Back to Journals » International Journal of Nanomedicine » Volume 17
Self-Assembled Micellar Glutaminase Allosteric Inhibitor for Effective Therapeutic Intervention
Authors Fang J ![]() , Chen Z
, Chen Z ![]() , Li J, Li D, Wang W, Ruan BH
, Li J, Li D, Wang W, Ruan BH ![]()
Received 3 November 2021
Accepted for publication 31 December 2021
Published 13 January 2022 Volume 2022:17 Pages 213—225
DOI https://doi.org/10.2147/IJN.S346596
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Farooq A. Shiekh
Jinzhang Fang,* Zhao Chen,* Jinxiu Li, Di Li, Wenxi Wang, Benfang Helen Ruan
IDD & CB, College of Pharmaceutical Science & Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, Zhejiang University of Technology, Hangzhou, 310014, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Benfang Helen Ruan; Wenxi Wang Email [email protected]; [email protected]
Introduction: Kidney-type glutaminase (KGA) has been an important anti-tumor drug target, and KGA allosteric inhibitors attracted much interest for their superior enzymatic specificity with good drug safety profiles. For glutaminase allosteric inhibitors such as BPTES, CB-839 and Selen derivatives, the low solubility remains as the main factor that limits in vivo efficacy. The 1,3,4-Selenadiazole compound CPD 23 showed improved in vivo efficacy but worse solubility; however, the graft polymer polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol (PVCap-PVA-PEG), Soluplus® (SOL) stood out as an excellent delivery carrier for CPD 23.
Methods: The CPD 23@SOL micelles were prepared, optimized and evaluated through on the basis of solubility improvement and loading capacity. Characterizations of particle size and Zeta potential by dynamic light scattering, morphology by transmission electron microscopy and solid state by X-ray powder diffraction were closely conducted. The biological studies included the tumor cell growth inhibition, blood and liver microsomal stability, in vivo pharmacokinetics and tissue biodistribution.
Results: At 1:20 ratio of CPD 23:SOL, CPD 23@SOL micelles were well-dispersed, spherical and stable, with size less than 200 nm with encapsulation efficiency of more than 90%. This SOL micellar system significantly increased the aqueous solubility of CPD 23 by 15,000 folds. Particularly, CPD 23@SOL micelles demonstrated higher stability in blood and liver microsomes, showing approximately 86% remaining at 2 h incubation and about 66% at 4 h, respectively. In addition, with or without micellar formulation, CPD 23 maintained essentially the same inhibitory activity in tumor cells. Interestingly, CPD 23@SOL micelles significantly improved the pharmacokinetic exposure, prolonged the in vivo circulation and dramatically changed tissue biodistributions of CPD 23.
Conclusion: The current work provided an encouraging and practical delivery system for novel Selenadiazoles and glutaminase allosteric inhibitors whose poor water-soluble characteristic has been a bottleneck for the field.
Keywords: glutaminase inhibitor, Selenadiazole compound, Soluplus®, polymeric micelles, stability, pharmacokinetics
Graphical Abstract:

Introduction
The kidney-type glutaminase (KGA or Glutaminase C, GAC) showed high distribution in kidney and brain and upregulation in tumors and was recognized as an important drug target for cancer therapeutics.1,2 Novel KGA inhibitors have been developed.3–5 For example, L-6-Diazo-5-oxonorleucine (DON) showed good in vivo efficacy but with high toxicity; a pro-drug of DON, HJU-083, was designed to overcome the toxicity.6,7 In addition, KGA allosteric inhibitors such as bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES) and CB-839 (Telaglenastat) featured a thiadiazole moiety have superior enzyme specificity and good drug safety profiles, but very low solubilization.8,9 The novel 1,3,4-Selenadiazole analogue (CPD 23, Table S1) has shown significantly enhanced in vivo anticancer activity but lower solubility.10 Water solubility has become the bottleneck for the further development of these potent KGA allosteric inhibitors.
For decades, researchers have taken pains to improve the solubility from perspectives of medicinal chemistry and drug formulation.11,12 Polymeric nanoparticles have been designed for BPTES and verified to show improvement in solubility, cellular uptake and tumor damage.13 A gold-polymer-CB-839 conjugate was produced as the delivery system and has increased the therapeutic efficacy of CB-839 in glioblastoma stem cells.14 In our previous work, the Hexylselen also showed great in vivo efficacy but poor solubility.15,16 However, by forming self-assembly micelles using the widely used polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol (PVCap-PVA-PEG, 13%-57%-30%, respectively) graft copolymer, named Soluplus® or SOL, the Hexylselen@SOL micelles have achieved significantly improved solubility, biological stability, in vivo bioavailability and antitumor efficacy.17 This indicated that the micelles are suitable as the delivery system for selenium-containing KGA allosteric inhibitors in practice.
Soluplus® can form nano-micelles at the critical micellar concentration (CMC) of 7.6 μg/mL with low toxicity and strong amphiphilic properties.18 As for the SOL polymer, hydrophobic moieties can interact with drugs with similar poor water-solubility to form hydrophobic cores for precipitation prevention, while the hydrophilic moieties extend to aqueous phase, consequently producing the stable micelles with hydrophobic drugs inside. Due to the high drug-loading capacity and surfactant properties,19 SOL is usually introduced to address solubilizing and anti-diluting problems for active pharmaceutical ingredients (APIs) especially in the Biopharmaceutical Classification System (BCS) group II.20,21 With multifunctional and superior characteristics, Soluplus® can be formulated into kinds of delivery systems such as nano-emulsion,22 nano-micelles,23,24 solid dispersion,25,26 transparent film27 and thermosensitive hydrogel28,29 to increase the solubility, bioavailability and efficacy of hydrophobic APIs. Recent reports have also demonstrated that delivery composites formed by SOL with materials such as Pluronic®,30 Solutol HS15,31 N-vinylpyrrolidone and vinyl acetate (PVPVA),32 D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS1000),33 and so on, have generated many formulations that have less toxicity, higher bioavailability and improved cancer site-targeting for antitumor drugs.
1,3,4-Selenadiazole CPD 23 has the structural feature of long hydrophobic chain and pyridazin derivative moiety, which contributes to high Calculated LogP of 4.58 and low water solubility at about 0.31 μg/mL (Table S1). As reported, excipients with moderate hydrophobic moiety can be promoted because their molecular interactions would be likely to produce considerable solubility.34 In this way, we tested amphiphilic polymers as delivery systems for CPD 23.
Here, we developed the optimized CPD 23@SOL micelles on the basis of solubility improvement and loading capacity. The optimized micelles were characterized to obtain particle size, Zeta potential, morphological identification and solid state. The improved biological activity was evaluated by the tumor cell growth inhibition, blood and mouse liver microsomal stability, pharmacokinetic study including in vivo exposure, blood circulation and tissue biodistributions. Importantly, the CPD 23@SOL micelles displayed 15,000-fold increase in solubility and markedly enhanced pharmacokinetic profiles with prolonged in vivo circulation throughout the organism.
