S, Brangsch H, et al. Determination of virulence-associated genes and antimicrobial resistance profiles in Brucella isolates recovered from humans and animals in Iran using NGS technology. Pathogens. 2023;12(1):82. doi:10.3390/pathogens12010082
61. Roop RM, Barton IS, Hopersberger D, Martin DW. Uncovering the hidden credentials of Brucella virulence. Microbiol Mol Biol Rev. 2021;85:1. doi:10.1128/MMBR.00021-19
62. Xiong X, Li B, Zhou Z, et al. The VirB system plays a crucial role in Brucella intracellular infection. Int J Mol Sci. 2021;22(24):13637. doi:10.3390/ijms222413637
63. Döhmer PH, Valguarnera E, Czibener C, Ugalde JE. Identification of a type IV secretion substrate of Brucella abortus that participates in the early stages of intracellular survival. Cell Microbiol. 2014;16(3):396–410. doi:10.1111/cmi.12224
64. Slavetinsky CJ, Hauser JN, Gekeler C, et al. Sensitizing Staphylococcus aureus to antibacterial agents by decoding and blocking the lipid flippase MprF. Elife. 2022:11. doi:10.7554/eLife.66376
65. Yang X, Wang Y, Li J, et al. Genetic characteristics of an amikacin-resistant Brucella abortus strain first isolated from Marmota himalayana. Microb Pathog. 2022;164:105402. doi:10.1016/j.micpath.2022.105402
66. Varani AM, Monteiro-Vitorello CB, Nakaya HI, Van Sluys MA. The role of prophage in plant-pathogenic bacteria. Annu Rev Phytopathol. 2013;51(1):429–451. doi:10.1146/annurev-phyto-081211-173010
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.