Materials and Experimental Methods
Materials and Chemical Reagents
The synthesized 1,3,4-Selenadiazole compound (CPD 23) powder has been previously reported.10 Soluplus® was a kind gift from BASF Ltd. (Ludwigshafen, Germany). Human non-small cell lung cancer (A549) and mouse hepatoma (H22) cell lines were purchased from Shanghai Institute of Biochemistry and Cell Biology of Chinese Academy of Sciences (Shanghai, China). RPMI1640 medium was purchased from M&C Gene Tech Inc. (Beijing, China) and fetal bovine serum (FBS) from Zhejiang Tianhang Biotech Inc. (Deqing, China). EZMTT detection reagents were obtained from JNF Bioscience Inc. (Hangzhou, China). ICR/CD1 mouse liver microsome was purchased from the Research Institute for Liver Diseases Inc. (Shanghai, China). Glucose-6-phosphate (G6P) and glucose-6-phosphate dehydrogenase (G6PDH) were purchased from Plant Cell Biotechnology Inc. (Beijing, China). NADPNa2 was provided from Shanghai Yuanye Biotech Inc. (Shanghai, China). Coumarin was purchased from Meryer Chemical Technology Inc. (Shanghai, China). ICR mice (SPF) were purchased from Zhejiang Institute of Medical Science (Hangzhou, China), and treated in compliance with ethical standards. All animal experimental procedures were strictly conducted under the institutional guidelines for the care and use of laboratory animals in Zhejiang University of Technology, Hangzhou, China, and conformed to the National Institutes of Health Guide for Care and Use of Laboratory Animals (Publication No. 85–23, revised 1996). Zhejiang University of Technology approved the experiments. Chromatographic grade methanol was obtained from Tedia Inc. (USA). Phosphate buffered saline (PBS) was purchased from Solarbio Inc. (Beijing, China). Purified water (Millipore, Bedford, MA, USA) was used throughout the study and all other reagents were of analytical grade.
Preparation and Formulation of CPD23@SOL Micelles
The thin film dispersion method was used to prepare CPD 23@SOL micelles according to the previous report.17 Briefly, CPD 23 and Soluplus® were first completely solubilized in mixture of dichloromethane and ethanol (1:4, v/v) by ultrasonication, then evaporated to remove the organic solvents under a vacuum of −0.1 MPa at 40–42 °C until dry, and the obtained thin layer was washed and hydrated with sterile saline (0.9% NaCl solution) to form CPD 23@SOL micelles. Several mass ratios of CPD 23 and Soluplus® were used for preparation, and the most stable formulation was selected by comparing the amount of prescription from micelles in 24 hours. The obtained micelles were filtered with a 0.45 μm acetate fiber membrane to remove the non-incorporated compound, which was then stored at 4 °C for the following experiments.
Particle Size and Morphology Characterizations
Using dynamic light scattering (DLS) in a Malvern system (ZEN-3600, Malvern Instruments, Worcestershire, UK), the average particle size, polydispersity index (PDI) and Zeta potential (ζ) of CPD 23@SOL micelles were determined. The morphology of CPD 23@SOL micelles and blank SOL micelles was observed by transmission electron microscopy (TEM, JEM-200CX, JEOL, Japan).
Encapsulation Efficiency and Loading Capacity
The encapsulation efficiency (EE%) was used here to mainly measure the entrapped compounds within the isolated compounds in micelles. Because of the poor aqueous solubility in saline or PBS (lower than 1 μg/mL), the CPD 23 concentration could be essentially neglected when compared with that encapsulated in micelles (more than 4 mg/mL). Here, the precipitated CPD 23 in micelles was removed by centrifugation at 3000 rpm for 10 min and followed by filtration with a 0.45 μm acetate fiber membrane. Then, 0.2 mL of the filtered micelles was disrupted by appropriate amount of methanol (HPLC grade) and syringed to a reversed phase high performance liquid chromatography (RP-HPLC) system for CPD 23 quantification.10 Because the free CPD 23 dissolved in aqueous solution can be ignored, Equations (1)-(2) were used to calculate the encapsulation efficiency (EE%) and drug loading (DL%) respectively. All samples were analyzed three times to get mean ± standard deviation (SD).
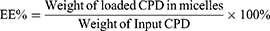
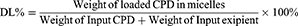
X-Ray Powder Diffraction
The CPD 23@SOL solid dispersion (SD) was obtained from of the lyophilized CPD 23@SOL micelles prepared above by thin film method and then ground to particles less than 200 mesh. The CPD 23 crystalline powder, CPD 23@SOL physical mixture and CPD 23@SOL SD were determined by X-ray powder diffraction (XRPD). On the disc of a diffractometer (D/Max-2400, Rigaku Instrument, Osaka, Japan) were the ground sample tiled and exposed to Cu-Kα radiation under 40 kV and 25 mA over the 2 theta (θ) range of 3–40 degree with a scanning speed of 0.03 degree/min. Diffractograms were collected and compared for state characterization.
Tumor Cell Growth Inhibition
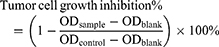
In this experiment, the H22 mouse hepatoma cells and A549 human non-small cell lung cancer cells (NSCLC) were regularly proliferated in the media of DMEM and RPMI with 10% (v/v) FBS and penicillin/streptomycin (100 U/mL for each) respectively at 37 °C in a humidified atmosphere containing 5% CO2. In a 96-well plate, 4 × 103/well of the cells were plated and treated with a series of dilutions of CPD 23 in PBS with 1% DMSO, CPD 23@SOL micelles (both CPD 23 topped at 2 mM) and blank SOL micelles for 5 days. The amount of NAD(P)H produced in viable cells was measured using the EZMTT reagents35,36 with 1 hour incubation, to collect OD values of the samples, control and blank wells at the wavelength of 450 nm. The inhibition of specimens to tumor cells was calculated by equation (3). The tests were repeated triplicate in at least two independent experiments.
Stability in Blood
Free CPD 23 was prepared in 1% DMSO, 5% ethanol, 5% Tween80, 10% PEG 400 and 3% poloxamer 188 with PBS buffer as the vehicle (it would be used in the following experiments).10 CPD 23@SOL micelles with the mass ratio CPD 23:SOL=1:20 were used. In several vials where 100 μL of fresh blood from ICR mouse with anticoagulative heparin sodium was preheated at 37 °C for about 5 min, 10 μL of CPD 23 vehicle or CPD 23@SOL micelles (both containing 1 mg/mL CPD 23) were then added, gently mixed and incubated continuously in 37 °C water bath. At the time points of 2 min, 30 min, 1 h and 2 h, the mixture in each vial was terminated with DMSO and extracted to get the organic layer. The collected DMSO extractions were lyophilized, then redissolved in methanol and applied to HPLC analysis for quantification. The percentages of CPD 23 retained in blood represented the blood stability of a compound, the tests were performed in triplicate.
The Partition in Plasma
As the blood stability was performed, another 10 μL CPD 23 vehicle or CPD 23@SOL micelles containing 1 mg/mL CPD 23 was mixed with 200 μL blood and incubated at 37 °C till the preset time points 0.033 h, 0.5 h, 1 h and 2 h. Each vial was gently centrifuged at 3000 rpm for 10 min to separate the plasma and rest cell pellets (mainly composed of red cells), both of which were terminated with DMSO. The collected DMSO extractions were then lyophilized and redissolved in methanol to quantify the partitioned amounts of CPD 23. For these samples, respective percentage of CPD 23 in plasma and red cell pellets were calculated and all experiments were performed in triplicate.
The Liver Microsomal Stability
According to the assay for the mouse liver microsome stability study,37 the CPD 23 vehicle and CPD 23@SOL micelles used in blood stability were used and compared, taking free coumarin as the control. Briefly, 50 μM CPD 23 (in vehicle, micelles or coumarin) was preincubated with mouse liver microsomes (0.5 mg/mL) in PBS (100 μL, pH 7.4) at 37 °C for 5 min. The reaction was then initiated by the addition of an NADPH-generating system (the solution containing 3 mM NADPH, 7.2 mM MgCl2, 6 mM G6P and 3 unit of G6PDH). After incubated at 37 °C in water bath for 0 min, 5 min, 30 min, 1 h, 2 h and 4 h, the resulting mixtures were terminated, extracted with DMSO and stored at −20 °C until analysis. The samples were stored at −20 °C and HPLC was further used to quantify the remaining amount of CPD 23 in liver microsomes. Half-life (t1/2) and clearance (CL) were calculated using Equations (4) and (5), respectively.
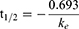
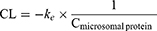
In the equations, ke is the slope of the relationship of lnC (input concentration of compound) and t (incubation time), and Cmicrosomal protein is the input concentration of microsomal protein. Experiments were performed in triplicate to obtain the mean and SD.
In vivo Pharmacokinetic Study
Healthy male ICR mice (6−8 weeks) were administered a single dose of 20 mg/kg CPD 23 vehicle or CPD 23@SOL micelles via intravenous injection route. At post-injected time points of 0.033, 0.5, 1, 2, 4, 12 and 24 h, three mice per time interval were euthanized, the blood was collected by cardiac puncture, and the organs including brain, heart, liver, spleen, lung and kidney were removed, washed in ice-cold physiological saline, sopped up with tissue, then weighted, and homogenized by a probe homogenizer (Dragon, BioSpec Products, China). Plasma was separated from the blood by centrifugation at 3000 rpm for 10 min, stored at −20 °C until the DMSO treatment and analysis by HPLC, in which the collected DMSO extractions were lyophilized, then redissolved in methanol and finally applied to HPLC to quantify CPD 23. For homogenized tissues, dichloromethane extraction and HPLC quantification were suited. After extracting the tissue homogenates at least three times, the mixture was centrifuged at 8000 rpm for 10 min to separate CH2Cl2 layer which was then collected, dried under nitrogen flow and redissolved in methanol. The obtained sample solutions were applied to HPLC assay for CPD 23 quantification. The amount of CPD 23 in plasma and organs at different time points was collected to draw the Area Under the Curves (AUC), and calculate PK parameters using DAS 2.0 (version 6.0) software.
High Performance Liquid Chromatography Assay
To quantify CPD 23 in in vitro and in vivo experiments, the RP-HPLC assay was performed on Welchrom S60 system equipped with an XB-C18 analytical column (250 mm × 4.6 mm, 5 µm; Welchrom).10 Under a detection wavelength of 254 nm, samples were injected (20 μL) using a mobile phase of methanol and water in a gradient elution program where methanol started from 65% for 2 min, increased to 100% for 12 min, and then maintained at 100% for 6 min at the flow rate of 1.0 mL/min. Quantification was based on external standard method, through the detected peak areas and comparison with the standard curve (A = 32619C + 13.292, R2 = 0.9999, A: peak area of CPD 23; C: CPD 23 concentration, in Methanol) of the corresponding pure CPD 23 (ranges of 0.004–0.25 mg/mL).
Data Analysis
All experiments were conducted triplicate and all data were presented as the mean with standard deviation. Statistical analysis of significance was performed by SPSS 19.0 statistical software (SPSS Inc., USA). The difference between two groups were evaluated using two-tailed Student’s t-test, and values of P < 0.05 (* or #), P < 0.01 (** or ##) and P < 0.01 (*** or ###) were considered as statistical significance, extreme significance and super significance respectively.
Results
Preparation of CPD 23@SOL Micelles
Using the thin film dispersion method, 1,3,4-Selenadiazole analogue (CPD 23) and Soluplus® (Figure 1A) were first completely dissolved in organic reagents, then evaporated to remove the solvent to produce the thin dried film and at last it was hydrated with sterilized physiological saline (scheme illustration in Figure 1B). Compared to water, the obtained CPD 23@SOL micelles were yellow brown opalescent liquid while the blank SOL micelles appeared with light blue transparent opalescence. In this preparation, the reddish brown CPD 23, featured with a high calculated LogP (CLogP) of 4.58 from the official tool of ALOGPS2.1 and low aqueous solubility, about 0.31 μg/mL (Table S1), was well encapsulated by Soluplus® to automatically assemble as micelles, indicating that the strong hydrophobic core of SOL showed high compatibility with CPD 23. Thus, SOL demonstrated greatly improved solubility of CPD 23.
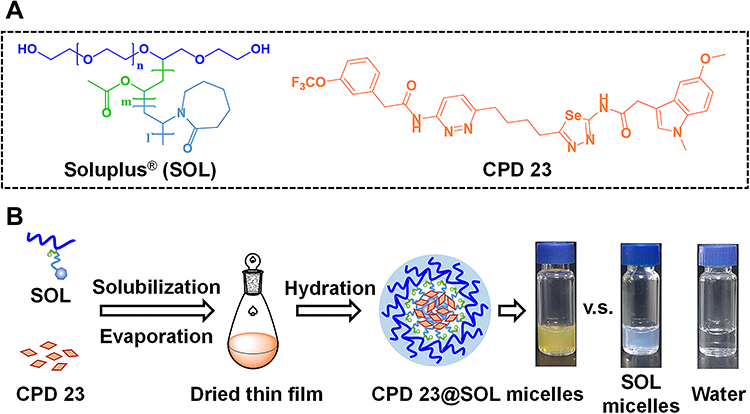 |
Figure 1 The structures of Soluplus® and CPD 23, and scheme illustration of CPD 23@micelles preparation. Notes: (A) the monomeric unit of Soluplus® and chemical structure of CPD 23; (B) scheme illustration of CPD 23@micelles preparation, and images of CPD 23@SOL micelles, blank SOL micelles and deionized water. |
In the following, CPD 23@SOL micelles with several mass ratios of CPD 23 and SOL were formulated and investigated. When the mass ratio of 1:10 (CPD 23:SOL) was used, the CPD 23@SOL micelles showed slight visible precipitation in yellow brown opalescent liquid with the encapsulating efficiency (EE) of 88.7 ± 2.8% (Table 1). However, little precipitation emerged in CPD 23@SOL micelles and the EE rose to over 93% when the ratio was adjusted to 1:20 and 1:30, with the decreased drug loading (DL) due to the increased amount of SOL. Coincidently, when incubated at 37 °C for 24 h, more than 90% CPD 23 was stable in CPD 23@SOL micelles at the CPD 23:SOL ratio of 1:20 while only about 47% retained in the micelles at the ratio of 1:10 (Figure S1). Thus, in the preparation, CPD 23@SOL micelles were firmly formed with moderate CPD 23:SOL ratios (1:20 and 1:30, for instance), otherwise less SOL would essentially result in more compound leakage and unstable micelles.
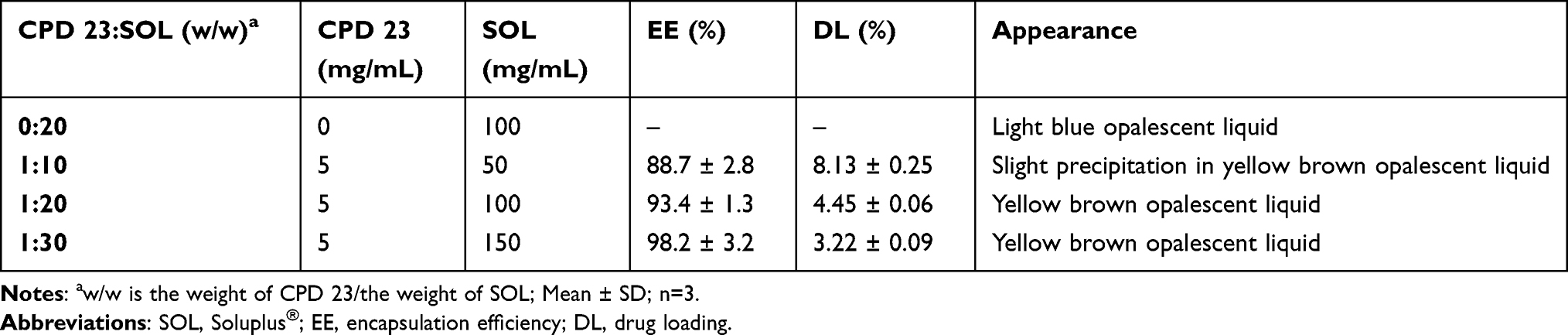 |
Table 1 The Encapsulation Capacity and Appearance of the Formulated CPD 23@SOL Micelles |
Accordingly, CPD 23 was significantly solubilized to achieve more than 4.5 mg/mL in aqueous solution, essentially due to the strong molecular interaction in hydrophobic core and good compatibility of SOL for CPD 23. To understand more about CPD 23@SOL micelles, the prepared micelles needed more characterizations and biological tests.
Particle Size and Morphology Characterizations
To further investigate the formulation and characterization, the CPD 23@SOL micelles with various input ratios of CPD 23 and SOL were prepared. From the images taken under TEM in Figure 2A and B, the CPD 23@SOL micelles showed spherically round and oval shapes, similar to the blank SOL micelles. In broad and narrow microscopic fields, both CPD 23@SOL micelles and blank SOL micelles were found evenly distributed with average particle size at almost 100 nm, which was consistent to the results obtained by dynamic light scattering (DLS) instrumentation (Figure 2C and D). With the input of 1–3 mg/mL CPD 23 at the 1:20 ratio of CPD 23:SOL, the average particle sizes of CPD 23@SOL micelles were less than 200 nm with a PDI of about 0.2 (Table 2). When the ratio was adjusted to 1:30 with 5 mg/mL CPD 23, the particle size grew to 200–300 nm, and PDI to over 0.5.
 |
Table 2 The Average Particle Sizes, PDI and Zeta Potentials of the Formulated CPD 23@SOL Micelles |
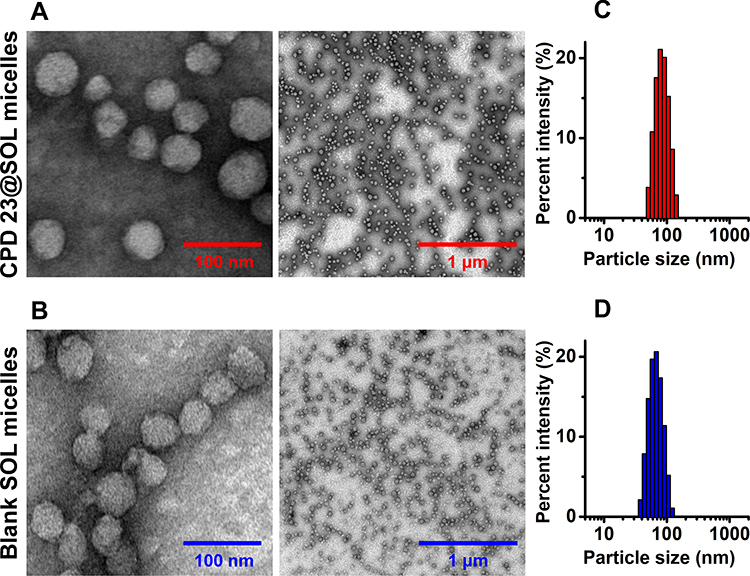 |
Figure 2 The particle size characterizations of CPD 23@SOL micelles and blank SOL micelles. Notes: The TEM images of CPD 23@SOL micelles (A) and blank SOL micelles (B) (bar scales: 100 nm and 1 μm); the particle size distribution of CPD 23@SOL micelles (C) and blank SOL micelles (D) by DLS method. |
From the results, the CPD 23:SOL ratio of 1:20 was an optimal ratio to modulate the particle size, because the excess amount of SOL might lead to self-aggregation of CPD 23@SOL micelles. Additionally, the Zeta potential floated from approximate −0.2 mV to 0.5 mV, indicating that both blank SOL micelles and CPD 23@SOL micelles were essentially free of charge (Table 2). The TEM and DLS results demonstrated that self-assembled CPD 23@SOL micelles had excellent micellar shapes and moderate particle sizes at liquid state, meanwhile the solid state was of interest to be characterized.
X-Ray Powder Diffraction
The prepared CPD 23@SOL micelles were characterized to be stable yellow brown opalescent liquid, and spherical shapes with particulate size of around 200 nm and good dispersity index. Further, X-ray powder diffraction (XRPD) was performed to identify the powder states of the CPD 23 powder, SOL powder, CPD 23@SOL physical mixture (PM) and CPD 23@SOL solid dispersion (SD). The crystalline CPD 23 showed a spectrum with multiple peaks while the amorphous SOL powder featured with two flat peaks as shown in Figure 3. For CPD 23@SOL PM, characteristic peaks at 2θ angles of 7.6302 °, 16.1027 ° and 18.8456 ° still appeared, which were similar to those sharp peaks in CPD 23 powder, demonstrating that CPD 23 kept crystalline state when it was only physically mixed with SOL, and also when given inadequate SOL as our previous report showed.17 Conversely, these prominent peaks essentially disappeared in CPD 23@SOL (1:20) SD after the formulating preparation and the resulting flat peaks were similar to those in SOL, suggesting that the crystalline CPD 23 was converted to the amorphous form.26 Therefore, CPD 23 was verified to show amorphous state with SOL in solid dispersion.
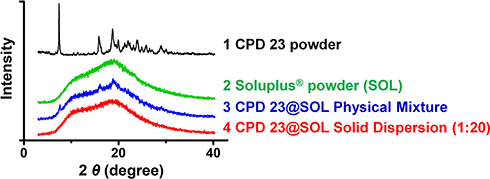 |
Figure 3 The identification of CPD 23 and CPD 23@SOL solid dispersion by XRPD. Notes: The diffractograms of CPD 23 powder (1), SOL powder (2), CPD 23@SOL physical mixture (PM) (3) and CPD 23@SOL solid dispersion (SD) (4) were produced by XRPD method. |
Tumor Cell Growth Inhibition
Apart from the chemical characterizations, the formulated CPD 23@SOL micelles in biological and pharmaceutical perspectives arouse more interest. In the following, we used the EZMTT assay to determine and compare the inhibition activities of CPD 23 (in PBS with 1% DMSO) and CPD 23@SOL micelles in H22 mouse hepatoma cells and A549 human non-small cell lung cancer cells (NSCLC).35 As shown in Figure 4 and Table S2, both CPD 23@SOL micelles and free CPD 23 achieved 60–80% growth inhibition to H22 and A549 cell lines, whereas the blank SOL micelles essentially showed no inhibition even at concentrations more than 30 μM. It demonstrated that blank SOL micelles displayed no toxicity to the tested tumor cells, neither interference to the inhibitory activity of CPD 23. The cell growth inhibition curves and IC50 values showed that the CPD 23@SOL micelles presented similar activity to that of free CPD 23 (IC50: 1.8–2.9 μM for H22 cells and 0.01–0.04 μM for A549 cells), which indicated that micelles did not reduce the potency of the CPD 23. Although the CPD 23@SOL micelles kept considerable cell growth inhibition, the biological compatibility and stability in animal blood or tissues were more important to be investigated for this CPD 23 micellar system.
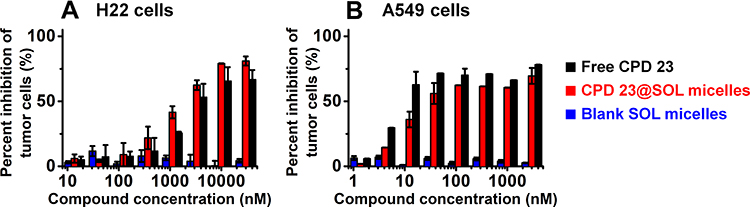 |
Figure 4 The in vitro inhibition of free CPD 23 and CPD 23@SOL micelles to (A) H22 mouse hepatoma cells and (B) A549 human non-small cell lung cancer cells by EZMTT assay. Abbreviation: EZMTT, 2-(3-(2-methoxy-4-nitrophenyl)-2-(4-nitrophenyl)-2H-tetrazol-3-ium-5-yl)benzenesulfonate sodium salt. Notes: Data were presented as mean ± SD; n=3. |
The Blood Stability and Plasma/Blood Partition
As blood plays the key role in delivering and distributing active chemicals to the organism, the stability of compounds exposed to blood is of great importance. According to the previous report, 1,3,4-Selenadizole structures showed moderate stability in blood.10 Here, the blood stability and plasma/blood cell pellets partition of CPD 23@SOL micelles were investigated with the DMSO extraction method and the RP-HPLC system for analysis and quantification. After 2 h incubation in blood, 86.2 ± 0.4% CPD 23@SOL micelles were found remaining, whereas the free CPD 23 only had 75.0 ± 1.1% CPD 23 left; the results indicated that the SOL micelles helped to keep CPD 23 more stable in blood (Figure 5A).
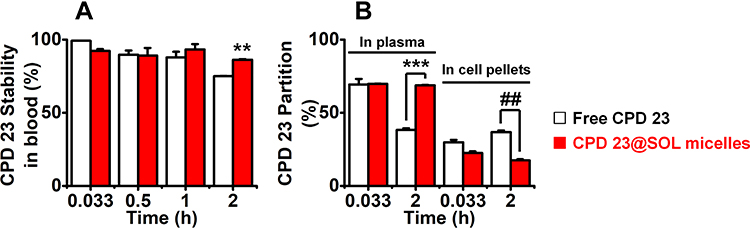 |
Figure 5 The stability of free CPD 23 and CPD 23@SOL micelles in blood and their partitions in plasma and blood cell pellets. Notes: Incubated with free CPD 23 and CPD 23@SOL micelles for 2 h, (A) the blood stability, (B) plasma partition and blood cell pellets partition of CPD 23 were determined; **P < 0.01, ***P < 0.001, CPD 23@SOL micelles significantly higher than free CPD 23; ##P < 0.01, CPD 23@SOL micelles significantly lower than free CPD 23, n=3. |
Additionally, there were some differences on the plasma and blood cell pellets partition of free CPD 23 and CPD 23@SOL micelles. In Figure 5B, about 70% free CPD 23 was found partitioned in plasma, but it almost fell by half after 2 hours incubation, while the rest 30% in blood cell pellets was slightly increased. Compared with this, CPD 23@SOL micelles stayed at about 70% distribution in plasma and essentially kept the same amount in blood cell pellets. The difference might be ascribed to the SOL micellar system, which protected the CPD 23 from exposure to blood red cells so that it maintained essentially unchanged plasma partition, but the free CPD 23 was exposed to the blood and metabolized faster. It can also be clearly seen that the partition of both free CPD 23 and CPD 23@SOL micelles in plasma was largely proportioned compared to that in blood cell pellets, indicating that the plasma concentration of CPD 23 could be used to evaluate the in vivo pharmacokinetic study. Moreover, different from the free CPD 23, the CPD 23@SOL micelles gained stronger storage and slower metabolization in plasma, which might contribute to efficient delivery to tissues or stable body circulation.
Liver Microsomal Stability
Taking coumarin as the positive control, the 4-hour mouse liver microsome experiments were carried out for CPD 23@SOL micelles. In Figure 6, after 4 hours incubation, the control coumarin level was dropped to 10.7 ± 2.2%, the level of free CPD 23 to 34.4 ± 9.5%, and interestingly CPD 23@SOL micelles 66.1 ± 7.6% of in liver microsome, demonstrating that CPD 23@SOL micelles showed significantly higher liver microsomal stability than free CPD 23. Further, 3 times longer half-life (t1/2) and over triple lower clearance of CPD 23@SOL micelles than those of free CPD 23 were obtained (Table 3). Taken together, SOL micelles showed increased stability of CPD 23 in liver microsomes, and was expected to make longer in vivo circulation and better exposure or bioavailability of CPD 23.
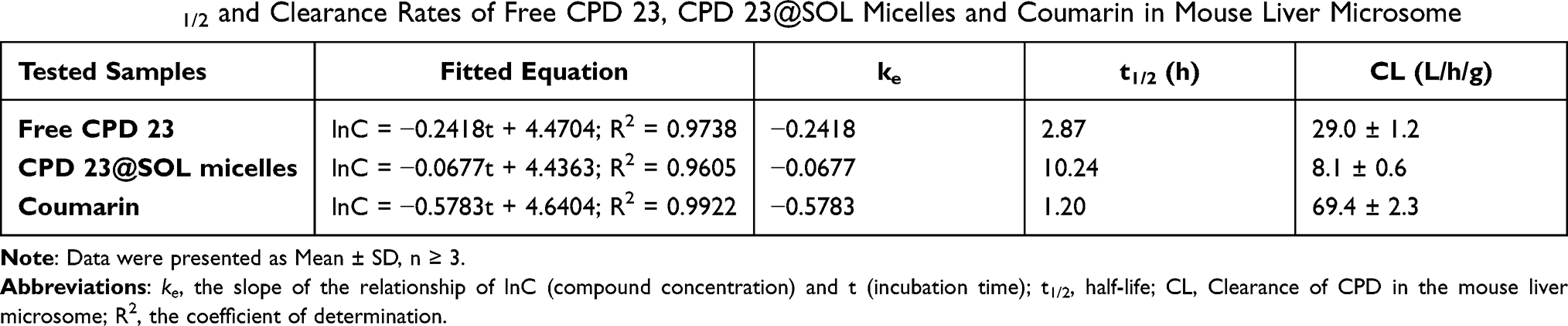 |
Table 3 The T1/2 and Clearance Rates of Free CPD 23, CPD 23@SOL Micelles and Coumarin in Mouse Liver Microsome |
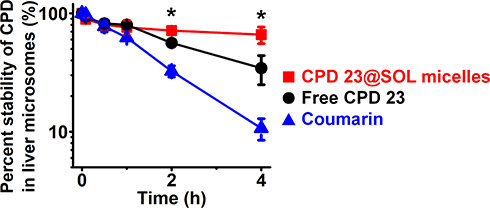 |
Figure 6 The stability of free CPD 23 and CPD 23@SOL micelles in mouse liver microsome. Notes: Coumarin was used as the positive control. *P < 0.05, CPD 23 versus CPD 23@SOL micelles; mean ± SD, n=3. |
In vivo Pharmacokinetic Study
The pharmacokinetic study was performed by intravenous (IV) administration to evaluate the CPD 23 vehicle and CPD 23@SOL micelles. AUC curves and pharmacokinetic parameters were shown in Figure 7 and Table 4. After tail vein injection with dose of 20 mg/kg (CPD 23) in 24 hours, the CPD 23 vehicle showed an AUC0-24 h of 6.6 ± 0.6 h·μg/mL, Cmax of 7.7 ± 0.3 μg/mL, and t1/2α of 1.22 ± 0.15 h, whereas CPD 23@SOL micelles displayed the increased AUC0-24 h, Cmax and t1/2α to 120.4 ± 15.3 h·μg/mL, 74.0 ± 1.2 μg/mL and 3.55 ± 0.25 h, respectively. The plasma concentration of CPD 23 vehicle decreased fast to negligible level, more swiftly than that of CPD 23@SOL micelles. Meanwhile, the CPD 23@SOL micelles exhibited over 18 times improved relative exposure in comparison with CPD 23 vehicle, whereas the clearance (CL) was significantly slowed down. Besides, the higher apparent volume of distribution (Vd) in CPD 23 vehicle suggested that it had lower plasma concentration of CPD 23 but larger distribution in tissues, while the lower Vd in CPD 23@SOL micelles indicated higher compound concentration in plasma and fewer tissue distribution.38 Accordingly, the PK results indicated that SOL not only significantly enhanced the exposure but also prolonged in vivo circulation of CPD 23, which promoted us for further investigation of the tissue biodistributions.
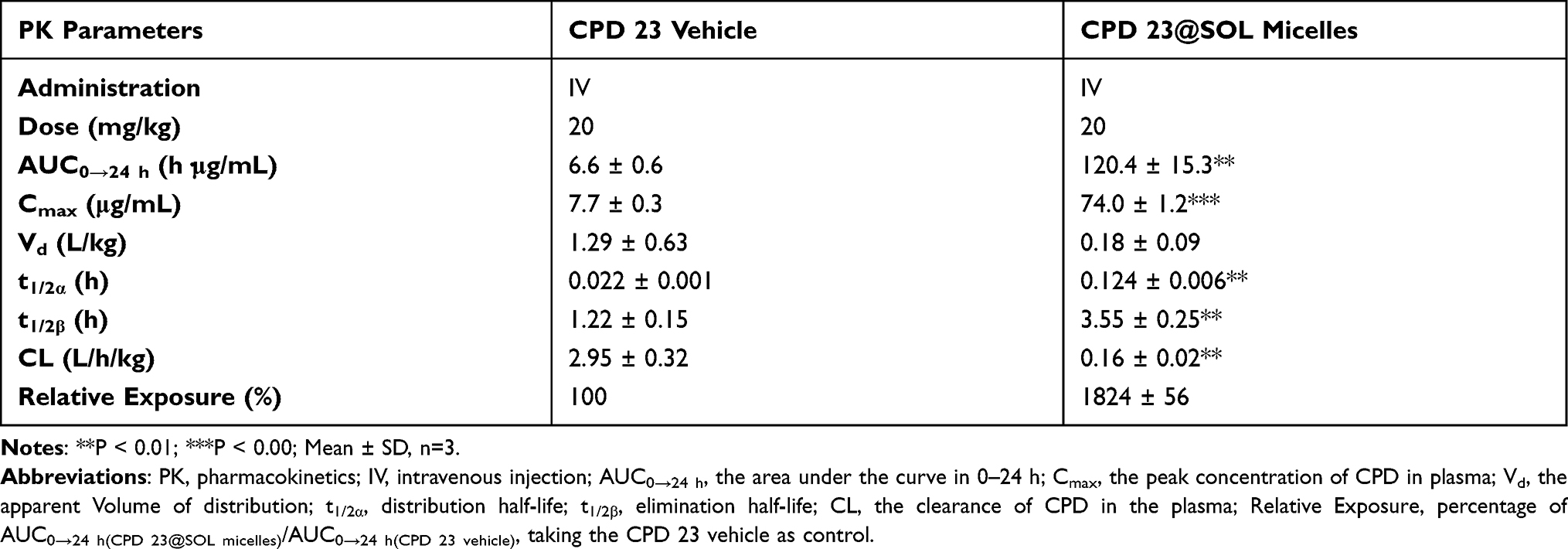 |
Table 4 The Pharmacokinetic Parameters of CPD 23 Vehicle and CPD 23@SOL Micelles |
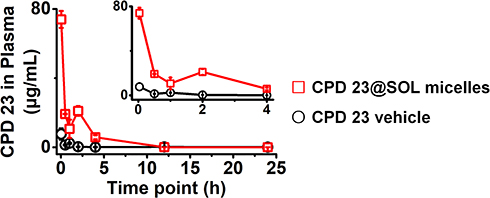 |
Figure 7 The AUC (Conc.-time) curves of in vivo pharmacokinetics of CPD 23 vehicle and CPD 23@SOL micelles via intravenous administration with dose of 20 mg/kg. Abbreviation: AUC, the area under curve from 0 h to 24 h. Notes: Conc.-time: the concentration of CPD 23 in plasma versus dosed time point; Data were presented as mean ± SD; n=3. |
In vivo Pharmacokinetic Biodistribution
Biodistribution is a critical PK parameter with important therapeutic significance. Therefore, we investigated the level of CPD 23 distributed in several tissues via IV administration at 20 mg/kg (CPD 23) dose of both CPD 23 vehicle and CPD 23@SOL micelles. The AUC curves based on CPD 23 level (the amount of CPD 23 versus tissue weight) versus dosed time were generated for pharmacokinetics evaluation.
As shown in Figures 8 and 9, CPD 23 vehicle was found at low level in brain, liver, kidney, at mild level in heart and spleen, but abundantly aggregated in lung within 24 hours. Surprisingly, for CPD 23@SOL micelles, the CPD 23 level in lung dropped by more than twenty folds, but significantly increased distribution in liver, spleen and kidney were observed.
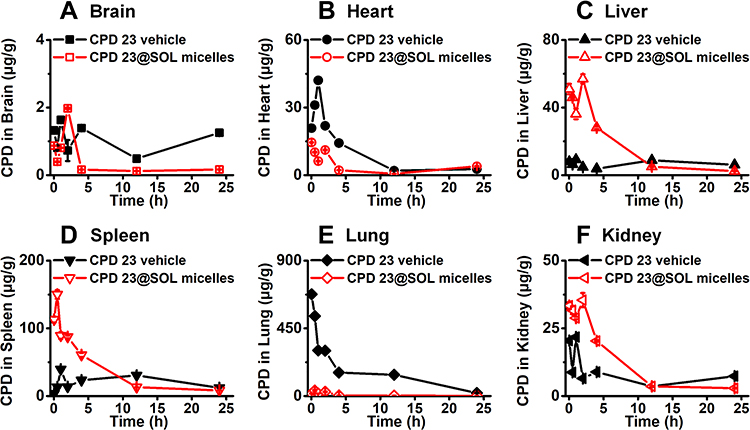 |
Figure 8 The biodistribution of CPD 23 vehicle and CPD 23@SOL micelles in tissues of (A) brain, (B) heart, (C) liver, (D) spleen, (E) lung and (F) kidney via intravenous administration at the dose of 20 mg/kg CPD 23. Notes: The retaining CPD 23 was quantified by HPLC and presented by the ratio of the CPD 23 amount versus the weight of tissue (mean ± SD; n=3). |
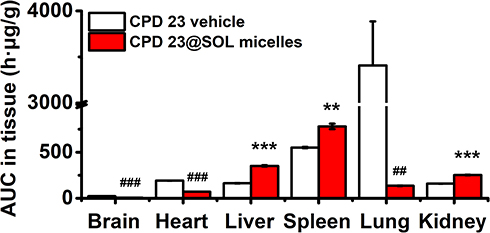 |
Figure 9 The tissue biodistributed AUC0-24 h of CPD 23 vehicle and CPD 23@SOL micelles by intravenous administration of 20 mg/kg dose (CPD 23). Abbreviation: AUC0→24 h, the area under the curve at 0–24 h. Notes: AUC0-24 h comparison: **P < 0.01, ***P < 0.001, CPD 23@SOL micelles significantly higher than CPD 23 vehicle; ##P < 0.01, ###P < 0.001, CPD 23 vehicle significantly higher than CPD 23@SOL micelles; mean ± SD; n=3. |
In terms of the total amount of CPD 23@SOL micelles in tissues, much higher level of CPD 23 in liver were also found than that in other tissues (Figure S2A), whereas the CPD 23 vehicle showed the highest in lung (Figure S2B). The difference between them might essentially be attributed to the different delivery carriers. Via the intravenous administration, CPD 23@SOL micelles with 200 nm particle size were primarily removed by the reticuloendothelial system (RES) and deposited by Kupffer cells in liver or spleen.39 However, the abundant CPD 23 vehicle retained in lung indicated that it had difficulty passing through the pulmonary vascular bed, in which some larger particulates (more than 500 nm) might be formed in the delivery process and essentially originated from the poor solubility and unsatisfactory stability.40,41 Therefore, CPD 23@SOL micelles enhanced solubility and improved stability produced encouraging pharmacokinetic biodistribution profiles for further formulation and efficacy investigation of CPD 23 and more potent glutaminolysis allosteric inhibitors.
Discussion and Conclusions
Poor water solubility has seriously limited the in vivo bioavailability and efficacy of the developed KGA allosteric inhibitors. Multiple drug delivery systems (DDS) and commonly-used excipients have been explored for potent allosteric inhibitors including the 1,3,4-Selenadiazole compound (CPD 23), but only limited solubility improvement has been achieved.10,17 Interestingly, Soluplus® has many advantageous and versatile properties,18 so we investigated SOL as the excipient to formulate nano-micelles to address the solubilization problem.
As is reported, SOL assembles micelles itself through the polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol grafted structure, within which the hydrophobic regions interact with lipophilic molecules to form strengthened hydrophobic core and the hydrophilic region contributes to prevent the compound incorporated inside the core from precipitation.20,42 Due to the high CLogP and hydrophobicity, CPD 23 showed strong compatibility with SOL and achieved 15,000-fold enhancement in water solubility as the micellar system. The prepared CPD 23@SOL micelles presented particle sizes at around 200 nm with good dispersity index, and in amorphous solid dispersion state as characterized by XRPD showing excellent compatibility.
Interestingly, the CPD 23@SOL micelles greatly changed the biophysical and pharmacological properties of CPD 23, even though both showed essentially the same activity in inhibiting the cancer cell proliferation. The CPD 23@SOL micelles showed excellent stability in mouse blood, and more steady partition in plasma than that of free CPD 23 due to the protection of the hydrophilic core-shell.43 In addition, the CPD 23@SOL micelles also showed longer half-life and lower clearance in liver microsome. In the in vivo pharmacokinetic study, CPD 23@SOL micelles markedly increased the in vivo exposure and prolonged circulation of CPD 23 in comparison with the CPD 23 vehicle. Importantly, CPD 23 vehicle massively aggregated in lung, whereas the CPD 23@SOL micelles accumulated in liver. This dramatic difference implied that their delivery pathways might be influenced by the formulated carriers with different characteristics, in which CPD 23@SOL showed superior solubility and stability than CPD 23 vehicle.39,40 These results indicated CPD 23@SOL micelles showed highly stable in blood and live microsome, improved relative exposure and liver targeting function.
In summary, Soluplus® demonstrated impressive capability to solubilize the novel hydrophobic 1,3,4-Selenadiazole CPD 23. CPD 23@SOL micelles not only increased the water solubility by 15,000 folds, but also improved the stability in blood and liver microsome. Importantly, the stable CPD 23@SOL micelles showed enhanced in vivo exposure, prolonged circulation and altered tissue targeting property. This represents an advantageous and practical method for improving the solubility and in vivo pharmacokinetics of potent hydrophobic allosteric glutaminase inhibitors.
Acknowledgments
The authors appreciate the financial supports from Zhejiang Qinglan plan grant, China (number H1160494) and Natural Science Foundation of Zhejiang Province/General Project, China (number LY19H300002). We also thank the senior engineer Hua Wang from Analysis Center of Agrobiology and Environmental Sciences, Zhejiang University, China for the kind help of transmission electron microscopy.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Yang L, Venneti S, Nagrath D. Glutaminolysis: a hallmark of cancer metabolism. Annu Rev Biomed Eng. 2017;19(1):
2. Xu X, Meng Y, Li L, et al. Overview of the development of glutaminase inhibitors: achievements and future directions. J Med Chem. 2019;62(3):1096–1115. doi:10.1021/acs.jmedchem.8b00961
3. Wise D, Thompson C. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35:427–433. doi:10.1016/j.tibs.2010.05.003
4. Erickson JW, Cerione RA. Glutaminase: a hot spot for regulation of cancer cell metabolism? Oncotarget. 2010;1:734–740. doi:10.18632/oncotarget.208
5. Li L, Meng Y, Li Z, et al. Discovery and development of small molecule modulators targeting glutamine metabolism. Eur J Med Chem. 2019;163:215–242. doi:10.1016/j.ejmech.2018.11.066
6. Gordon EB, Hart GT, Tran TM, et al. Targeting glutamine metabolism rescues mice from late-stage cerebral malaria. Proc Natl Acad Sci USA. 2015;112:13075–13080. doi:10.1073/pnas.1516544112
7. Leone RD, Zhao L, Englert JM, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. 2019;366(6468):1013–1021. doi:10.1126/science.aav2588
8. Krupa S, Ferraris DV, Thomas AG, et al. Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES) analogs as glutaminase inhibitors. J Med Chem. 2012;55(23):10551–10563. doi:10.1021/jm301191p
9. Gross MI, Demo SD, Dennison JB, Chen L, Bennett MK. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. 2014;13(4):890–901. doi:10.1158/1535-7163.MCT-13-0870
10. Chen Z, Li D, Xu N, et al. Novel 1,3,4-Selenadiazole-containing kidney-type glutaminase inhibitors showed improved cellular uptake and antitumor activity. J Med Chem. 2019;62(2):589–603. doi:10.1021/acs.jmedchem.8b01198
11. Soth MJ, Le K, Francesco MED, et al. Discovery of IPN60090, a clinical stage selective glutaminase-1 (GLS-1) inhibitor with excellent pharmacokinetic and physicochemical properties. J Med Chem. 2020;63(21):12957–12977. doi:10.1021/acs.jmedchem.0c01398
12. Vartak R, Patki M, Menon S, et al. β-cyclodextrin polymer/Soluplus® encapsulated Ebselen ternary complex (EβpolySol) as a potential therapy for vaginal candidiasis and pre-exposure prophylactic for HIV. Int J Pharmaceutics. 2020;589:119863. doi:10.1016/j.ijpharm.2020.119863
13. Elgogary A, Xu Q, Poore B, et al. Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc Natl Acad Sci USA. 2016;113(36):E5328–5336. doi:10.1073/pnas.1611406113
14. Giesen B, Nickel AC, Barthel J, Kahlert UD, Janiak C. Augmented therapeutic potential of glutaminase inhibitor CB839 in glioblastoma stem cells using gold nanoparticle delivery. Pharmaceutics. 2021;13:295. doi:10.3390/pharmaceutics13020295
15. Zhu M, Fang J, Zhang J, et al. Biomolecular interaction assays identified dual inhibitors of glutaminase and glutamate dehydrogenase that disrupt mitochondrial function and prevent growth of cancer cells. Anal Chem. 2017;89(3):1689–1696. doi:10.1021/acs.analchem.6b03849
16. Ruan JJ, Yu Y, Hou W, et al. Kidney-Type Glutaminase inhibitor Hexylselen selectively kills cancer cells via a three-pronged mechanism. ACS Pharmacol Transl Sci. 2019;2(1):18–30. doi:10.1021/acsptsci.8b00047
17. Fang J, Chen Z, Song J, et al. Biodegradable self-assembly micelles significantly enhanced the solubility, biological stability and in vivo antitumor efficacy of Hexylselen. RSC Chem Biol. 2021;2(6):1669–1681. doi:10.1039/D1CB00089F
18. Pignatello R, Corsaro R. Polymeric nanomicelles of soluplus as a strategy for enhancing the solubility, bioavailability and efficacy of poorly soluble active compounds. Curr Nanomed. 2019;09(3):184–197. doi:10.2174/2468187309666190314152451
19. Obata T, Suzuki Y, Ogawa N, et al. Improvement of the antitumor activity of poorly soluble sapacitabine (CS-682) by using Soluplus as a surfactant. Biol Pharm Bull. 2014;37(5):802–807. doi:10.1248/bpb.b13-00898
20. Linn M, Collnot EM, Djuric D, et al. Soluplus as an effective absorption enhancer of poorly soluble drugs in vitro and in vivo. Eur J Pharm Sci. 2012;45(3):336–343. doi:10.1016/j.ejps.2011.11.025
21. Sachan N, Bhattacharya A, Pushkar S, Mishra A. Biopharmaceutical classification system: a strategic tool for oral drug delivery technology. Asian J Pharm. 2009;3(2):76–81. doi:10.4103/0973-8398.55042
22. Quan G, Niu B, Singh V, et al. Supersaturable solid self-microemulsifying drug delivery system: precipitation inhibition and bioavailability enhancement. Int J Nanomed. 2017;12:8801–8811. doi:10.2147/IJN.S149717
23. Alopaeus JF, Hagesæther E, Tho I. Micellisation mechanism and behaviour of soluplus®–furosemide micelles: preformulation studies of an oral nanocarrier-based system. Pharmaceuticals. 2019;12(1):15. doi:10.3390/ph12010015
24. Zeng YC, Li S, Liu C, et al. Soluplus micelles for improving the oral bioavailability of scopoletin and their hypouricemic. Acta Pharmacol Sin. 2017;38:424–433. doi:10.1038/aps.2016.126
25. Nandi U, Ajiboye AL, Patel P, Douroumis D, Trivedi V. Preparation of Solid Dispersions of Simvastatin and Soluplus Using a Single-Step Organic Solvent-Free Supercritical Fluid Process for the Drug Solubility and Dissolution Rate Enhancement. Pharmaceuticals. 2021;14(9):846. doi:10.3390/ph14090846
26. Zhang Y, Liu Y, Luo Y, et al. Extruded Soluplus/SIM as an oral delivery system: characterization, interactions, in vitro and in vivo evaluations. Drug Deliv. 2016;23(6):1902–1911. doi:10.3109/10717544.2014.960982
27. Salawi A, Nazzal S. The physiochemical, mechanical, and adhesive properties of solvent-cast vitamin E Soluplus(R) films. Int J Pharmaceutics. 2018;552(1–2):378–387. doi:10.1016/j.ijpharm.2018.10.018
28. Salah I, Abou Shamat M, Cook MT. Soluplus solutions as thermothickening materials for topical drug delivery. J Appl Polym Sci. 2019;136(1):1–9. doi:10.1002/app.46915
29. Lee SY, Lee JJ, Park JH, et al. Electrosprayed nanocomposites based on hyaluronic acid derivative and soluplus for tumor-targeted drug delivery. Colloids Surf B Biointerfaces. 2016;45:267–274. doi:10.1016/j.colsurfb.2016.05.009
30. Ke Z, Zhang Z, Wu H, Jia X, Wang Y. Optimization and evaluation of oridonin-loaded Soluplus (R)-Pluronic P105 mixed micelles for oral administration. Int J Pharmaceutics. 2017;18(1–2):193–202. doi:10.1016/j.ijpharm.2016.12.047
31. Hou J, Sun E, Sun C, et al. Improved oral bioavailability and anticancer efficacy on breast cancer of paclitaxel via Novel Soluplus®-Solutol® HS15 binary mixed micelles system. Int J Pharmaceutics. 2016;12:186–193. doi:10.1016/j.ijpharm.2016.08.045
32. Zhu C, Gong S, Ding J, et al. Supersaturated polymeric micelles for oral silybin delivery: the role of the Soluplus–PVPVA complex. Acta Pharmaceutica Sinica B. 2019;9(1):107–117. doi:10.1016/j.apsb.2018.09.004
33. Dian L-H, Hu Y-J, Lin J-Y, et al. Fabrication of paclitaxel hybrid nanomicelles to treat resistant breast cancer via oral administration. Int J Nanomed. 2018;13:719–731. doi:10.2147/IJN.S150140
34. Ting JM, Porter WW, Mecca JM, Bates FS, Reineke TM. Advances in Polymer Design for Enhancing Oral Drug Solubility and Delivery. Bioconjugate Chem. 2018;29(4):339–952. doi:10.1021/acs.bioconjchem.7b00646
35. Zhang W, Zhu M, Wang F, et al. Mono-sulfonated tetrazolium salt based NAD(P)H detection reagents suitable for dehydrogenase and real-time cell viability assays. Anal Biochem. 2016;509:33–40. doi:10.1016/j.ab.2016.06.026
36. Rui J, Xu N, Yin J, et al. The EZMTT cell proliferation assay provides precise measurement for drug combinations and better correlation between in vitro and in vivo efficacy. Bioorg Med Chem Lett. 2020;30(11):127134. doi:10.1016/j.bmcl.2020.127134
37. Wang P, Zhao Y, Zhu Y, et al. Metabolism of dictamnine in liver microsomes from mouse, rat, dog, monkey, and human. J Pharm Biomed Anal. 2016;119:166–174. doi:10.1016/j.jpba.2015.11.016
38. Di L, Feng B, Goosen TC, et al. A perspective on the prediction of drug pharmacokinetics and disposition in drug research and development. Drug Metab Dispos. 2013;41(12):1975–1993. doi:10.1124/dmd.113.054031
39. Zeng YC, Li S, Liu C, et al. Soluplus micelles for improving the oral bioavailability of scopoletin and their hypouricemic effect in vivo. Acta Pharmacol Sin. 2017;38:424–433.
40. Dhand C, Prabhakaran PP, Beuerman RW, Lakshminarayanan R, Dwivedid N, Ramakrishna S. Role of size of drug delivery carriers for pulmonary and intravenous administration with emphasis on cancer therapeutics and lung-targeted drug delivery. RSC Adv. 2014;4:32673–32689. doi:10.1039/C4RA02861A
41. Razak SAA, Wahab HA, Fisol FA. Advances in Nanocarriers for Effective Delivery of Docetaxel in the Treatment of Lung Cancer: an Overview. Cancers. 2021;13(3):400. doi:10.3390/cancers13030400
42. Yang X, Zhu B, Dong T, et al. Interactions between an Anticancer Drug and Polymeric Micelles Based on Biodegradable Polyesters. Macromol Biosci. 2008;8(12):1116–1125. doi:10.1002/mabi.200800085
43. Gou J, Feng S, Xu H, et al. Decreased core crystallinity facilitated drug loading in polymeric micelles without affecting their biological performances. Biomacromolecules. 2015;16:2920–2929. doi:10.1021/acs.biomac.5b00826
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.